

Abstract
Background:
Ischemic preconditioning induces lateralization and dephosphorylation of Connexin 43 (Cx43). However, the Cx43 protein that remains at intercalated disks may be phosphorylated by casein kinase 1 (CK1) and protein kinase C (PKC), and both kinases provide cardioprotection from further ischemic injury. Here we explore the channel characteristics of a Cx43 mutant mimicking preconditioning by CK1 and PKC phosphorylation.
Materials and Methods:
Whole-cell patch-clamp recordings were performed in cells expressing the mutant Cx43pc (S325,328,330,368D, S365A-Cx43), and the connexin electrical behavior was analyzed at the single channel and macroscopic level.
Results:
Cx43pc hemichannels opened readily, whereas gap junctions channels displayed amplitudes between the wild-type and CK1 phosphorylated forms, and weaker voltage gating than either counterpart.
Conclusions:
Ischemic preconditioning and the ensuing phosphorylation of Cx43 by PKC may render junctional channels insensitive to transjunctional voltages, allowing the preservation of intercellular communication in ischemic conditions.
Keywords: Cx43, ischemia, preconditioning, CK1, PKC, patch clamp
Introduction
In the heart, the cell-to-cell gap junction channels (GJChs) that allow the propagation of electrical impulses are formed by Connexin 43 (Cx43) phosphorylated by casein kinase 1 (CK1) on serines 325, 328, and/or 330.1,2 Prolonged ischemia causes relocation of some percentage of the Cx43 protein from the intercalated disks to lateral membranes and dephosphorylation of these CK1 target sites.2,3 However, upon brief exposure to ischemia or during ischemic preconditioning, phosphorylation of Cx43 by protein kinase C (PKC) and potentially other kinases increases,2,4 and this phosphorylation by PKC provides a degree of resistance to further ischemic injury.5
Consistent with a role of CK1 phosphorylation in junctional plaque assembly,1 substituting nonphosphorylatable amino acids in the CK1 targets produces a cardiac phenotype prone to arrhythmias6 and cancels the beneficial effects of ischemic preconditioning.7 Some of these effects may be mediated by a decrease in the total levels of myocardial Cx43, activation of extracellular signal-regulated kinase, and interaction of Cx43 with hypoxia-inducible protein partners.8 In sheer contrast, and also consistent with the role of CK1 in Cx43 assembly,1 transgenic mice expressing a Cx43 mutant emulating CK1 phosphorylation are resistant to chronic pathological gap junction remodeling and induced ventricular arrhythmias.6
Phosphorylation by PKC is among the earliest reported post-translational modifications of Cx43, and it appeared to cause decreases on GJCh amplitude,9,10 macroscopic conductance, and permeability.11 The phosphorylation of Cx43 by PKC has been extensively studied in several systems and conditions. Thus, isoforms of PKC seem to phosphorylate Cx43 at several serines on its carboxyl terminus, including S255, S262, S365, S368, and S272/S373.12–14 The consequences of PKC phosphorylation of Cx43 are diverse,15,16 and chief among them is a cardiac phenotype resistant to arrhythmia and ischemia,12,17 not unlike the CK1–phosphomimetic Cx43 mutant.
We have been studying the function and regulation of Cx43 channels by phosphorylation.2,18,19 Summarizing available data from many laboratories and our own study, we noted that after ischemic preconditioning, the Cx43 protein at the junctional plaques displays a preserved overall phosphorylation and increased PKC phosphorylation on serine 368 (and 262), and that this is associated with reduced lateral relocation of Cx43 and delayed electrical uncoupling in a subsequent ischemia challenge.
Because the Cx43 protein remaining at the junctional plaques would thus be phosphorylated at CK1 and PKC sites, and both phosphomimetic mutants of these sites provide cardioprotection, we wanted to explore the electrophysiological characteristics of a Cx43 construct where all these serines were changed to the negatively charged aspartate and serine 365 was changed to alanine (Cx43-S325,328,330,368D, S365A), to emulate the phosphorylation pattern of a “preconditioned” form of Cx43, heretofore dubbed Cx43pc. Data suggest that PKC can phosphorylate other serine residues, including S262.12–14 However, for consistency with the body of our study and results from other laboratories,5,6 we did not modify other residues.
