

Abstract
Obesity is associated with elevated intracellular nitric oxide (NO) production, which promotes nitrosative stress in metabolic tissues such as liver and skeletal muscle, contributing to insulin resistance. The onset of obesity-associated insulin resistance is due, in part, to the compromise of hepatic autophagy, a process that leads to lysosomal degradation of cellular components. However, it is not known how NO bioactivity might impact autophagy in obesity. Here, we establish that S-nitrosoglutathione reductase (GSNOR), a major protein denitrosylase, provides a key regulatory link between inflammation and autophagy, which is disrupted in obesity and diabetes. We demonstrate that obesity promotes S-nitrosylation of lysosomal proteins in the liver, thereby impairing lysosomal enzyme activities. Moreover, in mice and humans, obesity and diabetes are accompanied by decreases in GSNOR activity, engendering nitrosative stress. In mice with a GSNOR deletion, diet-induced obesity increases lysosomal nitrosative stress and impairs autophagy in the liver, leading to hepatic insulin resistance. Conversely, liver-specific overexpression of GSNOR in obese mice markedly enhances lysosomal function and autophagy and, remarkably, improves insulin action and glucose homeostasis. Furthermore, overexpression of S-nitrosylation–resistant variants of lysosomal enzymes enhances autophagy, and pharmacologically and genetically enhancing autophagy improves hepatic insulin sensitivity in GSNOR-deficient hepatocytes. Taken together, our data indicate that obesity-induced protein S-nitrosylation is a key mechanism compromising the hepatic autophagy, contributing to hepatic insulin resistance.
Introduction
Autophagy plays important roles in protein quality control, organelle turnover, and the maintenance of basal energy balance (1). Failure to regulate autophagic processes is implicated in many aspects of obesity-associated metabolic diseases (1). In mouse models of obesity and diabetes, disruption of autophagy leads to increased food intake (2), insulin resistance (3), hepatic steatosis (3,4), hypoinsulinema (5), and muscle atrophy (6). Conversely, restoration of autophagy by either gain-of-function autophagy regulators (3) or treatment with chemical enhancers (7) ameliorates obesity-induced insulin resistance. Importantly, dysregulation of autophagy has been demonstrated in patients with metabolic disorders such as diabetes mellitus (8), obesity (9), fatty liver disease (10), and atherosclerosis (11). Despite this clear evidence that defects in autophagy play a critical role in chronic metabolic diseases, the physiological causes of this dysfunction of autophagy in obesity remain largely unknown.
Obesity-associated chronic inflammation is a major factor that contributes to insulin resistance (12). One hallmark of this obesity-associated inflammation is elevated nitric oxide (NO) production (13). NO regulates a wide range of cellular functions and signaling processes through protein S-nitrosylation (the covalent attachment of a nitrogen monoxide group to the thiol side chain of cysteine) by altering protein cellular localization, enzyme activity, protein stability, and protein complex formation (14). In cells, protein S-nitrosylation is regulated by both the NO synthase–mediated NO generation and dynamic removal of NO groups through cellular denitrosylation (15). S-nitrosoglutathione reductase (GSNOR), a major cellular denitrosylase (16), reduces GSNO (generated by transferring NO groups from S-nitrosylated protein to glutathione [GSH]) into GSH S-hydroxysulfenamide in a NADH-dependent manner (15).
In mouse models of obesity, S-nitrosylation of insulin receptor (IR), IR substrate, or Akt (protein kinase B) was shown to promote the onset of insulin resistance in skeletal muscle and adipose tissues (17). Chronic exposure to high energy and nutrient intake disturbs organelle homeostasis (12). Accumulating evidence shows that nitrosative modifications greatly influence organelle function, mediating the pathogenesis of human diseases. For example, S-nitrosylation of mitochondrial complex I positively regulates mitochondrial function at the reperfusion phase of myocardial infarction (18). In addition, Uehara et al. (19) established that S-nitrosylation of the redox enzyme protein disulfide-isomerase impairs endoplasmic reticulum (ER) function in mouse models of neurodegenerative disease. We recently demonstrated that S-nitrosylation of inositol-requiring enzyme 1 contributes to impaired ER homeostasis in mouse models of obesity (20). NO production also affects cellular homeostasis by modulating autophagy, although the directionality of this effect may be context dependent. Although it has been reported that NO suppresses starvation-induced autophagy in HeLa cells (21), NO signaling induces lipopolysaccharide-mediated autophagy in cardiomyocytes (22). Of note, lipid-induced impairment of autophagic flux in cardiomyocytes was implicated as being mediated by S-nitrosylation of a lysosomal ATPase (ATP6V1A1) (23).
Collectively, these findings indicate that NO and associated nitrosative modifications are critical to the regulation of organelle function, including the autophagic process. Therefore, it is plausible that nitrosative stress contributes to defective hepatic autophagy observed in obesity. In this study, we set out to examine the link between obesity-associated NO signaling and the autophagic process and to determine whether this underlies the emergence of insulin resistance and type 2 diabetes.
Research Design and Methods
Cell Culture and Reagents
Primary hepatocytes were isolated from wild-type (WT; C57BL/6J), GSNOR knockout (KO) mice, and WT mice transduced with either AAV8-thyroid hormone–binding globulin (TBG) green fluorescent protein (GFP) or AAV8-TBG GSNOR using collagenase type X (Wako Pure Chemical Industries, Tokyo, Japan) perfusion method (20). Earle’s balanced salt solution (EBSS; Sigma-Aldrich), 20 mmol/L ammonium chloride (Sigma-Aldrich), and 100 mmol/L leupeptin (Sigma-Aldrich) were used to treat cells for 4 h, and trehalose (100 mmol/L; Sigma-Aldrich) was used to treat cells for 16 h. For tests of insulin signaling, primary hepatocytes were stimulated with 5 nmol/L insulin (Sigma-Aldrich) for 10 min.
Mutagenesis, Adenovirus, and Adeno-Associated Virus Transduction
The hexosaminidase subunit β (HexB) and cathepsin B (CTSB) constructs were purchased from Sino Biological Inc. and OriGene, respectively, and the S-nitrosylation–resistant variants were generated by site-directed mutagenesis using the Q5 Site-Directed Mutagenesis Kit (New England BioLabs Inc.). Primary hepatocytes were transduced with the indicated adenoviruses (Ads) at a titer of 1 × 108 viral particles/mL. Adeno-associated virus 8 (AAV8)-TBG-icre, AAV8-TBG-eGFP, and AAV8-TBG-GSNOR were purchased from Vector Biolabs, and delivered into mice via retro-orbital injection at a titer of 1.25 × 1011 genome copy/mouse.
Western Blot Analysis
Proteins were extracted from cells or tissues and subjected to SDS-PAGE, as previously described (20). Membranes were incubated with anti-Atg7 (Cell Signaling Technology), anti-CTSB (Cell Signaling Technology), anti-GSNOR (Novus), anti-LC3(Novus), anti-p62 (Abnova), anti-Flag (Sigma-Aldrich), anti-HexB (Biorbyt), anti-pAKT (Cell Signaling Technology), anti-pIR (Calbiochem), anti-AKT (Santa Cruz Biotechnology), or anti-IR (Santa Cruz Biotechnology) antibody and then were incubated with the secondary antibody conjugated with horseradish peroxidase (Santa Cruz Biotechnology). Densitometry analyses of Western blot images were performed by using the Image Studio Software (LI-COR).
Quantitative Real-time RT-PCR
Total RNA was isolated using TRIzol reagent (Invitrogen) and reverse transcribed into cDNA using the iScript cDNA synthesis kit (Bio-Rad). Quantitative real-time RT-PCR analysis was performed using SYBR Green. The following primers are used: SCD1: forward 5′-TTCTTGCGATACACTCTGGTGC-3′, reverse 5′-CGGGATTGAATGTTCTTGTCGT-3′; SREBP-1a: forward 5′-GGCCGAGATGTGCGAACT-3′, reverse 5′-GGCCGAGATGTGCGAACTT-3′; SREBP-1c: forward 5′-GATGTGCGAACTGGACACAG-3′, reverse 5′-CATAGGGGGCGTCAAACAG-3′; SREBP 2: forward 5′-GCGTTCTGGAGACCATGGA-3′, reverse-5′-ACAAAGTTGCTCTGAAAACAAATCA-3′; FASN: forward 5′-AGAGATCCCGAGACGCTTCTA-3′, reverse 5′-GCCTGGTAGGCATTCTGTAGT-3′; PEPCK: forward 5′-CTGCATAACGGTCTGGACTTC-3′, reverse 5′-CAGCAACTGCCCGTACTCC-3′; G6Pase: forward 5′-CGACTCGCTATCTCCAAGTGA-3′, reverse 5′-GTTGAACCAGTCTCCGACCA-3′; Atg7: forward 5′-ATGCCAGGACACCCTGTGAACTTC-3′, reverse 5′-ACATCATTGCAGAAGTAGCAGCCA-3′; TFEB: forward 5′-CAAGGAGCGGCAGAAGAAAG-3′, reverse 5′- GCTGCTTGTTGTCATCTCC-3′; and 18S: forward 5′-AGTCCCTGCCCTTTGTACACA-3′, reverse 5′- CGATCCGAGGGCCTCACTA-3′.
Biotin Switch Assay
Biotin switch assays were performed as described by Jaffrey and Snyder (24) and Derakhshan et al. (25) with minor modifications (20). Biotinylated proteins were pulled down using NanoLink Streptavidin Magnetic Beads (Solulink), and the proteins of interest were examined using specific antibodies, as indicated. In situ detection of S-nitrosylated proteins was performed as described by Thibeault et al. (26). Biotinylated proteins were labeled using streptavidin conjugated with Alexa Fluor 488. The cells or liver sections were then subject to immunostaining for CTSB or HexB using anti-CTSB and anti-HexB antibodies and secondary antibodies conjugated with Alexa Fluor 568. The images were observed using a Zeiss 700 confocal microscope or a Leica Fluorescence Microscope. The images were quantified using ImarisColoc (Bitplane).
S-Nitrosylation Proteomics and Data Analysis
Briefly, liver tissues were lysed in HENS Buffer, free cysteines were blocked with methyl methanethiosulfonate, and S-nitrosylated cysteines were selectively labeled with iodoTMT (iodoacetyl Tandem Mass Tag) Reagent. TMT-labeled peptides were then enriched with anti-TMT antibody, and the multiplexed quantitative mass spectrometry data were collected using an Orbitrap Fusion Mass Spectrometer operating in MS3 mode using synchronous precursor selection for the MS2 to MS3 fragmentation (27). Tandem mass spectrometry (MS/MS) data were searched against the UniProt mouse database using the SEQUEST algorithm. The protein quantification values were normalized by quantile normalization followed by batch effect correction. Differential analysis was performed by using R package limma t test for two-group comparisons. The significant differentially expressed proteins (P < 0.05) were selected to conduct GO and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analyses by using hypergeometric testing with Benjamini-Hochberg procedure for multiple test adjustment and a minimum of two genes.
