

Abstract
Important bioactive natural products, including prostaglandin H2 and artemisinin, contain reactive endoperoxides. Known enzymatic pathways for endoperoxide installation require multiple hydrogen-atom transfers (HATs). For example, iron(II)- and 2-oxoglutarate-dependent verruculogen synthase (FtmOx1; EC 1.14.11.38) mediates HAT from aliphatic C21 of fumitremorgin B, capture of O2 by the C21 radical (C21•), addition of the peroxyl radical (C21-O-O•) to olefinic C27, and HAT to the resultant C26•. Recent studies proposed conflicting roles for FtmOx1 tyrosine residues, Tyr224 and Tyr68, in the HATs from C21 and to C26•. Here, analysis of variant proteins bearing a ring-halogenated tyrosine or (amino)phenylalanine in place of either residue establishes that Tyr68 is the hydrogen donor to C26•, while Tyr224 has no essential role. The radicals that accumulate rapidly in FtmOx1 variants bearing a HAT-competent tyrosine analog at position 68 exhibit hypsochromically shifted absorption and, in cases of fluorine substitution, 19F-coupled electron-paramagnetic-resonance (EPR) spectra. By contrast, functional Tyr224-substituted variants generate radicals with unaltered light-absorption and EPR signatures as they produce verruculogen. The alternative major product of the Tyr68Phe variant, which forms competitively with verruculogen also in wild-type FtmOx1 in 2H2O and in the variant with the less readily oxidized 2,3-F2Tyr at position 68, is identified by mass spectrometry and isotopic labeling as the 26-hydroxy-21,27-endoperoxide compound formed after capture of another equivalent of O2 by the longer lived C26•. The results highlight the considerable chemical challenges the enzyme must navigate in averting both oxygen rebound and a second O2 coupling to obtain verruculogen selectively over other possible products.
Keywords: enzyme mechanism, tyrosyl radical, fumitremorgin B endoperoxidase, hydrogen atom transfer, iron, 2-oxoglutarate, oxygenase, ferryl
Graphical Abstract:
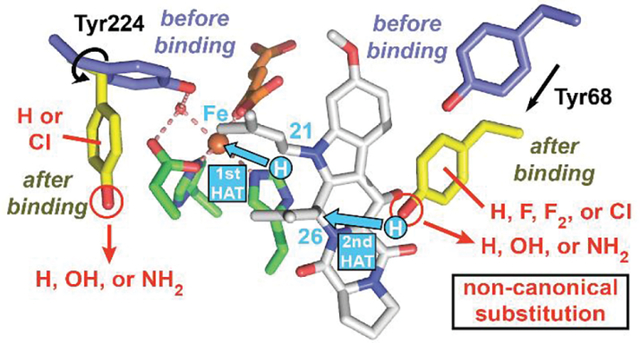
INTRODUCTION
The human hormones prostaglandin G2 and H2 (PGG2 and PGH2), the plant-derived malaria drug artemisinin, and several microbial natural products contain cyclic dialkylperoxide (endoperoxide) moieties that underpin their biological activities.1 Each case of enzymatic endoperoxide installation that has been studied to date involves capture of O2 between a carbon radical and nearby olefin of the substrate.2–4 The radical forms by hydrogen-atom (H•) transfer (HAT) from the substrate to an oxidized enzyme intermediate generated from or by an iron cofactor. The longest-known and most extensively studied endoperoxide-installing enzymes are the cyclooxygenases (COX), which convert arachidonic acid to PGG2 and PGH22 and are targets of non-steroidal anti-inflammatory drugs.5 Two more recently identified endoperoxide synthases belong to the iron(II)- and 2-oxoglutarate-dependent (Fe/2OG) enzyme family.3,4,6,7 Verruculogen synthase [also known as fumitremorgin B (ftnB) endoperoxidase or FtmOx1; EC 1.14.11.38] from Aspergillus fumigatus installs an endoperoxide between carbons 21 and 27 of ftnB (1 in Figure 1A), producing verruculogen (2 in Figure 1A) on the pathway to the tremor-inducing mycotoxin, fumitremorgin A.3 NvfI from Aspergillus novofumigatus IBT 16806 converts its substrate asnovolin A to fumigatonoid A by installing both (i) an endoperoxide between methyl (C13) and olefinic (C2’) carbons and (ii) a hydroxyl group on C3’ – reportedly in a single step.8
Figure 1. Structure and function of verruculogen synthase (FtmOx1).
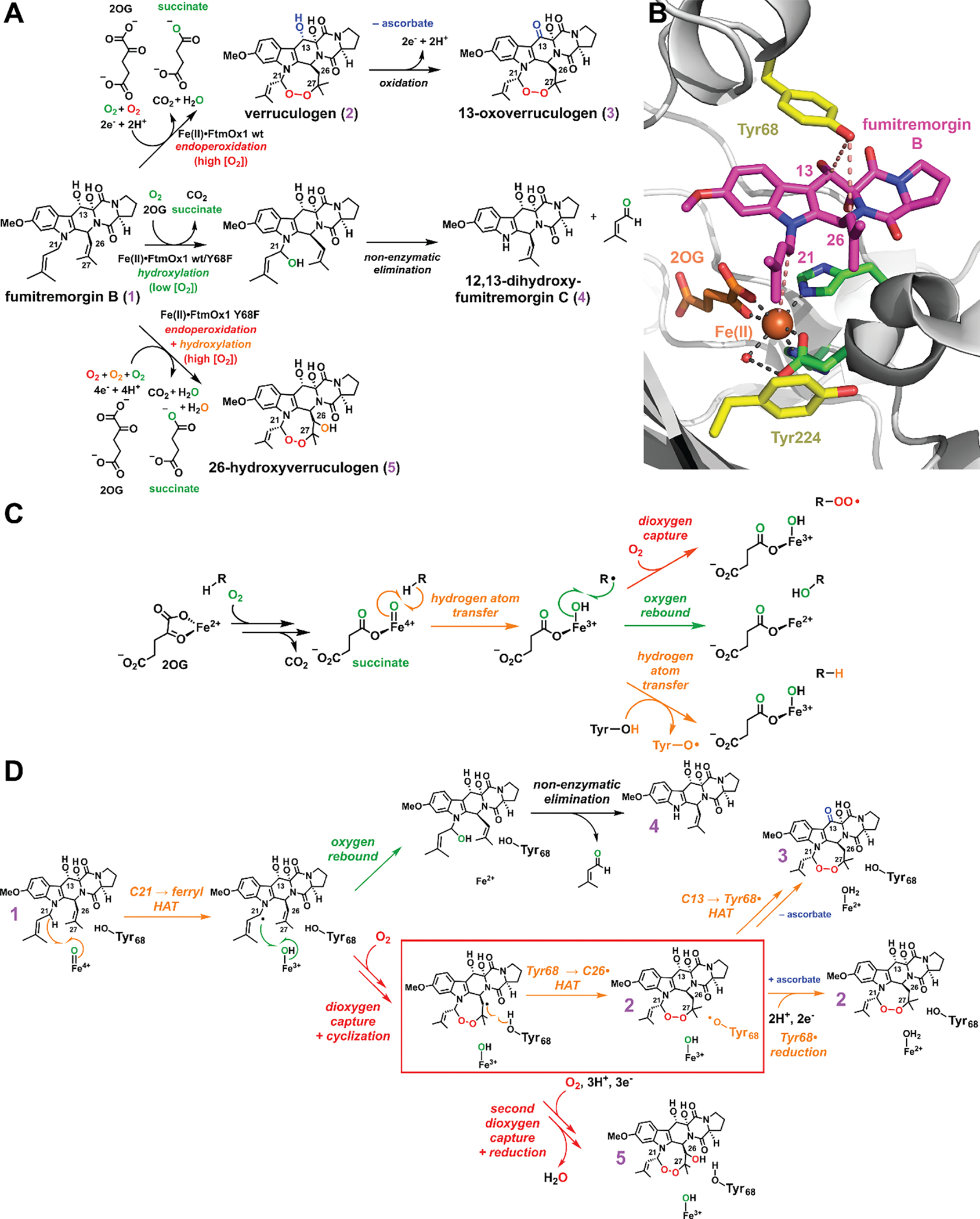
(A) Reaction(s) catalyzed by FtmOx1 and its Y68F variant, in which partitioning depends on [O2] and the presence or absence of reductant (ascorbate). (B) Blow-up of the active site in the FtmOx1 reactant complex (PDB entry: 7ETK).4 The facial triad is shown in green, iron(II) and 2OG are shown in orange, fumitremorgin B (1) is shown in magenta, the two contentious Tyr residues are highlighted in yellow,3,13 and distances relevant to the three HAT steps are shown with salmon dashed lines. The structure of the ternary complex3 is shown in Figure S1. (C) General fates of carbon-centered radicals of relevance to the FtmOx1 reactions. (D) Mechanisms proposed in reference 13 and this work for substrate endoperoxidation (orange arrows) and hydroxylation (green arrow) by FtmOx1. The key HAT step (Tyr68 → C26•) of interest is highlighted by a red box. A complete scheme relating the CarC-like and COX-like catalytic cycles is shown in Figure S2.
In the COX reaction, two HAT steps, from C13 and to a peroxyl radical on C15, initiate and terminate turnover.2 These steps bracket three C–O-bond-forming radical couplings that capture two O2 molecules to install the endo- and hydro-peroxides of the PGG2 product. A single tyrosine (Tyr or Y) mediates both HATs. This Tyr residue is oxidized to a quasi-stable radical (Tyr•) in a peroxidase-type reaction between the enzyme’s heme cofactor and the hydroperoxide of the initial PGG2 product, which is concomitantly reduced to the hydroxyl-bearing PGH2. The resulting Tyr• accepts H• from C13 and is regenerated by donating H• to the C15-O-O• intermediate.9 The reaction thus proceeds catalytically until and unless the Tyr• is adventitiously reduced, in which case it must be regenerated by another reaction of the heme with PGG2.
