

Abstract
Implementing a specific cloud resource to analyze extensive genomic data on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) poses a challenge when resources are limited. To overcome this, we repurposed a cloud platform initially designed for use in research on cancer genomics (https://cgc.sbgenomics.com) to enable its use in research on SARS-CoV-2 to build Cloud Workflow for Viral and Variant Identification (COWID). COWID is a workflow based on the Common Workflow Language that realizes the full potential of sequencing technology for use in reliable SARS-CoV-2 identification and leverages cloud computing to achieve efficient parallelization. COWID outperformed other contemporary methods for identification by offering scalable identification and reliable variant findings with no false-positive results. COWID typically processed each sample of raw sequencing data within 5 min at a cost of only US$0.01. The COWID source code is publicly available (https://github.com/hendrick0403/COWID) and can be accessed on any computer with Internet access. COWID is designed to be user-friendly; it can be implemented without prior programming knowledge. Therefore, COWID is a time-efficient tool that can be used during a pandemic.
Keywords: COVID-19, SARS-CoV-2, Illumina sequencing, cloud workflow, cloud repurposing, parallel computation
Graphical Abstract
Graphical Abstract.
INTRODUCTION
COVID-19 is the most recent global infectious disease event, with the event occurring after the emergence of the Middle East respiratory syndrome (MERS) epidemic that occurred in 2012 [1]. COVID-19 is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2, initially named 2019-nCoV), a novel coronavirus of probable bat origin [2] that was first reported to infect humans in Wuhan, China, in late December 2019 [3]. Within 3 years of its discovery, the virus has infected more than 650 million people worldwide [4], leading to COVID-19 being declared a pandemic by the World Health Organization in March 2020 [5]. To improve the effectiveness of the response to this pandemic, a means of accurately identifying SARS-CoV-2 infection must be identified.
Real-time reverse transcription-polymerase chain reaction (RT-PCR) is a widely used molecular diagnostic technique for detecting the presence of SARS-CoV-2 in clinical laboratory settings [6]. However, because the design of the assay necessitates the use of specific probe sequences, the RT-PCR technique has several limitations, including frequent false-negative results [7] and an inability to detect mutations in viral variants [8]. Modern sequencing technology can be used to analyze the entire length of a specific genome, which enables concurrent sequencing of millions of reads of nucleic acid bases. Such technology has extremely low per-base read error rates [9]. Thus, sequencing may be an appropriate method for simultaneously and accurately identifying SARS-CoV-2 and its variants. However, sequencing generates considerable amounts of raw, FASTQ read data for each sample, and therefore, additional analysis time is required for sequencing, which may delay the identification process. This drawback becomes particularly severe during the time of a pandemic, when speed is crucial. Typical desktop computers have a limited capacity to process raw sequencing data. However, cloud technology can facilitate the performance of data-intensive tasks on computers by providing access to numerous, more powerful computational resources through a network [10]. This can enable analytical results to be generated in a timely manner. Furthermore, some of these generated results (e.g., the consensus genomes of SARS-CoV-2 in the FASTA format) can be deposited on public repository databases, such as the Global Initiative on Sharing All Influenza Data (GISAID) [11] or GenBank [12], which is an online resource hosted by the National Center for Biotechnology Information (NCBI) [13], which enables other researchers around the world to utilize these results. As of December 2022, more than 20 million records of SARS-CoV-2 genome sequences derived using various sequencing technologies are available online; most of the sequences (~70%) are available on the GISAID (https://gisaid.org) and the remaining sequences are available on the NCBI GenBank (https://www.ncbi.nlm.nih.gov/sars-cov-2/). The majority of these sequences were identified using Illumina sequencing, which is a widely used sequencing technology in SARS-CoV-2 studies [14].
Direct utilization of cloud technology in clinical settings remains challenging because many clinical staff have little experience with this technology and lack expertise in computational fields, such as programming, systems design and systems administration. Several platforms for facilitating SARS-CoV-2 detection have been introduced. These include new public cloud platforms, such as IDseq (https://idseq.net) [15] and Serratus (https://serratus.io) [16], both of which were developed to facilitate detection of pathogens, including SARS-CoV-2. Existing platform capabilities have also been expanded to improve the platforms’ abilities to facilitate detection of COVID-19. The capabilities of the National Genomics Data Center platform (https://bigd.big.ac.cn) hosted by the China National Center for Bioinformation [17] were expanded to create the 2019 Novel Coronavirus Resource (2019nCoVR, https://bigd.big.ac.cn/ncov/) [18]. In addition, the capabilities of the Ensembl (https://www.ensembl.org) platform [19] of the European Bioinformatics Institute [20] were expanded to create Ensembl COVID-19 (https://covid-19.ensembl.org) [21]. Although these cloud-based resources are highly useful, their long-term development and maintenance necessitates the use of numerous resources, including infrastructure, financing, human capital and time. Therefore, such cloud-based platforms are unsustainable for use in developing or undeveloped countries that have limited resources and have also been affected by the global pandemic.
Seven Bridges Genomics (SBG) hosts the Cancer Genomics Cloud (CGC, https://cgc.sbgenomics.com) [22], a publicly accessible cloud-based platform that offers several services [23]. For example, it incorporates hundreds of built-in bioinformatics tools and workflows as part of its Software as a Service, provides programming environments and support through its Platform as a Service, and provides virtualized computational resources (i.e., processors, memory and storage) through Amazon Web Services (AWS) or the Google Cloud Platform under its Infrastructure as a Service. The CGC is a dedicated platform specifically designed to assist with cancer research. This platform improves the ease of access to and analysis of petabytes of genomics data related to cancer, like The Cancer Genome Atlas [24] that contains numerous human samples from various cancer types including variants associated with cancer [25], through its Data as a Service. Repurposing is widely employed in biomedicine. For example, the antiviral drugs remdesivir, molnupiravir and clevudine, which were originally used to treat Ebola, Venezuelan equine encephalitis, and hepatitis B, respectively [26], have been repurposed for treating COVID-19. Similarly, repurposing the CGC, which was originally developed to aid cancer research, to facilitate COVID-19 identification may be feasible because of (1) its considerable coverage of cloud services, which can be utilized to reduce development and maintenance costs, and (2) the shared features of cancer and COVID-19 in terms of the variants.
