

Abstract
Therapeutic interventions targeting secondary insults, such as delayed hypoxemia, provide a unique opportunity for treatment in severe traumatic brain injury (TBI). Erythropoietin (EPO) is a hypoxia-responsive cytokine with important roles in neurodevelopment, neuroprotection and neuromodulation. We hypothesized that recombinant human erythropoietin (rhEPO) administration would mitigate injury in a combined injury model of TBI and delayed hypoxemia. Utilizing a clinically relevant murine model of TBI and delayed hypoxemia, we characterized how ongoing rhEPO administration influenced neurogenesis, neuroprotection, synaptic density and, behavioral outcomes early after TBI, and the impact on long-lasting outcomes 6 months after injury. We employed novel object recognition (NOR) and fear conditioning to assess long-term memory. At 1-month post-injury, we observed a significant increase in cued-fear memory response in the rhEPO-injured mice compared with vehicle-injured mice. This was associated with neuroprotection and neurogenesis in the hippocampus and Mitogen-activated protein kinase (MAPK)/cAMP response element-binding protein (CREB) signaling activation and increased of excitatory synaptic density in the amygdala. Early rhEPO treatment after injury reduced neurodegeneration and increased excitatory synaptic density in the hippocampus and amygdala at 6 months post-injury. However at 6 months post-injury (4 months after discontinuation of rhEPO), we did not observe changes in behavioral assessments nor MAPK/CREB pathway activation. In summary, these data demonstrate that ongoing rhEPO treatment initiated at a clinically feasible time point improves neurological, cognitive, and histological outcomes after TBI in the setting of secondary hypoxemic insults.
Keywords: Traumatic brain injury, hypoxemia, erythropoietin, neuroprotection, fear conditioning, neurogenesis
Introduction
Approximately 1.7 million people experience traumatic brain injury (TBI) each year and over 5 million face TBI-related disabilities (Gardner and Zafonte, 2016). Systemic secondary insults such as hypoxemia and hypotension during the acute period may exacerbate injury severity (Davies et al., 2018; Parikh et al., 2016). Neuroprotective therapeutics targeting the primary injury have shown promise in preclinical studies but failed to translate in part due to longer intervals between injury and therapeutic administration in clinical trials (Arango and Bainbridge, 2008; Hutchison et al., 2008; Li et al., 2015; Winn et al., 2007). Targeting secondary insults after TBI in the intensive care setting provides a unique opportunity for therapeutic interventions with narrow temporal windows of efficacy.
The presence of erythropoietin (EPO) and the EPO receptor (EPOr) in the central nervous system (CNS) has prompted investigations into the therapeutic potential of exogenous EPO administration in the setting of CNS injury (Marti, 2004; Rabie and Marti, 2008). In preclinical studies, the neuroprotective effects of recombinant human EPO (rhEPO) have been proposed to be mediated through several mechanisms (Brines and Cerami, 2005) including reduction in glutamate toxicity (Kawakami et al., 2001), decreased apoptosis (Digicaylioglu and Lipton, 2001; Villa et al., 2003), antioxidant, and anti-inflammatory effects (Hellewell et al., 2013), increased neurogenesis (Hassouna et al., 2016) and angiogenesis (Marti et al., 2000), and mitochondrial protection (Rey et al., 2019). Limitations of previous reports include early drug administration in relation to injury (Grasso et al., 2007), lack of incorporation of systemic secondary insults, as well as the absence of long-term functional outcomes (Adamcio et al., 2008) reducing the translatability of preclinical studies to the clinical setting.
Clinical studies have showed inconclusive results of rhEPO efficacy in TBI patients (Gantner et al., 2018; Li et al., 2016; Nichol et al., 2015; Robertson et al., 2014). The EPO-TBI trial did not find improvement in the 6-month GOS-E in moderate and severe TBI patients treated with rhEPO(Nichol et al., 2015). However, there was a trend toward reduced 6 months mortality in patient receiving rhEPO. Sub-cohort analysis of this trial demonstrated 6 months mortality was reduced in patients receiving rhEPO classified as diffuse TBI and who did not receive a neurosurgical intervention (Skrifvars et al., 2018). Clinical trials have been limited to short courses of rhEPO (2–3 doses) and rudimentary 6 months outcomes measures. Animal studies with longer durations of rhEPO treatment with assessments of efficacy at similar long-term time points are needed to inform future clinical trial design.
rhEPO administration in rodents has been associated with functional improvements in hippocampal long-term potentiation and memory (Adamcio et al., 2008). Some of the functional effects of rhEPO in brain injury models have been speculated to be associated with stimulation of neuronal survival and neurogenesis (Lu et al., 2005; Mahmood et al., 2007; Wang et al., 2004). Mitogen-activated protein kinase (MAPK)/ cAMP response element-binding protein (CREB) signaling pathway activation has a well described role in memory consolidation and synaptic plasticity (Schafe et al., 2001). Therefore, we hypothesized that activation of the MAPK/CREB signaling pathway could serve as a possible mechanism by which rhEPO administration improves memory performance.
The present report sought to determine the biochemical, histological, and functional effects of rhEPO administration after TBI and delayed hypoxemia initiated at a clinically feasible time point. We hypothesized that rhEPO treatment would induce neuroprotective effects enhancing neuronal activation in the hippocampus-amygdala loop through MAPK/CREB signaling pathway.
Materials and methods
Traumatic brain injury and delayed hypoxemia
All procedures were approved by the Washington University Animal Studies Committee (Protocol 19–0864) and are consistent with the National Institutes of Health guidelines for the care and use of animals. Animals were housed 5/cage and had free access to water and food with a 12-hour light/dark cycle. C57BL/6J 6–8 week old male mice (Jackson Laboratory, Bar Harbor, ME) weighing 20–25 grams (g) were used. We performed controlled cortical impact (CCI) as previously described (Celorrio et al., 2021a; Celorrio et al., 2021b; Davies et al., 2018; Parikh et al., 2016). Briefly, mice were anesthetized with 5% isoflurane at induction, followed by maintenance at 2% isoflurane for the procedure’s duration. Buprenorphine sustained release (0.5 mg/kg subcutaneously) was administered before scalp incision. The head was shaved and ear bars were used to stabilize the head within the stereotaxic frame (MyNeurolab, St. Louis, MO). Then, a single 5-mm craniectomy was performed by an electric drill on the left lateral side of the skull centered 2.7 mm lateral from the midline and 3 mm anterior to lambda. Animals were randomized to sham or injury after craniectomy using computer-generated number randomization. For injured animals, the 3-mm electromagnetic impactor tip was then aligned with the craniectomy site at 1.2 mm left of midline and 1.5 mm anterior to the lambda suture. The impact was delivered at 2 mm depth (velocity 5 m/s, dwell time 100 ms). The ears bars were released immediately after the injury. All animals then received a loose fitting plastic cap secured over the craniectomy with Vetbond (3M, St. Paul, MN). The skin was closed with interrupted sutures and was treated with antibiotic ointment before the mouse was recovered from anesthesia on a warming pad. One day after surgery, animals who had undergone CCI experienced hypoxemia (8% O2, 4% CO2) for 60 min in a hypoxia chamber (Coy Laboratory, Grass Lake, MI). A mixture of N2, O2, and CO2 was utilized to maintain normocarbic hypoxemia (Parikh et al., 2016). Sham animals did not experience hypoxemia and were placed in cages directly next to the chamber while injured littermates were in the hypoxia chamber.
Recombinant human erythropoietin treatment
Male mice were injected intraperitoneally (i.p.) with 5000 U/kg rhEPO (Amgen, Thousand Oaks, CA), the most common dose utilized in murine models (Grasso et al., 2007; Lee et al., 2017). Initial doses at either 2 or 8 hours prior to hypoxia were used for acute experiments. For the 1-month (Fig. 2A) and 6-months (Fig. 5A) experiments, mice received a total of 6 and 9 doses of EPO, respectively, with the first dose of rhEPO administrated 8 h before hypoxemia. Daily doses were given for the next 2 consecutive days, followed by weekly rhEPO injections to model a previous clinical trial (Robertson et al., 2014). Injured animals were randomized to receive rhEPO or saline vehicle administration using computer-generated number randomization. All sham animals received EPO to assess for toxicity associated with long-term rhEPO exposure in the uninjured brain.
Fig. 2.