Materials and Methods
A description of plasmid construction and cell transfection has been previously published.19 In brief, the Cx43pc plasmid was constructed by mutating the mouse sequence to a rat sequence (N341S), then mutating the three CKI sites and lastly mutating the S365 and S368 sites. These mutations resulted in a rat amino acid sequence with all of the sites thought to contribute to a preconditioned state being fixed. All mutations were done with the QuickChange kit (Stratagene, San Diego, CA, USA), and sequencing was confirmed at the UAGC core facility.
The primers were as follows. For N341S: 5′ cga ttt ccc cga cga cag cca gaa tgc caa aaa ag 3′ and 5′ ctt ttt tgg cat tct ggc tgt cgt cgg gga aat cg 3′. For CKI (S325, 328, 330D): 5′ cat ggg gca ggc cgg aga cac cat cg acca cga tca cgc cca gcc gtt cg 3′ and 5′ cga acg cgt ggg cgt gat cgt tgt cga tgg tgt ctc cgg cct gcc cca tg 3′. For S365A, S368D: 5′ cca acg acc ttc cgc aag agc cga cag cc 3′ and 5′ ggc tgt cgg ctc ttg cgg aag gtc gtt gg 3′. The resultant construct S325,328,330,368D, S365A-Cx43 was labeled Cx43pc.
The plasmid pcDNA3 containing the mutations was amplified with the QIAgen Maxiprep kit (QIAgen, Hilden, Germany) to produce material for transfection. Rat insulinoma (Rin) cells that do not express Cx43 (parental line) were maintained under usual culture conditions, and transiently transfected with the Lipofectamine 2000 (Life Technologies, Grand Island, NY, USA) and pcDNA3 plasmid containing Cx43pc. Cells were used for electrophysiological measurements within a week of transfection.
Usage of mice was approved by the institutional animal care use committee (IACUC) for a study on Cx43 phosphorylation during ischemia (unpublished). Three-month-old wild-type C57Bl/6 mice were anesthetized until unresponsive to pain with a mix of isoflurane vapor and medical oxygen in a ventilated container connected to an anesthetic vaporizing machine and air filter for removal of circulating isoflurane.
Animals were immediately euthanized by cervical dislocation, their hearts promptly excised through abdominal and diaphragm incisions, and submerged in cold freshly prepared 1 × Krebs–Henseleit buffer solution (in mM: NaCl 118, KCl 4.7, MgSO4 1.2 mM, KH2PO4 1.2 mM, NaHCO3 25, CaCl2 1.25, glucose 11) to mimic global ischemia. Hearts were embedded in optimal cutting temperature medium (O.C.T. Compound, Sakura Finetek, Tissue-Tek, Item No. 4583), frozen, and sectioned in a cryostat. Heart slices of 8 μm thickness were prepared on glass slides for staining.
Immunostaining of Rin GVA, Rin cells stably transfected with wild-type rat Cx43 (WT Cx43), and of mouse heart subjected to 15′ of global ischemia2,18 were performed using primary antibodies against total Cx43 (tCx43) (Sigma-Aldrich, St. Louis, MO, USA), pS368-Cx43 (AbCam, Cambridge, MA, USA), and pS325/328/330-Cx43 (generous gift from Paul Lampe). Secondary antibodies labeled with Alexa488 or Alexa647 (Jackson ImmunoResearch, West Grove, PA, USA) were used for fluorescence detection. Heart slices were counterstained with wheat germ agglutinin to delineate cell borders.
