

Abstract
Delivery of self-amplifying mRNA (SAM) has high potential for infectious disease vaccination due its self-adjuvating and dose-sparing properties. Yet a challenge is the susceptibility of SAM to degradation and the need for SAM to reach the cytosol fully intact to enable self-amplification. Lipid nanoparticles have been successfully deployed at incredible speed for mRNA vaccination, but aspects such as cold storage, manufacturing, efficiency of delivery, and the therapeutic window would benefit from further improvement. To investigate alternatives to lipid nanoparticles, we developed a class of >200 biodegradable end-capped lipophilic poly(beta-amino ester)s (PBAEs) that enable efficient delivery of SAM in vitro and in vivo as assessed by measuring expression of SAM encoding reporter proteins. We evaluated the ability of these polymers to deliver SAM intramuscularly in mice, and identified a polymer-based formulation that yielded up to 37-fold higher intramuscular (IM) expression of SAM compared to injected naked SAM. Using the same nanoparticle formulation to deliver a SAM encoding rabies virus glycoprotein, the vaccine elicited superior immunogenicity compared to naked SAM delivery, leading to seroconversion in mice at low RNA injection doses. These biodegradable nanomaterials may be useful in the development of next-generation RNA vaccines for infectious diseases.
Keywords: self-amplifying mRNA, vaccine, polymer, nanoparticle, gene delivery
Graphical Abstract
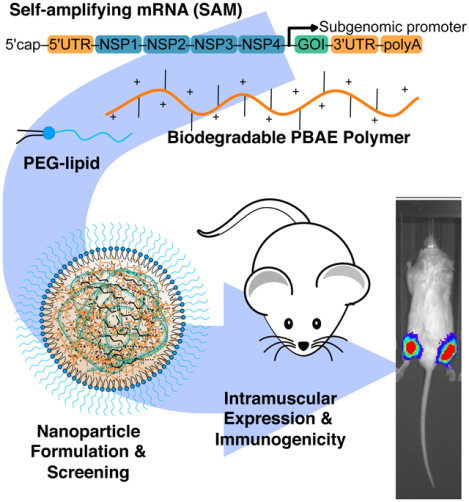
Biodegradable end-capped lipophilic poly(beta-amino ester)s (PBAEs) enable efficient delivery of self-amplifying mRNA (SAM) in vitro and in vivo. These nanomaterials yield up to 37-fold higher intramuscular (IM) expression of SAM compared to injected naked SAM. PBAE nanoparticle formulations deliver SAM encoding rabies virus glycoprotein as a vaccine and elicit seroconversion in mice at low RNA injection doses.
1. Introduction
Vaccines are one of the few strategic tools available for wide-spread utilization in combatting infectious disease epidemics and global pandemics. Yet, traditional vaccine development of live attenuated, inactivated or subunit vaccines typically requires years of development and production time for wide-spread distribution.[1] Genetic vaccines using mRNA to encode pathogenic antigens are one of the most promising advancements in vaccine development strategies, as they allow for rapid development of functional vaccine candidates as soon as the sequence of the desired protein target is known.[2] This approach has been adopted with great efficiency by Moderna and BioNTech amongst others in response to the COVID-19 pandemic with great efficacy.[3][4] Self-amplifying mRNA (SAM) technology is an innovative vaccine platform for high expression of a target antigen.[5] In addition to the antigen gene of interest, SAM also encodes four alphavirus derived non-structural proteins, which constitute the replication complex responsible for self-amplification of the original mRNA in the cytosol.[5,6] During the self-amplification process double-stranded RNA replication intermediates are formed and contribute to the induction of a robust Type I interferon response, that can be highly beneficial for eliciting a strong B cell response.[5] Due to its self-amplifying nature, SAM vaccines have the potential for high potency and dose-sparing, which could result in the production of a higher number of vaccine doses at an equivalent amount of mRNA when compared to conventional mRNA vaccines.[7][8] These features make SAM a platform optimal for vaccination and its utilization has been previously demonstrated to elicit protective immunity in different preclinical models, including mice and non-human primates against viral pathogens, including rabies[9,10], influenza[11], Ebola [11] and HIV.[12] The biggest challenge associated with SAM is to achieve effective cytosolic delivery, as mRNA is highly susceptible to nuclease damage, which would eliminate its ability to self-amplify.[10,13]
Delivery of SAM with gene delivery platforms including lipid nanoparticles[10,14], polymeric dendrimers[11], and cationic nanoemulsions[15] demonstrated varying degrees of success, but each strategy used to-date has drawbacks. Importantly, requirements for cold storage, and in some cases, the non-degradability of a mixture of synthetic components pose a challenge. Slow degradability, such as with polymeric dendrimers based on polyamidoamine modified with alkyl-epoxides, may also create issues such as inducing inflammation in vivo.[11] Certain end-capped poly(beta-amino ester) (PBAE) terpolymers have recently been developed for systemic delivery of mRNA, resulting in high efficacy of delivery to lung endothelial cells when administered intravenously in mice.[16] Compared to PBAEs lacking alkyl-amine side chain monomers, these amphiphilic PBAEs enabled highly improved mRNA complexation, protection and delivery and further allowed co-complexation with PEG-lipids to reduce rapid reticuloendothelial system (RES) clearance of cationic nanoparticles.[17] The alkyl chains of these PBAEs also enhanced structural stability of the nanoparticles by the hydrophobic effect[16] in contrast to canonical PBAE polymers which rely more on electrostatic interactions with nucleic acids to drive nanoparticle nucleation.[18] These features of amphiphilic PBAE terpolymers mimic much of what makes lipid nanoparticles highly effective for nucleic acid delivery, while also incorporating the primary benefits of PBAE polymers, including increased avidity compared to ionizable lipids, rapid ester degradation catalyzed by the tertiary amines in the backbone of the polymer[19,20], and structural end-cap monomer diversity enabling differential cell targeting.[21–23] Recent data has also highlighted that PBAE nanoparticles can be an order of magnitude more effective at endosomal escape to the cytosol compared to leading commercially available polymer and lipid-based transfection agents.[22]
Due to the self-amplifying nature of SAM in the cytosol, modified nucleosides replacement cannot be used to generate viable SAM with improved stability[24] and requires intact delivery of full-length SAM molecules to the cytosol for activity[2][5]; these features coupled with the long transcript length of SAM makes SAM especially susceptible to degradation by extracellular nucleases compared to other nucleic acid cargos. Thus, SAM vaccines or therapeutics can directly benefit by encapsulation into nanoparticles, both to increase intracellular delivery efficiency and to offer protection from degradation.
Here, we describe the development of a biodegradable cationic polyester for intramuscular delivery of SAM and demonstrate its efficiency in delivering SAM both in vitro to a myoblast mouse cell line and in vivo in mice following intramuscular administration. The resulting nanoparticles formed via a bulk-mixing and dialysis process were demonstrated to be consistent in size as well as stable following lyophilization. This work builds upon research utilizing similar polymers for systemic nucleoside modified mRNA delivery, where prior generation polymers have shown effectiveness for delivery to the lungs and spleen.[16,22] When used to deliver a rabies antigen encoding SAM, the new nanoparticles enabled protective immunity by eliciting neutralizing serum antibodies at doses of only 200 nanograms of RNA per mouse. Overall, this polymeric nanoparticle platform using rapidly biodegradable cationic esters holds promise as an effective delivery vehicle for intramuscular delivery of large RNA molecules, such as SAM. The hydrophobic PBAEs used here benefit from rapid polymer backbone degradation that limits cytotoxicity, have reduced complexity in the number of components required in the nanoparticle formulation, and as self-assembly is driven by electrostatic interactions with the nucleic acid cargo, can be manufactured flexibly from either bulk batch processes or fluidic mixing.[16,25,26] Additionally, PBAEs have high avidity to nucleic acids due to the repeating amine groups throughout the polymer, enhanced endosomal escape compared to leading commercially available polymer and lipid-based transfection agents[22], and the ability to precisely tune chemical structure to facilitate cell-targeting.[21] The polymer and RNA self-assemble to small, low polydispersity nanoparticles with high mRNA encapsulation efficiency, and that can be further surface shielded in a modular fashion with a sheddable PEG-lipid in the same manner as commercially approved lipid nanoparticles.