The 2019nCoVR has been integrated with a dedicated web-based analysis platform (https://bigd.big.ac.cn/ncov/online/tools) to enable analysis of raw sequencing reads of SARS-CoV-2 by using independent modules [27]. However, the utility of this platform is limited when large samples are being considered, as is the case during a pandemic, because the design of its default analytical pipeline only allows for one sample to be processed per execution. Analytical pipelines can be described as a form of workflow system [28]. These pipelines can be made scalable through parallelization, which enables the simultaneous performance of large-scale tasks to thereby enable analysis of multiple samples during a single execution. Parallelization can be achieved through multithread processing performed in a local system or batch processing performed in a cloud system [29].
Some studies have used the CGC for SARS-CoV-2 analysis. A study [30] used the CGC for viral identification by employing workflows built into the CGC to process SARS-CoV-2 sequencing data. Another study [31] optimized the workflows used in [30] and integrated them into a single workable workflow that enabled robust viral identification. Furthermore, a study [32] integrated several of the CGC’s built-in bioinformatics tools to develop a single workflow for use in variant identification. These three studies used the Common Workflow Language (CWL) [33], a language used to define workflows and that the CGC supports, to define their workflows. [32] used batch processing, whereas [30, 31] used multithread processing to achieve parallelization. A uniform workflow that integrates the capabilities of each workflow used in previous CGC-based studies must be developed to ensure the full potential of sequencing and cloud technology can be reached.
Herein, we present Cloud Workflow for Viral and Variant Identification (COWID), which can be used to identify SARS-CoV-2 by using Illumina sequencing data. COWID is a CWL-based workflow powered by the CGC that maximizes the potential of cloud-based sequencing and parallelization to complete identification. In addition, COWID builds upon the research of the aforementioned CGC-based studies by optimizing the parallelization of multiple threads and batches to complete viral and variant identification.
MATERIALS AND METHODS
Data selection
We used two open-access datasets listed on the public repository database of the NCBI BioProject [34]. The first dataset (accession no: PRJNA784038) was generated by a SARS-CoV-2 study considering Omicron, the SARS-CoV-2 variant of concern (VOC) that emerged in 2021. Omicron has 3.3-fold higher transmissibility than the Delta VOC [35] and was first reported in southern Africa [36]. The second dataset (accession no: PRJNA316178) was generated by a non-SARS-CoV-2 study of MERS coronavirus (MERS-CoV). MERS-CoV is closely related to SARS-CoV-2 at the genus level (Betacoronavirus) but not at the subgenus level—Merbecovirus is the subgenus for MERS-CoV [37] and Sarbecovirus is the subgenus for SARS-CoV-2 [38]. MERS-CoV and SARS-CoV-2 are the same enveloped positive single-strand ribonucleic acid (RNA) type of coronavirus.
We subsequently performed retrieval and assessment on the CGC platform to process the raw sequencing data of these two datasets. In total, 78 samples (70 in the first dataset and 8 in the second dataset) were selected on the basis of their Illumina sequencing data types and their high per-base sequence read quality; an assessment conducted using the CGC’s built-in FASTQC tool, which uses the FASTQC program (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) revealed no adapter sequences. The raw sequencing data were retrieved from the NCBI Sequence Read Archive (SRA) database [39] and transferred to the CGC platform by using a built-in CGC workflow named SRA Download and Set Metadata. In SRA Download and Set Metadata, SRA fasterq-dump from the SRA Toolkit is implemented, with the SRA metadata files of selected sample identifiers in listed TXT files used as input. In total, 156 paired-end FASTQ files were used. These files contained nearly 25 million raw sequencing reads amounting to more than 15 GB of data, which were used as input data for COWID (Figure 1 and Supplementary data in the Materials sheet).
Figure 1.
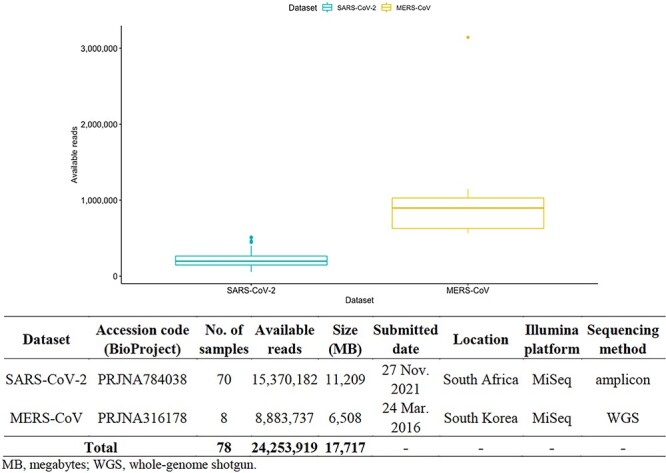
Summary of open-access datasets.
Data identification
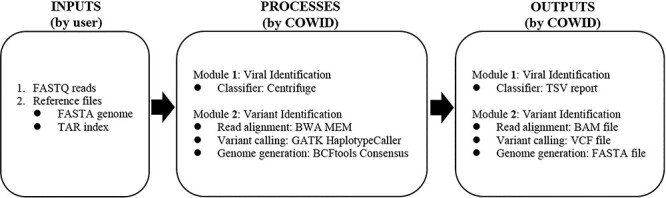
COWID was built using Rabix [40], a software environment that enables the development processes of coding, testing and debugging to be performed for CWL applications. The CWL application can be described as an independent tool or workflow. COWID is a workflow consisting of nodes, which may be inputs, tools, or outputs, and of edges. In COWID, data elements such as files or parameters flow between connected nodes. The nodes for inputs, tools and outputs, are represented by the icons  ,
,  and
and  , respectively. The edges are represented as gray lines connecting the nodes. COWID has three necessary inputs, six embedded tools and four generated outputs (Figure 2).
, respectively. The edges are represented as gray lines connecting the nodes. COWID has three necessary inputs, six embedded tools and four generated outputs (Figure 2).
Figure 2.