The first biophysical analysis of FtmOx1 proposed a COX-like mechanism invoking a similar catalytic role for a Tyr• – in this case generated from Tyr224 (Figure 1B) by the oxoiron(IV) (ferryl) intermediate characteristic of the Fe/2OG enzyme class (Figure S2, pink arrows).3 Evidence included an x-ray crystal structure of the enzyme in complex with 2OG, but lacking the prime substrate, 1 (Figure S1A), and transient ultraviolet-visible (UV-vis) absorption and electron paramagnetic resonance (EPR) spectra from the reaction of the complete FtmOx1•Fe(II)•2OG•1 complex with O2. Computational studies supported the feasibility of this mechanism but were handicapped by the absence of a structure of the full reactant complex, with 1 bound.10–12 A more recent experimental study13 posited HAT directly from C21 to the canonical ferryl complex – generated stoichiometrically in each turnover – and participation of a different tyrosine (Tyr68, Figure 1B) only as H• donor to complete production of 2 (Figures 1D and S2, orange arrows), a mechanism with analogy to that previously shown for the stereoinversion reaction catalyzed by carbapenem synthase (CarC).14 Incorporation of 18O from 18O2 into the prenal co-product from the conversion of 1 to 4 (Figure 1D, green arrow), which occurs in an [O2]-dependent partition with the primary 1 → 2 reaction, implied competitive “oxygen rebound”15 and therefore proximity of C21 to the iron cofactor (Figure 1A). The Tyr224Phe variant exhibited reaction kinetics and verruculogen yields similar to those of wild-type FtmOx1, whereas the Tyr68Phe variant failed to accumulate the initial Tyr• and generated an (unidentified) alternative product 5 along with very little verruculogen (Figure 1A). Very recently, while this study was in progress, an x-ray crystal structure of the FtmOx1•Fe(II)•2OG•1 complex was finally reported (Figure 1B).4 This structure showed (1) proximity of C21 to the cofactor (4.6 Å), (2) proximity of the Tyr68 phenolic oxygen (O7) to C26 (4.0 Å), and (3) greater separation of Tyr224 O7 from the cofactor (8.5 Å) than in the prior structures of the FtmOx1•Fe(II)•2OG ternary complex (4.2 Å).3,4,13 Computational analysis based on this structure led the authors to favor the CarC-like mechanism.
Here, we have used the methodology pioneered by Schultz and coworkers16 to replace either of the two tyrosines (Tyr68 and Tyr224, Figure 1B) with a non-canonical Tyr analog: 3-fluorotyrosine (3-FY), 2,3-difluorotyrosine (2,3-F2Y), 3,5-difluorotyrosine (3,5-F2Y), 3-chlorotyrosine (3-ClY), or 4-aminophenylalanine (4-NH2F) (Figures 2C and S3). UV-vis absorption and EPR spectra from the reactions of the variant proteins and analysis of the resultant organic products provide direct physical evidence that the Tyr• that forms concomitantly with 2 is on Tyr68 rather than Tyr224. Analysis of the other products, particularly those generated in the reactions of the Y68F and Y68(2,3-F2Y) variants and the wild-type enzyme in D2O (Figures 2D and 2E), reveals a second pathway for decay of the C26• – capture of an additional molecule of O2 (Figure 2F) – that can become competitive when the controlling HAT step is delayed. This step ultimately results in formation of a C26-hydroxylated endoperoxide-bearing product (5) analogous to PGH22 and fumigatonoid A.8 A simple guide to the different reactions demonstrated herein and the conditions under which each is favored is presented below (Figure 2) to aid in understanding of the experimental design.
Figure 2. Experimental strategies to characterize the key HAT step (Tyr68 → C26•) in the presence of the H• donor and the alternative product 5 that can form when this the key HAT step is impeded.
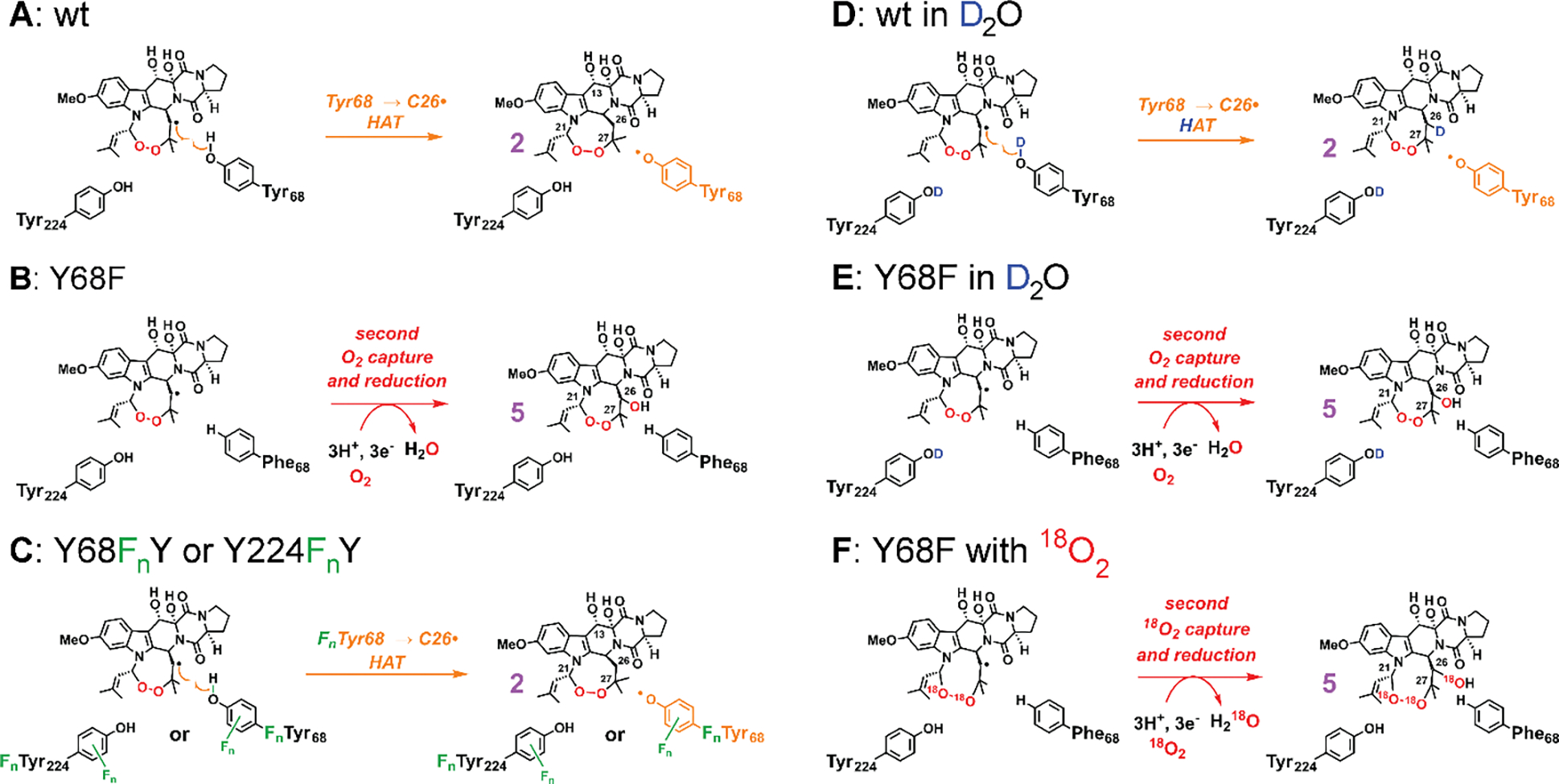
(A) Wild-type (wt) enzyme predominantly generates 2 accompanying Tyr68• under single-turnover conditions. (B) Replacement of Tyr68 with Phe diverts the reaction toward an alternative product 5 with no transient Tyr• detected.13 (C) Replacement of Tyr68 or Tyr224 with non-canonical tyrosine analogues (3-FY, 3,5-F2Y, 2,3-F2Y, 3-ClY, and 4-NH2F; only ring fluorination is shown in the scheme) allows for unambiguous identification of the radical harboring tyrosine (Tyr68). This strategy can also perturb the H•-donation ability of the residue and the partition between 2 and 5. (D and E) D2O leads to deuterium on the phenol, slowing donation. The partition ratio between 2 and 5 reflects competition between the HAT step (dominant in wt) and the second O2 capture (dominant in the Y68F variant). The isotopic distribution of 2 and 5 reinforces Tyr68 as the key donor and reveals the lack of newly formed C–D bond in 5. The solvent exchangeable sites in the products are shown with protia, as detected with LC-MS, in panels D and E. (F) Reaction of the Y68F variant with 18O2 generates 5 with three 18O atoms, and the LC-MS2 analysis reveals the identity of 5.
MATERIALS AND METHODS
Detailed procedures for construction of FtmOx1 expression vectors and aminoacyl-tRNA synthase plasmids, synthesis and characterization of fluorinated tyrosine analogs, expression and purification of FtmOx1 non-canonical variants, preparation of buffers in D2O, deoxygenation and oxygenation of proteins and buffers, use of UV-visible absorption spectroscopy to monitor 2OG binding, LC-MS (LC-MS2) analysis of FtmOx1 reaction products, and stopped-flow absorption, freeze-quench EPR, and chemical-quench LC-MS kinetics experiments are provided in the Supporting Information.
RESULTS
Tyr68 but not Tyr224 is the radical-harboring site and the hydrogen donor.
General properties of the ncAA-bearing proteins (Figure 2C).
Incorporation of 3-FY, 3,5-F2Y, 2,3-F2Y, 3-ClY, or 4-NH2F at position 68 of FtmOx1 yielded variants with the ability to direct endoperoxide installation upon 1 to form 2 (Figure 3). This competence contrasts with the previously noted13 redirection of the reaction to alternative product 5 by the canonical Y68F substitution (red trace in Figure 3A). This top-line observation confirmed our expectation that replacement of functional tyrosines with appropriate ncAAs would permit a finer dissection of the native reaction and roles of tyrosine residues therein.
Figure 3. LC-MS chromatograms from the reactions of wild-type (wt) FtmOx1 and its (A) Tyr68- and (B) Tyr224-substituted variants.