2. Results and Discussion
2.1. Biophysical properties/characterization of PBAE nanoparticles encapsulating SAM
To assess properties of PBAE terpolymers (Fig. 1) with SAM (Fig. 1A), we synthesized a library of PBAEs from small molecule monomers with structure denoted as described (Fig. 1B) following the naming scheme for B:base monomers, S:side-chain monomers, Sc:alkyl-side chain monomers and E:end-cap monomers (Fig. 1B) and using hyphens to separate classes of monomers.[27] While PBAE terpolymers enable co-complexation with nucleic acids using exclusively aqueous buffers for mixing, co-formulation with lipids and a mixing strategy using acidified ethanol and dialysis yielded similar nanoparticle diameters (Fig. 1C) with improved RNA encapsulation as previously observed in related structures.[16,22,28] Analysis of the PEG-lipid stabilized PBAE terpolymer eGFP SAM nanoparticles by transmission electron microscopy revealed dried nanoparticles of approximately 100 nm in diameter with a spherical shape (Fig. 1D). Inclusion of the alkyl-amine side chain monomer[29] was crucial to enable co-complexation of PEG-lipid molecules for surface shielding of the nanoparticles. We selected use of DMG-PEG2k as a saturated, 14-carbon diacyl lipid for sheddable PEGylation, which has been previously demonstrated to be useful for progressive shedding of the PEG-lipids in vivo to enable cell uptake.[30,31] In our experiments using acidified ethanol and dialysis to encapsulate SAM, admixing DMG-PEG2k at a mass percentage between 0–20% resulted in smaller, less polydisperse nanoparticles, with a neutral zeta potential for DMG-PEG2k content ≥5% by mass (Fig. 1E). This approach was applicable to PBAE terpolymers containing 12-carbon alkyl-amine side chain monomers of varied structure including for the two lead nanoparticles later identified (Fig. 1F), 7–90,c12–63, 50% Sc12 and 5–3,c12–39, 30% Sc12. Following screening of a polymer library with diverse structures, both lead nanoparticle formulations had a convergence of biophysical properties when assessed at a 30:1 weight PBAE : weight SAM ratio with the addition of 10% DMG-PEG2k by mass. At these conditions both types of nanoparticles had high encapsulation efficiency (≥94%), a particle size of approximately 115 nm, and neutral zeta potential.
Figure 1. Schematic of SAM delivery via polymeric nanoparticles.
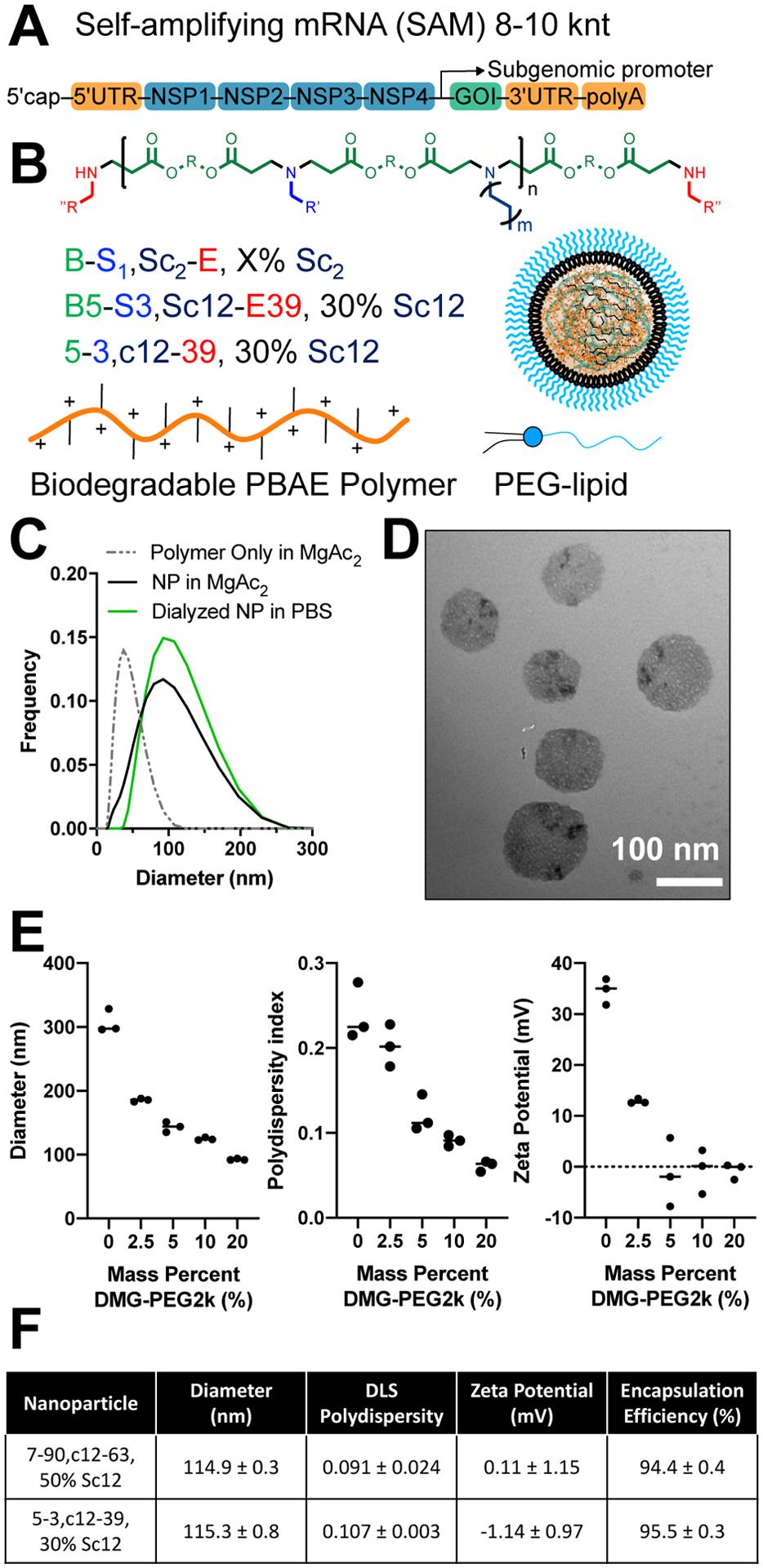
(A) SAM structure including a 5’ cap, 5’ untranslated region (UTR), non-structural protein genes 1–4 from alphavirus, GOI, 3’ UTR, and PolyA tail. (B) Generalized structure of PBAE polymer, naming scheme for 4-component polymer and cartoon of assembled nanoparticle with PEG-lipid (C) DLS measurement of polymeric nanoparticles with and without SAM. (D) TEM microscopy of SAM nanoparticles. (E) Effect of PEG-lipid inclusion on NP diameter, polydispersity and zeta potential. (F) Physiochemical properties and encapsulation efficiency of the two lead nanoparticle structures (mean ± SD of three measurements).
2.2. PBAE nanoparticle library synthesis and in vitro screening
To enable rapid screening of a diverse library of lipophilic 4-component PBAEs (Table S1) for SAM delivery, we adapted a semi-high-throughput strategy we previously employed for screening canonical 3-component PBAE polymers for plasmid DNA delivery.[32] This entailed synthesizing acrylate-terminated polymers in mid-size batches in parallel in glass vials and performing the final end-cap monomer reaction in parallel in 384-well plates.[32] For this approach, we synthesized sets of PBAEs to be acrylate-terminated (Table S2), characterized the polymers (Table S3), and then performed end-capping reactions with a set of previously identified high efficacy end-cap monomers (Fig. S1).[32] With this strategy we combinatorially synthesized a new library of 196 end-capped PBAEs with varying base monomer and side-chain hydrophobic monomer (horizontal on heatmap), and end-cap monomers (vertical on heatmap) (Fig. 2A). Each polymer was then complexed with SAM at weight:weight ratios of 30 and 60 and assessed for delivery efficiency in differentiated C2C12 murine myoblasts in 384-well plates. Differentiated C2C12 cells were utilized as an in vitro model system to better recapitulate the muscular microenvironment, given the eventual intramuscular delivery route (Fig. 2B–D).
Figure 2. Identification of nanoparticles effective for in vitro delivery of SAM to C2C12 murine myoblasts.
