

Abstract
Fluorinated oxindoles are frequently used building blocks in asymmetric synthesis and represent an important scaffold found in a variety of biologically relevant compounds. While it is understood that incorporation of fluorine atoms into organic molecules can improve their pharmacological properties, the impact on the configurational stability of chiral organofluorines is still underexplored. In this study, semipreparative HPLC enantioseparations of five oxindoles were carried out and the resulting enantiomerically enriched solutions were used to investigate base promoted racemization kinetics at room temperature. It was found that incorporation of fluorine at the chiral center increases the configurational stability, while substitutions on the aromatic ring, and at the lactam moiety also have significant effects on the rate of racemization, which generally follows reversible first-order reaction kinetics.
Keywords: Oxindoles, fluoride effect, enantioseparation, high performance liquid chromatography, configurational stability, enantioconversion
Graphical Abstract
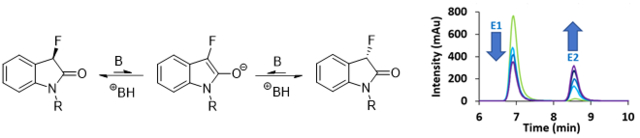
Introduction
The general impact of fluorinated chiral compounds across the chemical and health sciences has stimulated numerous drug development projects and directed increasing attention toward investigating the unique and often advantageous pharmacological and physicochemical properties that originate from the incorporation of carbon-fluorine bonds into organic molecules.1,2 These tasks are greatly facilitated by the steadily increasing pool of fluorinated chiral building blocks that are readily available for use in pharmaceutical discovery programs or other applications.1,2,3,4,5,6,7,8,9 The stereochemical integrity of organofluorines, however, is poorly understood and few studies that quantify the propensity to racemization and the role of fluorine when positioned at an enolizable chiral carbon center have been reported. To this end, we recently investigated the racemization kinetics of 2-aryl-2-fluoroacetonitriles and developed catalytic asymmetric allylic alkylation and stereodivergent Mannich reactions with this emerging class of compounds.10,11,12 Following our continuous interest in chiral organofluorines,13,14,15,16,17,18,19,20,21,22,23,24 we now wish to disclose the results of a racemization study with 3-fluorooxindoles which are frequently employed in catalytic asymmetric reaction developments and represent an important pharmacophore encountered in the potassium channel modulator Maxipost and other biologically active compounds.25,26
Semipreparative chiral HPLC enantioseparation of the oxindoles 1–5 shown in Figure 1 using a Whelk-O 1 or Chiralpak IB column produced highly enantioenriched samples that were subjected to catalytic or stoichiometric base amounts at room temperature. The change in the enantiomeric composition was monitored by chiral HPLC to determine reversible first-order racemization kinetics. Our analysis shows that the racemization rate of oxindoles is not only affected by the substituent at the chirality center but also quite sensitive to changes in the aromatic ring and at the nitrogen atom. Interestingly, the comparison of the racemization kinetics of compounds 2 and 5 obtained under the same conditions revealed that the fluorinated oxindole is configurationally more stable. In fact, we found that the replacement of the methyl group at C-3 by a fluoride atom results in a 2.5-fold decrease in the racemization rate.
Figure 1.
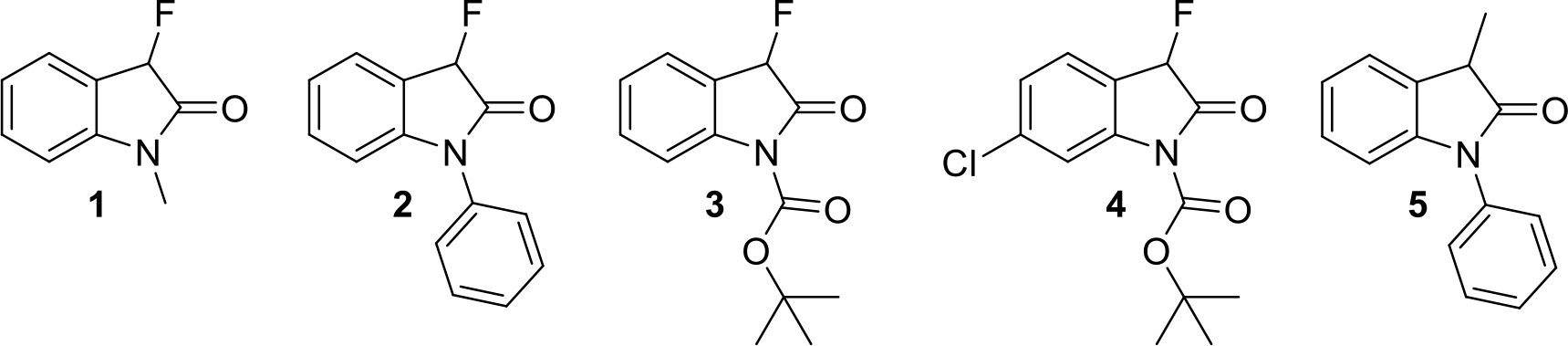
Structures of 3-fluorooxindoles 1–4 and the corresponding 3-methyl analog 5.
