

Abstract
Introduction:
Cryofibrinogen is an abnormal, cold-insoluble protein composed of a combination of fibrinogen, fibrin, and fibronectin. Cryofibrinogenemia can be essential (e.g. primary) or secondary to various conditions. While low levels of cryofibrinogen can be seen in asymptomatic healthy individuals without evidence of clinical features typical of cryofibrinogenemia, cryofibrinogenemia associated with clinical features is considered very rare. The clinical features of cryofibrinogenemia ranges from skin manifestations, including Raynaud’s phenomenon and livedo reticularis, to more severe organ-threatening manifestations such as tissue ischemia and gangrene.
Case description:
We report a case of a 48-year-old male who presented with blue finger and palpable purpura on his distal extremities. Laboratory workup was positive for anti-nuclear antibodies, anti-double-stranded DNA, anti-ribonucleoprotein, and rheumatoid factor, while antineutrophil cytoplasmic antibodies and cryoglobulins were negative. Testing for hypercoagulable states and infectious etiologies was unrevealing. Later, angiographic computed tomography showed multiple pulmonary embolisms and disruption of blood flow to the left fifth digit. As the aforementioned workup could not explain the presence of the thrombus by a thromboembolic cause, a search for an in situ cause other than antiphospholipid syndrome was initiated and concentrated mainly on cryofibrinogenemia. Blood samples collected using prewarmed anticoagulant containing tubes were sent to central lab familiar with performing the test. Two weeks later, a positive result for the presence of cryofibrinogen confirmed the diagnosis of cryofibrinogenemia. Due to the presence of multiple signs compatible with mixed connective tissue disease, he was diagnosed with cryofibrinogenemia secondary to mixed connective tissue disease, and treatment with prednisone, low-molecular-weight heparin, prostacyclin and hydroxychloroquine was initiaed with favorable outcome.
Conclusion:
Cryofibrinogenemia is a rare and underdiagnosed condition. Clinicians should be aware of this cryopathy especially in the cases of Raynaud’s phenomenon and ischemic ulcers not explained by other causes. Precautions must be taken during the diagnostic process, and therapy should be given as soon as possible.
Keywords: Cryofibrinogenemia, blue finger, ischemia, mixed connective tissue disease, purpura
Case description
A 48-year-old Arab patient presented to the emergency department with purpura on his distal extremities along with blue fifth digit of the left hand (Figure 1). His past medical history included hypertension treated with beta blockers and calcium channel blockers. At his presentation, vital signs were within normal limits. On physical examination, palpable purpura was evident on the distal lower extremities, along with Raynaud’s phenomenon (RP) of the digits of the hands with bluish discoloration on the left fifth digit (Figure 1). A lesion suspected for pitting scar was observed on the fifth digit (Figure 2). Ulnar and radial pulses were normally detected; a 3/6 systolic murmur was heard on auscultation, without other signs of infective endocarditis. No signs of photosensitivity, oral ulcers, puffy fingers, skin thickening, or active digital ulcers were noted. Complete blood count (CBC) showed normocytic anemia with hemoglobin of 12 g/dL, liver and kidney function were within normal limits, while the C-reactive protein (CRP) was mildly elevated at 10 mg/L (normal range: 0–5 mg/L). A 24-h urinary collection revealed proteinuria (710 mg/24 h). The beta-blocker treatment was stopped and intravenous prostacyclin was initiated.
Figure 1.

Blue-colored fifth digit of the left hand (a-c) and purpura on both feet (d).
Figure 2.
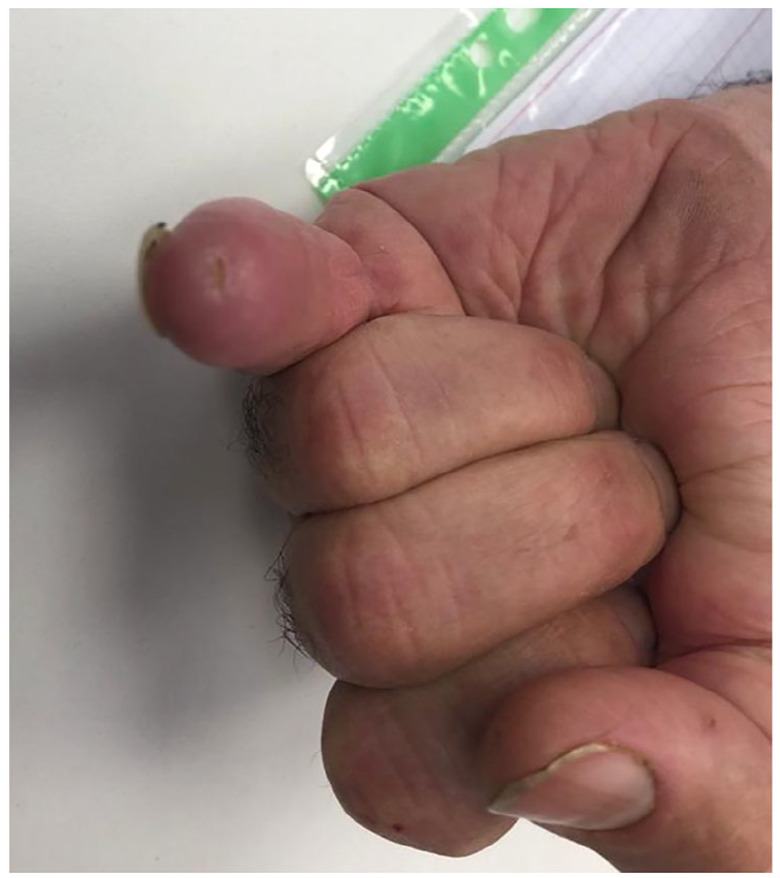
Lesion of the tip of the fifth digit suspicious for pitting scar.