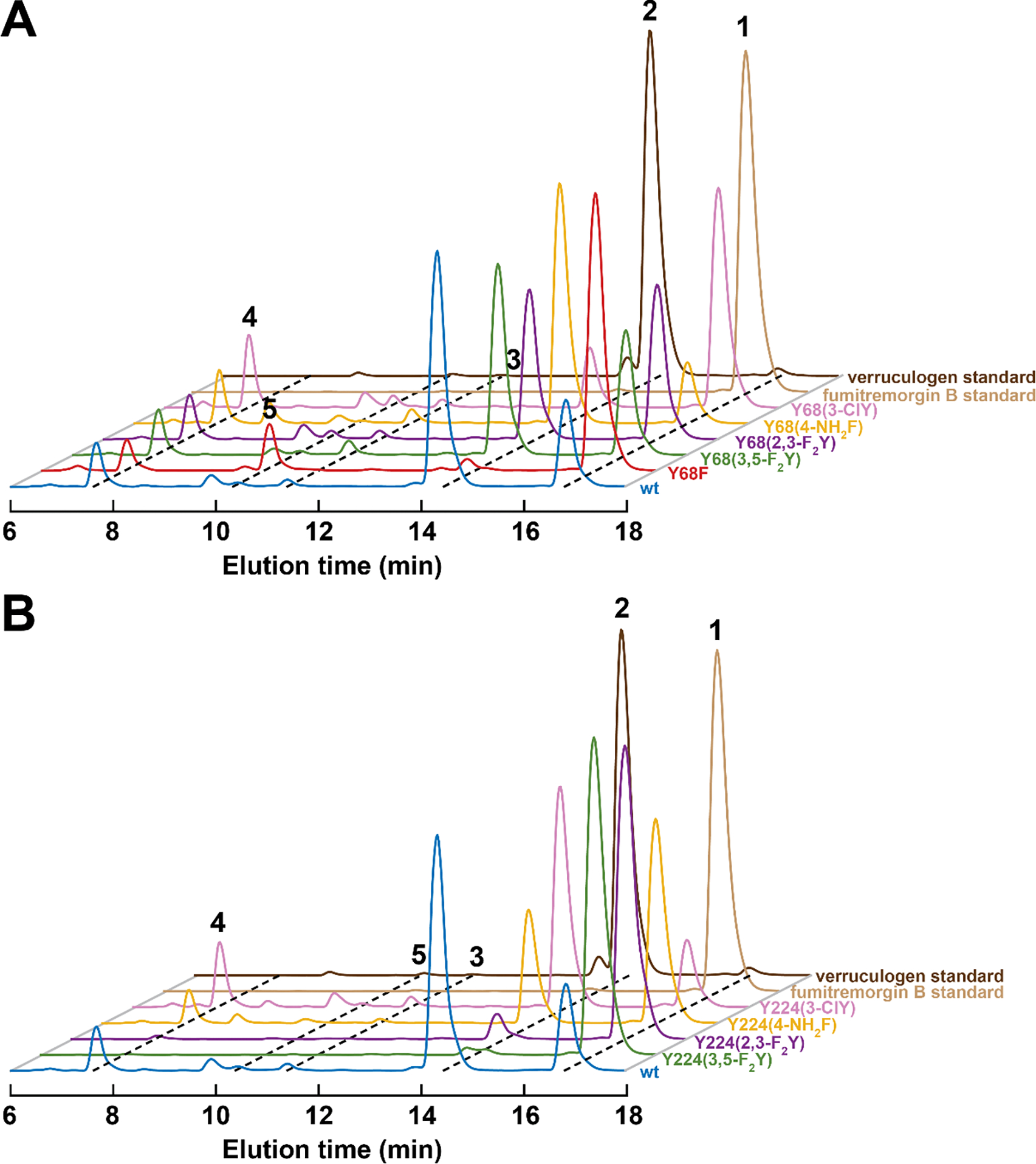
The traces are color-coded as in Figure S3 according to the tyrosine analog at position 68 or 224. The reactions contained 10 μM enzyme, 10 μM Fe(II), 1 mM 2OG, 1 mM ascorbate, 0.50 mM 1, and ~ 1.8 mM O2, pH 8.0 and were carried out at room temperature as described in Materials and Methods (Supporting Information).
For two of the Tyr224-substituted variants [Y224(3,5-F2Y) and Y224(2,3-F2Y)], however, we observed little or no product formation (green and purple traces in Figure 3B). On the basis of the inability of these variants to develop either the 520-nm metal-to-ligand charge-transfer (MLCT) band characteristic of the FtmOx1•Fe(II)•2OG ternary complex (Figure S4C) or the appropriate transient absorption features (Figures S5F and S5L) upon subsequent rapid mixing of the presumptive quaternary complex with O2, we attribute this inactivity to a deficiency in proper 2OG binding. Indeed, structural modeling indicates that the appended fluorines could cause significant steric clashes in the tight 2OG-binding pocket (Figure S4B). We also noted that the apo forms of these Tyr224 → fluorotyrosine variants were more prone to precipitation upon concentration (Figure S4A), suggesting that the substitutions unexpectedly impair the enzyme’s structural integrity.
Spectral-kinetic data on wild-type FtmOx1 for comparison (Figure 2A).
We next repeated aspects of the previously reported transient-state kinetic analysis of wild-type FtmOx1 to allow for the most direct comparison to the catalytic behavior of variants bearing a noncanonical Tyr analog. Mixing of the wild-type FtmOx1•Fe(II)•2OG•1 quaternary complex with an equal volume of O2-saturated buffer (~ 1.9 mM O2) resulted in development of the previously reported,3,13 broad, transient absorption band at ~ 417 nm (Figures 4A and S5A), which is significantly shifted from the usual ~ 410 nm peak of tyrosyl radicals. This feature reached its maximum intensity at 0.35 s (Figure S5A). The CW X-band EPR spectrum at 30 K of a sample freeze-quenched at this reaction time exhibits the broad g ≈ 2 signal seen in the earlier studies (Figure 4B). Product 2 was still readily detected in reactions containing ascorbate (Figure 3), which prevents Tyr• accumulation by rapidly reducing it (Figure S7). This result implies that the detected Tyr• is formed late in the reaction sequence and is not involved in the initiating HAT from C21 (orange arrows in Figures 1D and S2).13 This conclusion is further corroborated by the results of chemical quench experiments (Figures S8 and S9), which showed that the Tyr• develops as 2 forms. The temporal correspondence implies that the Tyr• is generated by HAT to C26• to complete 2 formation (Figure 2A). The chemical-quench results (Figures S8 and S9) also suggest that the previously reported subsequent dehydrogenation of 2 to 3 involves this Tyr•, as the decay of the radical was seen to correlate temporally with formation of 3 (Figures 1A and 1D). This conclusion is validated by the observation that the yield of 3 was vastly diminished in the presence of ascorbate (Figures 3 and S7).13 This sequence of reactions – namely, hydrogen donation followed by desaturation – is analogous to the sequence of steps mediated by CarC.14
Figure 4. SF-abs (left) and X-band FQ-EPR spectra (30 K, right) of the early radical intermediates in the reactions of wild-type (wt) FtmOx1 and variants that support their accumulation.
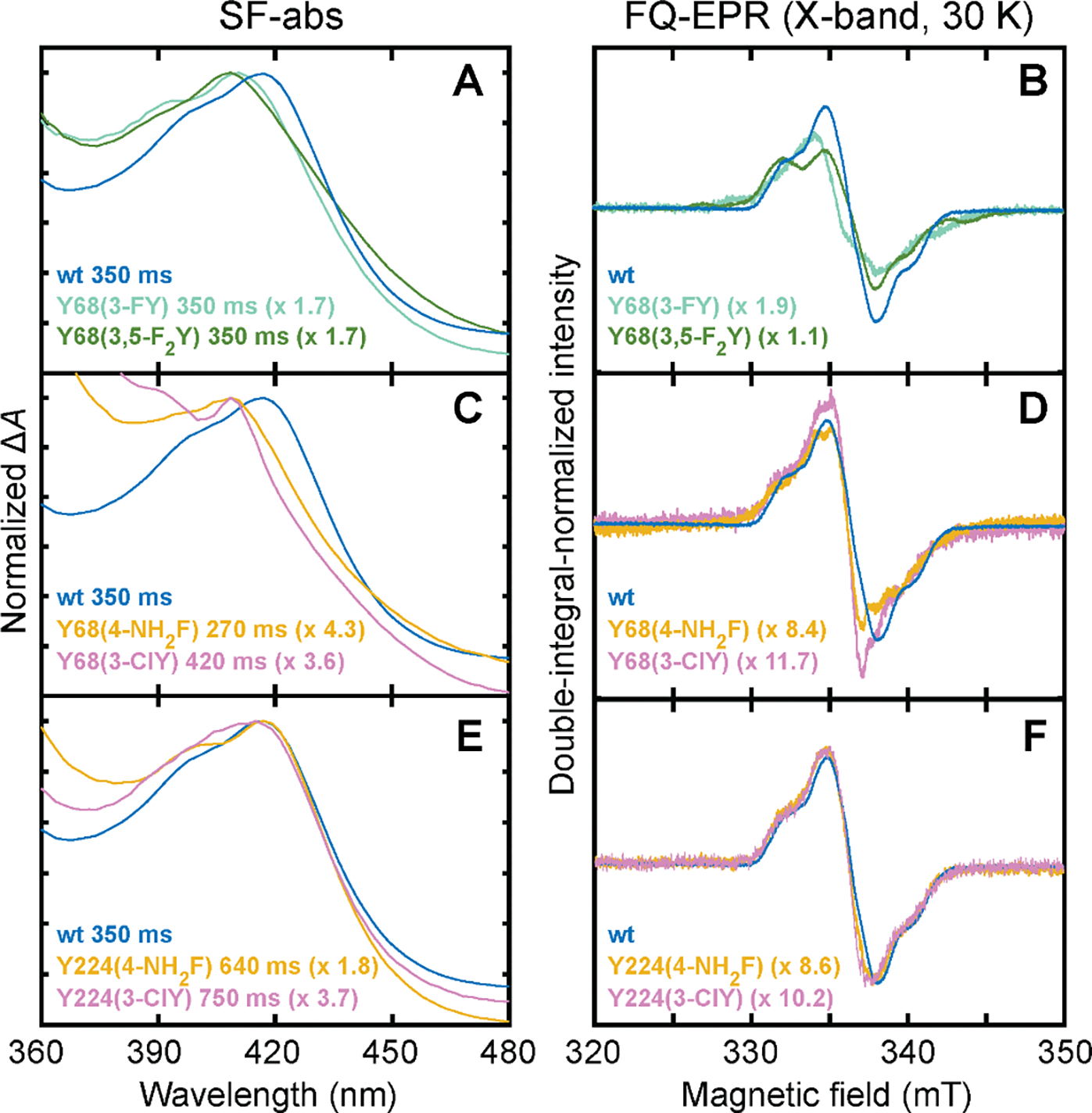
(A, B) Spectra from the reactions of the wt, Y68(3-FY), and Y68(3,5-F2Y) proteins, all at 350 ms. (C, D) Spectra from the reactions of the wt protein at 350 ms, the Y68(4-NH2F) variant at 270 ms, and the Y68(3-ClY) variant at 420 ms. (E, F) Spectra from the reactions of the wt protein at 350 ms, the Y224(4-NH2F) variant at 640 ms, and the Y224(3-ClY) variant at 750 ms. The reaction times for variants were selected to coincide with the time of maximal accumulation. The spectra from the wt FtmOx1 reaction are reproduced in each panel for ease of comparison. The absorption spectra have been scaled to coincide at the peak maxima, and the EPR spectra have been scaled to have equal double-integrated intensity; the scaling factors with respect to the wt spectra are noted in each panel. Quantification of the radical in the FQ samples from the reaction of wt FtmOx1 revealed that it accumulates to 25 (±1) % of the iron concentration at 350 ms (assuming a packing factor of 0.52 – 0.5524). The absorption was directly extracted from the same SF-abs data sets used in preparing Figure S5, and the reaction conditions in the FQ-EPR experiments were the same as in the stopped-flow experiments (see Figure S5 caption).