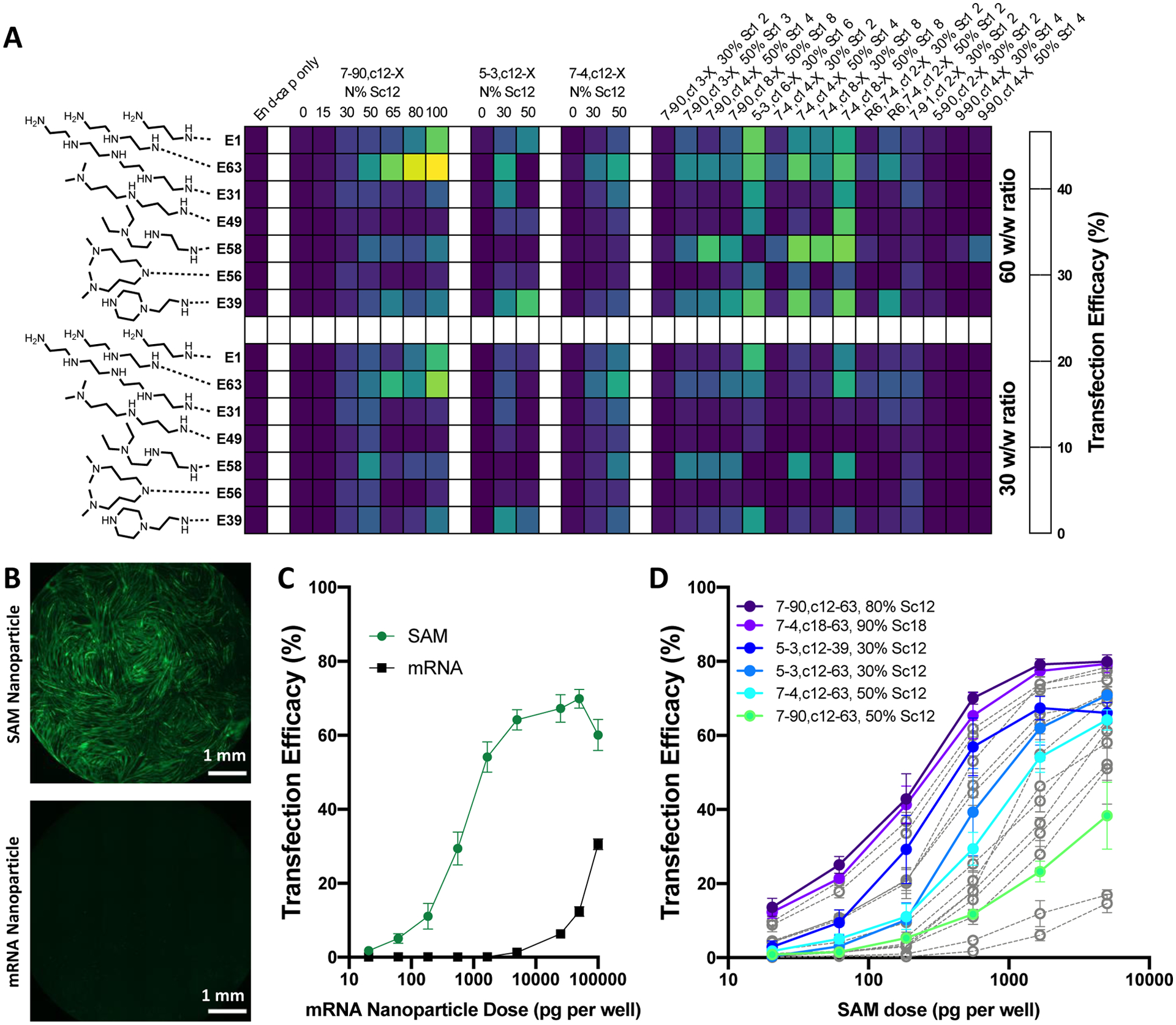
(A) A library of 196 PBAEs were synthesized combinatorially from 28 base acrylate terminated polymers and 7 end-cap monomers. Polymers were screened in 384-well plates in C2C12 cells for transfection at a dose of 1 ng of eGFP SAM per well at two w/w ratios. Each cell of the heatmap shows mean of two wells of a 384-well plate. Transfection of myoblasts using eGFP SAM was strongly improved compared to 5mou eGFP mRNA used at the same dose as visible by (B) Microscopy (25 ng per well) and (C) Quantified percent transfection and (D) Selected nanoparticles for follow-up dose-titration screening were potent down to 20.6 pg/well in a 96-well format.
The approach used for initial transfection screening of this PBAE library in C2C12 focused on differential polymer structures (without inclusion of DMG-PEG2k) and aqueous mixing of SAM and PBAE to best enable high-throughput nanoparticle self-assembly in parallel in multi-well plates and assessment of a greater number of polymer structures. In our method validation (Fig. S2), inclusion of DMG-PEG2k or use of the acidified ethanol dialysis encapsulation method did not improve transfection in vitro and similar trends in transfection efficacy were observed regardless of whether the nanoparticles were screened with or without the PEG-lipid. This is likely due to intracellular delivery and endosomal escape to the cytosol both being driven by the PBAE component, which makes up approximately 87–98% of the nanoparticle formulations evaluated by mass. With this approach to screening PBAE structures, we identified multiple PBAE structures effective for SAM delivery to differentiated C2C12 cells in vitro at extremely low doses of only 1 ng/50 μL media in each well of a 384-well plate equivalent to approximately 1 ng per 20,000 cells/well as assessed by nuclei counting. Alternative end-cap monomers previously effective for DNA delivery by PBAEs (including E6 and E7) were also included in an initial trial library of polymers but were not selected for our expanded 384-well screening library (Fig. S3). At these doses, in addition to being efficacious, the PBAE nanoparticles tested were also observed as non-cytotoxic in vitro, with negligible effects on C2C12 viability (Fig. S3).
With the structural variation included in the 384-well screening library, we identified multiple polymer characteristics that improved delivery efficacy. Specifically, increasing alkyl-amine Sc side chain mole fraction (see Fig 1B 7–90,c12-X, N% Sc12 series and 7–4,c12-X, N% Sc12 series) improved delivery efficacy in vitro even when alkyl-amine side chain mole fraction was increased up to ≥80%. Increasing alkyl-amine side chain length greater than 12 carbons was also beneficial for improving delivery efficacy in vitro. Finally, among the 12 end-cap monomers evaluated, the most effective end-caps across multiple polymers were E63, E58 and E39, which all possess three ionizable amines but differ by their hydrophobicity and ratio of their primary, secondary and tertiary amines following reaction to the acrylate terminated polymers.
To demonstrate increased efficiency of expression with SAM compared to mRNA, PBAE nanoparticles encapsulating eGFP SAM and 5mou-modified eGFP mRNA were prepared in parallel and used to transfect differentiated C2C12 myoblasts at doses across multiple orders of magnitude (Fig. 2B–C). Under these conditions, SAM yielded the same transfection efficacy at a 180-fold lower dose compared to 5-methoxyuridine-modified mRNA in vitro. Recapitulating this two-orders of magnitude level of efficiency improvement in vivo has the potential to enable dose-sparing that could dramatically reduce the supply constraints of mRNA vaccines for global vaccination, as encountered for SARS-CoV-2.
Top nanoparticle formulations identified by screening the 384-well library were evaluated with dose titration for transfection of differentiated C2C12 cells. Among these nanoparticles, lead polymers achieved >70% transfection of cells at a dose of 5 ng/well and >40% transfection at a dose of only 185 pg/well (Fig. 2D). To better understand the influence of the alkyl-amine side-chain monomer on in vitro transfection, we synthesized two series of polymers varying either the length of the alkyl-amine side-chain monomer or the mole fraction (increasing the total polymers in the library evaluated to >200) (Fig. S4). These experiments clearly demonstrated that both increased length of the alkyl-amine side-chain monomer and increased alkyl-amine side-chain mole fraction improved SAM transfection in vitro.
2.3. Intramuscular (IM) delivery of PEGylated SAM PBAE nanoparticles
To identify nanoparticles effective for intramuscular SAM delivery in vivo, we used a SAM construct encoding luciferase (FLuc) to rapidly assess overall SAM delivery efficiency when injected intramuscularly with live animal imaging. We first chose a single PBAE nanoparticle formulation and assessed the duration of expression when injected intramuscularly in BALB/c mice (Fig. S5A), observing that peak expression occurred at approximately 10 days post-injection and expression at that dose of SAM persisted through approximately 40 days post-injection. From this experiment, we selected 10 days as the time-point for live animal imaging studies for nanoparticle efficacy assessment in vivo. We further assessed optimal w/w ratio for a single PBAE nanoparticle formulation, evaluating 15, 30 and 60 w/w ratios with DMG-PEG2k included at a 10% mass fraction (Fig. 3A), selecting a 30 w/w ratio as optimal under these conditions. In contrast to in vitro assessment (Fig. S2), where inclusion of DMG-PEG2k moderately reduced transfection, PEGylation of the nanoparticle surface with DMG-PEG2k was shown to be required for efficacy following intramuscular injection of these polymeric nanoparticles (Fig. 3B), presumably by improving their extracellular transport properties. Particles not formulated with DMG-PEG2k yielded lower transfection intramuscularly than naked SAM injected under matched conditions and particles formulated with 10% mass DMG-PEG2k yielding the highest expression. This result was in some ways surprising, as cationic nanoemulsions[12,15], cationic liposomes[9] and cationic polyethyleneimine polyplex nanoparticles[33] have previously been demonstrated to be effective for intramuscular administration of nucleic acids despite their positively charged zeta potentials. Yet, the observation that cationic PBAE nanoparticles (even with alkyl-side chains for increased hydrophobicity) yielded no expression with intramuscular administration is consistent with prior observations of PBAE nanoparticles suppressing expression of plasmid DNA when injected intramuscularly[33] unlike when DNA or RNA containing PBAE nanoparticles are injected by other local routes such as intratumorally[34–36] or intraocularly[37], where there is high expression. This is most likely due to the extracellular environment of the muscle, perhaps by unPEGylated PBAE nanoparticles with a positive zeta potential sticking to the high concentration of extracellular matrix proteins in the intramuscular space and failing to reach cell plasma membranes to initiate endocytosis prior to the PBAE backbone beginning hydrolytic degradation.[38] Using polymers with different modes of degradation (like self-immolative oligo(alpha-amino ester)s) may be a possible pathway to improve efficacy without the use of PEG, as it was shown that that such charge-altering releasable transporters (CART) have demonstrated successful intramuscular mRNA delivery at injection doses of 7.5 μg mRNA.[39] In any case, mixing in 10% by mass DMG-PEG2k into the PBAE/SAM nanoparticles during formulation is an easy and modular way to functionalize them to be PEG-shielded for in vivo applications at low doses and the safety of using DMG-PEG2k has been demonstrated in humans via its inclusion in the Moderna COVID-19 vaccine.[4]
Figure 3. In vivo efficacy of intramuscular SAM delivery using luciferase SAM with expression assessed 10 days following injection.
