

Abstract
Adoptive transfer of genetically engineered chimeric antigen receptor (CAR)-T cell is emerging as a promising treatment option for hematological malignancies. However, T cell immunotherapies have largely failed in patients with solid tumors. Here, with a CRISPR-Cas9 pooled library, we performed an in vivo targeted loss-of-function screen and identified ST3 β-galactoside α−2,3-sialyltransferase 1 (St3gal1) as a key negative regulator of the cancer-specific migration of CAR-T cells. Analysis of glycosylated proteins reveals that CD18 is a major effector of St3gal1 in activated CD8 T cells. St3gal1-mediated glycosylation induces the spontaneous nonspecific tissue sequestration of T cells by altering lymphocyte function-associated antigen-1 (LFA-1) endocytic recycling. Engineered CAR-T cells with enhanced expression of βII-spectrin, a key LFA-1 associated cytoskeleton molecule, reversed St3gal1-mediated nonspecific T cell migration and significantly reduced tumor growth in mice by improving tumor-specific homing of CAR-T cells. These findings identify a role of the St3gal1- βII-spectrin axis as the key cell-intrinsic program for cancer targeting CAR-T cell migration and as a promising strategy for effective T cell immunotherapy.
Introduction
Adoptive transfer of therapeutic T cells has emerged as a promising approach with complete and durable responses in several disease conditions such as viral infection, autoimmune disease, atherosclerosis, and cancer1. For example, utilizing CAR-T cells has proven effective in treating hematological malignancies2. However, a major limitation to effective T cell immunotherapy is the physical access of immune cells to the target sites. Even if therapeutic T cells can recognize and kill target cells, they will fail in vivo if they cannot infiltrate into the target tissues.
After intravenous transfer into patients, most transferred T cells are observed at non-tumor-bearing tissues, with only a small percentage of the cells eventually reaching targeted tumor sites3. This is a clinically important issue because sequestration of ex vivo generated T cells at nonspecific tissue sites after intravenous transfer is often associated with impaired trafficking of T cells to the target tissue3 and the development of acute toxicity in patients4. Due to the inefficient trafficking of the transferred CAR-T cells to target tumor sites, significant clinical success of CAR-T cell immunotherapy has not yet been achieved for many solid tumors5. For the same reason, patients often have to receive a large number of highly activated engineered cells intravenously, and this further increases both cell manufacturing costs and the risk of T cell toxicity and systemic cytokine release syndrome (CRS).
The goal of this study is to understand why in vitro activated CD8 T cells get trapped in off-target tissue sites after being intravenously transferred. Here, we propose that autologous therapeutic T cells undergo significant molecular and cellular reprogramming during the ex-vivo manufacturing process, and that the intrinsic changes are important for the robust T cell activation and expansion but fail to derive T cell migration toward target sites and may serve to increase toxicity. We identified molecular mechanisms that could be harnessed to improve homing of T cells to target sites, thus increasing the efficacy of T cell immunotherapy, while reducing the risks associated with non-specific cytotoxicity6.
Results
CAR-T cells immediately accumulate in the lung after adoptive transfer.
Early clinical studies with CD19 CAR-T cells (CTL019) showed that after T cell infusion, there was an immediate and prolonged decline in circulating CTL019 cells in the patient blood for up to 7 days, which could be directly attributed to nonspecific distributions of CAR-T cells to random tissue sites7, 8. Similarly, after treatment with lympho-depleting chemotherapy followed by infusion of 1 – 2 × 108 viable CAR- positive T cells (Fig. 1A), all patients had undetectable circulating T cells for more than two days and significant T cell expansions did not occur until day 14 (Fig. 1B).
Figure 1. CAR-T cells are immediately accumulate in the lung after adoptive transfer.
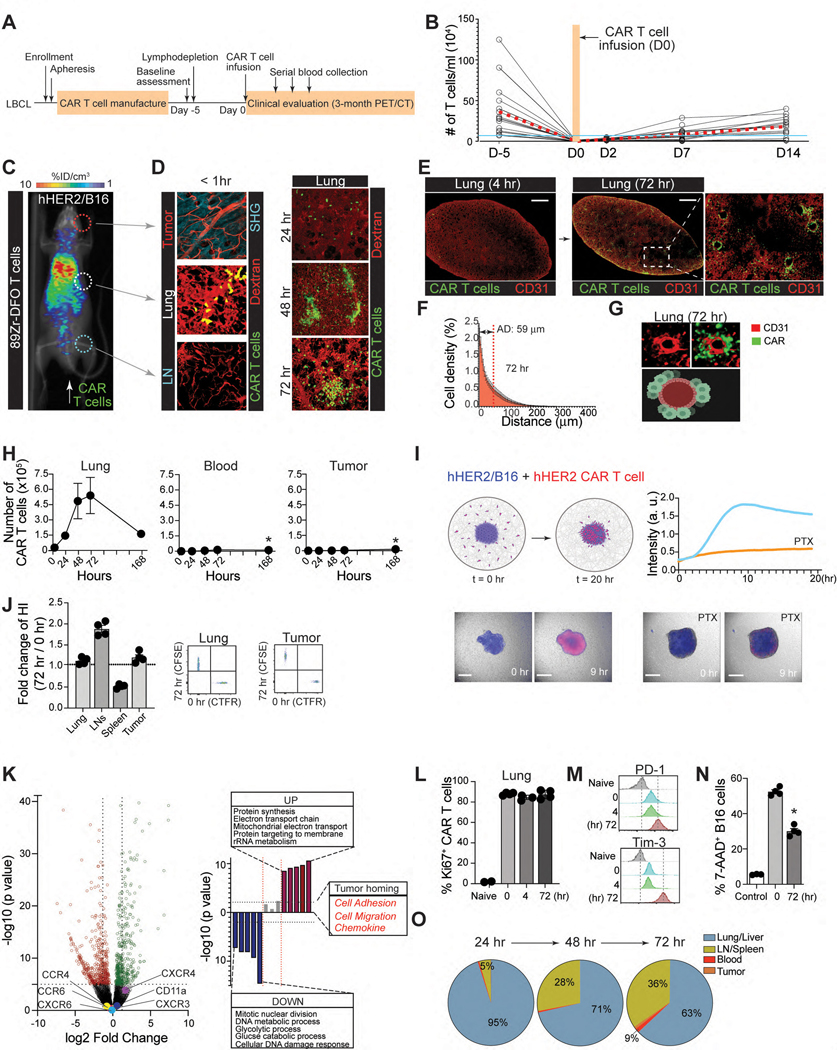
A. Clinical protocol for CAR T-cell infusion (CTL019). B. Blood T cell counts from patients (n = 15). Red dashed line, the mean ; blue line, total blood lymphocyte counts. C & D. Visualization of early sequestration (0–1 hr) of hHER2 CAR-T cells after adoptive transfer with PET/CT (89Zr-DFO-labeled T cells) (C) or IV-MPM (red, dextran-labeled vessels; green, GFP-expressing CAR-T cells; blue, SHG) (D). IV-MPM images from the lung at the indicated times post-injection (D). Scale bars, 50 μm. E. Representative 3D imaging of CAR-T cell accumulation in cleared lung 4 and 72 hr after i.v. injection (red, blood vessels; green, CAR-T cells). Scale bars, 1 mm. F. The cell density curve and histogram of the distribution of T cells extravasated from the nearest blood vessel (attraction distance, AD). G. 3D image of GFP-expressing CAR-T cells and CD31-labeled vessels. H. The number of injected CAR-T cells in the lung, blood, and tumor at the indicated times. I. in vitro migration of hHER2-CAR T cell (red) to hHER2-B16-BFP tumor spheroid (blue) and . Scale bars, 100 μm. J. CAR-T cells from the lung 72 hr after i.v. injection (T72; CFSE) and newly in vitro activated CAR-T cells (T0; CTFR) were co-transferred (1:1 mix) and the fold change in the homing index was analyzed 24 hr after injection (n = 4, 13 mice per experiment). K. Volcano plot and signaling pathway from RNA-seq analysis in CAR-T cells (T72 vs. T0). L. Percentage of Ki67+ cells in vitro activated CAR-T cells (0 hr) or CAR-T cells isolated from the lung after i.v. injection (4 and 72 hr). M. Flow cytometry results of PD-1 and Tim-3 expression levels on naïve and CAR-T cells isolated from the lung at indicated times post-injection. N. The cytotoxicity of CAR-T cells (PBS, T72, and T0) against B16-hHER2. O. The proportion of CAR-T cells distributed across different organs post-injection. Representative image of 5 mice (D-E). Representative data from three experiments with n = 3 per group (L-N) n = 3 mice per group (F, O). Results are shown mean ± SEM (H, J, L, N). The data were analyzed by ordinary one-way ANOVA with Tukey’s multiple comparison posttest (H) or two-tailed Student’s t test (N).
To further investigate the spatial and temporal fates of intravenously infused CAR-T cells in vivo, mouse CD8 T cells were retrovirally transduced with trastuzumab (4D5; anti-HER2)-based CAR9 at > 92% efficiency (Extended Data Fig. 1A). Coincubation of B16 mouse melanoma cell line that stably expresses human HER2 antigen (B16-hHER2) (Extended Data Fig. 1B) with hHER2-CAR expressing mouse CD8 T cells induced dramatic cytotoxicity but not in the control CD8 T cells (Extended Data Fig. 1C and Video 1), confirming that hHER2 is immunogenic in the B16 cell line. For in vivo assays, we used C57BL/6 human-HER2 (hHER2) transgenic (tg) mice that overexpress the human erbB-2 gene under the mouse mammary tumor virus promoter (obtained from Genentech10).
The current therapeutic T cell manufacturing process includes the selection and activation of T cells from patient apheresis products using CD3/CD28 antibody beads and large clinical scale T cell expansion with high concentrations of IL-211. Similarly, 1 × 107 CD8 T cells from hHER2 tg mice were expanded in vitro, transfected with the hHER2 CAR, and transferred to hHER2 tg mice bearing B16-hHER2 tumors in the ear (Fig. 1C). The migration and distribution of intravenously infused CAR-T cells were visualized by micropositron emission tomography/computed tomography (PET/CT) images (Fig. 1C) and intravital multiphoton-microscopy (IV-MPM) in different organs, such as the lung, lymph nodes, and tumors of live recipient mice simultaneously with the cell injection, and subsequently for 72 hr post-injection (Fig. 1D). Initially (< 1 hr to 24 hr post-injection), very few cells appeared in tumors or lymph nodes, with most CAR-T cells instantly migrating to the lung (Fig. 1C & 1D and Video 2). On days 2 and 3, we found that an increasing number of CAR-T cells accumulated and formed clusters within the lung (Fig. 1D and Video 3). Three-dimensional imaging of an optically cleared lung (Extended Data Fig. 1D) further confirmed that CAR-T cells actively accumulated in the lung 72 hr after adoptive transfer (Fig. 1E), mainly along the perivascular space (Fig. 1F & 1G, and Video 4).
Flow cytometric quantifications also revealed similar results, demonstrating the maximal accumulation of CAR-T cells peaking on day 3, followed by a steady decrease over time to day 7 (Fig. 1H). In contrast to those in the lung (but consistent with clinical data7), nearly all CAR-T cells were undetectable in the peripheral blood after intravenous infusion. 7 days later, a small (but significant) number of CAR-T cells was observed in the blood and at the tumor site (Fig. 1H). Importantly, in vitro generated CAR-T cells preserved their directional migration patterns toward the target tumors (B16-hHER2 spheroids), which was completely abolished by pretreatment of the cells with pertussis toxin (PTx), an inhibitor of Gαi heteromeric G proteins that blocks chemotactic T cell migration (Fig. 1I and Video 5). In addition, very few CAR-T cells were found to be localized within the vicinity of CD11c+ MHCII+ tissue resident antigen presenting cells (Extended Data Fig. 2A) and there was no sign of B16 micro-metastases in the lung during this period. Therefore, our data suggest that despite their tumor-specific trafficking ability following chemokine signals, the majority of intravenously transferred CAR-T cells quickly disappeared from the blood and homed to nonspecific peripheral tissues. Adoptively transferred OT-I T cells into B16-OVA bearing mice showed similar T cell accumulation in the lung (Extended Data Fig. 2B), suggesting that in vitro stimulated T cells migrate into the non-inflamed lung after adoptive transfer even in the absence of CAR expression.
Next, we investigated whether CAR-T cells gradually lose the nonspecific migration pattern and gain tumor-specific homing capability during retention in the lung. To test this, we first performed a competitive homing assay. hHER2 Tg mice bearing B16-hHER2 tumors received equal numbers (1:1) of freshly prepared hHER2 CAR-T cells (red; CART0) and CAR-T cells isolated from the lung 72 hr after adoptive transfer (green; CART72). The ratio of CART72/CART0 cells from various organs assessed one day post-transfer revealed that CART72 cells still readily re-entered non-tumor bearing lung tissue similar to CART0 cells, suggesting that lung retention for 72 hr did not diminish the nonspecific migration pattern (Fig. 1J). More importantly, CART72 cells did not show a greater ability to enter tumors than CART0 cells (Fig. 1J). Although the genome-wide transcriptional profiles showed substantial differences between CART0 vs. CART72 cells (Extended Data Fig. 2C), but the data demonstrated that the expression of genes controlling T cell adhesion, motility, and tumor-specific homing was not significantly changed during lung-retention (Fig. 1K).
In addition, the majority of CAR-T cells in the lung exhibited a higher frequency of Ki67 (Fig. 1L and Extended Data Fig. 2D–E) with diminished expression of inflammatory cytokines (Extended Data Fig. 2H). Although Ki67 is often used as an indication of the proliferative capability of T cells, it is also expressed on recently divided and terminally exhausted T cell populations with decreased effector functions12, 13. Indeed, both CAR-T cells (CART72) and OT-I T cells drastically upregulated PD-1 and Tim-3 expression after 72 hr lung retention, suggesting that these cells may have been functionally exhausted (Fig. 1M and Extended Data Fig. 2G). Furthermore, CART72 cells exhibited decreased cytotoxicity against B16-hHER2 cells (Fig. 1N). Therefore, during nonspecific retention in the lung, these CAR-T cells fail to functionally reprogram to raise their capacity to migrate toward target tumor sites, but upregulate exhaustion markers and decrease cytotoxic effector function. Notably, the number of CAR-T cells gradually increased in the lymph nodes (LNs) and spleen after 72 hr (Fig. 1O). CAR-T cells that arose in the LNs after 72 hr showed improved cytotoxicity against B16-hHER2 cells compared to CAR-T cells sequestered in the lung (Extended Data Fig. 2J). Furthermore, the enhanced effector functions of CAR-T cells in the LNs after 72 hr coincided with the downregulation of PD-1 and Tim-3 expression (Extended Data Fig. 2I). These results indicate a limited role of lung retention in CAR-T cell activation, and suggest that a shift in the dynamic equilibrium of CAR-T cell positions between non-tumor bearing tissues (such as the lung and liver) and the circulation (the blood/spleen and LN) after intravenous transfer may have important impacts on anti-cancer effector functions of CAR-T cells3, 14.
