

Abstract

This research describes the preparation of mixtures of new halogen-substituted phenol derivatives and their effects due to linkages with a fatty amide (pentanamide). The molecules were optimized using DFT, and the vibrational and electronic analysis was done subsequently. The energies of frontier molecular orbitals (FMOs) were used to estimate the global chemical reactivity parameters as we suggest that hydrogen-bonded networks may have contributed to the stability and reactivity of the compound. In addition to the experimental investigation, dielectric parameters were calculated. Fukui functions were analyzed to study the chemical reactivity. To get insight into interactions of σ → π* orbitals, natural bond orbital calculations were done. Additionally, surface analysis of the MEP and Hirshfeld charges were performed at the equivalent DFT levels. The research also indicated that both (interaction region indicator) IRI and (electron delocalize range) EDR would proficiently identify chemical-bonding and weak interaction regions, providing a significant advantage in exploring diverse chemical systems and reactions. This indicated that compounds could diffuse through noncovalent interactions, including intramolecular hydrogen bonding. Dielectric relaxation studies taken at five distinct molar ratios identified significant dielectric properties such as ε′, ε″, ε0, and ε∞. The PA with FP, CP, BP, and IP molecules has potential antiviral and antioxidant benefits for carbonic anhydrase, with favorable drug-like features and diverse biological benefits. Pharmacological effects were forecasted using the PASS server, and these molecules exhibited favorable pharmacokinetic properties.
1. Introduction
Novel halogen-substituted phenol derivatives (interaction between the hydrogen atom of the hydroxyl group (donor)) and pentanamide (oxygen atom of the carbonyl group (acceptor)) linkage are essential for novel drug development. The high interest in halogen-substituted phenols and pentanamides stems from their distinctive physical, chemical, and medicinal characteristics.1,2 Pentanamide, with the structure RCONH2, has applications in biologically active fields as an antiviral and antioxidant.
4-Fluorophenol is a water-soluble, colorless solid, and a polar solvent.3 4-Chlorophenol is manufactured as a predecessor to hydroquinone. It is a well-known precursor of quinizarin when reacting with phthalic anhydride.4 Quinizarin, a commercial dye, reacts with phthalic anhydride and then hydrolyzes the chloride. 4-Bromophenol is any organobromide of phenol that contains one or more covalently bonded bromine atoms. It plays various roles, including those of a mouse metabolite, a natural pollutant that persists over time, a human xenobiotic metabolite, a rat metabolite, a human urine metabolite, and a marine metabolite. The natural substance 4-bromophenol is found in Euglena gracilis and Ulva lactuca.(5) 4-Iodophenol is a mono-iodophenol and a colorless solid. Numerous coupling processes take place on 4-iodophenol.6 Additionally, it is used to enhance chemiluminescence for the detection of cancer cells and in the Eclox assay.7 The diazonium salt can be used to make 4-iodophenol from 4-aminophenol. A different production method includes decarboxylation after the salicylic acid and iodine reactions.
In investigations of biological systems, density functional theory (DFT) is a suitable and often employed method. Using DFT to predict a molecule’s geometry, electrical structure, and spectroscopic characteristics helps determine organic polar molecules’ structural behavior in a nonpolar solvent. The features of the PA with FP, CP, BP, and IP compounds were theoretically estimated and characterized using several local and global reactivity descriptors and thermodynamical parameters. The geometry obtained from DFT optimization and the molecular docking studies corresponds well with the NBO findings that support the existence of a hydrogen-bonding network.8 Using the TD-DFT method and IEFPCM (integral equation formalism polarizable continuum model), a comprehensive spectral analysis was done, which provided excited-state information.9 According to NBO calculations, the compound’s stability is due to the hyperconjugative interactions and hydrogen-bonding structure. To calculate the local reactivity of inhibitor compounds, condensed Fukui functions were utilized.10 The molecular electrostatic potential surface and Hirshfeld surface contour map were created to understand the PA with FP, CP, BP, and IP compound properties better. The interaction region indicator (IRI), a minor change of the RDG, is a unique real-space function that we propose. We observed that IRI could efficiently depict chemical bonding and weak interaction zones, which is particularly valuable in studying various chemical systems and events. IRI differs from previous algorithms because it has a simpler definition, a lower computing cost, and a higher graphical effect. Furthermore, chemical reactivity and biological activity were compared using global and local reactivity parameters.
The investigation of inter- and intramolecular interactions of molecules and the fluctuation of these parameters with temperature provided information on the physical behavior of molecules. Various studies11−13 investigated the connection of two polar molecules caused by hydrogen bonding using dielectric relaxation studies at microwave frequencies. These approaches have grown considerably more dependable in predicting the characteristics of molecules with high accuracy in recent times.
The goal of the study is to extensively use the DFT method to investigate the structure and electrical characteristics of a newly prepared ternary mixture halogen-substituted phenol derivative linkage with pentanamide. Further, analyses using natural bonding orbitals (NBOs), molecular electrostatic potential (MEP), Fukui index, Hirshfeld surface, interaction region indicator (IRI), and electron delocalize range (EDR) were carried out to anticipate any intramolecular charge transfer and redistributing electron energies in different orbitals. Due to its extreme sensitivity to structural changes, vibrational spectroscopy might provide essential details about the molecular potential for biological activity. Molecular docking analysis was done on the molecule and two control medicines, ampicillin and against protein receptors. Docking was used to keep ligand complexes rigid by changing all rotatable bonds to nonrotatable bonds. Lipophilicity, druggability, target identification, and pharmacokinetics are among the analyses performed, and they are based on previously recognized ADMET characteristics.14 The biological activity of the PA with FP, CP, BP, and IP compounds was predicted for the effect of the combination against different biological assays, which was employed using PASS.15 To present the experimental results, a microwave region (9.34 GHz) at 298 K was used to study the dielectric behavior of PA+FP, CP, BP, and IP with benzene at different molar ratios.16
2. Materials and Methods
2.1. Materials
The BDH Analar variety of 4-fluorophenol (FP), 4-chlorophenol (CP), 4-bromophenol (BP), and 4-iodophenol (IP) were distilled and then purified by recrystallization from diluted ethanol. Pentanamide was produced by condensing valeric acid and ammonia. The E. Merck variety of pentanamide (PA) was utilized without additional purification.
2.2. Methods
2.2.1. (a) Quantum Chemical (QC) Calculations
The compound’s entire geometry is analyzed from Chemdraw. Gaussian 16W was used to perform all quantum chemical calculations. The input files were generated using Chemdraw, and the output data was visualized using Gauss View 6.017,18
It is essential to utilize the provided equations precisely to obtain quantum chemical reactivity parameters. Density functional theory (DFT) calculations using B3LYP utilizing the LANL2DZ basis set were used to accomplish the whole compound optimization as shown in Figure 1, followed by analyses of the NBO,19 Fukui index, FMO, MEP, and vibrational frequencies.20−23 The band energy gap information was obtained from the highest occupied and lowest unoccupied molecular orbital representations (HOMO–LUMO). Global reactivity descriptors and thermodynamical parameters for different temperatures were computed and reported to expose the qualities of the PA with FP, CP, BP, and IP compounds. The methods employed here were DFT/B3LYP24 at the basis set of 6-311++G(d,p) and LANL2DZ. The complete vibrational assignments are made with the potential energy distribution (PED) calculation using VEDA425 software. Also, UV–vis spectra and electronic properties such as frontier molecular orbital analysis (HOMO–LUMO) were computed with the help of the time-dependent DFT (TD-DFT) method.
Figure 1.