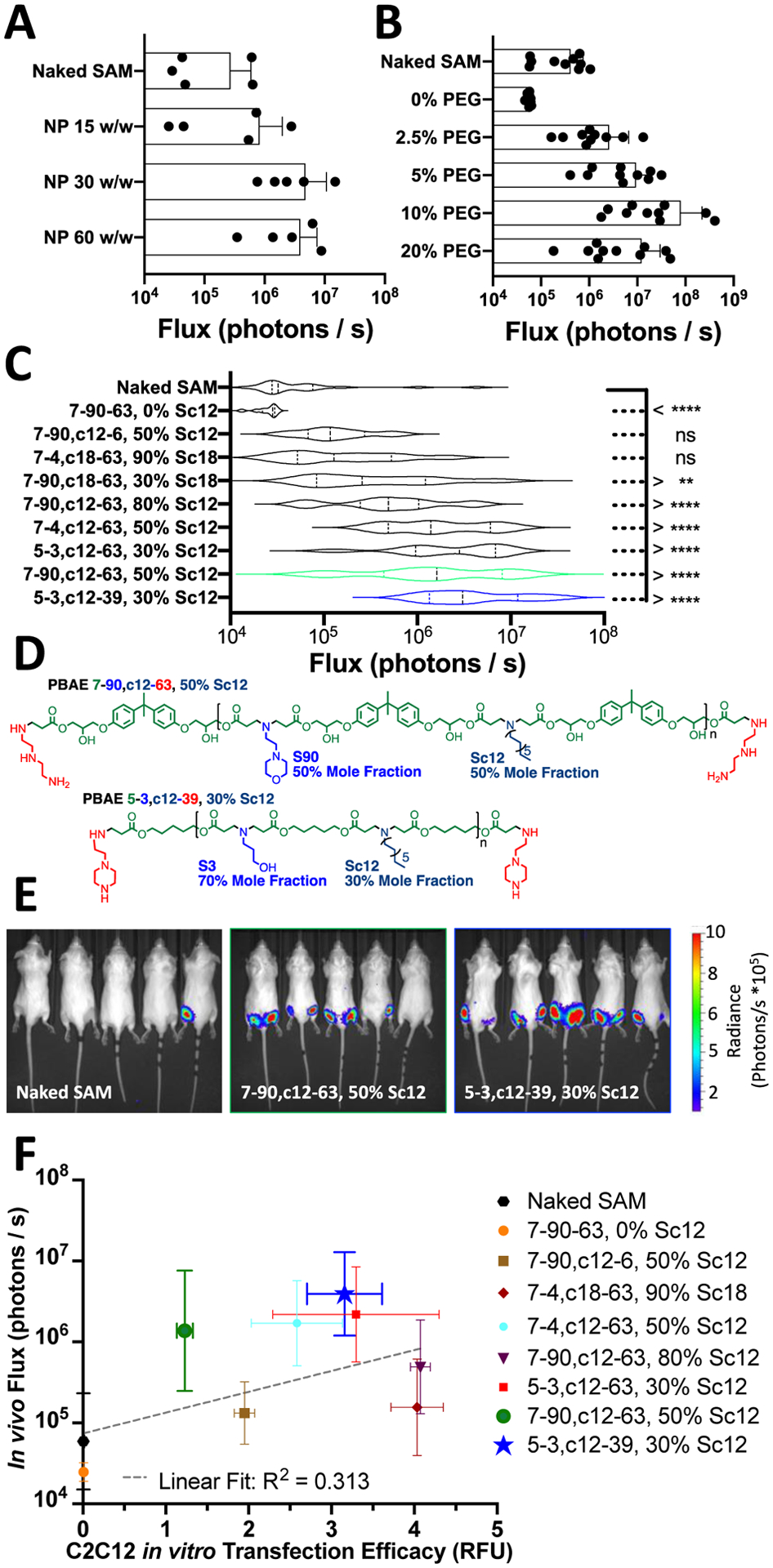
(A) Selection of polymer to SAM w/w ratio and (B) Particle mass fraction of DMG-PEG2k using nanoparticle 7–90,c12–63, 50% Sc12. (C) Violin plots of intramuscular luminescence measured at day 10 for nine PBAE NPs compared to naked SAM for a dose of 200 ng injected in 20 μL injection volume. N = 10 IM injections per polymer; assessed using one-way ANOVA corrected for multiple comparisons to naked SAM injection. (D) Structures of lead polymers for intramuscular administration. (E) Representative IVIS images with the top two nanoparticle formulations compared to naked SAM. (F) Relationship between in vitro transfection of C2C12 cells in 96-well plates at a dose of 5 ng/well and in vivo luminescence following intramuscular administration.
Intramuscular dosing of naked mRNA in mice can present a challenge in reproducibility and utility for assessing delivery efficacy, as murine quadriceps are quite small and naked mRNAs injected in buffer are capable of transfecting muscle cells primarily because of the high hydrostatic pressure achieved when injecting a relatively large volume, without intrinsic ability for the mRNA to safely reach the cytosol itself.[6,39–42] Naked nucleic acid expression following intramuscular injection in mice does not reproduce in human patients when typical intramuscular injection volumes in mice (20–50 μL) do not scale with either body surface area (228-fold higher in adult humans, equivalent of 5–11 mL) or body mass and routine intramuscular injection volumes in humans are limited to 500 μL (Fig. S5). The two primary mRNA-based vaccines brought to market for SARS-CoV-2 by BioNTech/Pfizer and Moderna use doses of 30 μg and 100 μg in injection volumes of 300 and 500 μL, respectively.[4,43] Scaling down from these doses and injection volumes to approximate a clinically relevant intramuscular injection in mice is not feasible due to the accuracy and precision constraints of injecting 5 μL intramuscularly. We evaluated using 7–90,c12–63, 50% Sc12 nanoparticles compared to naked SAM injected intramuscularly at a variety of doses between 7.5 to 0.05 μg and injection volumes between 50 μL and 5 μL (Fig. S5). Based on these results we selected a dose of 0.2 μg in 20 μL injection volume (0.01 μg/ μL) for all following experiments (Fig. S5).
Using this optimized dose and injection volume of SAM with intramuscular injections, we evaluated 9 different unique PBAE nanostructures prepared at a 30 w/w ratio with SAM and 10% by mass DMG-PEG2k (Fig. 3C). Among these polymers, 6 polymers led to significantly higher luciferase expression than naked SAM, with lead performing formulations of 7–90,c12–63, 50% Sc12 and 5–3,c12–39, 30% Sc12 yielding an increase of 23- and 37-fold higher luminescence over naked SAM (Fig. 3C,D). The structures of these lead polymers were considerably different from one another and demonstrate multiple structural possibilities for high levels of intramuscular delivery efficacy. The 7–90,c12–63 PBAE used a morpholino based ionizable side chain and bisphenol A (BPA) based diacrylate monomer, while the 5–3,c12–39 PBAE used an amino-alcohol based ionizable side chain and a pentanediol based diacrylate. In the context of vaccine development, the higher performing 5–3,c12–39 PBAE may also have a better-tolerability profile due to the avoidance of BPA.[44]
One of the primary roles associated with nanoparticle-based encapsulation of SAM and other mRNAs for intramuscular administration is to improve the potency and robustness of delivery over the injection of naked nucleic acids. For the 10 injections shown in representative IVIS images of two lead formulations and naked SAM, PBAE nanoparticle-based encapsulation yielded strongly detectable expression of luciferase 7–9 times out of 10 compared to naked SAM, where only 1/10 injections yielded a strong luminescent signal (Fig. 3E). Even at these low injection doses and volumes, the results show that the biodegradable polymeric nanoparticles can facilitate in vivo intracellular delivery in muscle. It is critical that the PBAE nanoparticles were able to induce significant SAM expression in muscle at modest doses because these small scales have the potential to be translatable to human patients in a manner that larger volume hydrodynamic injection cannot. The hydrodynamic effect by which naked nucleic acids injected intramuscularly in mice mediate effective cytosolic delivery has been demonstrated to have scaling challenges to larger animals like non-human primates.[45,46]
Many studies have assessed correlation between transfections in vitro and in vivo for screening different nanoparticle formulations, often revealing low correlation between the optimal materials identified for in vitro and in vivo based delivery. Notably, for lipid nanoparticles with delivery assessed using a barcoded, pooled library approach for delivery to macrophage cell lines in vitro and via intravenous administration in vivo, there was a linear correlation with R2<0.1.[47] In the context of intramuscular delivery, there have been strategies to improve in vitro models of muscle by using ECM mimicking substrates to influence the mechano-transduction of muscle cells and recapitulate endocytic pathways observed in vivo.[38] While the approach of creating more complex but applicable in vitro culture conditions for identifying nanoparticles effective for in vivo utilization is attractive, implementation can be difficult. For example, the low stiffness (10 kDa) hydrogel system developed by Bhosle et al. presents challenges for high throughput screening due to its limited applicability to a multiwell plate format.[38] In the current study, PBAE structures were identified by in vitro multiwell plate screening utilizing differentiated C2C12 myoblasts and further evaluated via in vivo intramuscular delivery. Expression following intramuscular administration was weakly positively correlated with in vitro transfection efficacy (R2=0.313, Fig. 3F). Differences between the in vitro and in vivo experiments, such as biodistribution, the effect of PEGylation on cellular uptake, and the nature of a 2D vs 3D environment may all contribute to lessening the strength of the correlation. None-the-less, formulations observed to have low transfection efficacy during the screening in vitro also had low transfection in vivo. In vitro screening of the PBAE system showed utility in that all the nanocarriers that achieved greater than 50% transfection of C2C12 myoblasts in vitro also had the capacity to successfully transfect in vivo following incorporation of DMG-PEG2k and IM injection.