Materials and Methods
Chiral HPLC
HPLC enantioseparations were carried out using an (S,S)-Whelk-O 1 or a Chiralpak IB column at room temperature with a flow rate of 1.0 mL/min. The chromatograms were recorded at 205, 214, 240, 254, and 260 nm with a diode array detector. The composition of the mobile phase was varied between 98:2 and 90:10 hexanes:isopropyl alcohol (IPA) ratios to optimize peak separation and retention times.
Synthesis of N-Methyl-3-fluorooxindole
N-Methyl-3-fluorooxindole, 1, was synthesized from commercially available N-methyl oxindole in 39% overall yield via trifluoroacetylation followed by fluorination using Selectfluor and base promoted removal of the trifluoroacetyl group as previously described by our laboratory, Scheme 1.27 1H NMR (400 MHz, CDCl3) δ 7.44 – 7.30 (m, 2H), 7.07 (m, 1H), 6.79 (d, J = 7.9, 1H), 5.60 (d, JF = 50.9, 1H), 3.13 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 171.1 (d, JC-F = 18.2 Hz), 144.7 (d, JC-F = 5.1 Hz), 131.4 (d, JC-F = 3.4 Hz), 126.0 (d, JC-F = 1.3 Hz), 123.2 (d, JC-F = 2.8 Hz), 122.8 (d, JC-F = 16.3 Hz), 108.7 (d, JC-F = 1.5 Hz), 85.5 (d, JC-F = 188.2 Hz), 26.2. 19F NMR (376 MHz, CDCl3) δ −193.36 (d, J = 51.1 Hz). The spectra were identical with previously published literature reports (Figures S2–S5).
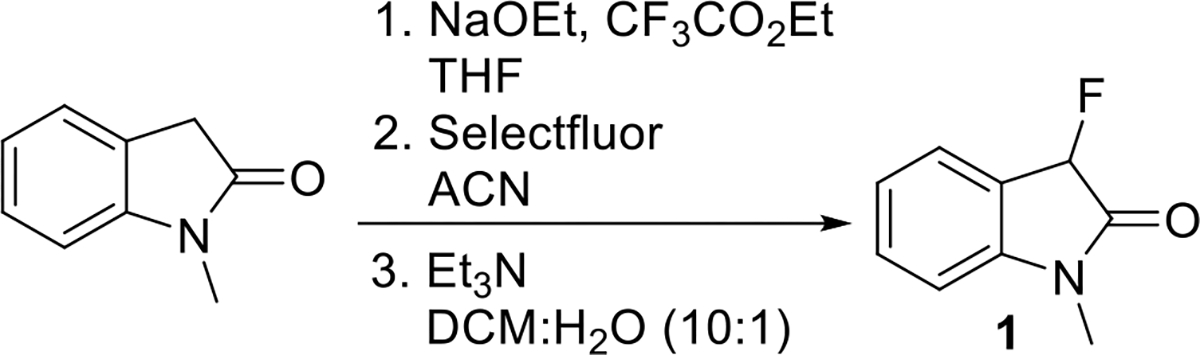
Scheme 1. Synthesis of 1.