Optimized geometrical structure of PA with (FP, CP, BP, and IP) compounds.
2.2.2. (b) Noncovalent Interaction (NCI) Analysis
A multidimensional wave function analysis tool, called Multiwfn,26,27 was used to perform IRI and EDR, while the VMD program was used to generate all isosurface maps. The Hirshfeld surface analysis of the experimental structure was performed using the Crystal Explorer 3.1 tool.28
2.2.3. (c) Docking Simulation
The optimized molecule was utilized to compare chemical reactivity and biological activity, and Autodock suite 4.2.1 was used to conduct molecular docking investigations. A detailed examination of the residues implicated in noncovalent interactions between the ligands and protein was carried out using Accelyrs Discovery Studio 4.129
2.2.4. (d) Dielectric Measurements
By combining PA with various molar ratios of FP, CP, BP, and IP in benzene (1:3, 1:2, 1:1, 2:1, and 3:1), a ternary mixture was proposed in a 10 mL solution (benzene). At a microwave frequency in the X-band of 9.34 GHz, the dielectric constant at an angular frequency (ε′) and the dielectric loss (ε″) were measured. With a dipole meter operating at 220 V and a WTW dipole meter DM 01 type, the static dielectric constant (ε0) was determined at 298 K.30 The derived values of ε′ and ε″ are accurate to within ±1 and ±5%, respectively. With Abbe’s refractometer, the refractive index was calculated.31 The measurement error for the refractive index is +0.003. Using a water-circulating thermostat, the temperature of all of these measurements was kept at 30°C. Thermostatically, the temperature was maintained within ±0.5°C. The density was determined using a 20 cm3 specific gravity container.
3. Results and Discussion
3.1. Structure Optimization
The centrosymmetric triclinic system with a P-1 space group was the crystallized structure of the chemical described in the PA with FP, CP, BP, and IP compounds;32 the unit cell parameters are a, b, c = 15.94Å and α, β, γ = 90°. This unit formula consisted of two halogen-substituted phenol derivative linkages with pentanamide (C11H17XN), where X is F, Cl, Br, I, and one benzene molecule. A relation of N–H/O–H···O hydrogen bonds connected these aspects. The parameters of molecular bonds are summarized here. According to the data presented in Table 1, using the DFT technique leads to the prediction of the NH (PA) - OC (FP) bond lengths to be around 1.40 Å. This finding implies a strong interaction within the PA with FP, CP, BP, and IP compounds.
Table 1. Optimized Geometrical Parameters for PA with FP, CP, BP, and IP Compound B3LYP/LANL2DZ Basis Set.
| symbol | bond (Å) [FP] | angle (°) | bond (Å) [CP] | angle(°) | bond (Å) [BP] | angle (°) | bond (Å) [IP] | angle (°) |
|---|---|---|---|---|---|---|---|---|
| C1 | ||||||||
| C2 | 2.565 | 2.565 | 2.565 | 2.564 | ||||
| C3 | 1.544 | 34.015 | 1.544 | 34.017 | 1.544 | 34.017 | 1.544 | 34.018 |
| N4 | 2.471 | 127.452 | 2.471 | 127.356 | 2.471 | 127.782 | 2.471 | 128.027 |
| O5 | 2.298 | 61.409 | 2.298 | 61.403 | 2.298 | 61.409 | 2.298 | 61.414 |
| C6 | 1.543 | 146.472 | 1.543 | 146.471 | 1.543 | 146.481 | 1.543 | 146.487 |
| C7 | 1.278 | 30.564 | 1.278 | 30.565 | 1.278 | 30.567 | 1.278 | 30.567 |
| H8 | 1.097 | 109.945 | 1.097 | 109.944 | 1.097 | 109.943 | 1.097 | 109.945 |
| H9 | 1.100 | 109.515 | 1.100 | 109.512 | 1.100 | 109.522 | 1.100 | 109.528 |
| H10 | 1.101 | 89.761 | 1.101 | 89.791 | 1.101 | 89.797 | 1.101 | 89.792 |
| H11 | 1.101 | 89.800 | 1.101 | 89.767 | 1.101 | 89.761 | 1.101 | 89.766 |
| H12 | 1.098 | 110.352 | 1.098 | 110.356 | 1.098 | 110.343 | 1.098 | 110.333 |
| H13 | 1.099 | 107.351 | 1.099 | 107.355 | 1.099 | 107.319 | 1.099 | 107.300 |
| H14 | 1.099 | 111.020 | 1.099 | 111.026 | 1.099 | 111.034 | 1.099 | 111.034 |
| H15 | 1.098 | 110.955 | 1.098 | 110.952 | 1.098 | 110.950 | 1.098 | 110.949 |
| H16 | 1.098 | 111.243 | 1.098 | 111.240 | 1.098 | 111.239 | 1.098 | 111.239 |
| H17 | 1.011 | 88.484 | 1.011 | 88.496 | 1.011 | 88.492 | 1.011 | 88.486 |
| H18 | 1.019 | 152.299 | 1.019 | 152.332 | 1.019 | 152.334 | 1.019 | 152.324 |
| H19 | 1.615 | 82.660 | 1.608 | 82.829 | 1.609 | 82.846 | 1.611 | 82.824 |
| H20 | 7.006 | 86.730 | 6.999 | 87.117 | 6.997 | 87.133 | 6.993 | 86.841 |
| H21 | 5.671 | 121.250 | 5.672 | 122.586 | 5.663 | 122.377 | 5.655 | 121.723 |
| H22 | 4.986 | 143.089 | 4.987 | 142.962 | 4.996 | 142.873 | 4.997 | 142.937 |
| H23 | 3.166 | 123.881 | 3.186 | 125.149 | 3.184 | 124.830 | 3.181 | 124.178 |
| C24 | 6.099 | 102.516 | 6.120 | 103.395 | 6.127 | 103.348 | 6.135 | 102.929 |
| C25 | 1.399 | 80.181 | 1.403 | 79.429 | 1.405 | 79.156 | 1.408 | 78.802 |
| C26 | 1.397 | 42.678 | 1.401 | 42.434 | 1.404 | 42.093 | 1.407 | 41.707 |
| C27 | 1.406 | 118.455 | 1.406 | 119.008 | 1.406 | 119.336 | 1.406 | 119.755 |
| C28 | 1.407 | 118.510 | 1.407 | 119.063 | 1.407 | 119.388 | 1.407 | 119.802 |
| C29 | 1.412 | 120.170 | 1.411 | 120.194 | 1.411 | 120.198 | 1.411 | 120.188 |
| O30 | 1.386 | 117.877 | 1.384 | 117.971 | 1.384 | 118.016 | 1.384 | 118.053 |
| X31 | 1.413 | 161.043 | 1.832 | 161.316 | 1.978 | 161.311 | 2.148 | 161.291 |
3.2. Quantum Chemical Analysis
The impact of the solvent environment on the electronic structure and energetics of the molecule is duly considered in the IEPCM. The IEPCM accurately accounts for the interaction between the solute and solvent molecules in the presence of a solvent. Calculations were performed in the presence of the solvent to analyze the energetic behavior.33 The calculated energy values for PA+FP, HOMO, and LUMO in the solvent were −6.065 and −0.511 eV. The energies and the pictorial illustration of HOMO, LUMO, and NBO are shown in Figures 2 and 3. The presence of red regions in the molecular orbitals indicates nucleophilic sites, which attract electrons. Green regions indicate electrophilic sites, which donate electrons. This color scheme helps identify the molecule’s reactivity and potential for chemical interactions, clearly representing its electronic properties.
Figure 2.

HOMO and LUMO surface maps of PA with FP, CP, BP, and IP compounds.
Figure 3.

Charge-transfer interactions by NBO studies of the PA with FP, CP, BP, and IP compounds.
Accordingly, the energy gap of HOMO–LUMO explains the eventual charge-transfer interaction within the mixture.34 Furthermore, in going from the FP to the BP, the increasing value of the energy gap suggests that the molecule is becoming more stable. Furthermore, a molecule with a small frontier orbital gap is easily polarizable and has strong chemical reactivity but low kinetic stability.35−37 Additionally, global chemical reactivity descriptors (GCRDs) are an important tool for understanding the chemical characteristics of a compound such as η, μ, S, χ, and ω. The GCRDs are obtained from HOMO–LUMO energies, under Koopman′s approximation HOMO as ionization potential (I) and LUMO as electron affinity (A), and the data are tabulated in Table 2. These characteristics are connected in the following equations:38 using the preceding equations, the chemical potential, hardness, and electrophilicity index have been determined, and their results were compared with two different basis sets, as displayed in Table 2 (Table S1). The ionization potential value indicates that an energy of 6.065 eV in the solvent phase is required to remove an electron from the HOMO. The lower value of electron affinity shows higher molecular reactivity with the nucleophiles. Higher hardness and lower softness values confirm the higher molecular hardness associated with the PA with FP, CP, BP, and IP compounds. The electrophilicity index helps in describing the biological activity of the mixture. Figure 4 shows the Mulliken charge distribution on 4N, 6C, and 30O atoms of PA with FP, CP, BP, and IP compounds and O atoms of the proton–acceptor molecule. Because of the existence of the N•••H-bond and O•••H-bond, the title chemical is a typical model of the class of molecules that form intermolecular H-bonds (Table 3).
Table 2. Calculated Energy Values of PA with FP, CP, BP, and IP Compounds in Benzene Solvent at 298 K in Its Ground States with Singlet Symmetry, First-Order Hyperpolarizability (β), Polarizability (α), and Dipole Moment at the DFT/LANL2DZ Method.
| LANL2DZ IEFPCM, solvent: benzene parameters | PA + FP | PA + CP | PA + BP | PA + IP |
|---|---|---|---|---|
| EHOMO | –6.051 | –6.065 | –6.008 | –5.930 |
| ELUMO | –0.546 | –0.541 | –0.511 | –0.830 |
| ionization potential | 6.051 | 6.065 | 6.008 | 5.930 |
| electron affinity | 0.546 | 0.541 | 0.511 | 0.830 |
| EHOMO – ELUMO | 5.505 | 5.523 | 5.497 | 5.100 |
| electron negativity (χ) | 3.298 | 3.303 | 3.260 | 3.380 |
chemical
potential (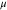 ) ) |
–3.298 | –3.303 | –3.260 | –3.380 |
absolute hardness ( ) ) |
2.752 | 2.762 | 2.749 | 2.550 |
| softness (S) | 0.182 | 0.181 | 0.182 | 0.196 |
| electrophilicity index (ω) | 1.976 | 1.975 | 1.933 | 2.240 |
| electronic charges (ΔNmax) | –0.600 | –0.598 | –0.593 | –0.663 |
| E(RB3LYP) (a.u) | –733.813 | –648.906 | –647.124 | –645.344 |
| dipole moment (Debye) | 8.082 | 8.390 | 8.146 | 7.781 |
| polarizability | 141.494 | 152.201 | 159.243 | 166.726 |
| hyperpolarizability | 290.374 | 183.069 | 224.850 | 325.979 |
| entropy (S) (cal/mol/K) | 139.218 | 144.317 | 152.384 | 146.783 |
| heat capacity (Cv) (cal/mol/K) | 58.746 | 59.600 | 65.892 | 60.089 |
Figure 4.