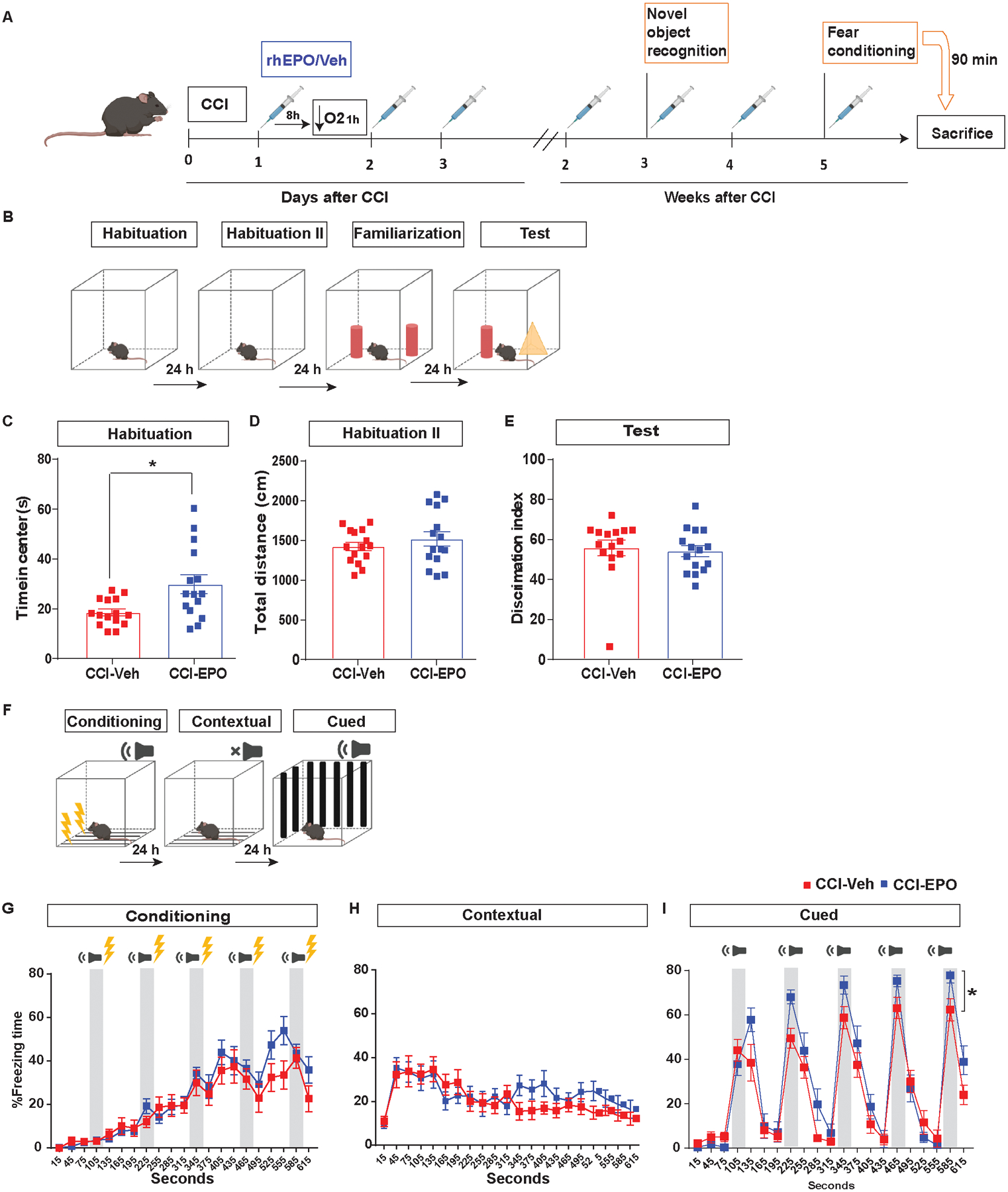
Ongoing rhEPO administration improved fear memory response but not novel memory. A, Experimental design performing NOR 15 days and fear conditioning 1 month post-injury. B, NOR paradigm. C, time in the center during the habituation day. Unpaired t-test *p< 0.05 (D) total distance during the second habituation day. (E) discrimination index during the test day. F, Fear conditioning scheme. Quantification of percentage of freezing time on G, conditioning, H, contextual memory I, cued memory. Two-way (Treatment and Tone) repeated measures ANOVA for tone freezing (n = 15/group). Tone F(4,112) = 31.11 p < 0.0001. Treatment*Tone interaction F(4,112) = 5.21 p = 0.0007 *p = 0.015. Abbreviations: CCI, controlled cortical impact; rhEPO, recombinant human erythropoietin; EPOr, erythropoietin receptor; Veh, vehicle.
Fig 5.
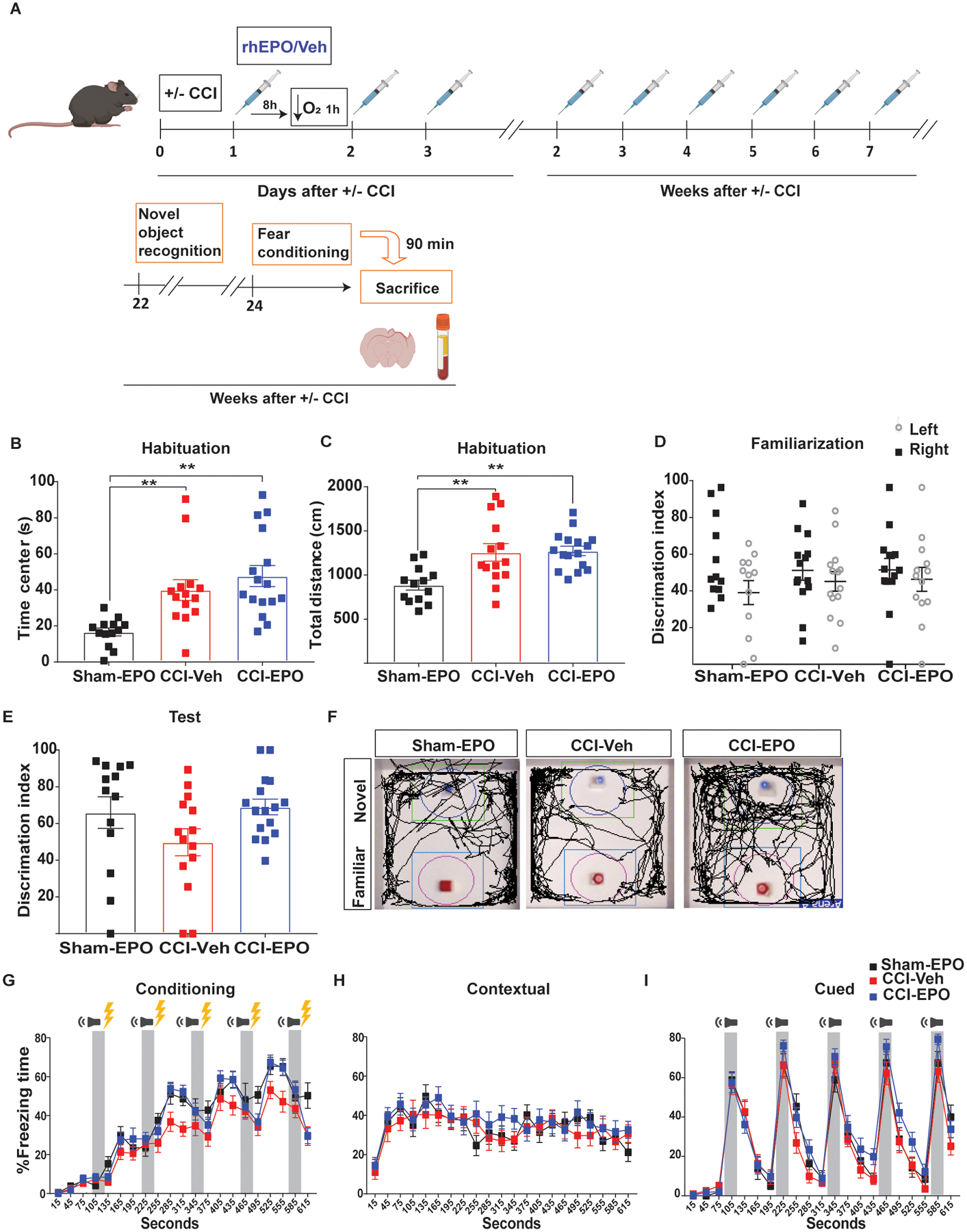
rhEPO treatment did not enhance behavioral improvement 6 months post-injury. A, Experimental design. NOR on day 1 quantification of (B) time in the center. One-way ANOVA followed by Tukey multiple comparison post hoc test F(2, 40) = 9.68, p = 0.0004. **p<0.01 (C) total distance. F(2, 40) = 8.91, p = 0.0006. **p<0.01. On day 3 (familiarization) quantification of (D) discrimination index. On day 4 (novel recognition test) quantification of (E) discrimination index. F, Open field arenas with exploration traces. Fear conditioning 3-day paradigm quantification of percentage freezing time of (G) conditioning, (H) contextual and (I) cued memory. n=13–16 mice per group. Abbreviations: CCI, controlled cortical impact; rhEPO, recombinant human erythropoietin; Veh, vehicle.
5-bromo-2’-deoxyuridine (BrdU) treatment
For the analysis of neurogenesis, mice received i.p. injections of 5-bromodeoxyuridine (BrdU, Sigma-Aldrich) 50 mg/kg for 10 consecutive days starting 24 h post-injury.
Enzyme-linked immunosorbent assay
Blood and brain EPO immunoassays were performed using an enzyme-linked immunosorbent assay (ELISA) in accordance with the manufacturers’ instructions (Quantikine® Mouse EPO immunoassay, R&D systems, Minneapolis, MN).
Hematological parameters
A 200-μl blood sample was collected retro-orbitally before sacrifice using EDTA-treated tubes. Blood samples were analyzed using xs-800i Sysmex (Kobe City, Japan).
Novel object recognition test
The Novel Object Recognition (NOR) was performed over a 4-day period (Fig. 2B). On day 1 and 2 (habituation phase), mice were placed in open-field arena and allowed to explore the space for 5 minutes (min). On day 3 (familiarization session), mice were placed in the open field arena with two identical objects (towers of Lego bricks) at 5 cm from the walls for 10 min. On day 4 (test day), mice were returned to the arenas where one of the objects was exchanged for a new object (small falcon tissue culture flask half-filled with mouse bedding). We calculated the Discrimination Index (DI), allowing discrimination between the novel and familiar objects, i.e., the exploration time for novel object (TN) was divided by the total amount of time interacting with the novel and familiar objects (TF): %DI = (100 × TN)/ (TN + TF). After every session, the open field box and objects were cleaned with 70% ethanol to minimize olfactory cues.
Contextual and cued fear conditioning
To study fear memory response in mice we used a fear conditioning protocol as previously described (Celorrio et al., 2021a; Celorrio et al., 2021b). Briefly, we used a 3-day fear conditioning experimental paradigm (Fig. 2F). On day 1 (conditioning), mice were placed in context A for 10.5 min and received 5 tone-shock pairings (30 s tone with 0.5 mA and shock during the last 2 s). Freezing time in 30 s epochs was measured by a blinded observer. On day 2 (contextual test), mice were placed in context A for 10.5 min and freezing time was measured to assess contextual fear memory. On day 3 (cued test), mice were placed in a novel context B (checkered walls and white hard cover on the floor) to eliminate any confounding interactions of contextual fear for 10.5 min and subsequently given five 30 s tones without any shocks. After the final tone-shock pairing (on day 1) or just tone (on day 3), mice remained in the conditioning chamber for 30 s before being returned to their home cages. Freezing was defined as the absence of visible movement except that required for respiration.