2.4. Immunization with PBAE nanoparticle delivered SAM improves rabies antibody titers in mice
Using a SAM construct encoding rabies virus glycoprotein, we assessed the potential of PBAE nanoparticles to deliver SAM to elicit neutralizing antibodies relative to injections of naked SAM.[10,13] Using a vaccination schedule of homologous prime/boost separated by three weeks, serum was collected two weeks following boost administration (day 35) (Fig. 4A). Neutralizing antibody titers against live rabies virus were then assessed via rapid fluorescent focus inhibition technique (RFFIT) assay.[48] The RFFIT assay directly assesses the presence of rabies virus neutralizing antibodies (RVnAbs) that can neutralize the rabies virus and prevent infection of healthy cells, providing better correlation for protection than anti-rabies virus glycoprotein binding antibody (bAb) titers measured by ELISA.[48] Comparison between naked SAM and both PBAE NPs tested (7–90,c12–63, 50% Sc12 and 5–3,c12–39, 30% Sc12) RVnAbs levels using the Mann-Whitney test demonstrated statistically significant antibody titer produced after initial immunization and booster injection. An antibody titer above the 0.5 IU/mL threshold (indicated by the dotted line) is considered protective by the assay[48], demonstrating at the low mRNA doses a seroconversion rate of 6/10 animals for both PBAE NP formulations and 2/10 for naked SAM (Fig. 4B). Thus, both biodegradable polymeric nanoparticle formulations improved the seroconversion of IM injected SAM compared to hydrodynamic injection of naked SAM. Delivery of SAM IM in various nanoparticle formulations has been previously reported in the literature.[49] Researchers have found that similar levels of titers can be achieved following IM dosing of 150 ng SAM in higher volume 50 μL injections (on day 0 and day 28) using 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) or dimethyldioctadecylammonium bromide (DDA) liposomes.[50] As polymeric PBAE NPs reached a neutralizing antibody level with low volume injections (10 μL), these polymeric NPs may be able to satisfactorily deliver SAM without relying on high hydrostatic pressure injection to facilitate intracellular delivery. Future research is needed with additional doses, volumes, and controls to fully elucidate the potential advantages of PBAE NPs for SAM delivery. These findings merit further investigation as higher doses and additional tuning of intracellular delivery parameters towards antigen-presenting cells could further boost titers. It is also noteworthy that as PBAE nanoparticles are capable of being lyophilized and stored in non-frozen conditions[51], future investigation may prove that they can be beneficial from a supply chain perspective and/or in the development of alternative routes of administration, including via microneedles, which has been demonstrated using PBAEs and plasmid DNA.[52]
Figure 4. Polymeric nanoparticles delivering SAM enable immunogenic expression of antigen greater than naked SAM.
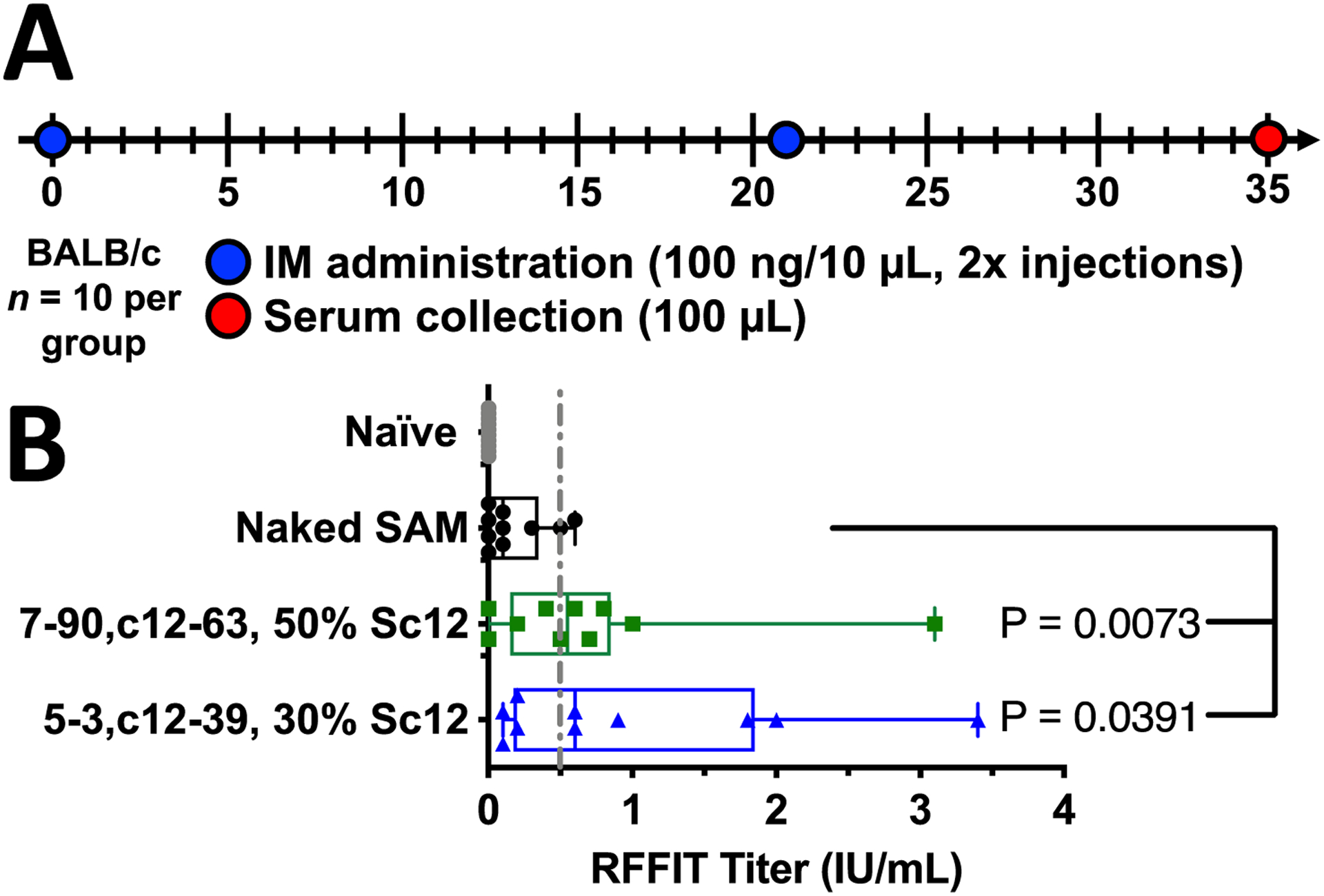
. (A) Schematic of FLuc-2A-rabies SAM dosing strategy (prime/boost). (B) Rabies Virus Neutralizing Antibody titers measured by RFFIT for top PBAE NP formulations 7–90,c12–63, 50% Sc12 and 5–3,c12–39, 30% Sc12 compared against naked SAM vaccinated and naïve serum. N=10 animals per group; Mann-Whitney test for statistical significance against naked SAM injection.
3. Conclusion
In this study, we developed biodegradable end-capped lipophilic poly(beta-amino ester) terpolymers to enable the delivery of SAM constructs both in vitro and in vivo for intramuscular vaccination. By screening a library of >200 PBAE terpolymers with varied base polymer structure, differential side chain hydrophobicity and varied end-cap monomers, we identified optimal structural properties to enable highly efficient SAM delivery to myoblasts at sub-nanogram doses per well. In particular, small changes to end-group structure and % of lipophilic side chain could make a profound difference to rates of transfection. Inclusion of alkyl side chains enabled admixing with DMG-PEG2k to yield nanoparticles with high encapsulation efficiency and neutral zeta potential for effective intramuscular administration. Among lead PBAE terpolymers further evaluated by intramuscular injection in mice, optimal PBAE formulations enabled up to 37-fold higher intramuscular expression of SAM compared to injected naked SAM constructs. In vitro screening with C2C12 myoblasts was found to be helpful in identifying polymeric nanocarriers with the capacity for successful intramuscular transfection. Delivery of SAM encoding rabies virus glycoprotein via two different PBAE nanostructures at low doses of mRNA led to protective seroconversion among most animals vaccinated and demonstrated higher humoral immunity compared to injection of naked SAM. This study reveals that biodegradable polymers, as a class of nanomaterials, can be promising delivery vehicles for next-generation mRNA-based vaccines. Although the field is dominated by lipid-based materials for non-viral mRNA delivery, biodegradable polymers have the potential benefits of a broader therapeutic window, ease in manufacturability, possibility for non-frozen supply chain, and efficiency of delivery without high hydrostatic pressure required. This motivates future work in further optimizing dosing, excipients, scale-up, and storage to better realize the potential of this class of nanomaterials.