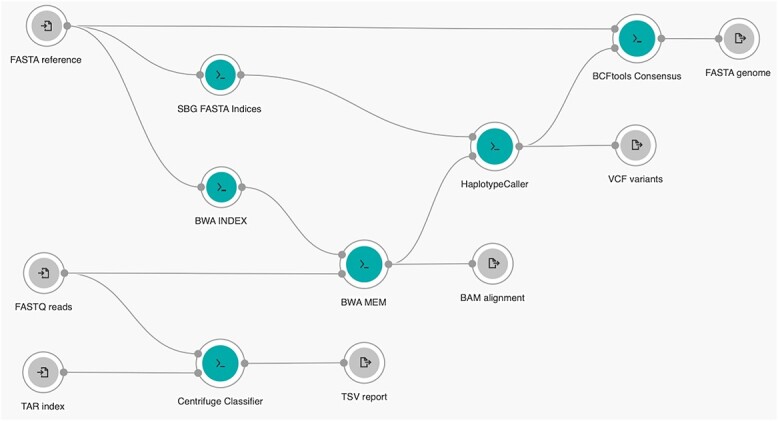
COWID graphical interface.
COWID integrates two parallel identification modules:
The viral identification module uses the Centrifuge algorithm [41] and comprises three components: Download, Build and Classifier. To improve efficiency, we included only the Classifier within COWID and retained the others within Reference Index Creation—a built-in Centrifuge workflow that is performed on the CGC before COWID is run. In doing so, we reduce redundancy in the creation of indexes for specific species, which might otherwise prolong the processing time when COWID is run in parallel. In this built-in workflow, some parameters of SARS-CoV-2 must be configured prior to execution. The taxonomic identifier (taxID) field must ‘2697049,’ which corresponds to the taxID of the SARS-CoV-2 sequences that are to be downloaded from the NCBI Taxonomy database [42]. The RefSeq category field should be set to ‘reference genome’ to filter the category of SARS-CoV-2 sequence data that are downloaded from the NCBI RefSeq database [43]. The term ‘viral’ must be entered into the domain and basename fields to specify the domain classification the SARS-CoV-2 sequence data should be obtained from and the name of the created reference index, respectively. The output reference index created using this built-in workflow is stored in the compressed TAR format (processable in 2 minutes for US$0.02) and can then be input into COWID. The Centrifuge Classifier tool (version 1.0.3) can then be used to identify SARS-CoV-2 in the input FASTQ reads. In the Centrifuge Classifier, a fast and memory-efficient full-text minute (FM) index based on space-optimized Burrows–Wheeler transform is used to assist in the classification of reads. This results in a balanced use of computational time and memory [44]. A final identification report is generated in the TSV format and contains the number of reads identified in the developed reference index.
The variant identification module incorporates aspects of the 2019nCoVR and Genome Analysis Toolkit (GATK) [45] best practice framework, which comprises three main stages. The first stage involves alignment with the Burrows–Wheeler Alignment Maximal Exact Matches (BWA MEM) tool (version 0.7.17) [46]. This tool is used to implement a data structure similar to that of the FM index used for the Centrifuge algorithm, which enables data to be rapidly aligned with reference genome data provided as another input in COWID after being processed by the BWA INDEX tool. The BWA MEM tool is bundled with the Biobambam2 sortmadup tool (version 2.0.87) [47], which is used to identify and eliminate duplicate reads. The second stage of the framework involves variant calling using the HaplotypeCaller tool (version 4.2.0.0) [48]. In this tool, the alignment output file, which is in the BAM format, is used to call a variant in a read that aligns with a reference genome that was processed using the SBG FASTA Indices tool. The third stage involves genome generation by using the BCFtools Consensus tool (version 1.9) [49]. This tool integrates the output file of the variant calling step, which is in the VCF format, with a reference genome file to generate a new consensus genome, with the resulting file in the FASTA format. We use a single nucleotide variation (SNV) or short insertion and deletion (indel) to define a variant as a mutation that caused a change in the original reference genome.
Data validation
We compared the original SARS-CoV-2 identification results available on the SRA database with the results we obtained using our viral identification system because the results were based on the same sample identifiers. The original identification results were obtained using the baseline method of the Sequence Taxonomic Analysis Tool (STAT) [50], a k-mer-based tool for classifying reads into taxonomic levels. Because the reference index was specifically built for SARS-CoV-2 identification, we only included the SARS-CoV-2 samples in the reference dataset when the STAT had been employed for SARS-CoV-2 identification. This ensured a fair comparison of the viral identification of COWID and the STAT. We visually inspected the normality distribution of the reads identified in both COWID and the STAT by using a density plot or, when only one variable was involved, the Shapiro–Wilk normality test. We employed a paired-sample significant parametric test if the data were normally distributed and a nonparametric test if the data were not normally distributed. Subsequently, a t-test was conducted for parametric data, and the Wilcoxon test was conducted for nonparametric data to determine the significance of the differences between the identification of the two methods that were identified in the normality tests [51]. The normality and significance tests were performed in R (version 4.2.0).
To validate the variant identification, we used three online resources with data on SARS-CoV-2 variants. The web-based Phylogenetic Assignment of Named Global Outbreak Lineages (Pangolin) tool (https://pangolin.cog-uk.io; version 4.2 with pangolin-data version 1.19; accessed on April 27, 2023) [52] was used to assign a lineage on the basis of the consensus genome sequence generated by COWID. Nextclade (https://clades.nextstrain.org; version 2.13.0, accessed on April 27, 2023) [53] was used to cross-validate the lineage assignment. Finally, the VOC tracker on the University of California Santa Cruz SARS-CoV-2 Genome Browser (https://genome.ucsc.edu/cgi-bin/hgTracks?db=wuhCor1; accessed on September 7, 2022) [54] was used to obtain a list of Omicron variants (B.1.1.529 lineage) from the 2022 version of the database [55], which incorporated genomics data from the GISAID. We also completed a comparison of variant identification on the 2019nCoVR platform by using the Fastq-to-Variants module and the same FASTQ input files. This comparison was conducted because the module also uses the GATK framework and a web-based interface to identify variants.
Technical settings
Application and execution settings must be applied to enable COWID to be run in parallel. For the application settings (Figure 3, top), the user must enable the batch option by selecting sample identifier metadata and defining sequencing FASTQ files as input reads, a downloaded FASTA file as a reference genome, and a prebuilt Centrifuge index TAR file as a reference index. For the execution settings (Figure 3, bottom), the user must select the spot instance setting on AWS rather than the normal on-demand instance setting to reduce execution costs. A custom compute optimized instance type of c5.2×large is recommended. This instance type which includes a configuration of 8 virtual central processing units (vCPUs) and 16 GB of random access memory (RAM) along with 32 GB of attached disk storage. This instance type typically costs US$0.34 per hour, which is considerably lower (~50% lower) than the cost of the on-demand type and has fewer interruptions (5% fewer) despite having the same configuration and service location (https://aws.amazon.com/ec2/spot/instance-advisor/).