In vivo CRISPR screen revealed a key regulator of CAR-T cell migration.
To identify genes that cause CAR-T cell migration to nonspecific sites in vivo, we used libraries of single-guide RNAs (sgRNAs) targeting 1,316 genes that are differentially expressed between in vitro activated vs. naïve CD8 T cells (Fig. 2A, Extended Data Fig. 3A & 3B). The sgRNA plasmid library prepared with 5 sgRNAs per gene and approximately 300 negative-control sgRNAs was verified for successful clones and a log-normal distribution (Extended Data Fig. 3C). The plasmid library was then packaged into retrovirus and expression of RFP+ signals was confirmed by flow cytometry to ensure adequate viral transfection in CD8 T cells isolated from OT-I/Rosa26LSL-GFPCas9 mice15 (Extended Data Fig. 3D). The double-positive populations of sgRNA (RFP+) and Cas9 (GFP+) OT-I cells (Extended Data Fig. 3D) were adoptively transferred into B16-OVA bearing congenic recipient mice (Fig. 2A). At 24 hr post-infusion, sgRNA+Cas9+ OT-I cells were isolated from different organs and evaluated. To ensure sufficient library coverage (> 500x), we limited the number of adoptively transferred Cas9-OT-I cells to 3.5×106 per mouse and 0.35×106 after taking from each organ compartment (~50 cells per sgRNA). For our screens, we predicted that infused sgRNA+Cas9+ OT-I cells were recruited into two different tissue compartments: T cells located in the non-tumor bearing lung and liver (termed Tsequestered) vs. T cells re-emerged in the circulation before their recruitment to the tumor site (termed Tmigratory). To interrogate the genetic landscape of CAR-T cell migration, we explored our dataset to identify genes with differential fitness effects in Tsequestered vs. Tmigratory populations relative to control cells using the drugZ algorithm16 (Fig. 2B, Extended Data Fig. 3E & 3F). This analysis revealed that 124 genes in Tsequestered population and 207 genes in Tmigratory population affect T cell migration in vivo (false discovery rate (FDR) < 5%) and identified key genes that may drive CAR-T cells to the non-tumor bearing tissues (Fig. 2B). Among these, St3gal1 and Spc24 were the strongest hits in repressing the Tsequestered population (Fig. 2C & 2D).
Figure 2. Discovery of a key regulator of CAR-T cell migration using a CRISPR screen.

A. Schematic for the development of a pooled in vivo CRISPR screening platform in mouse CD8 T cells (Created with BioRender.com) B. Rank-ordered norm z-score of the beta values for all three OT-I T cell distribution screens in B16-OVA bearing mice (yellow, migratory genes; blue, sequestered genes). The top three sequestered and the top five migratory genes are indicated. C. Genes rank based on the difference in beta scores across sequestered and migratory conditions. D. Sectored scatter plot of gene-level beta values from Tsequestered vs. Tmigratory cell populations isolated from B16-OVA bearing mice. Significant migratory genes and sequestered gene signatures (FDR < 5%) are colored yellow and blue, respectively. Ellipse (gray dashed lines), 5% FDR threshold delineated by normal ellipse fit to differential scores. E. qPCR of St3gal1 in mouse and human CD8 T cells (naïve vs. activated) as compared to three averaged housekeeping genes (n = 3–11). F. Expression levels of St3gal1 in activated CD8 T cells (from Days 0 to 5). Loading control: β-actin. G. qPCR of Spc24 in mouse and human CD8 T cells (naïve vs. activated) as compared to three averaged housekeeping genes (n = 3–4). H. Expression levels of Spc24 in activated CD8 T cells (from Days 0 to 5). Loading control: β-actin. I. Expression levels of Spc24 and St3gal1 in Cas-9 hHER2 CAR-T cells with sgSpc24 and sgSt3gal1, respectively. Loading control: β-actin. J. The cytotoxicity of CAR-T cells (PBS, sgSpc24 cells, and sgSt3gal1 cells) against B16-hHER2 (n = 3). K. The distribution of CAR-T cells with (WT; CTFR)/without (KO; CFSE) Spc24 or St3gal1 24 hr after injection (n = 3). The fold change in the homing index, as determined by (KOsample/WTsample)/(KOinput/WTinput). Results are shown mean ± SEM (E, G, J-K). The data reflect three independent experiments (J, K) The data were analyzed by two-tailed Student’s t test (E, G).
St3gal1 is a Golgi membrane-bound sialyltransferase that adds sialic acid residues in an α2,3 linkage to galactose-containing substrates (Gal β1,3 GalNAc)17. Importantly, St3gal1 regulates cell migration and lymphocyte differentiation18, 19, 20. Spc24 is a nuclear protein involved in the formation of the kinetochore complex, which mediates chromosome segregation during mitosis and cell cycle progression21. In in vitro activated CD8 T cells, qRT-PCR analyses revealed that the genes associated with St3gal1 and Spc24 expression were significantly increased in both human and mouse activated CD8 T cells (Fig. 2E & 2G). Consistently, the protein expression levels of both St3gal1 and Spc24 were also elevated during the activation of human and mouse CD8 T cells (Fig. 2F & 2H). To examine whether these proteins are directly involved in CAR-T cell migration, we cloned sgRNAs targeting St3gal1 and Spc24 into the retroviral CRISPR vector (sgSt3gal1 and sgSpc24). We then transduced Cas9-hHER2 CAR-T cells and confirmed successful knockout of the target proteins (Fig. 2I). Deletion of St3gal1 or Spc24 protein in CAR-T cells did not change CAR-mediated cytotoxicity (Fig. 2H), but it significantly reduced nonspecific migration patterns into the lung and liver (Fig. 2K). Notably, sgSt3gal1-infected Cas9 CAR-T cells, but not sgSpc24-infected CAR-T cells, dramatically improved their retention in the circulation (the spleen and LN) after intravenous transfer (Fig. 2K). These data suggest that St3gal1 may play an important role in regulation of both nonspecific sequestration and tumor-specific migration patterns of CAR-T cell.
St3gal1 regulates LFA-1 mediated CAR-T cell migration.
To further dissect the molecular mechanism by which St3gal1 regulates CAR-T cell migration, we first performed flow cytometry analyses. In vitro activated mouse and human CD8 T cells showed augmented cell surface binding of Maackia amurensis lectin-II (MAL-II), a plant lectin that preferentially binds α2,3-linked sialic acid22 (Fig. 3A). In addition, deletion of St3gal1 protein in activated CD8 T cells significantly reduced the cell surface binding of MAL-II, further confirming that the increased sialylated-glycan activity is associated with St3gal1 overexpression (Fig. 3B). To identify the T cell glycoproteins bearing St3gal1-specific α2,3-sialylated glycans, we immunoprecipitated MAL-II-binding glycoproteins from CD8 T cells. Analysis of MAL-II immunoprecipitates revealed that a prominent protein of ~90 kD was associated with MAL-II, and the protein signal was preferentially enriched in immunoprecipitates obtained from activated T cells compared to naïve T cells (Fig. 3C). The 90-kD species was identified as CD18, as determined by mass spectrometry (Fig. 3C, Extended Data Fig. 4A, and Supplementary Table 1). To further validate whether the interaction was specific to sialic acid, we treated the CD8 T cell lysates with neuraminidase to remove sialic acids. Neuraminidase treatment caused a detectable change in the electrophoretic mobility of CD18 in SDS-PAGE (Fig. 3D). The removal of N-glycans by PNGase F treatment further shifted CD18 to lower molecular weight (Fig. 3D). These findings suggested that sialylation of CD18 is likely an O-linked sialylated glycoprotein catalyzed by St3gal1. Consistent with this prediction, MAL-II pulldown showed that binding of MAL-II with CD18 was decreased after St3gal1 silencing, while total CD18 protein expression remained the same (Fig. 3E). To identify potential O-glycosylation sites, CD18 immunoprecipitates were treated with PNGase F (to remove N-glycans) and digested using trypsin, followed by LC–MS/MS analysis. The results of peptide mass spectra from CD18 revealed O-glycosylation on threonine 256 (T256) (Extended Data Fig. 4B and Supplementary Table 2). In the crystal structure of LFA-1 headpiece23, the O-glycan site locates within the βI domain, which plays a key role in determining ligand binding specificity and affinity of LFA-1 (Extended Data Fig. 4C)24. Therefore, our data suggest that overexpression of St3gal1 in activated T cells causes the early accumulation of in vitro activated T cells within the pulmonary circulation via glycosylation of LFA-1, the dominant CD18 integrin expressed on CD8 T cells.
Figure 3. St3gal1 regulates LFA-1 mediated CAR-T cell migration.

A & B. MAL-II binding to CD8 T cells (A) and St3gal1 KO CAR-T cells (B). C. Immunoprecipitation of MAL-II-binding proteins in CD8 T cells. D. Western blot of the activated CD8 T cell lysates after enzyme treatments. In the graph, each bar represents the calculated molecular weight of protein bands. E. Immunoprecipitation of MAL-II binding CD18 in activated CD8 T cells. F. Recruitment of CAR-T cells into lung tissue (n = 3 mice per group). G. The numbers of WT or LFA-1 (CD11a)-KO CAR-T cells 24 hr after CAR-T cells injection (n = 3). H. In vitro CD8 T cell migration on ICAM-1-coated plates. (n = 3, 13–38 individual cells per mouse). I. In vitro activated CD8 T cell migration on ICAM-1-coated plates ± CXCL12. (n = 3, 10–57 individual cells per mouse). J. LFA-1 FRET assay with CD11a-mYFP and CD18-mCFP. Scale bars, 5 μm. (n = 3, 20–40 individual cells per condition). K. The density of microvilli of CD8 T cells by SEM (n = 6–8). Scale bars, 1 μm. L. The ratio of the TIRFM signal from the LFA-1 accumulation of CD8 T cells at sites of contact with the surface (n=3). M. Changes in the endocytosis of LFA-1 in CD8 T cells after treatment with α2,3-neuraminidase (n = 3–4). N. Time course of surface and intracellular LFA-1 expression. O & P. Changes in the endocytosis (O) and cell surface expression (P) of LFA-1 in CAR-T cells. (n = 3). Q & R. The migration of activated CD8 T cells pretreated with Exo1 (Q) or transfected with Rab13 siRNA (R) (n =3, 20–68 individual cells per mouse). S. The cytotoxicity of CAR-T cells against B16-hHER2 (n = 3). T & U. The percentage of hHER2 CAR-T cells in peripheral tissue sites (T) and tumors (U) 24 hours after adoptive transfer (n=3 mice per group). Results are shown mean fluorescence intensities (MFI) ± SEM (A-B) and mean ± SEM (C, F-M, O-P). All data and representative images reflect three independent experiments. Statistical analyses were performed using one-way ANOVA with Bonferroni posttest (A-C, H-I, Q-R), two-sided Mann-Whitney test (F, T-U), two-sided, unpaired Student’s t test (G, J) or two-tailed Student’s t test (K-M, O-P). NS; not significant.
Indeed, the nonspecific accumulation of CAR-T cells in the lung was significantly reduced by an anti-LFA-1-blocking Ab (Fig. 3F). Compared to WT CAR-T cells, LFA-1 KO CAR-T cells also showed a marked decrease in lung accumulation with the majority CAR-T cells remaining in the spleen (Fig. 3G). Consistent with previous findings with T cell blasts25, 26, the accumulation of CAR-T cells in the non-inflamed lung was insensitive to PTx (Fig. 3F), suggesting “PTx-insensitive” and “LFA-1 dependent” accumulation of in vitro activated CAR-T cells in non-inflamed tissues. Note that the LFA-1 Ab and PTx concentrations used were determined based on previous publications27, 28 and from our studies in which they significantly blocked T cell entry into dLNs (Extended Data Fig. 5A).
In in vitro assays, the migration of naïve T cells was strictly dependent on chemokine stimulation (Fig. 3H). However, similar to the migration pattern observed in vivo, activated CD8 T cells migrated on ICAM-1 spontaneously in the absence of chemokines (Fig. 3H & Video 6). Key effector T cell chemokines, including CXCL12 and CXCL10, did not further enhance the migration of activated T cells (Extended Data Fig. 5B – 5D). Furthermore, neither PTx nor another pan-GPCR inhibitor, gallein (Gβγ-subunit inhibitor)29 blocked spontaneous T cell migration on ICAM-1 (Fig. 3H). Consistent with cell migration under static conditions, activated CD8 T cells adhered and migrated on ICAM-1 coated surface (Extended Data Fig. 5E – 5G and Video 7) or on a monolayer of primary mouse endothelial cells (Extended Data Fig. 5H) in a flow chamber under high shear conditions (1 dyne/cm2) that can remove any autocrine and/or paracrine chemotactic molecules released by migrating T cells. Therefore, our data further confirm that in vitro activated CD8 T cells can migrate on ICAM-1 using LFA-1 in the absence of chemokine stimulation. Selective depletion of St3gal1 in CD8 T cells resulted in a significant decrease in spontaneous and chemokine-independent T cell migration on ICAM-1, but more importantly, it allowed chemokine-dependent T cell migration on ICAM-1 (Fig. 3I), suggesting that there is a direct relationship between St3gal1 functions and “chemokine-independent/LFA-1-mediated” accumulation of CAR-T cell in nonspecific tissue sites.
St3gal1 does not directly activate T cell surface LFA-1.
We first addressed whether St3gal1 overexpression in activated CD8 T cell in vitro was associated with changes in LFA-1 affinity (conformation change). To determine whether in vitro T cell activation induces the constitutive activation of LFA-1 affinity, we performed LFA-1 fluorescence resonance energy transfer (FRET) assays with T cells isolated from LFA-1 FRET mice that express monomeric YFP and monomeric CFP tagged to the cytosolic tails of CD11a and CD18 respectively30 (Fig. 3J). When LFA-1 is in the high affinity state, the cytosolic tails are separated leading to low efficiency FRET. In this way, FRET efficiency is correlated with integrin affinity30, 31. The results suggest that LFA-1 conformation remains in the low affinity state under basal conditions in activated CD8 T cells (Fig. 3J).