Patch-clamp electrical recordings were performed as customarily.19 In brief, cells were placed in the stage of a microscope in a chamber superfused with an external solution (in mM: NaCl 142.5, KCl 4, MgCl2 1, glucose 5, Na-pyruvate 2, HEPES 10, CsCl 15, TEA-Cl 10, CaCl2 1). Patch-clamp pipettes filled with an internal solution (formulation in mM: 124 KCl, 3 MgCl2, glucose 5, HEPES 9, EGTA 9, CsCl 14, TEA-Cl 9, Na2-ATP 5, CaCl2 0.5) yielded electrode resistances of 4–10 MΩ. Using whole-cell voltage clamp mode, the membrane current (Im) of single cells or the junctional current (Ij) of cell pairs in response to voltage pulses of varying strength was recorded with switching clamp amplifiers and the pClamp10 software.20
Macroscopic junctional conductance (gj) was assessed with 2-s transjunctional voltage (Vj) pulses of ±10 mV. Voltage gating was induced with 5-s Vj pulses of increasing strength (±10 to ±100 mV), and gj was normalized using the initial 10 ms and the last 100 ms for each pulse. GJCh transition amplitudes were recorded at Vj of 40 and 80 mV after halothane-induced electrical uncoupling. Hemichannel (HCh) transitions were documented during minutes long pulses of Vm = +80 mV.
Analyses were performed with pClamp10 (Molecular Devices, LLC, San Jose, CA, USA) and Excel (Office 2010; Microsoft Corp., Redmond, WA, USA) as customarily.2,18 Values are reported as mean ± standard error of the mean (SEM). Graphics were created with SigmaPlot 2001 (Version 7.101; Systat Software, San Jose, CA, USA).
Results
Ischemia induces PKC phosphorylation of Cx43
To confirm that the Cx43 protein residing at junctional plaques can be phosphorylated at both CK1 and PKC sites, we used a polyclonal antibody recognizing tCx43, and two phospho-specific antibodies recognizing Cx43 phosphorylated at CK1 sites (pS235/328/330-Cx43) and PKC sites (pS368-Cx43). Because phospho-specific antibodies do not recognize phosphomimetic mutations, we used Rin cells expressing WT Cx43 in normal conditions to confirm the distribution of tCx43 and isoforms phosphorylated at the relevant sites. tCx43 was found both at cell contacts and at perinuclear spaces, whereas Cx43 phosphorylated by CK1 or PKC was concentrated at cell contacts (Fig. 1A).
FIG. 1.

Cx43 is phosphorylated by CK1 and PKC. (A, B) WT Cx43 in Rin GVA cells is abundantly expressed, and immunostaining with phosphospecific antibodies shows that plaque Cx43 (at cell-to-cell contacts) is phosphorylated by CK1 (A) and PKC (B) in cell-to-cell contacts). DIC image; colocalization of tCx43 and Cx43 phosphorylated at CK1 target sites (pCK1) shown in yellow at the merged image in a cell group. For pS368 Cx43, only that specific antibody was used, the merged image is the DIC and the fluorescence. (C) Mouse cardiac tissue after 15 min of global ischemia (see Materials and Methods section) displays lateral relocation of tCx43, and abundant phosphorylation at the CK1 and PKC sites of the protein remaining at the intercalated disks. WGA (blue) stain delineates cell membranes. Cx43 phosphorylated at CK1 and PKC sites appears transverse to the fibers' direction. CK1, casein kinase 1; DIC, differential interference contrast; PKC, protein kinase C; Rin, rat insulinoma; tCx43, total Cx43; WGA, wheat germ agglutinin; WT Cx43, wild-type Cx43.
In slices of mouse heart subjected to 15 min of global ischemia, tCx43 was found abundantly on the lateral sides of the cells, whereas CK1- and PKC-phosphorylated Cx43 remained at the intercalated disks (Fig. 1B), as previously reported.2,18 Although it cannot be assumed that every Cx43 molecule is phosphorylated by CK1, PKC, or both, it is possible that each GJCh has some of its 12 connexin units targeted by either or both kinases at the intercalated disks, but less so at the lateral membranes.
Cx43pc HChs open readily
In normal conditions, Rin GVA (constitutively expressing WT Cx43) displays rare openings of Cx43 HCh. In contrast, when held at Vm = +80 mV, Rin cells transfected with Cx43pc display abundant HChs, with current levels compatible with the expected conductance (∼180–240 pS) of Cx43 HCh (Fig. 2A). Interestingly, subconductive channel levels corresponding to a residual state were easily identified by analyzing expanded stretches of recordings (Fig. 2B). Notice that K+ channel blockers (cesium and tetraethyl ammonium) used in the external and internal solutions would, in addition to improving the definition of the recordings, inhibit K+ channel transitions.