Figure 3.

COWID setting interface for application (top) and execution (bottom).
RESULTS
Workflow performance
COWID enables batch processing, which allows for the simultaneous identification of all samples on the basis of the available instances in a single execution. When COWID was used, the cost and time required to identify one sample of paired sequencing data for the viral and variant forms of SARS-CoV-2 were typically US$0.01 and 5 minutes, respectively (Figure 4 and Supplementary data in the Computation sheet). In addition, with COWID, the analysis could be scaled up by inputting more samples when a sufficient number of instances could be accessed. In addition, unlike previous CGC-based studies (studies 1 to 3; [30–32]), which have used the default type of spot instance, in COWID, custom settings can be used to allocate computational resources. Retaining only the essential tools for viral and variant identification of previous CGC-based studies improved the performance of COWID in terms of both time and cost (Table 1).
Figure 4.

Results of COWID in a web-based interface for overall batch (top) and specific sample tasks (bottom).
Table 1.
Comparison between COWID and other CGC-associated studies
| Study | Identification | No. of samples | Computational resources | Time a | Cost b | ||
|---|---|---|---|---|---|---|---|
| vCPU c | Memory | Storage | |||||
| Study 1–Lim & Lee, 2020d | Viral | 062 | 16 | 122 | 1024 | 13 | 0.11 |
| Study 2–Lim et al., 2021d | Viral | 182 | 16 | 122 | 1024 | 10 | 0.08 |
| Study 3–Lim et al., 20221 | Variant | 055 | 08 | 015 | 1024 | 07 | 2.09e |
| COWID2 | Viral &Variant | 078 | 08 | 016 | 0032 | 05 | 1.17f |
aminutes.
bUS$.
cvirtual central processing units.
dr4.4xlarge Spot Instance (US$1.064/h); 1 c4.2xlarge Spot Instance (US$0.398/h); 2 c5.2xlarge Spot Instance (US$0.34/h).
eUS$0.04 per sample.
fUS$0.01per sample.
COWID facilitates multithread processing, enabling simultaneous viral and variant identification for each instance of each available sample. In our study, COWID typically required 5 minutes to analyze one sample, with approximately 1 minute spent initiating available computational resources and 4 minutes spent completing the main identification process (Figure 5A). During the main identification process, three tools were run in parallel (BWA INDEX for ~30 s, SBG FASTA Indices for ~60 s, and Centrifuge Classifier for ~40 s). These tools consumed more computational power. Subsequently, three additional tools were run (BWA MEM for ~20 s, HaplotypeCaller for ~100 s, and BCFtools Consensus for ~70 s). Rather than running each individual tool serially, which requires a longer amount of time, COWID runs some tools in parallel through multithread processing. Moreover, COWID enables these tools to run automatically, which further saves time. Our execution settings allocated the minimum configuration of computational resources for running COWID, ensuring suitable amounts of vCPU, RAM and disk storage were used (Figure 5B); an initial instance of c5.2×large with 8 vCPU and 16 GB of RAM was the most favorable of the tested configurations. This configuration is cost effective, and the attached storage usage was set to a minimum (32 GB) in accordance with the rule of the power of two (2n), which is a common binary system employed in the computational field.
Figure 5.

Computational performance of COWID. A, Tracking of COWID embedded tools. B, Monitoring process of COWID resources.
Viral identification
The ability of COWID to identify SARS-CoV-2 reads is dependent on the characteristics of the dataset. When a SARS-CoV-2 dataset was used, most of the reads were identified as SARS-CoV-2, resulting in a high identification rate. In contrast, few reads were identified as SARS-CoV-2 when the MERS-CoV dataset was used, resulting in a low identification rate (Figure 6A).
Figure 6.

Viral identification results obtained using COWID. A, Distribution of the identification rate. B, Distribution of identified reads obtained using both methods, with a dashed line indicating the mean. C, Visualization of the paired-sample significance test.
COWID outperformed the STAT in viral identification for every SARS-CoV-2 sample. Unlike the STAT, which considers many species in its identification, COWID focuses on only specific species of SARS-CoV-2, which enables robust read classification and a higher mean number of reads identified as SARS-CoV-2 (Figure 6B and Supplementary data in the Identification sheet). This study employed the nonparametric test of paired samples because (1) the identified reads of SARS-CoV-2 for both methods exhibited nonnormal distributions and (2) the normality test results indicated that the SARS-CoV-2 reads identified through the methods significantly differed, with P < 0.05 (P = 0.01178 for COWID and P = 0.009273 for the STAT). Furthermore, the results of the paired-sample Wilcoxon test revealed that the median number of identified reads obtained using COWID significantly differed from that obtained using the STAT (P < 0.05; Figure 6C).
Variant identification
The ability of COWID to identify SARS-CoV-2 variants is dependent on the characteristics of the dataset. Many variants were detected in the SARS-CoV-2 dataset, whereas fewer variants were detected in the MERS-CoV dataset (Figure 7A and Supplementary data in the Identification sheet). When we validated the consensus genome data for identified variants (Figure 7B), we observed that most of the samples in the SARS-CoV-2 dataset (~96%) were classified as Omicron (either B.1.1.529 or a BA sublineage); this finding is in accordance with the description of the dataset, which indicated that the dataset contained Omicron data. BA.1 was the dominant lineage in our SARS-CoV-2 dataset; this finding is consistent with that reported by Ou et al. [56], who indicated that BA.1 is a major circulating Omicron subtype. All samples in the MERS-CoV dataset were classified as the original ancestor lineage of SARS-CoV-2 (B lineage), indicating false-positive results. When we compared somewhat similar sequences (blastn) of the reference MERS-CoV genomes (GenBank accession no: NC_019843.3; 30,119 base pairs) with the SARS-CoV-2 genomes (GenBank accession no: NC_045512.2; 29,903 base pairs) by using the Nucleotide BLAST tool on the NCBI (https://blast.ncbi.nlm.nih.gov) [57], we identified 10 similar sequences, with one SARS-CoV-2 sequence (13,133–19,802 region) exhibiting a high identity alignment score (67%). All variants identified in the MERS-CoV dataset were located in the same region as that of this highly similar sequence (Figure 7C). These variants corresponded to a nonstructural protein (nsp) of RNA-dependent RNA polymerase (RdRp or nsp12) or helicase (nsp13) in open reading frame 1ab (ORF1ab), which are the two most conserved enzymes for positive-strand RNA viruses [58]. The similarity in the sequences of MERS-CoV and SARS-CoV-2 may explain why some reads identified as SARS-CoV-2 were detected in the MERS-CoV dataset in the results we obtained using our viral identification system; these reads may be located in those sequences.