The differential diagnosis at this stage included small-medium vessel vasculitis (including antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis, hypocomplementemic urticarial vasculitis, c1q deficiency, IgA vasculitis, and cryoglobulinemic vasculitis), autoimmune connective tissue diseases (systemic sclerosis (SSc), systemic lupus erythematosus (SLE), Sjogren’s syndrome, and inflammatory myopathies), hypercoagulable states (antiphospholipid syndrome (APS), heritable hypercoagulable states, intracardiac masses (e.g. atrial myxoma)), and infectious diseases (infective endocarditis).
Furthermore, laboratory workup was positive for anti-nuclear antibodies (ANA) (speckled pattern with titer of 1:640), anti-double-stranded DNA (dsDNA) (41, normal limits: 0–12), anti-ribonucleoprotein (RNP), and rheumatoid factor (RF) (76, normal limits: 0–14). Complement levels (C3 and C4) showed consumption with C4 below detectrd level. Urinalysis revealed mild proteinuria with a few red blood cells (RBCs) (three RBCs per high power field (HPF)), while 24-h urine collection demonstrated 710 mg of protein. Workup was negative for ANCA, cryoglobulins, as well as anti-Ro, anti-La, anti-topoisomerase, and anti-centromere. Testing for c1q deficiency was not readily available. Testing for hypercoagulable states was also unremarkable, including antiphospholipid antibodies (lupus anticoagulant (LAC), anticardiolipin, and β2-glycoprotein antibodies), antithrombin III mutation, protein S and C deficiency, factor V Leiden, homocysteine, and prothrombin mutation. Workup for endocarditis was unrevealing, including repeated negative blood cultures, negative ophthalmologic examination, and transesophageal echocardiography, showing no evidence of vegetations. Viral serologies including hepatitis B and C, human immunodeficiency virus (HIV), Q-fever, and Parvo B19 all were negative for acute infection. Skin biopsy was consistent with urticarial vasculitis (Figure 3(a)), while kidney biopsy was not consistent with SLE (Figure 3(b)).
Figure 3.
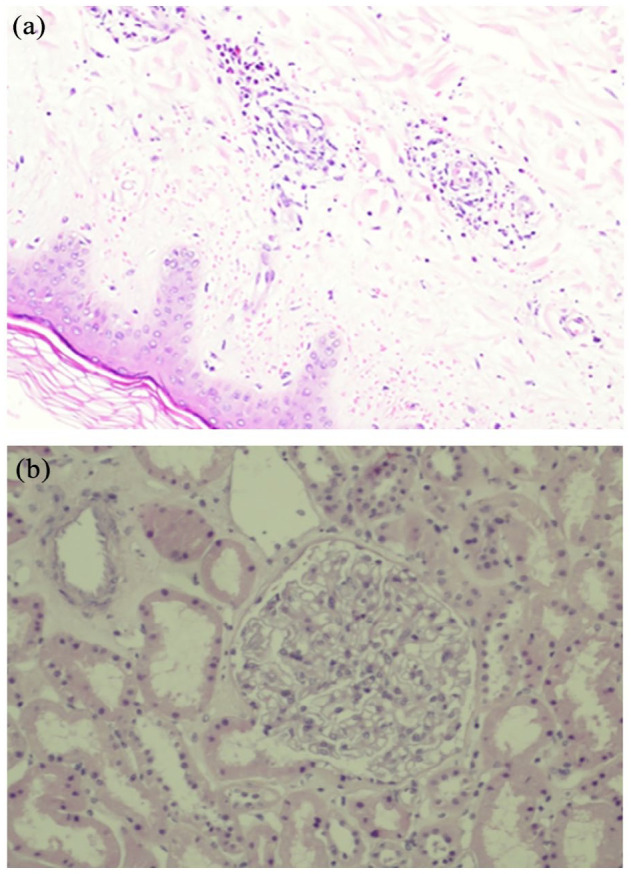
Biopsies of skin and kidney. (a) Biopsy of the skin showing urticarial vasculitis and (b) biopsy of the kidney showing normal glomeruli under light microscopy.