Mulliken charges of PA with FP, CP, BP, and IP compounds.
Table 3. Mullikan’s Atomic Charges of PA with FP, CP, BP, and IP Compounds.
| Mullikan’s atomic charges |
|||||
|---|---|---|---|---|---|
| s. no. | atoms (X = F, Cl, Br, I) | V + FP | V + CP | V + BP | V + IP |
| 1 | C | –0.452 | –0.452 | –0.452 | –0.442 |
| 2 | C | –0.300 | –0.300 | –0.300 | –0.300 |
| 3 | C | –0.307 | –0.307 | –0.307 | –0.307 |
| 4 | N | –0.634 | –0.632 | –0.633 | –0.641 |
| 5 | O | –0.367 | –0.368 | –0.368 | –0.404 |
| 6 | C | –0.657 | –0.657 | –0.657 | –0.653 |
| 7 | C | 0.299 | 0.300 | 0.301 | 0.322 |
| 8 | H | 0.210 | 0.210 | 0.210 | 0.211 |
| 9 | H | 0.182 | 0.182 | 0.182 | 0.183 |
| 10 | H | 0.177 | 0.177 | 0.177 | 0.176 |
| 11 | H | 0.180 | 0.181 | 0.181 | 0.180 |
| 12 | H | 0.197 | 0.198 | 0.198 | 0.199 |
| 13 | H | 0.223 | 0.223 | 0.223 | 0.225 |
| 14 | H | 0.198 | 0.198 | 0.198 | 0.197 |
| 15 | H | 0.201 | 0.201 | 0.201 | 0.201 |
| 16 | H | 0.205 | 0.205 | 0.205 | 0.204 |
| 17 | H | 0.339 | 0.339 | 0.339 | 0.339 |
| 18 | H | 0.370 | 0.369 | 0.369 | 0.385 |
| 19 | H | 0.401 | 0.401 | 0.402 | 0.406 |
| 20 | H | 0.255 | 0.253 | 0.250 | 0.249 |
| 21 | H | 0.254 | 0.252 | 0.249 | 0.249 |
| 22 | H | 0.251 | 0.252 | 0.252 | 0.249 |
| 23 | H | 0.254 | 0.255 | 0.255 | 0.250 |
| 24 | C | 0.312 | –0.119 | –0.242 | –0.356 |
| 25 | C | –0.335 | –0.179 | –0.124 | –0.136 |
| 26 | C | –0.331 | –0.173 | –0.117 | –0.128 |
| 27 | C | –0.319 | –0.343 | –0.344 | –0.340 |
| 28 | C | –0.341 | –0.369 | –0.372 | –0.363 |
| 29 | C | 0.294 | 0.299 | 0.298 | 0.324 |
| 30 | O | –0.517 | –0.513 | –0.513 | –0.555 |
| 31 | X | –0.242 | –0.082 | –0.060 | 0.074 |
3.3. TD-DFT Calculations
The molecule’s absorption spectrum exhibited one broad band, a long wavelength maximum across the 220–300 nm range, as shown in Table 4. These measured absorption wavelength maxima values 257.13, 257.86, 259.43, and 299.53 nm correspond to the calculated values.39,40 To investigate the system’s interactions, the IEFPCM model41 was employed. The electronic transition from the highest occupied molecular orbital, HOMO, to the lowest unoccupied molecular orbital, LUMO, accounts for 89, 87.8, 87.9, and 98.9% of the maximum absorption wavelength. The UV spectrum of PA with halogenated phenols is presented in Figure 5.
Table 4. Theoretical Electronic Absorption Spectral Values of PA with FP, CP, BP, and IP Compounds Using the TD-B3LYP/LANL2DZ Basis Set.
| compounds | energy (cm–1) | excitation energy (eV) | wavelength (nm) | oscillator strength (f) | coefficient % |
|---|---|---|---|---|---|
| F | 38891.22 | 4.8219 (state 1: 56 → 60, 57 → 58) | 257.13 | 0.0683 | 11.0, 89 |
| 40945.52 | 5.0766 (state 2: 57 → 59) | 244.23 | 0.0084 | 98.7 | |
| 45443.67 | 5.6343 (state 3: 55 → 59) | 220.05 | 0.0014 | 95.3 | |
| Cl | 38780.73 | 4.8082 (state 1: 55 → 59, 56 → 57) | 257.86 | 0.0522 | 11.8, 87.8 |
| 41076.18 | 5.0928 (state 2: 56 → 58) | 243.45 | 0.0113 | 98.2 | |
| 45472.71 | 5.6379 (state 3: 54 → 58, 56 → 60) | 219.91 | 0.0022 | 2.1, 96.3 | |
| Br | 38545.22 | 4.7790 (state 1:55 → 60, 56 → 57) | 259.43 | 0.0469 | 11.5, 87.9 |
| 39354.19 | 4.8793 (state 2: 56 → 59) | 254.10 | 0.0003 | 98.0 | |
| 40793.89 | 5.0578 (state 3: 56 → 58) | 245.14 | 0.0127 | 97.0 | |
| I | 33385.69 | 4.1393 (state 1:56 → 57) | 299.53 | 0.0003 | 98.9 |
| 38225.82 | 4.7394 (state 2: 55 → 60, 56 → 58) | 261.60 | 0.0408 | 11.0, 88.0 | |
| 40407.55 | 5.0099 (state 3: 56 → 59) | 247.48 | 0.0146 | 97.0 |
Figure 5.