Next, our scanning electron microscopy (SEM) of in vitro activated CD8 T cells confirmed a dramatic increase in the cell size with a significant increase in the microvilli density (Fig. 3K). Thus, we speculated that adhesion and migration of activated CD8 T cells on ICAM-1 in the absence of chemokines may be simply induced by St3gal1-mediated modification of LFA-1 topography and clustering (avidity) in the increased cell membrane surface31. Indeed, the total internal reflectance microscopy (TIRF)/epifluorescence (epi) ratio for LFA-1 (Extended Data Fig. 6A) was lower in activated cells prior to spreading, indicating the distal location of LFA-1 relative to the surface topography (Fig. 3L). As cells spread on ICAM-1, the membrane flattened, leading to high concentrations of LFA-1 being redistributed into the contact site with ICAM-1 substrate (a higher TIRF/epi ratio). Under this condition, however, we detected the same ratio of LFA-1 accumulation at the cell-ICAM-1 interface in both activated and naïve T cells (Fig. 3L and Extended Data Fig. 6B). These observations imply that topographical remodeling and clustering (avidity) of LFA-1 that occurs with T cell activation is unlikely to affect the spontaneous adhesion and migration of activated T cells on ICAM-1.
To screen potential mechanisms for the LFA-1-mediated spontaneous T cell migration on ICAM-1, we next turned our attention to integrin recycling. Importantly, desialylation of in vitro activated CD8 T cells by neuraminidase caused a significant increase in endocytosis of cell surface LFA-1 (Fig. 3M), suggesting that glycosylation of cell surface LFA-1 by St3gal1 may inhibit endocytic recycling of LFA-1, causing a sustained increase in cell surface expression level of LFA-1. Indeed, we observed that the surface level of LFA-1 increased 2.5-fold 48 hr after T cell activation and that total LFA-1 expression remained stable in the following days (Fig. 3N). In addition, the endocytosis of LFA-1 was significantly increased by genetically deleting St3gal1 protein (Fig. 3O), leading to a downregulation in cell surface LFA-1 expression level of activated CD8 T cells (Fig. 3P). Note that the enhanced endocytic recycling of LFA-1 in St3gal1 knockout CD8 T cells is closely associated with a decrease in spontaneous and chemokine-independent T cell migration but allows chemokine-dependent T cell migration on ICAM-1 (Fig. 3I). Consistent with these results, promoted endocytic trafficking of membrane LFA-1 by cell treatment with Exo-132 (an inhibitor of exocytic vascular transport) or siRNA-mediated downregulation of Rab13 expression33 (which can inhibit exocytic transport of LFA-1) resulted in a significant decrease in spontaneous and chemokine independent T cell migration on ICAM-1, but more importantly, it enhanced chemokine-dependent T cell migration on ICAM-1 (Fig. 3Q & 3R). Therefore, our data suggest that the inhibition of St3gal1 mediated protein glycosylation can normalize the LFA-1 endocytic recycling process and prevent nonspecific T cell migration in vivo, thus hold significant therapeutic potential to potentiate the tumor-specific migration and anti-tumor function of CAR-T cells.
To test whether reducing St3gal1 expression and/or inhibition of its functions can drive CAR-T cells to their desired tissue locations and improve the therapeutic efficacy, we first confirmed that treatment of hHER2-CAR-T cells with neuraminidase and sgSt3gal1 infection of Cas9 hHER2-CAR-T cells (St3gal1 KO) do not alter CAR-mediated cytotoxicity (Fig. 3S). As we expected, both deletion of St3gal1 expression in CAR-T cells and neuraminidase treatments suppressed nonspecific T cell retention in the lung and liver, and significantly improved CAR-T cell retention in the circulation (the spleen and LN) after intravenous transfer (Fig. 3T). However, unlike our prediction, St3gal1 knockout and pretreatment of CAR-T cells with neuraminidases prior to intravenous transfer did not promote more robust tumor-specific migration (Fig. 3U). This result suggests that the general inhibition of St3gal1-mediated cell surface sialylation may be a blunt tool, which not only reduces nonspecific tissue sequestration, but also negatively impacts the tissue specific homing of effector T cells17.
Spontaneous migration of T cell is associated with βII-spectrin.
LFA-1 binding to ICAM-1 initiates outside-in signals that reorganize the cell cytoskeleton to regulate adhesion turnover via integrin endocytosis and/or recycling34. Therefore, we hypothesize that the reduced LFA-1 endocytic process by St3gal1-mediated glycosylation can be counterbalanced by promoting cytoskeletal force that can drive greater LFA-1 endocytosis in in vitro activated T cells. To screen for potential cytoskeletal molecules mediating LFA-1 endocytosis and surface distribution upon ligand binding, LFA-1 immunoprecipitates obtained from ICAM-1-bound T cells (in the absence of chemokine stimulation) were analyzed by mass spectrometry. We discovered that three cytoskeletal molecules, αII- and βII-spectrin, and nonmuscle myosin heavy chain IIA (MyH9) were associated with LFA-1 in T cells (Fig. 4A). Western blot analysis revealed that these three LFA-1-associated molecules were highly expressed in naïve T cells (Fig. 4B). The expression levels of αII-spectrin and MyH9 remained constant after T cell activation; however, βII-spectrin expression was dramatically downregulated during in vitro activation of CD8 T cells (Fig. 4B).
Figure 4. St3gal1-βII-spectrin axis controls LFA-1 mediated T cell migration.

A. LFA-1 immunoprecipitates obtained from human T cells bound on ICAM-1 (IC-1)- or poly-L lysine (PLL)-coated cover glasses. LFA-1-associated proteins were identified by silver staining and mass spectrometry. B. Expression levels of αII-spectrin, βII-spectrin, and MyH9 in activated T cells (from Days 0 to 5). Loading control: β-actin. C. A confocal FRAP experiment with activated CD11a-mYFP CD8+ T cells (n = 3). Scale bars, 1 μm. D. & E. Representative TIRFM images of activated CD11a-mYFP CD8+ T cell adhesion on ICAM-1-coated plates (D) and the area and spreading speed calculated from zone including all TIRF signals for each cell (E). Scale bar, 20 μm. Each dot represents one cell (n = 3). Bar = mean. F. Naïve and activated CD8 T cell migration on ICAM-1- or ICAM-1+CXCL12-coated surfaces. (n = 3. 21–42 individual cells per mouse). G. Schematic illustrating the regulation of LFA-1 endocytic recycling via βII-spectrin or St3gal1 expression (Created with BioRender.com). H. βII-spectrin (GFP+) expression in T cells isolated from GFP-ROSAβII-spectrin mice (TβII-spectrin) after in vitro treatment with TAT-Cre recombinase. I. Cross sections image in the middle of hHER2-CAR T cell (WT and TβII-spectrin)-endothelial cell (bEND.3 cells) contacts by transmission EM. Black arrows, LFA-1 stainings at the cell-cell contacts. Scale bars, 2 μm and 0.1 μm. J. The ratio of the TIRFM signal from the LFA-1 accumulation of hHER2-CAR T cells (WT and TβII-spectrin) at sites of contact with the ICAM-1-coated surface (n = 3). K. Flow cytometry analysis of MAL-II binding to naïve and activated CD8 T cells (WT, TβII-spectrin, and TβII-spectrin + St3gal1-shRNA-treated cells) (n = 3). Representative images or data were collected from three independent experiments (A-B, F, H) or two independent experiments (I). Results are shown the mean ± SEM (C, F) and mean fluorescence intensities (MFI) ± SEM (K). Statistical analyses were performed by ordinary two-way ANOVA (C), two-sided, unpaired Student’s t test (E-F, J, K).
Spectrin regulates LFA-1 clustering in T cells during the formation of immunological synapses35. Although the dramatic downregulation of βII-spectrin during T cell activation hinted at a possible role for βII-spectrin in the LFA-1 functions of activated T cells, it was unclear whether there was a direct relationship between βII-spectrin and LFA-1 redistribution in T cells. To investigate this relationship, we transiently transfected activated CD8 T cells prepared from CD11a-mYFP knock-in mice36 with βII-spectrin. Fluorescent recovery after photobleaching (FRAP) assay revealed that restoration of βII-spectrin expression in activated CD8 T cells did not alter LFA-1 lateral mobility in the cell membrane (t1/2=91.3 ± 21.7 vs. t1/2=91.2 ± 23.3 with/without βII-spectrin expression) (Fig. 4C). Instead, the cell membrane LFA-1 FRAP analysis showed a significant decrease in the immobile LFA-1 fraction with βII-spectrin expression (33 ± 5% vs. 9 ± 9%). The fact that the membrane mobile fraction of LFA-1 increases without a change in the membrane lateral mobility suggests that forced βII-spectrin expression in in vitro activated CD8 T cells causes greater dynamic endocytic recycling of LFA-137, 38. Indeed, live video imaging and TIRF analysis of CD11a-mYFP expressing naïve and activated CD8 T cells binding to the ICAM-1 coated surface revealed that both LFA-1 mediated cell spreading rate (μm2/sec) and maximum contact area (μm2) on ICAM-1 were significantly increased upon T cell activation (Fig. 4D & 4E and Video 9), but when βII-spectrin expression was restored in activated CD8 T cells (by transfection), the spreading rate and total spreading area were significantly decreased on ICAM-1 coated dishes compared with WT activated CD8 T cells (Fig. 4D & 4E and Video 9). Indeed, transient transfection of activated CD8 T cells with βII-spectrin reduced chemokine-independent migration on ICAM-1 (Fig. 4F and Video 8), but more importantly, it preserved the chemokine-dependent (and PTx-sensitive) pattern of T cell migration on ICAM-1, which was comparable to that of naïve CD8 T cells (Fig. 4F).
In conclusion, our data suggest that the restoration of βII-spectrin expression in in vitro activated CD8 T cells normalizes overall cell adhesiveness to ICAM-1 and migration by reversing the St3gal1 induced downregulation of LFA-1 endocytic process (Fig. 4G). To further address the functions of βII-spectrin in LFA-1 regulation, we generated a conditional βII-spectrin transgenic mouse (GFP-ROSAβII-spectrin mouse) to restore βII-spectrin expression only in activated CD8 T cells under the control of the ROSA26 locus in a Cre recombinase-dependent manner (Extended Data Fig. 7A). Treatment of these T cells with the membrane-permeable TAT-Cre fusion protein conjugated with a nuclear localization sequence39 during in vitro activation led to significant expression of βII-spectrin in hHER2 CAR-T cells (Fig. 4H). The forced expression of βII-spectrin in hHER2 CAR-T cells did not alter their effector functions, including IFN-γ and TNF production or CAR-mediated cytotoxicity (Extended Data Fig. 7B – 7D). Consistent with our predicted model (Fig. 4G), transmission electron microscopy (EM) images of LFA-1 immunostaining at the contact between hHER2 CAR-T cells and endothelial cells revealed that the overexpression of βII-spectrin in T cells reduced LFA-1 accumulation at the contact site (Fig. 4I). The decreased LFA-1 distribution to the contact between ICAM-1 coated surface and βII-spectrin overexpressing hHER2 CAR-T cells was further confirmed quantitatively by measuring the cell surface topography of LFA-1 with TIRF/epifluorescence ratio signals (Fig. 4J). Notably, βII-spectrin overexpression was sufficient to decrease MAL-II binding on CAR-T cell surface and the combination of βII-spectrin overexpression and St3gal1 deletion further significantly reduced cell surface sialylated-glycan activity (Fig. 4K).
Interestingly, we observed chemokine independent migration only in activated CD8 T cells but not in CD4 T cells (Extended Data Fig. 7E). Unlike CD8 T cells, adoptively transferred in vitro activated CD4 T cells did not accumulate in the lung (Extended Data Fig. 7F). St3gal1 expression was low in naïve CD4 and CD8 T cells and similarly upregulated in both cell types after in vitro activation (Extended Data Fig. 7G). βII-spectrin was highly expressed in both naïve CD4 and CD8 T cells (Extended Data Fig. 7G). However, unlike in activated CD8 T cells, the expression level of βII-spectrin remained constant after CD4 T cell activation (Extended Data Fig. 7G). These results suggest that differential chemokine reliance for T cell migration may be linked to distinct βII-spectrin expression, leading to altered LFA-1 regulation after in vitro activation. Intriguingly, memory CD8 T cells showed a similar expression level of βII-spectrin as naïve T cells (Extended Data Fig. 7H).
Expression of βII-spectrin improves tumor-specific homing of CAR T cells.
We then performed in vivo competitive homing assays with T cells isolated from GFP-ROSAβII-spectrin mice (TβII-spectrin) and ROSAtdTomato (Tcontrol) mice. TAT-Cre treated TβII-spectrin (green) and Tcontrol (red) cells were transfected with hHER2-CAR and mixed 1:1 and then IV injected into hHER2 Tg mice bearing B16-HER2 tumors (Fig. 5A). During the early phase of adoptive transfer, a significantly lower number of CAR-TβII-spectrin cells were recruited to the lung than CAR-Tcontrol cells (Fig. 5B). Instead, CAR-TβII-spectrin cells showed a greater capacity to migrate to tumors, leading to a dramatic increase in intratumoral accumulation compared to CAR-Tcontrol cells (Fig. 5B & 5C). Compared to control hHER2 tumor-bearing mice into which CAR-Tcontrol cells were adoptively transferred, tumor-bearing mice in which CAR-TβII-spectrin cells were adoptively transferred showed pronounced tumor regression, as well as a significant improvement in long-term survival (Fig. 5D & 5E). Differences in tumor size were apparent as early as day 14 after adoptive T cell transfer and were associated with a marked increase in intratumoral and intralymph-nodal CAR-TβII-spectrin cell infiltration (Fig. 5F). These results suggest that the expression of βII-spectrin in CAR-T cells promotes their recruitment to target tumor sites and suppresses tumor growth.
Figure 5. βII-spectrin expression predicts patient response to CAR-T cell therapy.

A. Schematic showing the assessment of tissue homing of TβII-spectrin (green) and Tcontrol (red) cells transfected with hHER2-CAR in hHER2 Tg mice bearing B16-hHER2 tumors. B. TβII-spectrin (green) and Tcontrol (red) hHER2-CAR were cotransferred (1:1 mix) and the fold change in the homing index was analyzed at each time point (n = 5 mice per condition). C. Represented deep 3D imaging (1-mm thick) of CAR-T cell accumulation in a cleared mouse lung 72 hr after i.v. injection (blue; blood vessels [CD31], green; TβII-spectrin, red; Tcontrol). Scale bars, 100 μm. D. Tumor growth and final tumor size from hHER2 Tg mice bearing B16-hHER2 tumors infused with TβII-spectrin and Tcontrol cells transfected with hHER2-CAR (n = 10 mice per group). E. Mouse survival curves up to 50 d after tumor inoculations. F. Frequency of CAR-T cells over total lymphocytes in B16-hHER2 tumours, lung, or dLN as measured by flow cytometry on day 22. respectively (n = 4). G. βII-spectrin western blot analysis of CAR-T cell infusion products obtained from 23 BCL patients who showed a complete recovery (CR), partial recovery (PR), or progressive disease (PD). Representative male and female samples from each patient group are shown. H. Quantification of total βII-spectrin protein expression in CAR-T cell products from patients with CR, PR, or PD as determined by western blotting. I. Correlation between βII-spectrin expression in CAR-T cell products and patient neurotoxic effects and cytokine release syndrome. Pearson correlation calculations were performed for linear correlations. J. Luminex assay of the proinflammatory cytokines IFN-γ, IL-6 and IL-1β in patient serum before (Day −5) and after (Day 2) CAR-T cell infusion. The data represent the mean ± SEM (B, D, F, H). Statistical analyses were performed by ordinary two-way ANOVA (D), two-sided, unpaired Student’s t test (B, D, H) or two-sided log-rank Mantel–Cox test (E). Whiskers represent maximum and minimum values (D, F, H).