Angiographic computed tomography of the chest, abdomen, and peripheral arteries was performed and demonstrated multiple pulmonary embolisms (PEs), including segmental PE in the right lower lobe (RLL) and subsegmental PE in the right upper lobe (RUL) and the left lower lobe (LLL). Moreover, a disruption of blood flow to the left fifth digit was demonstrated in the distal artery of left fifth digit. Meanwhile, the patient was treated with hydroxychloroquine and prednisone due to the suspicion of underlying mixed connective tissue disease (MCTD). He was also treated with low-molecular-weight heparin (LMWH) due to the thrombotic events. As the aforementioned workup could not explain the presence of the thrombus by a thromboembolic cause, a search for an in situ cause other than APS was initiated and concentrated mainly on cryofibrinogenemia (CF).
Blood samples collected using prewarmed anticoagulant-containing tubes were sent to central lab familiar with performing the test. Two weeks later, a positive result for the presence of cryofibrinogen confirmed the diagnosis of CF.
Due to the presence of multiple signs compatible with MCTD (including RP, suspected pitting scar of fifth digit, and positive serologies for ANA, RF, anti-RNP, and anti-dsDNA), the most probable diagnosis was CF secondary to MCTD. Later, warfarin was initiated, while prednisone was tapered off. A few weeks later, the ischemia on the fifth digit was resolved (Figure 4). The patient continues to be treated with hydroxychloroquine and warfarin with continuous favorable response.
Figure 4.

Blue discoloration resolved after being treated with prednisone, hydroxychloroquine, prostacyclin and low-molecular-weight heparin.
Conclusion
CF is a rare disorder described first in 1955 by Korst and Kratochvil. 1 The prevalence of CF (e.g. cryofibrinogen in the blood) varies from 0% to 7% in healthy individuals and further increases to 8%–13% among hospitalized patients.2,3 However, most cases of CF, especially essential CF cases, are asymptomatic.4,5 CF can be classified as primary (e.g. essential CF) or secondary to other underlying disorders, including connective tissue diseases, vasculitis, neoplasms, and infections. 3 Recently, a high prevalence of CF was observed among patients presenting with chilblains after being infected with COVID-19, highlighting the fact that CF can be secondary to infectious causes. 6 The disease has a predilection for females and usually diagnosed during the sixth to seventh decade of life. 3 Skin manifestations are the most common features and present in 80% of cases, ranging from cold intolerance, RP, purpura, livedo reticularis, and urticaria to more severe manifestations such as tissue ischemia, skin necrosis, and gangrene leading to amputation. Acral areas are more susceptible to cold and thus are more often affected by ulcers. Systemic manifestations are also common and may include thrombotic events, myalgia, muscular weakness, arthralgia, arthritis, multi-neuritis, and renal involvement (whether glomerular, tubulointerstitial, or vascular).3,4 Arterial and venous thrombotic events are reported in up to 55% of cases and may lead to mesenteric, retinal, femoral, iliac, aortic, cerebral, or myocardial thrombosis. The occurrence of these thrombotic events is highly correlated with the amount of cryofibrinogen in the plasma. CF is associated with increased mortality, with sepsis secondary to gangrene being the most common cause of death. 3
The pathophysiology of CF is based upon a cryoprecipitate that forms at cold temperatures and is composed of fibrinogen, fibronectin, and fibrin, but may also contain various levels of albumin and immunoglobulins.3,4,7 Fibrinogen forms complexes with fibrin, while the fibronectin acts as a nucleus with multiple binding sites for fibrin–fibrinogen complexes. Precautions must be taken during the diagnostic process of cryofibrinogen. Cryofibrinogen can form within tubes that contain an anticoagulant: oxalate, citrate, or ethylenediaminetetraacetic acid (EDTA). Heparin tubes should not be used because of the likelihood of false-positive results. Blood samples must be taken and maintained using prewarmed tubes kept at 37 C to prevent autoabsorption of cryofibrinogen by RBCs. Samples should be transferred and analyzed in the laboratory within 1 h. Centrifugation of both sera (for cryoglobulin screening) and plasma (for cryofibrinogen screening) has to be done at 37 C. After centrifugation, the plasma and sera are cooled at 4°C for 3–8 days. Cryofibrinogen then appears as a precipitate, and dissolves when warmed to 37°C, but then reprecipitates when recooled to 4°C.3,4 Unlike cryoglobulin, which precipitated in the serum and plasma, cryofibrinogen precipitates only in the plasma. The characterization of purified cryoprecipitate components requires immunofixation or immunoblotting, while cryofibrinogen concentration can be measured using spectrophotometry.