GSH, GSNOR Activity, HexB Activity, CTSB Activity Assays, and Measurement of Lysosomal pH
GSH content was measured using a Glutathione Assay Kit (Cayman Chemical) (20). The GSNOR enzyme activity assay was performed as described previously (16). The HexB activity assay was performed as described by Vaidyanathan et al. (28). The CTSB assay was performed using a CTSB Detection Kit (ImmunoChemistry Technologies LLC), and lysosomal pH was measured using LysoSensor Green DND-189 (Thermo Fisher Scientific). The lysosomal fractions were prepared by using a Lysosome Isolation Kit (Sigma-Aldrich).
Mouse Models
Animal care and experimental procedures were performed with approval from the University of Iowa Institutional Animal Care and Use Committee. C57BL/6J (The Jackson Laboratory), GSNOR KO (provided by Dr. Jonathan S. Stamler, Case Western Reserve University, Cleveland, OH), red fluorescent protein (RFP)-GFP-LC3 (provided by Dr. Joseph A. Hill, University of Texas Southwestern Medical Center, Dallas, TX), and GSNOR KO;RFP-GFP-LC3 mice and littermate WT lean controls were kept on a 12-h light/dark cycle. Mice used in the diet-induced obesity (DIO) model were placed on a 60 kcal% fat high-fat diet (HFD; Research Diets), immediately after weaning for 16–24 weeks. The 2920X Teklad Global Diet was used as the control diet. Trehalose (Sigma-Aldrich) was administrated by intraperitoneal injections (2 g/kg) daily for 1 week. All tissues were harvested, frozen in liquid nitrogen, and kept at −80°C until processed.
Glucose Tolerance Tests, Insulin Tolerance Tests, and Insulin Infusions
Glucose tolerance tests were performed by intraperitoneal glucose injection (1 g/kg) after overnight fasting, insulin tolerance tests were performed by intraperitoneal insulin injection (0.75 international units [IU]/kg) after 6 h of food withdrawal during the day, and insulin infusion studies were performed in mice with 6 h of food withdrawal during the day (20).
Measurement of Serum Insulin Levels, Plasma Lipid Profile, Aspartate Aminotransferase, and Alanine Aminotransferase
Serum insulin levels were measured in mice after 6 h of food withdrawal, using an ELISA kit (Crystal Chemicals). The plasma lipid profile and alanine aminotransferase (ALT)/aspartate aminotransferase (AST) levels were measured using the Piccolo Lipid Panel Plus (Abaxis, Inc.).
Oil Red O Staining
Frozen liver sections were fixed with 10% formalin, stained with 0.3% Oil Red O solution, and observed under a Nikon Microscope (20×). The images were quantified using Fiji/ImageJ (National Institutes of Health).
Human Liver Sections
Human nondiabetic and diabetic liver sections were purchased from BioChain Institute Inc., and human steatotic liver samples were obtained from the Liver Tissue Cell Distribution System at the University of Minnesota (Minneapolis, MN). The use of human liver tissues was approved by the institutional review board as nonhuman subjects research.
Statistical Analysis
The results are expressed as the mean ± SEM; n represents the number of individual experiments. Statistical analysis was performed by Student t test, one-way ANOVA followed by the post hoc Tukey test, or two-way ANOVA followed by the post hoc Bonferroni test, using Prism, as indicated in the legends.
Results
Obesity Elevates S-Nitrosylation of Lysosomal Proteins in the Liver, Leading to Impaired Lysosomal Function
We previously demonstrated that obesity results in a general increase in protein S-nitrosylation in the liver (20). To characterize the targets of S-nitrosylation, we implemented an iodoTMT labeling strategy to map and quantify S-nitrosylation modifications in the liver from mice (C57BL/6J) placed on an HFD and on a control regular diet (RD) using an liquid chromatography–MS/MS-based proteomic approach. GO and KEGG pathway enrichment analysis identified ∼50 S-nitrosylated proteins that were altered in livers of HFD-fed mice compared with mice fed with RD (Fig. 1A and B). Notably, DIO increased the S-nitrosylation of two lysosomal enzymes: CTSB and HexB (Fig. 1C).
Figure 1.
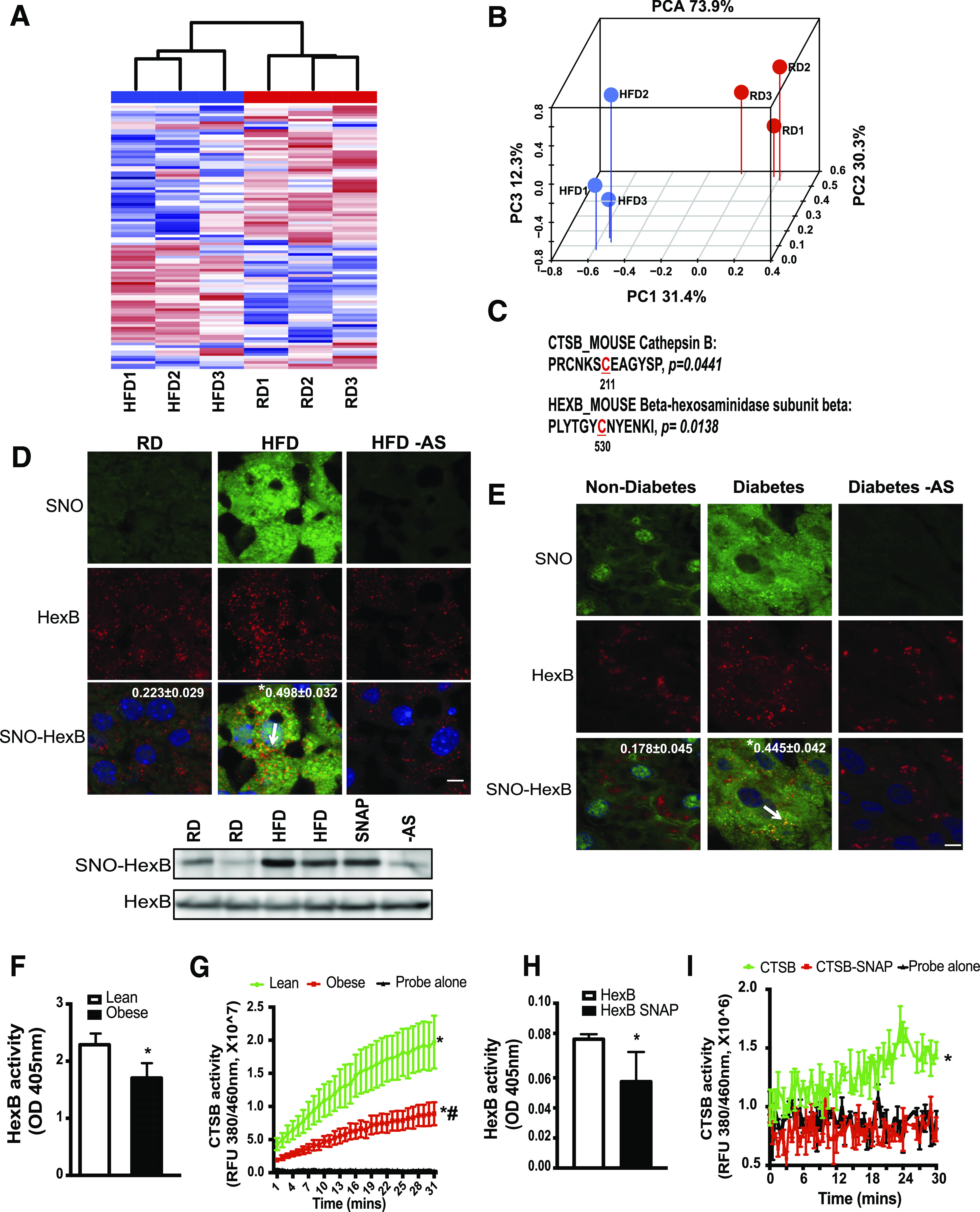
Obesity results in S-nitrosylation of lysosomal enzymes. A: Hierarchical clustering of S-nitrosylated (SNO) proteins in the livers of mice on the RD and HFD (n = 3, 16 weeks on HFD). S-nitrosylated proteins were labeled with iodoTMT and subjected to liquid chromatography–MS/MS-based proteomics. B: Top differential expressed 100 targets were selected for hierarchical clustering and principal component analysis (PCA). C: Lysosomal targets whose S-nitrosylation is significantly increased in the livers from mice fed with an HFD and the corresponding sites of S-nitrosylation. S-nitrosylated proteins that are deferentially expressed between the RD and HFD groups were used for GO and KEGG category classification. The sites of S-nitrosylation are indicated in red and numbered. D: top panel: Representative images (63×) of staining for S-nitrosylated HexB in the livers from mice fed with an RD or HFD (16 weeks on an HFD). S-nitrosylation staining was performed by a modified in situ biotin switch method. Blue, DAPI; green, S-nitrosylation; red, HexB. Arrow points to S-nitrosylated HexB. AS, ascorbate omitted (negative control for S-nitrosylation). Scale bar, 10 μm. Quantified colocalizations of S-nitrosylated HexB are shown on the top of each image. Data are shown as a Pearson correlation coefficient as the mean ± SEM. *Statistically significant difference relative to lean condition determined by Student t test (n = 3, P < 0.05). D, bottom panel: Representative Western blotting for S-nitrosylated HexB and input of HexB in livers. Each lane is a sample from an individual mouse. E: Representative images (63×) of staining for S-nitroyslated HexB in the livers from patients without diabetes and patients with diabetes. Blue, DAPI; green, S-nitrosylation; red, HexB. Arrow is S-nitrosylated HexB. Scale bar, 10 μm. Quantified colocalizations of S-nitrosylated HexB are shown on the top of each image. Data are shown as Pearson correlation coefficient as the mean ± SEM. *Statistically significant difference relative to nondiabetic condition determined by Student t test (n = 3, P < 0.05). F and G: HexB and CTSB activities in lysosomal fractions from lean and ob/ob mice (obese, 10 weeks old; n = 3). Same amount of liver tissues from lean and obese mice were used. Data are presented as the mean ± SEM. *Statistically significant difference relative to lean condition determined by Student t test (P < 0.05) (F). *Statistically significant difference relative to probe alone and #statistically significant difference relative to lean condition; analysis of the area under the curve (AUC) was performed by ANOVA with post hoc test (P < 0.05) (G). In vitro enzyme activity assays for HexB (H) and CTSB (I). Recombinant HexB (0.15 ng) or CTSB (10 ng) was used in the absence or presence of SNAP (10 mmol/L, 20 min). All data are presented as the mean ± SEM. *Statistically significant difference relative to samples without SNAP treatment, as assessed by Student t test (P < 0.05) (H), and analysis of the AUC was performed by ANOVA with post hoc test (P < 0.05) (I). OD, optical density; RFU, relative fluorescence units.