βII-spectrin expression predicts patient response to CAR-T cell therapy.
Our data suggest that dramatic downregulation of βII-spectrin is closely associated with nonspecific sequestration of the adoptively transferred T cells and may be coincident with reduced therapeutic outcomes and/or a delayed overall response, as well as an increase in cytokine signatures of systemic toxicity. To test this hypothesis in CAR-T cell therapy, we collected patient CAR-T cells and performed serial blood collections from patients on CD19 CAR-T cell therapy (axicabtagene ciloleucel) before (Day −5) and after treatment (Day 2 after the injection of CAR-T cells) (Fig. 1A & Supplementary Table 3). At the 100-day follow-up with PET/CT, 10 patients had progressive disease (PD) (42 %) and 14 showed partial response (PR) or complete response (CR) (58 %). Western blot analysis of the CD19 CAR-T cells revealed a significantly higher expression level of βII-spectrin in CAR-T cells within the infusion products of patients who had achieved PR or CR (Fig. 5G & 5H).
The two most commonly observed adverse events associated with CD19 CAR-T cell treatment are cytokine release syndrome (CRS) and neurotoxic effects40. Overall, the cytokine release syndrome of any grade occurred in 22 patients (92 %). Grade 1 – 2 neurotoxic effects were observed in 4 patients (17 %), grade 3 was observed in 7 (29 %), and grade 4 – 5 was observed in 4 (17 %). We found that there were similar inverse trends in the CAR T cell βII-spectrin expression levels vs. the severity grades of CRS and neurotoxic effects (P = 0.05) (Fig. 5I), suggesting that βII-spectrin expression may be an independent predictor of severe toxic effects in patients. We then explored whether the inverse correlation between βII-spectrin expression and severe toxic effects can be explained by serum cytokine levels. Interestingly, PD patients (with a significantly lower expression level of βII-spectrin in their CAR-T cell products (Fig. 5H)) showed a greater upregulation of cytokine expression in serum before (Day −5) vs. after (Day 2) CAR-T cell infusion (Fig. 5J), indicating that systemic inflammatory cytokine production may be dependent on βII-spectrin expression.
Discussion
Our study supports a new paradigm suggesting that the ability of in vitro-activated therapeutic T cells to home to target tissues may not be completely dictated by local chemokine signals but is additionally controlled by an important cell intrinsic program that is acquired by T cells during the ex vivo manufacturing process. This paradigm explains why adoptively transferred T cells predominantly localize to nonspecific tissue sites. We have shown that the St3gal1-βII-spectrin axis controls tissue-specific migration of intravenously transferred in vitro activated therapeutic T cells, and that suppression of St3gal1 function and/or restoration of βII-spectrin expression improves chemokine-dependent and tissue-specific migration of therapeutic T cells, including CAR-T cells, while reducing their nonspecific sequestration in non-inflamed tissues.
In an early clinical trial, a patient with metastatic colorectal cancer who received an infusion of HER2 CAR-T cells experienced acute respiratory distress and pulmonary edema and subsequently died9, 41. The toxicity detected in this patient was mainly caused by acute pulmonary sequestration of the intravenously administered HER2 CAR-T cells and massive release of inflammatory cytokines upon interaction with the HER2 antigen expressed in the normal lung endothelium and epithelial cells. Therefore, it is tempting to speculate that the prolonged and strong interaction of highly activated engineered T cells with the target antigen in non-tumor bearing tissues may be a driving factor for CRS in other CAR-T cell therapies.
βII-spectrin generally forms a heterodimer with α-spectrins. Interestingly, α-spectrins contain a highly conserved Src homology 3 domain (SH3), suggesting that it may mediate intracellular signals by recruiting or activating downstream molecules through the SH3 domain. We show that βII-spectrin restoration in activated CD8 T cells decreases spontaneous cell migration, while this overexpression preserves chemokine-dependent migration. We further confirmed that βII-spectrin expression had no effect on CD8 T cell activation and effector functions both in vitro and in vivo. How LFA-1 directly interacts with βII-spectrin is unknown and will be an important topic for future research. Spectrins can bind F-actin and may interact with integrins through adaptor proteins, such as talin and kindlin, or through other proteins involved in the association of F-actin with the membrane42, 43.
As seen with integrin independent migration in 3D tissue environments, it is also possible that chemokine insensitive migration can simply be another tool that T cells utilize to facilitate migration in conditions that are restrictive to traditional migration mechanisms. Random migration in the inflamed tissue may increase the likelihood that cells encounter chemokine signals that will direct them to the site of infections to elicit effector functions. Shulman et al.27 have shown that transmigration may be facilitated through intracellular chemokine stores that require direct T cell interaction. Therefore, T cell crawling along endothelial cells in the absence of extracellular chemokine signaling may facilitate discovery of these intracellular stores of chemokines to induce transmigration. It is important to note that chemokine insensitive migration does not mean that cells will not respond to chemokine stimulation. Instead, this should be considered a mechanism through which cells can find the general site of infection while chemokine signals play a key role in ensuring precise localization.
Materials and Methods
Antibodies and reagents
OneComp eBeads and PE-anti-Human IgG Fc secondary antibody were purchased from eBioscience. Anti-mouse CD16/CD32 Fc blocker, Antibodies against αII spectrin and βII spectrin for western blot and BD Cytofix/Cytoperm Fixation/Permeabilization Kit were purchased from BD Biosciences. Antibodies against LFA-1-APC (H155–78), CD45.1-FITC (A20), CD45.2-BV711 (104), CD8-PE/Cy7 (53–6.7), CD8-AF594 (53–6.7), CD11a-AF647 (M17/4), CD11a-AF488 (M17/4), CD31-PB (390), CD31-AF647 (390), IFN-γ-BV421 (XMG1.2), TNF-BV421 (MP6-XT22), Tim-3-BV605 (RMT3–23), PD-1-BV421 (29F.1A12), Granzyme B-APC (QA18A28), Ki67-APC (16A8), CD11c-BV711 (N418), I-A/I-E-BV711 (M5/114.15.2), anti-mouse IgG-AF647, anti-rat IgG-AF647, anti-CD11a (M17/4 or TS2/4), anti-CD18 (CBR LFA-1/2 or M18/2), anti-CD49d (R1–2) and anti-MyH9 (Poly19098), 7-AAD Viability Staining Solution (420403) were purchased from BioLegend. PNGase F and α−2,3 Neuraminidase were purchased from NEB. Recombinant human IL-2, recombinant mouse IL-7, and IL-15 were obtained from PeproTech. Maackia amurensis Lectin (MAA/MAL II) - Cy5, Maackia amurensis Lectin (MAA/MAL II) - Separopore 4B were purchased from glycoMatrix. TAT-Cre recombinase, Pertussis toxin (PTx) and ExtrAvidin-Peroxidase were purchased from Sigma. Biotinylated goat anti-rat IgG (H+L) was purchased from Vector Lab. Recombinant human ErbB2/Her2 Fc chimera protein and human ErbB2/Her2-Alexa Fluor-594 were obtained from R&D systems. Anti-St3gal1 antibody was obtained from LS bio. Anti-βII spectrin (F11) antibody for western blot and St3gal1-shRNA plasmid were purchased from Santa Cruz Biotechnology. Anti-β-Actin, anti-HER2/ErbB2, HRP–conjugated anti–rabbit or anti–mouse secondary antibodies were purchased from Cell signaling. RetroNectin was purchased from Takara Bio. Anti-CD18 antibody was obtained from Abcam. FTY720 was purchased from CaymanChem. Exo1 was purchased from Tocris. Lipofectamine 2000, CellTrace Far-Red Cell Proliferation Kit, CellTrace CFSE Cell Proliferation Kit, and DyLight/650-anti-Human IgG Fc secondary antibody were obtained from Invtrogen.
Mice
C57BL/6J, ROSA26-Cas9 KI, OT-I TCR, OT-II TCR, LFA-1 KO, B6-GFP and ROSAtdTomato mice were purchased from the Jackson Laboratory. CD11a-mYFP, CD18-mCFP, and GFP-ROSAβII-spectrin mice were generated at the Gene Targeting and Transgenic Core facility at the University of Rochester using similar gene targeting techniques previously used in our laboratory30, 36. LFA-1 Heterozygous mice were generated through breeding LFA-1 KO mice (Jackson) with CD11a-mYFP mice. Mice exhibited a 49–57% decrease in CD11a expression. Human-HER2 transgenic mice were obtained from Genentech10. Genotyping for each strain was performed according to the corresponding reference. All mice were further backcrossed with C57BL/6J mice for at least more than 12 generations. Seven- to 10-week-old female or male mice were used in this study. The mice were maintained under 12-hour light/dark cycles at 18–26°C and 30–70% humidity. Mice were fed with rodent diet 5010 (LabDiet) and given reverse osmosis-filtered water (HYDROPAC®). All animal experiments were performed in the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-accredited, specific pathogen-free facilities in the Division of Laboratory Animal Resources of the University of Rochester Medical Center (URMC). All animal experiments were approved by the University Committee on Animal Resources at the University of Rochester (Rochester, NY, USA). Mice were sacrificed at the indicated time points or when geometric mean of tumor burden was ≥ 2.0 cm3.
Cell culture
B16-OVA and B16-hHER2 cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin and 1 mg/mL G418 (Gibco). bEND.3 cells (ATCC) were cultured in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin. 293T Phoenix cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, 20 mM HEPES buffer (Gibco), 1% MEM Non-Essential Amino Acids (Gibco) and 50 μM ß-mercaptoethanol (Sigma-Aldrich). Platinum-E cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, 1 μg/ml of puromycin and 10 μg/ml of blasticidin S HCl. Purified mouse and human CD8+ T cells were cultured in RPMI 1640 supplemented with 10% FBS, 1% antibiotic-antimycotic (Gibco), 2 mM L-glutamine (Gibco), 20 mM HEPES buffer (Gibco), 1% MEM Non-Essential Amino Acids (Gibco), 50 μM ß-mercaptoethanol (Sigma-Aldrich) and 80 U/mL IL-2. All cells were maintained at 37°C in 5% CO2. For B16-hHER2 stable cell line, B16F10 mouse melanoma cells (ATCC) were transfected with human HER2 pcDNA3.1 mammalian expression plasmid (Addgene #16257) using Lipofectamine 2000 according to the manufacturer’s protocol. Antibiotic selection (G418, 1 mg/mL) was added to the media 24 hr post-transfection. Cells were stained with anti-human HER2 antibody and sorted for the top 5% of HER2 expressers. Sorted cells were plated in 96-well plates at 1 cell per well to generate single cell clones. When thawed, stable cells were cultured in the presence of G418, and used within 2 weeks.
T cell preparation and CAR-T cell generation
CD4+ or CD8+ T cells were prepared from negative selection via Dynabeads (Invitrogen) or CD8a+ T Cell Isolation kit (Miltenyi) with 99% purity. Purified T cells were activated with plate-bound 6 μg/mL CD3ε Ab and 1.6 μg/mL CD28 Ab for 1 day and cultured with IL-2. Human T cells were purified from peripheral blood of healthy donors or cancer patients treated with CAR-T therapy using the EasySep Direct Human T cell isolation kit (StemCell). CD8+ T cells isolated from healthy donors were activated with Dynabeads™ Human T-Activator CD3/CD28 (Gibco) for 2 days, followed by incubation with IL-2. For CAR-T study, activated mouse CD8+ T cells were transfected with the trastuzumab (4D5; anti-HER2)-based CAR. CAR sequence was first cloned into the pMSCV vector (631461, Clontech). Retrovirus was generated using the Platinum-E packaging cell line. Platinum-E cells were plated at 0.8×106 cells per well in a 6-well plate. The following day, Platinum-E cells were transfected with hHER2-CAR pMSCV vector and pCl-Eco packaging vector using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Media was swapped 6 hr post-transfection. Retroviral-containing supernatants were collected 48 and 72 hr post-transfection. For T cell transduction, 1×106 activated CD8+ T cells were plated on RetroNectin (10 μg/mL, Takara) coated 12-well plates with retrovirus-containing supernatants, supplemented with 80 U/mL IL-2, and spun twice at both 48 and 72 hr post-transfection at 2500 RPM. T cells are then detached from RetroNectin-coated plate at least 24 hr post transduction and analyzed for hHER2-CAR expression. GFP-ROSAβII-spectrin mice-derived CD8+ T cells were pretreated with TAT-Cre recombinase (4.4 μM, Sigma) for 30 min to induce βII-spectrin expression and then transduced with hHER2-CAR. To knockdown St3gal1 in GFP-ROSAβII-spectrin mice-derived CD8+ T cells, T cells were transfected with St3gal1-shRNA plasmid (Santacruz) after treatment with TAT-Cre recombinase.
T cell electroporation
Six to seven hours prior to transfection, CD8+ T cells were washed with lymphocyte separation media and allowed to rest in complete media; 2 × 106 cells were suspended in P3 primary cell transfection media (Lonza) and mixed with Rab13 siRNA (Thermo). For βII-spectrin, βII-spectrin-HA DNA was purchased from Addgene (Plasmid # 31070) and mixed with mRFP plasmids. T cell electroporation was performed using an Amaxa 4D-Nucleofector kit (Lonza) with the DN-100 protocol following the manufacturer’s instructions. Cells were incubated in mouse T cell Nucleofector media+IL-2 for 6 hr post-transfection before being transferred to complete media+IL-2. Cells were analyzed 24 hr post-transfection.
In vivo studies
2×105 B16-hHER2 cells in 10 μl PBS were intradermally injected into one ear pinna of recipient hHER2 Tg mice. For adoptive cell transfer experiments, CAR-T cells were pre-treated with anti-CD49d (200 μg; R1–2, Biolegend) anti-CD11a (200 μg; M17/4, Biolegend) or PTx (100 ng/mL, Sigma) for 1 hr or 6 hr respectively before injection. Pre-treated 5×106 T cells were injected intravenously via the tail vein. Organs including lungs, spleen and LNs were extracted 4 hr after injection and analyzed. For the egress assay, 5×106 T cells were injected intravenously via the tail vein and FTY720 (1 mg/kg, Cayman Chem) was injected into the peritoneal cavity 24 hr, and 48 hr after injection of CAR-T cells. Organs including lungs, spleen and LNs were extracted 72 hr after injection and analyzed.