FIG. 2.
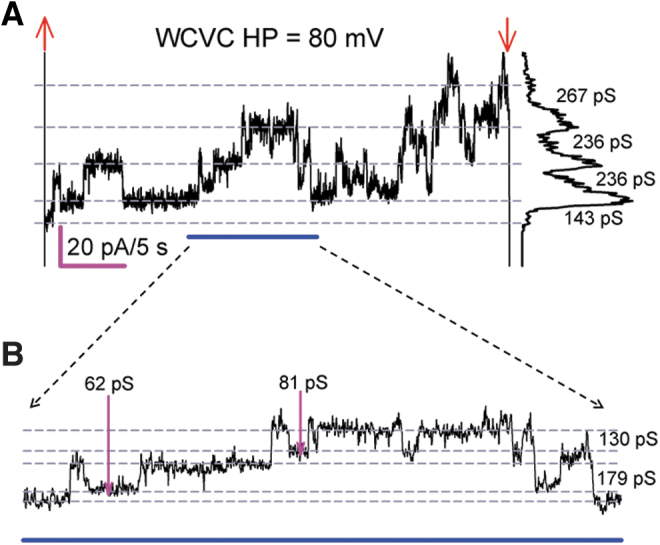
Cx43pc HChs open readily. (A) Representative Im sample (37 s) from a single Cx43pc-expressing Rin cell at +80 mV. The cell displayed frequent HCh openings twice the amplitude of GJCh transitions at sustained positive membrane voltage (A). Voltage pulse onset (upward red arrows) and offset (downward red arrows) are shown. The dotted lines indicate the various persistent current levels, and the plot at the right is the all-points histograms with transition conductance (g) values indicated. In (B), an expansion of the underlined (blue) region from (A) reveals the existence of subconductive channel opening (pink arrows). Extracellular Ca2+ = 1 mM.
GJChs of Cx43pc differ from those of WT Cx43 and Cx43–CK1D
Figure 3 shows GJCh activity from Cx43pc junctions at Vj of 40 and 80 mV, after halothane-induced uncoupling. Transitions from similar long recordings were manually measured to construct histograms of amplitude shown in Figure 4. At Vj = 40 mV, Cx43pc showed a range of transition amplitudes between 30 and 140 pS, with a wide peak at 70–100 pS, and a significant frequency of transitions <70 and >100 pS; at Vj = 80 mV, a clear shift toward smaller values (peak at ∼60–75 pS) occurred, and a second peak ∼25–35 pS (residual) appeared.
FIG. 3.
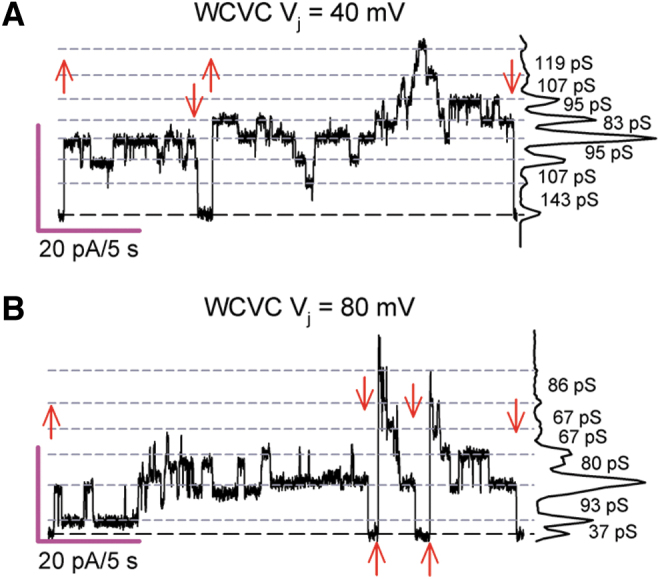
GJCh of Cx43pc display smaller transitions at larger Vj values. (A, B) Cx43pc GJCh transitions of variable amplitude suggest multiple conductive states at transjunctional voltage (Vj) of 40 mV (A) and 80 mV (B). Settings as shown in Figure 2. Notice that at Vj = 80 mV, channel transitions denote smaller conductance values than at Vj = 40 mV.