Tissue processing
Mice were anesthetized with isoflurane followed by transcardial perfusion with ice-cold 0.1 M heparinized phosphate-buffered saline (PBS, pH 7.4) followed by 4% paraformaldehyde (PFA, Sigma-Aldrich, St Louis, MO). Perfused brains were kept in 4% PFA at 4°C overnight, followed by equilibration in 30% sucrose for at least 48 h before sectioning. For biochemical techniques, hippocampus and amygdala were rapidly removed, dissected, and flash frozen.
Immunohistochemistry
Histological sections were examined to assess pathological changes in the hippocampus and amygdala. Brains were taken 90 min after the completion of the fear conditioning test for fluorescent labeling of pCREB. This time point was selected to accurately identify changes in pCREB expression, which have been shown to increase between 30 min and 120 min following fear memory (Chaaya et al., 2019). Fifty-μm thick serial sections were cut on a freezing microtome starting with a visibly complete corpus callosum and continuing caudally to bregma −3.08 mm. Sets of 12 cryosections spaced every 300 μm were used for immunohistochemical studies. Staining was performed on free-floating sections washed in Tris-buffered saline (TBS) between applications of primary and secondary antibodies. Endogenous peroxidase was blocked by incubating the tissue in TBS plus 0.3% hydrogen peroxide for 10 min. Normal goat serum (3%) in TBS with 0.25% Triton X (TBS-X) was used to block nonspecific staining for all antibodies. Slices were then incubated at 4°C overnight with the primary antibodies (Table 1). For colorimetric immunohistochemistry, antibody binding was detected by incubating sections with biotinylated secondary antibodies (Table 1) in TBS-X. Colorization was achieved using the VECTASTAIN Elite avidin-biotin complex (ABC)-HRP kit solution (Vector Laboratories, Burlingame, CA) followed by the application of 3–3’-diaminobenzidine (3–3’-DAB, Sigma-Aldrich). Sections were mounted on glass slides in TBS-X, dried and dehydrated in 50%, 70%, 95% and twice with 100% ethanol followed by Xylene (Sigma-Aldrich) before coverslipping with Dibutylphthalate Polystyrene Xylene (DPX, Sigma-Aldrich). For fluorescent immunohistochemistry, antibody binding was detected by incubating sections with Alexa fluorescence secondary antibody (Table 1) for 2 hours. Sections were mounted on glass slides in TBS-X, dried, and coverslipped with mounting medium for fluorescence with DAPI (Vector Laboratories, Burlingame, CA).
Table 1.
Overview of the primary antibodies used in the present study
| Antibody | Fluorophore | Species | Source | Product number |
|---|---|---|---|---|
| NeuN | Mouse monoclonal | Millipore | MAB377 | |
| NeuN | Rabbit polyclonal | Millipore | MAB377 | |
| DCX | Rabbit polyclonal | Abcam | Ab18723 | |
| BrdU | Rat monoclonal | Abcam | Ab6326 | |
| Iba1 | Rabbit polyclonal | Wako | 019–19741 | |
| PSD-95 | Rabbit polyclonal | Invitrogen | 51–6900 | |
| Synapsin | Guinea Pig | Synaptic systems | 106 004 | |
| β-actin | Mouse monoclonal | Sigma | ||
| pJAK2 | Rabbit polyclonal | Cell Signaling | 3776 | |
| JAK2 | Rabbit polyclonal | Cell Signaling | 3230 | |
| EPOr | Goat polyclonal | RandD system | P14753 | |
| MAPK | Rabbit polyclonal | Cell Signaling | 9101 | |
| pMAPK | Rabbit polyclonal | Cell Signaling | 4695 | |
| CREB | Rabbit polyclonal | Cell Signaling | 9197 | |
| pCREB | Rabbit polyclonal | Cell Signaling | 9198 | |
| Secondary antibody | AF598 | Donkey anti-goat | Thermo Fisher | A-11055 |
| Secondary antibody | AF594 | Donkey anti-rat | Thermo Fisher | A-21209 |
| Secondary antibody | AF647 | Donkey anti-mouse | Thermo Fisher | A-31571 |
| Secondary antibody | AF488 | Goat anti-guinea pig | Thermo Fisher | A-11073 |
| Secondary antibody | AF598 | Goat anti-Rabbit | Thermo Fisher | A-11011 |
| Secondary antibody | AF488 | Donkey anti-rabbit | Thermo Fisher | A-21206 |
| Secondary antibody | Biotinylated goat anti-rabbit | Vector Laboratories | BA-1000–1.5 | |
| Secondary antibody | IRDye® 800CW | Donkey anti-mouse | LI-COR | 925–32212 |
| Secondary antibody | IRDye® 800CW | Donkey anti-rabbit | LI-COR | 925–32213 |
Quantification of immunohistochemistry
Stereological analysis was performed as previously described (Celorrio et al., 2021a; Celorrio et al., 2021b). Fluorescence images were taken on a Zeiss Axio Imager Z2 Fluorescence Microscope (Zeiss) with ApoTome 2 optical sectioning grid imager with 20X objective. 20 μm z stacks with 1 μm interval were obtained of the ipsilateral dentate gyrus (DG). The DG of three serial dorsal hippocampal slices 300 μm apart from each mouse were counted for colocalization of BrdU with doublecortin (DCX) and NeuN (Table 1).
Immunoblotting
Nuclear, membrane, and cytosolic proteins were extracted from hippocampal and amygdala samples dissected from fresh tissue 90 min after fear conditioning (Chaaya et al., 2019). Each piece of tissue was homogenized in 5 volumes of lysis buffer containing 4 mM HEPES pH 7.4 (Sigma-Aldrich), 0.32 M sucrose (Sigma-Aldrich), 2 mM Na3VO4 (Sigma-Aldrich), 25 mM NaF (Sigma-Aldrich), and 1x protease inhibitors (Roche, Mannheim, Germany) using an overhead stirrer (Fisher Scientific, Waltham, MA). Samples were kept on ice for 20 min and centrifuged at 1500 g for 5 min at 4°C. The supernatant (S1) was recovered and spun at 16,000 g for 20 min to remove insoluble debris from the cytosolic fraction. The supernatant (S2) was stored at −20°C (cytosolic fraction). The pellet from the first centrifugation (P1) was resuspended in 5 mM PBS pH 7.4 containing 50 mM NaCl (Sigma-Aldrich), 150 mM sucrose (Sigma-Aldrich), 5 mM KCl (Sigma-Aldrich), 2 mM dithiothreitol (Sigma-Aldrich), 1 mM MgCl2 (Panreac), 0.5 mM CaCl2 (Sigma-Aldrich), 2mM Na3VO4 (Sigma-Aldrich), 25 mM NaF (Sigma-Aldrich) and, 1x protease inhibitors (Roche) and kept on ice for 1 minute. The samples were placed on the top of a sucrose layer (30% wt/vol sucrose and 10 mM NaCl in a 2.5 mM Tris-HCl pH 7.4 buffer) and centrifuged at 13,000 g for 5 min at 4°C. The supernatant was discarded and the pellet resuspended in 50 mM Tris-HCl pH 7.4 buffer containing 300 mM NaCl, 0.5% Triton X-100, and 1x protease inhibitors. Samples were incubated on ice for 20 min and centrifuged at maximum speed for 10 min at 4°C. The supernatant was stored at 20°C (nuclear fraction). Protein content was determined with the Thermo Scientific Pierce BCA Protein Assay Kit (Thermo scientific) following the manufacturer’s instructions. Protein samples from different experimental conditions were run in a randomized order on the same gels (Invitrogen) with the experimenter blinded to experimental condition. SDS-PAGE was performed on NuPAGE 4%−12% Bis-Tris polyacrylamide gels, and the proteins resolved were transferred on to nitrocellulose membranes. These membranes were blocked at room temperature for 1 h in Odyssey Blocking Buffer (PBS, LI-COR, Lincoln, NE). The membranes were then probed with the appropriate primary antibody at 4°C for 16 h (Table 1). Bound antibodies were detected using Highly Cross-Adsorbed IRDye Secondary Antibodies (LI-COR, Table 1).
Cresyl violet-staining
Cresyl violet staining was used for the detection of Nissl bodies in the cytoplasm of neurons on PFA sections in order to measure lesion and hippocampal volume. After 3 washes in TBS, tissue was mounted on charged slides and dried overnight. The following day, slides were put in a cylinder glass holder and incubated in FD cresyl violet solution (FD Neurothecnologies, Inc, Colombia, MD) for 10 min. The remaining cresyl violet solution was rinsed away with water for 20 min. Slides were then dried and dehydrated in 95% ethanol (10 min), twice in 100% ethanol placed in xylene (8 min) before being coverslipped with DPX (Sigma-Aldrich). The extent of tissue loss in the ipsilateral hemisphere for each animal was quantified using images of cresyl violet-stained slices acquired using a Zeiss Axio Scan Z1 Brightfield microscope (Zeiss, White Plains, NY). Tissue loss in the injured hemisphere was calculated as a percentage of the tissue volume in the contralateral hemisphere as previously described (Davies et al., 2018).
Quantification of synaptic loci
Quantification of synaptic loci was performed utilizing a semi-automated pipeline based on MATLAB (MathWorks, Portola Valley, CA) and Imaris 9.3.1 software (Bitplane, Concord, MA) as previously described (Reitz et al., 2021; Sauerbeck et al., 2020). Confocal images were obtained on a LSM 880 microscope with AiryScan detector (Zeiss) from the ipsilateral molecular layer of the DG. Spots were detected for each channel using an x-y size of 0.2 μm, a z size of 0.6 μm, and automated background subtraction. A 0.1-μm x-y and 0.3-μm z guard was applied to exclude spots intersecting the edge of the image volume. Synaptic loci were identified using previously developed MATLAB scripts to find the nearest neighbor based on the x-y-z centroid of the top 20% brightest puncta. A cutoff of 260 nm pre-to-postsynaptic separation was used to quantify synaptic loci.