Use of ncAA Tyr analogues to test for perturbed spectra and kinetics (Figure 2C).
To identify the site of the Tyr•, we tested variants bearing fluorinated tyrosines for the expected (1) hypsochromic shift in the sharp Tyr• absorption feature and (2) large hyperfine coupling (Az ≈ 150 MHz) from 19F ortho to the phenolic oxygen (Figure 2C).17–22 We anticipated that, as has been seen in other enzymes, the minimally perturbed oxidation potentials of the 3-FY and 3,5-F2Y analogs21,23 (Figure S3) would make these fluorotyrosines, in particular, ideal probes. In stopped-flow absorption (SF-abs) experiments on the Y68(3-FY) and Y68(3,5-F2Y) variants, the characteristic feature of the Tyr• developed with kinetics similar to those seen for the wild-type enzyme (Figures S5A–S5C and S6A). Relative to the spectrum arising from the transient Tyr• in the wild-type enzyme, the spectra of the radical in the Y68(3-FY) and Y68(3,5-F2Y) variants are indeed hypsochromically shifted to more usual maxima of 411 (Figures 4A and S5B) and 409 nm (Figures 4A and S5C), respectively. The direction and magnitudes of these shifts from 417 nm are comparable to those seen in comparing the corresponding fluorinated tyrosyl radicals to unsubstituted Tyr• in solution.21 For the two fluorotyrosine-bearing variants, the X-band EPR spectra at 30 K of samples freeze-quenched at a reaction time of 0.35 s exhibit prominent hyperfine couplings that are not seen with wild-type FtmOx1 (Figures 4B and S10). The spectrum of the protein harboring the difluorinated analog is the broadest and exhibits couplings from two 19F nuclei (Figures 4B and S10). These absorption and EPR spectra conclusively establish that the tyrosine (or fluorotyrosine) at position 68 donates hydrogen to form the accumulating radical.
To assess the possibility that Tyr224 can act as an alternative or secondary H• donor (Figure S4), we evaluated variants with 4-NH2F or 3-ClY in place of Tyr68 or Tyr224. Substitution of either residue with these ncAAs was seen to preserve function (Figure 3). In the reaction of the Y68(4-NH2F) variant, the radical accumulated maximally at ~ 260 ms (Figure S6A) and absorbed at ~ 409 nm (Figures 4C and S5E). This absorption maximum aligns with that arising from the radical derived from 4-methylaniline25 (Figure 4C). The EPR spectrum of samples freeze-quenched near the time of maximum 409-nm absorption has additional hyperfine splittings from both 14N (I = 1) and the proton (I = 1/2) it harbors26 (Figure 4D). To our knowledge, a simple anilino radical has not previously been characterized in a protein.27 By contrast to the clear perturbation of the accumulating radical in the reaction of the Y68(4-NH2F) variant, the Y224(4-NH2F) variant developed absorption (Figures 4E and S5I) and EPR spectra (Figure 4F) that matched exactly with their counterparts in the reaction of the wild-type enzyme (Figure S6C). The same observations were made in examination of the Y68(3-ClY) and Y224(3-ClY) variant enzymes. The chlorinated tyrosine caused the anticipated narrowing (a consequence of the diminished gyromagnetic ratios of 35Cl and 37Cl relative to 1H22) when incorporated at position 68 but not when it was incorporated at position 224 (Figures 4C–F, S5D, and S5H). These observations with non-canonical variants thus definitively establish that Tyr68 rather than Tyr224 is the H• donor in the FtmOx1 reaction, echoing the conclusions reached from investigation of the Y68F and Y224F variants13 the structure of the FtmOx1•Fe(II)•2OG•1 complex (Figures S11A and S12).4 The new data are consistent with and rationalize the prior observations that the Y224F variant is fully competent to produce 2 but the Y68F generates an alternative primary product, identified in experiments discussed below.
Impact of perturbing the H•-donor ability of residue 68 on the 2:5 partition ratio and identification of 5 (Figures 2B–2E).
General considerations.
With the ability to tune the properties of the position 68 residue now in hand, we used this tool to identify the alternative product, 5, of the Y68F variant (Figure 2B; red trace in Figure 3A) and probe determinants of its formation. Two perturbations were used in combination with mass spectrometry to provide evidence that the alternative product is 26-hydroxyverruculogen, resulting from a second O2 coupling step with the stabilized C26•. Incorporation of 2,3-F2Y at position 68 was anticipated to slow H• donation (Figure 2C), because the phenolic O–H bond of this Tyr analog is ~ 1 kcal/mol stronger than that of tyrosine itself (Figure S3). Similarly, use of 2H2O (D2O) as solvent to replace the phenolic protium with deuterium (Figures 2D and 2E) was expected to slow the HAT step as a result of the increased strength of the O–D bond and the diminished capacity of deuterium to undergo quantum-mechanical tunneling.
Incorporation of the poorer H• donor, 2,3-F2Y, at position 68 suppresses radical accumulation (Figure 2C).
In SF-abs experiments with the Y68(2,3-F2Y) variant (Figure 2C), we observed no transient absorption feature attributable to a Tyr• (Figure S5K). Indeed, its kinetic behavior was seen to mirror that of the Y68F variant (Figures S6A and S6B). In principle, the diminished pKa of this analog (~ 7.8 for the free amino acid) relative to that of tyrosine could cause it to deprotonate and lose competence for the quenching HAT step, but the full competence of 3,5-F2Y – with an even lower pKa (7.2) – at position 68 makes deprotonation an unlikely explanation for the altered kinetic behavior of the Y68(2,3-F2Y) variant. Indeed, LC-MS analysis of the products from reactions of this variant revealed formation of primarily the native product, 2, but with a significant quantity of the alternative product, 5 (Figures 3A and S19). Thus, the 2,3-F2Y analog is, unlike either tyrosine or phenylalanine, partly competent for the quenching HAT step. It appears that the more potently oxidizing 2,3-F2Y68• has a diminished lifetime relative to that of Tyr68•, causing it not to accumulate under the reaction conditions examined. The toolkit of fluorinated analogs at position 68 thus afforded a variant bridging the fully competent wild-type enzyme and incompetent Y68F variant, providing additional evidence that Tyr68 is the H• donor.
Reaction of wild-type FtmOx1 in 2H2O (D2O) gives perturbed product profile from delayed Tyr68 → C26• HAT (Figure 2D).
The second strategy to delay the quenching HAT step was replacement of the phenolic hydrogen atom with deuterium in the wild-type enzyme (Figure 2D). The substitution could be achieved simply by performing the reaction in D2O, owing to the fast 1H/2H exchange of solvent-exposed tyrosines (> 100 s−1).28 As anticipated, less 2 formed in D2O (Figure 5A, compare blue and cyan traces). Consistent with earlier work,13 the yield of 2 was also much less in the reaction of the Y68F variant (red and orange traces), with the reaction of the Y68F variant in D2O producing almost no verruculogen (Figure 2E). In all cases, the diminution in the yield of 2 was accompanied by an enhanced yield of 5 (see also Figure S19). The results with the Y68(2,3-F2Y) variant and wild-type FtmOx1 in D2O demonstrate that formation of 2 and 5 are competitive, with the former requiring the Tyr68-mediated HAT step and the latter proceeding via a different pathway.
Figure 5. LC-MS characterization of products derived from 1 in reactions of wild-type FtmOx1 and its Y68F variant in H2O (pH 8.0) and D2O (pD 8.0).
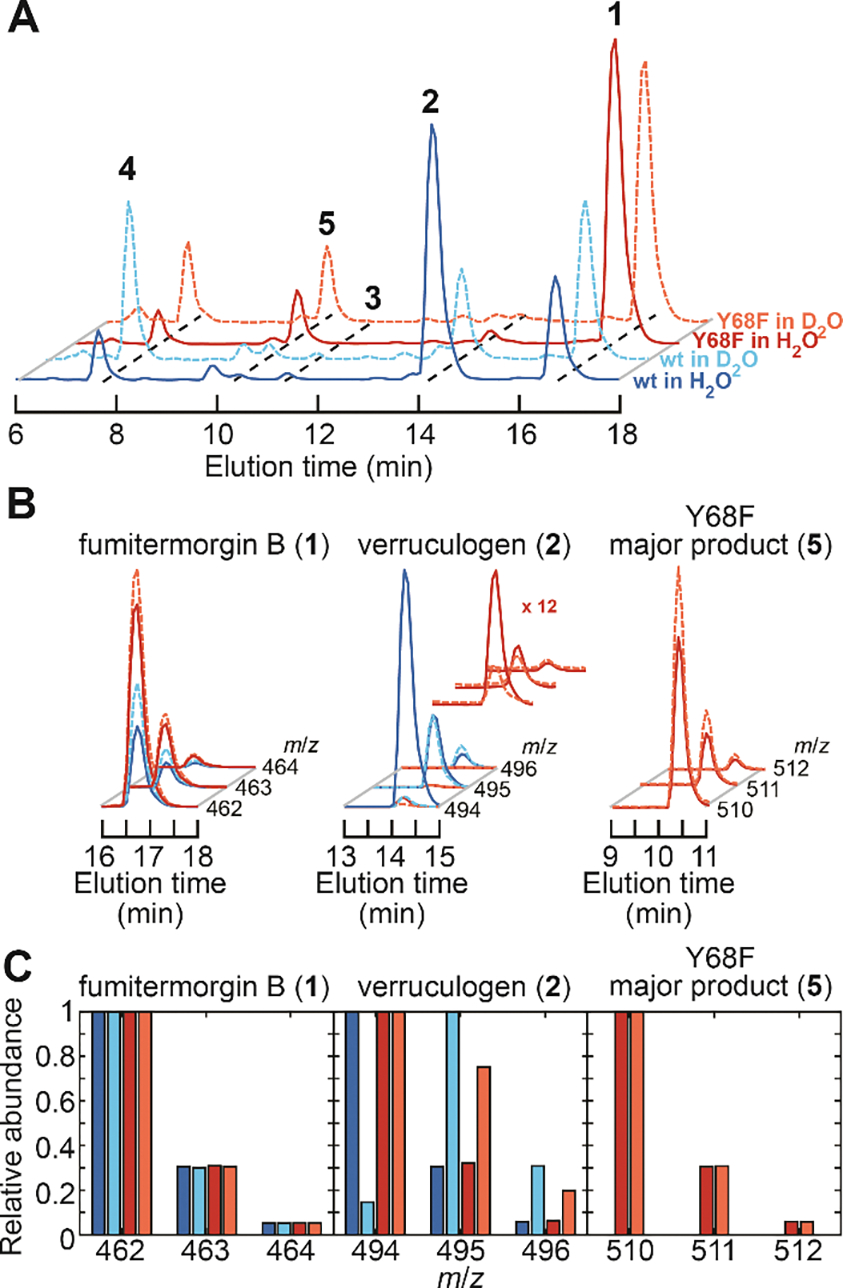
(A) Total-ion chromatograms (TICs) of products from the reactions of wt FtmOx1 in H2O (solid blue trace), wt enzymes in D2O (dashed cyan trace), the Y68F variant in H2O (solid red trace), and the Y68F variant in D2O (dashed orange trace). The color code also applies to panels B and C. (B) Isotope analysis from EICs of 1, 2, and 5 formed in the reactions of panel A. Traces of 2 from the reaction of the Y68F variant are magnified to better illustrate the isotopic distributions. (C) Relative abundances of the isotopologues of 1, 2, and 5 extracted from panel B. The reactions contained 10 μM enzyme, 10 μM Fe(II), 1 mM 2OG, 1 mM ascorbate, 0.50 mM 1, and ~ 1.8 mM O2, pH/pD 8.0 at room temperature.