FIG. 4.

Cx43pc GJCh transition amplitudes decrease at larger Vj values. (A) Histograms of transition amplitudes from Cx43pc gap junction channels. Channel conductive states (open, substates including a residual) and the nonconductive state (closed) are symbolized to represent the likely transitions between them. (B) Difference plot (transitions at 80 mV minus transitions at 40 mV). Visits to the residual and substates are more frequent, and transitions from the fully open to the fully closed state are less frequent at the higher Vj value.
This suggests that at large Vj values, fewer channels transit between the fully open and the fully closed state, and more visit the residual state or a substate before closing (see channel configuration symbols and difference plot, 4B).
We have previously reported that the mutant with aspartate substitution of only the CK1 sites (Cx43–CK1D) displayed multiple GJCh open states and a wide range of transition conductance values.19 In Figure 5, the Cx43–CK1D and WT Cx43 histograms of transitions are reproduced as all events histograms to compare with the Cx43pc data. On examination of all events recorded, at Vj = 40 mV, most Cx43–CK1D transitions amplitudes were between 60 and 180 pS, with a peak at 90–135 pS, and few events were 80 pS or less.
FIG. 5.

GJCh of Cx43pc displays smaller transitions than mutant Cx43–CK1D, and similar transitions to WT Cx43. (A, B) Comparisons of histogram of transitions amplitudes of Cx43pc and Cx43–CK1D show smaller range and smaller transitions for the “preconditioned” mutant, at both Vj = 40 (A) and 80 (B) mV. (C, D) Similar comparisons with WT Cx43 reveal a similar range of amplitudes, but more intermediate states for Cx43pc.
At Vj = 80 mV, the conductance range was reduced by the decrease of the largest transitions, the main peak shifted to smaller values (90–110 pS) and a second peak at ∼40 pS (residual) appeared. At both Vj values of 40 and 80 mV, the Cx43pc mutant displayed smaller transitions than Cx43–CK1D. However, compared with WT Cx43, the preconditioned mutant displayed a slight prevalence of 95–115 pS at 40 mV, and a lower frequency of substates and residual transitions.
At 80 mV, both WT and Cx43pc transitions shifted to smaller values, but the mutant showed a small peak of intermediate transitions (55–85 pS), in agreement with previous observations,18 but did not display as many residual transitions as the WT protein (difference plot in 5D). These data suggest that PKC phosphorylation causes GJChs to occupy a configuration intermediate between WT Cx43 and that phosphorylated by CK1 alone. Possibly, the residual state of Cx43pc is resistant to transitions to and from the closed state.
Cx43pc gap junctions display weak Vj-gating sensitivity.
Voltage-induced channel closure (or Vj-gating) is a thoroughly studied feature of connexin gap junctions.21 Although large transjunctional voltages are unlikely to develop in well-coupled tissues, Vj-gating is an informative biophysical parameter of the channels, which may reflect structural changes of the protein. Thus, Cx43 often displays a modest Vj sensitivity that can be modified by mutations in its carboxyl terminus.19 Accordingly, Figures 6 and 7 show that the Cx43pc mutant exhibits a weak Vj-gating compared with WT Cx43 or Cx43–CK1D.
FIG. 6.

Cx43pc gap junctions display weak or no Vj-gating sensitivity. (A, B) Two examples of junctional current (Ij) in response to a series of Vj pulses of increasing strength, from 10 to 100 mV, in 10 mV steps. The current at 100 mV is shown in black to accentuate the lack of Vj-gating (A) observed in the group and the only pair that showed Vj-gating (B). To ensure proper voltage control, only pairs with low gj values (mean ± SEM = 4.61 ± 0.63; n = 5) were used for this analysis. SEM, standard error of the mean.
FIG. 7.