For competitive homing assays, 1:1 mix of 5×106 CAR-T cells stained with CFSE or Far-red (Invtrogen) or expressing GFP or tdTomato fluorescence were injected intravenously through the tail vein. Several organs were harvested at specific times (4, 24, 48 or 72 hr) after i.v. injection and the distribution of cells was confirmed. The fold change of Homing Index (HI) was determined by (CFSE+sample/CTFR+sample) / (CFSE+input/CTFR+input) or (GFP+sample/tdTomato+sample) / (GFP+input/tdTomato+input). For tumour growth analysis, 5×105 B16-hHER2 cells were subcutaneously injected into a left hinge flank of recipient hHER2 Tg mice. On day 7 post tumour inoculation, 3×106 HER2 CAR-T cells were injected intravenously via the tail vein. Tumour growth was monitored every three days. Tumour volume was calculated using the formula, tumour volume = (width)2 × (Length)/2. On day 22, lungs, tumour dLNs, and tumour tissues were extracted and analyzed. As stipulated by IACUC, the survival experiment was carried out by setting the point when the tumor size reaches 2000 mm3 as the end point.
T Cells cytotoxicity assay
Target cells (B16-hHER2 cells or B16-Ova cells) were seeded to 24-well plate at 1×104 cells per well and effector cells (CAR-T cells or Cas9-OT-I T cells) expressing GFP were added to the target cells at 10:1 E:T ratio. After 24 hr of incubation at 37°C, 7-AAD dye was treated for 10 min and cells were analyzed on the BD LSR II flow cytometer.
In vitro migration assays
Prior to imaging, cells were treated for 6 hr prior to imaging with PTx (100 ng/mL) or gallein (10 μg/mL) for 30 min. The PTx concentration used was determined based on previous publications27, 28 and from our studies where it blocks more than 80% of naïve T cell migration. Cells were treated with apyrase for 30 min prior to imaging at the described concentrations and remained in solution throughout the imaging. Cells were treated with Exo1 (100 μm/mL) for 30 min prior to imaging and remained in solution throughout imaging. Cell migration chambers (Millicell EZ slide eight-well glass, Millipore; Delta T dishes (Bioptechs) were prepared by coating their glass bottom with 0.5 or 5 μg recombinant mouse ICAM-1 respectively (Sino Biological) with or without indicated chemokines (200 ng/mL unless otherwise noted). T cells were placed in L15 medium (Invitrogen) in the chamber at 37°C, and video microscopy was conducted using a TE2000-U microscope (Nikon) coupled to a CoolSNAP HQ CCD camera with a 20× objective (CFI Plan Fluor ELWD DM; Nikon) and 0.45 numerical aperture. For flow chamber assays, sticky-slide I0.6 Luer (Ibidi) with 22X60X1 coverslips (Fisherbrand) were prepared with ICAM-1 in a stage top incubator (Oko Labs) at 37°C. Fresh L-15 was pumped continuously through the slide at 1.0 dyne/cm2. Cells were allowed to adhere utilizing a flow rate of > 0.1 dyne/cm2 prior to imaging. To study T cell migration on the endothelial cell, bEND.3 cells were grown into a monolayer on I0.6 Luer (Ibidi) with 22 X 60 X 1 coverslips (Fisher) prior to the addition of T cells. WT or PTx treated T cells were stained with CFSE or Cell Proliferation Dye eFluor670 (eBioscience), respectively, and mixed at a 1:1 ratio. 2.5×105 mixed T cells were allowed to adhere utilizing a flow rate of > 0.1 dyne/cm2 prior to imaging. Fresh L-15 was then pumped continuously through the slide at 1.0 dyne/cm2 for the remainder of the experiment. Migration analysis was performed in Volocity software (PerkinElmer). Transwell chamber assays were performed using a 5 μm pore, 24-well system (Corning). Filter membranes were coated with Protein A (20 μg/mL) overnight at 4°C prior to ICAM-1 coating (5 μg) for 2 hr at room temperature. Lower chambers were filled with 500 μL of complete media supplemented with 200 ng/mL CXCL12 or 500 ng/mL CCL21, and 1×106 cells were seeded into the upper wells and allowed to migrate for 3 hr at 37°C, 5% CO2. The lower wells were collected, supplemented with 5 × 104 counting beads (Sphereotech) and counted using a flow cytometer. Each condition was run in triplicate. B16 spheroids were generated using the “hanging drop” method. Briefly, twenty 10 μl droplets containing 0.5×104 B16-hHER2-BFP tumor cells in complete DMEM were plated onto the lid of a 10 cm plate and inverted. 5 ml PBS was added to the 10 cm plate to prevent evaporation. After 96 hours, the spheroids were collected by gentle pipetting and allowed to settle in a 1.4 ml Eppendorf tube and excess media was aspirated. hHER2-CAR-T cells were stained with CellTrace-FarRed (Invitrogen) per manufacturers protocol. In PTx treatments, the stained CAR T cells were pre-treated with 1 μg/ml PTx (Sigma) for 6 hours. Stained CAR-T cells were resuspended in 50 μl of complete RPMI and mixed with the tumor spheroids. 50 μl of Matrigel (Corning) was added to the stained CAR-T cell/spheroid mixture and mixed thoroughly. The total volume (100 μl) pipetted to the center of a ΔT-dish (Bioptechs), and incubated at 37°C and 5% CO2 for 30 minutes to allow the gel to polymerize into a 3D matrix. Once solidified, 1 ml of Leibovitz’s L-15 media supplemented with 2 mg/ml glucose was added to the dish and live-imaging time-lapse microscopy was performed. Video microscopy was conducted in a 37°C chamber. Brightfield and fluorescent images were acquired every 60 seconds for up to 24 hours.
RT-qPCR
For gene expression analysis of mouse or human CD8+ T cells by RT-qPCR, cells were lysed in RLT buffer and RNA was isolated via the RNeasy Mini isolation kit (Qiagen), according to the manufacturer’s instructions. After RNA concentration and purity were checked using a nanodrop, 100 ng RNA per reaction was subsequently converted to cDNA using the SuperScript VILO cDNA synthesis kit (Invitrogen). Samples were assayed using
SsoAdvanced Universal SYBR Green Supermix (Bio-rad), and kinetic PCR was performed on a CFX Connect Real-Time PCR Detection System (Bio-rad). The indicated genes were analyzed by using the primers listed below. Data was normalized to the housekeeping gene Valosin-containing protein (VCP) and hypoxanthine guanine phosphoribosyltransferase (HPRT). Relative transcript levels were analyzed using the 2(− ΔΔCt) method. Target(Forward/Reverse);HuSt3gal1(GCATTTCTCTTTCCCACAGC/CTAATTCCCAGCCACCTTCA),HuSPC24(GGGATTATGAGTGTGAGCCAGG/ACTCCAGAGGTAGTCGCTGATG),HuVCP(AGGATGATCCAGTGCCTGAGAG/GGAATCTGAAGCTGCCAAAG),MoSt3gal1(GCAGACGTTGGGAGCCGGAC/GGCACGGGGACATAGGTGTGAG),MoSPC24(CAGTGGGATTATGAATGCGAGCC/GGTAGTCACTGATGAACTTCGGC),MoHPRT(GCTGGTGAAAAGGACCTCT/CACAGGACTAGAACACCTGC).
LFA-1 endocytosis assay
The protocol was modified from a previous study45. LFA-1 on the cell surface was labeled with anti-CD18 (CBR LFA-1/2, 10 μg/ml), anti-CD11a (TS2/4, 10 μg/ml) or anti-mouse CD18 (M18/2, 10 μg/ml) for 30 min on ice. After washing with cold migration buffer, cells were treated neuraminidase (200 ng/ml) for 3 hr at 37°C. After washing with cold migration buffer, the cells were incubated with AF647-anti-mouse IgG antibody (Poly4053, 2 μg/ml) or AF647-anti-rat IgG (Poly4053, 2 μg/ml) at 4°C for 30 min and fixed immediately in 3% PFA. The MFI (mean fluorescent intensity) was acquired by BD LSR II flow cytometer and analyzed via FlowJo.
Flow cytometry and Cell sorting
To detect CAR expression level on T cells, 1×106 CAR-T cells were pre-incubated with anti-mouse CD16/CD32 Fc blocker (1 μg/million cells in 100 μl, BD Bioscience) for 10 min at 4°C and incubated with recombinant human ErbB2/Her2 Fc chimera protein (R&D systems) at 4°C for 30 min. After washing, the cells were incubated with PE-anti-Human IgG Fc secondary antibody (eBioscience) or DyLight/650-anti-Human IgG Fc secondary antibody (Invitrogen) at 4°C for 30 min. These stained cells were detected by BD LSR II flow cytometer and analyzed via FlowJo. To detect MAL-II binding, cells were incubated with Cy5-MAL-II at 4°C for 1 hr in 1% BSA/PBS. For isolation of lymphocytes from organ tissues and tumors, mice were euthanized, and organs and tumors were extracted. Lungs, livers, and tumors were minced into small fragments and digested with 1 mg/ml collagenase D (Sigma) and 50 mg/mL DNase (Roche) at 37°C for 45 min, passed through a 40 μm cell strainer, and lymphocytes were isolated via Percoll gradient centrifugation. LNs and spleens were made single cells through 70 μm cell strainers. RBCs in single cells suspensions were lysed with ACK lysis buffer (Thermo), washed with DPBS and prepared for antibody staining. For in vitro T cells staining, they were re-stimulated with PMA/Ionomycin before staining. The obtained single cells were pre-incubated with anti-mouse CD16/CD32 Fc blocker (1 μg/million cells in 100 μl, BD Bioscience) for 10 min at 4°C and stained with the following antibodies at 4°C for 1 hr: Tim-3-BV605 (RMT3–23), PD-1-BV421 (29F.1A12), LFA-1-APC (H155–78), CD45.1-FITC (A20), CD45.2-BV711 (104), CD8-PE/Cy7 (53–6.7). Intracellular proteins or cytokines were stained with following antibodies at 4°C for 1 hr after fixation and permeabilization via BD Cytofix/Cytoperm™ Fixation/Permeabilization Kit: IFN-γ-BV421 (XMG1.2), TNF-BV421 (MP6-XT22), Granzyme B-APC (QA18A28) and Ki67-APC (16A8). These stained cells were detected by BD LSR II flow cytometer and analyzed via FlowJo. To analyze CAR-T cells in the lungs, CAR-T cells expressing GFP were sorted by BD FACSAria II cell sorter from single cell lymphocytes isolated from the mouse lungs.
Glycosidase treatment
20 μg of CD8+ T cell lysates were treated with 4 μl of α−2,3-Neuraminidase (P0743L, NEB) and/or 2 μl of PNGase F (P0704L, NEB) at 37°C for overnight or 2 hr. 4x Laemmli buffer was added and the samples were denatured by heating at 95°C. The samples were separated in 4–15% Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-rad), and western blots were performed as described below.
Western blot analysis
Cells were lysed in RIPA buffer and separated in 4–15% Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-rad) by SDS-PAGE. After transferring the proteins to the NC membrane, the membrane was blocked with 5% nonfat milk and 0.1% Tween-20 in TBS for 1 hr. Blots were incubated with 1:500 anti-HER2 (Cell signaling), 1:500 anti-St3gal1 (LS bio), 1:1000 anti-Spc24 (Novus bio), 1:1000 anti-βII spectrin (Santa Cruz), 1:500 anti-MyH9 (Biolegend), 1:1000 anti-αII spectrin (BD bioscience), 1:1000 anti-CD18 (Abcam) and 1:5000 anti-β-actin (Cell Signaling) overnight at 4°C. The membrane was then incubated with 1:3000 horseradish peroxidase–conjugated anti–rabbit or anti–mouse secondary antibody (Cell Signaling) for 1 hr at room temperature. Images were obtained using a ChemiDoc imaging system (Bio-Rad).
Immunoprecipitation
Immunoprecipitation was performed with the method we previously described46. Briefly, Cover glasses were coated with 6 μg/ml of ICAM-1 or 10 μg/ml poly- l lysine (PLL). T cells were then incubated at 37°C for 20 min on the coated cover glass in L-15 medium (Invitrogen) containing 2 mg/ml glucose or in 20 mM Hepes, 150 mM NaCl, 5 mM MgCl2, 1 mM EGTA, and 2 mg/ml glucose. Cell lysates were then precleared with 2 μg mouse IgG isotype control antibody and protein G agarose beads. Then, pre-cleared lysates were incubated with protein G agarose and TS2/4 or CBR LFA1/2 antibody. For immunoprecipitations with MAL-II, 30 μl of MAL-II agarose beads were mixed with 50 μg cell lysates and incubated for overnight at 4°C on a rotator. The samples were washed with IP buffer, and then eluted by boiling in 2x Laemmeli sample buffer. Equal volumes of immunoprecipitated samples were separated on 4–15% gradient gel (Bio-rad), and stained with SilverQuest™ Silver Staining Kit (Invitrogen) or SimplyBlue SafeStain (Invitrogen) or subjected to western blotting.
Liquid Chromatography Tandem Mass Spectrometry
The purified CD18 was excised from the coomassie-stained gel and in-gel digested by trypsin. The digested peptides were extracted by gradient acetonitrile (ACN) with 0.1% trifluoroacetic acid (TFA) from 10% to 100% ACN. The extracted peptides were pooled and speedvac dried. Then the peptides were desalted using Sep-Pak tC18 1 cc Vac cartridge (WAT054960). In brief, the columns were pre-equilibrated using 1 ml 100% ACN and washed with 0.1% TFA twice. The digested peptides were added to 200 μl with 0.1% TFA and load into the columns and washed with 1 ml 0.1% TFA twice, followed with 200 μl 0.5% acetic acid. The digested peptides were eluted using 75% ACN/0.5% acetic acid. The eluted peptides were dried in speedvac and then dissolved in 0.1% formic acid for nanoLC-MS/MS using an Orbitrap Fusion mass spectrometer (Thermo Scientific) equipped with a nanoelectrospray ion source (New Objective) and an UltiMate 3,000 RSLCnano system pump (Thermo Scientific Dionex). PMI-Byonic was used for the MS/MS-based protein and glycosylation site identification47. Methionine oxidation was enabled as a variable modification. O-glycopeptides were searched against 78 mammalian and 70 human databases.