4. Experimental Section
Materials:
Monomers were purchased from vendors listed in Table S1 and used without further purification. Acrylate monomers were stored with desiccant at 4°C, while amine monomers were stored with desiccant at room temperature. mRNA for eGFP (5-methoxyuridine, 5mou) was purchased from TriLink Biotechnologies (L-7201). All solvents were purchased from MilliporeSigma.
RNA Synthesis:
Self-amplifying mRNA (SAM) is an alphavirus derived mRNA, which comprises genes for nonstructural proteins (NSPs) and a gene of interest (GOI) whose expression was enabled via a subgenomic promoter (Fig. 1A). Three SAM constructs were prepared coding for eGFP, firefly luciferase and a dual firefly luciferase-2A-rabies antigen SAM separated by a 2A ribosomal skip site. RNAs were transcribed in vitro from template DNA constructs using T7 polymerase and purified as previously described[53], and RNA integrity was validated by agarose gel electrophoresis. Ability of the in vitro transcribed RNAs to self-amplify and express the target antigens was measured in BHK cells as previously described.[54]
Polymer Synthesis:
Poly(beta-amino ester)s (PBAEs) were synthesized as previously described and shown in Fig. S1 using a two-step Michael addition reaction with a combination of backbone, side chain, and end-cap monomers. Bioreducible monomer BR6 (2,2-disulfanediylbis(ethane-2,1-diyl) diacrylate) was synthesized according to Kozielski et al.[55] PBAE polymers were synthesized at the molar ratios of monomers specified in Table S2. The first Michael addition reaction between the backbone and side chain monomers occurred at 90°C for 48 hr with stirring producing acrylate-terminated base polymers. The second Michael addition reaction occurred in anhydrous tetrahydrofuran (THF) at room temperate for 1 hr with stirring resulting in end-capped PBAEs. These polymers were then precipitated into anhydrous diethyl ether with centrifugations at 3200 rcf and washing twice with anhydrous diethyl ether to purify the polymer. The PBAEs were then dried under vacuum for 48 hrs to eliminate residual diethyl ether, dissolved in anhydrous dimethyl sulfoxide (DMSO) at 100 mg/mL, and stored in aliquots at −20°C with desiccant to limit freeze/thaw cycles. PBAE nomenclature follows the numbering of diacrylate, hydrophilic amine, hydrophobic amine with percentage, and amine end-cap monomers shown in Fig. S1.
Polymer Characterization:
Prior to end-capping reactions, acrylate terminated polymers after the first Michael addition reaction were sampled and precipitated twice in anhydrous diethyl ether to yield a neat polymer that was then dissolved in a small amount of anhydrous DMSO-d6. The sampled acrylate-terminated polymers were dried under vacuum for 2 hrs then dissolved in additional DMSO for NMR spectrum analysis of acrylate peaks via Bruker 500 MHz NMR. Similar analysis was done with polymer samples post-end capping to confirm complete reaction by elimination of acrylate peaks between 5.5 and 6.5 ppm. Gel permeation chromatography (GPC) (Waters, Milford, MA) was also used to characterize the MN, MW and polydispersity index (PDI) relative to linear polystyrene standards for the sampled acrylate-terminated polymers as previously described.[56]
Encapsulation Efficiency Assay:
Loading efficiency of SAM loaded into the nanoparticles was analyzed using the commercial Invitrogen Ribogreen RNA analysis kit (ThermoFisher) as described previously.[28] Nanoparticles were complexed using two different PBAE formulations (7–90,c12–63, 50% Sc12 and 5–3,c12–39, 30% Sc12) to encapsulate SAM with 10m% DMG-PEG2k and underwent dialysis. Nanoparticles were then diluted to approximately 1 ng/μL SAM in PBS pH 7.4 buffer. Standards using the SAM molecules were between 0.125 and 2 ng/μL. Prepared nanoparticles were then mixed with either PBS buffer or 10 mg/mL heparin in TE buffer with the later disrupting the polymer binding allowing for the release of SAM. The nanoparticles were incubated in the buffer for 15 mins at 37°C and then diluted Ribogreen reagent was added and incubated for 3 mins at 37°C. Fluorescence of Ribogreen was measured using a plate reader (Biotek Synergy) at 500 nm / 525 nm excitation/emission according to the supplied protocol to determine encapsulation efficiency.
Nanoparticle Preparation:
Nanoparticles were prepared without incorporation of lipid-PEG for high-throughput screening of the polymers’ ability to facilitate intracellular delivery in vitro or by adding DMG-PEG2k as an extra component to neutralize surface charge followed by dialysis as previously described.[16] For transfections in 96-well plates, nanoparticles were formed by dissolving synthesized PBAE polymers in DMSO and eGFP SAM separately in 25 mM NaAc pH 5.0 buffer and combining them at a 1:1 volume ratio. The mixture was incubated at room temperature for 10 mins to allow for self-assembly into nanoparticles. For transfections in 384-well plates, nanoparticles were formed by resuspending synthesized PBAE polymers in 25 mM NaAc pH 5.0 buffer in parallel using a ViaFlo 384 (Integra Biosciences). Resuspended PBAE polymer was then mixed in parallel with SAM to yield a final nucleic acid concentration of 0.03 μg/μL in a 384 polypropylene nanoparticle source plate.
Nanoparticle Characterization:
For nanoparticle characterization via dynamic light scattering, SAM and PBAE polymer were prepared using DMG-PEG2k and dialysis.[16] The Z-average hydrodynamic diameters and zeta potential of the nanoparticle formulations in 25 mM NaAc and in six-fold dilution using isotonic 150 mM PBS pH 7.4 buffer at 25°C, respectively, were measured by dynamic light scattering (DLS) using a Zetasizer Pro (Malvern Instruments, Malvern, UK) with a 173° detection angle. For transmission electron microscopy (TEM), nanoparticles were prepared at 30 w/w ratio with 10 m% DMG-PEG2k using dialysis against PBS for 75 minutes. Twenty microliters of nanoparticles were used to coat a corona plasma-treated carbon film 400 square mesh TEM grid for 60 mins. Grids were then briefly washed in ultrapure water to eliminate excess dried salt crystals and dried under vacuum before acquiring images using a Philips CM120 (Philips Research, Briarcliffs Manor, New York).
Cell Culture:
C2C12 murine myoblast cells were purchased from ATCC (Manassas, VA, CRL-1772) and expanded in DMEM supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin. For differentiation to myotube-like cells, C2C12 cells were plated at a density of 31,250 cells/cm2 in tissue culture plates in DMEM supplemented with 2% horse serum, 1% insulin, selenium and transferrin (ITS, 41400045, ThermoFisher). For 96-well plate transfection efficacy experiments, cells were plated on CytoOne 96-well tissue culture plates (USA Scientific, Ocala, FL) 4 days prior to transfection with 12,000 cells/well in 100 μL complete differentiation media and media was changed on days 2 and 4. For noted 384-well plate transfection experiments, C2C12 cells were plated at 2,500 cells/well in 50 μL complete differentiation media in 384-well tissue culture plates (Santa Cruz, sc-206081) 2 days prior to transfection and media was replaced on the day of transfection. Cells were confirmed periodically to be mycoplasma negative via the MycoAlert test (Lonza).
In Vitro Transfection:
For 96-well transfections, 20 μL of SAM nanoparticle dilution with nucleic acid concentration between 1.03–250 pg/μL was added to each well of cells in 100 μL of complete media for a 2 hr incubation before changing media. Cell viability 24 hrs post-transfection was assessed using the MTS Celltiter 96 Aqueous One (Promega, Madison, WI) cell proliferation assay. For 384-well transfections, 5 μL of nanoparticle dilution was added to each well containing cells in 50 μL of complete media and left to incubate for 2 hr before a media change. Percent transfection efficiency was assessed after 48 hrs by staining nuclei with Hoechst stain and imaged for eGFP expression using a Cellomics Arrayscan VTI (Thermo Fisher Scientific, Laguna Hills, CA), an automated fluorescence-based high-content screening imaging system.