Racemization Study
Semipreparative enantioseparation of compounds 1–5 was achieved by repetitive injections on an (S,S)-Whelk-O 1 or Chiralpak IB column (250 × 4.6 mm) under optimized conditions (see Figures S1 and S6–S10 in the Supporting Information for details). The combined fractions containing 50% of the originally injected racemic compound amount in highly enantioenriched form were dried under reduced vacuum and dissolved at the desired concentration (e.g. 185 nM). The enantiomeric purity of the collected fractions was verified by the same chiral HPLC method. To these solutions, 0.1–2 equivalents of either diisopropylethylamine (DIPEA) or 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) were added using diluted stock solutions and the samples were stirred at room temperature in a sealed vial. The decrasing enantiomeric excess (ee) was monitored by the same HPLC method using reaction aliquots without further dilution to determine the extent of racemization versus time. All racemization studies were analyzed according to reversible first-order reactions kinetics described in Equation 1, where is the percentage of the starting enantiomer at , is the percentage of the enantiomer at time and is 50% as determined from the ratio of the areas under the peaks (Figures S11–S74 and Tables S1–S7 in the SI):
| Eq 1 |
Results and Discussion
We expected that the presence of a hydrogen bond acceptor and an aromatic ring close to the chirality center would allow semipreparative chiral HPLC enantioseparation of the oxindoles 1–5 on the Whelk-O 1 column.28,29,30,31 While this was indeed the case with the 3-fluorooxindoles 1–4 the enantiomers of the 3-methyl analog 5 were best separated on a cellulose column.32,33,34
Chiral oxindoles are configurationally stable under neutral conditions at room temperature. Deprotonation, however, forms an achiral enolate and therefore results in racemization. While the high electronegativity of fluorine may be expected to increase the oxindole acidity, the deprotonation generates a negative charge in close proximity to the three lone electron pairs at the halide atom which is expected to destabilize the enolate form. We conducted racemization experiments with compounds 1–5 using 0.1–0.2 equivalents of DBU or excess of DIPEA. As demonstrated with N-methyl-3-fluorooxindole, 1, the racemization of the enantiomerically enriched sample was monitored using chiral HPLC analysis (Figure 2).