Statistical analysis
All data for each animal was entered and tracked utilizing a REDCap database to maintain data integrity (Harris et al., 2009). For initial short-term studies, experiments were powered to detect a 30% difference between injured-mice treated with rhEPO (CCI-EPO) and injured mice receiving vehicle (CCI-Veh). For long-term behavior studies, experiments were powered to detect a 33% improvement in performance of CCI-EPO compared with CCI-Veh. Sham mice treated with EPO (Sham-EPO) were utilized to assess consistency between cohorts at 6 months experiments. Data was assessed for normal distribution with Shapiro Wilk test. Student t test or Mann Whitney U test were used for histological data and behavioral data when appropriate. For fear memory tests, data was found to have normal distribution and repeated measures two-way ANOVA was performed. All analysis was performed with Statistica v13.3 (TIBCO software. Palo Alto, CA).
Results
Delayed hypoxemia increased endogenous EPO and EPOr synthesis in the hippocampus following TBI
EPO expression is canonically induced by hypoxia-inducible factor (HIF) in the kidneys and in the brain (Chandel et al., 1998). Two hours post-hypoxia (26 h post-injury, Fig. 1A) we observed a 3-fold increase in the plasma EPO concentration of injured animals compared with sham (Fig. 1B); however, there were no differences in brain EPO levels between the groups (Fig. 1C). Additionally, hypoxemia increased EPOr expression in the injured hippocampus at 32 h (Fig. 1D and E) and 48 h after injury compared with sham (Fig. 1F). However, biochemical evidence for increased activation of the EPOr signaling pathway (EPOr/JAK2) after hypoxemia was not observed (Fig. 1G and H), providing evidence that in our model the endogenous production of EPO is insufficient to activate the EPOr pathway within 32 h after injury.
Fig. 1.
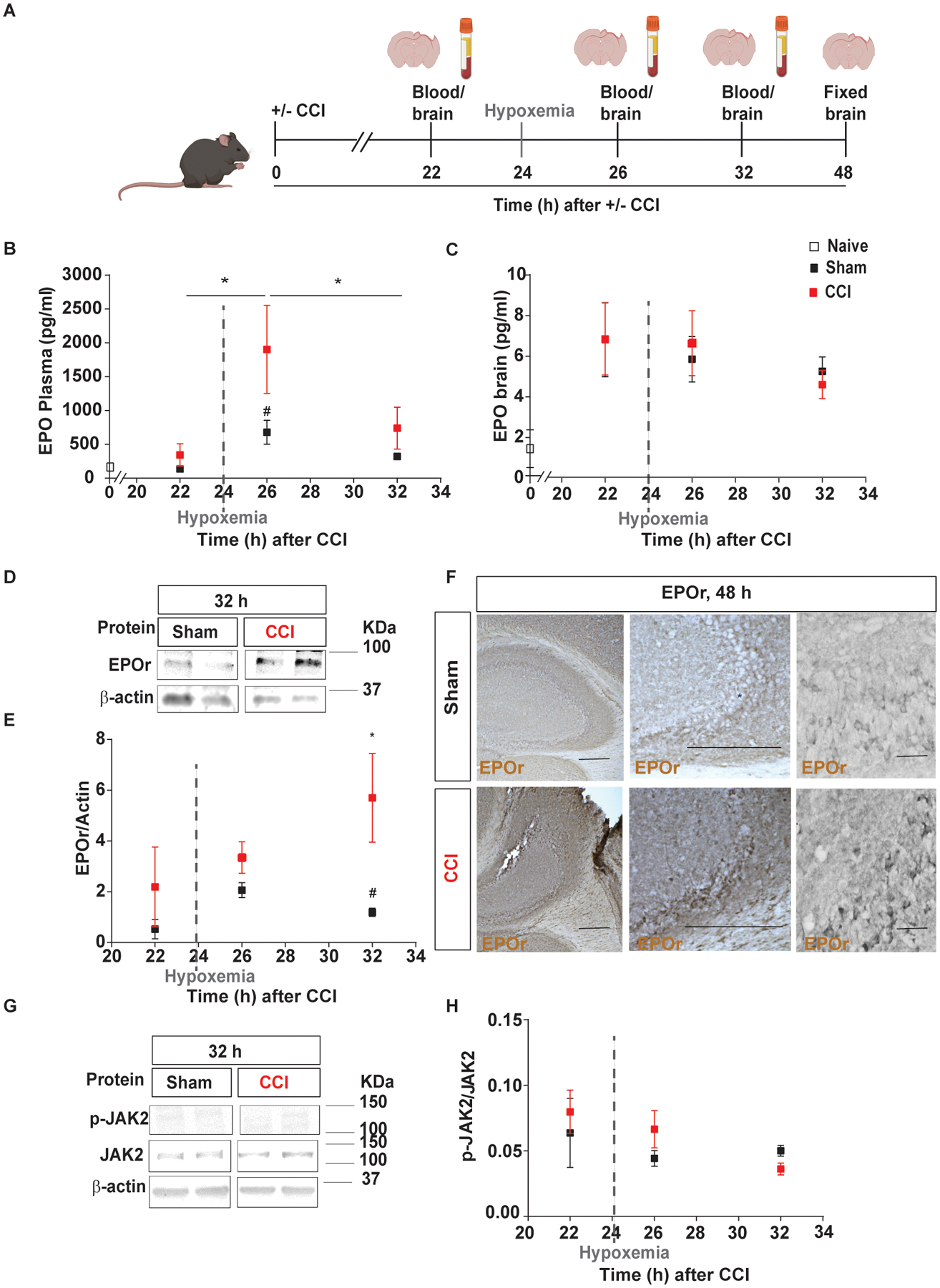
Delayed hypoxemia increased EPO and EPOr synthesis in the blood and brain, respectively, following TBI. A, Experimental design: brain and blood collection 22h, 24h, 26h, 32h and 48h after injury inducing 1h of hypoxemia at 24h post-injury. B, Endogenous mouse EPO in plasma over time analyzed by ELISA after TBI with delayed hypoxemia. C, Endogenous mouse EPO in the brain over time analyzed by ELISA after TBI with delayed hypoxemia. D, Representative western blot of the hippocampus membrane fraction probed for EPOr and (E) densitometric analysis. F, Representative image of EPOr-positive cells in CA3 region of injured hippocampi. G, Representative western blot of the hippocampus cytoplasm fraction probed for pJAK2 and (H) densitometric analysis. β-actin was used as a loading control. For western blot images, 2 samples per condition always from the same gel, using the same two samples throughout the figure. Two-way (Time and hypoxemia) ANOVA were used to determine statistical differences for hypoxemia effect followed by Tukey multiple comparison post-hoc test were used to determine statistical differences, n=6–7 mice per group. Time F(5,57) = 3.424 p = 0.009. Hypoxemia F(1,57) = 10.53 p < 0.0020. Scale bar: 20 μm. Abbreviations: CCI, controlled cortical impact; EPO, erythropoietin, EPOr, erythropoietin receptor.
rhEPO improved fear memory response, hippocampal neuroprotection, and neurogenesis
A single i.p. injection of rhEPO (5000 U/kg) at 2 or 8 h before hypoxia had no impact on hippocampal neuronal density and lesion volume (data not shown). We then examined the impact of chronic rhEPO administration throughout the first month post-injury (Fig. 2A). As expected, rhEPO treated mice had significant increases in red blood cell production (Table 2). During the habituation period of NOR testing (Fig. 2B) at 15 days post-injury, CCI-EPO spent more time in the center of the arena compared with CCI-Veh. Increased time spent in the center of the arena represents a decrease in anxiety-like behavior (Fig. 2C). We did not find any differences in total distance traveled between the groups (Fig 2D). During the test day, we did not find novel memory differences between the groups (Fig. 2E). However, at 1 month post-injury, CCI-EPO increased freezing time during cued-fear memory compared with CCI-Veh (Fig. 2I).
Table 2.
Hematological parameter 1 month post-injury.
| Hematological parameter | CCI-Veh | CCI-EPO |
|---|---|---|
| WBC (K/uL) | 6.79* | 4.2 |
| RBC (M/uL) | 9.17 | 10.72* |
| HCT (% red cells) | 38.79 | 46.29* |
| PLT (K/uL) | 726.87 | 723.47 |
WBC, white blood cell: RBC, red blood cells; HCT, hematocrit; PLT, platelet.
p < 0.05 by Mann-Whitney U test for comparison of mean between injured groups.
Remarkably, reduction in neuronal loss (Fig. 3B and C) and lesion volume (Fig. 3D) were found in CCI-EPO compared with CCI-Veh. rhEPO has been shown to stimulate dentate DG neurogenesis (Mahmood et al., 2007). rhEPO administration in injured animals resulted in increased neuronal proliferation in the ipsilateral DG as measured by summation of the total lineage cells per area of DG (Fig. 3E and F). rhEPO treatment has been shown to improve synaptic density in the uninjured CNS in both normoxic and hypoxic conditions (Adamcio et al., 2008; Wakhloo et al., 2020). We next investigated the impact of rhEPO treatment on synaptic density in the ipsilateral DG and central amygdala (CeA, Fig. 3G), regions involved in hippocampus-amygdala circuitry. Although we did not find differences in synaptic density in DG between groups (Fig. 3H), we observed a significant increase in synaptic density in CeA region in CCI-EPO compared with CCI-Veh (Fig. 3I). Together, these data demonstrate that chronic rhEPO administration after TBI and secondary hypoxemia improved cued-fear memory recall with associated increases in hippocampal neuronal survival, neurogenesis and, synaptic connectivity.
Fig. 3.