Lysosomes are acidic catabolic organelles containing around 60 soluble hydrolases that degrade extracellular and intracellular components sequestered by endocytosis or autophagy to facilitate the degradation and recycling of internal and external substrates (29). To validate that obesity elevates S-nitrosylation of these two candidates in vivo, we assessed S-nitrosylation in situ in liver sections of RD and HFD mice using a modified in situ biotin switch assay (26). Indeed, obesity promoted S-nitosylation of HexB and CTSB (Fig. 1D and Supplementary Fig. 1A and B) in the liver. To further quantify the S-nitrosylation of HexB, we performed a biotin switch assay in the liver lysates from HFD and RD mice. As shown in the Fig. 1D, obesity significantly increased S-nitrosylation of HexB in the liver. Of note, S-nitrosylation of HexB and CTSB was also elevated in the liver sections from patients with diabetes (Fig. 1E and Supplementary Fig. 1C) and patients with a high level of steatosis (Supplementary Fig. 1D).
To determine the effect of S-nitrosylation on the function of these enzymes, we first assessed HexB and CTSB activities in the lysosomal fractions isolated from the livers of lean and ob/ob mice. As shown in Fig. 1F and G, obesity resulted in decreased HexB and CTSB enzyme activities, indicating an impaired lysosomal function. We then performed an in vitro enzyme activity assay to examine the direct effects of S-nitrosylation on the functions of HexB and CTSB, using recombinant HexB and CTSB proteins. As shown in Fig. 1H and I, treatment with S-nitroso-N-acetylpenicillamine (SNAP; a chemical NO donor) impaired HexB and CTSB activities. Collectively, these results suggest that obesity-associated protein S-nitrosylation impairs the activities of lysosomal enzymes.
Obesity Results in Decreased Cellular Denitrosylation Capacity
GSNOR plays an important role in reversing the S-nitrosylation of various proteins, thereby influencing many pathophysiological processes (30,31). To determine whether GSNOR dysfunction contributes to the increased nitrosative stress in the liver in obesity, we first examined the GSNOR expression level in the livers from mice fed the RD and HFD. Although obesity did not significantly alter the protein expression level or the transcriptional regulation of GSNOR (Fig. 2A–C), it suppressed GSNOR activity in the livers of HFD mice (Fig. 2D). Similar results were observed in genetically obese (ob/ob) mice (Fig. 2E–H). GSH is required for GSNOR-mediated denitrosylation reactions (15). As shown in Supplementary Fig. 1E, HFD feeding reduced the level of GSH in the liver, which could contribute to a decreased GSNOR activity. Importantly, GSNOR activity was also attenuated in livers of human patients with diabetes (Fig. 2I and J), and there was a decrease in the expression of GSNOR in the livers of patients with a high level of steatosis (Supplementary Fig. 2). Together, these results demonstrate that obesity and diabetes impair GSNOR activity leading to decreased protein denitrosylation capacity and elevated nitrosative stress in the liver.
Figure 2.
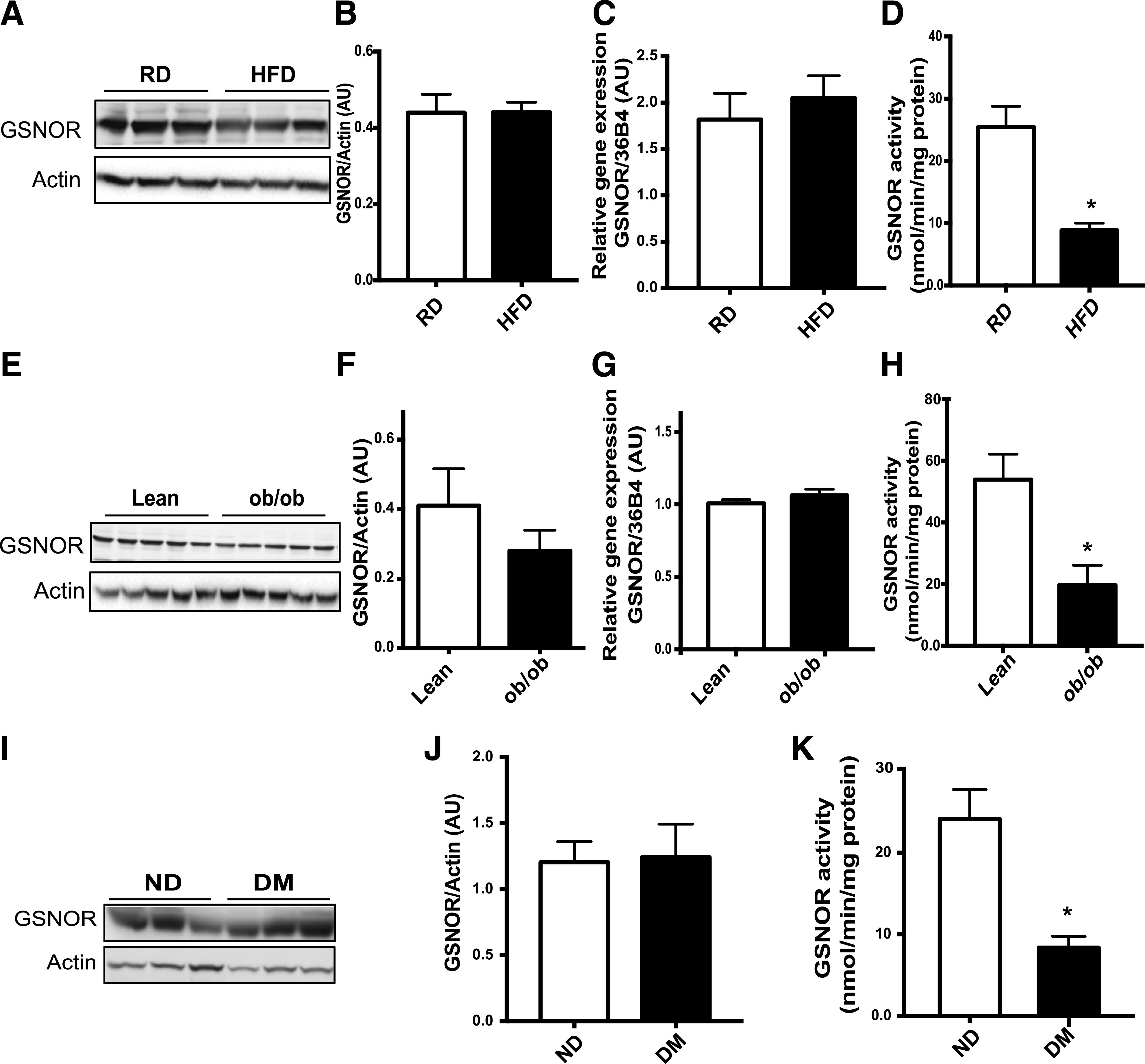
Obesity and diabetes are associated with decreased GSNOR-mediated denitrosylation. A: Western blotting of GSNOR protein in the livers of mice on the RD or HFD (n = 3, 16 weeks). B: Quantification of GSNOR protein in A, normalized to actin (n = 6). GSNOR mRNA (C) and activity (D) in livers from the mice in A (n = 6). *Statistically significant difference relative to lean control mice and analysis of area under curve was performed by Student t test (P < 0.05). Ten micrograms of liver lysate was used to measure the kinetics of GSNO-dependent NADH consumption in the absence or presence of 100 μmol/L GSNO. E: Western blotting of GSNOR protein and gene expression levels in ob/ob mice (10 weeks). F: Quantification of GSNOR protein in E. Quantification of GSNOR mRNA (G) and enzyme activity (H) were examined in the livers from these mice (n = 5). All data are presented as the mean ± SEM. *Indicates statistically significant difference relative to lean control mice in each group by Student t test (P < 0.05). Western blotting of GSNOR protein expression level (I), quantification of GSNOR protein (G), and enzyme activity (K) were examined in the livers from patients without diabetes (ND) and with diabetes (DM) (n = 4). All data are presented as the mean ± SEM. *Indicates statistically significant difference relative to the nondiabetic condition determined by Student t test (P < 0.05). AU, arbitrary units.
GSNOR Regulates Hepatic Lysosomal Function and Autophagy
To determine whether the reduction in GSNOR activity in obesity contributes directly to lysosomal nitrosative stress, HexB and CTSB S-ntirosylation was compared in the livers of WT and germline GSNOR KO mice fed either an RD or an HFD. In GSNOR KO mice, hepatic GSNOR activity was significantly reduced regardless of diet (Fig. 3A). Importantly, GSNOR deletion promoted general S-nitrosylated proteins in the liver and increased S-nitrosylation of HexB (Figs. 3B and 6E). To address whether the loss of GSNOR alters lysosomal function, we isolated primary hepatocytes from GSNOR KO and WT mice and induced the autophagy-lysosomal pathway by treating these cells with EBSS. GSNOR deletion not only led to impaired activities of CTSB and HexB (Fig. 3C and D) and an increase in lysosomal pH (Fig. 3E), but also exacerbated the obesity-associated downregulation of lysosomal enzyme activities and functions (Fig. 3C and E).
Figure 3.
