

Abstract
Gram-negative bacteria are uniquely equipped to defeat antibiotics. Their outermost layer, the cell envelope, is a natural permeability barrier that contains an array of resistance proteins capable of neutralizing most existing antimicrobials. As a result, its presence creates a major obstacle for the treatment of resistant infections and for the development of new antibiotics. Despite this seemingly impenetrable armor, in-depth understanding of the cell envelope, including structural, functional and systems biology insights, has promoted efforts to target it that can ultimately lead to the generation of new antibacterial therapies. In this article, we broadly overview the biology of the cell envelope and highlight attempts and successes in generating inhibitors that impair its function or biogenesis. We argue that the very structure that has hampered antibiotic discovery for decades has untapped potential for the design of novel next-generation therapeutics against bacterial pathogens.
Keywords: Gram-negative bacteria, antimicrobial resistance, cell envelope, bacterial membrane, peptidoglycan, lipopolysaccharide, extracytoplasmic protein, antibiotic discovery, antibiotic target, antibiotic adjuvant
1. INTRODUCTION
Antimicrobial resistance is a silent pandemic that imposes significant logistical and financial burdens on healthcare systems worldwide. The continuous emergence of resistant bacterial strains combined with the dwindling antibiotic discovery pipeline led many to suggest that we are at the dawn of the ‘post-antibiotic era’. If this prediction materializes, common medical procedures, like surgery and chemotherapy, will become unviable due to the possibility of infections that cannot be treated. Gram-negative pathogens, and in particular multidrug-resistant strains of Escherichia coli, Acinetobacter baumannii, and Klebsiella pneumoniae, were found to be responsible for the majority of the 1.3 million deaths directly attributed to antimicrobial resistance in 2019 (Murray et al., 2022). Alarmingly, for some of these bacterial species, such as A. baumannii and Burkholderia cenocepacia, many strains of which are pan-resistant (Karakonstantis, Kritsotakis, & Gikas, 2019), the bleak ‘post-antibiotic era’ scenario is already at our doorstep. The biggest challenge for the development of antibiotics that target Gram-negative bacteria is to bypass their outermost layer, a highly organized tripartite structure composed of the outer membrane, the cell wall, and the plasma membrane, which encloses the periplasm and is collectively termed “the cell envelope” (Figure 1).
Figure 1. Overview of the structure and the key components of the Gram-negative cell envelope.
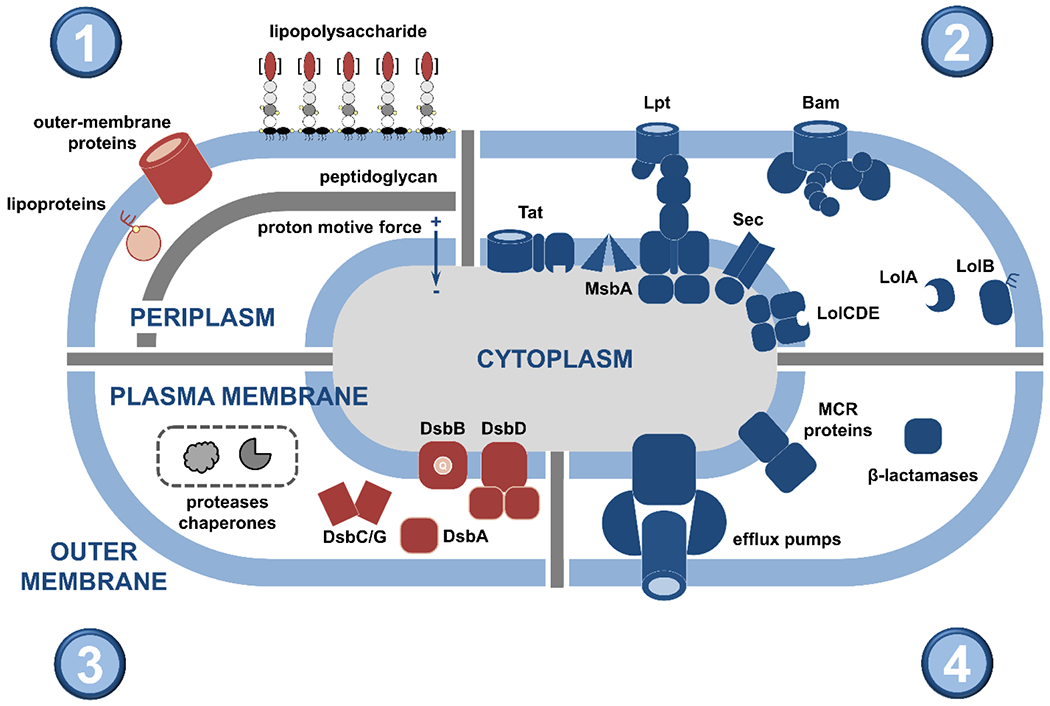
This review article discusses the potential of the constituents and processes depicted here as targets for the generation of novel strategies against bacterial pathogens. (1; top left) The cell envelope is composed of the outer membrane, the peptidoglycan layer, and the plasma membrane. The asymmetric outer membrane is composed of glycolipid lipopolysaccharide and phospholipids that are located in its extracellular and periplasmic leaflets, respectively. Embedded within this lipidic environment, β-barrel proteins aid nutrient transport and act as receptors, and lipoproteins perform a range of functions often linked to the biogenesis of the cell envelope. The rigid peptidoglycan layer is attached to the outer membrane, helping the cell retain its shape against a variety of stresses. Separating the periplasm from the cytoplasm, the phospholipid-rich plasma membrane is a platform for numerous cellular processes that connect the cell interior with the extracellular environment, many of which are energized by the proton motive force. (2; top right) Cell envelope biogenesis and function rely on the translocation of newly synthesized proteins across the plasma membrane by two translocation systems, the general secretory pathway (Sec) and the twin arginine pathway (Tat). Their substrates are inserted into the plasma membrane, released into the periplasm, extruded to the extra-cellular space, or passed on to other protein partners for further processing. The latter includes the Lol pathway, which is involved in the biogenesis of lipoproteins, and the Bam pathway that is responsible for the localization and folding of outer-membrane β-barrels. Generation and transport of the lipopolysaccharide occurs separately and relies on specialized cytoplasmic machinery, the flippase MsbA (which moves the lipopolysaccharide precursor to the periplasmic leaflet of the plasma membrane) and the Lpt pathway. (3; bottom left) Cell envelope biogenesis, maintenance, and homeostasis rely on complex quality control networks. Major players include soluble chaperones, involved in shuttling proteins and lipids across the periplasm to their final destinations, proteases that degrade misfolded and aggregated proteins, and members of the disulfide bond formation pathway (DsbA-D shown here) that perform oxidative protein folding. (4; bottom right) Many broad-acting antimicrobial resistance determinants are located in the cell envelope, such as β-lactamase enzymes that break down antibiotics like penicillin, mobile colistin resistance (MCR) proteins, which protect the cell against last-resort polymyxin drugs, and efflux pumps, responsible for extruding many different classes of antibiotics.
The glycolipid outer membrane of the cell envelope acts as a selective barrier that prevents cellular entry of a wide range of molecules. In addition, the rigid peptidoglycan cell wall, attached to the periplasmic leaflet of the outer membrane, offers bacteria protection against mechanical stresses. Between these structures and the plasma membrane lies the periplasm, home to a plethora of stress response pathways, virulence factors, and antimicrobial resistance determinants that support bacterial pathogenesis and survival. The cell envelope is, therefore, likely the primary biological reason for the lack of progress in the discovery of new antibiotics against Gram-negative bacteria. That said, this compartment and its components also represent an attractive assortment of unexploited targets for the development of first-in-class antibiotics or adjuvant and potentiator compounds that could help safeguard and prolong the lifespan of existing antimicrobials. This review aims to provide an overview of these targets and discuss the state of the art in discovering inhibitors against them (Figure 1); the approaches we discuss here are focused on Gram-negative species, however for select strategies that have also been successful against critical Gram-positive pathogens, we expand on such successes.
2. OUTER AND PLASMA MEMBRANES
2.1. Targeting the outer membrane
One of the distinguishing features of Gram-negative bacteria is their asymmetric outer membrane, which contains glycolipids in its outer leaflet and phospholipids in the inner leaflet (Silhavy, Kahne, & Walker, 2010). This lipidic platform supports many structures that are essential for nutrient uptake and bacterial survival, including proteins (lipoproteins and β-barrel proteins), oligosaccharides, and species-specific surface receptors. Correct assembly and maintenance of the outer membrane relies on the activity and interactions of several membrane-embedded and periplasmic proteins, which jointly provide an extensive landscape for the discovery of novel therapeutics. This section reviews methods that are already in use or hold promise for targeting the outer membrane, either through inhibition of key proteins or simply through its permeabilization. Many of these strategies rely on direct interactions between the anti-infective and the bacterial surface. While compounds that disrupt the cell envelope from the inner, periplasmic leaflet of the outer membrane are also theoretically useful, they have the same drawbacks as therapeutics that target processes in the cell interior, starting with the challenge of crossing the outer membrane.
2.1.1. Lipopolysaccharide (LPS).
The main component of the outer glycolipid leaflet is the LPS, a charged lipopolysaccharide that forms a permeability barrier for many small molecules and serves as an intrinsic resistance mechanism against antimicrobials and host defense systems (Bertani & Ruiz, 2018). The prototypical LPS molecule consists of lipid A, which forms the hydrophobic layer of the outer membrane, the oligosaccharide sugar core, and the polysaccharide O-antigen component attached to the most distal end of the LPS from the bacterial surface; part of the lipid A component, the core, and the O-antigen are all surface exposed (Raetz & Whitfield, 2002). Its localization at the surface of the bacterium, along with its key role in maintaining cellular integrity, makes LPS a highly coveted target against Gram-negative pathogens. Several avenues can be exploited to that effect, including direct LPS binding as well as inhibition of its synthesis and assembly (for the latter see section 3.1).
Approved antibiotics.
FDA-approved compounds such as the polypeptides gramicidin or the polymyxins interact with LPS to permeabilize the outer membrane (C. H. Chen & Lu, 2020). A prototypical example of such drugs is the last-resort antibiotic colistin, a polymyxin compound that is attracted to the negative charge of lipid A on the cell surface. This charge interaction allows colistin to insert its lipophilic tail into the outer membrane, disrupt this first cell envelope barrier and enter the periplasm (Andrade, Silva, Rodrigues, & Pina-Vaz, 2020). There, it targets newly synthesized lipid A in the plasma membrane and lyses the cell (Sabnis et al., 2021). Naturally, modifications of the lipid A charge can disrupt this interaction of LPS with colistin and lead to resistance (see section 6.2) (Manioglu et al., 2022; Sabnis et al., 2021). Similar changes in the charge or structure of the outer membrane inactivate many LPS-binding drugs and present a major challenge in the development of new LPS-targeting antimicrobials. Multidrug combination therapies can be employed to bypass this problem and to prolong the usability of some of these last-resort therapeutics. For example, administration of colistin in conjunction with the glycopeptide vancomycin, which is normally only effective against Gram-positive bacteria, clears A. baumannii that is resistant to both monotherapies in a Galleria mellonella infection model, likely due to colistin allowing vancomycin to traverse the outer membrane (O’Hara et al., 2013). Exploitation of similar strategies is being considered for other promising compounds that target processes in the cell interior but are not efficient in crossing the outer membrane, so as to expand their efficacy and decrease the potential for resistance evolution.
Antimicrobial peptides (AMPs).
Another avenue for outer-membrane disruption is offered by antimicrobial peptides. Their hydrophobic, amphipathic, and often positively charged nature promotes strong interactions with the membrane, rapidly resulting in defects and pores that cause periplasmic leakage (Cesare, Cristy, Garsin, & Lorenz, 2020). Their efficient mode of action along with their broad structural and functional diversity (Moretta et al., 2021; Y. Sun & Shang, 2015), make resistance to AMPs harder to evolve, a highly desirable attribute in novel therapeutic approaches (Bechinger & Gorr, 2017; Rodríguez-Rojas, Moreno-Morales, Mason, & Rolff, 2018). Many naturally occurring peptides are active against Gram-negative bacteria (e.g., colimycin-methosulfate, α- and β-defensin, tachyplesin, bactenecin, cecropin A, indolicidin, and nisin (Cesare et al., 2020) and, encouragingly, pexiganan is currently progressing through Phase III clinical trials for use in diabetic foot ulcers (Barreto-Santamaría, Arévalo-Pinzón, Patarroyo, & Patarroyo, 2021; Gottler & Ramamoorthy, 2009). While most AMPs are positively charged, select anionic peptides can also achieve membrane penetration by interacting with the outer membrane through salt bridges mediated by cationic metals, such as Zn2+ (Brogden, 2005; Jeżowska-Bojczuk & Stokowa-Sołtys, 2018). On this front, combination of the anionic peptide GMAP2 with G. mellonella lysozyme affects cell shape, decreases the cellular turgor and increases membrane permeabilization in E. coli and K. pneumoniae (Zdybicka-Barabas et al., 2012); similar effects have also been reported with hemocidin (Meister et al., 2006) as well as HD-1, −2, and −3 (Schnapp & Harris, 1998).
It should be noted that despite their potential, a downside of peptide-based antimicrobials is that they are often cytotoxic. For example colistin, although clinically used, is nephrotoxic (Gai, Samodelov, Kullak-Ublick, & Visentin, 2019), and the promising experimental AMP murepavidin, which is co-administered with standard of care drugs, failed Phase III clinical trials due to safety concerns (Prasad, Seiple, Cirz, & Rosenberg, 2022) (clinical trial numbers NCT03582007 and NCT03409679, https://clinicaltrials.gov/). Notably, an inhaled formulation of murepavidin is currently undergoing Phase I trials as a treatment against Pseudomonas aeruginosa, and the first-in-human trials are showing promising safety results (https://spexisbio.com/impv/). There are ongoing efforts to decrease AMP off-target effects through judicious use of protective adjuvants like cilastatin (Gai et al., 2019) or through development of less toxic derivatives, like the nonapeptide SPR2067 that is expected to soon enter Phase II clinical trials (Reig, Gouellec, & Bleves, 2022; Spero). Somewhat counterintuitively several AMPs, including epinecidin-I (EPI-I), have been implicated in the dampening of LPS-associated adverse effects, for example exaggerated immune system responses due to host-LPS interactions (Chee et al., 2019). Although these peptides cannot be used as anti-infectives, their properties might be beneficial in combination therapies, whereby use at lower concentrations could harness positive pharmacokinetic effects without unacceptable toxicity levels, as was hoped for during the first unsuccessful murepavidin clinical trials.
2.1.2. Outer-membrane β-barrel proteins.
Outer-membrane proteins, like OmpA, OmpF, OmpT, FepA, or PhoE (Fairman, Noinaj, & Buchanan, 2011; Galdiero et al., 2012), have β-barrel structures that are comprised of 8 to 24 β-strands and form water-filled channels lined with charged amino acids. This allows them to differentiate molecules of a certain size, shape, or charge (Achouak, Heulin, & Pagès, 2001), thus modulating molecule uptake and extrusion, cell adhesion, and membrane biogenesis (for the latter see section 3.3) (Fairman et al., 2011). Sub-optimal function of these proteins deregulates the cell envelope and leads to cell death (Imai et al., 2019; Kaur et al., 2021). In some cases though, it can also cause antibiotic resistance through reduced drug access into the bacterium, as seen in Pseudomonas spp. where mutations in or deletion of the OprD porin block entry of carbapenem β-lactam antibiotics (Ochs, McCusker, Bains, & Hancock, 1999). In similar way, mutations in the major non-specific porin OmpK36 of K. pneumoniae support the function of the β-lactamase KPC-2 and afford increased resistance to carbapenems, while causing a significant fitness cost for the resistant mutants (Wong et al., 2019). Although β-barrel diversity complicates their categorization, it also highlights their critical role in cell function and survival and justifies the fact that their inhibition is considered a promising strategy for the generation of novel antimicrobials.
Inhibition of β-barrel proteins.