FRET and FRAP assay
FRET imaging was conducted with a dual-view image splitter (Photometrics) and CFP/YFP dual-band filter set (Chroma) by the sensitized emission method as we previously described30. Data analysis was performed with the Autoquant imaging algorithm. FRET cells were resuspended in L15 medium with 2 mg/ml glucose. Cells were placed in an uncoated delta T dish and equilibrated for 10 min at 37°C. Then each image of CFP, YFP, and DIC of each cell was acquired for 0.5 s with 2 × 2 binning through a ×60 oil 1.49 aperture immersion objective lens without a ND filter. For membrane vs. cytosol experiments, a region of interest (ROI) of the membrane and cytosol were selected for each cell. The FRET efficiency of each was then compared to generate a ratio of LFA-1 conformation between the two. FRAP was performed using an Olympus FV1000 laser scanning confocal microscope housed at the URMC Confocal and Conventional Microscopy Core. Cells were imaged live using a 60× 1.4 NA oil immersion objective at 37°C. FRAP was performed on βII-spectrin expressing cells or WT control cells 24 hr post transfection. Photo-bleaching was performed with the SIM scanner capabilities using the 488-nm laser line set to 100% power for 2 s and images collected every three seconds. FRAP analysis was performed using NIS Elements software. Two ROI’s, one of the total cell membranes and one of the photobleached area were collected following background correction. All fluorescent ROI values were reported as relative fluorescence units (RFU). Florescent values were normalized using the ratio of photobleached ROI to that of the control ROI. The recovery of fluorescence signals was calculated from the fluorescence of the bleached and non-bleached membrane areas during the time series. t1/2 values were calculated by fitting data to non-linear regression curves. The immobile fraction was calculated by subtracting the post-bleached membrane fluorescent intensity from the pre-bleached intensity.
Mobile fraction (Fm) = (IE - I0) / (II - I0)
Immobile fraction (Fi) = 1 - Fm
IE: The recovered fluorescence intensity
I0: The post-bleach fluorescence intensity
II: The pre-bleach fluorescence intensity
TIRF microscopy
CD11a-mYFP CD8+ T cells in L15 medium (Invitrogen) were allowed to settle on No. 11/2 coverslips (Corning) coated with ICAM-1 (5 μg, Sino) with or without CXCL12 (200 ng/mL, R&D biological). hHER2-CAR T cells (wt and βII-spectrin) in L15 medium (Invitrogen) were allowed to settle on No. 11/2 coverslips (Corning) coated with ICAM-1 (5 μg, Sino). TIRF was performed with the Andor Dragonfly TIRFM module coupled to a Nikon Ti-E microscope equipped with a 60x NA 1.49 APO TIRF oil immersion objective, and with images being captured by a Zyla 4.2 sCMOS camera (Andor). mYFP was excited with a fiber coupled OBIS LX 488 nm wavelength solid state laser (Coherent) line, passed through a dichroic and 450/50 nm bandpass emission filter. All live cell imaging was performed at 37°C with 5% CO2 with/without 20μg anti-CD11a (BD biological), maintained by a stage top incubator (Okolab) throughout the experiment. Images were acquired every second for 6–10 min. Image acquisition was performed using Fusion (Andor). All image processing was performed with Matlab (Mathworks). For LFA-1 surface topography assay, cells were labeled with Alexa Flour 488 anti-mouse CD11a (clone I21/7, BioLegend). For controls, cells were labeled with Alexa Flour 488 in 1 ml of HBSS with 10mM HEPES containing Alexa Flour 488 carboxyl acid, tetraflourophenyl ester (component A from the Protein Labeling Kit (Invitrogen)). Cells were spreading onto ICAM-1 or BSA coated coverslips. BSA coated, as well as uncoated coverslips, were served as controls. The cells were allowed to equilibrate for at least 20 minutes. A series of a DIC, epifluorescence (Epi), and TIRF images were acquired and saved for offline analysis. To avoid the potential artifacts in fluorescence due to changes in the cell shape as it interacted with the surface, TIRF signal was normalized by Epi fluorescence over a 2.0 μm diameter at the center of the contact zone. The normalized TIRF signal was calculated based on four regions of interest for each image: the Epi image at the center of the cell (), the Epi image over the area, where no cells were present for the Epi background fluorescence measurement (), TIRF image at the center of the cell (), and TIRF over the area, where no cells were present for the TIRF background fluorescence measurement ():
Electron microscopy
The bEND.3 cells were seeded in cell migration chambers (Millicell EZ slide eight-well glass, Millipore). 1 × 106 CD8+ T cells were added on the chambers for 1 hr and fixed with 4% PFA for 2 hr. The samples were incubated with 1:100 anti-LFA-1 (H155–78, biolegend) for overnight at 4°C. The samples were then incubated with 1:200 biotinylated goat anti-rat IgG (H+L) (Vector Lab) for 1 hr at room temperature and then, the samples were incubated with 1:150 ExtrAvidin-HRP (E2886 Sigma-Aldrich) enzyme conjugate. The samples were presoaked in TRIS buffer and diaminobenzidene-HCl (DAB) for 20 min and H2O2 was added for 7 minutes. Labelled cells were washed and post-fixed in 2% glutaraldehyde overnight prior to both silver and gold enhancement and post-fixation in 1.0% OsO4. The labelled cells on the slides were dehydrated in a graded series of ethanol up to 100% and infiltrated with Spurr’s epoxy resin overnight. Size 3 BEEM capsules were filled with epoxy resin and set up on top of the slides over the labelled cells for the “Pop-off” technique. Rethen polymerized at 60°C for overnight. The capsules were “popped off” the slides by dipping in liquid nitrogen to break the surface tension between the glass and the polymerized blocks containing the labeled cells. The blocks were trimmed to areas of cells and thin sectioned using a diamond knife at 70 nm onto formvar/carbon coated slot grids. The grids were examined and photographed using a Hitachi 7650 transmission electron microscope and a Gatan Erlangshen 11 megapixel digital camera system.
RNA-sequencing
RNA-sequencing was performed as we previously described. Briefly, total RNA (1 ng) was pre-amplified with the SMARTer Ultra Low Input kit v4 per manufacturer’s recommendations (Clontech). We used the Qubit Fluorometer (Life Technologies) and the Agilent Bioanalyzer 2100 to measure the quantity and quality of the subsequent cDNA. of cDNA (150 pg) was used to generate Illumina compatible sequencing libraries using the NexteraXT library preparation kit per manufacturer’s protocols (Illumina). We then hybridized the amplified libraries to the Illumina single end flow cell and amplified using the cBot (Illumina). Single end reads (100 nt) were generated. After filtering out genes with zero reads, genes between conditions were compared based on adjusted p values and log2 fold change. DESeq2–1.22.1 within R-3.5.1 was used to perform data normalization and differential expression analysis with an adjusted p-value threshold of 0.05 on each set of raw expression measures. The heat map, volcano plot were created using pheatmap (version 1.0.12) and pcaExplorer (versuib 2.8.0) with the DESeq2 normalized data and results. The Enrichr gene database was used to obtain categories of signaling pathways related to genes that were downregulated and upregulated in CART72 compared to CART0. The listed categories were ranked by the likelihood that genes fall into the group as a function of P value. Principle PCA) plots, volcano plot and heat map representations were produced using RStudio (Version 1.3.1093, 2009–2020) or Graphpad Prism (v9).
Intravital microscopy
Intravital microscopy was performed as we previously described48. Briefly, in vivo imaging was performed using an Olympus FluoView FVMPE-RS Twin-Laser Gantry system with a x25 water immersion objective (NA 1.05 Olympus) and two lasers, the Spectra-Physics InsightX3 and MaiTai HP DeepSee Ti:Sapphire (University of Rochester Multiphoton Research Core Facility). Mice were anesthetized by intraperitoneally injecting pentobarbital sodium-salt (65 mg kg−1). To restraint and to avoid psychological stress and pain during imaging, mice were further maintained by isoflurane inhalation. For the lung image, non-recovery surgery on the lung and surrounding tissue was performed. First, three left anterior ribs of mice were resected and a small portion of the left lung was exposed. 20–25 mm Hg of suction was carefully applied to immobilize the part of lung within the thoracic suction window. The microscope objective was then placed over the thoracic suction window for live imaging. Depth of anesthesia was confirmed by monitoring of pain reflexes (pedal reflex), monitoring of respiratory rate before intubation, monitoring of respiratory pressure through ventilator after intubation, and monitoring of heart rate and blood oxygenation via infrared pulse -oximetry. An anesthesia record / surgery log was maintained throughout the procedure, and the paralytic was administerd after the depth of anesthesia was confirmed. A cannula was put into place once the trachea was exposed and prepared. The cannula was attached to the ventilator once secured49. For draining lymph node, hair on the right hind leg and inguinal area of the mouse was removed by an electric trimmer. Then a small 2 to 3 mm incision with scissors was made at the right knee to expose the extensor tendon and then the right popliteal fossa. Subsequently, the right popliteal lymph node of the anesthetized mice was surgically exposed on the custom designed platform. For ear (tumour) imaging, mice were anesthetized and laid flat on the custom designed plate to expose the ear tumour under the cover slip onto microscopy stage. To visualize blood vessels, PB- or AF647-conjugated CD31 antibody was i.v. injected before the start of imaging. Images were obtained using FluoView software (Olympus) at a resolution of 512 × 512 (16 bit). Each z-stack was 2.5–3.5 μm apart at a frame rate of 1.5 s for total 40 frames. Fluorescent signals were excited as follow; tdTomato: 975 nm, mRFP: 1,100 nm, Alexa Fluor 647: 12,00 nm, FITC/GFP: 975 nm (GFP) or 820 nm (FITC), BFP: 820 nm, and second-harmonic generation (SHG): 850 nm. We also used the following filter cubes: Red/fRed (575–630 nm/645–685 nm, DM650), SHG/Blue/Green (420–460 nm/495–540 nm, DM485). We used Volocity (PerkinElmer), Imaris (Bitplane), Arivis Vision 4D (Arivis) and ImageJ softwares to process raw imaging data. For micro-PET/CT image of CAR T cell distribution, 1 × 107 T cells washed 3x with PBS. After the final wash, cells were resuspended in100 μl 0.1M HEPES (pH 8) and incubate with SCN-Bz-Df for 30–60 min at 37 °C. Then, cells were washed with pH 7.4 PBS 3x. After the final wash, cells were resuspended in 400 μl PBS (pH 6.5) and incubated with 20 μl of 89Zr solution (in 1M oxalic acid/1M sodium carbonate) + 80 μl of 0.5 M HEPES at RT for 1 hr. After the incubation, cells were washed 3x with PBS (pH 7.4).
Tissue clearing
ScaleCUBIC Reagent-1 [25 wt% urea, 25 wt% Quadrol and 15 wt% Triton X-100 in dH2O] and ScaleCUBIC Reagent-2 [50 wt% sucrose, 25 wt% urea, and 10 wt% triethanolamine in dH2O] were prepared for tissue clearing50. To visualize blood vessels, PB-CD31 antibody was i.v. injected before lung extraction. After lung perfusion with cold PBS, whole-lung tissues were extracted and fixed with 4% PFA overnight. Whole-lung tissues were washed with PBS with shaking at room temperature and incubated in ScaleCUBIC Reagent-1 for 3 days at 37°C with gentle shaking. After PBS washing, the tissues were stored in ScaleCUBIC Reagent-2 for 1 day at room temperature. For immunofluorescent staining, the tissues were stained with Alexa Fluor 594-hHER2, BV711-CD11c, or BV711-MHCII antibodies for 3 days at room temperature after tissue clearing with Reagent-1. After PBS washing, the tissues were incubated with ScaleCUBIC Reagent-2 for 1 day at room temperature. Images were acquired Olympus FluoView FVMPE-RS Twin-Laser Gantry system with a x10 water immersion objective (NA 1.05 Olympus). Raw imaging data were processed and analyzed with Volocity (PerkinElmer), Arivis Vision 4D (Arivis) and ImageJ.
CRISPR Library construction
5 sgRNAs were designed against individual genes. The sgRNA oligos were synthesized (Cellecta, CA) and pooled in equal molarity. The pooled sgRNA oligos were then cloned into pMSCVURP-U6-sg-HTS6MC-UbiC-TagRFP-2A-Puro. We performed a deep-sequencing analysis and verified that > 95% of the designed sgRNAs were cloned in the vector and > 95% of individual sgRNA constructs was within 5-fold of the mean. Transformation was performed using Invitrogen DH5a MAX efficiency cells following the manufacturers protocol. 10ng of library DNA was used in each reaction, and three reactions were performed in parallel. To calculate transformation efficiency, 10 μL was taken from the pooled reactions and serially diluted to 1:50, 1:100, and 1:1000 in SOC media. Transformation efficiency was calculated by dividing the number of colonies counted on a given dilution plate by the number of micrograms used in the transformation and then multiplying by the dilution factor. We required a transformation efficiency 500x the number of constructs in our library in order to proceed. Gylcerol stocks were made from successfully transformed competent cells. In each 1mL aliquot, there was at minimum 500x library coverage based on cell number as determined by OD600 of the overnight liquid culture. Cells were frozen in fresh culture media with 25% glycerol by volume.
Mouse experimental workflow for in vivo CRISPR screen
Retroviral virus was produced using 293T Phoenix cells. To concentrate virus, Takara’s RetroX Concentrator was used as per manufacturers protocol. To titrate virus, three different volumes of thawed viral concentrate were used to infect 1 million T cells each. After 48hrs, transduction efficiency was assessed using flow cytometry (RFP). The effective viral titer was calculated by multiplying the number of cells at the time of infection by the fraction of infected cells and dividing the product by the milliliters of virus used for infection. This process was repeated for each new batch of virus. CD8+ T cells were isolated from Cas9/OT-I donor mice, then seeded on plates coated with anti-CD3/CD28 and cultured with the addition of IL-2 at 20 IU/mL (Day 0). On Day 2, the cells were transduced with retroviral libraries and cultured with mouse IL-7 at 2.5 ng/mL, mouse IL-15 at 25 ng/mL and human IL-2 at 20 IU/mL for 2 days. The transducer cells were selected with 3 mg/mL for another 3 days. On Day 7, transduction was assessed by flow cytometry and 3.5×106 cells were injected into recipient mice (B16-OVA tumor bearing mice) intravenously. For all adoptive transfer experiments, CD8+ T cells from Cas9/OT-I mice were infected at an MOI of 0.3 to achieve approximately 20% transduction efficiency using the techniques described above. On the day of transfer, an aliquot of infected cells totaling 500x the library coverage was saved as an “input” for comparison during later analysis of sgRNA enrichment. After 24 hr, about 7×105 Cas9/OT-I/sgRNA T cells were recovered from the lung/liver and spleen/lymph nodes/blood (1.5×105 cells/per group). T cells from 5 −6 mice were pooled. We repeated this sample three times (n=3, 5 – 6 recipient mice per group).