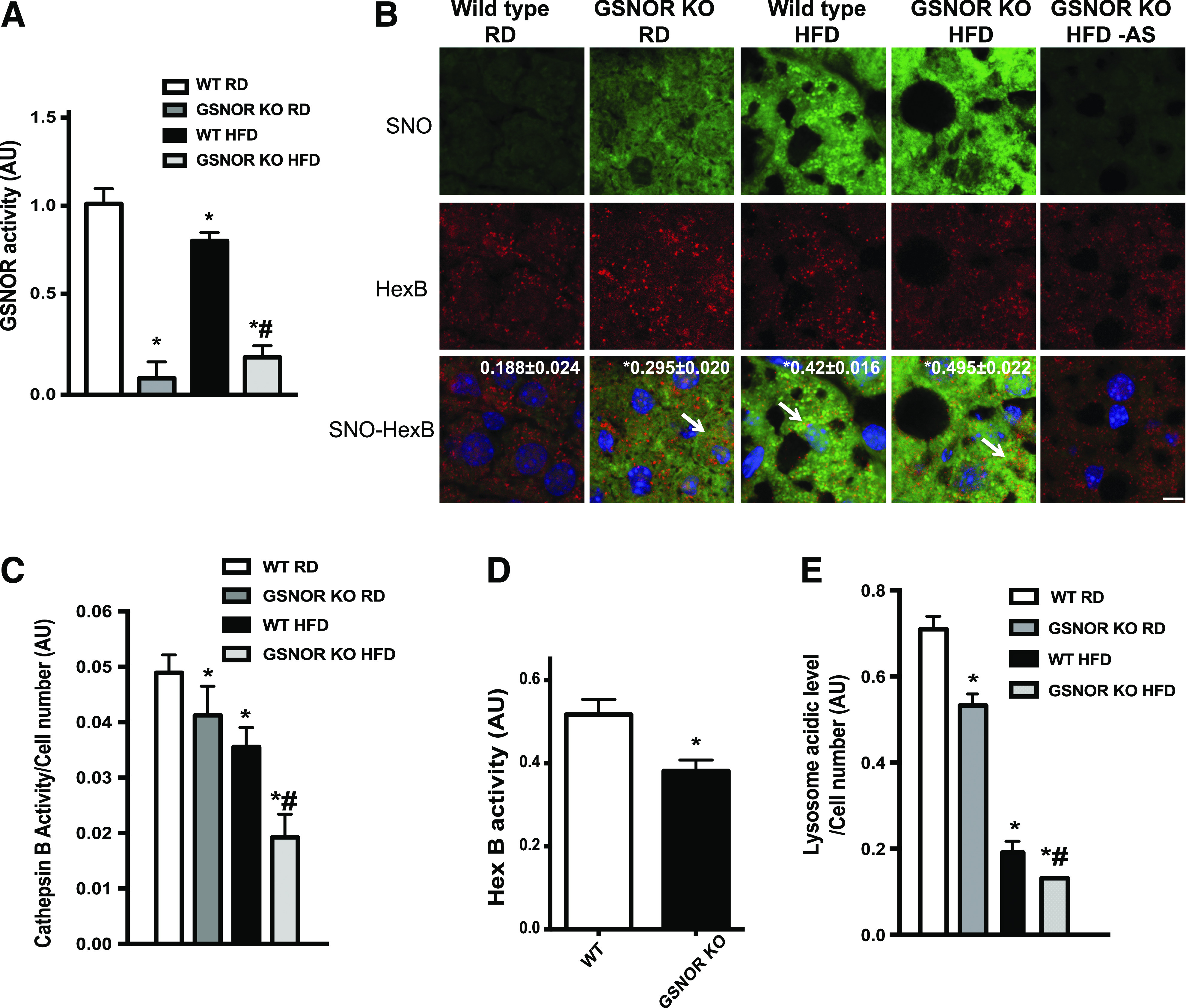
GSNOR deficiency results in impaired lysosomal function. A: GSNOR activities in the livers (n = 3) of WT and GSNOR KO mice on the RD or HFD (16 weeks). The resulting GSNOR activity was first normalized to the area under the curve of samples without GSNO then was normalized to those for the WT RD group; and results are presented as the mean ± SEM. *Significant difference relative to WT RD; #statistically significant difference between WT and GSNOR KO groups on the HFD determined by ANOVA followed by post hoc test (P < 0.05). B: Representative confocal images (63×) of staining for S-nitrosylation in liver sections from WT and GSNOR KO mice. Blue, DAPI; green, S-nitrosylation; red, HexB. Arrows are S-nitrosylated (SNO) HexB. Scale bar, 10 μm. Quantified colocalizations of S-nitrosylated HexB are shown on the top of each image. Data are shown as a Pearson correlation coefficient as the mean ± SEM. *Indicates statistically significant difference relative to WT RD determined by ANOVA followed by post hoc test (P < 0.05). C: CTSB activity measured in primary hepatocytes isolated from WT and GSNOR KO mice (n = 3; 16 weeks on HFD). Autophagy was induced by EBSS (4 h). *Indicates statistical significance compared with EBSS treatment in WT RD; #indicates statistical significance within WT and GSNOR KO groups on the HFD (determined by ANOVA followed by post hoc test) (P < 0.05). D: HexB activities in the primary hepatocytes isolated from WT and GSNOR KO mice on the RD (n = 3). All data are presented as the mean ± SEM. *Indicates statistical significance compared with WT group by Student t test (P < 0.05). E: Lysosomal acidity in live primary hepatocytes from WT and GSNOR KO mice on the RD or HFD (n = 3; 16 weeks on the HFD). Autophagy was induced by EBSS (4 h). *Indicates statistical significance compared with EBSS treatment in WT mice on the RD; #indicates statistical significance within WT and GSNOR KO groups on the HFD determined by ANOVA followed by post hoc test (P < 0.05). AU, arbitrary units; AS, ascorbate omitted.
Figure 6.
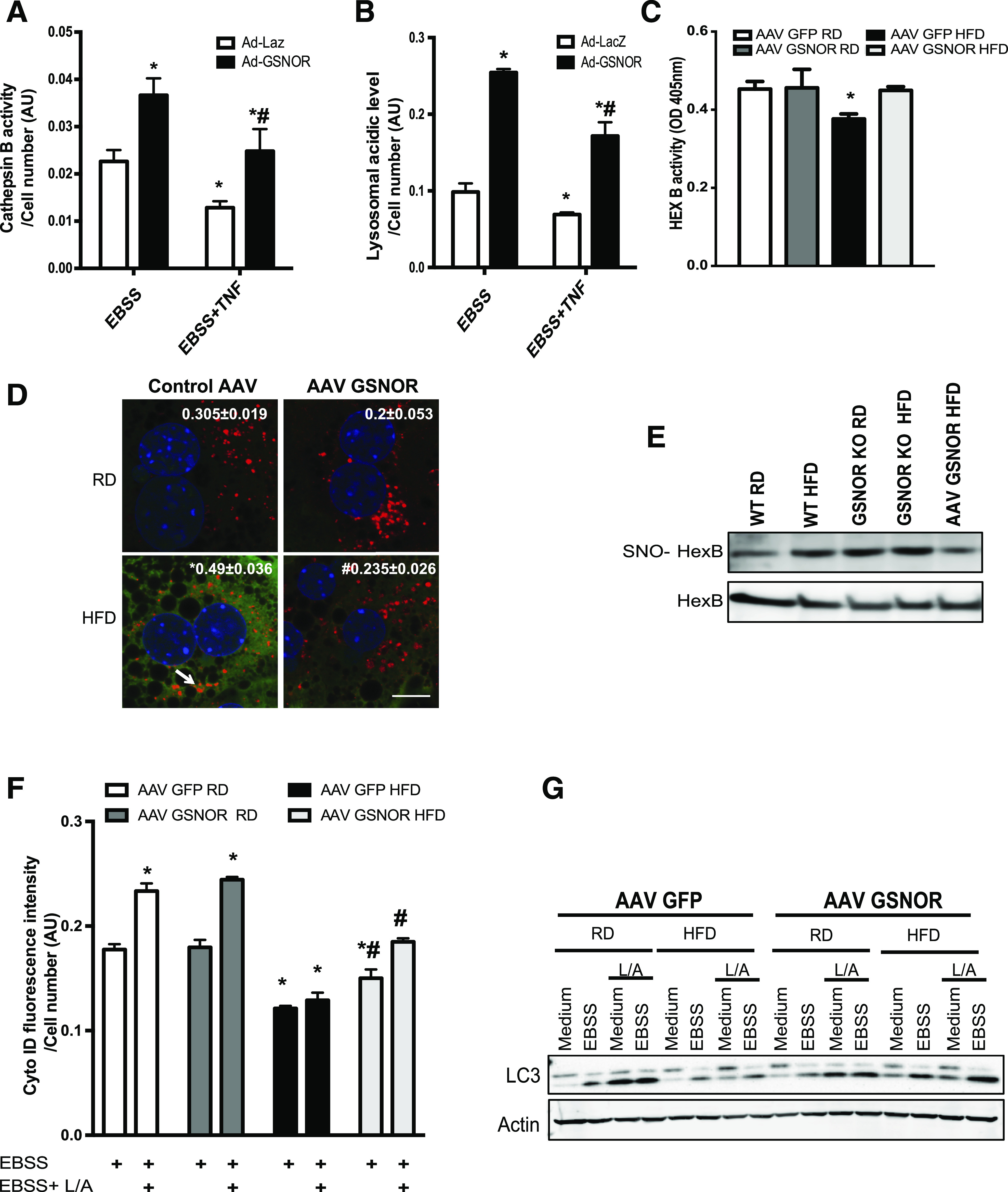
Amelioration of nitrosative stress by GSNOR improves hepatic lysosomal function and autophagy in obesity. CTSB activity (A) and lysosomal acidity (B) in live primary hepatocytes isolated from GSNOR KO mice (n = 3, fed on RD) transduced with control (Ad-LacZ) or Ad-GSNOR. Cells were treated with EBSS with or without pretreatment of tumor necrosis factor (10 ng/mL, 16 h). All data are presented as the mean ± SEM. *Indicates statistical significance compared with Ad-LacZ group determined by ANOVA followed by post hoc test (P < 0.05); #indicates statistical significance between Ad-GSNOR groups determined by ANOVA followed by post hoc test (P < 0.05). C: HexB activity in the primary hepatocytes isolated from WT mice transduced with AAV GSNOR vs. control virus and raised on an RD or an HFD (n = 3, 12 weeks on HFD). All data are presented as the mean ± SEM. *Indicates statistical significance compared with AAV GFP RD, determined by ANOVA with post hoc test (P < 0.05). D: Representative confocal images (63×) of staining for S-nitrosylation in primary hepatocytes from WT mice transduced with AAV GSNOR. Red, HexB; green, S-nitrosylation; blue, DAPI. Arrow points to S-nitrosylated HexB. Scale bar, 10 μm. Quantified colocalizations of S-nitrosylated (SNO) HexB are shown on the top of each image. Data are shown as Pearson correlation coefficient as the mean ± SEM. *Indicates statistically significant difference relative to AAV GFP RD; #indicates statistical significance between HFD groups determined by ANOVA followed by post hoc test (P < 0.05). E: S-nitrosylation of HexB in livers from WT mice, GSNOR KO mice, and WT overexpressing GSNOR and raised on the RD or HFD. Each lane used a mixture of protein lysates from three mice. F: Autophagic vacuoles in live primary hepatocytes from WT mice transduced with AAV GSNOR or AAV GFP and raised on the RD or HFD (n = 3) for 12 weeks, as detected using a Cyto-ID Kit. Cells were treated with EBSS (4 h); 20 mmol/L ammonium chloride and 100 mmol/L leupeptin (L/A; 4 h) were used to inhibit lysosomal degradation. All data are presented as the mean ± SEM. *Indicates statistical significance compared with AAV GFP RD in EBSS treatment; #indicates statistical significance between HFD groups determined by ANOVA followed by post hoc test (P < 0.05). G: LC3 conversion (arrow, LC3-II) in the primary hepatocytes from livers from WT mice with GSNOR overexpression. EBSS (4 h) was used to induce autophagy; 20 mmol/L ammonium chloride and 100 mmol/L leupeptin (4 h) were used to inhibit lysosomal degradation. Each lane contains a mixture of protein lysates from three mice. AU, arbitrary units; OD, optical density.