Cx43pc junctional conductance displays lower Vj-gating than WT Cx43 of Cx43–CK1D. (A, B) Normalized junctional conductance (Gj)/Vj curves from Cx43pc experiments shown in Figure 5 were not amenable to fit with a Boltzmann function (A), in comparison with Cx43wt (black lines; mean fit of published data), or a phosphomutant of only the CK1 sites (B; Cx43–CK1D; see Ref.19).
This suggests that PKC phosphorylation decreases the sensitivity of Cx43 GJCh to transjunctional voltage gradients. Together with the channel data, one can imagine that Cx43 phosphorylation by CK1D alone produces large conductance channel conformations that are amenable to transitioning from fully open to fully closed, while Cx43 phosphorylation by PKC causes intermediate conductance channel conformations that are less prone to complete closure.
Discussion
After brief ischemia or ischemic preconditioning of the heart, a population of Cx43 channels remains at the intercalated disks. These channels are phosphorylated by CK12 and PKC18 (see Fig. 1). Here, a Cx43 mutant mimicking phosphorylation by these two kinases, Cx43pc, expressed in a cell system without other connexins, displayed GJChs with transitions of smaller amplitude than the Cx43–CK1D phosphomimetic alone. Cx43pc gap junctions also showed weaker Vj-gating than Cx43–CK1D. In addition, Cx43pc HCh openings were readily observed in single cells.
The CK1 phosphomimetic Cx43-CK1D (aspartate for serine) or the dephosphomimetic Cx43–CK1A (alanine for serine) was previously studied in Rin cells.19 Compared with WT Cx43, both of these mutants displayed multiple GJCh open states, a macroscopic conductance resistant to acidification, and a tendency to open in HCh configuration in normal external calcium. Important differences were also noted, as the Cx43–CK1D mutant gap junctions exhibited variable permselectivity (similar to WT Cx43), stronger voltage dependence, and better regulation by pH. Cx43–CK1D also displayed HCh with better defined transitions than Cx43–CK1A.19
Focusing only on the electrical characteristics of the junctional channels, it would seem that either phosphorylation or dephosphorylation of the CK1 sites would yield gap junctions resistant to ischemic injury. However, the CK1 dephosphorylated form of Cx43 migrates to the lateral sides of cells,2 where it may not form gap junctions but instead open HChs. In contrast, the CK1 phosphorylated form of Cx43 remains at the intercalated disks, where HCh opening may have less damaging potential. Accordingly, a Cx43–CK1 dephosphomimetic is prone to ischemic damage and arrhythmia, whereas a Cx43–CK1 phosphomimetic is resistant to ischemic challenges and arrhythmia.6
Likewise, Cx43 phosphorylation by PKC is a well-known cardioprotective event,12,17 and the Cx43 protein that remains at the intercalated disks after brief ischemia is phosphorylated by PKC.2,18 All these data suggest that CK1 and PKC phosphorylation may be synergistic at providing cardioprotection, the former, plausibly by increasing the amount of available Cx43 channels,6–8 the latter by modifying characteristics of the channels themselves. Previously, a higher dye permeability was associated with intermediate Cx43 GJCh transitions, whereas a lower dye permeability was linked to fully open Cx43 GJCh.18
Cx43pc and WT Cx43 displayed a similar range of GJCh transitions, but the PKC–phosphomimetic mutant showed a peak of intermediate transitions at large Vj gradients. More intermediate GJCh transitions were also seen in Cx43pc than in the CK1D phosphomimetic. Possibly, preconditioned Cx43 channels have higher permeability to large molecules.18 It is also possible that WT Cx43 and our Cx43pc mutant are phosphorylated at other PKC sites12–14 during ischemia. Although this does not invalidate the results presented here, in which mimicking S368 phosphorylation produces measurable changes in channel behavior, the role of those alternative kinase targets remains to be determined.
In normal circumstances, it seems unlikely that well-coupled cells within a tissue would display transjunctional voltage gradients sufficient to close gap junctions. However, if most junctional channels are abruptly closed or become unavailable, the resistance between cells would increase, and voltage gradients due to depolarization of damaged cells may arise. The data shown here could mean that Cx43 GJCh phosphorylated by PKC, independent of their conductance, is resistant to voltage gradients between ischemic and nonischemic cardiac zones. Junctional channels with weak Vj-gating might provide for a degree of electrical and metabolic coupling (high permeability) and the eventual survival of cells in an infarcted/ischemic area.