Analysis of pooled CRISPR screen
Analysis was performed as previously described51. Raw fastq files were cleaned and trimmed using cut adapt version 1.18 and aligned using bowtie version 1.2.2. Read counts for each sgRNA barcode were identified using feature Counts version 1.6.2. To identify hits from the screen, we used the MAGeCK software version 0.5.9 to quantify and test for guide enrichment52. Each assay was repeated three times. Then, we ran MAGeCK-MLE with three replicates (n=3, 5 – 6 mice per group) for each organ compartment (Lung/Liver (termed TSequestered), Sp/LN (termed Tmigratory), and baseline (non-infused, termed T0)). The p-values and FDR of the beta scores shown in Figure 2 were calculated using these replicated data sets for each compartment based on MAGeCK-MLE’s maximum likelihood estimate 53. The beta value for each gene represents the extent of selection for that gene in a particular condition (a positive beta value means the gene is positively selected in that condition, and a negative beta value suggests negative selection). MAGeCK-MLE negative binomial distribution is used to also estimate a mean and z-score for the estimated beta value. Fig. 2B is a visualization of the z-scores of the beta values for each of the target genes in the Sp/LN and Lung/Liver tissues using MEGeCk-MLE. The genes are ranked by the z-scores of their beta values within each condition and plotted. The x-axis represents the rank of the genes based on their z-scores, and the y-axis represents the actual z-scores. Genes with more negative z-scores suggest strong negative selection in the reported tissue. The highlighted genes in the figure have a false discovery rate (FDR) <= 0.05 (Wald test). The size of each highlighted points is scaled based on the -log10(FDR). Points with smaller FDR are larger in size. MAGeCK-MLE is able to model complex experimental designs with multiple screening conditions. In our case, we used three screening conditions: Lung/Liver (termed TSequestered), Sp/LN (termed Tmigratory), and baseline (non-infused, termed T0). T0 was used to MAGeCK-MLE’s model building and for all sgRNA efficiency calculations. A positive beta score indicating positive selection, and a negative beta score indicating negative selection. The p-values and FDR of the beta scores were estimated using both a permutation test and a Wald test within the MAGeCK-mle module. MAGeCK analysis for sgRNA and gene level enrichment and normalized and raw count files can be found in Source Data.
Preparation of genomic DNA for next-generation sequencing
Amplification and barcoding of sgRNAs was performed following the manufacturer’s specifications for constructing CRISPR pooled lentiviral sgRNA libraries (Cellecta, Mountain View, CA). Briefly, an initial round of 25 cycles of PCR was performed using 100ng genomic DNA to amplify sgRNA sequences. Amplicons from the first PCR were used in a second PCR of 9 cycles to add index, P5, and P7 sequences for Illumina sequencing. The final sequencing libraries were quantified using the Qubit dsDNA assay (ThermoFisher) and assessed for size using the Tapestation D1000 assay (Agilent). The pooled sequencing libraries were then size-selected for the size range of 340–425bp using the PippinHT (Sage Science). Libraries were sequenced on a NextSeq550 High-output flowcell (Illumina) to obtain single-end 50-nt reads.
Patient samples
Peripheral blood mononuclear cells (PBMCs) and plasma samples are being collected from patients with B-cell lymphoma that received axicabtagene ciloleucel (2 × 106 cells/kg). Patients consent for blood collection under the University of Rochester Medical Center Wilmot Cancer Center’s lymphoma research program tissue collection protocol ULAB-03012 in accordance with the Institutional Review Board. Peripheral blood was obtained in sodium heparin tubes before treatment and after axicabtagene ciloleucel infusion from forty patients. Plasma is obtained by centrifugation of heparinized blood. Plasma samples are stored at Wilmot Cancer Center of the University of Rochester. All stored aliquots of plasma were re-labeled to a random, non-linked code, before any analysis. These same patients were undergoing CAR-T cell therapy treatment. CAR-T cells are prepared commercially and returned to the Wilmot Cancer Center for infusion. After infusion, some cells typically remain in the infusion tubing and storage bag. These cells are usually discarded but were recovered and used in the current studies. After completion of CAR-T cell infusion, the infusion bag was washed with PBS to obtain residual cells. Patient’s plasma was also collected to measure IFNγ, IL-6, and IL-1β levels using xMAP® Human Cytokine-Chemokine Magnetic Bead Panel kit (HCYTOMAG-60K, EMD Millipore) followed manufacturer’s instructions. Briefly, patient’s plasma samples were thawed on the ice one hour prior to the experiment. The standards or plasma samples were pipetted (in duplicate) into 96-well Luminex plate. Next, 25 μl plasma samples were mixed and incubated with 25 μl of specific color-coded antibody-linked magnetic beads for IFNγ, IL-6, and IL-1β at RT on a shaker (800 rpm) for 120 min. After adding biotinylated detection antibodies, the sample was incubated with streptavidin-phycoerythrin. Then, the plate was washed twice using BioPlex Pro magnetic wash station. Beads were resuspended in 125 μl of Bio-Plex assay buffer before reading on a calibrated Bio-Plex 200 system (Bio-Rad, Marnes-la-Coquette, France). A five-parameter logistic curve was used and data were analyzed with Bio-Plex Manager 6.0 software.
Statistical analysis
All statistical tests were performed with GraphPad Prism (v9). Statistical analysis was performed using Mantel-Cox test for survival curves, Two-way ANOVA, ordinary One-Way ANOVA with a Tukey’s multiple-comparison post-test, unpaired t-test, and Mann– Whitney test when appropriate. Differences were considered significant when P values were <0.05. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications48. Data distribution was assumed to be normal but this was not formally tested. In all in vivo experiments, C57BL/6 mice and transgenic mice with male or female mice aged 7–10 weeks were age- and gender- matched and randomly assigned to each group. For the in vitro assays, samples and cell conditions were randomly distributed across treatment groups. For in vivo experiments, there was no blinding in collection and analysis of the experimental data due to requirements for cage labeling and staffing needs. For in vitro experiments, all the treatments were performed in a parallel manner without the risk of bias in interpretation. All data were acquired and analyzed by softwares with object standard. Therefore, blinding was not relevant to the study. No data were excluded from the study.
Extended Data
Extended Data Figure 1. hHER2 CAR-T cell assays and in vivo distribution.

A. Gating strategy for detection of hHER2-CAR-T cells. A median of > 90% of mouse CD8 T cells transduced with the hHER2-CAR retrovirus were positive for the transgene as judged by flow cytometry analysis. B. B16F10 cells were transfected with mammalian human HER2 construct with lipofectamine. Cells were grown in the presence of G418 and sorted twice to generate single cell clones. C. CAR-T-mediated B16-HER2 cell killing assay. Apoptotic cells were stained for Annexin V and analyzed by flow cytometry. D. Whole mouse lung before (left) and after (right) CUBIC clearing.
Extended Data Figure 2. hHER2 CAR-T cell distribution.

A. Left: Deep 3D imaging (1 mm thick) of CAR-T cell accumulation in the cleared mouse lung after 72 hr after i.v. injection (red; CD11c (top) or MHCII (bottom), green; CAR-T cell (GFP)). Right: Graph shows colocalization of red signal versus GFP (CAR-T cells). Pearson coefficient was generated as Green (CAR-T cell) /Red (anti-CD11c or anti-MHCII). Data are presented as mean ± SEM; n = 3 mice/group. B. Flow cytometric analysis of OT-I T cells in the lung, blood, and tumor after i.v. injection (4, 24, 48, and 72 hr; mean ± s.e.m., n = 3 mice per group). C. PCA of differentially expressed genes in CAR-T cells (T72 vs. T0). D. Gating strategy for detection of hHER2-CAR-T cells from organs (lungs) E. Expression of Ki67+ in CAR-T cells isolated from the lung after i.v. injection. F. Relative expression of IFN-γ and TNF-α in CAR-T cells isolated from lung at indicated times after i.v. injection. Data are presented as mean ± SEM; n = 3 mice/group. G. Representative flow cytometry results show cell surface PD-1 and Tim-3 expression levels in OT-I naïve T cells, freshly activated in vitro (0 hr), or isolated from the lung after 72 hr post-injection. H. hHER2-CAR-T cells isolated from the lung or dLN after 72 hr i.v. injection were cocultured with B16-hHER2 for 24 hr. Dead B16 cells were stained with 7-amino-actinomycin D (7-AAD). The data reflect 3 independent experiments (the mean ± SEM, n = 3). The data were analyzed by two-tailed Student’s t test (*P = 0.002). I. Representative flow cytometry results showing cell surface PD-1 and Tim-3 expression levels in naïve and CAR-T cells isolated from the lung at the indicated times post-injection.
Extended Data Figure 3. In vivo T cell CRISPR screen.

A. Volcano plot shows comparison of differentially expressed genes in naïve vs. in vitro activated CD8 T cells. Red denotes genes increased and blue denotes genes decreased in T cells. P values were adjusted using the Benjamini–Hochberg method. B. Heatmap of RNA-sequencing data from naïve vs. in vitro activated CD8 T cell samples (n=3). Heatmap represents top 1,316 genes chosen from highest 0.25% p-value summary. C. Retroviral construct used for sgRNA delivery. D. A schematic of the experimental design showing timeline of T cell stimulation, transduction, enrichment, and flow analysis. T cells were purified by FACS sorting at day 7 and i.v injected into recipient tumor bearing mice. E. Scatter plot of the enrichment of candidates (n = 5 sgRNAs per gene) showing that sgRNAs targeting essential genes (left) were significantly reduced in the samples after two representative in vivo screens, but not the non-targeting guides (right). F. Genes with positive beta score (Tmigratory) and negative beta score (TSequestered) ( >2 standard deviations from the mean (red)) are highlighted in yellow and blue, respectively.
Extended Data Figure 4. Identification of CD18.

A. The spectrum of one of the CD18 peptides obtained by nanospray-ion trap tandem mass spectrometry. B. The deconvoluted MS/MS spectrum of tryptic peptide NVTR with 3 HexNAc. M: the parent ion of NVTR-HexNAc(3) at m/z 549.760 Da. M-18: loss one H2O in NVTR-HexNA(3). a2: the observed m/z 186.1218 Da matched to the a2 ion. y1: the observed m/z 175.1261 Da matched to the y1 ion. C. The crystal structure of LFA-1 headpiece (5E6U.pbd). The region encircled by dots from the left view is enlarged (right), representing the O-glycosylation on Thr256 in βI-domain.
Extended Data Figure 5. Activated T cell migration in vitro.

A. Recruitment of CAR-T cells into dLN was blocked by LFA-1-blocking Ab and PTx treatments. Percentages of total transferred CD8+ T cells are shown. B & C. Activated CD8+ T cell migration on ICAM-1-coated plates ± CXCL12 (B) or CXCL10 (C) with varying concentrations as indicated. Data were collected from 3 independent experiments (n = 3, 11–20 individual cells per mouse). Data represent mean ± SEM. Statistical analyses were performed using one-way ANOVA with Bonferroni post-test. D. Representative Transwell assay of in vitro activated CD8+ T cell migration in response CXCL12 (2 μg/ml). Data were collected from 3 independent experiments (1 × 106 cells/well). Data represent mean ± SEM. Statistical analyses were performed using two-sided, unpaired Student’s t test. E. Schematic depicting the flow chamber assay. F & G. Activated CD8+ T cell migration on ICAM-1 coated plates with flow. Cells were treated with PTx (6 h) (F) and/or gallein (30 min) (G) ± CXCL12 (G) where indicated. Cells were allowed to settle for 10 minutes prior to 1 dyne/cm2 of fresh media being run across the surface of the plate. Data were collected from 3 independent experiments (n = 3, 25–45 individual cells per mouse). Data represent mean ± SEM. Statistical analyses were performed using one-way ANOVA with Bonferroni post-test. H. Activated CD8+ T cell migration on bEND.3 coated plates. bEND.3 cells were allowed to grow to a monolayer for at least 12 h prior to imaging. CD8+ were treated with PTx (6 h) where indicated. T cells were allowed to settle for 10 minutes prior to 1 dyne/cm2 of fresh media being run across the surface of the plate. Data were collected from 3 independent experiments (n = 3, 20–39 individual cells per mouse). Data represent mean ± SEM. Statistical analyses were performed using two-sided, unpaired Student’s t test.
Extended Data Figure 6. TIRFM measurements of LFA-1 distribution.

A. Schematic of TIRFM measurements. Molecules near the surface fluoresce more brightly than molecules between microvilli and those farther from the substrate. B. Human CD8 T cells labeled for LFA-1 spreading on glass, BSA, or ICAM-1 coated substrate. Contrast and brightness have been adjusted for visibility, but the original gray values are indicated in the scale bars to the right of each image. All images in the same row are at the same magnification. Scale bars, 5 μm. Representative images from > five independent experiments are shown.
Extended Data Figure 7. βII-spectrin expression in T cells.

A. Generation of GFP-ROSAβII-spectrin mouse model (Rosa26tm(CAG-LSL-Sptbn1-IRES-GFP)). Mouse Sptbn1 cDNA was inserted into the CAG-STOP-GFP-Rosa targeting vector, CTV, between a floxed Stop cassette and the internal ribosome entry site (IRES) followed by the enhanced Green Fluorescent Protein gene (eGFP). Transcription is under control of the CAG promoter. The targeting vector contained Rosa26 homology arms (1 kb 5′ and 3.8 kb 3′), so that the entire loxP-stop-loxP-Tmc2-IRES-GFP transcriptional cassette was inserted into the first intron of Rosa26 gene on chromosome 6. B & C. Flow cytometry analysis of IFNγ and TNFα expression in hHER2-CAR transfected T cells from GFP-ROSAβII-spectrin mouse (TβII-spectrin) after treated with PBS or Tat-Cre Recombinase. Cells were co-cultured with B16-HER2 cells. Data represent mean ± SEM. n = 9. D. CAR-mediated B16-HER2 cell killing assay with TβII-spectrin cells after treated with PBS or Tat-Cre Recombinase. Cell death was stained for NucSpot. Data represent mean ± SEM. n = 9. E. Activated CD4+ T cell (OT-II) migration on ICAM-1 coated plates ± CXCL12. Cells were treated with PTx (6 hr) or Gallein (30 min) where indicated. Data were collected from 2 independent experiments (n = 2, 17–34 individual cells per mouse). Data represent mean ± SEM Statistical analyses were performed using one-way ANOVA with Bonferroni post-test. *P = 0.007. F. The pie charts depict the proportion of CD4 T cells distributed in the tumour, blood, LN/spleen, or lung/liver 72 h post-injection. G. Expression levels of βII-spectrin and St3gal1 in human CD4 and CD8 T cells (before and after activation). Loading control: β-actin. Representative western blot images from three independent experiments are shown. H. Expression levels of βII-spectrin in human CD8 memory T cells (CD8+CD45RO+CD45RA–CD56–CD57–). Loading control: β-actin. Representative western blot images from three independent experiments are shown.
Supplementary Material
Movie S1.