TD-DFT of PA with FP, CP, BP, and IP compounds.
3.4. Analysis of Natural Bond Orbitals (NBOs)
NBO can be used to explain filled orbitals, unoccupied orbitals, and second-order stabilization energy. One of the most well-known methods for investigating inter- and intramolecular bonding interactions, along with interpreting conjugative interactions and charge transformation processes in the system under study, is the natural bond orbital (NBO) technique. In an NBO investigation, the second-order Fock matrix helps evaluate the reciprocal interaction between the donor and acceptor. The analysis of the Fock matrix using second-order perturbation theory reveals many hyperconjugative interactions, as shown in Table 5, and E(2) exhibits a simple association with the size of interactions between the donor and acceptor.
Table 5. Donor–Acceptor Interactions of PA with FP, CP, BP, and IP Compounds.
| type | donor NBO (i) | type | acceptor NBO (j) | E(2) kcal/mol | E(j) – E(i) a.u. | F(i,j) a.u. |
|---|---|---|---|---|---|---|
| PA + FP | ||||||
| σ | H19–O30 | σ* | C1–C7 | 0.12 | 1.12 | 0.01 |
| σ | H19–O30 | σ* | N4–H18 | 0.15 | 1.22 | 0.01 |
| σ | H19–O30 | σ* | O5–C7 | 0.11 | 0.80 | 0.01 |
| σ | H19–O30 | π* | O5–C7 | 0.47 | 1.19 | 0.02 |
| LP(1) | O30 | σ* | N4–H18 | 0.56 | 1.00 | 0.02 |
| PA + CP | ||||||
| σ | H19–O30 | σ* | C1–C7 | 0.11 | 1.12 | 0.01 |
| σ | H19–O30 | σ* | N4–H18 | 0.14 | 1.22 | 0.01 |
| σ | H19–O30 | σ* | O5–C7 | 0.10 | 0.80 | 0.01 |
| σ | H19–O30 | π* | O5–C7 | 0.48 | 1.19 | 0.02 |
| LP(1) | O30 | σ* | N4–H18 | 0.50 | 0.99 | 0.02 |
| PA + BP | ||||||
| σ | H19–O30 | σ* | C1–C7 | 0.11 | 1.12 | 0.01 |
| σ | H19–O30 | σ* | N4–H18 | 0.14 | 1.22 | 0.01 |
| σ | H19–O30 | σ* | O5–C7 | 0.10 | 0.80 | 0.01 |
| σ | H19–O30 | π* | O5–C7 | 0.49 | 1.20 | 0.02 |
| LP(1) | O30 | σ* | N4–H18 | 0.52 | 0.99 | 0.02 |
| PA + IP | ||||||
| σ | H19–O30 | σ* | C1–C7 | 0.17 | 1.10 | 0.01 |
| σ | H19–O30 | σ* | N4–H18 | 0.23 | 1.20 | 0.02 |
| σ | H19–O30 | σ* | O5–C7 | 0.07 | 0.73 | 0.01 |
| σ | H19–O30 | π* | O5–C7 | 0.66 | 1.14 | 0.03 |
| LP(1) | O30 | σ* | C1–C7 | 0.05 | 0.89 | 0.01 |
| LP(1) | O30 | σ* | N4–H18 | 1.96 | 0.98 | 0.04 |
3.5. Vibrational Investigations
Figure 6 shows the IR spectra, and Table 6 (Tables S2 and S3) provides summaries of theoretically estimated frequencies, relative intensities, likely assignments, and potential energy distribution (PED) of two different basis sets for PA+(FP, CP, BP, IP). The compound consists of 31 atoms and has 87 fundamental vibrational modes. The possible energy distributions (PEDs) provide ≥10% of the assignments. Regarding the structure, benzene is similar to heterocyclic aromatic compounds and its derivatives. With a PED contribution of 69%, the IR band was found at 3241.15, 3238.59, 3235.43, and 3232.13 cm–1 and its assigned to CH. With a PED contribution of 30%, the IR band was detected at 1519.73, 1519.72, 1519.74, and 1519.70 cm–1 assigned to CN. The NH stretching modes generally contain vibrational bands in analyzing the level above 3000–3500 cm–1. Observed at 3432 cm–1, this band is in the IR spectra. They have a theoretically determined harmonic wavenumber at 3723.59 and 3535.94 cm–1 with an 82% PED contribution. The C=O peak in the current study should theoretically arise at wavelengths of 1651.64 cm–1. Although the C=C stretching modes in the compound’s benzene rings are seen at around 1642.93 cm–1 in IR spectra, the C=C structural stretching vibrations in the aromatic rings typically emerge at 1430–1625 cm–142,43 It may also be noted in the IR spectra that it is at 1665.64 cm–1. Stretching modes identified at 1444.57 cm–1 in IR spectra are assigned to C–N vibrations with a 30% PED contribution. The FP (O–H) stretching vibration mode with a PED contribution of 93% is given to the absorption band in IR spectra centered at 2997.75 cm–1 at high wavenumbers.
Figure 6.

Theoretical IR spectra of the PA with FP, CP, BP, and IP compounds.
Table 6. FT-IR Spectra of PA with FP Compounds with Two Different Basis Setsa.
| PA + FP (B3LYP/LANL2DZ) |
PA + FP (B3LYP/6-311G++(d,p)) |
||||
|---|---|---|---|---|---|
| assignments and freq. scaled | IR (cm–1) | assignments and freq. scaled | IR (cm–1) | ||
| γNH | 3723.59 | 107.66 | γNH | 3702.07 | 113.94 |
| γNH | 3535.94 | 202.57 | γNH | 3551.09 | 146.98 |
| γOH | 3246.48 | 2.35 | γOH | 3335.42 | 1697.09 |
| γCH | 3241.15 | 7.64 | γCH | 3084.39 | 91.67 |
| γOC | 1665.64 | 3.13 | γOC | 1693.86 | 510.23 |
| γCC | 1642.93 | 14.75 | γCC | 1639.10 | 62.93 |
| βHOC | 1328.3 | 7.06 | βHOC | 1263.62 | 216.76 |
| βCCC | 844.68 | 1.93 | βCCC | 1263.62 | 216.76 |
| τHNHO | 749.27 | 12.09 | τHNHO | 1477.56 | 33.38 |
| τHCCC | 676.52 | 50.06 | τHCCC | 1395.15 | 113.39 |
Scaling factor: 0.961; γ, stretching; β, in-plane-bending; α, out-of-plane bending; ρ, scissoring; ω, wagging; r, rocking; t, twisting; τ, torsion.
3.6. Molecular Electrostatic Potential (MEP)
In this part, the surface of molecular electrostatic potential (MEP) is created to predict and analyze the molecular reactive behavior of a studied molecule. The mapping was done using a DFT/B3LYP method. It is related to the electronic density and is very useful for studying hydrogen-bonding interactions and for determining electrophilic and nucleophilic attack sites. In terms of color grading, electrostatic potential is used to depict the molecular and electrostatic potentials.44Figure 7 displays the potential as a multicolored MEP map. These mappings’ color codes vary from −8.060 a.u to 8.060 a.u. Donor atoms are shown in blue regions with positive potential; localized hydrogen atoms represent these strongest positive electrostatic potential (nucleophilic site). Acceptor atoms with a negative potential in red areas have the strongest electronegative potential (electrophilic sites), mainly located over anionic groups. The green or yellow color indicates the neutral zone. Electrophilic sites are near hydrogen atoms, whereas nucleophilic sites are near oxygen atoms. The MEP surface is a donor–acceptor interaction that confirms the formation of hydrogen bonds.45 The negative region mainly forms a hydrogen bond interaction with the protein. This is highly corroborated by molecular docking and molecular dynamics studies.
Figure 7.

Surface distance projection map by MEP of PA and FP compounds.
3.7. Hirshfeld Surface Evaluation
The molecular Hirshfeld surface,46−48 curvedness, de, di, fragment, dnorm, shape index, and hydrogen-bonding interaction (as dashed lines) for the PA+(FP, CP, BP, IP) compounds are depicted in Figure 8a–g and are mapped on dnorm—ranges (0.7568–9.7750), shape index ranges (1.000–1.000), curvedness (4.000–0.400), and de ranges (1.6541–12). The dnorm values were mapped on the Hirshfeld surface by the use of red, blue, and white color plots; the red region appear for the region of close contacts and blue longer contacts. These contacts have been observed on the de map where the large orange spots are attributed to the N–H...O hydrogen bonds, while the small blue spots correspond to the H...H, F...H, and H...F contacts. The absence of red circles on the shape index map and the small flat segments delimited by red circles on the curvedness map include the presence of π–π and C–H···π interactions in our crystal structure.49 Two-dimensional (2D) map fingerprint plots examine the whole range of intermolecular interactions (Figure 9). The compound’s characteristic is stimulated by hydrogen-bonding and intermolecular interactions inside the crystal. The intermolecular H•••F, H•••H, and F•••H interactions, with total Hirshfeld areas of around 30.7, 57.1, and 7.5%, respectively, are shown to be the primary contacts in the 2D fingerprints shown in Figure 9.
Figure 8.

Hirshfeld surfaces of individual compounds of PA with FP showing (a) curvedness, (b) de, (c) di, (d) fragment, (e) dnorm, (f) shape index, and (g) hydrogen-bonding interactions (as dashed lines).
Figure 9.

Fingerprint plots of pentanamide with fluorophenol.
3.8. Interaction Region Indicator (IRI)
The vdW interaction marked by sync between the PA and FP may also be located using the IRI map. The six-membered carbon rings’ IRI isosurface maps in Figure 10a show the steric effect as red dots within the rings. IRI is beneficial for understanding chemical interactions because it may reveal a change in the strength of chemical bonds and weak connections. Figure 10 presents two chemical systems, which not only contain chemical bonds but also involve intramolecular or intermolecular interactions. In the case of PA and FP, it can be seen that IRI nicely exhibits the interaction between hydroxyl and carbonyl groups. At suitable locations, the IRI isosurfaces’ color is gradually blue. IRI isosurfaces demonstrate the presence of the noncovalent C=O···H interaction during the reaction. Green to yellow isosurfaces indicate the electron delocalize range (EDR) in Figure 10b, which shows the EDR with an intermolecular contact between the PA of the carbonyl group and the FP of the hydroxyl group. Belonging to H-bond interactions, one spike is found in the region at −1.70. In addition, two spikes are found in the region between −1.70 and −4.80, which belong to vdW interactions and spikes belonging to steric effects that are located in 7.0. Figure 10a,b depicts title structures that include chemical-bonding and intramolecular or intermolecular interactions.
Figure 10.