Use of D2O provides evidence for altered pathway to verruculogen (2) in the Y68F variant (Figure 2E).
Prior work13 showed, and the results above (Figure 5A, red traces) confirm, that the Y68F variant still produces a detectable quantity of 2, implying that H• can be provided (inefficiently) to C26• by an additional source when Tyr68 is absent. The results of comparative mass-spectrometric analysis of 2 generated in D2O by the wild-type and Y68F proteins provides support for this conclusion. To verify the isotopic distribution of substrate 1 (which should be nearly identical to that of the product 2 formed in H2O), the commercial compound was subjected to MS analysis and shown to conform to the predicted distribution (m:m+1:m+2 = 1:0.3:0.05; Figures 5B,C left), which arises primarily from the 1.1% natural abundance of 13C. A similar mass distribution was observed for 2 produced by either the wild-type or Y68F enzyme in H2O buffer (m:m+1:m+2 = 1:0.3:0.06; Figures 5B,C middle). By contrast, the mass spectrum observed for 2 produced by wild-type FtmOx1 in D2O buffer is dominated by the second lightest isotopologue (m/z = 495 after dehydration; m:m+1:m+2 = 1:6.9:2 or ~ 70%; Figures 5B,C middle). This difference demonstrates that the H• donated to C26• is exchangeable with solvent, as would be expected for the phenolic hydrogen of a solvent accessible tyrosine such as Tyr68. The miniscule quantity of 2 produced by the Y68F variant in D2O buffer was also found to have more than the natural abundance of its m+1 isotopologue (Figures 5B,C middle), but, in this case, the heavier species was quantitatively much less prevalent (1:0.75:0.2, or ~ 38%) than in the corresponding reaction of the wild-type protein. The diminished deuterium incorporation in the reaction of the Y68F variant could originate from a less solvent-exposed hydrogen donor that incompletely exchanges with the solvent, a larger deuterium kinetic isotope effect (2H-KIE) that selects to a greater extent for residual protium in the buffer (< 4% 1H), or a combination of these effects. Regardless of the explanation, the differential deuterium incorporation implies that the Y68F variant utilizes a different H• donor to generate 2 than does wild-type FtmOx1, again consistent with the assigned role of Tyr68.
Mass of the major product (5) from the Y68F variant (Figures 2E,F).
Unexpectedly, we also discovered that the alternative product, 5, generated by either wild-type FtmOx1 or its Y68F variant in D2O buffer conforms to the natural isotopic abundance (Figure 5B,C right), indicating either that a deuterium is not incorporated in forming 5 or that any incorporated deuterium remains solvent exchangeable (Figure 2E). In other words, no new C–H/D bond is formed in production of 5. In assays with ascorbate, the 12,13-dihydroxy moiety is protected from further oxidation by Tyr68• (Figures S7 and S8), and its presence renders 5 also highly susceptible to dehydration during ionization, as previously found for both 1 and 2. Product 5 exhibits a Δ(m/z) of +48 relative to 1 and +16 relative to 2, suggesting that it might harbor both the endoperoxide and an additional oxygen. One pathway by which such a product could form is by rebound from the Fe(III)-OH form of the cofactor to the longer-lived C26•, a mechanism that would be directly analogous to that recently proposed for NvfI.8 However, the much greater separation between C26 and the cofactor in the recent FtmOx1•Fe(II)•2OG•1 structure (7.1 Å) (Figure S13) than between C3’ of asnovolin A and the Fe(II) cofactor in the Nvfl structure (4.2 Å) (Figure S11C) provides argument against this possibility and suggests an alternative pathway. Instead, a COX-like capture of a second O2 equivalent by C26• followed by reduction of the C26 peroxyl radical (C26–O-O•) to the alcohol seems more probable, and it would also provide a rationale for the observation that the presence of the reductant ascorbate substantially enhances formation of 5 in the reaction of the Y68F variant.13
Incorporation of three 18O atoms from 18O2 into 5 (Figure 2F).
To test this mechanistic hypothesis, we carried out enzymatic assays with 18O2 substrate (Figures 2F and 6). To ensure minimal 16O2 contamination during the assay, we also evaluated the isotopic composition of the succinate co-product, into which one O-atom from O2 is obligatorily incorporated (Figure 1C).29 The consistent ~99% 18O incorporation efficiency for succinate (Figure S14) across all samples confirmed proper exclusion of atmosphere from the reactions. Product 2 generated by either wild-type FtmOx1 or the Y68F variant incorporated two 18O atoms (81 ± 1% and 69 ± 5%, respectively; Figures 6B,C middle), as anticipated from the endoperoxide bridge.6,30 A possible explanation for the modestly diminished incorporation efficiency is the unavoidable introduction of ambient 16O2 during product extraction (see Materials and Methods in the Supporting Information), during which some product bearing endoperoxide with natural isotopic composition could have formed. By contrast, the corresponding analysis of 5 produced by the Y68F variant (Figures 6B,C right; see also Figure S15) shows that much more of this product bears three 18O atoms (91.4 ± 0.5%) than bears two 18O atoms (~ 1%) (m:m+2:m+4:m+6:m+8 = 0.08:0009:0.007:1:0.06). These observations support the assignment of 5 as 26-hydroxyverruculogen and strongly favor a second O2-capture step over oxygen rebound as being responsible for incorporation of the third O2-derived oxygen.
Figure 6. LC-MS characterization of products derived from 1 in reactions of wild-type FtmOx1 and its Y68F variant with 18O2.
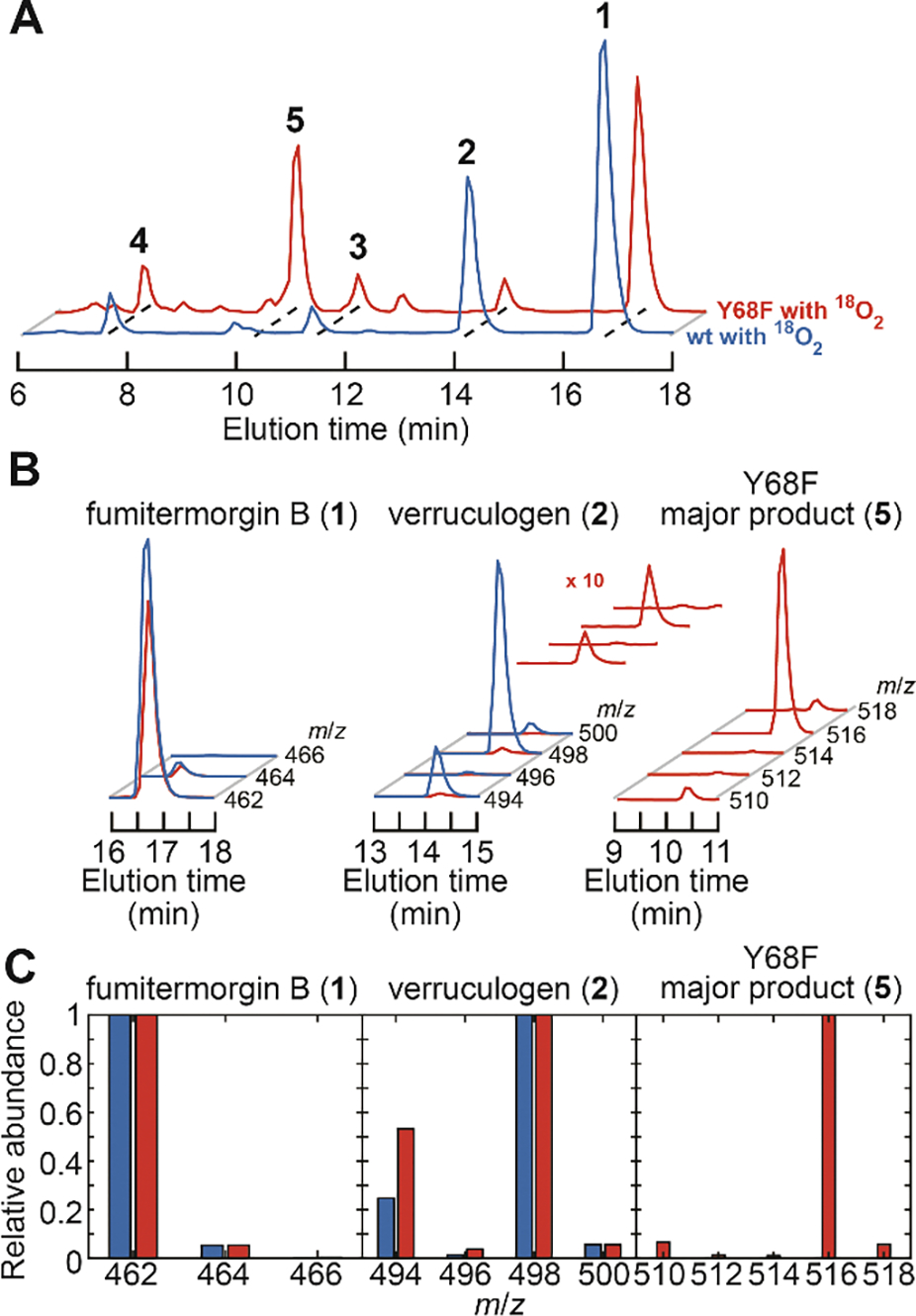
(A) TICs from the reaction of the wt (blue trace) and Y68F (red trace) enzymes. The same color code applies to panels B and C. (B) Isotope analysis from the EICs in A of 1, 2, and 5 produced in these reactions. Traces for 2 from the Y68F variant are magnified to better illustrate the isotope distributions. (C) Relative abundances for isotopologues of 1, 2, and 5 extracted from panel B. The reactions contained 510 μM enzyme, 510 μM Fe(II), 2.5 mM 2OG, 250 μM ascorbate, 0.25 mM 1, and ~ 1.0 mM 18O2, pH 8.0 at room temperature.