The well characterized, non-specific porin OmpA is a non-covalent peptidoglycan anchor (Samsudin, Ortiz-Suarez, Piggot, Bond, & Khalid, 2016) with a key role in virulence, biofilm formation, and host-adhesion in several Gram-negative pathogens (Choi & Lee, 2019). Its inhibition with the cyclic hexapeptide AOA-2 impedes A. baumannii, P. aeruginosa, and E. coli adhesion and improves survival in a murine infection model (Vila-Farrés et al., 2017). Moreover, ompA gene suppression through the use of the repressor 62520 sterilizes A. baumannii infections (Na et al., 2021). Similarly, polyamine compounds (e.g., spermine, spermidine, and cadaverin) block the E. coli OmpF and OmpC porins in vitro (delaVega & Delcour, 1995; Iyer & Delcour, 1997). Furthermore, targeting the largely conserved structure of β-barrel porins (Chaturvedi & Mahalakshmi, 2017) might be a promising route for inhibiting multiple cell envelope pathways. This strategy has been shown to be possible through the discovery of a protegrin I peptidomimetic, JB-95, which shows activity against two β-barrel proteins involved in membrane biogenesis (LptD and BamA; see section 3.1.4 and 3.3, respectively) (Urfer et al., 2016). Despite these efforts, no porin inhibitor has been developed into a clinically used therapeutic to date.
Promoting uptake through β-barrel proteins.
The activity of many current antibiotics relies on porin-mediated outer membrane crossing. However, for critical pathogens like P. aeruginosa and A. baumannii mutations affecting porin function occur readily due to several non-specific porins being dispensable, and this leads to reduced drug uptake and resistance (Ude et al., 2021). As such, in addition to strategies blocking porin function (see previous section), efforts are also underway to promote porin-mediated uptake in order to increase the efficacy of antibiotics (Pandeya et al., 2021). To that effect, small molecule potentiators could be designed to enhance drug uptake of already approved antibiotics. In addition, carboxylic acid groups, that are often included in antibiotic scaffolds to aid solubility have been found to impede porin transport (Ude et al., 2021), and selection of another ‘solubility enhancing’ moiety should be considered during the design of future antimicrobials in order to aid the transport of novel drugs across the membrane (Ballatore, Huryn, & Smith, 2013).
Overall, current efforts indicate that porin inhibitors are unlikely to find generalized use in combination therapy. Any potential monotherapy uses will require thorough optimization to achieve clinically relevant inhibition levels for pathogens that are known to fare well without functional porins (Zgurskaya & Rybenkov, 2020). Notably, porins like NspA, which is involved in host immune system evasion in Neisseria spp., might also be suitable species-specific targets for vaccine development (Lewis, Rice, & Ram, 2019).
2.1.3. Outer-membrane lipoproteins.
Lipoproteins represent the second type of protein found in the outer membrane and are anchored to the lipid bilayer through a characteristic lipophilic tail. They perform a diverse set of functions and, along with β-barrel proteins, are accessible proteinaceous targets for novel therapies (Galdiero et al., 2012; Rayes, Rodríguez-Alonso, & Collet, 2021). More than 90, predominantly outer-membrane, lipoproteins have been described in E. coli (Okuda & Tokuda, 2011). Some of these are required for the biogenesis of the cell envelope, such as the peptidoglycan anchoring protein Lpp (section 3.2), BamB and BamD that are involved in β-barrel assembly (section 3.3), or LptE, which is required for the correct assembly of the essential outer-membrane β-barrel LptD (section 3.1) (Chimalakonda et al., 2011). Their inhibition can indirectly disrupt outer membrane integrity and lead to cell death; for clarity, strategies against these proteins will be discussed within their respective biochemical pathways (see section 3).
2.1.4. Molecules that facilitate outer-membrane disruption through unknown mechanisms.
There are a few noteworthy examples of species-specific antimicrobial approaches that are generally related to outer membrane perturbation, but whose mechanism of action remains unclear. For example, a quinazolone thiazole compound was found to compromise E. coli and P. aeruginosa membranes and was used successfully as an antibacterial in conjunction with the fluoroquinolone norfloxacin (J. Wang, Ansari, & Zhou, 2021). Similarly, topical application of 2-phenoxyethanol, potentiated by the preservative ethylhexylglycerin, exhibits promising bactericidal properties linked to membrane disruption (Langsrud et al., 2016).
Evidently, direct inhibition of key outer-membrane components or disruption of the membrane barrier have generated invaluable clinical therapies and continue to provide a basis for the development of future antimicrobials against Gram-negative pathogens. Nonetheless, antibacterial strategies against the outer membrane still face major challenges. For example, target accessibility is often an issue and can be limited either by steric inhibition or incompatibility between the biophysical properties required for optimal drug uptake versus attributes that drive target inhibition. Additional problems stem from rapid target evolution or drug toxicity in in vivo trials. Some of these issues can be addressed through combination therapies that have the advantage of increased antibacterial activity achieved through inhibition of multiple targets, decreased toxicity, and lower potential for resistance evolution.
2.2. Targeting the plasma (inner) membrane
The bacterial plasma membrane separates the cytoplasm from the periplasm and is similar to the inner leaflet of the outer membrane in that it is composed primarily of phosphatidylglycerol (PG), phosphatidylethanolamine (PE) and cardiolipin (CL) (Epand, Walker, Epand, & Magarvey, 2016). Additional lipid types, including phosphatidyl serine or polyisoprenoid carriers, are also found in the plasma membrane, but in smaller amounts and, often, only in specific bacterial species (Epand et al., 2016). In addition to the lipids, the plasma membrane contains many other antibiotic targets, like protein complexes involved in cell envelope biogenesis and protein translocation (see section 3) or in the generation of membrane potential.
2.2.1. Plasma-membrane phospholipids.
Permeabilization of the plasma membrane can be achieved by the same compounds that disrupt the outer membrane. For example, cationic AMPs target anionic lipids like PG and CL (Epand & Epand, 2011), while colistin interacts with the lipid A molecule that is newly synthesized in the plasma membrane as it awaits transport to the outer membrane (Sabnis et al., 2021). However, targeting of the lipids in the Gram-negative plasma membrane is not straightforward because of the protective presence of the outer membrane (see section 2.1), and thus has been successfully used mostly against Gram-positive species. More recent strategies have exploited the use of siderophore-antibiotic conjugates to potentiate classically Gram-positive drugs, such as daptomycin (Ghosh et al., 2017), by circumventing the outer membrane to allow use against Gram-negative pathogens.
Phosphoatidylethanolamine (PE).
PE is the major E. coli phospholipid accounting for ~75% of the cell envelope lipid content (Bogdanov et al., 2020; Langley, Hawrot, & Kennedy, 1982). It localizes asymmetrically, favoring the periplasmic leaflets, where it forms the PE-enriched inner leaflet of the outer membrane or acts as a substrate for lipoprotein acylation and lipid A ethanolamine modification in the plasma membrane (Bogdanov et al., 2020; Henderson et al., 2015). PE content in the plasma membrane is in equilibrium with other lipid components. As a result, an increase in PE leads to lower levels of cardiolipin and vice versa, making PE an appropriate target for disrupting lipid homeostasis and, therefore, the integrity of the plasma membrane (Bogdanov et al., 2020). Accordingly, PE has been highlighted in the literature for its high affinity for cationic AMPs, including the class I type B lantibiotic cinnamycin (Sahl, Jack, & Bierbaum, 1995) and duramycin (Fredenhagen et al., 1990).
Cardiolipin (CL).
CL is commonly organized in anionic microdomains that localize at the cell poles and the division sites of prokaryotic and eukaryotic plasma membranes (Khoury et al., 2017; T.-Y. Lin & Weibel, 2016). CL microdomain formation is associated with the correct function of the respiratory chain and other membranous protein complexes (Khoury et al., 2017; T.-Y. Lin & Weibel, 2016). Localization of CL on the less accessible, cytoplasmic side of eukaryotic membranes opens the possibility of selectively targeting the periplasmically accessible CL on bacterial membranes (Epand et al., 2016). The potential of this strategy is demonstrated by the finding that 3’,6-dinonyl neamine interacts with the cardiolipin headgroups and changes the morphology of the P. aeruginosa plasma membrane and, in particular, decreases its fluidity while increasing its permeability (Khoury et al., 2017). Other compounds that promote CL-dependent cell death in Gram-negative bacteria include the phospholipid alcohol sphingosine (Verhaegh, Becker, Edwards, & Gulbins, 2020) and cyclic R-, W- rich hexapeptides (Junkes et al., 2008; Scheinpflug, Krylova, Nikolenko, Thurm, & Dathe, 2015). Targeting CL naturally disrupts the function of the respiratory chain, deregulates the chemical proton gradient, or affects other important processes, such as cell wall synthesis, through CL associated proteins; collectively these pleiotropic effects have been highlighted as a great advantage of strategies that exploit the central role of CL for the development of broad-spectrum antibacterials (T.-Y. Lin & Weibel, 2016; Swain et al., 2018).
Phosphatidylglycerol (PG).
In contrast to Gram-positive bacteria, PG is less prevalent in Gram-negative plasma membranes, and as such, its inhibition has not been as thoroughly investigated. It has, however, been identified as the source of acyl chains in the plasma membrane and recognized as critical for cell survival (Sankaran & Wu, 1994). The Gram-positive antibiotic daptomycin interacts with anionic PG to stall peptidoglycan synthesis, causing membrane rearrangement and depolarization (Grein et al., 2020; Silverman, Perlmutter, & Shapiro, 2003). This mode of action has also been exploited in a siderophore-daptomycin conjugate that inhibits growth of multidrug-resistant A. baumannii (Ghosh et al., 2017).
Finally, although mechanisms behind the biogenesis and maintenance of the plasma membrane have yet to be fully elucidated, known key players might be valuable targets for the design of future antimicrobials. For more information on these, as well as a more in-depth review on the extensive functions of plasma-membrane lipids in essential cellular processes, we refer the reader to Lin & Weibel (T.-Y. Lin & Weibel, 2016) and Sohlenkamp & Geiger (Sohlenkamp & Geiger, 2016).
2.2.2. Proton motive force (PMF).
The proton motive force, comprising the membrane electric potential (ΔΨ) and the transmembrane chemical proton gradient (ΔpH), powers several essential plasma-membrane-embedded protein apparatuses (Strahl & Hamoen, 2010). Coordination between ΔΨ and ΔpH is crucial for maintaining the constancy of the PMF under stress, therefore protecting the bacterial cell from metabolic perturbations and growth inhibition that would eventually lead to cell death (Bakker & Mangerich, 1981; Farha, Verschoor, Bowdish, & Brown, 2013). Directly targeting the PMF has long been considered undesirable, because such strategies could result in eukaryotic cell toxicity or generate pleiotropic effects that confound the mechanisms of action of PMF-targeting compounds (Gentry et al., 2010; Singh, 2006), as seen with the ionophore carbonyl cyanide-m-chlorophenylhydrazone (CCCP) that was long thought to be an efflux inhibitor (Novo, Perlmutter, Hunt, & Shapiro, 2000; Sekyere & Amoako, 2017; Thakur, Uniyal, & Tiwari, 2021). Nonetheless, disruption of energy generation has been identified as one of the few ways to successfully target and eradicate bacterial persister cells (Hurdle, O’Neill, Chopra, & Lee, 2011). This attribute of PMF inhibitors which is rarely achieved by other antibiotics, together with the PMF-dissipating properties of many already approved antibiotics (e.g., aminoglycosides (Bruni & Kralj, 2020) or trimethoprim (Feng et al., 2015)), have highlighted the PMF as an underexploited target and opened it to antibiotic discovery (Farha et al., 2013; Hurdle et al., 2011). It should be noted that compounds responsible for membrane depolarization can often concurrently disrupt membrane permeability; for example the lipid II binder telavancin inhibits cell wall synthesis, while also causing rapid membrane depolarization (Higgins et al., 2005). Conversely, membrane hyperpolarization also has bactericidal effects, like in the case of antibiotics that inhibit protein synthesis, such as aminoglycosides, which cause hyperpolarize the membrane, resulting in metabolic arrest, pore formation, increased antibiotic uptake and eventually cell death (Bruni & Kralj, 2020).
Membrane electric potential (ΔΨ).
Compounds targeting the PMF have been identified accidentally during high-throughput screens against other plasma-membrane targets, including the twin arginine (Tat) protein translocation system or efflux pumps (Bageshwar et al., 2016). This includes the molecule tetrandrine, that was identified as an inhibitor of mobile colistin resistance proteins (Yi et al., 2022), or the hypertension drug verapamil (Pohl, Krylov, Block, & Pohl, 1998), that is approved for use against Mycobacterium tuberculosis (C. Chen et al., 2018) and has been thought to be a multidrug and toxin extrusion pump inhibitor (S. Gupta et al., 2014; Radchenko, Symersky, Nie, & Lu, 2015; A. Sharma, Gupta, & Pathania, 2019). Recent research on verapamil showed that its interaction with plasma-membrane lipids causes concentration-dependent structural and electrical perturbations that result in dissipation of the membrane potential (C. Chen et al., 2018; Meier, Blatter, Seelig, & Seelig, 2006). As maintenance of energized membranes is critical for the survival of M. tuberculosis, the rapid loss of membrane potential is likely responsible for cell death and potentiation of several M. tuberculosis-specific drugs (C. Chen et al., 2018; Hurdle et al., 2011). This mechanism of action is in agreement with previous observations of verapamil-liposome interactions, while efflux inhibition is likely a downstream effect caused by the dissipation of the PMF (Pohl et al., 1998). To date, verapamil is one of the very few FDA-approved drugs directly targeting the PMF.
Electron transport chain.
The membrane potential can also be disrupted through targeting enzymes or cofactors that are critical components of the electron transport chain. For example, species-specific type II NADH dehydrogenase enzymes catalyze the essential transfer of electrons from NADH to quinone acceptors. Seven compounds targeting these enzymes and acting as indirect PMF inhibitors have been discovered for E. coli (Bageshwar et al., 2016), along with phenothiazine compounds with specific activity against M. tuberculosis enzymes (Weinstein et al., 2005). Terminal oxidases have also been proposed as promising targets as seen with the combination of Telacebec and ND-011992 that jointly inhibit two redundant cytochrome bd-type oxidases of M. tuberculosis (B. S. Lee et al., 2021) or with the previously mentioned outer-membrane disruptor gramicidin. Along the same line, disruption of the PMF through targeting of membrane-embedded cofactors results in rapid cell lysis, as seen with lysocin E that binds menaquinone (Hamamoto et al., 2015), Ro-48-8071(a high cholesterol treatment) which inhibits ManA in the first step of menaquinone synthesis (Dhiman et al., 2009), and GSK1733953A (DG70), a molecule that interrupts the final menaquinone synthesis step through interaction with MenG (Pujari, Rozman, Dhiman, Aldrich, & Crick, 2022).
Transmembrane chemical proton gradient (ΔpH).
Unlike the electron-reliant component of the PMF, its chemical gradient element uses protons to generate energy. DCAP disrupts this PMF component and causes mislocalization of essential, membrane-associated proteins involved in cell division, such as MinD and FtsA (Eun et al., 2012). Like other PMF-targeting molecules, DCAP is successful in eradicating bacterial cells in deep stationary phase or persisters, and in sterilizing biofilms. Conveniently, DCAP does not exhibit the classical cytotoxicity profile usually associated with other PMF inhibitors, but further research is required to overcome bacterial efflux activity that is already reported during it use against E. coli and P. aeruginosa (Eun et al., 2012). Another non-toxic and highly promising inhibitor of the proton gradient, IITR08027, was initially identified as an efflux inhibitor (section 6.3) and fluoroquinolone potentiator in A. baumannii (Bhattacharyya, Sharma, Akhter, & Pathania, 2017). Moreover, recently, improved understanding of neural network models enabled in silico identification of novel chemical scaffolds and allowed reassessment of FDA-approved drugs for potential antibacterial or adjuvant properties. This process led to the investigation of halicin, an approved drug for diabetes that showed potent activity against both Gram-positive and Gram-negative bacteria and was effective in murine infection models for A. baumannii and C. difficile (Stokes et al., 2020). Halicin’s antimicrobial properties were found to stem from dissipation of the proton gradient, which explained its broad-spectrum of activity. Finally, the most successful chemical gradient inhibitors identified to date are diarylquinoline compounds that target the proton pump adenosine triphosphate synthase with bedaquiline (TMC207) now approved for use against multidrug-resistant M. tuberculosis (Matteelli, Carvalho, Dooley, & Kritski, 2010; Worley & Estrada, 2014).