(a) Interaction region indicator and (b) electron delocalized range of PA and FP compounds.
3.9. Selectivity of Active Sites
Current descriptors, such as dual descriptor Δf(r), an abbreviated version of Δf(r) ΔSk, were computed for better comprehension.50 If Δf(r) > 0, the dual descriptor Δf(r) is provided by
This formula is Δf(r) times the molecular softness (ΔSk)51 in a more condensed form
The explanation is related to Δf(r). Table 7 (Tables S4–S6) provides each atom’s Fukui functions and local softness values for the studied PA and FP molecules.
Table 7. Calculated Fukui Functions for the Studied PA and FP Molecules.
| atoms | f– | f+ | f0 | dual descriptor | hardness (au) | W– (eV) | W+ (eV) |
|---|---|---|---|---|---|---|---|
| C1 | 0 | 0.0142 | 0.0071 | 0.0142 | –0.0001 | 0 | 0.0135 |
| C2 | 0 | 0.0075 | 0.0037 | 0.0075 | –0.0001 | 0 | 0.0071 |
| C3 | 0 | 0.0187 | 0.0094 | 0.0187 | –0.0001 | 0 | 0.0178 |
| N4 | 0 | 0.0084 | 0.0042 | 0.0084 | –0.0001 | 0 | 0.008 |
| O5 | 0 | 0.3212 | 0.1606 | 0.3212 | –0.0025 | 0 | 0.3049 |
| C6 | 0 | 0.0003 | 0.0001 | 0.0003 | 0 | 0 | 0.0002 |
| C7 | 0 | 0.6243 | 0.3122 | 0.6243 | –0.0048 | 0 | 0.5926 |
| H8 | 0 | 0.0022 | 0.0011 | 0.0022 | 0 | 0 | 0.0021 |
| H9 | 0 | 0.0002 | 0.0001 | 0.0002 | 0 | 0 | 0.0002 |
| H10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H11 | 0 | 0.0001 | 0 | 0.0001 | 0 | 0 | 0.0001 |
| H12 | 0 | 0.0001 | 0.0001 | 0.0001 | 0 | 0 | 0.0001 |
| H13 | 0 | 0.0007 | 0.0003 | 0.0007 | 0 | 0 | 0.0006 |
| H14 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H15 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H16 | 0 | 0.0001 | 0 | 0.0001 | 0 | 0 | 0.0001 |
| H17 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H18 | 0 | 0.0001 | 0.0001 | 0.0001 | 0 | 0 | 0.0001 |
| H19 | 0 | 0.0012 | 0.0006 | 0.0012 | 0 | 0 | 0.0011 |
| H20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H21 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H22 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H23 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| C24 | 0.0122 | 0 | 0.0061 | –0.0122 | 0.0031 | 0.0116 | 0 |
| C25 | 0.4806 | 0 | 0.2403 | –0.4806 | 0.1226 | 0.4562 | 0 |
| C26 | 0.0001 | 0 | 0.0001 | –0.0001 | 0 | 0.0001 | 0 |
| C27 | 0.4943 | 0 | 0.2472 | –0.4943 | 0.1261 | 0.4692 | 0 |
| C28 | 0.0002 | 0.0001 | 0.0002 | –0.0001 | 0 | 0.0002 | 0.0001 |
| C29 | 0.0124 | 0.0002 | 0.0063 | –0.0122 | 0.0032 | 0.0118 | 0.0002 |
| O30 | 0.0001 | 0.0003 | 0.0002 | 0.0003 | 0 | 0.0001 | 0.0003 |
| F31 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
The maximum threshold mainly determines the nucleophilic and electrophilic behaviors of different sites of the compounds fk+ and fk values. When a molecule accepts electrons, the changes in electron density are measured by the fk+ values, while the fk values give the measures of the changes in electron density when the molecules lose the electrons. The high fk+ and fk values imply the molecules’ high electron acceptance and donation capabilities, respectively. From the calculated Fukui indices (Table 6), it can be observed that the most susceptible sites for electron acceptance or donation are the O5 heteroatoms, followed by C7 and C27 atoms of the aliphatic compound. The favorable centers for electron acceptance, i.e., O5 and C7 atoms having fk+ 0.3212 and 0.6243, respectively, are nucleophilic attacks. On the other hand, the susceptible sites for electrophilic attacks, i.e., electron donation, are C27 atoms having fk 0.4943. In addition, it is also observed that the calculated Fukui indices are in good accord with the electron density distribution in the HOMO and LUMO of the studied molecules. Hence, these analyses signify the same reactive sites for the corresponding D–A-type interaction on the metallic surfaces.
3.10. Dielectric Relaxation Analysis
The current investigation is being done to identify noncovalent interactions between PA and halophenol in benzene. This work uses the Higashi technique52 to validate Debye’s single-frequency equation. The average relaxation time (τ1), total relaxation time (τ2), and geometric meantime (τ0) are determined by the Debye equation, which defines slopes a’ and a″. The relaxation time (τ0) values for the title molecule were measured at room temperature for five distinct mole ratios: 1:3, 1:2, 1:1, 3:1, and 2:1. The relaxation time was determined to be the maximum at a 1:1 molar ratio. Due to the H bonding between the halogenated phenols hydroxyl hydrogen and the N atom in the PA, NH group, the formation is like (PA) HNδ−.... δ+HO. An increase in the effective size of the molecular spinning unit might explain the increased relaxation time.53 The relaxation time appears to decrease for various mole ratios. The value of relaxation time is a 1:1 ratio; the resulting value agrees with Higashi’s method. Using the Higashi approach for an equal mole ratio, the title compounds’ value was determined and is shown in Table 8.
Table 8. Dielectric Parameter Values of PA with (FP, CP, BP, and IP) in Benzene (X-Band) at 298 K for Various Weight Fractions.
| weight fraction | relaxation time (ps) |
|||||||
|---|---|---|---|---|---|---|---|---|
| ratio | w2 | ε0 | ε’ | ε″ | ε∞ | τ1 | τ2 | τ0 |
| PA + FP | ||||||||
| 1:3 | 0.03745 | 6.1342 | 2.9875 | 0.9717 | 2.6399 | 21.34 | 21.91 | 21.62 |
| 1:2 | 0.03714 | 6.1423 | 2.9887 | 0.9704 | 2.6341 | 20.87 | 21.09 | 20.98 |
| 1:1 | 0.03652 | 6.1678 | 3.0130 | 0.9779 | 2.6410 | 21.67 | 22.42 | 22.04 |
| 2:1 | 0.03589 | 6.1479 | 2.9856 | 0.9701 | 2.6378 | 20.79 | 20.83 | 20.81 |
| 3:1 | 0.03558 | 6.1371 | 2.9864 | 0.9712 | 2.6387 | 21.42 | 21.47 | 21.45 |
| PA + CP | ||||||||
| 1:3 | 0.04168 | 6.1401 | 2.9928 | 0.9753 | 2.6411 | 21.16 | 21.20 | 21.18 |
| 1:2 | 0.04092 | 6.1502 | 2.9969 | 0.9762 | 2.6392 | 20.78 | 20.96 | 20.87 |
| 1:1 | 0.03933 | 6.1790 | 3.1013 | 0.9792 | 2.6480 | 21.35 | 22.09 | 21.72 |
| 2:1 | 0.03777 | 6.1599 | 2.9926 | 0.9788 | 2.6396 | 20.34 | 20.69 | 20.51 |
| 3:1 | 0.03699 | 6.1407 | 2.9943 | 0.9723 | 2.6409 | 21.14 | 21.11 | 21.12 |
| PA + BP | ||||||||
| 1:3 | 0.05310 | 6.1463 | 3.0993 | 0.9771 | 2.6439 | 21.05 | 21.04 | 21.04 |
| 1:2 | 0.05105 | 6.1545 | 3.0600 | 0.9794 | 2.6409 | 20.63 | 20.78 | 20.71 |
| 1:1 | 0.04695 | 6.1824 | 3.1641 | 0.9812 | 2.6508 | 21.21 | 21.72 | 21.46 |
| 2:1 | 0.04284 | 6.1620 | 3.0893 | 0.9802 | 2.6440 | 20.23 | 20.39 | 20.31 |
| 3:1 | 0.04079 | 6.1508 | 3.0499 | 0.9785 | 2.6428 | 20.98 | 20.96 | 20.97 |
| PA + IP | ||||||||
| 1:3 | 0.06517 | 6.1509 | 3.1410 | 0.9897 | 2.6473 | 20.75 | 20.84 | 20.79 |
| 1:2 | 0.06178 | 6.1647 | 3.1036 | 0.9841 | 2.6445 | 20.32 | 20.45 | 20.38 |
| 1:1 | 0.05499 | 6.1862 | 3.2024 | 0.9862 | 2.6563 | 21.19 | 21.34 | 21.26 |
| 2:1 | 0.04821 | 6.1670 | 3.1075 | 0.9819 | 2.6460 | 19.87 | 20.12 | 19.99 |
| 3:1 | 0.04482 | 6.1542 | 3.0994 | 0.9875 | 2.6487 | 20.79 | 20.49 | 20.64 |
3.11. Molecular Docking
The crystalline structure of Staphylococcusaureustyrosyl-tRNA synthetase ligase (Uniprot = Q15631 for Translin), which has antibacterial and antifungal properties, was obtained from PDB.54 Chemsketch was used to generate ligand molecules. The molecules were rendered in 3D using the Babel software. The Protein Preparation Wizard Module55 was used to produce the receptor structures, and the ligand models were created using Schrodinger’s ligprep module to ensure that they were chemically precise. Molecular docking was performed using the Glide ligand docking module.56 The glide receptor grid generation module was used to build receptor grids. For the prepared proteins, grids were built. Ciprofloxacin-centered grids were formed for the S. aureusgyrase complex and SB-239629 ligand-centered grids for the S. aureusTyrRS complex. The boundary box was set to its default size, 14 Å × 14 Å × 14 Å, which was large enough to include the binding area by the ligand.57 The COACH-D server,58 which uses the Autodock Vina method,59,60 was also used to conduct docking between the ligands and the S. aureustyrosyl-tRNA synthetase protein structure (PDB id: 1JIJ). The protein–ligand interactions were classified into four types such as hydrophobic interactions, ionic bonding, hydrogen bonding, and disulfide linkages. The latter is commonly grouped, such as acceptor, donor, backbone donor, and backbone acceptor categories. They play a crucial role in ligand–protein binding and are essential to consider when designing new drugs. The interactions identified Arg-12 and Leu-274 as the H-bond interacting amino acid residues for the 1J1J complex. As discovered, pi-interactions involving the ligand PA with FP, CP, BP, and IP compounds in all of the examined systems, as shown in Table 9 and Figure 11(Figures S1 and S3), and many biologically relevant factors were evaluated. Table 10 and Figures 12 and 13 mentioned in the analyses include lipophilicity, druggability, solubility, and pharmacokinetics based on ADMET (absorption, distribution, metabolism, excretion, and toxicity).61,62 All components were computationally screened for possible biological effects; PASS63 was employed to calculate quantitative structure–activity connections by analyzing chemical structures into 2D and 3D descriptors and then generating models from bioactive ligands.64 Results of predictions were reported as a proportion of probable activity (Pa) and probable inactivity (Pi) (Pi). Pa and Pi values for four of the top moves with Pa > Pi and Pa > 0.700 were considered in this assessment.
Table 9. Affinity and Hydrogen Bond Interaction of PA with FP, CP, BP, and IP Compounds.
| residue type | interaction distance | binding energy/coordinates | donor type | interactions |
|---|---|---|---|---|
| 1JIJ-F | ||||
| ARG12 | 2.44 Å | –6.30 kcal/mol −13.23, 21.25, 110.35 | oxygen | H-Bond |
| ARG12 | 2.93 Å | oxygen | H-Bond | |
| LEU274 | 2.48 Å | oxygen | H-Bond | |
| 1JIJ-Cl | ||||
| ARG12 | 2.45 Å | –6.30 kcal/mol −13.25, 21.21, 110.36 | oxygen | H-Bond |
| ARG12 | 2.81 Å | oxygen | H-Bond | |
| LEU274 | 2.38 Å | oxygen | H-Bond | |
| 1JIJ-Br | ||||
| ARG12 | 2.52 Å | –6.40 kcal/mol −13.31, 21.21, 110.42 | oxygen | H-Bond |
| ARG12 | 2.86 Å | oxygen | H-Bond | |
| LEU274 | 2.43 Å | oxygen | H-Bond | |
| 1JIJ-I | ||||
| ARG12 | 3.14 Å | –6.40 kcal/mol −13.27, 21.22, 110.38 | oxygen | H-Bond |
| ARG12 | 3.87 Å | oxygen | H-Bond | |
| LEU274 | 3.12 Å | oxygen | H-Bond | |
Figure 11.