In vitro CAR T cell killing assay. WT CD8 T cells or hHER2 CAR T cells were coincubated with B16-hHER2 cells (green) in the presence of propidium iodide (red). Scale bar = 20 μm.
Movie S2.
In vivo CAR T cell migration. hHER2 CAR T cells (green) was i.v. injected by the tail vein of the B12-hHER2 bearing mouse, and intravital imaging was simultaneously performed to visualize T cells in the lung, lymph node, and tumour. The blood vessels were labeled with anti-CD31 Ab (red). Time is marked as min:sec. Scale bar = 50 μm (Part 1, 3, and 4), 20 μm (Part 2).
Movie S3.
CAR T cell accumulation. hHER2 CAR T cells (green) was i.v. injected by the tail vein of the B12-hHER2 bearing mouse, and intravital imaging was performed 48 h and 72 j post-infusion to visualize T cells in the lung. The blood vessels were labeled with anti-CD31 Ab (red). Time is marked as min:sec. Scale bar = 50 μm
Movie S4.
CAR T cell accumulation. Deep 3D imaging (1-mm thick) of CAR-T cell accumulation in a cleared mouse lung 72 hr after i.v. injection (red, blood vessels [CD31], green; CAR-T cells [GFP]).
Movie S5.
In vitro CAR T cell migration to tumor spheroid. Long-term time-lapse imaging (9 hr) of hHER2 CAR T cells (red) embedded in 3D Matrigel around hHER2-B16 (blue) spheroid +/− PTX pre-treatment. Scale bars, 100 μm. Time in hr:min.
Movie S6.
In vitro CD8 T cell migration. Migration of naïve and activated (day 5) mouse CD8+ T cells on ICAM-1 ± CXCL12 coated plates. Time is marked as min:sec. Scale bar = 50 μm
Movie S7.
In vitro CD8 T cell migration. Migration of activated (day 5) mouse CD8+ T cells on ICAM-1 coated plates with (1 dyne/cm2) and without (0 dyne/cm2) flow. Scale bar = 50 μm
Movie S8.
In vitro CD8 T cell migration with βII spectrin expression. Activated CD8 T cell were transfected with GFP only (Mock) or βII-spectrin cDNA, and migrated on ICAM-1 ± CXCL12 coated plates. Time is marked as min:sec. Scale bar = 20 μm
Movie S9.
LFA-1 mediated CD8 T cell spreading. TIRFM images of activated CD11a-mYFP CD8+ T cell adhesion on ICAM-1± CXCL12 -coated plates with and without βII spectrin expression. LFA-1 intensity (YFP) is shown on a pseudocolor scale (from low [blue] to high [red]). Each frame is 15 sec. Scale bar = 10 μm
Table 1.
Mass spectrometry data
Table 2.
LC–MS/MS analysis data
Table 3.
CD19 CAR-T cell patient information
Acknowledgments
We thank the High Content Image Core (University of Rochester) for help with fluorescent imaging and use of the Dragonfly spinning disc Confocal. We thank Yurong Gao and Philip Rock for their technical assistance on the manuscript. We especially thank Dr. Moonsoo Jin (Weill Cornell) for the anti-HER2 CAR DNA construct and for his help with micro-PET/CT image, Dr. Lin Gan (Augusta University) for help with βII-spectrin transgenic mouse generation, Dr. Richard Burack and Dr. Jozal Moore for patient sample collections and data review, Drs John Ashton, Dalia Ghoneim, and Jeffrey Malik for help with CRISPR transcriptomic analyses and depositing RNA-sequencing data, and all members of the Kim Laboratory for their comments during the course of these studies and input during preparation of the manuscript. We obtained hHER2 tg mice from Genentech.
Funding:
National Institutes of Health grant AI102851 (MK & PWO), National Institutes of Health grant AI147362 (MK & REW), National Institutes of Health grant T32AI007285 (AMA & CJS), National Science and Technology Council in Taiwan MOST 111–2314-B-182A-069 & MOST 111–2321-B-182–002 (TCF), and K-GRC GO! KRICT project BSF23–113 (KDK)
Footnotes
Competing interests
The authors declare no competing financial interests.
Data availability
All data analyzed during the present study are included in this article and its supporting information files. Source data are provided in this paper. PDB accession code is 5E6U. Signaling pathway analysis was performed with Enrichr using NCI-Nature 2016 database for pathway enrichment analysis (http://amp.pharm.mssm.edu/Enrichr). Sequencing datasets have been deposited in the Gene Expression Omnibus database under accession codes GSE227429 (CRISPR screen), GSE227509 (CD8 T cell bulk RNA-seq) and GSE227430 (CAR-T cell bulk RNA-seq) and are publicly available.
References
- 1.Aghajanian H, Rurik JG & Epstein JA CAR-based therapies: opportunities for immuno-medicine beyond cancer. Nat Metab 4, 163–169 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lizee G. et al. Harnessing the power of the immune system to target cancer. Annual review of medicine 64, 71–90 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Bobisse S. et al. Reprogramming T lymphocytes for melanoma adoptive immunotherapy by T-cell receptor gene transfer with lentiviral vectors. Cancer research 69, 9385–9394 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Junghans RP The challenges of solid tumor for designer CAR-T therapies: a 25-year perspective. Cancer gene therapy 24, 89–99 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Hernandez-Lopez A, Tellez-Gonzalez MA, Mondragon-Teran P. & Meneses-Acosta A. Chimeric Antigen Receptor-T Cells: A Pharmaceutical Scope. Front Pharmacol 12, 720692 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fowell DJ & Kim M. The spatio-temporal control of effector T cell migration. Nature reviews 21, 582–596 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mueller KT et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 130, 2317–2325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee DW et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan RA et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18, 843–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finkle D. et al. HER2-targeted therapy reduces incidence and progression of midlife mammary tumors in female murine mammary tumor virus huHER2-transgenic mice. Clin Cancer Res 10, 2499–2511 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Amitrano AM et al. Optical Control of CD8(+) T Cell Metabolism and Effector Functions. Front Immunol 12, 666231 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paley MA et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.You R. et al. Active surveillance characterizes human intratumoral T cell exhaustion. The Journal of clinical investigation 131 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donnadieu E, Dupre L, Pinho LG & Cotta-de-Almeida V. Surmounting the obstacles that impede effective CAR T cell trafficking to solid tumors. Journal of leukocyte biology 108, 1067–1079 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Platt RJ et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmermann M. et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hobbs SJ & Nolz JC Regulation of T Cell Trafficking by Enzymatic Synthesis of O-Glycans. Front Immunol 8, 600 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pietrobono S. et al. ST3GAL1 is a target of the SOX2-GLI1 transcriptional complex and promotes melanoma metastasis through AXL. Nat Commun 11, 5865 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Priatel JJ et al. The ST3Gal-I sialyltransferase controls CD8+ T lymphocyte homeostasis by modulating O-glycan biosynthesis. Immunity 12, 273–283 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Giovannone N. et al. Human B Cell Differentiation Is Characterized by Progressive Remodeling of O-Linked Glycans. Front Immunol 9, 2857 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen S. et al. Increased SPC24 in prostatic diseases and diagnostic value of SPC24 and its interacting partners in prostate cancer. Exp Ther Med 22, 923 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geisler C. & Jarvis DL Effective glycoanalysis with Maackia amurensis lectins requires a clear understanding of their binding specificities. Glycobiology 21, 988–993 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sen M. & Springer TA Leukocyte integrin alphaLbeta2 headpiece structures: The alphaI domain, the pocket for the internal ligand, and concerted movements of its loops. Proceedings of the National Academy of Sciences of the United States of America 113, 2940–2945 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walling BL & Kim M. LFA-1 in T Cell Migration and Differentiation. Front Immunol 9, 952 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galkina E. et al. Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. The Journal of clinical investigation 115, 3473–3483 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walch JM et al. Cognate antigen directs CD8+ T cell migration to vascularized transplants. The Journal of clinical investigation 123, 2663–2671 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shulman Z. et al. Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nature immunology 13, 67–76 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Cyster JG & Goodnow CC Pertussis toxin inhibits migration of B and T lymphocytes into splenic white pulp cords. J. Exp. Med 182, 581–586 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Surve CR, Lehmann D. & Smrcka AV A chemical biology approach demonstrates G protein betagamma subunits are sufficient to mediate directional neutrophil chemotaxis. The Journal of biological chemistry 289, 17791–17801 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim M, Carman CV & Springer TA Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301, 1720–1725 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Kim M, Carman CV, Yang W, Salas A. & Springer TA The primacy of affinity over clustering in regulation of adhesiveness of the integrin {alpha}L{beta}2. The Journal of cell biology 167, 1241–1253 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng Y. et al. Exo1: a new chemical inhibitor of the exocytic pathway. Proceedings of the National Academy of Sciences of the United States of America 100, 6469–6474 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishikimi A. et al. Rab13 acts downstream of the kinase Mst1 to deliver the integrin LFA-1 to the cell surface for lymphocyte trafficking. Sci Signal 7, ra72 (2014). [DOI] [PubMed] [Google Scholar]
- 34.De Franceschi N, Hamidi H, Alanko J, Sahgal P. & Ivaska J. Integrin traffic - the update. Journal of cell science 128, 839–852 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meissner JM et al. alphaII-spectrin in T cells is involved in the regulation of cell-cell contact leading to immunological synapse formation? PLoS One 12, e0189545 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Capece T. et al. A novel intracellular pool of LFA-1 is critical for asymmetric CD8+ T cell activation and differentiation. The Journal of cell biology 216, 3817–3829 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomsen P, Roepstorff K, Stahlhut M. & van Deurs B. Caveolae are highly immobile plasma membrane microdomains, which are not involved in constitutive endocytic trafficking. Molecular biology of the cell 13, 238–250 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fritzsche M. & Charras G. Dissecting protein reaction dynamics in living cells by fluorescence recovery after photobleaching. Nature protocols 10, 660–680 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Lingel H. et al. CTLA-4-mediated posttranslational modifications direct cytotoxic T-lymphocyte differentiation. Cell Death Differ 24, 1739–1749 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park JH et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. The New England journal of medicine 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heslop HE Safer CARS. Mol Ther 18, 661–662 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beaty BT et al. Talin regulates moesin-NHE-1 recruitment to invadopodia and promotes mammary tumor metastasis. The Journal of cell biology 205, 737–751 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Medina E. et al. Crumbs interacts with moesin and beta(Heavy)-spectrin in the apical membrane skeleton of Drosophila. The Journal of cell biology 158, 941–951 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gossez M. et al. Evaluation of a novel automated volumetric flow cytometer for absolute CD4+ T lymphocyte quantitation. Cytometry B Clin Cytom 92, 456–464 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Potrzebowska K, Lehtonen J, Samuelsson M. & Svensson L. Flow Cytometry Assay for Recycling of LFA-1 in T-lymphocytes. Bio Protoc 8, e3104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morin NA et al. Nonmuscle myosin heavy chain IIA mediates integrin LFA-1 de-adhesion during T lymphocyte migration. The Journal of experimental medicine 205, 195–205 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bern M, Kil YJ & Becker C. Byonic: advanced peptide and protein identification software. Curr Protoc Bioinformatics Chapter 13, 13 20 11–13 20 14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim K. et al. In situ neutrophil efferocytosis shapes T cell immunity to influenza infection. Nature immunology 21, 1046–1057 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Looney MR et al. Stabilized imaging of immune surveillance in the mouse lung. Nature methods 8, 91–96 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Susaki EA et al. Advanced CUBIC protocols for whole-brain and whole-body clearing and imaging. Nature protocols 10, 1709–1727 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Shifrut E. et al. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971 e1915 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li W. et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li W. et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol 16, 281 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1.
In vitro CAR T cell killing assay. WT CD8 T cells or hHER2 CAR T cells were coincubated with B16-hHER2 cells (green) in the presence of propidium iodide (red). Scale bar = 20 μm.
Movie S2.
In vivo CAR T cell migration. hHER2 CAR T cells (green) was i.v. injected by the tail vein of the B12-hHER2 bearing mouse, and intravital imaging was simultaneously performed to visualize T cells in the lung, lymph node, and tumour. The blood vessels were labeled with anti-CD31 Ab (red). Time is marked as min:sec. Scale bar = 50 μm (Part 1, 3, and 4), 20 μm (Part 2).
Movie S3.
CAR T cell accumulation. hHER2 CAR T cells (green) was i.v. injected by the tail vein of the B12-hHER2 bearing mouse, and intravital imaging was performed 48 h and 72 j post-infusion to visualize T cells in the lung. The blood vessels were labeled with anti-CD31 Ab (red). Time is marked as min:sec. Scale bar = 50 μm
Movie S4.
CAR T cell accumulation. Deep 3D imaging (1-mm thick) of CAR-T cell accumulation in a cleared mouse lung 72 hr after i.v. injection (red, blood vessels [CD31], green; CAR-T cells [GFP]).
Movie S5.
In vitro CAR T cell migration to tumor spheroid. Long-term time-lapse imaging (9 hr) of hHER2 CAR T cells (red) embedded in 3D Matrigel around hHER2-B16 (blue) spheroid +/− PTX pre-treatment. Scale bars, 100 μm. Time in hr:min.
Movie S6.
In vitro CD8 T cell migration. Migration of naïve and activated (day 5) mouse CD8+ T cells on ICAM-1 ± CXCL12 coated plates. Time is marked as min:sec. Scale bar = 50 μm
Movie S7.
In vitro CD8 T cell migration. Migration of activated (day 5) mouse CD8+ T cells on ICAM-1 coated plates with (1 dyne/cm2) and without (0 dyne/cm2) flow. Scale bar = 50 μm
Movie S8.
In vitro CD8 T cell migration with βII spectrin expression. Activated CD8 T cell were transfected with GFP only (Mock) or βII-spectrin cDNA, and migrated on ICAM-1 ± CXCL12 coated plates. Time is marked as min:sec. Scale bar = 20 μm
Movie S9.
LFA-1 mediated CD8 T cell spreading. TIRFM images of activated CD11a-mYFP CD8+ T cell adhesion on ICAM-1± CXCL12 -coated plates with and without βII spectrin expression. LFA-1 intensity (YFP) is shown on a pseudocolor scale (from low [blue] to high [red]). Each frame is 15 sec. Scale bar = 10 μm
Data Availability Statement
All data analyzed during the present study are included in this article and its supporting information files. Source data are provided in this paper. PDB accession code is 5E6U. Signaling pathway analysis was performed with Enrichr using NCI-Nature 2016 database for pathway enrichment analysis (http://amp.pharm.mssm.edu/Enrichr). Sequencing datasets have been deposited in the Gene Expression Omnibus database under accession codes GSE227429 (CRISPR screen), GSE227509 (CD8 T cell bulk RNA-seq) and GSE227430 (CAR-T cell bulk RNA-seq) and are publicly available.