Figure 2.
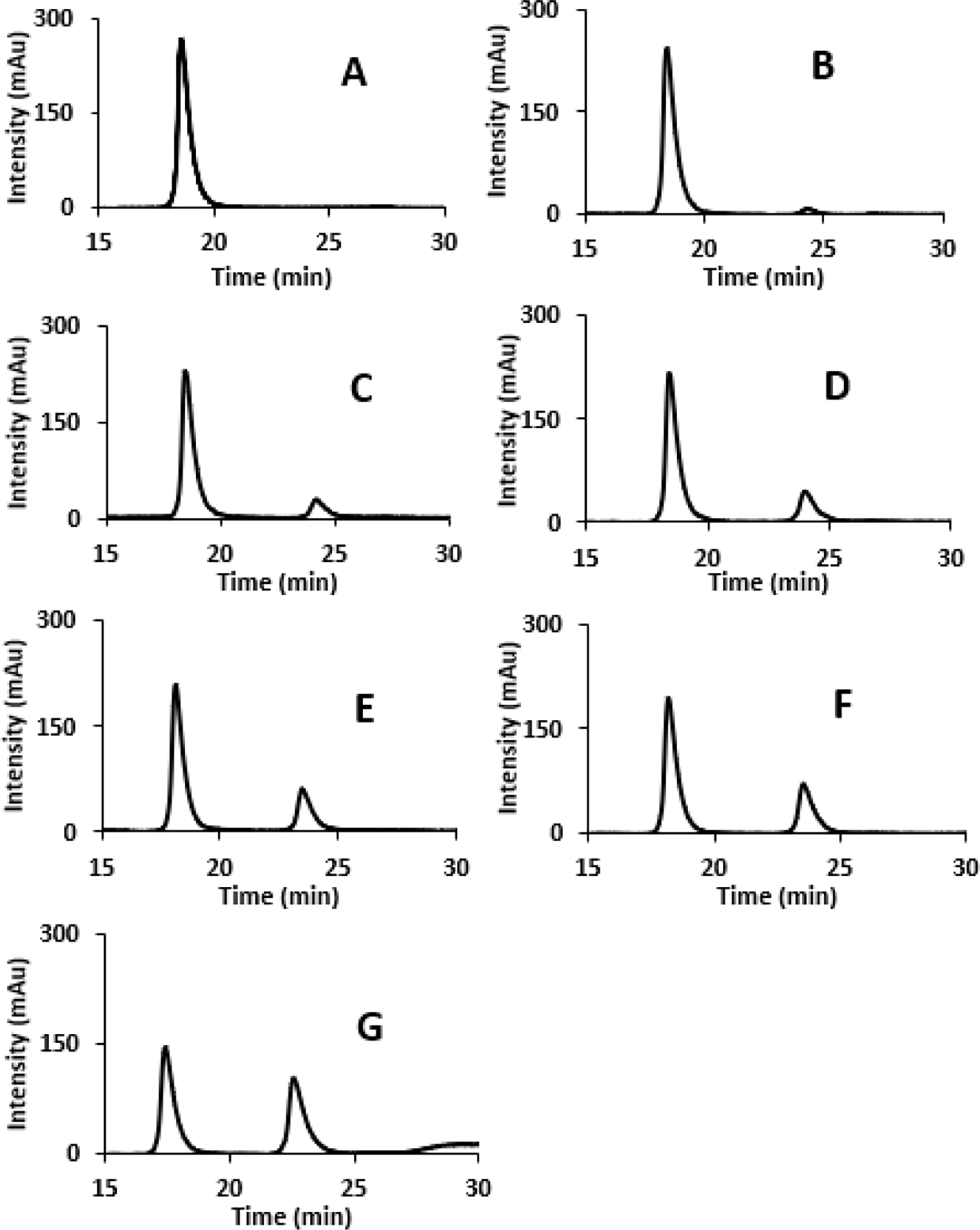
HPLC chromatograms of compound 1 at 0 (A), 60 (B), 180 (C), 300 (D), 420 (E), 540 (F), and 1582 minutes (G) following semipreparative enantioseparation and treatment of the enantiopure solution with 0.1 equivalents of DBU in 90:10 hexanes:IPA (Whelk-O 1, 1 mL/min).
We found that 1 racemizes considerably more slowly than 2–5. We were surprised to observe a profound effect on the rate of racemization when the lactam protecting group was varied. The replacement of a methyl group with a phenyl ring resulted in more than a 5-fold decrease in the half-life time from 427.8 minutes to 82.5 minutes (Table 1). The slower racemization of the N-methyl derivative 1 can be attributed to the increased involvement of the carbonyl group in the lactam resonance which reduces the oxindole acidity as shown in Figure 3 while the N-phenyl ring more effectively favors enolate formation. The Boc-protected fluorooxindole 3 is slightly more stable to racemization than 2 and we obtained a half-life time of 96.3 minutes in the presence of 10 mol% of DBU.. These measurements demonstrate the possibility of mild catalytic fluorooxindole racemization and the considerable impact of N-protecting groups on the configurational stability at the chiral center. A comparison of the half-lives of 1–3 obtained using 10 mol% of DBU as base reveals that incorporation of electron-withdrawing phenyl or carbamoyl groups at the oxindole nitrogen atom increases the racemization rate by approximately five times. Interestingly, the presence of a fluorine atom in 2 results in an increase in the half-life from 33.0 to 82.5 min, a 2.5-fold change in the racemization rate compared to the methylated analog 5.
Table 1.
Results of the racemization study with compounds 1–5.
| Compound | Base (equiv.) | krac (s−1) | t1/2 (min) |
|---|---|---|---|
| 1 | DBU (0.1 eq.) | 2.7 x 10−5 | 427.8a |
| 2 | DBU (0.1 eq.) | 1.4 x 10−4 | 82.5a |
| 3 | DBU (0.1 eq.) DBU (0.2 eq.) |
1.2 x 10−4 4.5 x 10−4 |
96.3a 25.7 |
| 4 | DBU (0.1 eq.) DIPEA (2 eq.) |
1.1 x 10−3 3.8 x 10−5 |
10.5a 303.9 |
| 5 | DBU (0.1 eq.) | 3.5 x 10−4 | 33.0a |
Oxindole concentration was 185 nM in hexanes:IPA mixtures. See SI for more details.
Figure 3.