In late-stage autophagy, the autophagosome fuses with a lysosome to degrade its sequestered contents by lysosomal enzymes (32). To evaluate the effect of GSNOR-mediated lysosomal nitrosative stress on autophagy, we examined LC3 conversion and the accumulation of p62 in livers from WT and GSNOR KO mice. As shown in Fig. 4A, obesity blunted the fasting-induced LC3 conversion and elevated p62 levels in the livers of WT mice. This fasting-induced autophagy was almost abolished in the livers from lean and obese GSNOR KO mice. Furthermore, we bred GSNOR KO mice with RFP-GFP-LC3 transgenic mice (provided by Dr. Joseph Hill, University of Texas Southwestern Medical Center, Dallas, TX) and evaluated fasting-induced autophagic flux in the liver by monitoring the loss of GFP fluorescence in the tandem mRFP-GFP-LC3 reporter (33). As shown in Fig. 4B and C, obesity resulted in a decrease in GFP degradation, which was augmented by GSNOR deletion. In addition, we isolated primary hepatocytes from WT and GSNOR KO mice and transduced these cells with adeno-RFP-GFP-LC3. As shown in Fig. 4D, HFD feeding impaired the autophagic flux, which is augmented by GSNOR deletion regardless of diet. The same results were observed by quantifying autophagic vacuoles in these cells (Fig. 4E). Collectively, these results demonstrate that GSNOR plays an important role in modulating hepatic autophagy via regulating the lysosome-mediated autophagy process. However, the LC3 conversion is also suppressed in the GSNOR-deficient mice (Fig. 4A). It has been demonstrated that the S-nitrosylation of c-Jun N-terminal kinase (JNK) and IκB kinase (IKK) (21) and that of Bcl2 (34) impairs autophagy. We did not detect S-nitrosylated JNK or IKK (Fig. 4F) or a downregulation of the expression of genes encoding several key regulators of autophagy (Fig. 4G) in the livers of mice fed the HFD. Thus, GSNOR-mediated regulation of the early stages of autophagy appears to involve an unknown mechanism.
Figure 4.
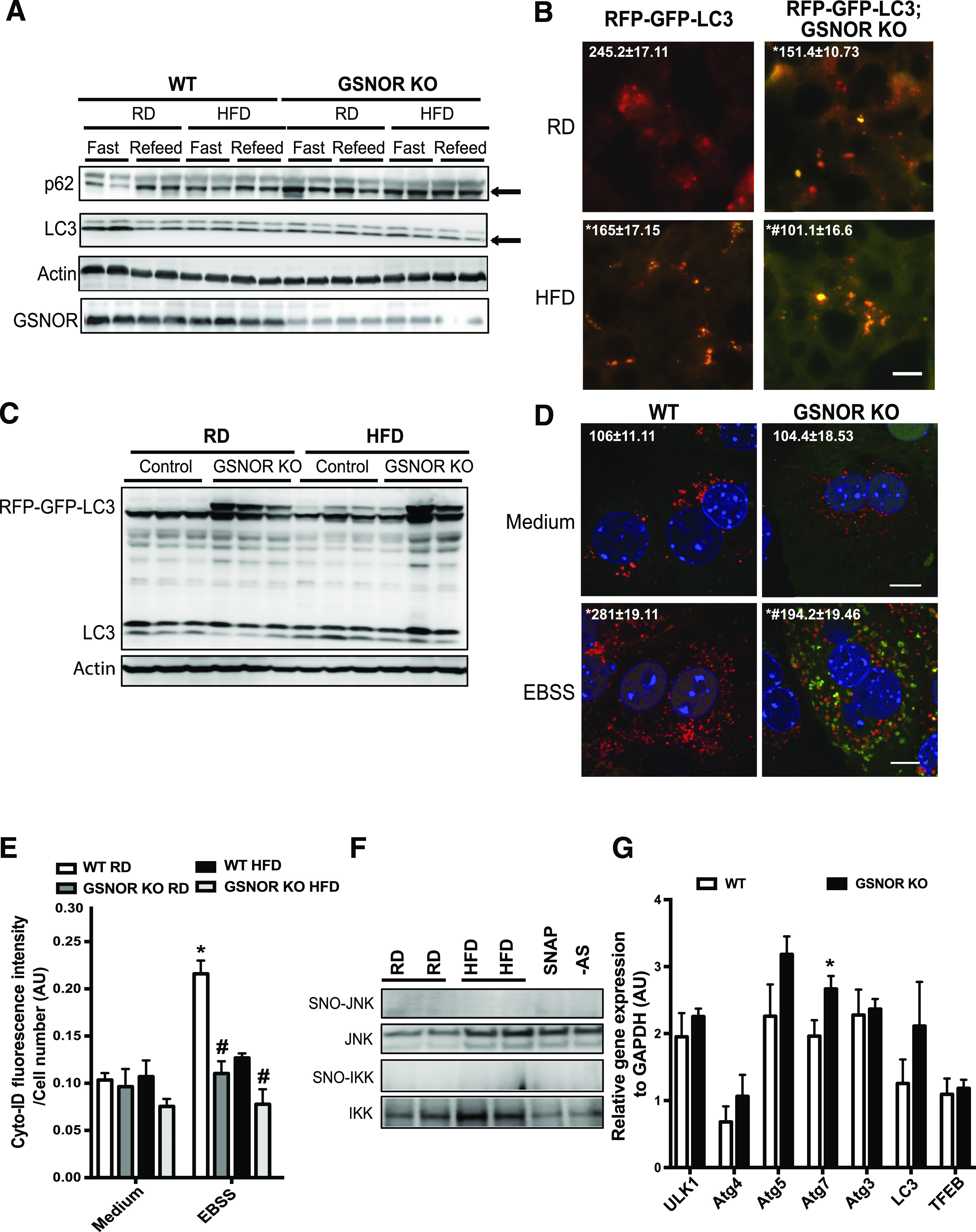
GSNOR deficiency contributes to defective autophagy. A: LC3 conversion (arrow indicated LC3-II) and p62 expression (arrow) in livers of WT and GSNOR KO HFD or RD mice (16 weeks). Mice were fasted for 16 h with or without refeeding for 4 h before being sacrificed. B: Representative images (40×) of RFP-GFP-LC3 puncta in the livers of RFP-GFP-LC3 and RFP-GFP-LC3;GSNOR KO mice fed the RD or HFD (16 weeks on HFD, fasted for 16 h). Quantified numbers of the red LC3 puncta/field are shown on the top of each image. Data are shown as the mean ± SEM. *Indicates statistically significant difference relative to RFP-GFP-LC3 RD. #Indicates statistical significance between HFD groups determined by ANOVA followed by post hoc test (P < 0.05). Scale bar, 10 μm. C: LC3 expression in livers from RFP-GFP-LC3 and RFP-GFP-LC3;GSNOR KO mice fed the RD or HFD (16 weeks on HFD, fasted for 16 h). D: Representative confocal images (63×) of primary hepatocytes isolated from WT or GSNOR KO mice (n = 3) transduced with ade-mRFP-GFP-LC3 (multiplicity of infection = 2). Cells were treated with EBSS (4 h), with the number of the red LC3 puncta/field on the top of image. All data are presented as the mean ± SEM. *Indicates statistical significance compared with WT medium; #indicates statistical significance between EBSS groups determined by ANOVA followed by post hoc test (P < 0.05). Scale bar, 10 μm. E: Quantification of autophagic vacuoles in live primary hepatocytes from WT and GSNOR KO mice (n = 3) using a Cyto-ID kit. Cells were treated with EBSS (4 h). All data are presented as the mean ± SEM. *Indicates statistical significance compared with WT RD group in medium treatment; #indicates statistical significance within WT and GSNOR KO groups on same diet determined by ANOVA followed by post hoc test (P < 0.05). F: Representative Western blots of S-nitrosylated (SNO) JNK and IKKβ in livers of mice fed with RD and HFD. AS, ascorbate omitted (negative control for biotin switch assay); SNAP, sample treated with SNAP, positive control. G: Levels of mRNAs encoding genes involved in autophagy regulation in livers of WT mice and GSNOR KO mice (fasted for 16 h), as assessed by quantitative RT-PCR. Data are presented as the mean ± SEM. *Indicates statistical significance compared with WT group, determined by Student t test (P < 0.05). AU, arbitrary units.
GSNOR Deletion Impairs Hepatic Insulin Sensitivity
To determine the physiological relevance of impaired cellular denitrosylation in the liver of obese mice, we evaluated the metabolic phenotype of germline GSNOR KO mice in lean and obese conditions. GSNOR KO mice have a similar glucose tolerance and insulin tolerance compared with littermate WT control mice in both RD- and HFD-fed conditions (Supplementary Fig. 3A and B) but have a slight decrease in body weight at the late time course of HFD (Supplementary Fig. 3C). In addition, there was no difference in the serum insulin levels between GSNOR KO and WT mice on either diet (Supplementary Fig. 3D). However, the GSNOR KO mice exhibited significantly diminished hepatic insulin action regardless of diet, as evidenced by a significant reduction in insulin-stimulated phosphorylation of IR β-subunit and phosphorylation of Akt (Fig. 5A and Supplementary Fig. 3E). This is associated with elevated lipid accumulation, and serum levels of cholesterol, triglyceride, LDL, ALT, and AST (Fig. 5B and C). We reasoned that the decreased hepatic insulin action but unaltered systemic glucose tolerance in the GSNOR KO mice might be due to compensation from other tissues, such as skeletal muscle, in the GSNOR KO mice (Supplementary Fig. 3F).
Figure 5.