Overall, PMF-targeting compounds seem to be a promising class of next-generation therapeutics. Their utility has been expanded by recent observations of the critical role of active PMF maintenance in bacterial species that exhibit phenotypic and transient antibiotic resistance (M. Wang, Chan, Wan, Wong, & Chen, 2021). For example, induced adaptive resistance of P. aeruginosa to aminoglycosides can be ameliorated by triclosan, which disrupts the membrane potential (Maiden & Waters, 2020). The promise of PMF inhibition is further supported by studies showing that disruption of the membrane potential and the proton gradient independently, through use of two synergizing compounds, decreases eukaryotic toxicity and reduces resistance selection (Bakker & Mangerich, 1981; Farha et al., 2013). This progress in PMF inhibition, combined with the fact that most antibiotics fail to eradicate persisters, slow-growing bacterial cells exhibiting transient resistance phenotypes, as well as pathogens with fully active PMF-dependent resistance determinants (e.g., efflux pumps; see section 6.3), makes the PMF a highly desirable target for future antibiotic, adjuvant, and potentiator therapies.
2.2.3. Cellular turgor.
The capacity of bacteria to adapt to drastically different environments relies on their ability to control the osmolarity in their cell’s interior, so as to maintain periplasmic and cytoplasmic homeostasis (Rojas & Huang, 2018). Changes in extracellular osmolarity are counteracted at the plasma membrane through strict regulation of the transport of potassium ions, glutamate, betaine, and proline (Robin Wray, Wang, Blount, & Iscla, 2022). Under osmotic stress, high internal turgor is corrected by a gated channel, MscL, that allows rapid release of molecules of up to 30Å from the cytoplasm (Balleza & Gómez-Lagunas, 2008; Blount & Iscla, 2020; Robin Wray et al., 2022). The low tolerance of MscL to structural changes (Iscla, Levin, Wray, & Blount, 2007; Iscla, Wray, & Blount, 2008; Robin Wray et al., 2015), combined with its constitutive expression, high conservation, and absence of a known mammalian orthologue (Barh et al., 2011; Booth & Blount, 2012), make this transporter a promising target; its inhibition would dysregulate bacterial growth, division and integrity, and potentiate existing antibiotics (Robin Wray, Herrera, Iscla, Wang, & Blount, 2019; Robin Wray et al., 2015; Robin Wray, Iscla, Kovacs, Wang, & Blount, 2019; Robin Wray et al., 2022; Robin Wray, Wang, Iscla, & Blount, 2020).
Several compounds targeting MscL have been developed, including the small molecule 011A which forces the channel open (Robin Wray, Herrera, et al., 2019; Robin Wray, Iscla, et al., 2019), the bacteriocin sublancin 168 (Kouwen et al., 2009), and the first-in-class drug ramizol that is expected to enter Phase I clinical trials for the treatment of C. difficile (Rao et al., 2016). In addition, a few approved and clinically used antibiotics have now been shown to use MscL to reach their cytoplasmic targets (Robin Wray, Herrera, et al., 2019), including the aminoglycoside antibiotic streptomycin; this explains the previously confounding observation of rapid decrease in potassium ion concentration during streptomycin treatment (Dubin, Hancock, & Davis, 1963; Iscla, Wray, Wei, Posner, & Blount, 2014). This observation prompted the development of dual-function compounds (similar to cefiderocol, see section 4.1.2) and led to SCH79797, a folate pathway inhibitor with activity against both Gram-positive and Gram-negative bacteria. This molecule contains a cumene moiety, which allows it to access the cytoplasm through MscL (Martin et al., 2020). Further development to improve the cytotoxicity profile of SCH79797 resulted in the generation of a derivative, Iresistin-16 (IRS-16) (Martin et al., 2020), and opened new avenues for future targeting of both the plasma membrane and the cytoplasm. In addition to allowing antibiotic access to the cytoplasm, research shows that forcing MscL open disrupts the membrane potential (Walton, Idigo, Herrera, & Rees, 2015; R. Wray, Iscla, & Blount, 2021), something that would enhance the activity of antibacterial compounds using it for access. Finally, a smaller plasma-membrane channel, MscS, plays a minor role in cellular turgor correction similar to MscL (Levina et al., 1999; Robin Wray et al., 2022). This partial functional redundancy makes compounds targeting such channels more suitable as antibiotic potentiators or as components of dual-function drugs, rather than bactericidal agents.
3. BIOGENESIS OF THE CELL ENVELOPE
Targeting of the membranous components of the cell envelope has led to the discovery of invaluable antibiotics and antibacterial strategies. Cell envelope biogenesis pathways, responsible for the processing, modification, and transport of the constituent components of this compartment, also provide a wide range of promising antibiotic targets. Key proteins and lipids making up the cell envelope need to be transported from the cytoplasm to the periplasm or integrated into the plasma and outer membranes. For proteins, crossing of the plasma membrane is driven by two highly conserved systems, the general secretory (Sec) system and the twin arginine (Tat) pathway (see section 3.4). The transport of lipids across the plasma membrane barrier is performed by separate dedicated machineries (and discussed in more detail in multiple sections of this review that are pertinent to their biogenesis). After translocation into the periplasm, precursors of outer-membrane components are safely shuttled through a network of chaperones and other accessory proteins to their destination. This section will provide a brief overview of the enzymes involved in the biogenesis and transport of cell envelope components, i.e., the LPS, lipoproteins and outer-membrane proteins, and discuss their potential as druggable antibiotic targets. The role of the chaperone and protease network in these processes is explored separately in section 5.
3.1. Targeting the biogenesis of the LPS
A schematic overview of the processes discussed in this section can be found in Figure 2.
Figure 2. Overview of the lipopolysaccharide (LPS) biogenesis pathway.
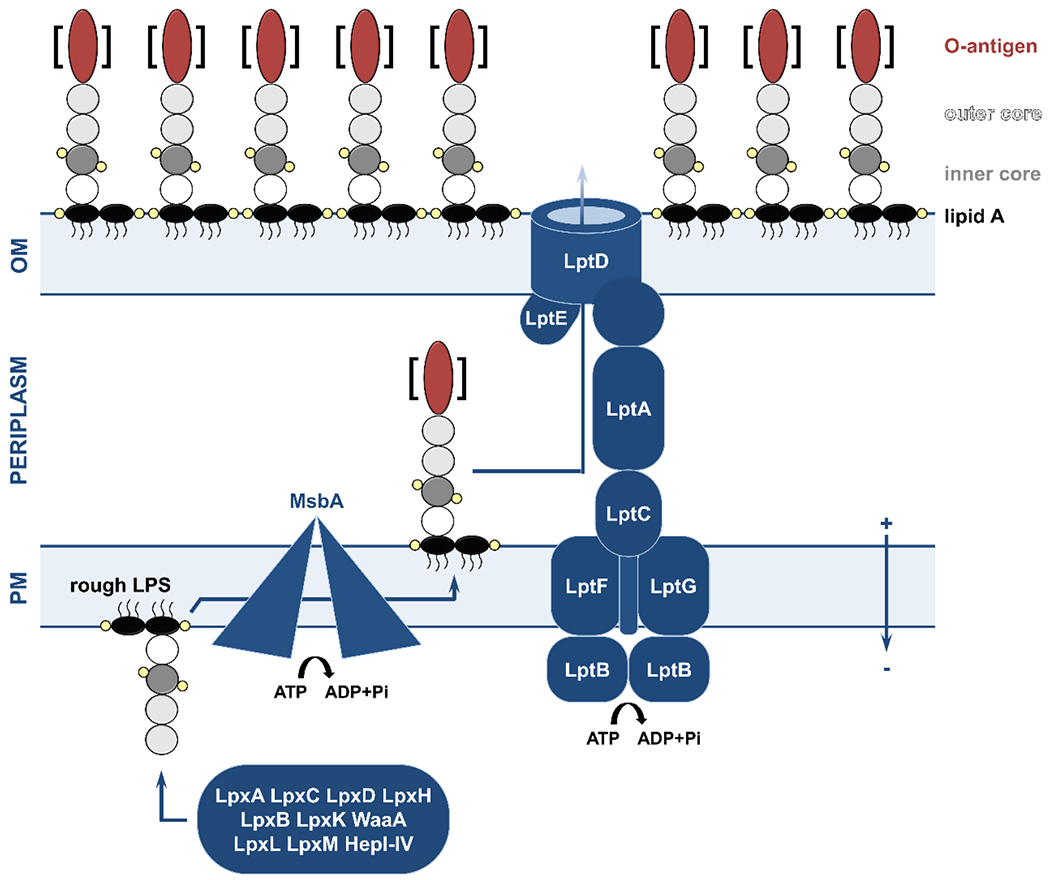
LPS synthesis begins in the cytoplasm, where the successive function of the acetylase-deacetylase pairs LpxA-LpxC and LpxD-LpxH, the disaccharide synthase LpxB, and the lipid kinase LpxK produce a lipid A precursor, lipid IVA (black ovals; small yellow circles indicate phosphate groups). Once inserted in the membrane, WaaA, LpxL, and LpxM attach the Kdo sugars (white circle), while the heptosyltransferases HepI-IV add the outer core component (dark and light gray circles, respectively). The rough LPS molecule is then flipped to the periplasmic leaflet of the plasma membrane by the ATP-dependent MsbA transporter. In some species, the cytoplasmically synthesized O-antigen part (red oval, synthesis pathway is omitted for clarity) is attached by WaaL (the brackets denote that some bacterial strains lack the O-antigen). Translocation of the LPS molecule across the periplasm is performed by the LptB2FGC ATP-dependent complex which pushes the LPS across an LptA bridge to the outer membrane LptDE translocon complex. LPS is then incorporated into the extracellular leaflet of the outer membrane. OM, outer membrane; PM, plasma membrane.
3.1.1. Synthesis of lipid A precursors at the plasma membrane.
The biogenesis of the LPS starts at the inner leaflet of the plasma membrane with the generation of the lipid A precursor, lipid IVA as part of the Raetz pathway. This process involves the formation of lipid X from a UDP-GlcNAc precursor via two conserved acylation-deacetylation steps (Whitfield & Trent, 2014). The first step is catalyzed by the LpxA (Anderson & Raetz, 1987) - LpxC (Jackman, Raetz, & Fierke, 1999) cytoplasmic complex, while the second step is performed by the cytoplasmic protein LpxD (Bartling & Raetz, 2008) and its membrane-associated partner LpxH (Babinski, Ribeiro, & Raetz, 2002). A disaccharide precursor is then formed by the introduction of a β-1’-6 glycosidic linkage connecting Lipid X to a UDP-2,3-diacylGlcN molecule, carried out by the peripheral membrane protein LpxB (Radika & Raetz, 1988; Simpson & Trent, 2019; Whitfield & Trent, 2014). The last step of this process relies on a protein that is embedded in the plasma membrane, LpxK, which phosphorylates the disaccharide at the 4’ position to produce lipid IVA (Garrett, Kadrmas, & Raetz, 1997).
The acetyltransferases LpxA and LpxD.
Targeting the acetylation steps of LPS synthesis has historically been hampered by insufficient mechanistic understanding of this process and lack of straightforward activity assays for monitoring the efficacy of putative inhibitor molecules. Despite these limitations, several compounds capable of competitive and non-competitive LpxA binding have been described (W. Han et al., 2020; M. D. Ryan et al., 2021), while LpxD has been validated as the target of the small-molecule inhibitor 6359-0284 (X. Ma et al., 2020). Small molecule- and peptide-based dual-action inhibitors that target both acetyltransferases of P. aeruginosa (Jenkins & Dotson, 2012; Kroeck et al., 2019) and A. baumannii (Fereshteh et al., 2022) have also been discovered, and this strategy has been highlighted to have increased efficacy and decreased mutational resistance.
The metallo-deacetylases LpxC and LpxH.
The zinc-dependent protein LpxC is widely conserved across Gram-negative species and is, therefore, a target worthy of consideration. Concentrated efforts have led to the identification of numerous small-molecule inhibitors against this enzyme with activity against several critical pathogens, like E. coli, P. aeruginosa, and Enterobacter cloacae (Erwin, 2016; Kalinin & Holl, 2017; Mdluli et al., 2006; Onishi et al., 1996). Great promise was reported for hydroxamic acid-containing compounds that bind on the LpxC active-site zinc metal center. The most advanced of these molecules, ACHN-975, entered and, unfortunately, failed Phase I clinical trials (Achaogen, 2013; Kalinin & Holl, 2017; Mansoor et al., 2011). Despite this setback, another zinc-binding molecule, the biphenylacetylene-based inhibitor LPC-233, is currently being investigated as a candidate for Phase I trials (RePORT). Finally, an indirect LpxC intervention strategy through the disruption of the YciM/FtsH complex, which is responsible for LpxC degradation (Mahalakshmi, Sunayana, SaiSree, & Reddy, 2014), has been proposed, and another LpxC inhibitor, TP0586532, was shown to potentiate β-lactam antibiotics through partial permeabilization of the outer membrane (Yoshida, Takata, Fujita, Takashima, & Sugiyama, 2022).
In contrast to LpxC, which is highly conserved, there is a significant variation in the second deacetylation step of LPS synthesis across Gram-negative bacteria. Its catalysis can be driven by the Mn2+-dependent protein LpxH (β- and γ-proteobacteria), the Mn2+/Mg2+-dependent enzyme LpxI (α-proteobacteria) (Whitfield & Trent, 2014), and LpxG (Chlamydiae) (Young et al., 2016). Loss of these late-stage deacetylase enzymes results in accumulation of toxic lipid A precursors in the plasma membrane, offering a highly promising avenue against Gram-negative species (Babinski, Kanjilal, & Raetz, 2002). This is especially applicable for bacteria where the general loss of LPS is not associated with lethality (e.g., A. baumannii and Neisseria meningitidis) and where activity of LpxA, LpxC, or LpxD is not essential (Moffatt et al., 2010; Steeghs et al., 2001). Only one LpxH inhibitor has been identified to date, the sulfonyl piperazine AZ1, which binds in the acyl substrate pocket of this enzyme (J. Cho et al., 2020; Nayar et al., 2015), whereas, to the best of our knowledge, no inhibitors of LpxI or LpxG have been generated.
The lipid A disaccharide synthetase LpxB.
Very little is known about the structure and mechanism of action for LpxB, making inhibitor design challenging. Nonetheless, since LpxB is essential in the extremely drug-resistant pathogen A. baumannii, where its absence is known to lead to toxic precursor accumulation similar to that reported in lpxH mutants, targeting of LpxB has been pursued for this bacterium. Silencing of lpxB in A. baumannii using peptide-nucleic acid conjugates results in bactericidal effects and synergy with colistin (Babinski, Kanjilal, et al., 2002; Martínez-Guitián et al., 2019).
The lipid kinase LpxK.
The metal-dependent (Mg2+/Mn2+/Co2+) LpxK kinase is the last essential component of lipid IVA synthesis, however no inhibitors against this protein have been identified so far (Garrett, Que, & Raetz, 1998).
3.1.2. Synthesis of the oligosaccharide core and transport to the periplasm.
After the generation of lipid IVA, the Raetz pathway is concluded by the attachment of two Kdo sugar residues by WaaA (forming Kdo2-lipid IVA) (Simpson & Trent, 2019; Whitfield & Trent, 2014), followed by the addition of two terminal acyl chains by LpxL and LpxM (Vorachek-Warren, Ramirez, Cotter, & Raetz, 2002), ultimately yielding Kdo2-lipid A. Despite the key role of these three enzymes in the synthesis of the first LPS component that can be transported to the outer membrane, they have yet to be successfully inhibited (P. Zhou & Zhao, 2017).