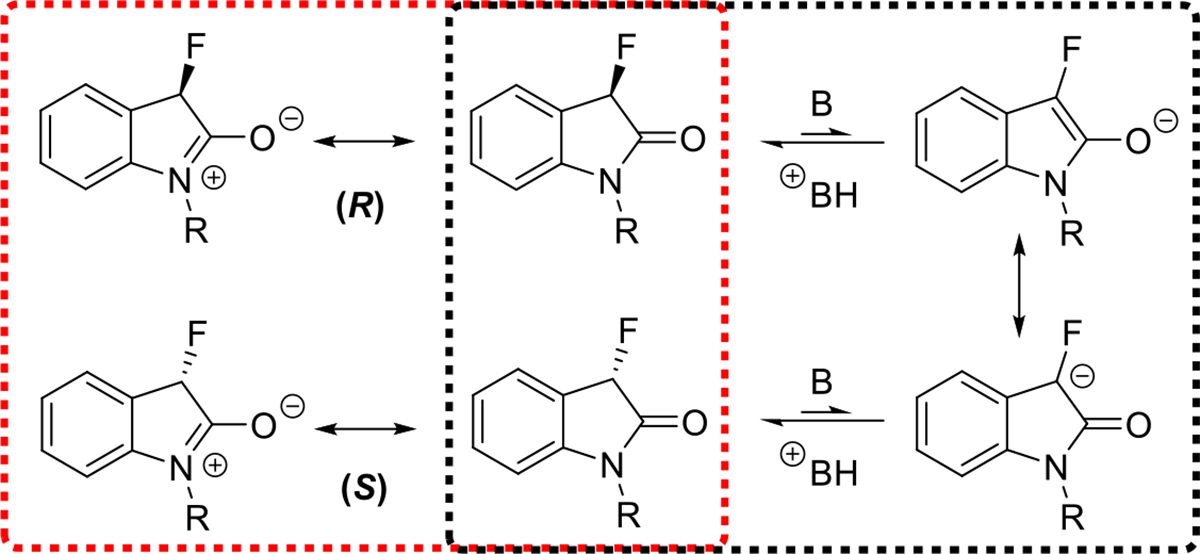
Base promoted racemization mechanism (black) and competing lactam resonance (red).
Substitutions on the aromatic ring also affect the rate of racemization, as evidenced by the introduction of a chlorine atom in compound 4. Under the same conditions used in the study with compound 3, the chloride-substituted analog fully racemized ~10 times faster (Table 1). In the presence of two equivalents of DIPEA, the half-life time was determined as 303.9 minutes which highlights that these compounds may be configurationally stable for much longer times when weaker bases are used. In fact, the racemization of compound 2 took over a week when it was treated with one equivalent of DIPEA (see Supporting Information). The change in the enantiomeric composition and the ee decline of 1–5 are shown in Figure 4.
Figure 4.
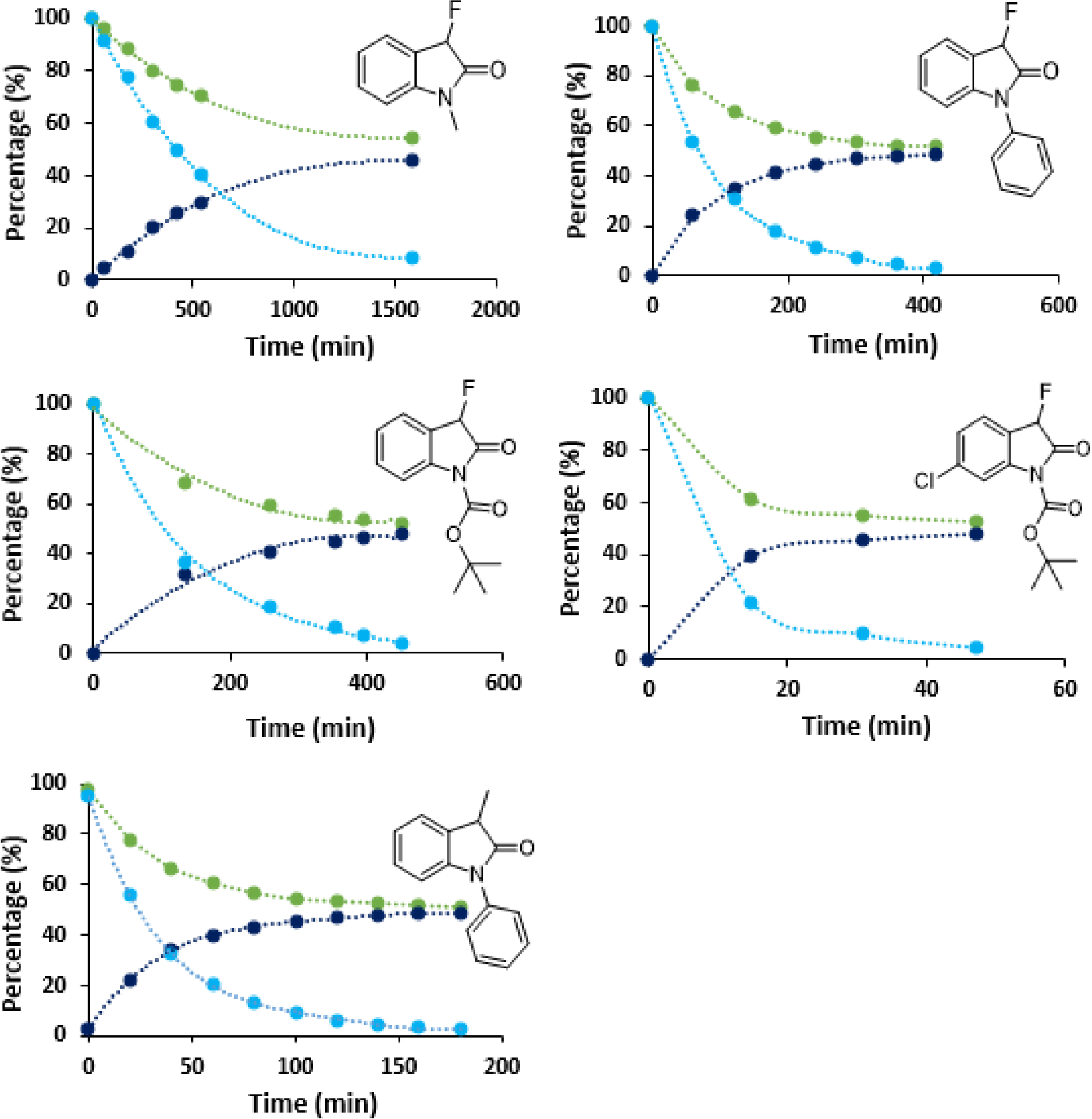
Change in the enantiomeric composition of 1–5 during base promoted racemization of samples at 185 nm in the presence of 10% DBU (% ee is light blue, percentages of each enantiomer are shown in green and dark blue). See SI for experimental details.
Conclusion
In summary, we have examined the configurational stability of five 3-substituted chiral oxindoles using HPLC with the widely available Whelk-O 1 and Chiralpak IB columns. Semipreparative separations produced highly enantioenriched solutions that were employed in racemization studies with catalytic or stoichiometric amounts of DBU or DIPEA at room temperature. Reaction monitoring and data analysis according to reversible first-order reaction kinetics showed that closely related oxindoles can have quite different half-life times and that the presence of fluorine at the chiral center increases the configurational stability relative to its 3-methyl analogue. It was found that the lactam protecting groups have a large influence on the rate of racemization, with replacement of a methyl group by a phenyl ring resulting in more than a 5-fold decrease in the half-life time. Substitutions in the aromatic ring are also important and we observed almost complete racemization of N-Boc-6-chloro-3-fluorooxindole in approximately 1 hour compared to over 7 hours for its non-chlorinated analog in the presence of 10 mol% of DBU. It is expected that the results of the racemization study with the chiral oxindoles 1–5 will become important considerations in future pharmaceutical developments and asymmetric reaction optimization projects.
Supplementary Material
Acknowledgment
We gratefully acknowledge financial support from the U.S. National Institutes of Health (GM106260) and the U.S. National Science Foundation (CHE1764135). EN thanks the Henry Luce Foundation for a Clare Boothe Luce Graduate Fellowship.
Footnotes
Supporting Information
Additional supporting information including HPLC chromatograms and details of the kinetic studies may be found in the online version of this article at the publisher’s website.
Data Availability Statement
The data that support the findings of this study are available in the supporting information of this article.