MS2 fragmentation analysis provides further evidence for assignment of 5.
The commercial substrate 1 and its derived products (2, 3, and 5) formed with substrates and solvent of natural isotopic abundance were all further structurally characterized by mass-spectrometric fragmentation analysis (MS2). Even though the dominant ions for 3 and 5 were found to share the same nominal mass (m/z = 510), their fragmentation patterns are distinct (Figure S16). This observation, along with their different elution times, establish that they are different compounds.13 Interestingly, even though 2 and 5 were expected to differ by only one oxygen atom, the absence of a simple correlation of their fragment ions implies quite different fragmentation pathways. To facilitate fragment assignments, we utilized the isotopically labeled 2 and 5 produced in the reactions in D2O or with 18O2 to ascertain which atoms were retained (Figures S17 and S18). The assignments are listed in Table S1 and are consistent with a previous MS2 study of 2.31 The results suggest that, while 2 retains at least one of the position 26 hydrogens (and C26 by association), 5 must have a heteroatom at C26 that promotes α-cleavage of the C3–C26 bond (numbering shown in Figure S18) and loss of C26 in the primary fragment ions. This pattern of fragmentation adds substantial weight to the assignment of 5 as 26-hydroxyverruculogen.
DISCUSSION
Multiple roles and conformational heterogeneity of Tyr68.
Through non-canonical substitutions of Tyr68 and Tyr224 in FtmOx1 and subsequent biophysical characterization (Figure 2C), we unequivocally assigned Tyr68 rather than Tyr224 as the radical-harboring site and the critical H• donor (Figure 1D). The recently published structure of the FtmOx1•Fe(II)•2OG•1 quaternary complex reveals sufficient proximity (4.6 Å) between Fe(II) and C21 to enable direct hydrogen abstraction by the ferryl moiety (Figures S11A).4 This structure implies that, associated with binding of 1, Tyr224 unexpectedly twists away from Fe(II) (Figure S1). Its location in the reactant complex disfavors its participation as initial H• donor to the ferryl complex in a COX-like mechanism. By contrast, as in CarC,14 the loop on which the actual H• donor resides (Tyr68 in FtmOx1 and Tyr165 in CarC) becomes more ordered (partially due to crystal packing, vide infra) and moves toward the active site upon substrate binding (Figure S1). The close approach (4.0 Å) of the oxygen of Tyr68 to C26 of 1 would be expected to enable the terminating HAT step of the proposed CarC-like mechanism (Figures S11A and S12). The offline arrangements of 2OG and the Fe(II)-bound water in the active site of FtmOx1 are also reminiscent of those seen in the structure of the CarC quaternary complex (Figures S11A and S11B).
Intriguingly, Tyr68 also approaches C13 of 1 to within 3.7 Å (Figures S11A and S12), consistent with initiation of the ensuing oxidation of 2 to 3 by the Tyr68• intermediate (Figures S8 and S9). Tyr68• decay likely proceeds, at least in part, through HAT from C134 to generate a ketyl radical on the pathway to 3. However, given that 2 is not quantitatively oxidized during decay of Tyr68• (Figures S8 and S9), other decay pathways must exist. Further inspection of the FtmOx1•Fe(II)•2OG•1 structure suggests that, even though the electron density implies a single Tyr68 conformation, in which the phenol stacks upon the indole ring of 1, this apparent homogeneity may be artificially enforced by constraints on the conformation of the Tyr68-harboring segment by crystal contacts with symmetry-related monomers (Figure S22). By contrast, in the structure of the FtmOx1•Fe(II)•2OG ternary complex, the surface loop harboring Tyr68 is less well ordered (Figure S21). Indeed, simulations of the dynamics of the quaternary complex suggest conformational flexibility of Tyr68•.4 In fact, if Tyr68• were to assume a single conformation in the apparently optimal position for HAT to C13, as the crystal structure suggests (Figures S11A, S12, S22A, and S22B), further oxidation of 2 by Tyr68• might be faster than the observed decay of the radical (0.4 s−1; Table S2), and other decay pathways, such as the implied reduction by ascorbate, might not compete. One might even expect HAT from C13 of 1 to Tyr68• to be sufficiently rapid to prevent accumulation of Tyr68• during a single turnover. In this regard, as a solvent-exposed redox-active residue, Tyr68 brings the remaining oxidizing equivalent to the protein surface after product formation, where the resulting radical can readily be reductively quenched.32 This situation is reminiscent of cofactor assembly in the radical generating subunit (β) of the class Ia ribonucleotide reductase, in which a conserved near-surface tryptophan (Trp48 in the E. coli homolog) plays an analogous role.24
The proposed conformational heterogeneity of Tyr68• is consistent with its broad and poorly resolved EPR signature (Figures 4 and S10), which does not coincide with the spectrum predicted for a Tyr• with any single dihedral angle33 but rather resembles the spectra of free tyrosyl radicals in frozen glasses, in which heterogeneity is expected.21 This situation contrasts with that in CarC, in which the unusually broad EPR lineshape of Tyr165• observed at 10 K was attributed to efficient relaxation by dipolar coupling to the paramagnetic Fe(III) center,14 an assignment confirmed by the observation of the underlying hyperfine structure [which matched that predicted according to the dihedral angle of Tyr165 modeled in the crystal structure (Figure S11B)] when the EPR spectrum was measured at 80 K. Indeed, acquisition of spectra of Tyr68• in FtmOx1 at higher temperatures led only to general broadening; it did not reveal the defined hyperfine couplings to the C3 proton(s) expected of a single well-defined side-chain conformation (Figure S23). This disparate behavior of the two radicals supports the notion that conformational heterogeneity, rather than dipolar coupling, is the dominant cause of the unresolved hyperfine coupling in the spectrum of Tyr68• in FtmOx1. The lesser importance of dipolar broadening is consistent with the longer Fe(II)-to-phenolic-oxygen distance in FtmOx1 (10.6 Å)4 than in CarC (8.6 Å)14 seen in the crystal structures. This heterogeneity could also be responsible for the broad UV-vis absorption band of Tyr68•. It has been reported that the UV-vis absorption of a Tyr• in a cryptochrome is red-shifted (417 nm) due to π–π stacking with a nearby tryptophan,34 reminiscent of the interaction between Tyr68 and 1 seen in the recent crystal structure.4 Heterogeneity in the conformation of Tyr68• would render π–π stacking less effective in some fraction of the population and thus broaden the absorption peak. The narrowing of both the absorption and EPR features of the 3-ClY68• compared to its unsubstituted counterpart could be attributed to a diminished conformational heterogeneity due to the steric bulk of chlorine (Figures 4C and 4D).
Tyr224• as the second radical after one catalytic cycle.
For the Y224(3-ClY) variant, an absorption maximum of 404 nm was seen in the later-accumulating species (Figures S5H and S6D), suggesting that Tyr224 could indeed harbor the second radical detected in our previous study.13 This assignment could imply another pathway by which FtmOx1 manages its remaining oxidizing equivalent (the Tyr68•) in order to avoid deleterious effects.32 Alternatively, the long reaction time at which the second Tyr• develops could reflect its formation in a second O2-activation event (e.g., triggered by product 3) that proceeds through a different pathway and leads to oxidation of Tyr224 rather than Tyr68.3,35
Hydrogen donation from Tyr68 determines the fate of the substrate radical.
By interrogation of the reactions of wild-type FtmOx1 in D2O (Figure 2D) and the Y68(2,3-F2Y) variant (Figure 2C), we showed that production of 5 and 2 compete, with formation of the alternative product not involving the final HAT to C26•. The demonstrated incorporation of three O2-derived oxygen atoms into 5 is a telling clue as to the mechanism of its formation (Figure 2F). It has been demonstrated that the 18O-labeled Fe(III) hydroxo ligand formed in the C–H cleavage step can be, in some Fe/2OG enzymes, susceptible to solvent exchange.29 Given the long lifetime of the hydroxo-Fe(III) state in the FtmOx1 reaction (as long as seconds in the wild-type enzyme3), it is expected that the hydroxo group should have undergone significant solvent exchange in the case that the formation of the alternative product competes effectively with formation of 2 by HAT to the C26• intermediate. Consequently, even if the C26 radical were ultimately resolved by oxygen rebound, 5 would not be expected to harbor three O2-derived oxygen atoms in its predominant isotopologue. The more likely pathway to 5 is for C26• to capture another O2 molecule when the HAT step is slowed (red arrow in Figure 7), as in the cases of both prostaglandin G2 formation catalyzed by COX2 and non-enzymatic free radical lipid peroxidation.36 In this most probable pathway, the resulting peroxyl radical would then be reduced by ascorbate (or another source in its absence) to yield the C26 hydroxyl of 5.
Figure 7. Origins of the dominant 1-derived products (except for 3, which is shown in Figure 1D) and the fate of the substrate radical.