The assembly of the LPS progresses through extension of Kdo2-lipid A by heptosyltransferase enzymes that add the remainder of the oligosaccharide core to the LPS precursor (Whitfield & Trent, 2014). This rough LPS molecule can then be transported from the cytoplasmic leaflet of the plasma membrane to the periplasmic leaflet by an ABC transporter, MsbA, which initializes the journey of the LPS to the outer membrane (Voss & Trent, 2018; Whitfield & Trent, 2014).
Heptosyltransferase enzymes.
There are four heptosyltransferases that can catalyze the sequential addition of heptose molecules onto Kdo2-lipid A. HepI (also known as WaaC or RfaC) and HepII are highly conserved and add the first two sugar moieties onto the Kdo inner core, whereas HepIII and HepIV are species specific and are responsible for the formation of the outer core (Cote & Taylor, 2017). The only confirmed inhibitor of these enzymes to date targets HepI and it is a non-cleavable analogue of the enzyme’s natural substrate, ADP-2F-heptose (Grizot et al., 2006). Moreover, HepI appears to be a secondary target of the aminoglycoside antibiotics kanamycin (competitive inhibition) and streptomycin (non-competitive inhibition), and modified derivatives of these antibiotics might allow HepI specific inhibition. Overall, HepI-targeting compounds are likely only suitable for use in combination therapies, since LPS truncations result in cell sensitivity to extracellular stresses, like exposure to macrolide and β-lactam antibiotics (Coleman & Leive, 1979; Milicaj et al., 2022).
The flippase MsbA.
The universally conserved MsbA protein binds the nascent rough LPS at the cytoplasmic side of the plasma membrane and employs an ATP-dependent ‘trap and flip’ mechanism to release the molecule through its side into the periplasmic leaflet (Voss & Trent, 2018). The potential of MsbA as an antimicrobial target was first noted when molecules abrogating its ATPase activity were shown to inhibit the growth of A. baumannii (G. Zhang et al., 2018). Later, a series of quinolone compounds targeting the lipid binding site showed promising bactericidal activity against E. coli (Alexander et al., 2018; Whitfield & Trent, 2014). Preclinical development of these compounds by Genentech was later stopped due to opposing properties required for cell entry (hydrophilic) and MsbA inhibition (lipophilic) (Verma et al., 2022). Notably, the mechanism of action of MsbA is similar to the way the outer membrane LptDE complex transfers the mature LPS from the periplasmic leaflet of the outer membrane to the extracellular leaflet (see section 3.1.4) (Botos et al., 2016; Dong et al., 2014; Voss & Trent, 2018), and the similarity of these processes may open the possibility of dual-function inhibitor strategies in the future.
3.1.3. Synthesis and attachment of the O-antigen.
In some Gram-negative species, the extremity of the LPS that is away from the cell surface is additionally glycosylated through a variable polysaccharide structure, the O-antigen. O-antigens are first assembled in the cytoplasm by Wzy-dependent, ABC-transporter-associated, or synthase-reliant pathways. They are then transported to the periplasmic leaflet of the plasma membrane through an undecaprenyl-pyrophosphate carrier, and finally attached to the LPS by the O-antigen-polysaccharide ligase WaaL (Whitfield & Trent, 2014). For more information on these pathways we refer the reader to the excellent reviews by Samuel & Reeves (Samuel & Reeves, 2003) and Whitfield, Williams & Kelly (Whitfield, Williams, & Kelly, 2020). Although the biogenesis process of this LPS component relies on enzymes that could serve as antimicrobial targets, albeit species-specific ones, no efforts to inhibit them have been reported to date; the exception is the inhibition of the recycling of the undecaprenyl-pyrophosphate carrier, which we discuss briefly in section 4.1.1.
Finally, in addition to the broadly conserved enzymes that generate the constituents of the LPS, a wide range of species- and strain-specific variations of this pathway may enable the development of species-, genus- or infection-condition-specific therapeutic approaches. These targeted strategies may also be considered for use against bacteria with augmented LPS, and, in particular, against bacteria where LPS modifications via addition of sugars, acyl chains, and other moieties (e.g., 4-aminoarabinose or phosphoethanolamine; see section 6.2) have been shown to affect antibiotic resistance (Raetz, Reynolds, Trent, & Bishop, 2007).
3.1.4. Transport of the LPS to the outer membrane.
After MsbA-mediated flipping of rough LPS into the periplasmic leaflet of the plasma membrane (followed by O-antigen attachment, when applicable), the Lpt pathway proteins create a bridge spanning the entirety of the cell envelope that allows transport of the mature LPS to its final destination on the surface of the cell. The activity of the Lpt system has been likened to the PEZ candy dispenser (known as the ‘PEZ’ model), whereby continuous stacking of LPS molecules at the bottom of the transport apparatus in the plasma membrane, eventually leads to them exiting at the extra-cellular leaflet of the outer membrane (Okuda, Sherman, Silhavy, Ruiz, & Kahne, 2016). Consequently, all Lpt proteins are essential for LPS transport and their loss, which also results in loss of the LPS, compromises the stability of the entire complex (Martorana et al., 2021). The key role of each of these proteins, combined with the fact that they are widely conserved across Gram-negative bacteria, makes the Lpt pathway an ideal target for novel therapeutics.
LptB2FGC.
The transmembrane ABC transporter complex LptB2FGC extracts the LPS from the lipidic environment of the plasma membrane through the activity of the ATPase LptB (Okuda, Freinkman, & Kahne, 2012; Sherman et al., 2018). Several LptB-binding compounds have been identified through screens on the purified enzyme (Gronenberg & Kahne, 2010). A 4-phenylpyrrolocarbazole derivative of these molecules was later optimized and reported to have bactericidal activity against a permeable E. coli variant, validating both LptB and the Lpt pathway as antimicrobial targets (Sherman, Okuda, Denny, & Kahne, 2013). Unfortunately, no activity was observed for cells with intact outer membranes, an issue that is often a challenge for candidate antimicrobials against Gram-negative species. It should be noted that ATPases and ABC-type transporters are commonly found in the plasma membrane. Such proteins are often key components of bacterial efflux machineries and depend on an active PMF. For this reason, compounds targeting efflux (see section 6.3) or dissipating the PMF (see section 2.2.2) may also affect LptB activity.
LptA.
The periplasmic protein LptA connects the LptB2FGC complex in the plasma membrane to the integral outer-membrane β-barrel LptD (Sherman et al., 2018). Two inhibitors targeting the interaction between LptA and other Lpt proteins have been described to date. IMB-88 blocks the LptA-LptC interaction and has been shown to inhibit the growth in E. coli and P. aeruginosa strains, including in several multidrug-resistant clinical isolates (X. Zhang et al., 2019). Similarly, an insect-derived antimicrobial peptide, thanatin, and its variants have inhibitory activity against numerous Gram-negative pathogens in vitro and in vivo (Dash & Bhattacharjya, 2021; B. Ma et al., 2016; Vetterli et al., 2018). The structural similarity of the LptA-LptC and LptA-LptD interfaces suggests that thanatin may be effective at disrupting the Lpt pathway at both ends of the cell envelope (Fiorentino et al., 2021; Vetterli et al., 2018).
LptDE.
The outer-membrane translocon complex, composed of the LptD β-barrel and the lipoprotein LptE, ensures that LPS is efficiently inserted into the extra-cellular leaflet despite the concentration gradient that works against this localization (Okuda et al., 2012; Sherman et al., 2018). The most promising inhibitor of LptD to date, murepavidin (POL7080), reached Phase III clinical trials, but unexpectedly failed on account of nephrotoxicity; no other inhibitors have been identified since (Srinivas et al., 2010).
It should be noted that targeting the Lpt pathway is complicated because of the structural organization of its proteinaceous components around the transported LPS molecules. The stacks of nascent LPS chains across this trans-envelope bridge minimize the number of potential drug interaction sites and limit the accessibility of the Lpt protein targets. As a result, it might be more useful to target this process by focusing on non-competitive inhibition strategies or on the disruption of the biogenesis of the Lpt proteins themselves.
3.2. Targeting the localization of lipoproteins (Lol) pathway
A schematic overview of the processes discussed in this section can be found in Figure 3.
Figure 3. Overview of the plasma-membrane translocation pathways and of the lipoprotein biogenesis process.
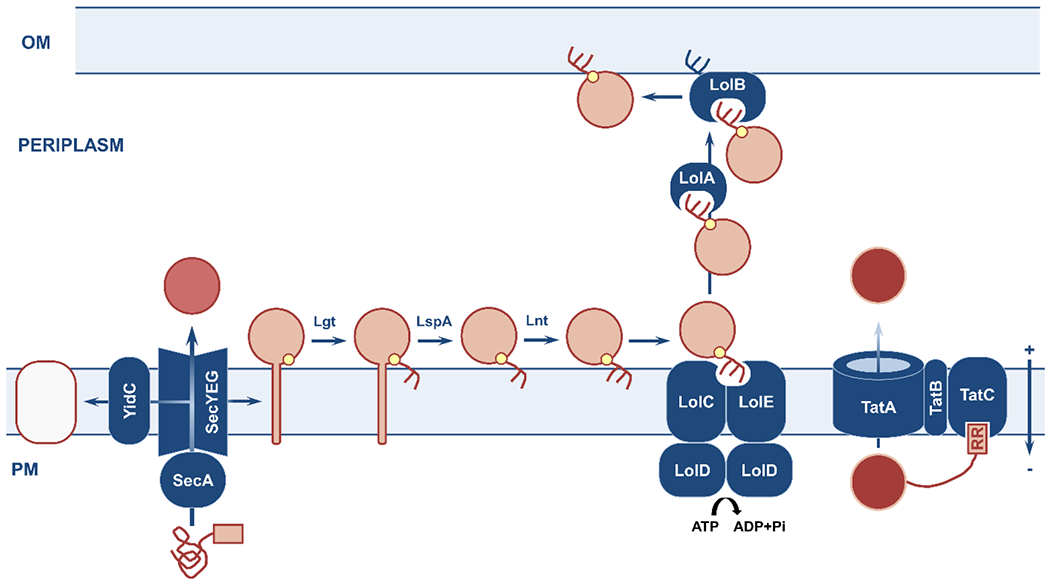
Nascent polypeptides with specific N-terminal signal sequences (pink box) are delivered to the Sec and Tat machineries for translocation across the plasma membrane; lipoproteins require further processing after Sec-dependent translocation into the cell envelope. (left) The Sec translocon handles unfolded polypeptides destined for insertion into the plasma membrane (white protein substrate; aided by YidC), release into the periplasm (dark pink protein substrate), or maturation into lipoproteins (light pink protein substrate). (center) Sec-translocated lipoprotein precursors are recognized by the presence of an invariable lipobox. They are acylated at a conserved cysteine residue (small yellow circle) by Lgt, their signal peptide is cleaved by LspA, and their acylated cysteine is further acylated by Lnt. The mature lipoprotein interacts with the membrane-embedded LolCDE complex and is carried to the outer membrane by a dedicated, soluble chaperone, LolA. Interaction with the acceptor lipoprotein LolB results in insertion into the outer membrane. (right) The Tat translocation apparatus selectively handles folded proteins (red protein substrate) that are recognized by their twin-arginine signal sequence. OM, outer membrane; PM, plasma membrane.
3.2.1. Acylation of lipoprotein precursors at the plasma membrane.
Proteins intended for insertion into the outer membrane are usually first translocated across the plasma membrane via the Sec system (see section 3.4.1) and then recognized by an invariable lipobox in their signal sequence (Hayashi & Wu, 1985; Sugai & Wu, 1992). At the periplasmic side of the plasma membrane, a diacylglycerol moiety is attached to the conserved cysteine residue of the lipobox by Lgt (Sankaran & Wu, 1994), before the translocation signal is cleaved by a type II signal peptidase, LspA (Inouye, Franceschini, Sato, Itakura, & Inouye, 1983; Vogeley et al., 2016). This allows the addition of a third acyl chain by Lnt (Wiktor et al., 2017), which fulfils the final requirement for the mature lipoprotein to be moved to the outer membrane. All these plasma-membrane proteins are essential components of the lipoprotein biogenesis process and are conserved across Gram-negative bacteria, making them suitable antibiotic targets. Strategies against Lgt are described in this section, while LspA will be discussed in section 3.4.2; to date, no inhibitor has been identified against the trans-acylase Lnt (Okuda & Tokuda, 2011).
The diacylglyceryl transferase Lgt.
Insufficient acylation of the lipobox due to Lgt depletion causes morphology changes and cell death in E. coli. This is due to accumulation of lipoproteins, like Lpp, in the plasma membrane and erroneous linkage of the peptidoglycan layer to the plasma instead of the outer membrane, which eventually causes cell lysis (Legood, Seng, Boneca, & Buddelmeijer, 2022). Two inhibitor molecules, palmitic acid (Mao et al., 2016) and the macrocyclic peptide G2824, have demonstrated the possibility of targeting Lgt, and disrupted the growth of E. coli and A. baumannii (Diao et al., 2021). Notably, unlike other inhibitor strategies discussed in this section, G2824-mediated inhibition does not give rise to Lpp precursor accumulation, which is commonly associated with resistance emergence through lpp deletion; the mechanism behind this difference remains unclear (Diao et al., 2021).
3.2.2. Transport of lipoproteins to the outer membrane.
Following acylation, mature lipoproteins interact with LolE (Mizutani et al., 2013), which is part of the LolCDE complex in the plasma membrane, while LolC recruits the diffusible periplasmic chaperone LolA (discussed later in this section and in section 5.1.1) (Okuda & Tokuda, 2009; Okuda, Watanabe, & Tokuda, 2008). ATP binding and hydrolysis by LolD leads to lipoprotein extraction from the membrane and its transfer to LolA (Ito, Kanamaru, Taniguchi, Miyamoto, & Tokuda, 2006). The solubilized lipoprotein is then shuttled across the periplasm to the outer-membrane-anchored protein LolB, which inserts it into the periplasmic leaflet of the outer membrane (Matsuyama, Yokota, & Tokuda, 1997). Although many lipoproteins are intended to remain in the periplasmic leaflet of the outer membrane, several key examples, like RcsF in E. coli and SLAM in Neisseria spp. (Rayes et al., 2021), require further processing for display on the cell surface. The mechanism behind this final translocation step is not yet understood, but its elucidation could yield novel and easily accessible antimicrobial targets in the future.
LolCDE.
The LolCDE complex is completely conserved across Gram-negative bacteria and is responsible for the recognition of lipoproteins destined for the outer membrane. The separation of Lol dependent lipoproteins, from those intended to remain in the plasma membrane, is driven by the recognition of a Lol avoidance signal, otherwise known as the ‘+2 rule’ of the E. coli Lol pathway, whereby the presence of an aspartic acid two positions after the conserved cysteine of the lipobox sequence leads to retention of a lipoprotein in the plasma membrane instead of its export to the outer membrane (S. Sharma et al., 2021). High-throughput cell-wall reporter assays have led to the discovery of pyrazole inhibitors postulated to affect LolCDE (Lorenz, Dougherty, & Lory, 2016; Nayar et al., 2015), while a pyridineimidazole inhibitor of this complex, identified by phenotypic screening, exhibited promising activity against E. coli and Haemophilus influenzae (McLeod et al., 2015). A study involving the use of efflux deficient, permeable E. coli also highlighted the antibacterial potential of a pyrrolopyrimidinedione compound that stimulated the ATPase activity of LolD, with the promise of this lead compound being further supported by a basic derivative screen (Nickerson et al., 2018).
The acceptor protein LolB.
Recently, procyanidin, stevioside, troxerutin and rutin, which are all natural products approved for human use, have been highlighted as potential LolB inhibitors in Vibrio parahaemolyticus, highlighting the druggability of this outer-membrane-associated component (J. Liu et al., 2022).
The caveat to targeting LolA and LolB.