In Vivo Experiments:
Animal work was performed in compliance with an approved protocol by the Johns Hopkins University Animal Care and Use Committee (ACUC). Female BALB/c mice, 6–8 weeks old were purchased from The Jackson Laboratory and maintained in accordance with the JHH animal care facility. For in vivo transfection analysis, nanoparticles were made with luciferase (FLuc) SAM and PBAE polymers and injected intramuscularly in mice at 0.2 μg dose for bioluminescent luciferase expression assessment at specified time points. For assessment of luciferase expression, mice were injected intraperitoneally (i.p.) with 100 μL of 150 mg/kg d-luciferin (potassium salt solution in 1× PBS; Cayman Chemical Company, Ann Arbor, MI). After 7 min, mice were anesthetized using isoflurane and imaged using an In Vivo Imaging System (IVIS Spectrum; PerkinElmer, Shelton, CT) to measure bioluminescence. For the rabies vaccination studies, 6–8 weeks old female BALB/c mice were each injected intramuscularly with nanoparticles carrying SAM encoding both rabies virus glycoprotein antigen and the luciferase reporter protein (0.1 μg in 10 μL in opposite quadriceps for 0.2 μg total dose) on day 0 followed by a booster on day 21. Serum was then collected from the mice at day 35, 14 days after the booster vaccination.
Neutralizing Antibody Titer Assay:
Serum samples were analyzed for rabies virus neutralizing antibody (RVNA) titer using a rapid fluorescent foci inhibition test (RFFIT) at the Kansas State University Rabies Laboratory.[48] Serum was first diluted five-fold and then serially five-fold before incubating with live rabies virus. Cultured cells were then combined with the serum dilutions with virus to test for protection resulting from RVNA presence via a titer value calculated from the percent of infected cells. KSU Rabies Laboratory and the World Health Organization (WHO) report a titer of 0.5 international units per millimeter (IU/mL) as a protective response resulting from rabies vaccination and this level is indicated on data plots with a dotted line.
Data Analysis and Statistics:
Cellomics HCS Studio (Thermo Fisher) was used for image acquisition-based in vitro transfection analysis as previously described.[32] Polymer structures were characterized in ChemDraw (Perkin Elmer, Boston, MA) and Marvin (ChemAxon, Cambridge, MA) to determine logP and logD values. Prism 8 (GraphPad, La Jolla, CA) was used for all statistical analyses and curve plotting. Unless otherwise specified, statistical tests were performed with a global alpha value of 0.05. Unless otherwise stated, absence of statistical significance markings where a test was stated to have been performed signified no statistical significance. Statistical significance was denoted as follows: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Supplementary Material
Acknowledgements
The authors thank the lab of Dr. Donald Zack and the Wilmer Equipment Core for use of the Cellomics Arrayscan for quantification of C2C12 transfection experiments.
Funding.
The authors thank the following sources for funding support: NSF Graduate Research Fellowships DGE-0707427 to DRW, DGE-1232825 to YR and DGE-1746891 to SYN; sponsored research agreement with GlaxoSmithKline Biologicals SA; the Bloomberg~Kimmel Institute for Cancer Immunotherapy; and support in part by the NIH (P41EB028239, R01CA228133, R37CA246699, R01EY031097, and the Wilmer Core Grant P30EY001765).
Footnotes
Supporting Information
Supporting figures and tables include chemical properties, polymer synthesis conditions, polymer properties and optimization figures.
Conflict of Interest
JLF, JM, GM, RJa and RJo are, or were at the time of the study employees of the GSK group of companies. GM owns shares in GSK. DW, ST, YR, SN, KL, and JG are co-inventors on a pending patent related to this manuscript (63/274,361). Any potential conflicts of interests for Johns Hopkins personnel are managed by the Johns Hopkins Committee on Outside Interests.
Contributor Information
David R. Wilson, Department of Biomedical Engineering, Institute for NanoBioTechnology, and the Translational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA.
Stephany Y. Tzeng, Department of Biomedical Engineering, Institute for NanoBioTechnology, and the Translational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA.
Yuan Rui, Department of Biomedical Engineering, Institute for NanoBioTechnology, and the Translational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA.
Sarah Y. Neshat, Department of Biomedical Engineering, Institute for NanoBioTechnology, and the Translational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA.
Marranne J Conge, Department of Biomedical Engineering, Institute for NanoBioTechnology, and the Translational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA.
Kathryn M. Luly, Department of Biomedical Engineering, Institute for NanoBioTechnology, and the Translational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA
Ellen Wang, Department of Biomedical Engineering, Institute for NanoBioTechnology, and the Translational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA.
Jessica L. Firestone, GSK Vaccines, Rockville, MD 20850, USA
Josie McAuliffe, GSK Vaccines, Rockville, MD 20850, USA.
Giulietta Maruggi, GSK Vaccines, Rockville, MD 20850, USA.
Rashmi Jalah, GSK Vaccines, Rockville, MD 20850, USA.
Russell Johnson, GSK Vaccines, Rockville, MD 20850, USA.
Joshua C. Doloff, Department of Biomedical Engineering, Institute for NanoBioTechnology, and the Translational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA
Jordan J. Green, Department of Biomedical Engineering, Institute for NanoBioTechnology, and the Translational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA Departments of Chemical & Biomolecular Engineering, Materials Science & Engineering, Neurosurgery, Oncology, and Ophthalmology, Sidney Kimmel Comprehensive Cancer Center and Bloomberg~Kimmel Institute for Cancer Immunotherapy, Johns Hopkins University, Baltimore, MD 21231, USA.
Data Availability Statement
The data that supports the findings of this study are available in the article and the supplementary material of this article. Any additional information regarding the findings of this study are available from the corresponding author upon reasonable request.
References:
- [1].V Parums D, Med Sci Monit 2021, 27, e932915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Blakney AK, Ip S, Geall AJ, Vaccines (Basel) 2021, 9, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, v Kalina W, Cooper D, Frenck RW, Hammitt LL, Türeci Ö, Nell H, Schaefer A, Ünal S, Tresnan DB, Mather S, Dormitzer PR, Şahin U, Jansen KU, Gruber WC, C4591001 Clinical Trial Group, N Engl J Med 2020, 383, 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Baden LR, el Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, McGettigan J, Khetan S, Segall N, Solis J, Brosz A, Fierro C, Schwartz H, Neuzil K, Corey L, Gilbert P, Janes H, Follmann D, Marovich M, Mascola J, Polakowski L, Ledgerwood J, Graham BS, Bennett H, Pajon R, Knightly C, Leav B, Deng W, Zhou H, Han S, Ivarsson M, Miller J, Zaks T, New England Journal of Medicine 2021, 384, 403.33378609 [Google Scholar]
- [5].Brito LA, Kommareddy S, Maione D, Uematsu Y, Giovani C, Berlanda Scorza F, Otten GR, Yu D, Mandl CW, Mason PW, Dormitzer PR, Ulmer JB, Geall AJ, Self-Amplifying mRNA Vaccines, Vol. 89, Elsevier Ltd, 2015. [DOI] [PubMed] [Google Scholar]
- [6].Geall AJ, Verma A, Otten GR, Shaw CA, Hekele A, Banerjee K, Cu Y, Beard CW, Brito LA, Krucker T, O’Hagan DT, Singh M, Mason PW, Valiante NM, Dormitzer PR, Barnett SW, Rappuoli R, Ulmer JB, Mandl CW, Proceedings of the National Academy of Sciences 2012, 109, 14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vogel AB, Lambert L, Kinnear E, Busse D, Erbar S, Reuter KC, Wicke L, Perkovic M, Beissert T, Haas H, Reece ST, Sahin U, Tregoning JS, Molecular Therapy 2018, 26, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pardi N, Hogan MJ, Weissman D, Curr Opin Immunol 2020, 65, 14. [DOI] [PubMed] [Google Scholar]
- [9].Blakney AK, McKay PF, Yus BI, Aldon Y, Shattock RJ, Gene Ther 2019, 26, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lou G, Anderluzzi G, Schmidt ST, Woods S, Gallorini S, Brazzoli M, Giusti F, Ferlenghi I, Johnson RN, Roberts CW, O’Hagan DT, Baudner BC, Perrie Y, Journal of Controlled Release 2020, 325, 370. [DOI] [PubMed] [Google Scholar]