There are no established diagnostic criteria for CF.3,4 However, a compatible clinical presentation along with a positive test for cryofibrinogen should be sufficient to make the diagnosis.3,4 Pathological and histological tests may be pursued if the diagnosis remains uncertain despite the above. The diagnosis of essential CF requires the exclusion of the secondary CF.3,4 Thus, in cases of ischemic ulcer, workup should cover vascular causes (including vasculitis), embolic causes, hypercoagulable states, and other rare causes. In cases of negative workup for all the above, a suspicion of CF should be raised. Due to the rarity of CF, the low awareness of clinicians for this condition, and the challenging laboratory examination with possible false positives and negatives, CF remains an underdiagnosed condition and a challenging diagnosis.
The mainstay of treatment of CF relies on avoidance of cold exposure, smoking cessation, avoidance of sympathomimetic drugs, and, in the cases of secondary CF, treatment of the underlying cause. Pharmaceutical management of CF relies on two approaches: the anti-ischemic and the immunosuppressive approach. Fibrinolytic agents, alteplase and streptokinase, have been successfully used to treat CF. However, its long-term use is limited because of its intravenous administration. Stanozolol, an oral drug with fibrinolysis-enhancing effect, has been used to efficiently treat CF. It is usually used as a maintenance therapy. The use of anticoagulant drugs (heparin or warfarin) seems to be limited to cases of venous thrombotic events with various degrees of success. Plasmapheresis may be used although it mostly has a poor outcome. If severe skin necrosis develops, antibiotics and/or amputation should also be considered.
Corticosteroids have been previously used, usually in combination with low-dose aspirin or another immunosuppressive drug for non-severe symptoms (cutaneous, arthralgia, and/or superficial thrombophlebitis). However, relapses are frequent after stopping or decreasing corticosteroid treatment during the first 6 months. Immunosuppressive drugs may include azathioprine and chlorambucil, while cyclophosphamide monotherapy seems less effective. Regular follow-ups are essential because of the possibility for recurrence and the increased prevalence of lymphoma among patients with CF.8,9
In conclusion, CF is a rare and underdiagnosed condition. The aim of this case-based review is to increase the awareness to this condition. Clinicians should be able to recognize this cryopathy especially in the cases of RP and ischemic ulcers not explained by other causes. Precautions must be taken during the diagnostic process, and therapy should be given as soon as possible. Close monitoring is essential due to the risk of recurrence.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iDs: Fadi Hassan  https://orcid.org/0000-0002-9648-1130
https://orcid.org/0000-0002-9648-1130
Helana Jeries
https://orcid.org/0000-0001-8624-6332
References
- 1. Korst DR, Kratochvil CH. Cryofibrinogen in a case of lung neoplasm associated with thrombophlebitis migrans. Blood 1955; 10: 945–953. [PubMed] [Google Scholar]
- 2. Stathakis NE, Karamanolis D, Koukoulis G, et al. Characterization of cryofibrinogen isolated from patients plasma. Haemostasis 1981; 10: 195–202. [DOI] [PubMed] [Google Scholar]
- 3. Michaud M, Pourrat J. Cryofibrinogenemia. J Clin Rheumatol 2013; 19: 142–148. [DOI] [PubMed] [Google Scholar]
- 4. Moiseev S, Luqmani R, Novikov P, et al. Cryofibrinogenaemia-a neglected disease. Rheumatology 2017; 56(9): 1445–1451. [DOI] [PubMed] [Google Scholar]
- 5. Smith SB, Arkin C. Cryofibrinogenemia: incidence, clinical correlations, and a review of the literature. Am J Clin Pathol 1972; 58: 524–530. [DOI] [PubMed] [Google Scholar]
- 6. Gómez-Fernández C, López-Sundh AE, González-Vela C, et al. High prevalence of cryofibrinogenemia in patients with chilblains during the COVID-19 outbreak. Int J Dermatol 2020; 59(12): 1475–1484. [DOI] [PubMed] [Google Scholar]
- 7. Amdo TD, Welker JA. An approach to the diagnosis and treatment of cryofibrinogenemia. Am J Med 2004; 116(5): 332–337. [DOI] [PubMed] [Google Scholar]
- 8. Saadoun D, Elalamy I, Ghillani-Dalbin P, et al. Cryofibrinogenemia: new insights into clinical and pathogenic features. Am J Med 2009; 122: 1128–1135. [DOI] [PubMed] [Google Scholar]
- 9. Belizna C, Loufrani L, Subra JF, et al. A 5-year prospective follow-up study in essential cryofibrinogenemia patients. Autoimmune Rev 2011; 10: 559–562. [DOI] [PubMed] [Google Scholar]