The importance of LolA and LolB for cell viability is restricted to a subset of Gram-negative bacteria that express Lpp and OsmB lipoproteins (section 4.1.5) (Grabowicz, 2019; Grabowicz& Silhavy, 2017). The loss of either LolA or LolB in species without these lipoproteins causes decreased efficiency of the Lol system and results in indiscriminate insertion of other lipoproteins into both the plasma and outer membranes, but is not necessarily lethal (Grabowicz, 2019; Grabowicz & Silhavy, 2017). While there are no clinical inhibitors against LolA and LolB yet (for studies investigating LolA as a target see section 5.1.1), it is important to remember that targeting these proteins can only generate species-specific antibacterial strategies that will depend on the simultaneous development of appropriate diagnostics. Additional variations in the components and mechanism of the Lol pathway have been reported across Gram-negative organisms and may offer other species-specific cell envelope targets related to this pathway; for an overview of those we refer the reader to reviews by Amaout & Soulimane (Amaout & Soulimane, 2019) and Grabowicz (Grabowicz, 2019).
3.3. Targeting the β-barrel assembly machinery (Bam)
The biogenesis of outer-membrane β-barrel proteins relies on the orchestrated function of several chaperones, including SurA, Skp, and FkpA that shuttle the protein precursors across the periplasm (see section 5), and on the β-barrel assembly machinery (Bam), which mediates their insertion into the outer membrane (Costello, Plummer, Fleming, & Fleming, 2016; Lundquist, Billings, Bi, Wellnitz, & Noinaj, 2021) (see Figure 4). Bam substrates are recognized by the outer-membrane β-barrel BamA and the lipoprotein BamD, through a distinct, species-specific C-terminal β-strand of the substrate (β-signal) (J. Lee et al., 2018; Robert et al., 2006). Binding of the substrate is proposed to cause a conformational change in BamA that, in turn, leads to localized destabilization and thinning of the membrane along the lateral seam of the β-barrel (Costello et al., 2016; Gessmann et al., 2014; J. Lee et al.). Two models for the insertion process have been proposed; substrates are thought to either use the BamA β-barrel as a template for their own folding (budding model) or to insert spontaneously through the weakened membrane (assisted model) (Lundquist et al., 2021). This process is further supported by BamB, which likely mediates formation of Bam complex clusters at the cell’s midbody to support β-barrel insertion (Gunasinghe et al., 2018), and the surface-exposed proteins BamC and BamE that promote stability and folding of the substrate (K. R. Ryan, Taylor, & Bowers, 2010; Sikora et al., 2018; Sklar et al., 2007; Webb et al., 2012).
Figure 4. Overview of outer-membrane protein biogenesis by the Bam pathway.
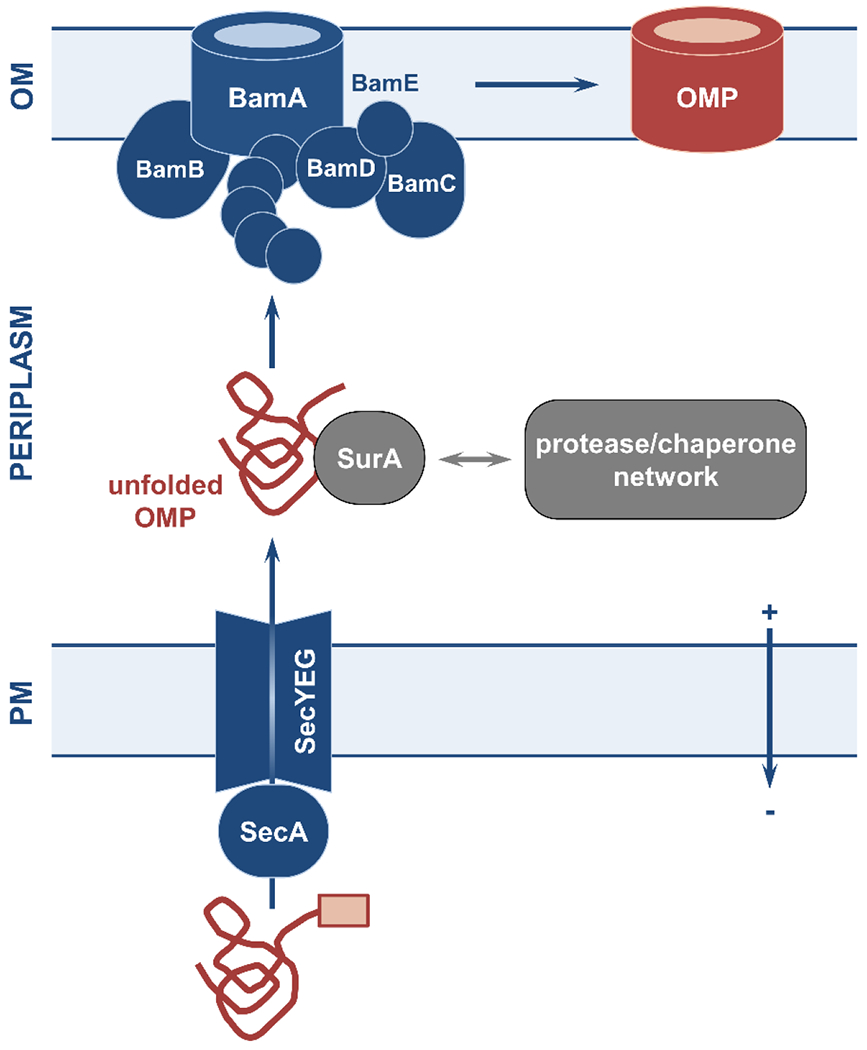
Unfolded outer-membrane protein (OMP) precursors are exported to the periplasm via the Sec translocon and traverse the periplasm helped by the soluble chaperone SurA or by other components of the chaperone/protease network. Once near the outer membrane, unfolded OMPs are recognized by the transporter BamA and the lipoprotein BamD and are threaded into the membrane, where they fold into their β-barrel structure. The activity of BamA and BamD is supported by the ancillary proteins BamB, BamC, and BamE. OM, outer membrane; PM, plasma membrane.
BamAD.
BamA and BamD are conserved across all Gram-negative bacteria and are essential for outer-membrane assembly and bacterial survival. Their conservation, key role, and easily accessible cellular location at the outer membrane, has made them highly sought after antimicrobial targets (Malinverni et al., 2006; T. Wu et al., 2005). Numerous inhibitors have been identified over the past decade against these Bam components, although none have made it into clinical use (Steenhuis, van Ulsen, Martin, & Luirink, 2021). The most promising compound is the small molecule darobactin, which mimics the β-signal and blocks the lateral seam of BamA, preventing substrate insertion (Imai et al., 2019; Kaur et al., 2021). The promise of this strategy is showcased by the fact that darobactin inhibits bacterial growth and is effective in in vivo infection models, as well as the fact that in silico studies were successful in identifying even more potent darobactin derivatives (Böhringer et al., 2021). An interesting attribute of BamA and BamD inhibition is that Bam function can be abrogated from different cellular locations. For example, the inhibitor MRL-494 exploits the accessibility of the Bam complex from outside the outer membrane and blocks β-barrel formation in Gram-negative species (Hart et al., 2019), while IMB-H4 targets the BamA-BamD interaction achieving the same result from the periplasmic side (Y. Li et al., 2020). Finally, because of its accessibility, inhibition of the Bam has also been achieved using antibodies and nanobodies, such as MAB1 (Storek et al., 2018) and NanoE6/NanoF7 (Kaur et al., 2019). For a more comprehensive review of Bam inhibitors, we refer the reader to Steenhuis et al (Steenhuis, van Ulsen, et al., 2021).
BamBCE.
At least two of these Bam accessory proteins are required for cell survival, which suggests that some of these surface-displayed components of the Bam are essential for successful β-barrel generation (Lundquist et al., 2021; Malinverni et al., 2006; T. Wu et al., 2005). That said, with limited insight into the function of these proteins, inhibitors have yet to be identified.
Recent research has demonstrated that β-barrel assembly is linked to the biogenesis of the peptidoglycan layer (Mamou et al., 2022). More specifically, it was shown that binding of the mature peptidoglycan at the Bam complex site suppresses Bam function at the cell poles. This is in agreement with the previous observation that poor binding of nascent peptidoglycan at the cell division site promotes outer-membrane protein insertion at that location (Gunasinghe et al., 2018). Thus, targeting either of these pathways may result in general disruption of the homeostasis of the cell envelope.
3.4. Targeting the transport across the plasma membrane
Translocation of extracytoplasmic proteins across, or their insertion into, the plasma membrane is driven by the Sec and Tat machineries (see Figure 3) and relies on the recognition of specific signal sequences at the N-terminus of the nascent polypeptide chain. Even though these recognition sequences are largely similar, ones containing a twin arginine motif delineate proteins intended for Tat processing (Palmer & Berks, 2012). Regardless of the translocation pathway, signal sequences are cleaved at the last step of protein translocation by a type I signal peptidase (e.g., LepB, discussed in section 3.4.2) or a type II lipoprotein signal peptidase (e.g., LspA, also discussed in section 3.4.2). The supporting cytoplasmic chaperone network that helps direct the nascent polypeptides to the appropriate translocation machinery is complex and beyond the scope of this review; for an introduction into this expansive topic we refer the reader to the review by Castanié-Cornet, Bruel & Genevaux (Castanié-Cornet, Bruel, & Genevaux, 2014).
3.4.1. Sec pathway.
The Sec system translocon comprises a highly conserved SecYEG transmembrane channel and a SecDF accessory complex (Natale, Brüser, & Driessen, 2008; Nijeholt & Driessen, 2012). Unfolded substrates are delivered to the Sec machinery by the SecA ATPase (either in its membrane-associated or soluble form), a process that, depending on the substrate, can also be aided by the cytoplasmic chaperone SecB, the signal recognition particle, and/or the trigger factor. Proteins intended for plasma membrane insertion, often undergo co-translational transport, whereby continued translation at the expense of GTP pushes the unfolded polypeptide into the SecYEG channel. This, along with the membrane chaperone YidC and polypeptide-membrane interactions, achieves protein insertion into the plasma membrane (Urbanus et al., 2001). By contrast, post-translationally modified peptides rely mostly on SecA-mediated ATP hydrolysis for transport (Nijeholt & Driessen, 2012). In all cases, Sec function depends on the PMF (Nijeholt & Driessen, 2012).
SecA.
The most studied Sec system target is the ATPase SecA, which is completely unique to prokaryotes (Puyenbroeck & Vermeire, 2018). This protein has also been implicated in other key cytoplasmic processes relating to efflux and ribosomal function; for their overview we defer the reader to the review by Jin et al. (Jin et al., 2018). The majority of identified SecA inhibitors target its ATP binding site. For example, a derivative of the fungal antibiotic equisetin, CJ-21058, exhibits activity against Gram-positive species like Staphylococcus aureus and Enterococcus faecalis (Sugie et al., 2002), while two fluorescein analogues prevent the translocation of pre-OmpA in E. coli in a non-competitive (Rose Bengal) and competitive (erythrosin B) fashion (C.V, Waelheyns, Economou, & Anné, 2014; Y. J. Huang et al., 2012). In silico approaches to SecA inhibition estimated the binding energies of 60,000 candidate compounds and identified over 30 potential hits (Waelheyns et al., 2015). Further optimization, led to a thiouracil derivative with high potency against E. coli (Waelheyns et al., 2015). Similar results were reported in a separate study that developed triazolo-thiadiazole thiouracil derivatives (Cui et al., 2017; Waelheyns et al., 2015). Unfortunately, most SecA-inhibiting compounds have failed on grounds of high cytotoxicity, low antimicrobial activity, or inability to cross the outer membrane (Waelheyns et al., 2015).
YidC.
The membrane protein YidC is essential for E. coli growth, since its depletion dissipates the PMF (Patil, Sharma, Srivastava, Navani, & Pathania, 2013). Essential oil extracts eugenol and carvacrol have been proposed to inhibit YidC, although their actual mechanism of action has not been fully elucidated (Patil et al., 2013). Moreover, since mitochondria express two homologues of YidC, the proteins Oxa-1 and -2, more work needs to be done to exclude the possibility of off-target effects of YidC inhibitors on these mammalian counterparts (C.V et al., 2014).
SecYEG.
The translocation channel SecYEG has been traditionally considered an unsuitable target; it does not perform an enzymatic function that would allow efficient inhibitor screening, while the existence of a conserved eukaryotic homologue, Sec61, necessitates very selective SecYEG targeting. Nonetheless, optimization of the Sec61 inhibitor eyarestatin 1 (ES1), identified in a 2020 anti-cancer screen (Steenhuis, Koningstein, et al., 2021), yielded ES24, which causes protein mislocalization and growth defects and impairs Sec secretion in E. coli, S. aureus and E. faecalis (Gamayun et al., 2019; Steenhuis, Koningstein, et al., 2021; Wojdyla et al., 2015). Additional investigation showed that ES24 depends on reduction by nitroreductases A and B, similar to the FDA-approved antibiotic nitrofurantoin, and exhibits comparable toxicity at the required pharmaceutical dose (Steenhuis, Koningstein, et al., 2021). The successful optimization of ES24 suggests that not only can selectivity for the bacterial SecYEG channel be achieved, but also that previously discovered Sec61 inhibitors could be repurposed or used as templates for the development of specific SecYEG-targeting compounds. This includes molecules like the irreversible inhibitor mycolactone from Mycobacterium ulcerans or the fungal cyclic decadepsipeptide decatransin (Hall et al., 2014; Junne et al., 2015).
SecDF.
Targeting the accessory SecDF complex is another potential strategy to abrogate Sec system activity. However, research shows that because of the high absolute amounts of the Sec components, bacteria can perform essential tasks even at low Sec functionality or in SecYEG-depleted conditions and, thus, decreasing the Sec translocation efficiency, which is what SecDF targeting would achieve, may not be sufficient for growth inhibition (Crowther et al., 2015; B.-R. Lin, Hsieh, Jiang, & Tai, 2012).
3.4.2. TAT pathway.
The Tat system plays an important role in processes such as energy metabolism and pathogenesis. It operates in parallel to the Sec pathway but is responsible for the transport of folded proteins that require cofactor or metal binding in the cytoplasm (Sargent et al., 1998; Urrutia et al., 2018; Vasil, Tomaras, & Pritchard, 2012). With no known mammalian homologue, the Tat pathway is a prime target for decreasing cell viability and virulence (Buck, Lammertyn, & Anné, 2008). The minimal operational unit of this system is TatAC, with a second TatA-like protein, TatB, involved in protein translocation in most Gram-negative and some Gram-positive species (Palmer & Stansfeld, 2020). Upon interaction of the substrate with the membrane protein TatC (or with the TatBC complex), PMF-dependent TatA multimerization occurs, resulting in channel formation and/or weakening of the phospholipid bilayer, ultimately achieving protein translocation (Natale et al., 2008; Palmer & Stansfeld, 2020). A few accessory proteins are involved in this process, including the nuclease TatD and the putative chaperone TatF (Nijeholt & Driessen, 2012).
Despite continuous efforts and several high-throughput screens, only a couple of Tat inhibitors have been described to date, NPM and Bat 11-7082, both of which function in P. aeruginosa (Bageshwar et al., 2016; Vasil et al., 2012). Neither of these compounds, however, shows activity against the E. coli machinery and both exhibit high cytotoxicity levels (Bageshwar et al., 2016; Vasil et al., 2012). Another accidentally discovered inhibitor of Tat-dependent translocation, N,N′-Dicyclohexylcarbodiimide (DCCD, DCC), interferes with the insertion of TorA signal sequence into the TatBC complex (Blümmel et al., 2017) and thus prevents TorA secretion in E. coli (Panahandeh, Maurer, Moser, DeLisa, & Müller, 2008). Overall, our lack of knowledge around the structure and the mechanism of the Tat proteins, and the fact that activity assays specific to Tat translocation are arduous, are hindering inhibitor development, and more optimization is required for this pathway to be fully validated as a promising antimicrobial target.
Both the Sec and the Tat translocation pathways, as well as other membrane biogenesis systems like the Bam and Lol pathways, can be disrupted by the inhibition of signal peptidase enzymes that recognize conserved signal sequence motifs and catalyze the release of translocated substrates at the periplasmic leaflet of the plasma membrane. Here we discuss the two most common signal peptidase enzymes, and we refer the reader to the comprehensive review by Paetzel et al. for a more detailed overview (Paetzel, Karla, Strynadka, & Dalbey, 2002).