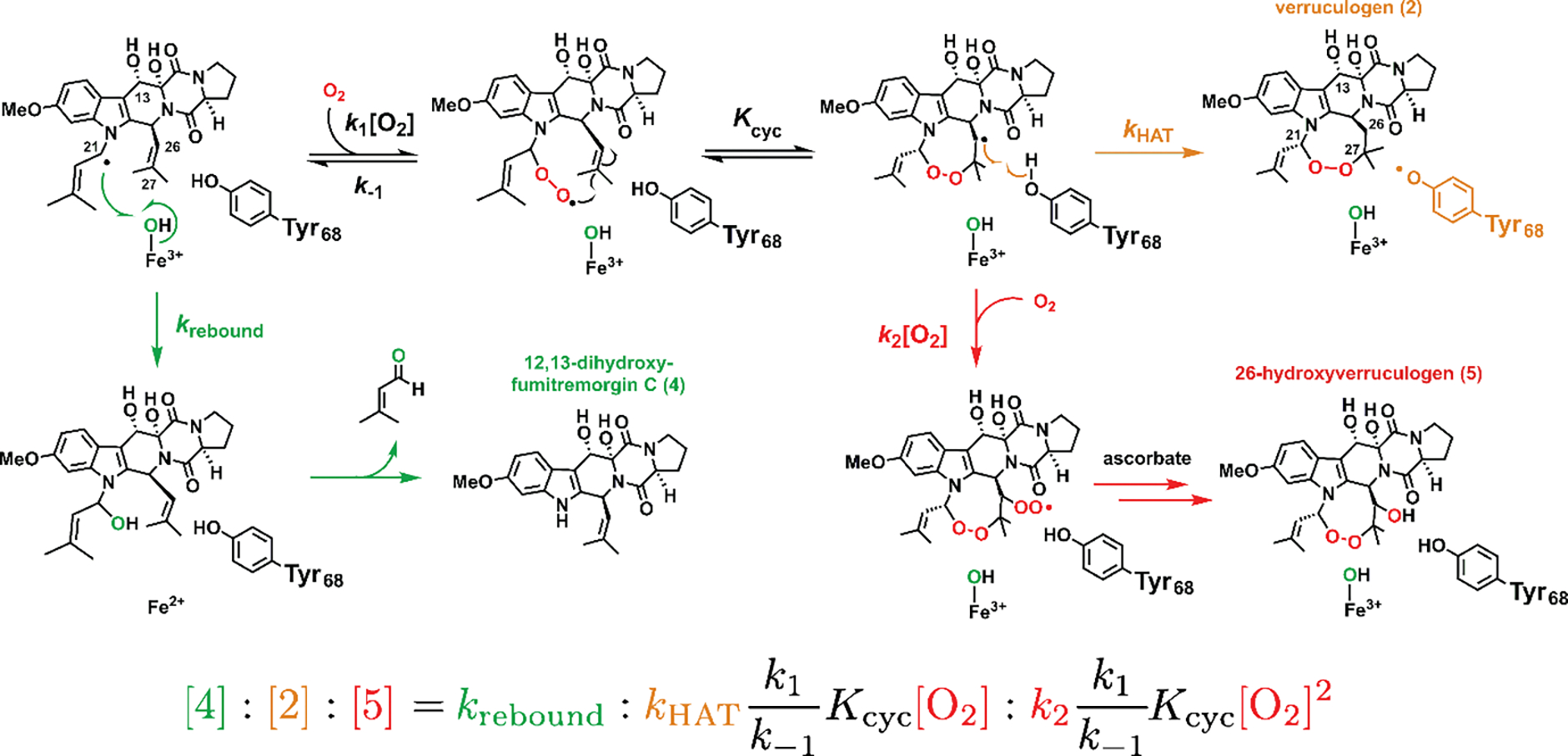
The rate constants and equilibrium constant are labeled at each critical step, and the predicted product ratio is governed by the oxygen concentration [O2], the rebound rate constant (krebound), the HAT rate constant (kHAT), and various rate constants for oxygen addition, dissociation, and cyclization. The equation assumes rapid equilibria among the radical intermediates (see text).
It is also informative that, for the reactions of both wild-type FtmOx1 and its Y68F variant (Figures 2D and 2E), the yield of 4 produced by oxygen rebound (green arrow in Figure 7) and non-enzymatic elimination (Figure 1A) is greater in D2O than in H2O (Figure 5A). This observation implies that each step between the HATs from C21 to the ferryl complex and from Tyr68 to the C26• must be readily reversible (Figures 1D and 7), highlighting the importance of an efficient HAT step to irreversibly fix the endoperoxide moiety and avert oxygen rebound. The reversibility of O2 addition to and dissociation from allylic radicals has been noted in the literature on lipid peroxidation,36 inferred from interconversion of thermodynamically and kinetically controlled products. The secondary aliphatic C26• formed upon olefinic addition of the peroxyl radical is expected to be less stable than the allylic C21•, such that even a modestly slowed HAT step (orange arrow in Figure 7) could be enough to favor the rebound product. However, the capture of another O2 equivalent by C26• (yielding 5, red arrow in Figure 7) competes with the reversion to the rebound-susceptible C21• (green arrows in Figure 7). The efficiency of C26• ↔ O2 coupling explains why a modest quantity of 5 is produced in reactions in which HAT to C26• is only subtly impeded, such as in the reaction of either wild-type FtmOx1 in D2O (Figure 5A) or the Y68(2,3-F2Y) variant in H2O (Figure 3A, see also Figure S19 for MS2 analysis of 5 from these sources). It is curious, then, that the perturbation expected to most profoundly impede HAT to C26• – namely, the Y68F substitution – overwhelmingly favors production of 5 over 4. As Tyr68 appears ideally poised in the wild-type quaternary complex4 to donate H• to C26• in the intermediate state, the preferred outcome of the Y68F variant might be attributable to an enlarged protein pocket favoring access of O2 to the C26• intermediate (Figure S13). The more structurally conservative nature of the modestly HAT-impeding perturbations (solvent deuteration and the non-canonical tyrosine) do not have this secondary impact on O2 access to the key product radical, making formation of 5 less favored.
It thus appears that the multiple substrate radical species on the FtmOx1-catalyzed pathway to 2 conform to the behavior of chemically similar species in non-enzymatic contexts.36 It is the presence of Tyr68 and its ability to efficiently donate H• to the C26• intermediate that engenders selectivity for production of 2 over formation of 4 or 5 by oxygen rebound or C26• ↔ O2 coupling, respectively. To frame this scheme quantitatively, we can assign a rate constant (or an equilibrium constant) to each step in the progression from 1 to 2 (Figure 7). For 2 to predominate in the reaction of wild-type FtmOx1, two branch points, corresponding to the radical residing on C21 and C26, must be navigated. Successful navigation requires, first, that the effective first-order rate constant for O2 addition to C21• (k1[O2]) be greater than the first-order rate constant for oxygen rebound, krebound. The second-order rate constant for O2 addition to allylic radicals, corresponding to k1, has been determined to be ~ 109 M−1 s−1 in non-enzymatic systems,36 and so one expects O2 addition to C21• to occur on the microsecond timescale in our experiments ([O2] ~ 1 mM). By comparison, oxygen rebound can be very fast (1010 – 1011 s−1) in heme-dependent hydroxylases.37,38 For wild-type FtmOx1, the rebound step competes effectively and even dominates under low [O2],13 which might seem to suggest only modest proficiency in control. However, to be able even to form 2, krebound must be significantly slowed – by a mechanism that is not yet known – relative to the aforementioned heme-dependent hydroxylases.39–41 Second, HAT to C26• has to be faster than the second O2 capture and ring opening, which are also estimated to have rate constants of ~ 106 s−1 from studies of non-enzymatic lipid peroxidation.36 The same competition between HAT (usually by α-tocopherol, another phenol-based hydrogen donor) and further O2 capture has also been interrogated in lipid peroxidation.2,36,42 It would not be surprising for kHAT in FtmOx1 to be of similar magnitude, given that formation of 4 by rebound is drastically favored in D2O. Unfortunately, direct extraction of kHAT from Figure S6 is not possible from the existing data. The fast equilibria between C21•, the peroxyl radical, and C26• are also estimated to occur on the microsecond timescale,42 which are well within the deadtime of our stopped-flow experiments, and so it is not surprising that the observed rate of Tyr68• formation is directly modulated by [O2] (Figure S8 in ref. 13) and is convolved with all the prior elementary steps before HAT (O2 addition to the cofactor, 2OG decarboxylation, etc.). Studying the product distribution (equation in Figure 7) as a function of [O2] might provide a means of estimating kHAT, or at least of extracting trends thereupon, in the reactions of variants bearing non-canonical tyrosine analogs.
CONCLUSIONS
Biophysical analysis of FtmOx1 variants with non-canonical tyrosine analogs incorporated in place of Tyr68 has established that this residue, harbored on a flexible loop that allows it to approach C26 of the substrate, donates H• to this carbon to complete installation of the endoperoxide bridge of verruculogen. Modulation of the proficiency of this HAT step by use of ncAAs or deuterated solvent has revealed that, even after averting the normally facile “oxygen rebound” to the C21 radical generated by the ferryl complex in favor of O2 capture, the enzyme must still suppress two potentially competing steps to arrive at verruculogen (Figures 1D and 7): (1) reversal of the C21• ↔ O2 and subsequent C27–O coupling steps, leading to additional opportunities for subversion of endoperoxidation by rebound, and (2) coupling of another O2 molecule rather than H• at the secondary radical site, C26•, leading to incorporation of both the C1–O–O–C27 endoperoxide and a hydroxyl group on C26. The structural architecture approximating Tyr68 optimally for HAT to C26 and appropriate phenolic O–H homolytic bond dissociation energy are thus crucial for outcome control in FtmOx1.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Noah Dunham for helpful discussions. C.-Y.L. was supported by a grant from The Jane Coffin Childs Memorial Fund for Medical Research. This work was supported by grants from the National Institutes of Health (GM113106 to J.M.B./C.K., GM127079 to C.K., and GM119707 to A.K.B.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
This information is available free of charge on the ACS Publications website. Detailed procedures, complete scheme relating proposed mechanisms for FtmOx1 and COX, structural analyses of FtmOx1 and related Fe/2OG oxygenases, stopped-flow traces and kinetic analyses, chemical-quench results, 2OG binding analysis for variants, further EPR and MS data, NMR spectra for fluorotyrosines, SDS-PAGE analysis and ESI-MS spectra for variants, and assignments of MS2 product fragments.