(a) Aromaticity, (b) charges, (c) H-bond, and panels (d, e) show the best pose, which corresponds to the lowest energies of the ligands-FP in the 1J1J protein and 2D interactions.
Table 10. ADMET of PA with FP, CP, BP, and IP Compounds.
| molecule | molecule 1 | molecule 2 | molecule 3 | molecule 4 |
|---|---|---|---|---|
| formula | C11H17FNO2 | C11H17ClNO2 | C11H17BrNO2 | C11H17INO2 |
| MW | 213.25 | 229.7 | 274.15 | 321.15 |
| #heavy atoms | 15 | 15 | 15 | 15 |
| #aromatic heavy atoms | 6 | 6 | 6 | 6 |
| #rotatable bonds | 3 | 3 | 3 | 3 |
| #H-bond acceptors | 3 | 2 | 2 | 2 |
| #H-bond donors | 3 | 3 | 3 | 3 |
| MR | 58.59 | 63.64 | 66.33 | 71.35 |
| TPSA | 66.48 | 66.48 | 66.48 | 66.48 |
| iLOGP | 0 | 0 | 0 | 0 |
| XLOGP3 | 2.5 | 3.03 | 3.09 | 3.74 |
| MLOGP | 2.37 | 2.51 | 2.65 | 2.79 |
| silicos-IT log P | 0.95 | 1.17 | 1.21 | 1.5 |
| consensus log P | 1.66 | 1.86 | 1.93 | 2.11 |
| ESOL log S | –2.84 | –3.27 | –3.58 | –4.29 |
| ESOL solubility (mg/mL) | 3.12 × 10–1 | 1.23 × 10–1 | 7.14 × 10–2 | 1.66 × 10–2 |
| ESOL solubility (mol/L) | 1.46 × 10–3 | 5.36 × 10–4 | 2.60 × 10–4 | 5.18 × 10–5 |
| silicos-IT class | soluble | soluble | moderately soluble | moderately soluble |
| GI absorption | high | high | high | high |
| BBB permeant | yes | yes | yes | yes |
| Pgp substrate | no | yes | yes | yes |
| CYP1A2 inhibitor | no | no | no | no |
| CYP2C19 inhibitor | no | no | no | no |
| CYP2C9 inhibitor | no | no | no | no |
| CYP2D6 inhibitor | no | no | no | no |
| CYP3A4 inhibitor | no | no | no | no |
| bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 |
| synthetic accessibility | 1.2 | 1.21 | 1.33 | 1.84 |
Figure 12.

BOILED-egg predictive model for PA with FP, CP, BP, and IP compounds.
Figure 13.