Type I signal peptidase, LepB.
E. coli LepB is the most characterized type I signal peptidase and is an unusual, constitutively expressed serine-lysine enzyme that is essential for cell viability (Paetzel, Dalbey, & Strynadka, 2000). The structural and mechanistic differences between LepB and the eukaryotic type I signal peptidase, makes this bacterial protein a promising antimicrobial target that could be inhibited without mammalian cross-reactivity (Paetzel et al., 2002). Several different types of LepB inhibitors have been discovered, including penem compounds (Harris, Powers, & Romesberg, 2009), peptides that mimic the signal sequence (Buzder-Lantos, Bockstael, Anne, & Herdewijn, 2009; Kaderbhai, Khan, & Kaderbhai, 2008), and arylomycins that bind the catalytic residue of this enzyme (Smith et al., 2018). The most promising inhibitor to date has been the arylomycin derivative G0775 generated by Genentech, which engages the catalytic lysine residue of LepB and exhibits in vivo activity against E. coli, K. pneumoniae, and A. baumannii (Smith et al., 2018). The potential of this target is further demonstrated by the fact that a second LepB inhibitor, RG6319, is currently in early development by Genentech (Genentech, 2023). Notably, Gram-positive type I signal peptidases, such as the SpsB, from S. aureus, perform a similar function and are expected to be affected by LepB inhibitors (Harris et al., 2009; Kaderbhai et al., 2008; Paetzel et al., 2000).
Type II signal peptidase, LspA.
A key lipoprotein of the plasma membrane, the type II signal peptidase LspA, is involved in the cleavage of the signal sequences of, among others, the Lol pathways substrates (see section 3.2). LspA inhibition is of profound interest due to its role in lipoprotein biogenesis, whereby loss of LspA leads to lipoprotein mislocalization, and because of its association with the Sec and Tat translocation machineries (described in this section). Several inhibitors have been identified including Globomycin, which targets the LspA binding site (J. S. Lai et al., 1981; Omoto, Suzuki, & Inouye, 2006) and myxovirescin, which inhibits LspA signal sequence cleavage through an unknown mechanism of action (Xiao, Gerth, Müller, & Wall, 2012). A compound named “inhibitor 99” also shows non-competitive binding of LspA when used in conjunction with the outer-membrane-permeabilizing polymyxin B nonapeptide (Arnaout & Soulimane, 2019; Kitamura, Owensby, Wall, & Wolan, 2018).
Finally, an attribute of targeting signal peptidases is that their inhibition has knock-on effects on other important bacterial processes, like virulence, cell envelope biogenesis and homeostasis, which can, in turn, enhance the antibacterial efficacy of strategies developed against them. Nonetheless, this approach might not be as effective in pathogens like P. aeruginosa or Listeria monocytogenes, which express more than one type I signal peptidase homologue; for these species better characterization of the structures, mechanisms, and substrates of signal peptidase proteins will be critical if inhibition were to be pursued (Bonnemain et al., 2004; Waite et al., 2012).
4. PEPTIDOGLYCAN CELL WALL
At the interface of the Gram-negative periplasm and the outer membrane sits a single rigid peptidoglycan layer. This structure acts as physical barrier, provides an anchor for cell envelope components, supports the cell shape, and plays a critical role in cell growth and division (Dramsi, Magnet, Davison, & Arthur, 2008; Neuhaus & Baddiley, 2003). It is primarily composed of a crosslinked mesh of polymerized [N-acetylglucosamine (GlcNAc)-N-acetylmuramic acid (MurNAc)] disaccharides that are attached to the outer membrane through Lpp and OmpA. This is in stark contrast to Gram-positive bacteria that have a thicker, multi-layered peptidoglycan cell wall, threaded with molecules like teichoic and lipoteichoic acid, which make it more resistant to the stresses of the extracytoplasmic environment (Neuhaus & Baddiley, 2003). The Gram-positive peptidoglycan also allows the attachment of plasma-membrane and plasma-membrane-associated proteins, which will be discussed in more detail in section 4.2 (Dramsi et al., 2008; Silhavy et al., 2010). In both Gram-negative and Gram-positive bacteria, the formation and maintenance of the peptidoglycan cell wall is a continuous process that can be targeted by compounds that are often effective against both types of organisms. The following section overviews the key components driving peptidoglycan biogenesis (see Figure 5), as well as approaches aiming to target it that can lead to potent, broad-spectrum therapeutics. For a more comprehensive review of the peptidoglycan architecture, synthesis, and remodeling we refer the reader to the reviews by Egan, Errington & Vollmer and Vollmer, Blantot & Pedro (Egan, Errington, & Vollmer, 2020; Vollmer, Blanot, & Pedro, 2008).
Figure 5. Overview of peptidoglycan formation in Gram-negative bacteria.
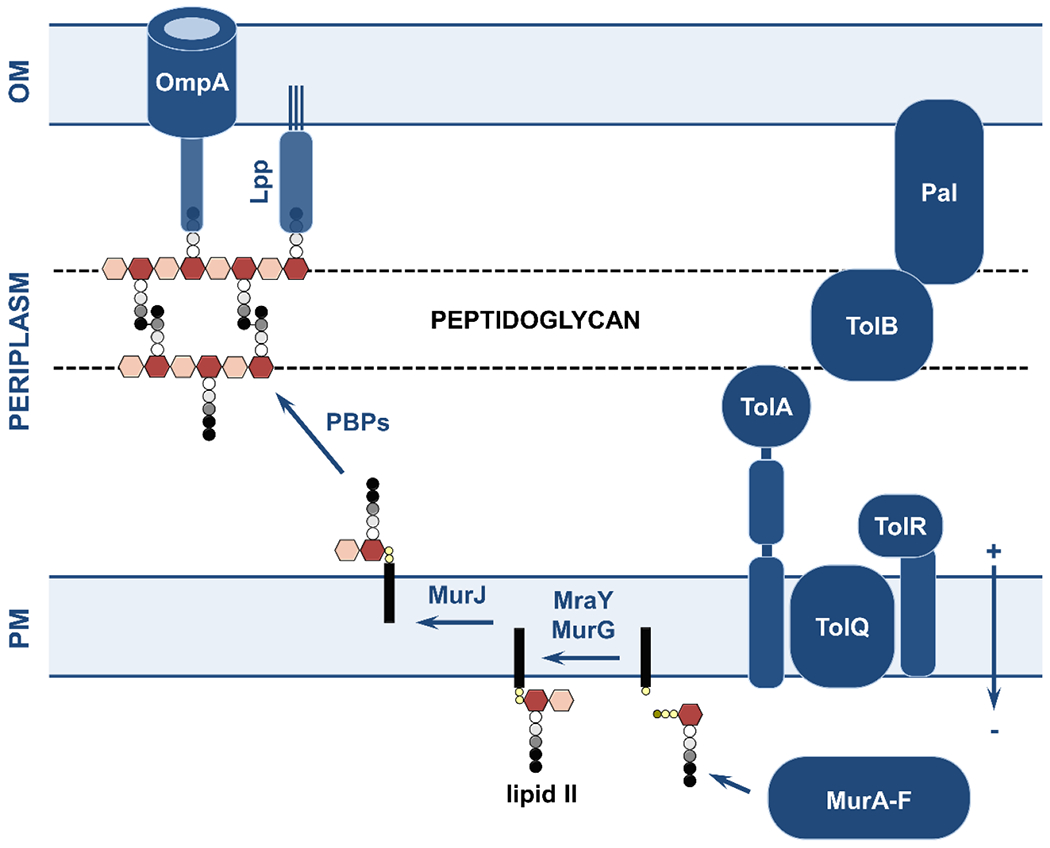
(left). UDP-MurNAc-pentapeptide precursor monomers (the dark pink hexagon represents the MurNAc sugar, the grayscale circles the pentapeptide, and the small brown and yellow circles the uridine and phosphate moieties, respectively) are synthesized in the cytoplasm by MurA-F. Their attachment to a lipid carrier (black rectangle) by MraY and addition of a GlcNAc monomer (light pink hexagon) catalyzed by MurG, results in lipid II. The disaccharide is then flipped to the periplasmic leaflet of the plasma membrane by the proton-motive-force-dependent MurJ protein. The nascent peptidoglycan components are then polymerized and crosslinked into the pre-existing peptidoglycan matrix either by bifunctional class A penicillin binding proteins (PBPs) or through the synchronized function of a monofunctional class B PBP and a glycosyltransferase enzyme. The peptidoglycan matrix is covalently attached to the outer membrane through the lipoprotein Lpp and associated with OmpA through electrostatic interactions. (right) The Tol-Pal system (TolR, TolQ, TolA, TolB, and Pal) is responsible for coordination of cell division and peptidoglycan synthesis. OM, outer membrane; PG, peptidoglycan; PM, plasma membrane.
4.1. Targeting peptidoglycan synthesis in Gram-negative bacteria
4.1.1. Peptidoglycan synthesis in the cytoplasm.
Monomeric peptidoglycan precursors are synthesized in the cytoplasm by the concerted action of numerous proteins, including MurA-F, MurI, Alr, DadX and DdlA (Egan et al., 2020). These monomers are then anchored to the cytoplasmic leaflet of the plasma membrane by MraY, which facilitates the linkage of each UDP-MurNAc-pentapeptide precursor onto an undecaprenyl phosphate lipid carrier to form lipid I (Egan et al., 2020). Subsequently, the glycosyltransferase MurG completes the cytoplasmic part of the synthesis process by linking lipid I to UDP-GlcNAc to form lipid II. This is then flipped into the periplasmic leaflet of the plasma membrane by the essential integral membrane protein MurJ in a PMF-dependent manner (Egan et al., 2020). Strategies aiming to disrupt the cytoplasmic components of peptidoglycan synthesis include the inhibition of MurA using Fosfomycin (Silver, 2017) or the use of monoclonal antibodies and mureidomycin against MraY (Cao et al., 2017). The recycling step of the lipid carrier is also a worthwhile target (Torrens et al., 2019) and cyclic peptides like bacitracin or amphomycin have been shown to successfully block the activity of the undecaprenyl pyrophosphate phosphatase UppP that is essential for this process (Rubinchik et al., 2011; Stone & Strominger, 1971); inhibition of this protein is advantageous in that it may also disrupt the synthesis of the LPS (see section 3.1.3)
4.1.2. Peptidoglycan synthases.
On the periplasmic side of the plasma membrane, lipid II subunits are polymerized by glycosyltransferase enzymes and crosslinked into the existing peptidoglycan structure by DD-transpeptidases. These polymerization and insertion steps either occur simultaneously, catalyzed by the bifunctional class A penicillin binding proteins (PBPs), or stepwise through monofunctional class B transpeptidases PBP2 or PBP3 and a monofunctional glycosyltransferase (MgtA) (Typas, Banzhaf, Gross, & Vollmer, 2012). For a more comprehensive review on peptidoglycan synthase enzymes we refer the reader to Garde, Chodisetti & Reddy (Garde, Chodisetti, & Reddy, 2021).
DD-transpeptidases.
DD-transpeptidase enzymes are vital for cell growth and division (Egan et al., 2020). The activity of bifunctional class A proteins PBP1A and PBP1B is partially redundant, with only one class A enzyme required for bacterial survival at any given time. PBP1A is activated by the lipoprotein LpoA during cell elongation and produces short, lightly-crosslinked peptidoglycan (Bertsche, Breukink, Kast, & Vollmer, 2005; Born, Breukink, & Vollmer, 2006; Paradis-Bleau et al., 2010). While PBP1B is induced by LpoB during cell division and interacts with FtsN and the division-specific PBP3 enzyme (also known as FtsI) to form longer, highly-crosslinked peptidoglycan (Bertsche et al., 2005; Born et al., 2006; Typas et al., 2012). The third class A enzyme, PBP1C, has been implicated in host cell colonization (Budd, Blandin, Levashina, & Gibson, 2004). Monofunctional class B PBP enzymes are solely responsible for the peptidoglycan chain polymerization, with PBP2 activity being essential for cell elongation and PBP3 for cell division. Despite the apparent complexity and functional redundancy of this protein network, the inhibition of these enzymes using substrate β-lactam mimics is arguably the biggest success story of drug discovery and development; β-lactam antibiotics are the most prescribed antibiotics worldwide due to their broad spectrum, high efficacy, and low toxicity (Versporten et al., 2018).
β-lactam antibiotics, such as penicillins, cephalosporins and carbapenems, emulate the highly specific D-Ala-D-Ala moiety of the nascent peptidoglycan chain terminus, and competitively bind into the active site of transpeptidase enzymes (Tipper & Strominger, 1965). The four-membered β-lactam ring is opened through the formation of a covalent and irreversible bond with the catalytically active serine residue of the transpeptidase and this attachment permanently blocks the entry of the native peptidic substrates, thus inhibiting the crosslinking reaction. Recently, a novel β-lactam conjugate antibiotic, cefiderocol, carrying both a strained β-lactam ring and a siderophore domain, which ensures uptake through the outer membrane, was approved for clinical use against multi-drug resistant Gram-negative pathogens (Taheri et al., 2021; J. Y. Wu, Srinivas, & Pogue, 2020).
Glysosyltransferases.
Similar to β-lactam antibiotics, phosphoglycolipid-based moenomycins imitate the disaccharide pyrophosphate moiety of lipid II and competitively inhibit the glycosyltransferase reaction (Huber & Nesemann, 1968; Lovering, Castro, Lim, & Strynadka, 2007). Other antibiotic classes that interfere directly with the lipid II precursor include glycopeptide antibiotics like vancomycin that bind to the D-Ala-D-Ala moiety (Sarkar, Yarlagadda, Ghosh, & Haldar, 2017), or the pyrophosphate- and MurNac-binding cyclic peptide teixobactin (Ling et al., 2015), the lipoglycopeptide telavancin (Higgins et al., 2005), and the cyclic lipopeptide daptomycin (Grein et al., 2020). Additionally, class I and II bacteriocins have been shown to interact with several lipid II components (Chatterjee, Paul, Xie, & Donk, 2005).
4.1.3. Peptidoglycan hydrolases.
Peptidoglycan hydrolases break down glycan polymers and peptidic crosslinks during cell division and cell wall maintenance (Egan et al., 2020), processes during which up to 50% of the cell’s peptidoglycan is removed, replaced, and recycled (Park & Uehara, 2008). In E. coli, 13 highly redundant peptidoglycan hydrolases have been described to date, which can be categorized into three main groups: glycosidases, amidases and peptidases (A. K. Sharma, Kumar, K, Dhakan, & Sharma, 2016). For example, DD-carboxypeptidases, like PBP5/DacA, trim the peptide stems of the newly inserted glycan chains of the peptidoglycan layer and their loss leads to changes in cell morphology; absence of these proteins combined with simultaneous loss of class A or B PBPs causes more severe defects in the structure of the cell (Potluri et al., 2010; Suvorov, Vakulenko, & Mobashery, 2007). In addition to their native role in peptidoglycan maintenance, hydrolase enzymes are found as effectors of systems involved in interbacterial competition. For example, class III bacteriocin lysostaphin from Staphylococcus simulans cleaves peptidoglycan crosslinks at the third and fourth glycine residues and causes cell lysis in S. aureus (Schindler & Schuhardt, 1964), while enterolysin A from E. faecalis shows similar activity against other Enterococci (Nilsen, Nes, & Holo, 2003). These enzymes, previously highlighted for their potential use in food preservation (Callewaert, Walmagh, Michiels, & Lavigne, 2011), could serve both as potential targets for inhibiting peptidoglycan maintenance and as novel antibiotic scaffolds.
4.1.4. Tol-Pal.
The Tol-Pal system is an ensemble of protein complexes that spans the entire cell envelope and coordinates cell division and peptidoglycan synthesis. Mutations in components of the Tol-Pal system have been shown to disrupt outer membrane integrity and to prevent cell separation during division, a phenotype linked to the dysregulation of peptidoglycan hydrolase activity (Hirakawa, Suzue, & Tomita, 2022). Although originally identified in E. coli, orthologues of the proteins of the Tol-Pal system are found in other Gram-negative species, and, for example, in the pathogen K. pneumoniae they are essential for growth and virulence (Hirakawa et al., 2022). As such, components of this pathway have been proposed as promising targets for vaccine, antibacterial and adjuvant strategies. Given the expansive role of the Tol-Pal proteins in the viability of Gram-negative bacteria, we refer the reader to the review by Hirakawa, Suzue & Tomita (Hirakawa et al., 2022) for a more comprehensive review.