We should note here that the resiliency provided by PKC phosphorylation may involve mechanisms beyond the regulation of junctional channels.5,12,14,22 Thus, in addition to reducing ischemic damage and increasing cardiomyocyte survival, ischemic preconditioning was reported to preserve Cx43 phosphorylation, endothelial nitric oxide synthase, and PKCɛ expression, and to decrease chemical coupling through gap junctions during a subsequent ischemia/reperfusion injury.5 A transgenic mouse line with a constitutively active PKC isoform displayed cardiac protection against ischemia/reperfusion injury,17 and it was suggested that this was explained by mitochondrial resiliency to anoxia–reoxygenation.16
Finally, it was also suggested that the cardioprotection provided by ischemic preconditioning may be due to the inhibition of Cx43 HCh opening, which prevented edema under simulated ischemic conditions.22
HCh opening was observed in the Cx43 mutants CK1D and CK1A,19 as here in Cx43pc-expressing cells. Although such openings appeared under the artificial condition of long +80 mV pulses, they reveal an overall increase in the HCh open probability. If these openings were to occur in normally polarized cells, they could cause depolarization, and at the same time allow molecular exchanges with the extracellular space (e.g., entrance of calcium ions and release of bioactive agents).23 Cx43 HCh opening may be one of the pathological events after ischemia.24
Consistent with this idea, upon activation of the sodium/calcium exchanger (NCX) by caffeine, unitary events identified as Cx43 HCh appeared in cardiomyocytes expressing WT Cx43 or the Cx43 mutant with dephosphomimetic substitutions of the MAPK sites, but the frequency of these events was low in the CK1 sites null-mutation (Cx43CK1mut: S325A-S328Y-S330A), and almost absent in PKC sites null-mutation (Cx43PKCmut: S368A).7 Although single-channel conductance values were not reported, the study's results are compatible with the data shown here.
Whether the opening of HChs is beneficial or detrimental remains to be firmly established. For instance, the opening of a few HChs could support calcium signaling or other secondary signaling, whereas massive HCh opening could collapse the membrane potential and perhaps support re-entrant arrhythmias. Probably, as for all the cellular changes induced by ischemia, the final outcome will depend on the strength and duration of the challenge.
Conclusion
A mutant Cx43 emulating a preconditioned phosphoform of the protein displayed intermediate GJCh conductance and weak gating by transjunctional voltage. It is possible that concurrent phosphorylation of Cx43 by CK1 and PKC yields GJChs resistant to closure by large transjunctional voltages. These channels could sustain the electrical and metabolic communication between ischemic and nonischemic cells and may constitute an important factor explaining the resilience of preconditioned hearts.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
National Heart, Lung and Blood Institute, R01HL058732 and R01HL131712 to J.M.B.