Bioavailability radar of PA with FP, CP, BP, and IP compounds that correspond to an optimum range for each property is shown by the pink area.
4. Conclusions
The study mainly focused on noncovalent interactions of PA with FP, CP, BP, and IP compounds. IR spectra were utilized to assign frequencies and validate the computational approach through excellent agreement with data from the literature. The HOMO and LUMO energy changes within the molecule were found to facilitate charge-transfer interactions. Through the solvation effect using TD-DFT/B3LYP with the IEFPCM model, the reactive sites were predicted with the aid of MEP and Hirshfeld charges. Additionally, the compound was subjected to the DFT method to test its chemical and thermal stability. The effectiveness of IRI analysis for characterizing changes in covalent and noncovalent interactions in ab initio molecular dynamics simulations is highly promising. The relaxation time of the PA with FP, CP, BP, and IP compounds was determined using the Higashi technique, and it revealed that the complex formation is maximal at a 1:1 ratio due to hydrogen bonding between PA and halogen-substituted phenol derivatives. The drug-likeness of the molecule and ligand–protein interactions, as with molecular docking simulations, and pharmacokinetics were evaluated to investigate the potentiality of these therapeutics. The in silico docking simulations showed several interactions between ligand and protein clusters with high binding energy scores, supporting the biological efficacy of the four novel ternary mixture compounds against various bacterial infections. Further, in silico pharmacokinetics analyses indicated that the pharmacological compounds (PA•••FP, PA•••CP, PA•••BP, and PA•••IP) have significant inhibitory effects, implying that these promiscuous molecules may further be studied for cancer therapy.
Acknowledgments
The authors thank the Management of Islamiah College (Autonomous) for the facilities provided to carry out the research work in the Department of Physics & Chemistry, Islamiah College (Autonomous), Vaniyambadi.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c04710.
Evaluated quantum chemical properties of the PA+FP, CP, BP, and IP compounds by the DFT/B3LYP/6-311++G(d,p) method; assignments of theoretical wavenumbers of PA+FP, CP, BP, and IP compounds with the 6-311++G(d,p) basis set; and reactivity and selectivity of the Fukui functions and the figures of molecular docking stimulation of the PA+FP, CP, BP, and IP compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Allen C. L.; Williams J. M. Metal-catalysed approaches to amide bond formation. Chem. Soc. Rev. 2011, 40, 3405–3415. 10.1039/c0cs00196a. [DOI] [PubMed] [Google Scholar]
- Montaño M.; Gutleb A. C.; Murk A. J. Persistent toxic burdens of halogenated phenolic compounds in humans and wildlife. Environ. Sci. Technol. 2013, 47, 6071–6081. 10.1021/es400478k. [DOI] [PubMed] [Google Scholar]
- Muller F.; Caillard L.. Chlorophenols.; Ullmann’s Encyclopedia of Industrial Chemistry, 2000. [Google Scholar]
- Reynolds H. H.; Bigelow L. A. A STUDY OF THE PREPARATION OF QUINIZARIN1. J. Am. Chem. Soc. 1926, 48, 420–422. 10.1021/ja01413a017. [DOI] [Google Scholar]
- Bien H. S.; Stawitz J.; Wunderlich K.. Anthraquinone Dyes and Intermediates; Ullmann’s Encyclopedia of Industrial Chemistry, 2000. [Google Scholar]
- Rappoport Z.Handbook Tables for Organic Compound Identification, 3rd ed.; CRC press, 1984. [Google Scholar]
- Dains F. B.; Eberly Floyd. ″p-Iodophenol″. Org. Synth. 1935, 15, 39. 10.15227/orgsyn.015.0039. [DOI] [Google Scholar]
- Wong B. M.; Hsieh T. H. Optoelectronic and excitonic properties of oligoacenes: substantial improvements from range-separated time-dependent density functional theory. J. Chem. Theory Comput. 2010, 6, 3704–3712. 10.1021/ct100529s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik S. A.; Cui X. Y.; Ringer S. P.; Stampfl C. TDDFT Study of the Optical Excitation of Nucleic Acid Bases-C60 Complexes. J. Phys. Chem. A 2017, 121, 9058–9063. 10.1021/acs.jpca.7b07442. [DOI] [PubMed] [Google Scholar]
- Parr R. G.; Yang W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. 10.1021/ja00326a036. [DOI] [Google Scholar]
- Sharma V.; Thakur N.; Sharma D. R.; Rangra V. S.; Negi N. S.. Dielectric relaxation studies of binary mixtures of ethyl alcohol and tetramethylurea in the benzene solution from microwave absorption data. 2006 10.1524/zpch.2006.220.3.325. [DOI]
- Kumar R.; Rangra V. S. Dielectric relaxation studies of binary mixtures of N-methylacetamide and acetonitrile in benzene solutions using microwave absorption studies. Z. Phys. Chem. 2005, 219, 169–180. 10.1524/zpch.219.2.169.57304. [DOI] [Google Scholar]
- Thenappan T.; Sankar U. Study of correlation factors and dipolar excess free energies of esters in benzene. J. Mol. Liq. 2006, 126, 23–28. 10.1016/j.molliq.2004.09.004. [DOI] [Google Scholar]
- Mhadhbi N.; Dgachi S.; Ben Ahmed A.; Issaoui N.; Nasr S.; Badraoui R.; Naïli H. Vibrational Spectroscopies, Global Reactivity, Molecular Docking, Thermodynamic Properties and Linear and Nonlinear Optical Parameters of Monohydrate Arsonate Salt of 4-Aminopyridine. Chem. Afr. 2023, 6, 1897–1912. 10.1007/s42250-023-00620-8. [DOI] [Google Scholar]
- Filimonov D. A.; Lagunin A. A.; Gloriozova T. A.; Rudik A. V.; Druzhilovskii D. S.; Pogodin P. V.; Poroikov V. V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. 10.1007/s10593-014-1496-1. [DOI] [Google Scholar]
- Basha A. A.; Khan F. L. A.; Hussain B. K. Dielectric relaxation and thermodynamical parameters of hydrogen bonded complexes for Heptanamide and Pentanamide with halogenated phenols in benzene. J. Mol. Liq. 2022, 363, 119853 10.1016/j.molliq.2022.119853. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb Ma.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson Ga.; Nakatsuji H.; Li X.; Caricato M.; Marenich aV.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov aF.; Sonnenberg J. L.; Williams F.; Ding F.; Lipparini F.; Egidi J.; Goings B.; Peng A.; Petrone T.; Henderson D.; Ranasinghe V. G.; Zakrzewski J.; Gao N.; Rega G.; Zheng W.; Liang M.; Hada M.; Ehara K.; Toyota R.; Fukuda J.; Hasegawa M.; Ishida T.; Nakajima Y.; Honda O.; Kitao H.; Nakai T.; Vreven K.; Throssell Ja.; Montgomery Jr J. E.; Peralta F.; Ogliaro M. J.; Bearpark J. J.; Heyd E. N.; Brothers K. N.; Kudin V. N.; Staroverov Ta.; Keith R.; Kobayashi J.; Normand K.; Raghavachari aP.; Rendell J. C.; Burant S. S.; Iyengar J.; Tomasi M.; Cossi J. M.; Millam M.; Klene C.; Adamo R.; Cammi J. W.; Ochterski R. L.; Martin K.; Morokuma O.; Farkas J. B.; Foresman D. J.; Fox. Gaussian 16, Revision C.01, G16_C01; Gaussian, Inc.: Wallin., 2016.
- Frisch A.; Hratchian H. P.; Dennington R. D. II; Keith T. A.; Millam J.; Nielsen B.; Hiscocks J.. GaussView, Version 5.0. 8; Gaussian. Inc.: Wallingford, CT, USA, 2009.
- Glendening E. D.; Badenhoop J. K.; Reed A. D.; Carpenter J. E.; Weinhold F.. Theoretical Chemistry Institute, NBO 3.1; University of Wisconsin: Madison, WI, 1996.
- Perdew J. P.; Ernzerhof M.; Burke K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. 10.1063/1.472933. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Wadt W. R.; Hay P. J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. 10.1063/1.448800. [DOI] [Google Scholar]
- Kansız S.; Azam M.; Dege N.; Ermiş N.; Al-Resayes S. I.; Alam M. Supramolecular assembly in designing co-crystals of fumaric acid and pyrimidine/picolinate derivatives. Green Chem. Lett. Reviews 2022, 15, 825–836. 10.1080/17518253.2022.2130016. [DOI] [Google Scholar]
- Becke A. D. Density-functional thermochemistry III, the role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Jamroz M. H.Vibrational Energy Distribution Analysis; VEDA 4 Program Warsaw: Poland, 2004.
- Azam M.; Sahoo P. K.; Mohapatra R. K.; Kumar M.; Ansari A.; Moon I. S.; Biswal S. K. Structural investigations, Hirsfeld surface analyses, and molecular docking studies of a phenoxo-bridged binuclear Zinc (II) complex. J. Mol. Struct. 2022, 1251, 132039 10.1016/j.molstruc.2021.132039. [DOI] [Google Scholar]
- Kansız S.; Tolan A.; Azam M.; Dege N.; Alam M.; Sert Y.; İçbudak H. Acesulfame based Co (II) complex: Synthesis, structural investigations, solvatochromism, Hirshfeld surface analysis and molecular docking studies. Polyhedron 2022, 218, 115762 10.1016/j.poly.2022.115762. [DOI] [Google Scholar]
- Turner M. J.; McKinnon J. J.; Wolff S. K.; Grimwood D. J.; Spackman P. R.; Jayatilaka D.. CrystalExplorer17, Explorer17, C, University of Western Australia; 2017.
- Discovery Studio . Release 4.5; Accelerys Software Inc.: San Diego, 2016. [Google Scholar]
- Thenappan T.; Devaraj A. P. Dielectric studies on binary polar mixtures of propanoic acid with esters. J. Mol. Liq. 2006, 123, 72–79. 10.1016/j.molliq.2005.04.010. [DOI] [Google Scholar]