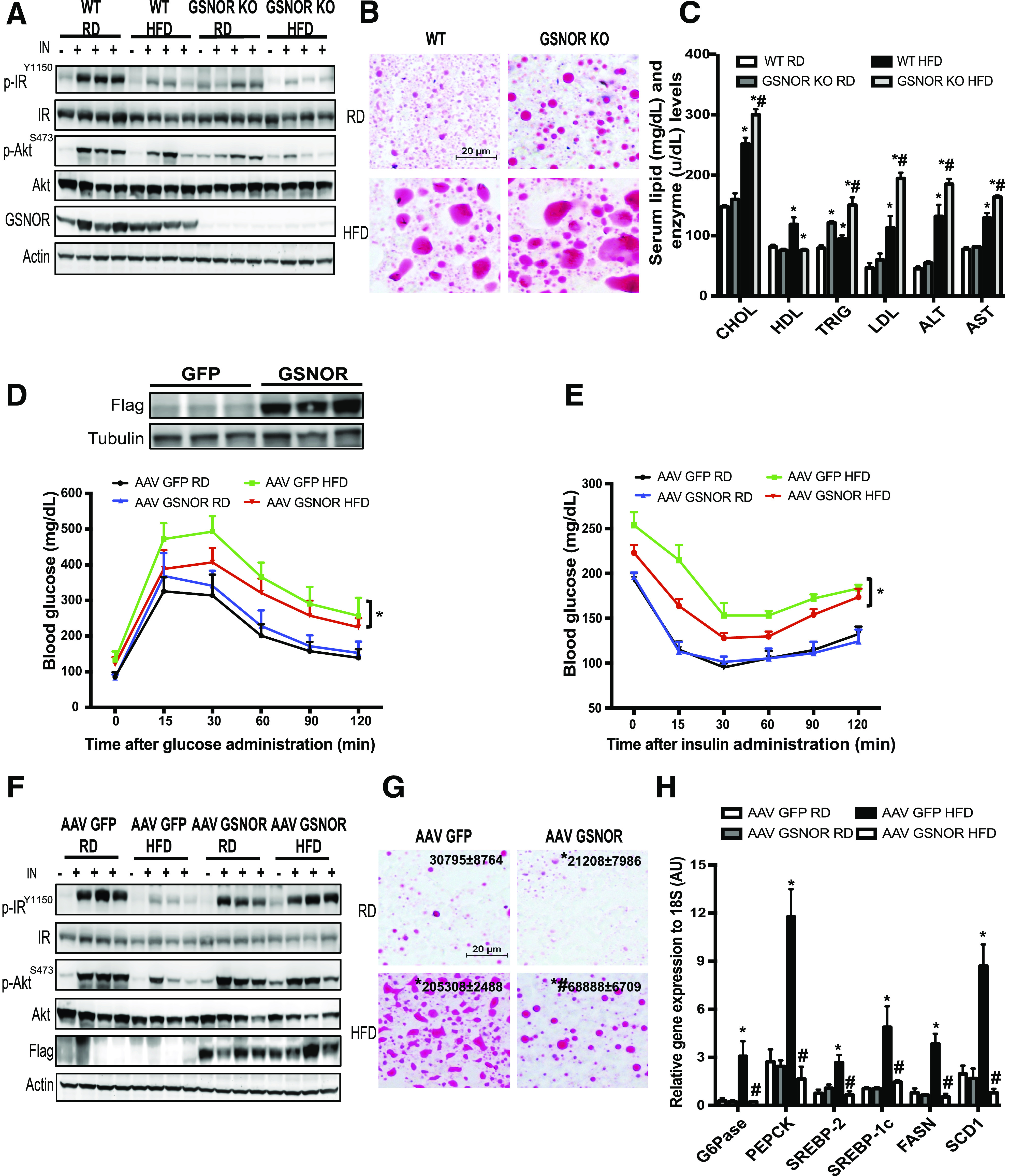
GSNOR regulates hepatic insulin sensitivity. A: Hepatic insulin action in the livers from WT and GSNOR KO mice (16 weeks on HFD). IN, insulin (0.75 IU/kg for 3 min); p-AKT, Aktser473; p-IR, IRtyr1150/1151. Data are representative of two individual cohorts of mice. B: Representative images (20×) of Oil Red O staining of liver sections of WT and GSNOR KO mice, as in A. C: Serum lipid profiles and levels of AST and ALT in these WT and GSNOR KO mice reared on the RD and HFD (n = 3, samples were collected after 6 h of food withdrawal). Data are presented as the mean ± SEM. *Indicates statistically significant difference relative to WT RD group; #indicates statistically significant difference between HFD groups determined by ANOVA followed by post hoc test (P < 0.05). D: Glucose tolerance in WT mice transduced with AAV8-TBG-GFP or AAV8-TBG-GSNOR and fed with RD or HFD (n = 10; on HFD for 8 weeks). GSNOR expression in the liver is shown in embedded panel. Data are representative of two individual cohorts of mice. E: Insulin tolerance in the mice shown in D. Data are presented as the mean ± SEM. *Indicates statistical analysis of the area under the curve between HFD groups performed by two-way ANOVA with post hoc test (P < 0.05). F: Hepatic insulin action in livers from WT mice transduced with AAV8-TBG eGFP or AAV8-TBG GSNOR (12 weeks on HFD). Each lane represents a mouse. Data are representative of two individual cohorts of mice. G: Representative images (20×) of Oil Red O staining of liver sections of WT mice transduced with AAV8-TBG-GFP or AAV8-TBG-GSNOR and fed with RD or HFD. *Indicates statistically significant difference relative to AAV GFP RD; #indicates statistical significance between the HFD groups determined by ANOVA followed by post hoc test (P < 0.05). H: Levels of mRNAs encoding gluconeogenesis and lipogenesis genes in livers of WT mice transduced with AAV GSNOR or control virus (n = 4–6), as assessed by quantitative RT-PCR. Data are presented as the mean ± SEM. *Indicates statistical significance compared with AAV GFP RD group; #indicates statistical significance between HFD groups determined by ANOVA followed by post hoc test (P < 0.05). AU, arbitrary units; CHOL, cholesterol; p, phosphorylated; TRIG, triglycerides.
GSNOR Overexpression Improves Glucose Homeostasis and Insulin Sensitivity in Obese Mice and Restores Hepatic Autophagy
Although GSNOR deletion led to hepatic insulin resistance both in vivo and in vitro, these experiments did not provide evidence that defects in GSNOR-mediated denitrosylation are the cause of the abnormal insulin action observed in obesity. To determine whether enhancing the hepatic denitrosylation capacity might improve insulin sensitivity in obese mice, we developed an AAV8-TBG-GSNOR vector, and transduced it to WT mice fed an RD and an HFD. AAV8 has greater liver transduction efficiency than other serotypes, and TBG is a hybrid promoter that incorporates sequences from the human TBG promoter and the macroglobulin/bikunin enhancer. Together they promote liver-specific transgene expression. AAV-mediated overexpression of GSNOR in the liver significantly enhanced GSNOR activities in the livers of WT mice (Supplementary Fig. 4A). Although overexpression of GSNOR did not alter glucose homeostasis in mice fed with an RD, GSNOR liver-specific overexpression significantly improved glucose tolerance (Fig. 5D), insulin tolerance (Fig. 5E), and hepatic insulin action (Fig. 5F and Supplementary Fig. 4C) in mice fed with an HFD. In agreement with enhanced hepatic insulin action, hepatic steatosis was attenuated and the expression of genes involved in gluconeogenesis and lipogenesis was suppressed by overexpression of GSNOR in livers from DIO mice (Fig. 5G and H). However, the overexpression of GSNOR in the liver did not affect serum insulin levels in either the RD or HFD mice (Supplementary Fig. 4B), nor did it affect GSNOR expression in muscle and white adipose tissues (Supplementary Fig. 4D and E).
To address whether the metabolic benefits of GSNOR are due to the regulation of hepatic autophagy, we first examined lysosomal function in hepatocytes isolated from GSNOR KO mice with GSNOR reconstitution. As shown in Fig. 6A and B, the restoration of GSNOR significantly enhanced CTSB activity and lysosomal acidity level in GSNOR KO hepatocytes. Moreover, the overexpression of GSNOR in DIO mice resulted in increased HexB activity in the liver (Fig. 6C). To determine whether the GSNOR-mediated enhancement of lysosomal function in obesity is through the attenuation of lysosomal nitrosative stress, we examined S-nitrosylated HexB in primary hepatocytes with GSNOR overexpression, and quantified S-nitrosylated HexB in the livers from WT mice, GSNOR KO mice, as well as from mice with GSNOR overexpression. In hepatocytes from obese mice, GSNOR overexpression lowered HexB S-nitrosylation (Fig. 6D). Moreover, the obesity-associated increases in HexB S-nitrosylation were augmented by GSNOR deletion but dampened by GSNOR overexpression (Fig. 6E). We also assessed starvation-induced (with EBSS) autophagy in hepatocytes isolated from RD and HFD mice with or without GSNOR overexpression. As shown in Fig. 6F and G and Supplementary Fig. 4F, GSNOR overexpression significantly improved starvation-induced autophagy in hepatocytes from DIO mice. Hence, experiments in both cellular systems and whole animals demonstrate the important role of GSNOR in sustaining hepatic autophagy in obesity.
Dysfunction of GSNOR-Mediated Autophagy Contributes to Hepatic Insulin Resistance
To link GSNOR-mediated lysosomal nitrosative stress with defective autophagy, we generated S-nitrosylation–resistant HexB (C530A) and CTSB (C211A) (Fig. 7A and Supplementary Fig. 5A). We found that the overexpression of S-nitrosylation–resistant HexB improved HexB activity and autophagy in GSNOR-deficient hepatocytes (Fig. 7A and B). Similar results were observed in GSNOR-deficient hepatocytes with S-nitrosylation–resistant CTSB overexpression (Supplementary Fig. 5B and C). Previously, we demonstrated that the restoration of Atg7 (a key autophagy regulator) in the liver improves autophagy and hepatic insulin sensitivity in obese mice (3). To determine the contributions of GSNOR-mediated autophagy to hepatic insulin resistance, we examined insulin action in GSNOR KO primary hepatocytes transduced with adeno-Atg7. Overexpression of Atg7 improved hepatic insulin resistance (Fig. 7C and Supplementary Fig. 5D). Similar results were observed in primary hepatocytes and livers of GSNOR-deficient mice that were administered an autophagy enhancer, trehalose (35) (Fig. 7D and E and Supplementary Fig. 5E and F). Together these data indicate that GSNOR sensitizes hepatic insulin signaling in part through modulating autophagy.