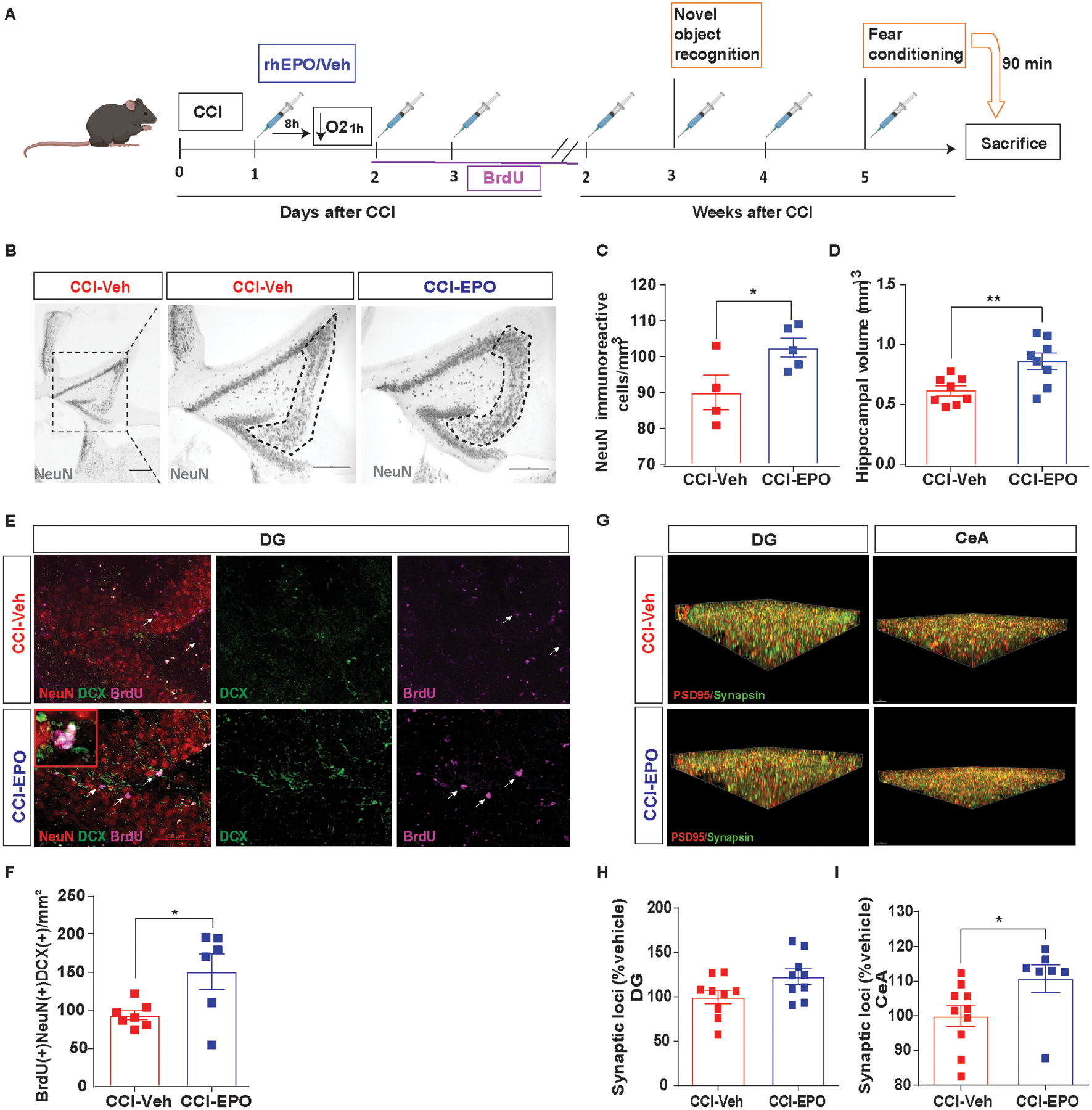
Ongoing rhEPO administration induced neuroprotection, increased neurogenesis in hippocampus and, increased synaptic density in amygdala. A, Experimental design. B, Representative image of NeuN+ cells in CA3 region of injured hippocampi (indicated by the dotted line) and (C) stereological quantification. D, Hippocampal volume quantification. E, Representative immunofluorescence image of the SGZ in the DG of the hippocampus labeled with NeuN (red), DCX (green) and BrdU (magenta) with a zoomed in insert. White arrows indicate BrdU-positives cells. F Summation of total neuronal lineage cells per area of hippocampi. G, Puncta detection of PSD95 (red) synapsin (green) images in DG (left) and CeA (right). H, Quantification of synaptic loci (% vehicle) in DG and (I) CeA. Unpaired t-test *p< 0.05, **p< 0.01, n=4–8 mice per group. Scale bar: 50 μm. Abbreviations: CCI, controlled cortical impact; rhEPO, recombinant human erythropoietin; Veh, vehicle; SGZ, subgranular zone; DG, dentate gyrus; DCX, doublecortin; BrdU, 5-bromo-2’-deoxyuridine. CeA, central amygdala.
rhEPO administration activates the MAPK/CREB pathway
MAPK/CREB signaling pathway activation in amygdala has a well described role in memory consolidation and synaptic plasticity during fear conditioning (Schafe et al., 2001). Mice were euthanized 90 min after cued-fear memory to capture MAPK/CREB signaling pathway activation in the hippocampus and amygdala (Fig. 4A). We measured the expression of non-phosphorylated and phosphorylated MAPK1 and 2 and CREB in the amygdala (Fig. 4B–F) and hippocampus (Fig. 4I–M) via western blot. In the amygdala cytoplasm fraction, we found increases in pMAPK2 in CCI-EPO compared with CCI-Veh (Fig. 4B and D). Consequently, we found pCREB increased in CCI-EPO compared with CCI-Veh by both western blot (Fig. 4E and F) and immunohistochemistry (Fig. 4G and H). Surprisingly, we did not find changes in MAPK/CREB pathway activation in the hippocampus (Fig. 4I and O).
Fig. 4.
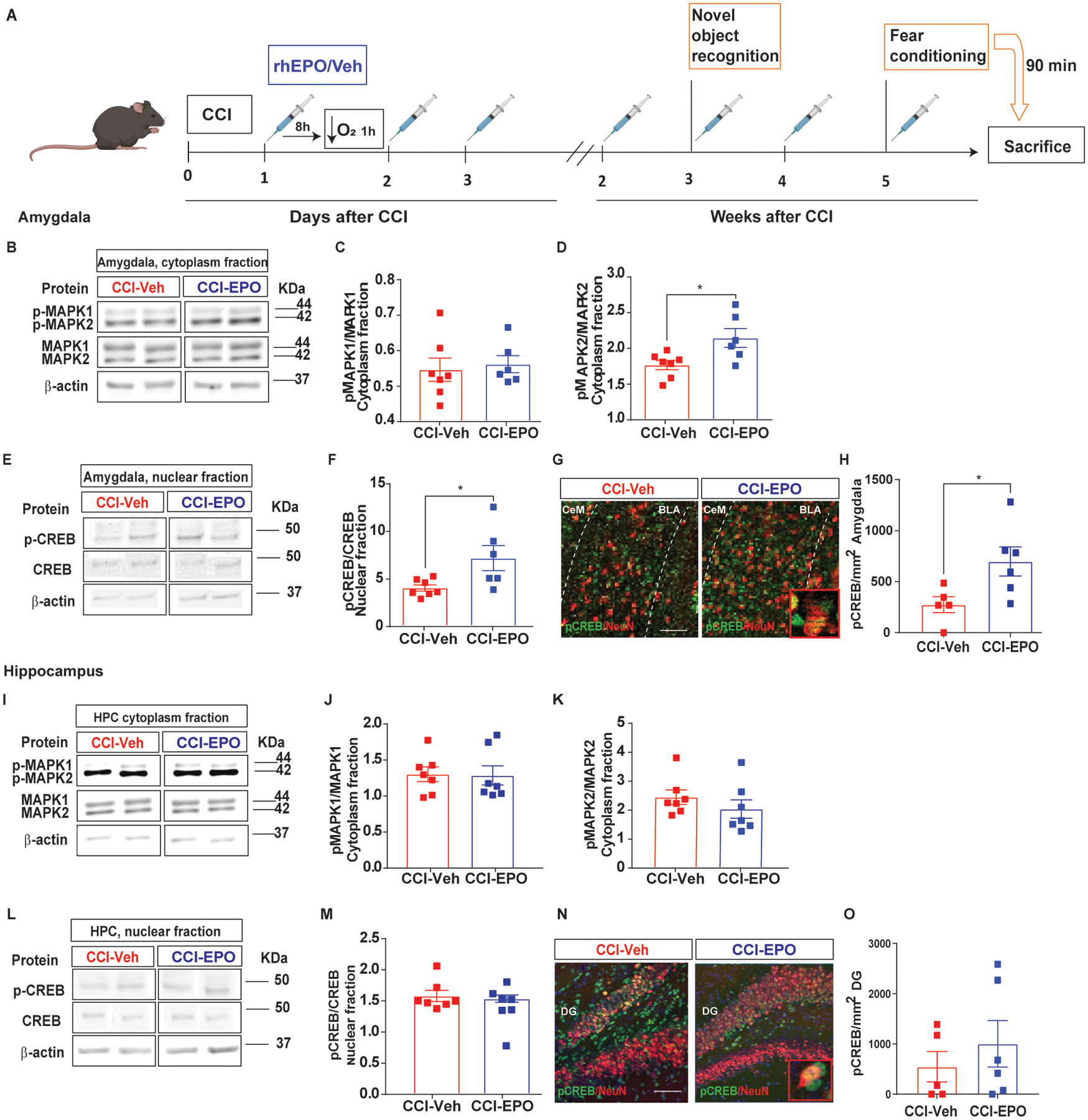
MAPK/CREB signaling pathway activation after rhEPO treatment in amygdala but not in hippocampus. A, Experimental design. B, Representative western blot of the amygdalar cytoplasm fraction probed for total and phosphorylated MAPK1 and 2. C, Densitometric analysis of pMAPK1 expression normalized by total MAPK1. D, Densitometric analysis of pMAPK2 expression normalized by total MAPK2. E, Representative western blot of the amygdalar nuclear fraction probed for total and phospho-CREB. F, Densitometric analysis of pCREB expression normalized by total CREB. β-actin was used as a loading control. G, Representative immunofluorescence image of the BLA labeled with pCREB (green) and NeuN (red) with a zoomed in insert. H, Quantification of pCREB density expression in neurons in the BLA. I, Representative western blot of the hippocampal cytoplasm fraction probed for total and phosphorylated MAPK1 and 2. J, Densitometric analysis of pMAPK1 expression normalized by total MAPK1. K, Densitometric analysis of pMAPK2 expression normalized by total MAPK2. L, Representative western blot of the hippocampal nuclear fraction probed for total and phospho-CREB. M, Densitometric analysis of pCREB expression normalized by total CREB. β-actin was used as a loading control. For western blot images, 2 samples per condition always from the same gel, using the same two samples throughout the figure. N, Representative immunofluorescence image of the BLA labeled with pCREB (green) and NeuN (red) with a zoomed in insert. O, Quantification of pCREB density expression in neurons in the BLA. Mean values are plotted ± SEM, One-way ANOVA followed by Tukey multiple comparison post hoc test were used to determine statistical differences; *p<0.05, n=5–7 mice per group. Mean values are plotted ± SEM, unpaired t-test *p<0.05, n=5–6 mice per group. Scale bar: 50 μm. Abbreviations: CCI, controlled cortical impact; rhEPO, recombinant human erythropoietin; Veh, vehicle; pCREB, phosphorylated cAMP-response element binding protein; pMAPK, phosphorylated mitogen-activated protein kinase; BLA, basolateral amygdala; CeA, central amygdala.