- [11].Chahal JS, Khan OF, Cooper CL, McPartlan JS, Tsosie JK, Tilley LD, Sidik SM, Lourido S, Langer R, Bavari S, Ploegh HL, Anderson DG, Proceedings of the National Academy of Sciences 2016, 113, E4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bogers WM, Oostermeijer H, Mooij P, Koopman G, Verschoor EJ, Davis D, Ulmer JB, Brito LA, Cu Y, Banerjee K, Otten GR, Burke B, Dey A, Heeney JL, Shen X, Tomaras GD, Labranche C, Montefiori DC, Liao HX, Haynes B, Geall AJ, Barnett SW, Journal of Infectious Diseases 2015, 211, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Anderluzzi G, Lou G, Gallorini S, Brazzoli M, Johnson R, O’Hagan DT, Baudner BC, Perrie Y, Vaccines (Basel) 2020, 8, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Geall AJ, Verma A, Otten GR, Shaw CA, Hekele A, Banerjee K, Cu Y, Beard CW, Brito LA, Krucker T, O’Hagan DT, Singh M, Mason PW, Valiante NM, Dormitzer PR, Barnett SW, Rappuoli R, Ulmer JB, Mandl CW, Proceedings of the National Academy of Sciences 2012, 109, 14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Archer J, Barnett SW, Lilja A, Singh M, Carsillo T, Mason PW, Otten GR, Valiante NM, Brito LA, Schaefer M, Mandl CW, Seubert A, Chan M, Dormitzer PR, Geall AJ, Beard CW, Dey AK, Shaw CA, Hekele A, Ulmer JB, O’Hagan DT, Molecular Therapy 2014, 22, 2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kaczmarek JC, Kauffman KJ, Fenton OS, Sadtler K, Patel AK, Heartlein MW, DeRosa F, Anderson DG, Nano Lett 2018, 18, 6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Karlsson J, Rhodes KR, Green JJ, Tzeng SY, Expert Opin Drug Deliv 2020, 17, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Anderson DG, Lynn DM, Langer R, Angew Chem Int Ed Engl 2003, 42, 3153. [DOI] [PubMed] [Google Scholar]
- [19].Wilson DR, Suprenant MP, Michel JH, Wang EB, Tzeng SY, Green JJ, Biotechnol Bioeng 2019, 116, 1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sunshine JC, Akanda MI, Li D, Kozielski KL, Green JJ, Biomacromolecules 2011, 12, 3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim J, Sunshine JC, Green JJ, Bioconjug Chem 2014, 25, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rui Y, Wilson DR, Tzeng SY, Yamagata HM, Sudhakar D, Conge M, Berlinicke CA, Zack DJ, Tuesca A, Green JJ, Sci Adv 2022, 8, eabk2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shmueli RB, Sunshine JC, Xu Z, Duh EJ, Green JJ, Nanomedicine 2012, 8, 1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Erasmus JH, Archer J, Fuerte-Stone J, Khandhar AP, Voigt E, Granger B, Bombardi RG, Govero J, Tan Q, Durnell LA, Coler RN, Diamond MS, Crowe JE, Reed SG, Thackray LB, Carnahan RH, Van Hoeven N, Mol Ther Methods Clin Dev 2020, 18, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wilson DR, Mosenia A, Suprenant MP, Upadhya R, Routkevitch D, Meyer RA, Quinones-Hinojosa A, Green JJ, J Biomed Mater Res A 2017, 105, 1813. [DOI] [PubMed] [Google Scholar]
- [26].Hu Y, Zhu Y, Sutherland ND, Wilson DR, Pang M, Liu E, Staub JR, Berlinicke CA, Zack DJ, Green JJ, Reddy SK, Mao HQ, Nano Lett 2021, 21, 5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tzeng SY, Guerrero-Cázares H, Martinez EE, Sunshine JC, Quiñones-Hinojosa A, Green JJ, Biomaterials 2011, 32, 5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kaczmarek JC, Patel AK, Kauffman KJ, Fenton OS, Webber MJ, Heartlein MW, DeRosa F, Anderson DG, Angew Chem Int Ed Engl 2016, 55, 13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Eltoukhy AA, Chen D, Alabi CA, Langer R, Anderson DG, Advanced Materials 2013, 25, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ryals RC, Patel S, Acosta C, McKinney M, Pennesi ME, Sahay G, PLoS One 2020, 15, e0241006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhu X, Tao W, Liu D, Wu J, Guo Z, Ji X, Bharwani Z, Zhao L, Zhao X, Farokhzad OC, Shi J, Theranostics 2017, 7, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mishra B, Wilson DR, Sripathi SR, Suprenant MP, Rui Y, Wahlin KJ, Berlinicke CA, Green JJ, Zack DJ, Regen Eng Transl Med 2019, 6, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Anderson DG, Peng W, Akinc A, Hossain N, Kohn A, Padera R, Langer R, Sawicki JA, Proceedings of the National Academy of Sciences 2004, 101, 16028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Vaughan HJ, Zamboni CG, Radant NP, Bhardwaj P, Lechtich ER, Hassan LF, Shah K, Green JJ, Mol Ther Oncolytics 2021, 21, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tzeng SY, Patel KK, Wilson DR, Meyer RA, Rhodes KR, Green JJ, Proc Natl Acad Sci U S A 2020, 117, 4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lopez-Bertoni H, Kozielski KL, Rui Y, Lal B, Vaughan H, Wilson DR, Mihelson N, Eberhart CG, Laterra J, Green JJ, Nano Lett 2018, 18, 4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shen J, Kim J, Tzeng SY, Ding K, Hafiz Z, Long D, Wang J, Green JJ, Campochiaro PA, Sci Adv 2020, 6, eaba1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bhosle SM, Loomis KH, Kirschman JL, Blanchard EL, Vanover DA, Zurla C, Habrant D, Edwards D, Baumhof P, Pitard B, Santangelo PJ, Biomaterials 2018, 159, 189. [DOI] [PubMed] [Google Scholar]
- [39].McKinlay CJ, Vargas JR, Blake TR, Hardy JW, Kanada M, Contag CH, Wender PA, Waymouth RM, Proc Natl Acad Sci U S A 2017, 114, E448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Greig JA, Peng H, Ohlstein J, Medina-Jaszek CA, Ahonkhai O, Mentzinger A, Grant RL, Roy S, Chen SJ, Bell P, Tretiakova AP, Wilson JM, PLoS One 2014, 9, e112268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gehling AM, Kuszpit K, Bailey EJ, Allen-Worthington KH, Fetterer DP, Rico PJ, Bocan TM, Hofer CC, J Am Assoc Lab Anim Sci 2018, 57, 35. [PMC free article] [PubMed] [Google Scholar]
- [42].Johanning FW, Conry RM, LoBuglio AF, Wright M, Sumerel LA, Pike MJ, Curiel DT, Nucleic Acids Res 1995, 23, 1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Walsh EE, Frenck RW, Falsey AR, Kitchin N, Absalon J, Gurtman A, Lockhart S, Neuzil K, Mulligan MJ, Bailey R, Swanson KA, Li P, Koury K, Kalina W, Cooper D, Fontes-Garfias C, Shi P-Y, Türeci Ö, Tompkins KR, Lyke KE, Raabe V, Dormitzer PR, Jansen KU, Şahin U, Gruber WC, New England Journal of Medicine 2020, 383, 2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ramos JG, Varayoud J, Kass L, Rodriíguez H, Costabel L, Muñoz-de-Toro M, Luque EH, Endocrinology 2003, 144, 3206. [DOI] [PubMed] [Google Scholar]
- [45].Wells DJ, Molecular Therapy 2004, 10, 207. [DOI] [PubMed] [Google Scholar]
- [46].Itaka K, Osada K, Morii K, Kim P, Yun S-H, Kataoka K, Journal of controlled release 2010, 143, 112. [DOI] [PubMed] [Google Scholar]
- [47].Paunovska K, Sago CD, Monaco CM, Hudson WH, Castro MG, Rudoltz TG, Kalathoor S, Vanover DA, Santangelo PJ, Ahmed R, Bryksin A. v., Dahlman JE, Nano Lett 2018, 18, 2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bahloul C, Taieb D, Kaabi B, Diouani MF, Ben Hadjahmed S, Chtourou Y, B’chir BI, Dellagi K, Epidemiol Infect 2005, 133, 749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Miao L, Zhang Y, Huang L, Mol Cancer 2021, 20, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Anderluzzi G, Lou G, Gallorini S, Brazzoli M, Johnson R, O’Hagan DT, Baudner BC, Perrie Y, Vaccines (Basel) 2020, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Guerrero-Cázares H, Tzeng SY, Young NP, Abutaleb AO, Quiñones-Hinojosa A, Green JJ, ACS Nano 2014, 8, 5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Qu M, Kim H-J, Zhou X, Wang C, Jiang X, Zhu J, Xue Y, Tebon P, Sarabi SA, Ahadian S, Nanoscale 2020, 12, 16724. [DOI] [PubMed] [Google Scholar]
- [53].Geall AJ, Verma A, Otten GR, Shaw CA, Hekele A, Banerjee K, Cu Y, Beard CW, Brito LA, Krucker T, O’Hagan DT, Singh M, Mason PW, Valiante NM, Dormitzer PR, Barnett SW, Rappuoli R, Ulmer JB, Mandl CW, Proc Natl Acad Sci U S A 2012, 109, 14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Maruggi G, Mallett CP, Westerbeck JW, Chen T, Lofano G, Friedrich K, Qu L, Sun JT, McAuliffe J, Kanitkar A, Arrildt KT, Wang K-F, McBee I, McCoy D, Terry R, Rowles A, Abrahim MA, Ringenberg MA, Gains MJ, Spickler C, Xie X, Zou J, Shi P-Y, Dutt T, Henao-Tamayo M, Ragan I, Bowen RA, Johnson R, Nuti S, Luisi K, Ulmer JB, Steff A-M, Jalah R, Bertholet S, Stokes AH, Yu D, Mol Ther 2022, 30, 1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kozielski KL, Tzeng SY, Green JJ, Chem Commun (Camb) 2013, 49, 5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wilson DR, Rui Y, Siddiq K, Routkevitch D, Green JJ, Mol Pharm 2019, 16, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that supports the findings of this study are available in the article and the supplementary material of this article. Any additional information regarding the findings of this study are available from the corresponding author upon reasonable request.