Figure 7.
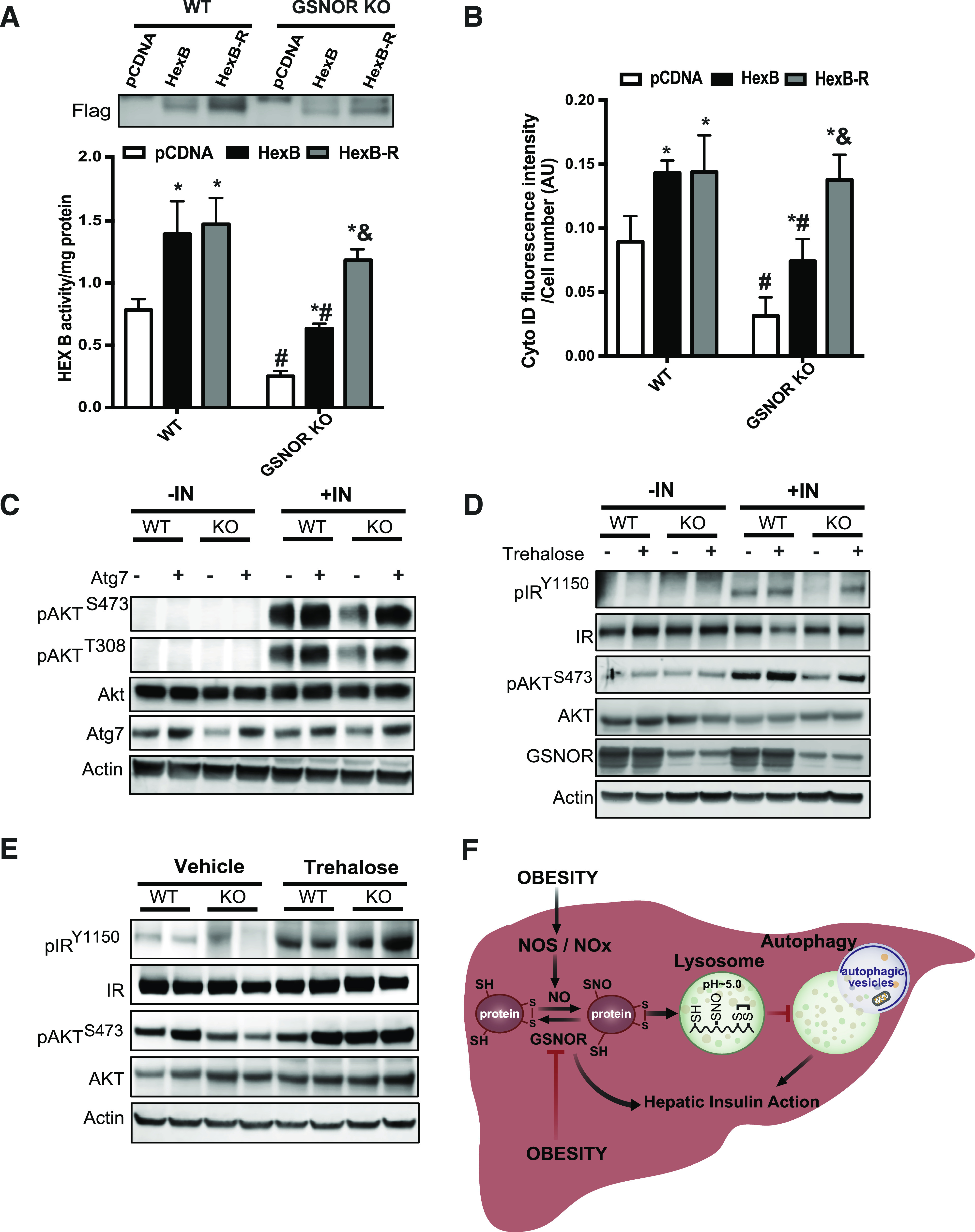
GSNOR-mediated lysosomal nitrosative stress contributes to impaired hepatic autophagy and insulin resistance. HexB activity (A) and autophagic vacuoles (B) in primary hepatocytes from WT and GSNOR KO mice (n = 3; 8 weeks on the RD) with EBSS treatment (4 h). HexB-R, S-nitrosylation (SNO)–resistant HexB; pcDNA, control plasmid. Data are presented as the mean ± SEM. *Indicates statistical significance compared with pcDNA in same mouse line; #indicates statistical significance between the WT and GSNOR KO groups with the same treatment; &indicates statistical significance between HexB and HexB-R groups in the same mouse line, determined by ANOVA followed by post hoc test (P < 0.05). C: Hepatic insulin action in primary hepatocytes isolated from WT and GSNOR KO mice transduced with Ad-Atg7 or control virus (Ad-lacZ). Each lane contains a mixture of protein lysates from 3 mice. IN, insulin, 5 nmol/L for 10 min. D: Hepatic insulin action in primary hepatocytes isolated from WT and GSNOR KO mice treated with trehalose (16 h, 100 mmol/L). For each lane, a mixture of protein lysates from three mice was used. E: Hepatic insulin signaling in the livers from WT and GSNOR KO mice (8 weeks on RD) treated with trehalose (2 g/kg, daily for 1 week). F: Working model of this study. Obesity results in impaired GSNOR-mediated denitrosylation of proteins in the liver, leading to elevated S-nitrosylation of lysosomal enzymes, defective autophagy, and impaired hepatic insulin action. AU, arbitrary units; NOx: nitrogen oxides; SH: free thiols.
Discussion
Obesity is characterized by a defect in hepatic autophagy. Multiple mechanisms have been implicated including hyperinsulinemia (36), the hyperactivation of mammalian target of rapamycin (37), lipid-induced blockage of the fusion of autophagosomes and lysosomes (38), and the downregulation of autophagy regulators (3). Although chronic inflammation is a well-established feature of obesity, thus far there is no evidence of cross talk between inflammatory pathways and autophagy regulation. Our study suggests that GSNOR deficiency plays a central role in the autophagic defect and provides a molecular basis for aberrant S-nitrosylation of key lysosomal enzymes. More specifically, in obesity, the loss of GSNOR results in lysosomal nitrosative stress, impairing autophagic flux and contributing to the development of insulin resistance and diabetes (Fig. 7F).
Obesity elevates nitrosative stress in the liver, skeletal muscle, and adipose tissue (17), contributing to the obesity-associated metabolic abnormities. Here, we identified a set of proteins whose S-nitrosylation state is significantly altered in the livers of mice with DIO (Fig. 1A) and confirmed the effect of S-nitrosylation on the functions of two lysosomal enzymes, HexB and CTSB. Previous studies have demonstrated that CTSB is subject to S-nitrosylation both in vitro (39) and in vivo (40) but did not evaluate the physiological consequences. We show, for the first time, the consequences of S-nitrosylation of CTSB and HexB in the setting of obesity; namely, the impairing of enzyme activity, lysosomal function, and autophagic flux, dampening hepatic insulin sensitivity. This is in agreement with a recent study showing that CTSB activity is suppressed in the livers of ob/ob mice (41).
NO plays important roles in the physiology and pathophysiology of the liver. Previously we demonstrated that obesity promotes S-nitrosylation of an ER protein, thereby contributing to disturbed ER function and glucose homeostasis (20). Our latest findings reveal that NO-mediated signaling also inhibits autophagic flux both in vitro and in vivo (Fig. 4). This is consistent with accumulating evidence that autophagy may be regulated by NO at multiple loci. For example, it was reported that NO inhibits autophagy induction by S-nitrosylation of JNK (21) and Bcl2 (34), whereas, NO can enhance autophagic flux by activating ataxia telangiectasia–mutated protein in breast cancer cells (42). Although we did not detect S-nitrosylated JNK and IKK in the liver (Fig. 4F), we did find that GSNOR-mediated signaling interfered with autophagy at an early stage (Figs. 4A and 6G). Further studies are required to establish the autophagic S-nitrosylation proteome as well as the temporal and spatial regulation of autophagy by inflammatory signals in the context of obesity.
The nitrosylation state of a protein is determined by the balance between nitrosylation and denitrosylation. GSNOR is a major cellular denitrosylase that removes NO groups from nitrosylated proteins (15). Although it has been shown that GSNOR deficiency contributes to impaired adipogenesis (43) and nonalcoholic steatohepatitis (44), the role of GSNOR in the pathogenesis of obesity and insulin resistance is unknown. Using a GSNOR KO mouse model and an AAV-mediated hepatic GSNOR overexpression approach, we have provided new insights into the effects of GSNOR on glucose and insulin metabolism in obesity. Although GSNOR KO mice show no overt changes in systemic glucose or insulin tolerance, a trend toward lower body weight and improved insulin sensitivity in the skeletal muscle was apparent in GSNOR KO mice fed the HFD (Supplementary Fig. 3). Nevertheless, hepatic overexpression of GSNOR in WT mice by AAV-mediated gene delivery clearly demonstrated the important role of GSNOR in the regulation of hepatic insulin action and whole-body glucose homeostasis (Fig. 5D–H). We do not exclude the possibility that the benefits of GSNOR overexpression might be due in part to improvements in other signaling pathways. In fact, it has been shown that S-nitrosylation of fatty acid synthase (45) and GSNOR deficiency impair DNA stability (46). It is possible that GSNOR-mediated signals regulate a transcriptional program that involved hepatic gluconeogenesis and lipogenesis. Nevertheless, the findings that both the gain and loss of GSNOR function alters hepatic autophagy (Figs. 4 and 6) and that enhancing autophagy improves hepatic insulin action in GSNOR KO cells or livers (Fig. 7C–E) indicates that the dysfunction of GSNOR-mediated autophagy is one factor that contributes to the defects in insulin signaling in obesity.
The finding that protein denitrosylation regulates hepatic autophagy provides a new perspective on obesity-associated insulin resistance. In line with our data, Montagna et al. (47) reported that GSNOR deletion impairs exercise-induced mitophagy in skeletal muscle (although this result contrasts with data showing that GSNOR deficiency enhances skeletal muscle strength) (48). Therefore, future studies should pay attention to the role of nitrosative stress in the regulation of autophagy activation and autophagic flux in a tissue and stress signaling–specific manner. Using the DIO mouse model, we demonstrated for the first time that obesity is associated with decreased hepatic GSNOR activity and showed that similar downregulation occurs in the livers of patients with diabetes and steatosis. These findings are consistent with the notion that GSNOR expression is significantly decreased in patients with hepatocellular carcinoma (46). However, whereas GSNOR inhibition has a genetic basis in hepatocellular carcinoma, we propose that in obesity reduced hepatic GSH and cellular NADH/NADPH levels and the dysfunction of the thioredoxin system (15) may be responsible. Moreover, our finding that inducible nitric oxide synthase deletion significantly improved GSNOR activity in the livers of obese mice (Supplementary Fig. 1F) suggests that obesity-associated inflammatory stress might lead to GSNOR inactivation.