To verify the glial and/or neuronal MAPK/CREB activation, we analyzed pCREB expression in neurons and astrocytes in the amygdala (Zhou et al., 2009). rhEPO treatment increased pCREB expression in neurons compared with CCI-Veh 90 min after cued-fear memory (Fig. 4N and O). No significant co-expression of GFAP and pCREB was found in amygdala (data not shown). One possible explanation is that MAPK/CREB pathway activation in the amygdala was enhanced by rhEPO activation of EPO/EPOr pathway. However, we found no differences in EPOr expression and activation between CCI-EPO and CCI-Veh in amygdala (Supplementary fig. 1). With no evidence for increased EPOr expression or activation (via the EPOR/JAK2 signaling pathway) by rhEPO, our results support the hypothesis that the impact of ongoing rhEPO administration on cued-fear memory is mediated by neuronal MAPK/CREB pathway activation independently of EPOr activation.
rhEPO administration does not improve long-term behavioral outcomes after TBI and delayed hypoxemia
Identification of rhEPO neuropathological and functional evidence of efficacy for short-term neuroprotection is promising. However to improve the translatability of our preclinical findings, we performed additional studies assessing the impact of rhEPO administration on behavior and histological outcomes 6 months after injury (Fig. 5A). Mice were randomized to sham-EPO, CCI-Veh, CCI-EPO (N = 14–16/group). Two mice did not survive to the six month time point (one sham-EPO and one CCI-Veh). No differences in the hematological measurements were found between the groups at 6 months (Table 3). We observed hyperactivity during open field testing in both injured groups compared with sham-EPO (Fig. 5B and C). Discrimination index during NOR testing between CCI-EPO and CCI-Veh (Fig. 5E and F) did not reach statistical significance. We performed contextual and cued fear conditioning testing. Again we did not observe differences between CCI-EPO and CCI-Veh in contextual or cued fear memory 6 months after injury (Fig. 5G–I). No biochemical evidence were found for changes in MAPK/CREB signaling pathway activation in hippocampus and amygdala 90 min after fear memory at this time point (Supplementary fig. 2). Therefore, these results highlighted the importance for ongoing rhEPO administration to chronically improve behavior and activation of MAPK/CREB signaling pathway.
Table 3.
Hematological parameters 6 months post-injury.
| Hematological parameter | Sham-EPO | CCI-Veh | CCI-EPO |
|---|---|---|---|
| WBC (K/uL) | 8.00 | 8.33 | 8.53 |
| RBC (M/uL) | 9.18 | 8.86 | 9.91 |
| HCT (% red cells) | 13.38 | 13.39 | 13.40 |
| PLT (K/uL) | 886.17 | 833.70 | 877.30 |
WBC, white blood cell: RBC, red blood cells; HCT, hematocrit; PLT, platelet.
rhEPO administration provides long-term neuroprotection
Stereological quantification of NeuN positive cells in the CA3 region at 6 months after injury (Fig. 6A) revealed reduced neuronal loss (Fig. 6B and C) and increased hippocampal volumes (Fig. 6D and E) in CCI-EPO compared with CCI-Veh. In addition, we explored glutamatergic synaptic density in the ipsilateral DG and CeA using synaptic evaluation and quantification by imaging nanostructure (SEQUIN) (Reitz et al., 2021; Sauerbeck et al., 2020) (Fig. 6F–H). Remarkably, we observed a reduction in TBI-induced synaptic loss in both DG and CeA 4 months after the last dose of rhEPO. Therefore, early rhEPO’s administration induced neuronal preservation and increased excitatory synaptic density in the hippocampus and amygdala but did not induce behavioral improvements.
Fig 6.
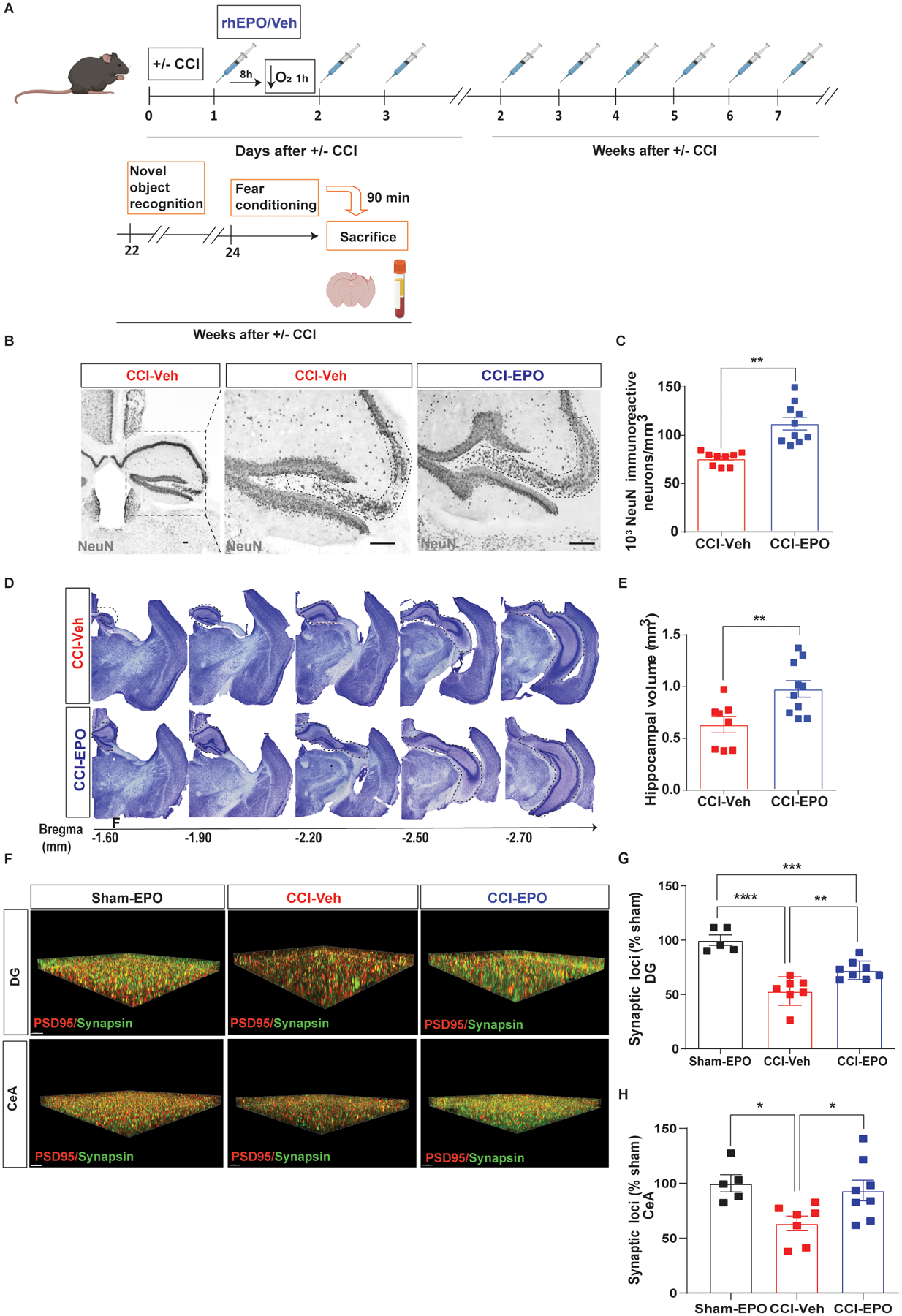
rhEPO reduces neurodegeneration and improves excitatory synaptic plasticity 6 months post-injury. A, Experimental design. B, Representative image of NeuN+ cells in CA3 region of injured hippocampi (indicated by the dotted line) and (C) stereological quantification. Scale bar: 20 μm. D, Representative image of cresyl violet in the injured side of the brain. E, Hippocampal volume quantification. F, Top images, puncta detection of PSD95 (red) synapsin (green) in DG and, bottom images, in CeA. Scale bar: 5 μm. G, Quantification of synaptic loci (% sham-EPO) in DG. One-way ANOVA followed by Tukey multiple comparison post hoc test. F(2,17) = 27.1, p < 0.0001. ****p < 0.0001, ***p < 0.001, **p<0.01. H, Quantification of synaptic loci (% sham-EPO) in CeA. F(2,17) = 5.16, p = 0.018, *p<0.05, n=5–8 mice per group. Abbreviations: CCI, controlled cortical impact; rhEPO, recombinant human erythropoietin; Veh, vehicle; DG, dentate gyrus. CeA, central amygdala.
Discussion
Our study provides new evidences for ongoing rhEPO’s mechanism of action for neuroprotection, and enhancement of short-term (1-month post-injury) fear memory after experimental TBI with delayed hypoxemia. Ongoing rhEPO treatment induced increased neurogenesis associated with improvements in memory performance through neuronal MAPK/CREB pathway activation in an amygdala-dependent matter. We further demonstrated how rhEPO administration impacted hippocampal neuronal survival and increasing synaptic density in both hippocampus and amygdala but no changes behavioral performance in CCI-EPO compared with CCI-Veh were observed at 6 months post-injury. These data support that chronic rhEPO administration in the setting of TBI has dual effects reducing neuronal degeneration and modulating functional performance.