4.1.5. Peptidoglycan anchoring to the outer membrane.
Peptidoglycan-associated lipoproteins have been linked to virulence in several bacterial species and are thought to underpin acute host inflammatory responses (Agar et al., 2009; Dennehy et al., 2017; Hernández et al., 2015; Sha et al., 2004). The most important one is Lpp, which is numerically the most abundant protein in E. coli (G. W. Li, Burkhardt, Gross, & Weissman, 2014). Also known as Braun’s lipoprotein, Lpp crosslinks the peptidoglycan to the outer membrane (Mathelié-Guinlet, Asmar, Collet, & Dufrêne, 2020; Winkle et al., 2021), essentially defining the size of the periplasm (Mathelié-Guinlet et al., 2020). Disruption of Lpp anchoring results in increased susceptibility of Gram-negative bacteria to vancomycin, suggesting an outer-membrane defect (Mathelié-Guinlet et al., 2020). Conversely, mislocalization of Lpp to the plasma membrane results in cell death, due to crosslinking of the peptidoglycan to the inner rather than the outer membrane (Lehman, Smith, & Grabowicz). While no Lpp-specific inhibitors have been discovered to date, two pyridineimidazole LolCDE inhibitors successfully inhibit Lpp localization at the outer membrane, leading to significant growth defects and increased antibiotic susceptibility, thus highlighting the potential of targeting Lpp transport as an antimicrobial strategy (McLeod et al., 2015).
4.2. Targeting peptidoglycan synthesis in Gram-positive bacteria
Enzymes required for the synthesis of peptidoglycan in Gram-positive bacteria are mostly similar to those discussed in section 4.1 for Gram-negative species. The thick Gram-positive cell wall differs in that it also contains teichoic acids and proteins that are attached to this structure by sortase enzymes. These additional peptidoglycan constituents have a role in the recognition of host extracellular matrix components, iron acquisition, and virulence (Clarke & Foster, 2006).
Sortases.
Prior to the nascent peptidoglycan chains being incorporated into the existing cell wall structure, sortase enzymes integrate other proteins through recognition of a guide terminal signal sequence (DeDent, Bae, Missiakas, & Schneewind, 2008). These additional peptidoglycan proteins have a variety of functions that can broadly be categorized into surface protein display (class A), iron uptake (class B), pilus assembly (class C), sporulation (class D), or others (class E and F) (Spirig, Weiner, & Clubb, 2011). Limited understanding of substrate specificity of sortases along with variability among bacterial species, complicates their exploitation as antimicrobial targets. Nonetheless, efforts have been made on this front and several covalent inhibitors were recently identified as potential scaffolds against sortase A from S. aureus (Jaudzems et al., 2020). For an overview of sortase enzymes, we refer the reader to Spirig, Weiner & Clubb (Spirig et al., 2011).
Teichoic acids.
These surface polymers are attached to either the Gram-positive peptidoglycan (wall teichoic acids) or embedded in the plasma membrane (lipoteichoic acids), and their interaction with cationic molecules modulates the cell wall rigidity and porosity (Campbell et al., 2011). While teichoic acids are not essential for bacterial survival, loss of their biogenesis pathways leads to defects in cell division, morphological changes, and cell death (Campbell et al., 2011), suggesting that inhibition of their formation might have some potential as an antimicrobial strategy (Pasquina, Maria, & Walker, 2013).
4.3. Targeting peptidoglycan synthesis during cell division.
Peptidoglycan synthesis plays a critical role in cell division, during which it is controlled by the assembly of early (driven by FtsZ, FtsA, ZipA) and late divisomes (orchestrated by FtzK, FtsQ, FtsL, FtsB, FtsW, FtsN, FtsI - also known as PBP3, AmiC) at the plasma membrane (Erickson, Anderson, & Osawa, 2010). For a comprehensive review on this process, we refer the reader to Macheboeuf et al. (Macheboeuf, Contreras-Martel, Job, Dideberg, & Dessen, 2006).
Cell division can be inhibited through the clinically used monocyclic β-lactam antibiotic aztreonam, which targets peptidoglycan formation when that process is catalyzed by the division-specific PBP3 protein (S. Han et al., 2010); encouragingly, a novel monobactam compound, LYS228, is currently in clinical trials (Osborn et al., 2019). Peptidoglycan formation during cell division can also be interrupted by the small-molecule inhibitor divin (Eun et al., 2013), which disrupts the association of FtsQ with FtsB (Kureisaite-Ciziene et al., 2018), or through targeting of the tubulin homologue FtsZ (Nogales, Downing, Amos, & Löwe, 1998), a strategy that has been demonstrated to be effective in a study using the compound PC190723 against Staphylococcus spp (Haydon et al., 2008). Notably, TXA6101, an analogue of the latter FtsZ inhibitor, also exhibits activity against K. pneumoniae (Rosado-Lugo et al., 2022), while a related lead candidate, TXA709 (generated by Taxis Pharmaceuticals) successfully passed Phase I clinical trials for use against methicillin-resistant S. aureus.(Taxis Pharmaceuticals, 2023) These candidate drugs highlight the promise of targeting essential proteins or protein complexes involved in cell division and peptidoglycan synthesis for the development of new-in-class antibiotics (Egan et al., 2020; Lock & Harry, 2008).
While the components and the biogenesis of the peptidoglycan cell wall offer many tractable targets for antibiotic development, as demonstrated by the extraordinary success of β-lactam and glycopeptide drugs, resistance against such compounds is widespread through the bacterial phylogeny, posing an imminent threat to many invaluable antibiotics. Improved understanding of the early stages of peptidoglycan synthesis, and of the permutations of this process in different pathogens, could open new avenues for drug development that are immune to current mechanisms of resistance or for the inception of novel adjuvant strategies.
5. PROTEIN HOMEOSTASIS
5.1. Targeting periplasmic chaperones and proteases
Figure 4 and Figure 6 encompass some of the pathways described in this section.
Figure 6. Overview of the cell envelope chaperone/protease network and of the oxidative protein folding pathway.
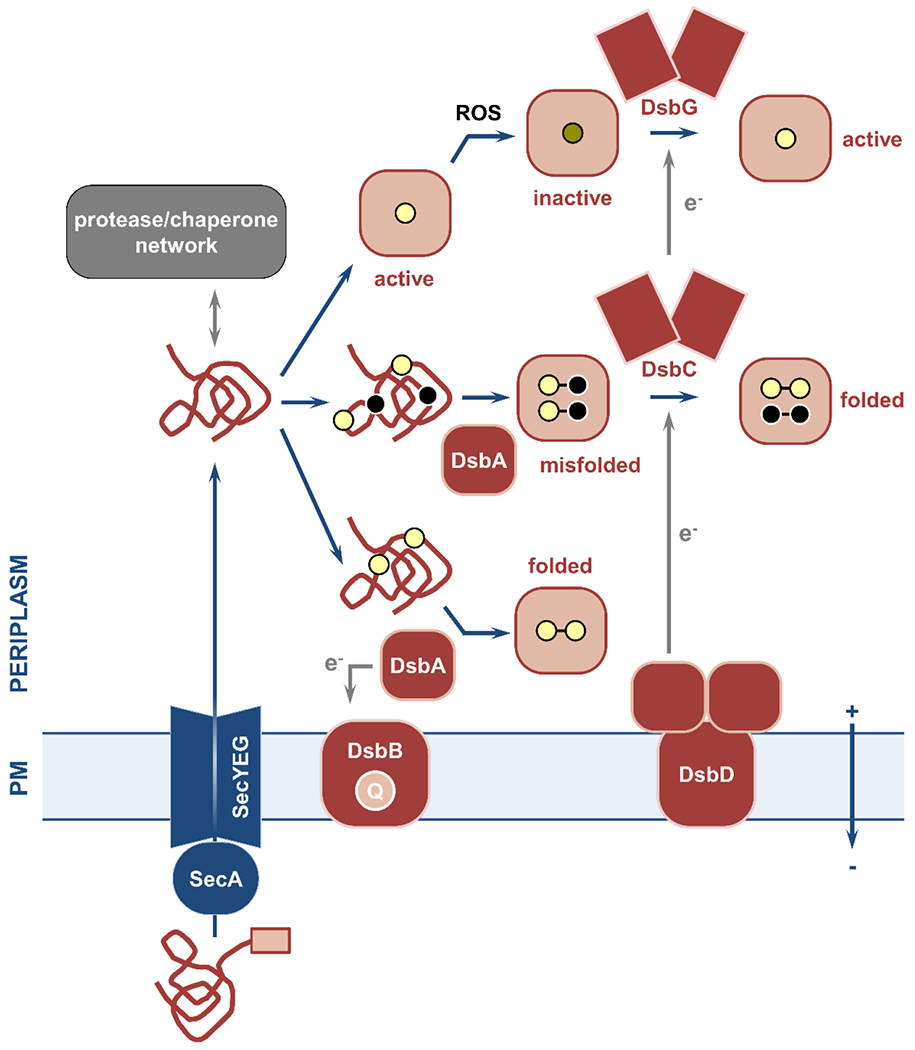
Nascent polypeptides with specific N-terminal signal sequences (pink box) are delivered to the Sec translocon and transported across the plasma membrane. (left) The periplasmic quality control network of chaperones and proteases ensures safe passage of these polypeptides to their final destinations and degradation of misfolded and aggregated proteins, respectively. (right) Oxidative protein folding in Gram-negative bacteria is carried out by the disulfide bond formation system. This process relies on the introduction of disulfide bonds between two consecutive cysteine residues (yellow circles) immediately after Sec-mediated protein translocation. Disulfide formation is catalyzed by the primary oxidase DsbA, which is then reinstated in its active oxidized form by its partner, the quinone (Q)-binding integral membrane protein DsbB. For proteins with only two cysteines (bottom) the introduction of a single disulfide bond results in correct folding. For substrates with more than two cysteines (middle), introduction of bonds in a sequential manner by DsbA may lead to the formation of non-native linkages and protein misfolding. In this case, the isomerase DsbC and its membrane-embedded partner DsbD use cytoplasmic reducing equivalents (e−) to aid refolding of the misfolded protein into its active form. The disulfide bond formation system also includes the reductase DsbG that reverses oxidative damage caused by reactive oxygen species (ROS) to proteins with single cysteines (top). PM, plasma membrane.
All the cell envelope biogenesis processes discussed in the previous sections rely on a wide network of chaperones and proteases that safeguard the integrity of this compartment against the harsh conditions of the extracytoplasmic space, namely low pH, high salt, and presence of oxidants and toxic byproducts of cellular metabolism. Chaperones direct their protein substrates to their final destinations, by identifying and handling misfolded proteins, whereas proteases manage the terminal cases by degrading protein aggregates. This section aims to provide a brief overview of the role and complex interactions of the main cell envelope quality control players, and to discuss their potential as druggable targets in novel antibiotic therapies. For a comprehensive overview of such pathways in Gram-negative bacteria we refer the reader to reviews by Goemans, Denoncin & Collet (Goemans, Denoncin, & Collet, 2014) and Stull, Betton & Bardwell (Stull, Betton, & Bardwell, 2018).
5.1.1. Periplasmic chaperones.
Transport and folding of outer-membrane proteins is primarily supported either by SurA or by the complementary Skp/DegP pathway (note that DegP functions primarily as a protease and will be discussed further in the following section). Partial complementarity between these two systems means that concurrent loss of members from both pathways is poorly tolerated and has deleterious effects (simultaneous loss of Skp and DegP) or leads to cell death (simultaneous loss of SurA and Skp, or SurA and DegP) (Ge et al., 2014; Goemans et al., 2014). The function of these primary systems is complemented by the ancillary chaperones Spy, HdeA/B, PpiA and FkpA in response to induction of the σE, Cpx, Bae, and RcsB stress responses (Stull et al., 2018).
SurA.
Arguably the most important periplasmic chaperone under normal growth conditions, SurA ensures the safe passage of Bam protein precursors to the outer membrane, a process essential for cell survival in stationary phase and important for growth in other cell cycle stages (Goemans et al., 2014). Moreover, like many other periplasmic chaperones, SurA has a secondary peptidyl-prolyl-cis-trans isomerase activity (Goemans et al., 2014). Absence of SurA causes changes in the composition of the outer membrane, reduction in virulence, and increased susceptibility to complement and antibiotic killing in E. coli, Salmonella and Pseudomonas (Bell, Zheng, & Ryno, 2018; Goemans et al., 2014). These effects are likely due to build up of unfolded proteins in the periplasm and are linked to increased dependence on the secondary Skp/DegP/FkpA pathway (Bell et al., 2018). In silico screening identified a potential, high-affinity SurA binder, Fmoc-β-(2-quinolyl)-D-alanine which, along with two derivatives of this molecule, opened up the possibility of generating other inhibitors against this chaperone (Bell et al., 2018).
Skp and FkpA.
The chaperones and holdases Skp and FkpA, together with SurA, rapidly unravel and refold outer-membrane porin aggregates and ensure their transport through the aqueous environment of the periplasm (Goemans et al., 2014). Both proteins also directly aid SurA with the folding of the essential protein LptD and the FhuA receptor; notably FkpA function in this process is indispensable since it cannot be complemented by either of the other two chaperones. (Schwalm, Mahoney, Soltes, & Silhavy, 2013; Thoma, Burmann, Hiller, & Müller, 2015). FkpA is also critical for counter-acting high temperature stress, whereby it rescues a double surA/skp mutant (Schwalm et al., 2013). Under stress conditions FkpA becomes the most important periplasmic chaperone, something that could be exploited if it were to be considered as a target for future antimicrobials. Finally, FkpA, like SurA, exhibits peptidyl-prolyl cis-trans isomerase activity, which has been successfully inhibited (Ramm & Plückthun, 2001). To our knowledge, no inhibitors have yet been identified for Skp.
HdeA and HdeB.
The cell envelope proteome is protected against low pH by the joint forces of the essential acid-activated chaperones HdeA and HdeB, which are supported by the activity of the protease DegP (Fu et al., 2018; Stull et al., 2018). Exposure to extremely low pH (pH:1-3) induces a conformational change in HdeA that enables binding and refolding of a wide variety of substrates, including other periplasmic chaperones (like DegP, DegQ, SurA, FkpA, LolA, DsbC, DsbG), thus preventing their denaturation (Stull et al., 2018; S. Zhang et al., 2016). Less extreme pH conditions (pH: 4-5) activate the partner protein of HdeA, HdeB. Denatured proteins that cannot be salvaged by either HdeA or HdeB are degraded by DegP (S. Zhang et al., 2016).
Other stress-induced chaperones include Spy, that protects the cell from protein aggregation in cases of chemical stress or under cell-wall damaging conditions, or the peptidyl-prolyl cis-trans isomerase PpiA, which plays a role in biofilm and motility regulation (Goemans et al., 2014; Pandey et al., 2016; Skagia et al., 2017). Overall, beyond SurA, which is a promising target, inhibition of secondary or stress-induced chaperones may have limited utility, for example in specific infection conditions or if simultaneous inhibition of multiple quality control proteins was possible.
Finally, several dedicated proteins link the plasma-membrane translocation systems to the soluble periplasmic chaperone network. For example, the widely conserved plasma-membrane-anchored chaperone YfgM and the peptidyl-prolyl-cis-trans isomerase PpiD are responsible for the transport of Sec-translocated proteins from the translocon to other periplasmic partners (Fürst, Zhou, Merfort, & Müller, 2018; Götzke et al., 2014; Jauss et al., 2019), while LolA transports lipoproteins across the cell envelope to the outer membrane (see section 3.2.2) (Goemans et al., 2014). These dedicated shuttles might be interesting antimicrobial targets, as shown by the work done on LolA (see below).
LolA.