References
- 1. Cooper CD, Lampe PD. Casein kinase 1 regulates connexin-43 gap junction assembly. J Biol Chem 2002;277(47):44962–44968. [DOI] [PubMed] [Google Scholar]
- 2. Lampe PD, Cooper CD, King TJ, et al. Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci.2006;119(Pt 16):3435–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beardslee MA, Lerner DL, Tadros PN, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res 2000;87(8):656–662. [DOI] [PubMed] [Google Scholar]
- 4. Srisakuldee W, Jeyaraman MM, Nickel BE, et al. Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc Res 2009;83(4):672–681. [DOI] [PubMed] [Google Scholar]
- 5. Rong B, Xie F, Sun T, et al. Nitric oxide, PKC-epsilon, and connexin43 are crucial for ischemic preconditioning-induced chemical gap junction uncoupling. Oncotarget 2016;7(43):69243–69255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Remo BF, Qu J, Volpicelli FM, et al. Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ Res 2011;108(12):1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hirschhauser C, Lissoni A, Gorge PM, et al. Connexin 43 phosphorylation by casein kinase 1 is essential for the cardioprotection by ischemic preconditioning. Basic Res Cardiol 2021;116(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Solan JL, Marquez-Rosado L, Lampe PD. Cx43 phosphorylation-mediated effects on ERK and Akt protect against ischemia reperfusion injury and alter the stability of the stress-inducible protein NDRG1. J Biol Chem 2019;294(31):11762–11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moreno AP, Saez JC, Fishman GI, et al. Human connexin43 gap junction channels. Regulation of unitary conductances by phosphorylation. Circ Res 1994;74(6):1050–1057. [DOI] [PubMed] [Google Scholar]
- 10. Kwak BR, Hermans MM, De Jonge HR, et al. Differential regulation of distinct types of gap junction channels by similar phosphorylating conditions. Mol Biol Cell. 1995;6(12):1707–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kwak BR, Jongsma HJ. Regulation of cardiac gap junction channel permeability and conductance by several phosphorylating conditions. Mol Cell Biochem 1996;157(1–2):93–99. [DOI] [PubMed] [Google Scholar]
- 12. Jeyaraman MM, Srisakuldee W, Nickel BE, et al. Connexin43 phosphorylation and cytoprotection in the heart. Biochim Biophys Acta 2012;1818(8):2009–2013. [DOI] [PubMed] [Google Scholar]
- 13. Alstrom JS, Stroemlund LW, Nielsen MS, et al. Protein kinase C-dependent regulation of connexin43 gap junctions and hemichannels. Biochem Soc Trans 2015;43(3):519–523. [DOI] [PubMed] [Google Scholar]
- 14. Leithe E, Mesnil M, Aasen T. The connexin 43 C-terminus: A tail of many tales. Biochim Biophys Acta Biomembr 2018;1860(1):48–64. [DOI] [PubMed] [Google Scholar]
- 15. Vondriska TM, Zhang J, Song C, et al. Protein kinase C epsilon-Src modules direct signal transduction in nitric oxide-induced cardioprotection: complex formation as a means for cardioprotective signaling. Circ Res 2001;88(12):1306–1313. [DOI] [PubMed] [Google Scholar]
- 16. McCarthy J, McLeod CJ, Minners J, et al. PKCepsilon activation augments cardiac mitochondrial respiratory post-anoxic reserve—A putative mechanism in PKCepsilon cardioprotection. J Mol Cell Cardiol 2005;38(4):697–700. [DOI] [PubMed] [Google Scholar]
- 17. Cross HR, Murphy E, Bolli R, et al. Expression of activated PKC epsilon (PKC epsilon) protects the ischemic heart, without attenuating ischemic H(+) production. J Mol Cell Cardiol 2002;34(3):361–367. [DOI] [PubMed] [Google Scholar]
- 18. Ek-Vitorin JF, King TJ, Heyman NS, et al. Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ Res 2006;98(12):1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ek-Vitorin JF, Pontifex TK, Burt JM. Cx43 channel gating and permeation: Multiple phosphorylation-dependent roles of the carboxyl terminus. Int J Mol Sci. 2018;19(6):1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ek-Vitorin JF, Burt JM. Quantification of gap junction selectivity. Am J Physiol Cell Physiol 2005;289(6):C1535–C1546. [DOI] [PubMed] [Google Scholar]
- 21. Gonzalez D, Gomez-Hernandez JM, Barrio LC. Molecular basis of voltage dependence of connexin channels: An integrative appraisal. Prog Biophys Mol Biol 2007;94(1–2):66–106. [DOI] [PubMed] [Google Scholar]
- 22. Wang W, Zheng D, Li H, et al. Hemichannel-mediated volume regulation contributes to IPC-induced cardiomyocyte protection. Exp Ther Med 2019;17(3):1847–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mandal A, Shahidullah M, Delamere NA. Calcium entry via connexin hemichannels in lens epithelium. Exp Eye Res 2015;132:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shintani-Ishida K, Uemura K, Yoshida K. Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am J Physiol Heart Circ Physiol 2007;293(3):H1714–H1720. [DOI] [PubMed] [Google Scholar]