Acute rhEPO treatment improves memory and learning in mice (Adamcio et al., 2008) and humans (Miskowiak et al., 2008). These observations led to more in-depth studies aiming to characterize the pleiotropic effects of rhEPO and its behavioral potential benefits in neurodegenerative diseases such as Alzheimer’s disease (Esmaeili Tazangi et al., 2015; Lee et al., 2012; Rey et al., 2019) and brain injury such as stroke and TBI (Lu et al., 2005; Schober et al., 2014; Shein et al., 2008). The hippocampus-amygdala circuitry is a central nucleus from the limbic system for the processing of fear-associated memory (Ehrlich et al., 2009; Fidan et al., 2016; Rodrigues et al., 2004; Schafe et al., 2001). More concretely, cued memory only involves the amygdala to form and retrieve cued-fear association whereas contextual-fear memory calls upon both the hippocampus and amygdala to retrieve contextual-fear association. Moreover, other reports highlighted the MAPK/CREB pathway activation in amygdala as being required for cued-fear conditioning consolidation (Rodrigues et al., 2004; Schafe et al., 2001). In line with these findings, in our model of delayed hypoxemia after TBI, we have found that rhEPO treatment for 1 month influenced cued-fear memory response through neuronal MAPK/CREB signaling activation in the amygdala.
Synaptic plasticity in amygdala neurons is essential for associative fear learning and memory storage (52). Although optical stimulation of lateral amygdala pyramidal neurons produced some freezing behavior, the stimulation of the CeA is directly connected with the fear memory behavioral output pathways (53). By using synapse quantification with SEQUIN, at 1-month post injury we found an increase of synaptic density in the CeA following fear memory improvements in CCI-EPO. However, at 6 months post injury we found an increase of synaptic density in CCI-EPO compared with vehicle but not followed by significant behavior improvement or MAPK/CREB signaling activation. Thus, we deduced that early rhEPO administration induced short and long-term neuronal survival and increased synaptic density; however, ongoing rhEPO administration was required for activation of MAPK/CREB signaling and memory enhancement. These results suggest that rhEPO has a crucial role on the activation of MAPK/CREB signaling pathway to enhance long-term fear memory consolidation associated with synaptogenesis (Ota et al., 2008; Schafe et al., 2001; Schafe et al., 2008).
Animal studies have identified sex and age-related differences in responses to injury and recovery (Islam et al., 2021; Kolb and Cioe, 1996). They have shown that manipulation of gonadal hormones as well as age significantly influence injury recovery (Islam et al., 2021; Kolb and Cioe, 1996). One limitation in our investigations was the absence of experiments exploring the impact of sex or age on rhEPO treatment efficacy. Previous work have reported confounding results related to sex-dependent differences in the brain’s response to rhEPO. A mouse model of neonatal stroke demonstrated greater benefit in female mice (Wen et al., 2006). However an intermittent hypoxia neonatal murine model reported a greater response to rhEPO in male mice (Laouafa et al., 2019). Previous investigations of sex-dependent differences in murine models of TBI have not shown sex differences in histological or behavior in response to rhEPO (Xiong et al., 2007). These studies suggest that future investigations of rhEPO administration should include the interaction of age and sex on the candidate therapeutic evaluation. Our studies did not include a sham group without rhEPO limiting our ability to understand the long-term effects of rhEPO on the uninjured brain, nevertheless our results provide promising evidence for a beneficial effect in the traumatically injured brain. We did not evaluate rhEPO’s effect on different TBI models or in gyrencephalic animal models, limiting rapid translation of our findings to the clinical setting.
Conclusions
Taken together, our data demonstrates the rhEPO’s functional and neuroprotective efficacy in targeting delayed hypoxemia after TBI. Mimicking the effects of endogenous neuronal EPO, ongoing rhEPO treatment induced neuronal survival, neurogenesis, synaptogenesis and, enhanced fear memory response associated with neuronal activation of MAPK/CREB signaling pathway in amygdala. rhEPO treatment reduced neurodegeneration at 6 months post-injury and was accompanied with increasing of excitatory synaptic density in the hippocampus and amygdala but not followed by behavioral improvement or MAPK/CREB signaling activation. Further investigation of chronically rhEPO administration may be crucial for MAPK signaling activation within hippocampus-amygdala circuitry to improve fear response 6 months after TBI with delayed hypoxemia. Studies on characterizing the ongoing rhEPO impact on inhibition and/or excitation balance in the brain will be essential to understanding the mechanism underlying how rhEPO modulates neuronal activation and behavioral outcomes after injury.
Supplementary Material
Funding Statement
This work was supported by the National Institutes of Health (R01NS097721). Fluorescent imaging was performed on a Zeiss Axio Imager Z2 Fluorescence Microscope with ApoTome 2 optical sectioning grid imager through the use of Washington University Center for Cellular Imaging (WUCCI) supported by Washington University School of Medicine, The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2015-505 and CDI-CORE-2019-813) and the Foundation for Barnes-Jewish Hospital (3770 and 4642). Confocal imaging was generated on a Zeiss LSM 880 Airyscan Confocal Microscope which was purchased with support from the Office of Research Infrastructure Programs (ORIP), a part of the NIH Office of the Director under grant OD021629.
Abbreviations:
- TBI
traumatic brain injury
- EPO
erythropoietin
- rEPO
erythropoietin receptor
- CCI
controlled cortical impact
- NOR
novel object recognition
- MAPK/CREB
mitogen-activated protein kinase/cAMP response element-binding protein
- CeA
central amygdala
- SEQUIN
synaptic evaluation and quantification by imaging nanostructure
Footnotes
Conflict of Interest Statement
The authors have no conflicts of interest to disclose.
Declaration of Interest: None
References
- Adamcio B, Sargin D, Stradomska A, Medrihan L, Gertler C, Theis F, Zhang M, Muller M, Hassouna I, Hannke K, Sperling S, Radyushkin K, El-Kordi A, Schulze L, Ronnenberg A, Wolf F, Brose N, Rhee JS, Zhang W, Ehrenreich H, 2008. Erythropoietin enhances hippocampal long-term potentiation and memory. BMC Biol. 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arango MF, Bainbridge D, 2008. Magnesium for acute traumatic brain injury. Cochrane Database Syst Rev. CD005400. [DOI] [PubMed] [Google Scholar]
- Brines M, Cerami A, 2005. Emerging biological roles for erythropoietin in the nervous system. Nat Rev Neurosci. 6, 484–94. [DOI] [PubMed] [Google Scholar]
- Celorrio M, Abellanas MA, Rhodes J, Goodwin V, Moritz J, Vadivelu S, Wang L, Rodgers R, Xiao S, Anabayan I, Payne C, Perry AM, Baldridge MT, Aymerich MS, Steed A, Friess SH, 2021a. Gut microbial dysbiosis after traumatic brain injury modulates the immune response and impairs neurogenesis. Acta Neuropathol Commun. 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celorrio M, Rhodes J, Vadivelu S, Davies M, Friess SH, 2021b. N-acetylcysteine reduces brain injury after delayed hypoxemia following traumatic brain injury. Exp Neurol. 335, 113507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaaya N, Jacques A, Belmer A, Beecher K, Ali SA, Chehrehasa F, Battle AR, Johnson LR, Bartlett SE, 2019. Contextual Fear Conditioning Alter Microglia Number and Morphology in the Rat Dorsal Hippocampus. Front Cell Neurosci. 13, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT, 1998. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 95, 11715–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M, Jacobs A, Brody DL, Friess SH, 2018. Delayed Hypoxemia after Traumatic Brain Injury Exacerbates Long-Term Behavioral Deficits. J Neurotrauma. 35, 790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digicaylioglu M, Lipton SA, 2001. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 412, 641–7. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A, 2009. Amygdala inhibitory circuits and the control of fear memory. Neuron. 62, 757–71. [DOI] [PubMed] [Google Scholar]
- Esmaeili Tazangi P, Moosavi SM, Shabani M, Haghani M, 2015. Erythropoietin improves synaptic plasticity and memory deficits by decrease of the neurotransmitter release probability in the rat model of Alzheimer’s disease. Pharmacol Biochem Behav. 130, 15–21. [DOI] [PubMed] [Google Scholar]
- Fidan E, Lewis J, Kline AE, Garman RH, Alexander H, Cheng JP, Bondi CO, Clark RS, Dezfulian C, Kochanek PM, Kagan VE, Bayir H, 2016. Repetitive Mild Traumatic Brain Injury in the Developing Brain: Effects on Long-Term Functional Outcome and Neuropathology. J Neurotrauma. 33, 641–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner DC, Bailey M, Presneill J, French CJ, Nichol A, Little L, Bellomo R, Investigators, E.-T., the, A.C.T.G., 2018. Erythropoietin to Reduce Mortality in Traumatic Brain Injury: A Post-hoc Dose-effect Analysis. Ann Surg. 267, 585–589. [DOI] [PubMed] [Google Scholar]
- Gardner AJ, Zafonte R, 2016. Neuroepidemiology of traumatic brain injury. Handb Clin Neurol. 138, 207–23. [DOI] [PubMed] [Google Scholar]
- Grasso G, Sfacteria A, Meli F, Fodale V, Buemi M, Iacopino DG, 2007. Neuroprotection by erythropoietin administration after experimental traumatic brain injury. Brain Res. 1182, 99–105. [DOI] [PubMed] [Google Scholar]
- Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG, 2009. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 42, 377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassouna I, Ott C, Wustefeld L, Offen N, Neher RA, Mitkovski M, Winkler D, Sperling S, Fries L, Goebbels S, Vreja IC, Hagemeyer N, Dittrich M, Rossetti MF, Krohnert K, Hannke K, Boretius S, Zeug A, Hoschen C, Dandekar T, Dere E, Neher E, Rizzoli SO, Nave KA, Siren AL, Ehrenreich H, 2016. Revisiting adult neurogenesis and the role of erythropoietin for neuronal and oligodendroglial differentiation in the hippocampus. Mol Psychiatry. 21, 1752–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellewell SC, Yan EB, Alwis DS, Bye N, Morganti-Kossmann MC, 2013. Erythropoietin improves motor and cognitive deficit, axonal pathology, and neuroinflammation in a combined model of diffuse traumatic brain injury and hypoxia, in association with upregulation of the erythropoietin receptor. J Neuroinflammation. 10, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison JS, Ward RE, Lacroix J, Hebert PC, Barnes MA, Bohn DJ, Dirks PB, Doucette S, Fergusson D, Gottesman R, Joffe AR, Kirpalani HM, Meyer PG, Morris KP, Moher D, Singh RN, Skippen PW, Hypothermia Pediatric Head Injury Trial, I., the Canadian Critical Care Trials, G., 2008. Hypothermia therapy after traumatic brain injury in children. N Engl J Med. 358, 2447–56. [DOI] [PubMed] [Google Scholar]
- Islam M, Davis B.T.t., Kando MJ, Mao Q, Procissi D, Weiss C, Schwulst SJ, 2021. Differential neuropathology and functional outcome after equivalent traumatic brain injury in aged versus young adult mice. Exp Neurol. 341, 113714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami M, Sekiguchi M, Sato K, Kozaki S, Takahashi M, 2001. Erythropoietin receptor-mediated inhibition of exocytotic glutamate release confers neuroprotection during chemical ischemia. J Biol Chem. 276, 39469–75. [DOI] [PubMed] [Google Scholar]
- Kolb B, Cioe J, 1996. Sex-related differences in cortical function after medial frontal lesions in rats. Behav Neurosci. 110, 1271–81. [DOI] [PubMed] [Google Scholar]
- Laouafa S, Iturri P, Arias-Reyes C, Marcouiller F, Gonzales M, Joseph V, Bairam A, Soliz J, 2019. Erythropoietin and caffeine exert similar protective impact against neonatal intermittent hypoxia: Apnea of prematurity and sex dimorphism. Exp Neurol. 320, 112985. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kam EH, Kim SY, Cheon SY, Kim EJ, Chung S, Jeong JH, Koo BN, 2017. Erythropoietin Attenuates Postoperative Cognitive Dysfunction by Shifting Macrophage Activation toward the M2 Phenotype. Front Pharmacol. 8, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ST, Chu K, Park JE, Jung KH, Jeon D, Lim JY, Lee SK, Kim M, Roh JK, 2012. Erythropoietin improves memory function with reducing endothelial dysfunction and amyloid-beta burden in Alzheimer’s disease models. J Neurochem. 120, 115–24. [DOI] [PubMed] [Google Scholar]
- Li W, Bai YA, Li YJ, Liu KG, Wang MD, Xu GZ, Shang HL, Li YF, 2015. Magnesium sulfate for acute traumatic brain injury. J Craniofac Surg. 26, 393–8. [DOI] [PubMed] [Google Scholar]
- Li ZM, Xiao YL, Zhu JX, Geng FY, Guo CJ, Chong ZL, Wang LX, 2016. Recombinant human erythropoietin improves functional recovery in patients with severe traumatic brain injury: A randomized, double blind and controlled clinical trial. Clin Neurol Neurosurg. 150, 80–83. [DOI] [PubMed] [Google Scholar]
- Lu D, Mahmood A, Qu C, Goussev A, Schallert T, Chopp M, 2005. Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. J Neurotrauma. 22, 1011–7. [DOI] [PubMed] [Google Scholar]
- Mahmood A, Lu D, Qu C, Goussev A, Zhang ZG, Lu C, Chopp M, 2007. Treatment of traumatic brain injury in rats with erythropoietin and carbamylated erythropoietin. J Neurosurg. 107, 392–7. [DOI] [PubMed] [Google Scholar]
- Marti HH, Bernaudin M, Petit E, Bauer C, 2000. Neuroprotection and Angiogenesis: Dual Role of Erythropoietin in Brain Ischemia. News Physiol Sci. 15, 225–229. [DOI] [PubMed] [Google Scholar]
- Marti HH, 2004. Erythropoietin and the hypoxic brain. J Exp Biol. 207, 3233–42. [DOI] [PubMed] [Google Scholar]
- Miskowiak K, Inkster B, Selvaraj S, Wise R, Goodwin GM, Harmer CJ, 2008. Erythropoietin improves mood and modulates the cognitive and neural processing of emotion 3 days post administration. Neuropsychopharmacology. 33, 611–8. [DOI] [PubMed] [Google Scholar]
- Nichol A, French C, Little L, Haddad S, Presneill J, Arabi Y, Bailey M, Cooper DJ, Duranteau J, Huet O, Mak A, McArthur C, Pettila V, Skrifvars M, Vallance S, Varma D, Wills J, Bellomo R, Investigators, E.-T., Group, A.C.T., 2015. Erythropoietin in traumatic brain injury (EPO-TBI): a double-blind randomised controlled trial. Lancet. 386, 2499–506. [DOI] [PubMed] [Google Scholar]
- Ota KT, Pierre VJ, Ploski JE, Queen K, Schafe GE, 2008. The NO-cGMP-PKG signaling pathway regulates synaptic plasticity and fear memory consolidation in the lateral amygdala via activation of ERK/MAP kinase. Learn Mem. 15, 792–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh U, Williams M, Jacobs A, Pineda JA, Brody DL, Friess SH, 2016. Delayed Hypoxemia Following Traumatic Brain Injury Exacerbates White Matter Injury. J Neuropathol Exp Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabie T, Marti HH, 2008. Brain protection by erythropoietin: a manifold task. Physiology (Bethesda). 23, 263–74. [DOI] [PubMed] [Google Scholar]
- Reitz SJ, Sauerbeck AD, Kummer TT, 2021. SEQUIN: An imaging and analysis platform for quantification and characterization of synaptic structures in mouse. STAR Protoc. 2, 100268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey F, Balsari A, Giallongo T, Ottolenghi S, Di Giulio AM, Samaja M, Carelli S, 2019. Erythropoietin as a Neuroprotective Molecule: An Overview of Its Therapeutic Potential in Neurodegenerative Diseases. ASN Neuro. 11, 1759091419871420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CS, Hannay HJ, Yamal JM, Gopinath S, Goodman JC, Tilley BC, Epo Severe, T.B.I.T.I., Baldwin A, Rivera Lara L, Saucedo-Crespo H, Ahmed O, Sadasivan S, Ponce L, Cruz-Navarro J, Shahin H, Aisiku IP, Doshi P, Valadka A, Neipert L, Waguspack JM, Rubin ML, Benoit JS, Swank P, 2014. Effect of erythropoietin and transfusion threshold on neurological recovery after traumatic brain injury: a randomized clinical trial. JAMA. 312, 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE, 2004. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 44, 75–91. [DOI] [PubMed] [Google Scholar]
- Sauerbeck AD, Gangolli M, Reitz SJ, Salyards MH, Kim SH, Hemingway C, Gratuze M, Makkapati T, Kerschensteiner M, Holtzman DM, Brody DL, Kummer TT, 2020. SEQUIN Multiscale Imaging of Mammalian Central Synapses Reveals Loss of Synaptic Connectivity Resulting from Diffuse Traumatic Brain Injury. Neuron. 107, 257–273 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Nader K, Blair HT, LeDoux JE, 2001. Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci. 24, 540–6. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Swank MW, Rodrigues SM, Debiec J, Doyere V, 2008. Phosphorylation of ERK/MAP kinase is required for long-term potentiation in anatomically restricted regions of the lateral amygdala in vivo. Learn Mem. 15, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober ME, Requena DF, Block B, Davis LJ, Rodesch C, Casper TC, Juul SE, Kesner RP, Lane RH, 2014. Erythropoietin improved cognitive function and decreased hippocampal caspase activity in rat pups after traumatic brain injury. J Neurotrauma. 31, 358–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shein NA, Grigoriadis N, Alexandrovich AG, Simeonidou C, Spandou E, Tsenter J, Yatsiv I, Horowitz M, Shohami E, 2008. Differential neuroprotective properties of endogenous and exogenous erythropoietin in a mouse model of traumatic brain injury. J Neurotrauma. 25, 112–23. [DOI] [PubMed] [Google Scholar]
- Skrifvars MB, French C, Bailey M, Presneill J, Nichol A, Little L, Durantea J, Huet O, Haddad S, Arabi Y, McArthur C, Cooper DJ, Bellomo R, 2018. Cause and Timing of Death and Subgroup Differential Effects of Erythropoietin in the EPO-TBI Study. J Neurotrauma. 35, 333–340. [DOI] [PubMed] [Google Scholar]
- Villa P, Bigini P, Mennini T, Agnello D, Laragione T, Cagnotto A, Viviani B, Marinovich M, Cerami A, Coleman TR, Brines M, Ghezzi P, 2003. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J Exp Med. 198, 971–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakhloo D, Scharkowski F, Curto Y, Javed Butt U, Bansal V, Steixner-Kumar AA, Wustefeld L, Rajput A, Arinrad S, Zillmann MR, Seelbach A, Hassouna I, Schneider K, Qadir Ibrahim A, Werner HB, Martens H, Miskowiak K, Wojcik SM, Bonn S, Nacher J, Nave KA, Ehrenreich H, 2020. Functional hypoxia drives neuroplasticity and neurogenesis via brain erythropoietin. Nat Commun. 11, 1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhang Z, Wang Y, Zhang R, Chopp M, 2004. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke. 35, 1732–7. [DOI] [PubMed] [Google Scholar]
- Wen TC, Rogido M, Peng H, Genetta T, Moore J, Sola A, 2006. Gender differences in long-term beneficial effects of erythropoietin given after neonatal stroke in postnatal day-7 rats. Neuroscience. 139, 803–11. [DOI] [PubMed] [Google Scholar]
- Winn HR, Temkin NR, Anderson GD, Dikmen SS, 2007. Magnesium for neuroprotection after traumatic brain injury. Lancet Neurol. 6, 478–9. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Lu D, Qu C, Goussev A, Schallert T, Chopp M, 2007. Role of gender in outcome after traumatic brain injury and therapeutic effect of erythropoietin in mice. Brain Res. 1185, 301–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, Neve R, Poirazi P, Silva AJ, 2009. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci. 12, 1438–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.