- Basha A. A.; Khan F.. Dielectrics relaxation studies of acrylamide and acetanilide with halogenated phenols in benzene, AIP Conference Proceedings; AIP Publishing, 2023.
- Basha A. A.; Ali Khan; F L.; Mohamed Imran P.; Kubaib A. Valeramide and halo-phenol in a non-polar liquid: DFT based characterization and reactivity, non-covalent interaction, and dielectric relaxation studies. Polycyclic Aromat. Compd. 2023, 1–26. 10.1080/10406638.2023.2169475. [DOI] [Google Scholar]
- Jomaa I.; Noureddine O.; Gatfaoui S.; Issaoui N.; Roisnel T.; Marouani H. Experimental, computational, and in silico analysis of (C8H14N2) 2 [CdCl6] compound. J. Mol. Struct. 2020, 1213, 128186 10.1016/j.molstruc.2020.128186. [DOI] [Google Scholar]
- Foster M. E.; Wong B. M. Nonempirically tuned range-separated DFT accurately predicts both fundamental and excitation gaps in DNA and RNA nucleobases. J. Chem. Theory Comput. 2012, 8, 2682–2687. 10.1021/ct300420f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issaoui N.; Ghalla H.; Muthu S.; Flakus H. T.; Oujia B. Molecular structure, vibrational spectra, AIM, HOMO–LUMO, NBO, UV, first order hyperpolarizability, analysis of 3-thiophenecarboxylic acid monomer and dimer by Hartree–Fock and density functional theory. Spectrochim. Acta, Part A 2015, 136, 1227–1242. 10.1016/j.saa.2014.10.008. [DOI] [PubMed] [Google Scholar]
- Lewis D. F. V.; Ioannides C.; Parke D. V. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: Computer analysis of structure—activity relationships. Xenobiotica 1994, 24, 401–408. 10.3109/00498259409043243. [DOI] [PubMed] [Google Scholar]
- Issaoui N.; Ghalla H.; Brandán S. A.; Bardak F.; Flakus H. T.; Atac A.; Oujia B. Experimental FTIR and FT-Raman and theoretical studies on the molecular structures of monomer and dimer of 3-thiopheneacrylic acid. J. Mol. Struct. 2017, 1135, 209–221. 10.1016/j.molstruc.2017.01.074. [DOI] [Google Scholar]
- Issa T. B.; Sagaama A.; Issaoui N. Computational study of 3-thiophene acetic acid: Molecular docking, electronic and intermolecular interactions investigations. Comput. Biol. Chem. 2020, 86, 107268 10.1016/j.compbiolchem.2020.107268. [DOI] [PubMed] [Google Scholar]
- Abu Eittah R. H.; Hamed M. M.; Mohamed A. A. Electronic absorption spectra of some nicotinamides and nicotinic acids. Molecular orbital treatment. Int. J. Quant. Chem. 1997, 64, 689–701. . [DOI] [Google Scholar]
- Fathima Rizwana B.; Prasana J. C.; Abraham C. S.; Muthu S. Spectroscopic investigation, hirshfeld surface analysis and molecular docking studies on antiviral drug entecavir. J. Mol. Struct. 2018, 1164, 447–458. 10.1016/j.molstruc.2018.03.090. [DOI] [Google Scholar]
- Klamt A.; Moya C.; Palomar J. A Comprehensive Comparison of the IEFPCM and SS(V)PE Continuum Solvation Methods with the COSMO Approach. J. Chem. Theory Comput. 2015, 11, 4220–4225. 10.1021/acs.jctc.5b00601. [DOI] [PubMed] [Google Scholar]
- Durgun M.; Ceylan Ü.; Yalçın Ş. P.; Türkmen H.; Özdemir N.; Koyuncu I. Synthesis, molecular structure, spectroscopic characterization, NBO, NLO and NPA analysis and in vitro cytotoxicity study of 3-chloro-N-(4-sulfamoylphenethyl) propanamide with experimental and computational study. J. Mol. Struct. 2016, 1114, 95–107. 10.1016/j.molstruc.2016.02.062. [DOI] [Google Scholar]
- Sert Y.; Karakaya M.; Çırak Ç.; Eskiyurt B.; Kürekçi M. Structural optimization and vibrational analysis of an antidiabetic drug: tolbutamide. J. Sulfur Chem. 2015, 36, 450–461. 10.1080/17415993.2015.1050397. [DOI] [Google Scholar]
- Sagaama A.; Noureddine O.; Brandán S. A.; Jarczyk-Jędryka A.; Flakus H. T.; Ghalla H.; Issaoui N. Molecular docking studies, structural and spectroscopic properties of monomeric and dimeric species of benzofuran-carboxylic acids derivatives: DFT calculations and biological activities. Comput. Biol. Chem. 2020, 87, 107311 10.1016/j.compbiolchem.2020.107311. [DOI] [PubMed] [Google Scholar]
- Mohan N.; Suresh C. H. A molecular electrostatic potential analysis of hydrogen, halogen, and dihydrogen bonds. J. Phys. Chem. A 2014, 118, 1697–1705. 10.1021/jp4115699. [DOI] [PubMed] [Google Scholar]
- Azam M.; Sahoo P. K.; Mohapatra R. K.; Kumar M.; Ansari A.; Moon I. S.; Biswal S. K. Structural investigations, Hirsfeld surface analyses, and molecular docking studies of a phenoxo-bridged binuclear Zinc (II) complex. J. Mol. Struct. 2022, 1251, 132039 10.1016/j.molstruc.2021.132039. [DOI] [Google Scholar]
- Çakmak S.; Kansiz S.; Azam M.; Ersanlı C. C.; İdil O.; Veyisoğlu A.; Chutia A. Synthesis, Structural Investigation, Hirshfeld Surface Analysis, and Biological Evaluation of N-(3-Cyanothiophen-2-yl)-2-(thiophen-2-yl) acetamide. ACS Omega 2022, 7, 11320–11329. 10.1021/acsomega.2c00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Çakmak S.; Kansiz S.; Azam M.; Veyisoglu A.; Yakan H.; Min K. Synthesis, Spectroscopic Characterization, Single-Crystal Structure, Hirshfeld Surface Analysis, and Antimicrobial Studies of 3-Acetoxy-2-methylbenzoic Anhydride. ACS Omega 2022, 7, 17192–17201. 10.1021/acsomega.2c00879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatfaoui S.; Issaoui N.; Roisnel T.; Marouani H. Synthesis, experimental and computational study of a non-centrosymmetric material 3-methylbenzylammonium. J. Mol. Struct. 2020, 1225, 129132 10.1016/j.molstruc.2020.129132. [DOI] [Google Scholar]
- Morell C.; Grand A.; Toro-Labbé A. New dual descriptor for chemical reactivity. J. Phys. Chem. A 2005, 109, 205–212. 10.1021/jp046577a. [DOI] [PubMed] [Google Scholar]
- Chattaraj P. K.; Maiti B.; Sarkar U. Philicity: a unified treatment of chemical reactivity and selectivity. J. Phys. Chem. A 2003, 107, 4973–4975. 10.1021/jp034707u. [DOI] [Google Scholar]
- Basha A. A.; Khan F. L. A.; Muzammil P.; Fasiuddin G. S. Dielectric relaxation and dipole moment studies of hydrogen bonded complexes for enanthamide and valeramide with halogenated phenols using J-band microwave frequency. Mater. Res. Express 2022, 9, 075303 10.1088/2053-1591/ac7ea2. [DOI] [Google Scholar]
- Basha A. A.; Khan F. L. A.; Muthu S.; Imran P. M.; Kubaib A. Dielectric relaxation, dipole moment, electronic characterization and non-covalent interaction behavior of valeramide and halo-phenol in non-polar liquid: A density functional theory-based approach. J. Mol. Liq. 2023, 370, 121027 10.1016/j.molliq.2022.121027. [DOI] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Bourne P. E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavi Sastry G.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Schrödinger L. L. C.Schrödinger release 2019-4: protein preparation wizard; Epik; impact; prime; glide; LigPrep; induced fit docking protocol, New York, NY. 2019.
- Ammarah U.; Kumar A.; Pal R.; Bal N. C.; Misra G. Identification of new inhibitors against human Great wall kinase using in silico approaches. Sci. Rep. 2018, 8, 4894 10.1038/s41598-018-23246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q.; Peng Z.; Zhang Y.; Yang J. COACH-D: improved protein–ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 2018, 46, W438–W442. 10.1093/nar/gky439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floris S.; Fais A.; Rosa A.; Piras A.; Marzouki H.; Medda R.; Era B. Phytochemical composition and the cholinesterase and xanthine oxidase inhibitory properties of seed extracts from the Washingtonia filifera palm fruit. RSC Adv. 2019, 9, 21278–21287. 10.1039/C9RA02928A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra R. K.; Dhama K.; El–Arabey A. A.; Sarangi A. K.; Tiwari R.; Emran T. B.; Abdalla M. Repurposing benzimidazole and benzothiazole derivatives as potential inhibitors of SARS-CoV-2: DFT, QSAR, molecular docking, molecular dynamics simulation, and in-silico pharmacokinetic and toxicity studies. J. King Saud Univ., Sci. 2021, 33, 101637 10.1016/j.jksus.2021.101637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badraoui R.; Rebai T.; Elkahoui S.; Alreshidi M.; Veettil N.; V Noumi E.; Snoussi M. Allium subhirsutum L. as a potential source of antioxidant and anticancer bioactive molecules: HR-LCMS phytochemical profiling, in vitro and in vivo pharmacological study. Antioxidants 2020, 9, 1003. 10.3390/antiox9101003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed A. E.; Alghamdi S. S.; Alharbi N. K.; Alshehri F.; Suliman R. S.; Al-Dhabaan F.; Alharbi M. Limoniastrum monopetalum–Mediated Nanoparticles and Biomedicines: In Silico Study and Molecular Prediction of Biomolecules. Molecules 2022, 27, 8014. 10.3390/molecules27228014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneria M.; Parmar J.; Rakholiya K. Molecular docking and drug design of phytoconstituents from Couroupita guianensis–An in silico perspective. J. Pharmacogn. Phytochem. 2019, 8, 53–60. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.