REFERENCES
- 1.Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Acena JL, Soloshonok VA, Izawa K, Liu H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016;116: 422–518. [DOI] [PubMed] [Google Scholar]
- 2.Smith BR, Eastman CM, Njardarson JT. Beyond C, H, O, and N Analysis of the Elemental Composition of U.S. FDA Approved Drug Architectures. J. Med. Chem. 2014;57:9764–9773. [DOI] [PubMed] [Google Scholar]
- 3.Liang T, Neumann CN, Ritter T. Introduction of Fluorine-containing Functional Groups. Angew. Chem. Int. Ed. 2013;52:8214–8264. [DOI] [PubMed] [Google Scholar]
- 4.Yang X, Wu T, Phipps RJ, Toste FD Advances in Catalytic Enantioselective Fluorination, Mono‑, Di‑, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 2015;115:826–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu Y, Han J, Wang J, Shibata N, Sodeoka M, Soloshonok VA, Coelho JAS, Toste FD. Modern Approaches for Asymmetric Construction of Carbon−Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018;118:3887–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paladhi S; Park SY; Yang JW; Song CE Asymmetric Synthesis of α-Fluoro-β-Amino-oxindoles with Tetrasubstituted C–F Stereogenic Centers via Cooperative Cation-Binding Catalysis Org. Lett 2017;19:5336–5339. [DOI] [PubMed] [Google Scholar]
- 7.Jin Y; Chen M; Ge S; Hartwig JF Palladium-Catalyzed, Enantioselective α-Arylation of α-Fluorooxindoles. Org. Lett 2017;19:1390–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.B.-Y. Li.; Du D-M Chiral Squaramide-Catalyzed Asymmetric Mannich Reactions for Synthesis of Fluorinated 3,3′-Bisoxindoles. Adv. Synth. Catal 2018;360:3164–3170 [Google Scholar]
- 9.Liu Y-L; Wang X-P; Wei J; Li Y Synthesis of Oxindoles Bearing a Stereogenic 3-Fluorinated Carbon Center from 3-Fluorooxindoles. Org. Biomol. Chem 2022;20:538–552. [DOI] [PubMed] [Google Scholar]
- 10.Steber SE; Pham ANDL; Nelson E; Wolf C Enantioseparation and Racemization of α-Aryl-α-Fluoroacetonitriles. Chirality 2021;3:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sripada A; Wolf C Catalytic Asymmetric Allylic Alkylation with Arylfluoroacetonitriles. J. Org. Chem. 2022;87:11880–11887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding R, De los Santos ZA, Wolf C. Catalytic Asymmetric Mannich Reaction of α-Fluoronitriles with Ketimines: Enantioselective and Diastereodivergent Construction of Vicinal Tetrasubstituted Stereocenters. ACS Catal. 2019;9:2169–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu H, Wolf C. Synthesis of Chiral Tertiary Trifluoromethyl Alcohols by Asymmetric Nitroaldol Reaction with a Cu(II)-Bisoxazolidine Catalyst. Chem. Commun. 2010;46:8026–8028. [DOI] [PubMed] [Google Scholar]
- 14.Wolf C, Zhang P. Asymmetric Friedel-Crafts Reaction of Indoles with Ethyl Trifluoropyruvate Using a Copper(I)-Bisoxazolidine Catalyst. Adv. Synth. Catal. 2011;353:760–766. [Google Scholar]
- 15.Zhang P, Wolf C. Catalytic Enantioselective Difluoroalkylation of Aldehydes. Angew. Chem. Int. Ed. 2013;52:7869–7873. [DOI] [PubMed] [Google Scholar]
- 16.Balaraman K, Moskowitz M, Liu Y, Wolf C. Detrifluoroacetylative Generation of Halogenated Enolates: Practical Access to Perhalogenated Ketones and Alkenes. Synthesis 2016;2376–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding R, Wolf C. Catalytic Insertion of Aldehydes into Dihalonitroacetophenones via Sequential Bond Scission-Aldol Reaction-Acyl Transfer. Chem. Commun. 