Figure 7.

Variant identification results obtained using COWID. A, Distribution of identified variants. B, Summary of assigned lineages. C, Summary of identified variants with their corresponding amino acid mutation in the MERS-CoV dataset. D, Summary of identified variants with their corresponding amino acid mutation in the SARS-CoV-2 dataset.
COWID identified all 61 Omicron-related variants that are primarily located on the spike (S) protein as either SNV variants or, less frequently, as indel variants. It identified a similar proportion of variants to that identified by 2019nCoVR for the same SARS-CoV-2 dataset, indicating COWID is reliable with respect to variant identification (Figure 7D). Syed et al. [59] demonstrated that some variations in the structural proteins of SARS-CoV-2 were crucial to the high transferability of Omicron. Six variations in the receptor binding domain of the S protein (K417N, N440K, G446S, G496S, Q498R and N501Y, which correspond to G22813T, T22882G, G22898A, G23048A, A23055G and A23063T on our list) can reduce the neutralization capabilities of the antibodies induced by vaccination. In addition, two variants in the serine/arginine–rich region of the N protein (R203K and G204R, which correspond to G28881A and G28883C on our list) play a vital role in viral packaging and cell entry efficiency, which enhance viral infectivity. Moreover, a mutation in the E protein (T9I, which corresponds to C26270T on our list) and three mutations in the M protein (D3G, Q19E and A63T, which correspond to A26530G, C26577G and G26709A on our list) may reduce viral assembly and fitness. Three Omicron variants (G22813T, T22882G and G22898A) had considerably low incidence rates (7%) relative to other variants, which may be because different primers were used when they were sequenced. A version of a particular primer (ARTIC v4) was reported to be insensitive to these three variants [60]. In addition, a sample assigned to lineage B in the SARS-CoV-2 dataset was suspected to be a false positive because it consistently had the lowest rate of identification (1.83%) and of variant identification (12). Moreover, this sample did not contain any of the four signature variants of SARS-CoV-2, namely C241T, C3037T, C14408T and A23403G [61], which correspond to the 5′ untranslated region, ORF1ab polyprotein F924F (or nsp3 F106F) and P4715L (or RdRp P323L), and S protein D614G mutations.
DISCUSSION
In this study, we developed and applied COWID to analyze publicly available SARS-CoV-2 sequencing data. COWID is mainly built on the CGC, which is a publicly accessible cloud platform, and comprises two main analytical modules: viral identification and variant identification. Each module is constructed using widely used bioinformatics tools that are publicly available on the CGC, and publicly available SARS-CoV-2 reference genome is used as its input. COWID generates consensus genomes, which can be deposited in public repository databases, such as the GISAID or NCBI GenBank. We demonstrated that identification must be completed for both viruses and variants to maximize the potential of sequencing for characterization purposes. If it is not completed for both, identification results may be unreliable; moreover, variant and consensus genomes derived from variant identification results would not be able to be detected on the basis of the viral identification rate, which could increase the risk of false positives. We validated the viral and variant identification results we obtained using COWID by comparing them with those available on online resources. Finally, reducing the time required for and complexity of bioinformatics analysis through the parallelization of identification processes and multithread processing of sequencing sample batches can improve the effectiveness of using metagenomics for pathogen surveillance [62].
COWID is designed to be accessible and implementable in resource-limited settings; to implement COWID, only a computer and an Internet connection are required. Its interface can be accessed on a browser by anyone, regardless of their level of programming knowledge. Several studies have developed dedicated computational workflows for analyzing SARS-CoV-2 sequencing data in parallel through the use of different workflow management systems, including COVseq [63] in Snakemake [64], poreCov [65] and viralrecon [66] in Nextflow [67], and ViReflow [68] in Reflow (https://github.com/grailbio/reflow). These workflows have different analytical purposes: COVseq is used to generate large-scale primer sequences for SARS-CoV-2 sequencing libraries; poreCov and viralrecon have comprehensive, end-to-end viral and variant identification capabilities; and ViReflow can generate consensus genomes on a large scale. Regarding input data, only data from Illumina sequencing can be used in COVseq and ViReflow, only data from nanopore sequencing can be used in poreCov, and both forms of data can be used in viralrecon. Only ViReflow, which has a graphical interface, is user-friendly; the other workflows can only be implemented if the user has prior programming knowledge because the user must understand the code syntax when running the analysis. In addition, these workflows all involve multithread processing, which requires a high configuration of computational resources and therefore may require the use of expensive services if parallelization is implemented. Compared with these workflows and 2019nCoVR, COWID is easier to use; it has a web-based interface and efficient parallel processing capabilities (Table 2). In addition, unlike other workflows (COVseq, poreCov, viralrecon and ViReflow) that require the prior installation of all dependencies to set up the workflow environment (e.g., a workflow manager and a software container), COWID leverages the containerized software tools available in Docker [69] and the embedded CWL workflow management system predefined on the CGC, which eliminates the need for additional installation. This enables researchers to complete robust identification without being required to complete an installation process. Users can directly implement COWID by copying and pasting its open-source code (available at https://github.com/hendrick0403/COWID), which is available in the machine-readable JSON format, on the CGC platform. The simplicity of its implementation ensures that individuals with no programming expertise can effectively utilize COWID for identification without being required to manually run code-based computational tools. The user-friendly COWID interface may be a solution for ease-of-use problems that were reported for CWL when it was implemented on other platforms [70]. In addition, COWID is suitable for use during a pandemic, when scientists are likely to be working remotely [71], because COWID enables analyses to be run online.
Table 2.