Obesity-associated inflammation has been demonstrated to contribute to abnormal mitochondrial function (49) and disturbed ER homeostasis (20). Lysosomes are acidic catabolic organelles that degrade both extracellular materials by endocytosis and intracellular components sequestered by autophagy. Although the importance of lysosomes in inflammation was demonstrated in acute tissue injury (50), autoimmune disease (51), and infectious disease (52), how lysosomes respond to the nitrosative stress of metabolic tissues in obesity remains unresolved. Our study provides the first evidence that obesity-associated nitrosative stress directly targets the lysosome through S-nitrosylation–mediated modification of lysosomal proteins, contributing to defective hepatic autophagy. Indeed, a recent study (53) demonstrated that in primary macrophages extremely low doses of the inflammatory agent lipopolysaccharide inhibit fusion between lysosomes and autophagosomes.
In summary, our study demonstrates that the malfunction of GSNOR in obesity promotes lysosomal nitrosative stress and suppresses hepatic autophagy, contributing to the obesity-associated insulin resistance. Our study provides important insights into the molecular mechanisms that underlie the fine-tuning of the autophagy process by nitrosative signaling pathways. In addition, our study will also have a broader impact on the understanding of the interactions between inflammatory signaling and the lysosomes relevant to type 2 diabetes, fatty liver disease, cardiovascular diseases, and lysosomal storage diseases. It remains an intriguing possibility that small-molecule modulators of GSNOR activity and autophagy could be exploited for therapeutic interventions in obesity and type 2 diabetes.
Article Information
Acknowledgments. The authors thank Drs. Dale Abel (University of Iowa) and Vitor A. Lira (University of Iowa) for scientific discussions and insights. The authors also thank Drs. Tiangang Li (University of Kansas Medical Center), Steven Weinman (University of Kansas Medical Center), and Joseph Hill (University of Texas Southwestern Medical Center) for providing materials for the preliminary experiments and Dr. Gökhan Hotamisligil, Dr. Karen Inouye, and Alexandra Lee (Harvard T.H. Chan School of Public Health, Boston, MA) for technical support.
Funding. L.Y. is supported by American Heart Association Scientist Development Grant 15SDG25510016.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. Q.Q. designed the study, performed the experiments, and analyzed the data. Z.Z. performed the experiments and analyzed the data. A.O., S.C., R.C.K., and N.R.L.L. performed the experiments. W.-X.D. and J.S.S. provided critical reagents and scientific suggestions on the manuscript. Y.X. performed statistical analysis. L.Y. designed the study, performed the experiments, analyzed the data, conceived and supervised the study, and wrote the manuscript. L.Y. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db17-0223/-/DC1.
See accompanying article, p. 180.
References
- 1.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013;368:1845–1846 [DOI] [PubMed] [Google Scholar]
- 2.Kaushik S, Rodriguez-Navarro JA, Arias E, et al. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab 2011;14:173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010;11:467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jung HS, Chung KW, Won Kim J, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 2008;8:318–324 [DOI] [PubMed] [Google Scholar]
- 6.Masiero E, Agatea L, Mammucari C, et al. Autophagy is required to maintain muscle mass. Cell Metab 2009;10:507–515 [DOI] [PubMed] [Google Scholar]
- 7.Arai C, Miyake M, Matsumoto Y, et al. Trehalose prevents adipocyte hypertrophy and mitigates insulin resistance in mice with established obesity. J Nutr Sci Vitaminol (Tokyo) 2013;59:393–401 [DOI] [PubMed] [Google Scholar]
- 8.Masini M, Bugliani M, Lupi R, et al. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009;52:1083–1086 [DOI] [PubMed] [Google Scholar]
- 9.Kovsan J, Blüher M, Tarnovscki T, et al. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab 2011;96:E268–E277 [DOI] [PubMed] [Google Scholar]
- 10.Fukuo Y, Yamashina S, Sonoue H, et al. Abnormality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease. Hepatol Res 2014;44:1026–1036 [DOI] [PubMed] [Google Scholar]
- 11.Martinet W, De Meyer GR. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res 2009;104:304–317 [DOI] [PubMed] [Google Scholar]
- 12.Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006;444:860–867 [DOI] [PubMed] [Google Scholar]
- 13.Zahedi Asl S, Ghasemi A, Azizi F. Serum nitric oxide metabolites in subjects with metabolic syndrome. Clin Biochem 2008;41:1342–1347 [DOI] [PubMed] [Google Scholar]
- 14.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 2005;6:150–166 [DOI] [PubMed] [Google Scholar]
- 15.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol 2009;10:721–732 [DOI] [PubMed] [Google Scholar]
- 16.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2001;410:490–494 [DOI] [PubMed] [Google Scholar]
- 17.Kaneki M, Shimizu N, Yamada D, Chang K. Nitrosative stress and pathogenesis of insulin resistance. Antioxid Redox Signal 2007;9:319–329 [DOI] [PubMed] [Google Scholar]
- 18.Chouchani ET, Methner C, Nadtochiy SM, et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med 2013;19:753–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uehara T, Nakamura T, Yao D, et al. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006;441:513–517 [DOI] [PubMed] [Google Scholar]
- 20.Yang L, Calay ES, Fan J, et al. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 2015;349:500–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarkar S, Korolchuk VI, Renna M, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell 2011;43:19–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan H, Perry CN, Huang C, et al. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol 2009;296:H470–H479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaishy B, Zhang Q, Chung HS, et al. Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity. J Lipid Res 2015;56:546–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 2001;2001:pl1. [DOI] [PubMed] [Google Scholar]
- 25.Derakhshan B, Wille PC, Gross SS. Unbiased identification of cysteine S-nitrosylation sites on proteins. Nat Protoc 2007;2:1685–1691 [DOI] [PubMed] [Google Scholar]
- 26.Thibeault S, Rautureau Y, Oubaha M, et al. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol Cell 2010;39:468–476 [DOI] [PubMed] [Google Scholar]
- 27.McAlister GC, Nusinow DP, Jedrychowski MP, et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 2014;86:7150–7158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaidyanathan VV, Puri N, Roche PA. The last exon of SNAP-23 regulates granule exocytosis from mast cells. J Biol Chem 2001;276:25101–25106 [DOI] [PubMed] [Google Scholar]
- 29.Journet A, Chapel A, Kieffer S, Roux F, Garin J. Proteomic analysis of human lysosomes: application to monocytic and breast cancer cells. Proteomics 2002;2:1026–1040 [DOI] [PubMed] [Google Scholar]
- 30.Beigi F, Gonzalez DR, Minhas KM, et al. Dynamic denitrosylation via S-nitrosoglutathione reductase regulates cardiovascular function. Proc Natl Acad Sci U S A 2012;109:4314–4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Que LG, Yang Z, Stamler JS, Lugogo NL, Kraft M. S-nitrosoglutathione reductase: an important regulator in human asthma. Am J Respir Crit Care Med 2009;180:226–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001;2:211–216 [DOI] [PubMed] [Google Scholar]
- 33.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010;140:313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright C, Iyer AK, Kulkarni Y, Azad N. S-Nitrosylation of Bcl-2 Negatively Affects Autophagy in Lung Epithelial Cells. J Cell Biochem 2016;117:521–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 2007;282:5641–5652 [DOI] [PubMed] [Google Scholar]
- 36.Liu HY, Han J, Cao SY, et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem 2009;284:31484–31492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komatsu M. Liver autophagy: physiology and pathology. J Biochem 2012;152:5–15 [DOI] [PubMed] [Google Scholar]
- 38.Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J 2010;24:3052–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stamler JS, Simon DI, Osborne JA, et al. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A 1992;89:444–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci Signal 2013;6:rs1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inami Y, Yamashina S, Izumi K, et al. Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem Biophys Res Commun 2011;412:618–625 [DOI] [PubMed] [Google Scholar]
- 42.Tripathi DN, Chowdhury R, Trudel LJ, et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci U S A 2013;110:E2950–E2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao Y, Gomes SA, Rangel EB, et al. S-nitrosoglutathione reductase-dependent PPARγ denitrosylation participates in MSC-derived adipogenesis and osteogenesis. J Clin Invest 2015;125:1679–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goto M, Kitamura H, Alam MM, et al. Alcohol dehydrogenase 3 contributes to the protection of liver from nonalcoholic steatohepatitis. Genes Cells 2015;20:464–480 [DOI] [PubMed] [Google Scholar]
- 45.Choi MS, Jung JY, Kim HJ, Ham MR, Lee TR, Shin DW. S-nitrosylation of fatty acid synthase regulates its activity through dimerization. J Lipid Res 2016;57:607–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei W, Yang Z, Tang CH, Liu L. Targeted deletion of GSNOR in hepatocytes of mice causes nitrosative inactivation of O6-alkylguanine-DNA alkyltransferase and increased sensitivity to genotoxic diethylnitrosamine. Carcinogenesis 2011;32:973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montagna C, Di Giacomo G, Rizza S, et al. S-nitrosoglutathione reductase deficiency-induced S-nitrosylation results in neuromuscular dysfunction. Antioxid Redox Signal 2014;21:570–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moon Y, Cao Y, Zhu J, et al. GSNOR deficiency enhances in situ skeletal muscle strength, fatigue resistance, and RyR1 S-nitrosylation without impacting mitochondrial content and activity. Antioxid Redox Signal 2017;26:165–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384–387 [DOI] [PubMed] [Google Scholar]
- 50.Yang M, Cao L, Xie M, et al. Chloroquine inhibits HMGB1 inflammatory signaling and protects mice from lethal sepsis. Biochem Pharmacol 2013;86:410–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Persellin RH. Role of lysosomes in the pathogenesis of rheumatoid arthritis. Med Clin North Am 1968;52:635–641 [PubMed] [Google Scholar]
- 52.Weissmann G. The role of lysosomes in inflammation and disease. Annu Rev Med 1967;18:97–112 [DOI] [PubMed] [Google Scholar]
- 53.Baker B, Geng S, Chen K, et al. Alteration of lysosome fusion and low-grade inflammation mediated by super-low-dose endotoxin. J Biol Chem 2015;290:6670–6678 [DOI] [PMC free article] [PubMed] [Google Scholar]