2016;52:3576–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cook AM, Wolf C. Efficient Access to Multifunctional Trifluoromethyl Alcohols through Base-Free Catalytic Asymmetric C−C Bond Formation with Terminal Ynamides. Angew. Chem. Int. Ed. 2016;55:2929–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding R, Bakhshi PR, Wolf C. Organocatalytic Insertion of Isatins into Aryl Difluoronitromethyl Ketones. J. Org. Chem. 2017;82:1273–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balaraman K, Wolf C. Catalytic Enantioselective and Diastereoselective Allylic Alkylation with Fluoroenolates: Efficient Access to C3-Fluorinated and All-Carbon Quaternary Oxindoles. Angew. Chem. Int. Ed. 2017;56:1390–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balaraman K, Ding R, Wolf C. Stereoselective Synthesis of 3,3’-Bisindolines by Organocatalytic Michael Additions of Fluorooxindole Enolates to Isatylidene Malononitriles in Aqueous Solution. Adv. Synth. Catal. 2017;359:4165–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding R, Wolf C. Organocatalytic Asymmetric Synthesis of α-Oxetanyl and α-Azetidinyl Tertiary Alkyl Fluorides and Chlorides. Org. Lett. 2018;20:892–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moskowitz M, Balaraman K, Wolf C. Organocatalytic Stereoselective Synthesis of Fluorinated 3,3’-Linked Bisoxindoles. J. Org. Chem. 2018;83:1661–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balaraman K, Moskowitz M, Wolf C. Organocatalytic Decarboxylative Cyanomethylation of Difluoromethyl and Trifluoromethyl Ketones. Adv. Synth. Catal. 2018;360:4705–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gribkoff VK; Starrett JE Jr.; Dworetzky SL; Hewawasam P; Boissard CG; Cook DA; Frantz SW; Heman K; Hibbard JR; Huston K; Johnson G; Krishnan BS; Kinney GG; Lombardo LA; Meanwell NA; Molinoff PB; Myers RA; Moon SL; Ortiz A; Pajor L; Pieschl RL; Post-Munson DJ; Signor LJ; Srinivas N; Taber MT; Thalody G; Trojnacki JT; Wiener H; Yeleswaram K; Yeola SW Targeting Acute Ischemic Stroke with a Calcium-sensitive Opener of Maxi-K Potassium Channels. Nat. Med. 2001;7:471–477. [DOI] [PubMed] [Google Scholar]
- 26.Hewawasam P; Gribkoff VK; Pendri Y; Dworetzky SI; Meanwell NA; Martinez E; Boissard CG; Post-Munson DJ; Trojnacki JT; Yeleswaram K; Pajor LM; Knipe J; Gao Q; Perrone R; Starrett JE Jr. The Synthesis and Characterization of BMS-204352 (MaxiPost) and Related 3-Fluorooxindoles as Openers of Maxi-K Potassium Channels. Bioorg. Med. Chem. Lett. 2002;12:1023–1026. [DOI] [PubMed] [Google Scholar]
- 27.Balaraman K; Wolf C Catalytic Enantioselective and Diastereoselective Allylic Alkylation with Fluoroenolates: Efficient Access to C3-Fluorinated and All-Carbon Quaternary Oxindoles. Angew. Chem. Int. Ed. 2016;1390–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pirkle WH, Welch CJ, Lamm B. Design, Synthesis, and Evaluation of an Improved Enantioselective Naproxen Selector. J. Org. Chem. 1992;57:3854–3860. [Google Scholar]
- 29.Wolf C Dynamic Stereochemistry of Chiral Compounds, RSC Publishing, Cambridge, 2008, pp. 136–179. [Google Scholar]
- 30.Wolf C, Pirkle WH. Synthesis and Evaluation of a Copolymeric Chiral Stationary Phase. J. Chromatogr. A 1998;799:177–184. [Google Scholar]
- 31.Welch CJ. Evolution of Chiral Stationary Phase Design in the Pirkle Laboratories. J. Chromatogr. A 1994;666:3–26. [Google Scholar]
- 32.Okamoto Y, Kaida Y. Resolution by High-performance Liquid Chromatography Using Polysaccharide Carbamates and Benzoates as Chiral Stationary Phases. J. Chromatogr. A. 1994;666:403–419. [Google Scholar]
- 33.Okamoto Y, Yashima E. Polysaccharide Derivatives for Chromatographic Separation of Enantiomers. Angew. Chem., Int. Ed. 1998;37:1021–1043. [DOI] [PubMed] [Google Scholar]
- 34.Okamoto Y, Ikai T. Chiral HPLC for Efficient Resolution of Enantiomers. Chem. Soc. Rev. 2008;37:2593–2608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the supporting information of this article.