Comparison of available computational workflows of SARS-CoV-2
| Category | Feature | 2019nCoVR | COVseq | poreCov | viralrecon | ViReflow | COWID |
|---|---|---|---|---|---|---|---|
| Design | Interface | web-based | CLI | CLI | CLI | GUI | web-based |
| Workflow systems | No | Snakemake | Nextflow | Nextflow | Reflow | CWL | |
| Cloud-based | No | Optional | Optional | Optional | Yes (AWS) | Yes (CGC) | |
| Parallelization | No | Yesa | Yesa | Yesa | Yesa | Yesb,c | |
| Sequencing platform | Illumina | Illumina | Nanopore | Illumina, Nanopore | Illumina | Illumina | |
| Function | Viral identification | No | No | Yes | Yes | No | Yes |
| Variant identification | Yes | No | Yes | Yes | No | Yes | |
| Genome generation | Yesc | Yesd | Yesd | Yesc,d | Yesc,d | Yesd |
CLI, command line interface; GUI, graphical user interface; CWL, Common Workflow Language; AWS, Amazon Web Services; CGC, Cancer Genomics Cloud.
amultithread.
bbatch.
cassembly.
dalignment.
COWID provides a uniform yet standardized bioinformatics workflow that integrates several well-established bioinformatics tools to minimize analytical bias when identification is being completed for multiple samples. COWID can reduce human error that may occur during large-scale identification, such as that that may be conducted during a pandemic. COWID generates intermediate files of alignment reads in the BAM format and a list of variants in the VCF format, both of which can be used for advanced downstream analysis. COWID is powered by the CGC platform, which supports the programming languages R and Python. These languages are widely used by data scientists and life scientists because they can be used to implement many packages that are used for biological research. In addition, COWID can be used to analyze user-provided sequencing data. However, to ensure the reliability of results, users should ensure the quality of reads before running COWID. Users can browse and select files for uploading to the CGC from a local system. Furthermore, the core identification modules of COWID can be applied for identification of pathogen species other than SARS-CoV-2 if genome and index references for the species are available.
COWID was developed to adhere to the principles of Findability, Accessibility, Interoperability and Reusability [72]. The COWID code is available online (findability) and are publicly accessible (accessibility). COWID can be used to obtain consensus genome results, which are interoperable with other online resources (interoperability). Moreover, users can use their own data rather than solely open-access data (reusability). Because COWID was built using a code specific to the CGC platform to enable the CGC’s bioinformatics tools and computational resources to be accessed, it might not function on other cloud platforms. Therefore, users are required to register online to gain access to the CGC platform prior to running COWID. Nevertheless, COWID can serve as a model for achieving scalable identification of SARS-CoV-2 on other cloud platforms because the tools used to construct the COWID workflow are open-sourced. Moreover, users can employ other tools when their source codes are available. Because our workflow operates on a cloud system, Internet of a sufficient speed is necessary for its use, especially during the time-consuming process of uploading sequencing data from a local system. Additionally, the simultaneous batch processing of COWID has a default limit of 80 instances, necessitating a request to the SBG team when a user wishes to run the workflow in parallel in more instances. The current COWID version accepts only input files from the Illumina sequencing platform, which uses sequencing-by-synthesis technology to generate short read data types [73]. We expect to enable COWID to accept long read sequencing data from Nanopore in the future to enable comprehensive identification using any sequencing data type. In addition, because our workflow focuses on only identification, earlier preprocessing steps must be performed using other tools or workflows. The integration of preprocessing tools or workflows with COWID may enhance its end-to-end viral and variant identification capabilities.
CONCLUSION
In this study, we present a proof of concept for repurposing the CGC from its original purpose of use for cancer research to the purpose of use for COVID-19 research through our customizable workflow, COWID. Using the existing cloud platform to build COWID was an effective solution to resource-setting limitations because building a cloud resource for SARS-CoV-2 identification from scratch would have required considerable effort. COWID leverages the CGC’s capabilities in that it is capable of parallel processing of instances through multithread processing, which enables it to process multiple sample batches and simultaneously perform viral and variant identification. Moreover, running COWID with a custom spot instance type ensures both affordability and low execution time. We demonstrated that SARS-CoV-2 identification must be performed for both viruses and variants to reduce the risk of false-positive results and enhance variant identification. Our simple tool with a low computational cost can be applied by people with or without prior programming knowledge in their normal workspace, even when the workspace has limited resources, to perform viral and variant identification of SARS-CoV-2.
Key Points
We present COWID, a novel genomics workflow based on the Common Workflow Language (CWL), that addresses the CWL’s ease-of-use problem that occurs when it is implemented on other platforms because of its dependence on command lines. COWID’s web-based interface is suitable for individuals with or without prior programming knowledge.
COWID was developed on the basis of the cloud repurposing concept. It is suitable for countries with limited resources. Repurposing a cloud-based database required fewer resources than constructing a cloud resource for SARS-CoV-2 identification from scratch would have.
COWID enables parallelization through batch multithreading, ensuring an efficient and scalable analysis and that execution times and costs remain low, for the completion of reliable viral and variant identification of SARS-CoV-2.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Seven Bridges Genomics bioinformatics team for providing the tools used in our workflow and for their technical support when we executed our workflow. We also thank the authors and the laboratories that generated the SARS-CoV-2 and MERS-CoV datasets on the NCBI BioProject and SARS-CoV-2 reference genome data on the NCBI GenBank and those who contributed to the GISAID, which served as a source in our cross-validation of our variant identification results. We also thank Daniel Pickren Chamberlin, Wallace Academic Editing, and the Office of Research and Development at Taipei Medical University for their assistance with editing our English manuscript.
Author Biographies
Hendrick Gao-Min Lim, PhD, graduated in Yuan-Chii Gladys Lee’s group at the Graduate Institute of Biomedical Informatics, Taipei Medical University. He is currently working as a research staff member at Tzu Chi Hospital in Pantai Indah Kapuk, Indonesia. His research interests include biological data analysis, cloud computing and bioinformatics tools development.
Yang C. Fann is a director of the Intramural IT and Bioinformatics Program at the National Institute of Neurological Disorders and Stroke. His research interests are computational biology, clinical research informatics and application of IT to advance translational biomedical research.
Yuan-Chii Gladys Lee is an associate professor at the Graduate Institute of Biomedical Informatics, Taipei Medical University. Her research focuses on bioinformatics, cancer genome biology and next generation sequencing data analysis.