REFERENCES
- (1).Liu D-Z; Liu J-K Peroxy natural products. Nat. Prod. Bioprospect 2013, 3, 161–206. [Google Scholar]
- (2).Tsai A-L; Kulmacz RJ Prostaglandin H synthase: Resolved and unresolved mechanistic issues. Arch. Biochem. Biophys. 2010, 493, 103–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Yan P; Song H; Song F; Guo Y; Wu C-H; Her AS; Pu Y; Wang S; Naowarojna N; Weitz A; Hendrich MP; Costello CE; Zhang L; Liu P; Zhang YS Endoperoxide formation by an α-ketoglutarate-dependent mononuclear non-haem iron enzyme. Nature 2015, 527, 539–543. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (4).Wu L; Wang Z; Cen Y; Wang B; Zhou J Structural insight into the catalytic mechanism of endoperoxide synthase FtmOx1. Angew. Chem Int. Ed. 2022, 61, e202112063. [DOI] [PubMed] [Google Scholar]
- (5).Rouzer CA; Marnett LJ Structural and chemical biology of the interaction of cyclooxygenase with substrates and non-steroidal anti-inflammatory drugs. Chem. Rev. 2020, 120, 7592–7641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Steffan N; Grundmann A; Afiyatullov S; Ruan H; Li S-M. FtmOx1, a non-heme Fe(II) and α-ketoglutarate-dependent dioxygenase, catalyses the endoperoxide formation of verruculogen in Aspergillus fumigatus. Org. Biomol. Chem. 2009, 7, 4082–4087. [DOI] [PubMed] [Google Scholar]
- (7).Matsuda Y; Bai T; Phippen CBW; Nødvig CS; Kjærbølling I; Vesth TC; Andersen MR; Mortensen UH; Gotfredsen CH; Abe I; Larsen TO Novofumigatonin biosynthesis involves a non-heme iron-dependent endoperoxide isomerase for orthoester formation. Nat. Commun. 2018, 9, 2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mori T; Zhai R; Ushimaru R; Matsuda Y; Abe I Molecular insights into the endoperoxide formation by Fe(II)/α-KG-dependent oxygenase NvfI. Nat. Commun. 2021, 12, 4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Christov CZ; Lodola A; Karabencheva-Christova TG; Wan S; Coveney PV; Mulholland AJ Conformational effects on the pro-S hydrogen abstraction reaction in cyclooxygenase-1: An integrated QM/MM and MD study. Biophys. J. 2013, 104, L05–L07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang X; Su H; Liu Y Insights into the unprecedented epoxidation mechanism of fumitremorgin B endoperoxidase (FtmOx1) from Aspergillus fumigatus by QM/MM calculations. Phys. Chem. Chem. Phys. 2017, 19, 7668–7677. [DOI] [PubMed] [Google Scholar]
- (11).Ji J-N; Chen S-L Asymmetric abstraction of two chemically-equivalent methylene hydrogens: Significant enantioselectivity of endoperoxide presented by fumitremorgin B endoperoxidase. Phys. Chem. Chem. Phys. 2018, 20, 26500–26505. [DOI] [PubMed] [Google Scholar]
- (12).Miłaczewska A; Borowski T On the reaction mechanism of an endoperoxide ring formation by fumitremorgin B endoperoxidase. The right arrangement makes a difference. Dalton Trans. 2019, 48, 16211–16221. [DOI] [PubMed] [Google Scholar]
- (13).Dunham NP; Del Río Pantoja JM; Zhang B; Rajakovich LJ; Allen BD; Krebs C; Boal AK; Bollinger JM Jr. Hydrogen donation but not abstraction by a tyrosine (Y68) during endoperoxide installation by verruculogen synthase (FtmOx1). J. Am. Chem. Soc. 2019, 141, 9964–9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chang W.-c.; Guo Y; Wang C; Butch SE; Rosenzweig AC; Boal AK; Krebs C; Bollinger JM Jr. Mechanism of the C5 stereoinversion reaction in the biosynthesis of carbapenem antibiotics. Science 2014, 343, 1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Groves JT Key elements of the chemistry of cytochrome P-450: The oxygen rebound mechanism. J. Chem. Educ. 1985, 62, 928–931. [Google Scholar]
- (16).Young DD; Schultz PG Playing with the molecules of life. ACS Chem. Biol. 2018, 13, 854–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Minnihan EC; Young DD; Schultz PG; Stubbe J Incorporation of fluorotyrosines into ribonucleotide reductase using an evolved, polyspecific aminoacyl-tRNA synthetase. J. Am. Chem. Soc. 2011, 133, 15942–15945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Oyala PH; Ravichandran KR; Funk MA; Stucky PA; Stich TA; Drennan CL; Britt RD; Stubbe J Biophysical characterization of fluorotyrosines probes site-specifically incorporated into enzymes: E. coli ribonucleotide reductase as an example. J. Am. Chem. Soc. 2016, 138, 7951–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rappaport F; Boussac A; Force DA; Peloquin J; Brynda M; Sugiura M; Un S; Britt RD; Diner BA Probing the coupling between proton and electron transfer in photosystem II core complexes containing a 3-fluorotyrosine. J. Am. Chem. Soc. 2009, 131, 4425–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yokoyama K; Smith AA; Corzilius B; Griffin RG; Stubbe J Equilibration of tyrosyl radicals (Y356•, Y731•, Y730•) in the radical propagation pathway of the Escherichia coli class Ia ribonucleotide. J. Am. Chem. Soc. 2011, 133, 18420–18432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Seyedsayamdost MR; Reece SY; Nocera DG; Stubbe J Mono-, di-, tri-, and tetra-substituted fluorotyrosines: New probes for enzymes that use tyrosyl radicals in catalysis. J. Am. Chem. Soc. 2006, 128, 1569–1579. [DOI] [PubMed] [Google Scholar]
- (22).Yu Y; Lv X; Li J; Zhou Q; Cui C; Hosseinzadeh P; Mukherjee A; Nilges MJ; Wang J; Lu Y Defining the role of tyrosine and rational tuning of oxidase activity by genetic incorporation of unnatural tyrosine analogs. J. Am. Chem. Soc. 2015, 137, 4594–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ravichandran KR; Zong AB; Taguchi AT; Nocera DG; Stubbe J; Tommos C Formal reduction potentials of difluorotyrosine and trifluorotyrosine protein residues: Defining the thermodynamics of multistep radical transfer. J. Am. Chem. Soc. 2017, 139, 2994–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Baldwin J; Krebs C; Ley BA; Edmondson DE; Huynh BH; Bollinger JM Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 1. Evidence for a transient tryptophan radical. J. Am. Chem. Soc. 2000, 122, 12195–12206. [Google Scholar]
- (25).Jonsson M; Lind J; Eriksen TE; Merenyi G Redox and acidity properties of 4-substituted aniline radical cations in water. J. Am. Chem. Soc. 1994, 116, 1423–1427. [Google Scholar]
- (26).Adamo C; Subra R; Di Matteo A; Barone V Structure and magnetic properties of benzyl, anilino, and phenoxyl radicals by density functional computations. J. Chem. Phys. 1998, 109, 10244. [Google Scholar]
- (27).Morris JL; Reddington SC; Murphy DM; Jones DD; Platts JA; Tippmann EM Aryl azide photochemistry in defined protein environments. Org. Lett. 2013, 15, 728–731. [DOI] [PubMed] [Google Scholar]
- (28).Takeda M; Jee J; Ono AM; Terauchi T; Kainosho M Hydrogen exchange rate of tyrosine hydroxyl groups in proteins as studied by the deuterium isotope effect on Cζ chemical shifts. J. Am. Chem. Soc. 2009, 131, 18556–18562. [DOI] [PubMed] [Google Scholar]
- (29).Pan J; Wenger ES; Matthews ML; Pollock CJ; Bhardwaj M; Kim AJ; Allen BD; Grossman RB; Krebs C; Bollinger JM Jr. Evidence for modulation of oxygen rebound rate in control of outcome by iron(II)- and 2-oxoglutarate-dependent oxygenases. J. Am. Chem. Soc. 2019, 141, 15153–15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kato N; Suzuki H; Takagi H; Uramoto M; Takahashi S; Osada H Gene disruption and biochemical characterization of verruculogen synthase of Aspergillus fumigatus. ChemBioChem 2011, 12, 711–714. [DOI] [PubMed] [Google Scholar]
- (31).Fornal E; Parfieniuk E; Czeczko R; Bilinska-Wielgus N; Frac M Fast and easy liquid chromatography–mass spectrometry method for evaluation of postharvest fruit safety by determination of mycotoxins: Fumitremorgin C and verruculogen. Postharvest Biol. Tec. 2017, 131, 46–54. [Google Scholar]
- (32).Gray HB; Winkler JR Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 10920–10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Svistunenko DA; Cooper CE A new method of identifying the site of tyrosyl radicals in proteins. Biophys. J. 2004, 87, 582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Oldemeyer S; Mittag M; Kottke T Time-resolved infrared and visible spectroscopy on cryptochrome aCRY: Basis for red light reception. Biophys. J. 2019, 117, 490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ryle MJ; Liu A; Muthukumaran RB; Ho RYN; Koehntop KD; McCracken J; Que L Jr.; Hausinger RP O2− and α-ketoglutarate-dependent tyrosyl radical formation in TauD, an α-keto acid-dependent non-heme iron dioxygenase. Biochemistry, 2003, 42, 1854–1862. [DOI] [PubMed] [Google Scholar]
- (36).Yin H; Xu L; Porter NA Free radical lipid peroxidation: mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [DOI] [PubMed] [Google Scholar]
- (37).Huang X; Groves JT Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C–H activation. J. Biol. Inorg. Chem. 2017, 22, 185–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Huang X; Groves JT Oxygen activation and radical transformations in heme proteins and metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wong SD; Srnec M; Matthews ML; Liu LV; Kwak Y; Park K; Bell CB III; Alp EE; Zhao J; Yoda Y; Kitao S; Seto M; Krebs C; Bollinger JM Jr.; Solomon EI Elucidation of the Fe(IV)=O intermediate in the catalytic cycle of the halogenase SryB2. Nature 2013, 499, 320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Mehmood R; Qi HW; Steeves AH; Kulik HJ The protein’s role in substrate positioning and reactivity for biosynthetic enzyme complexes: The case of SyrB2/SyrB1. ACS Catal. 2019, 9, 4930–4943. [Google Scholar]
- (41).Chaturvedi SS; Ramanan R; Lehnert N; Schofield CJ; Karabencheva-Christova TG; Christov CZ Catalysis by the non-heme iron (II) histone demethylase PHF8 involves iron center rearrangement and conformational modulation of substrate orientation. ACS Catal. 2020, 10, 1195–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Tallman KA; Pratt DA; Porter NA Kinetic products of linoleate peroxidation: rapid β-fragmentation of nonconjugated peroxyls. J. Am. Chem. Soc. 2001, 123, 11827–1182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.