Contributor Information
Hendrick Gao-Min Lim, Graduate Institute of Biomedical Informatics, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan 11031; Department of Medical Research, Tzu Chi Hospital Indonesia, Pantai Indah Kapuk, Greater Jakarta, Indonesia 14470.
Yang C Fann, IT and Bioinformatics Program, Division of Intramural, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland, USA 20892.
Yuan-Chii Gladys Lee, Graduate Institute of Biomedical Informatics, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan 11031.
FUNDING
This work was supported by the National Science and Technology Council of the Taiwanese government (MOST 111-2221-E-038-027 to Y-C.G.L.) and the Seven Bridges Cancer Research Data Commons Cloud Resource, which is funded in whole or in part by federal funds from the National Cancer Institute, National Institutes of Health (HHSN261201400008C), and ID/IQ agreement no. 17X146 (HHSN261201500003I and 75N91019D00024). Part of the funding was provided by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health of Bethesda, Maryland, USA (ZIC NS009443-01 to Y.C.F.).
DATA AVAILABILITY
The COWID source code is available online at the GitHub repository (https://github.com/hendrick0403/COWID). A simple walkthrough of using COWID on the CGC platform has been attached along with the index reference we created prior to running the code. COWID can be implemented on the Cancer Genomics Cloud platform of Seven Bridges Genomics (https://cgc.sbgenomics.com). The open-access data used in this study were downloaded from the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject/) under accession no. PRJNA784038 for the SARS-CoV-2 dataset and PRJNA316178 for the MERS-CoV dataset. The reference genome data for SARS-CoV-2 can be downloaded from NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under accession no. NC_045512.2.
AUTHORS’ CONTRIBUTIONS
H.G.-M.L. prepared the manuscript, including the figures and supplementary data; developed and executed the workflow; and conducted main analysis. Y.C.F. contributed to the discussion of the study design, acquired sequencing data, and assisted data analysis and interpretation. Y-C.G.L. validated the biological results, provided computational resources, and supervised the study. All authors have read and agreed to the final version of the manuscript.
REFERENCES
- 1. Zaki AM, van Boheemen S, Bestebroer TM, et al. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012;367:1814–20. [DOI] [PubMed] [Google Scholar]
- 2. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 2020;382:727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis 2020;20:533–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mahase E. Covid-19: WHO declares pandemic because of "alarming levels" of spread, severity, and inaction. BMJ 2020;368:m1036. [DOI] [PubMed] [Google Scholar]
- 6. Corman VM, Landt O, Kaiser M, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 2020;25:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xiao AT, Tong YX, Zhang S. False negative of RT-PCR and prolonged nucleic acid conversion in COVID-19: rather than recurrence. J Med Virol 2020;92:1755–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ascoli CA. Could mutations of SARS-CoV-2 suppress diagnostic detection? Nat Biotechnol 2021;39:274–5. [DOI] [PubMed] [Google Scholar]
- 9. Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol 2008;26:1135–45. [DOI] [PubMed] [Google Scholar]
- 10. Langmead B, Nellore A. Cloud computing for genomic data analysis and collaboration. Nat Rev Genet 2018;19:208–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data - from vision to reality. Euro Surveill 2017;22:30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sayers EW, Cavanaugh M, Clark K, et al. GenBank. Nucleic Acids Res 2022;50:D161–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sayers EW, Bolton EE, Brister JR, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res 2022;50:D20–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Knyazev S, Chhugani K, Sarwal V, et al. Unlocking capacities of genomics for the COVID-19 response and future pandemics. Nat Methods 2022;19:374–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalantar KL, Carvalho T, de Bourcy CFA, et al. IDseq-an open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. Gigascience 2020;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Edgar RC, Taylor J, Lin V, et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 2022;602:142–7. [DOI] [PubMed] [Google Scholar]
- 17. Members C-N, Partners . Database resources of the National Genomics Data Center, China National Center for bioinformation in 2022. Nucleic Acids Res 2022;50:D27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song S, Ma L, Zou D, et al. The global landscape of SARS-CoV-2 genomes, variants, and haplotypes in 2019nCoVR. Genomics Proteomics Bioinformatics 2020;18:749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cunningham F, Allen JE, Allen J, et al. Ensembl 2022. Nucleic Acids Res 2022;50:D988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cantelli G, Bateman A, Brooksbank C, et al. The European bioinformatics institute (EMBL-EBI) in 2021. Nucleic Acids Res 2022;50:D11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Silva NH, Bhai J, Chakiachvili M, et al. The Ensembl COVID-19 resource: ongoing integration of public SARS-CoV-2 data. Nucleic Acids Res 2022;50:D765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lau JW, Lehnert E, Sethi A, et al. The cancer genomics cloud: collaborative, reproducible, and democratized-a new paradigm in large-scale computational research. Cancer Res 2017;77:e3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Navale V, Bourne PE. Cloud computing applications for biomedical science: a perspective. PLoS Comput Biol 2018;14:e1006144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang KL, Huang KL, Mashl RJ, et al. Pathogenic germline variants in 10,389 adult cancers. Cell 2018;173:355–370.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cully M. A tale of two antiviral targets - and the COVID-19 drugs that bind them. Nat Rev Drug Discov 2022;21:3–5. [DOI] [PubMed] [Google Scholar]
- 27. Gong Z, Zhu JW, Li CP, et al. An online coronavirus analysis platform from the National Genomics Data Center. Zool Res 2020;41:705–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perkel JM. Workflow systems turn raw data into scientific knowledge. Nature 2019;573:149–50. [DOI] [PubMed] [Google Scholar]
- 29. Strozzi F, Janssen R, Wurmus R, et al. Scalable workflows and reproducible data analysis for genomics. Methods Mol Biol 2019;1910:723–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lim HG, Lee YG. Empowering cloud technology for SARS-CoV2 identification. F1000Research 2020;9:858 (poster). [Google Scholar]
- 31. Lim HG, Hsiao SH, Lee YG. Orchestrating an optimized next-generation sequencing-based cloud workflow for robust viral identification during pandemics. Biology (Basel) 2021;10:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lim HG, Hsiao SH, Fann YC, Lee YCG. Robust mutation profiling of SARS-CoV-2 variants from multiple raw Illumina sequencing data with cloud workflow. Genes (Basel) 2022;13:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Amstutz P, Crusoe MR, Tijanić N, et al. Common Workflow Language, v1.0. Figshare 2016. [Google Scholar]
- 34. Barrett T, Clark K, Gevorgyan R, et al. BioProject and BioSample databases at NCBI: facilitating capture and organization of metadata. Nucleic Acids Res 2012;40:D57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Balint G, Voros-Horvath B, Szechenyi A. Omicron: increased transmissibility and decreased pathogenicity. Signal Transduct Target Ther 2022;7:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Viana R, Moyo S, Amoako DG, et al. Rapid epidemic expansion of the SARS-CoV-2 omicron variant in southern Africa. Nature 2022;603:679–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Boheemen S, de Graaf M, Lauber C, et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio 2012;3:10–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020;395:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Katz K, Shutov O, Lapoint R, et al. The sequence read archive: a decade more of explosive growth. Nucleic Acids Res 2022;50:D387–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kaushik G, Ivkovic S, Simonovic J, et al. Rabix: an open-source workflow executor supporting Recomputability and interoperability of workflow descriptions. Pac Symp Biocomput 2017;22:154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim D, Song L, Breitwieser FP, et al. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res 2016;26:1721–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schoch CL, Ciufo S, Domrachev M, et al. NCBI taxonomy: a comprehensive update on curation, resources and tools. Database (Oxford) 2020;2020:baaa062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O'Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 2016;44:D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ye SH, Siddle KJ, Park DJ, et al. Benchmarking metagenomics tools for taxonomic classification. Cell 2019;178:779–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint 2013:1303.3997.
- 47. Tischler G, Leonard S. Biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol Med 2014;9:13. [Google Scholar]
- 48. Poplin R, Ruano-Rubio V, MA DP, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018:201178.
- 49. Danecek P, Bonfield JK, Liddle J, et al. Twelve years of SAMtools and BCFtools. Gigascience 2021;10:giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Katz KS, Shutov O, Lapoint R, et al. STAT: a fast, scalable, MinHash-based k-mer tool to assess sequence read archive next-generation sequence submissions. Genome Biol 2021;22:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Winters R, Winters A, Amedee RG. Statistics: a brief overview. Ochsner J 2010;10:213–6. [PMC free article] [PubMed] [Google Scholar]
- 52. O'Toole A, Scher E, Underwood A, et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol 2021;7:veab064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aksamentov I, Roemer C, Hodcroft EB, et al. Nextclade: clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw 2021;6:3773. [Google Scholar]
- 54. Fernandes JD, Hinrichs AS, Clawson H, et al. The UCSC SARS-CoV-2 genome browser. Nat Genet 2020;52:991–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee BT, Barber GP, Benet-Pages A, et al. The UCSC genome browser database: 2022 update. Nucleic Acids Res 2022;50:D1115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ou J, Lan W, Wu X, et al. Tracking SARS-CoV-2 omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct Target Ther 2022;7:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Boratyn GM, Camacho C, Cooper PS, et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res 2013;41:W29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ziebuhr J. The coronavirus replicase: insights into a sophisticated enzyme machinery. Adv Exp Med Biol 2006;581:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Syed AM, Ciling A, Taha TY, et al. Omicron mutations enhance infectivity and reduce antibody neutralization of SARS-CoV-2 virus-like particles. Proc Natl Acad Sci U S A 2022;119:e2200592119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Illumina . Guidelines for detecting the SARS-CoV-2 Omicron variant using the Illumina COVIDSeqTM Test (RUO Version), 2022.
- 61. Yang HC, Chen CH, Wang JH, et al. Analysis of genomic distributions of SARS-CoV-2 reveals a dominant strain type with strong allelic associations. Proc Natl Acad Sci U S A 2020;117:30679–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ko KKK, Chng KR, Nagarajan N. Metagenomics-enabled microbial surveillance. Nat Microbiol 2022;7:486–96. [DOI] [PubMed] [Google Scholar]
- 63. Simonetti M, Zhang N, Harbers L, et al. COVseq is a cost-effective workflow for mass-scale SARS-CoV-2 genomic surveillance. Nat Commun 2021;12:3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Koster J, Rahmann S. Snakemake--a scalable bioinformatics workflow engine. Bioinformatics 2012;28:2520–2. [DOI] [PubMed] [Google Scholar]
- 65. Brandt C, Krautwurst S, Spott R, et al. PoreCov-an easy to use, fast, and robust workflow for SARS-CoV-2 genome reconstruction via Nanopore sequencing. Front Genet 2021;12:711437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Patel H, Varona S, Monzón S, et al. nf-core/viralrecon: nf-core/viralrecon v2.5 - Manganese Monkey. Zenodo 2022. [Google Scholar]
- 67. Di Tommaso P, Chatzou M, Floden EW, et al. Nextflow enables reproducible computational workflows. Nat Biotechnol 2017;35:316–9. [DOI] [PubMed] [Google Scholar]
- 68. Moshiri N, Fisch KM, Birmingham A, et al. The ViReflow pipeline enables user friendly large scale viral consensus genome reconstruction. Sci Rep 2022;12:5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Merkel D. Docker: lightweight Linux containers for consistent development and deployment. Linux J 2014;239:2. [Google Scholar]
- 70. Wratten L, Wilm A, Goke J. Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers. Nat Methods 2021;18:1161–8. [DOI] [PubMed] [Google Scholar]
- 71. Korbel JO, Stegle O. Effects of the COVID-19 pandemic on life scientists. Genome Biol 2020;21:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wilkinson MD, Wilkinson MD, Dumontier M, et al. The FAIR guiding principles for scientific data management and stewardship. Sci Data 2016;3:160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 2016;17:333–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The COWID source code is available online at the GitHub repository (https://github.com/hendrick0403/COWID). A simple walkthrough of using COWID on the CGC platform has been attached along with the index reference we created prior to running the code. COWID can be implemented on the Cancer Genomics Cloud platform of Seven Bridges Genomics (https://cgc.sbgenomics.com). The open-access data used in this study were downloaded from the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject/) under accession no. PRJNA784038 for the SARS-CoV-2 dataset and PRJNA316178 for the MERS-CoV dataset. The reference genome data for SARS-CoV-2 can be downloaded from NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under accession no. NC_045512.2.