High-throughput screening identified a potential, small-molecule LolA inhibitor, MAC13243, which increases the permeability of E. coli (Muheim et al., 2017) and a multi-drug resistant P. aeruginosa (Pathania et al., 2009) to vancomycin. It should be noted that while MAC13243 was confirmed to be a LolA inhibitor in an independent study (Kaplan, Greene, Jepson, & Koronakis, 2022), a recent publication proposed that the true target of this compound (and of another closely related molecule) is the plasma-membrane-embedded chaperone MreB, which is essential for peptidoglycan synthesis (Lehman et al., 2022); this discrepancy has not yet been resolved. Despite disagreement on the target of MAC13243, these studies demonstrate the potential of targeting chaperones as a means of inhibiting cell envelope biogenesis.
5.1.2. Periplasmic proteases.
Periplasmic chaperone function is supported by the activity of proteases that are responsible for the degradation of misfolded and aggregated proteins that cannot be rescued.
DegP.
DegP (also known as HtrA) is a key oligomeric serine protease that is essential for bacterial survival at high temperatures (>42 °C), but switches to having ATP-dependent chaperone function at low temperatures (<28 °C) (Spiess, Beil, & Ehrmann, 1999). DegP activity has also been linked to biofilm formation and pathogenesis in Salmonella enterica and P. aeruginosa (Xue et al., 2021). Loss of DegP increases sensitivity to acid and temperature stress (Fu et al., 2018; Goemans et al., 2014; He, Zhang, Liu, Xie, & Chen, 2019), while its overexpression results in growth arrest due to excessive proteolysis, as shown through the use of compounds that cause its hyperactivation (H. Cho et al., 2020). Although true inhibitors are yet to be developed, the central role of DegP in cell envelope proteostasis, in addition to its conservation even in Gram-positive species, make it a promising target for drug development (Jones, Bolken, Jones, Zeller, & Hruby, 2001; Wessler, Schneider, & Backert, 2017).
DegS.
DegS is an essential plasma-membrane-anchored serine protease (Alba & Gross, 2004; Waller & Sauer, 1996) that activates the σE periplasmic stress response through the identification of misfolded or mislocalized outer-membrane proteins (Bongard et al., 2019; Hasselblatt et al., 2007; Waller & Sauer, 1996). This, in turn, controls the expression of other chaperones, proteases, and lipid A biosynthesis genes that all help maintain the integrity of the cell envelope (Alba & Gross, 2004). Absence of DegS results in cell death via disruption of outer-membrane protein synthesis, making it a good antimicrobial target. Inhibitors include the commercially available broad-spectrum compound batimastat, which indirectly disrupts DegS signaling (Konovalova et al., 2018), and several small molecules that bind DegS non-covalently,synergize with colistin and have been shown to be active in vivo (Bongard et al., 2019).
DegQ.
The oligomeric serine protease DegQ is a partial homologue of DegP (Waller & Sauer, 1996) and is involved in the awakening of persister cells through degradation of the HokB toxin. Considering that eradication of persisters is challenging, DegQ might offer a promising target for development of therapeutics against these infections (Wilmaerts et al., 2019).
Prc.
Prc is part of a large family of specialized C-terminal-processing proteases and is responsible for the proteolytic turnover of the peptidoglycan hydrolase MepS and the transpeptidase PBP3 (Su et al., 2017). As such, loss of Prc leads to increased periplasmic leakage, sensitivity to osmotic stress, greater susceptibility to complement killing, and lower pathogenesis in uropathogenic E. coli (W.-C. Huang et al., 2020; Seoane, Sabbaj, McMurry, & Levy, 1992; C.-Y. Wang et al., 2012). These effects were shown to potentiate the activity of multiple antibiotics, although somewhat counterintuitively, there is no synergy reported for β-lactam drugs (G. C. Lai, Cho, & Bernhardt, 2017; Seoane et al., 1992). Despite the key role of Prc on the regulation of cell envelope formation and its promise as an antibacterial target, only one potential inhibitor, an eight-amino-acid peptide, is described in the literature (Beebe & Pei, 1998).
Another way to exploit periplasmic proteases for the generation of novel antibacterial strategies is to use them for the activation of conjugate drugs connected via a protease-cleavable linker (Boyce et al., 2020). These conjugates function akin to a prodrug; after entry into the periplasm the protease promotes the dissociation of the two parts of the compound and the release of the antibiotic component in the interior of the cell. Daptomycin-siderophore conjugates, where the siderophore promotes cell entry, were shown to sensitize A. baumannii to daptomycin, an antibiotic that normally is active only against Gram-positive bacteria (Boyce et al., 2020). Yet another application of proteases in this space was highlighted by the deployment of acyldepsipeptides that upregulate the cytoplasmic protease ClpP, which degrades the essential division protein FtsZ (Sass et al., 2011). Together, these strategies showcase the potential uses of both periplasmic and cytoplasmic proteases against the very bacteria that produce them.
In summary, the periplasmic protein quality control network likely provides a useful arena for the disruption of several pathways that are essential to cell envelope biogenesis (Justice et al., 2005). The complexity of this network, including the codependence, redundancy, and condition-specific activity of some of its members, is the reason behind its potential not being apparent. Although gaps in our understanding of the complex relationships between periplasmic chaperones and proteases need to be addressed before these proteins can form the basis for next-generation antibacterial strategies, their inherent redundancy denotes their importance for bacterial survival.
5.2. Targeting oxidative protein folding
Oxidative protein folding occurs in the periplasm and plasma membrane of Gram-negative bacteria and involves the formation and isomerization of covalent bonds between two spatially proximal cysteine amino acids. Formation of these linkages assists the folding and promotes the structural integrity of hundreds of cell envelope proteins that contain two or more cysteines and transverse the plasma membrane through the Sec system (Dutton, Boyd, Berkmen, & Beckwith, 2008; B. Heras, Kurz, Shouldice, & Martin, 2007; Vertommen et al., 2008). Despite disulfide bond formation being a seemingly simple reaction, requiring only the removal of two protons and two electrons, this process is catalyzed by a dedicated set of enzymes, jointly known as the disulfide bond formation (DSB) system (B. Heras et al., 2007). The prototypical Gram-negative DSB system, which is extensively characterized in E. coli K-12 (Landeta, Boyd, & Beckwith, 2018), is composed of complementary oxidative and the reductive pathways that control the introduction and isomerization of disulfides, respectively (see Figure 6).
5.2.1. The oxidative pathway.
Upon entering the periplasm, the first N-terminal cysteine of a Sec-translocated polypeptide forms a mixed-disulfide bond with the soluble periplasmic thiol oxidase DsbA. This intermediate complex is quickly resolved via disulfide transfer from DsbA to the polypeptide, leaving the oxidase to be restored in its active state by its partner, the inner-membrane quinone-binding protein DsbB (Messens & Collet, 2006). Electrons from the DsbA-DsbB thiol-disulfide exchange reaction are then shuttled through the DsbB quinone cofactor to the electron transport chain (Messens & Collet, 2006). Interestingly, there is a remarkable diversity in the DSB systems of Gram-negative bacteria. Extended DSB protein repertoires are present in pathogens, like N. meningitidis, P. aeruginosa, and S. enterica, where additional analogues, like DsbI/DsbL or SrgA, have specialized functions directly linked to virulence, host colonization, and intracellular survival (Begoña Heras et al., 2009; Yu, 1998). For this reason, DsbA and DsbB have been proposed as promising targets for the generation of broad-acting anti-virulence therapies (Begoña Heras et al., 2009; Yu, 1998). Recently, disulfide bond formation was discovered to also underpin antimicrobial resistance in Gram-negative bacteria (see section 6.4), highlighting the promise of targeting the DSB proteins for the development of resistance breakers (Furniss et al., 2022).
Given that the structure of the DsbA-DsbB complex had been determined at high resolution (Inaba et al., 2009; Inaba et al., 2006), peptides and peptidomimetics inhibiting its formation were pursued to inhibit DSB system function. Targeted design combined with virtual screening identified both covalent and non-covalent binders that could be used as scaffolds for further optimization (Duprez, Bachu, et al., 2015; Duprez, Premkumar, et al., 2015). Attempts to identify small-molecule inhibitors also led to several candidates that disrupted DsbB binding to its quinone cofactor or to DsbA (Früh et al., 2010; Halili et al., 2015). More recently, this work was expanded to DSB system inhibition in pathogens encoding multiple DsbA analogues through the use of phenylthiophene- and phenoxyphenol-based inhibitors with varied success (Totsika et al., 2018). Finally, high-throughput screening for DsbB inhibitors, led to the discovery of some of the most potent DSB-targeting compounds with activity against Enterobacteriaceae, P. aeruginosa, and M. tuberculosis (Landeta et al., 2015; Landeta et al., 2019).
5.2.2. The reductive pathway.
DsbA-mediated oxidative protein folding is limited to the linkage of consecutive cysteines and can, therefore, result in the formation of non-native disulfide bonds in polypeptides that contain more than two cysteines. A soluble thiol-disulfide isomerase, DsbC, and its dedicated plasma-membrane-embedded partner protein, DsbD, prevent protein misfolding and aggregation by reducing and correcting these erroneous linkages (Messens & Collet, 2006). Moreover, DsbC plays an integral role in copper detoxification (Hiniker, Collet, & Bardwell, 2005), a process for which dedicated thioredoxin systems have evolved in other species, like the proteins encoded by the scsABCD operon of S. enterica (Shepherd et al., 2013; Subedi et al., 2019). The reductive pathway also includes a DsbC homologue, DsbG, which acts as an early intermediate folding catalyst and protects single thiols from irreversible oxidation by reactive oxygen species. This is especially important for safeguarding catalytically active cysteine residues in transpeptidase enzymes, such as YbiS and ErfK (Depuydt et al., 2009; Shao, Bader, Jakob, & Bardwell, 2000). It should be noted though that in the absence of DsbC, or under stress conditions, the upregulation of DsbA, which belongs to the same protein superfamily as DsbC and DsbG, enables it to perform disulfide isomerization (Shouldice et al., 2010) which makes the proteins of the reductive pathway a less promising target for novel antimicrobials.
Although the DSB system is a classical Gram-negative cell envelope pathway, presence of its constituents is not limited to Gram-negative bacteria. For bacteria without a true periplasm, species-specific thiol-disulfide oxidation proteins localize at the interface between the plasma membrane and the peptidoglycan-teichoic/mycolic-acid cell wall. Examples include the MtDsbA-VKOR pair of M. tuberculosis and the Bdb system of Bacillus subtilis (Bolhuis, Venema, Quax, Bron, & Dijl, 1999; Landeta et al., 2018). Optimization of several small molecule-inhibitors has led to compounds capable of selective inhibition of these non-prototypical Gram-positive enzymes (Landeta et al., 2019). Overall, the current body of literature demonstrates that development of broad-acting inhibitors against the DSB system is challenging, while generation of species-specific inhibitors requires better understanding of the roles of DSB system polymorphisms and the simultaneous deployment of suitable diagnostics. That said, the broad involvement of DsbA in virulence and antimicrobial resistance (see section 6.4) highlight the potential of this non-essential system for next-generation therapies.
6. ANTIMICROBIAL RESISTANCE
A schematic overview of the processes discussed in this section can be found in Figure 7.
Figure 7. Overview of antimicrobial resistance determinants in the cell envelope.
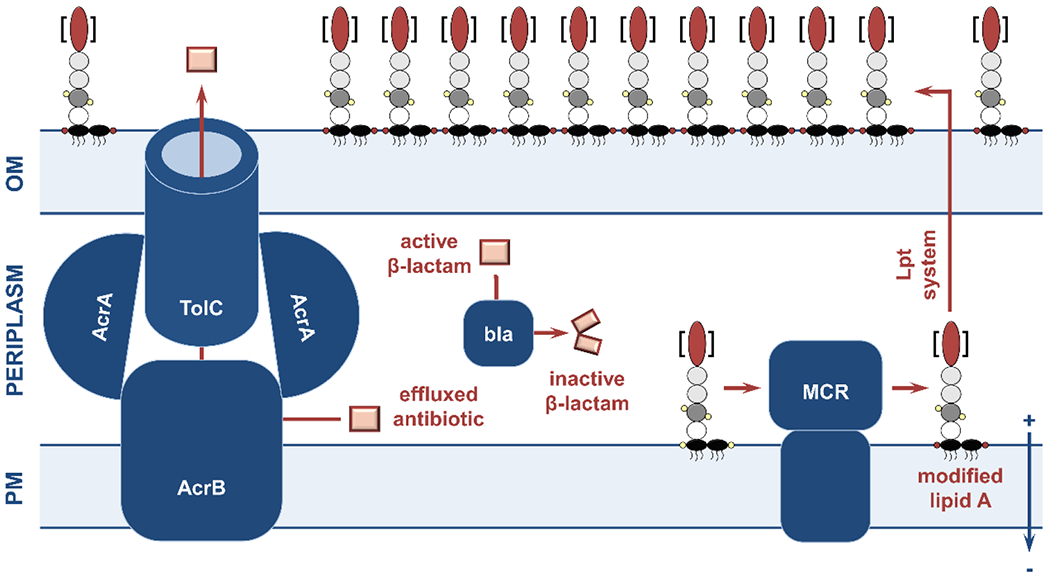
(left) Expression or upregulation of efflux pumps results in resistance to multiple antibiotic classes. The E. coli tripartite AcrAB-TolC pump depicted here is a typical Resistance-Nodulation-Division (RND) pump that extrudes fluoroquinolones, macrolides, and β-lactams from the periplasm to the extracellular milieu. (center) Soluble β-lactamase enzymes competitively bind β-lactam antibiotics in the periplasm and hydrolyze their essential four membered β-lactam ring, therefore preventing inhibition of peptidoglycan synthesis. (right) During LPS synthesis, the phosphate moieties (small yellow circles) of lipid A (black ovals) are modified by mobile colistin resistance (MCR) proteins (phosphoethanolamine addition is depicted by red circles on the lipid A component). This, in turn, reduces the overall charge of the outer membrane and obstructs the activity of last-line polymyxin antibiotics. OM, outer membrane; PM, plasma membrane.
6.1. Countering resistance to β-lactams
β-lactamases are, most commonly, soluble extra-cytoplasmic enzymes that catalyze the hydrolysis of the strained, and critical for activity, four-membered ring of β-lactam antibiotics. The diversity and evolvability of these hydrolases, as well as their prevalence across the bacterial phylogeny makes them a major threat to the longevity of all β-lactam antibiotics. β-lactamases are broadly categorized according to their hydrolytic activities into narrow-spectrum (e.g., SHV-1, TEM-1; hydrolyze first-generation penicillins and early cephalosporins), extended-spectrum (e.g. CTX-M-15, OXA-10; hydrolyze cephalosporins and monobactams), and carbapenemase (e.g. KPC-3, L1-1; hydrolyze last-resort carbapenems) enzymes. They are further classified based on their structures (Ambler classification) or using a combination of criteria pertaining to both their structural features and activity profiles (Bush-Jacoby-Medeiros classification) (Bush & Jacoby, 2010). For a more comprehensive overview of β-lactamase enzymes and their classification we refer the reader to the reviews by Tooke et al., Papp-Wallace, or Bush & Bradford (Bush & Bradford, 2019; Papp-Wallace, 2019; Tooke et al., 2019). For the purpose of this review, the simpler and more widely used Ambler classification system will be followed.
Briefly, Ambler class A, C and D β-lactamases rely on a conserved catalytic serine residue in their active site and have arisen from the structurally similar PBPs (see section 4.1.2) (Matagne, Lamotte-Brasseur, & FrÉRe, 1998). By contrast, class B β-lactamases are metallo-hydrolases that depend on one or two zinc ions for activity (Hong et al., 2015; Papp-Wallace, 2019) and are responsible for resistance in some of the most severe nosocomial infections (Boyd, Livermore, Hooper, & Hope, 2020). While archetypical narrow-spectrum enzymes are often found on chromosomal loci, many more recently discovered extended-spectrum β-lactamases and carbapenemases are harbored by plasmids or on mobile elements (Kurittu et al., 2021; Mahmud et al., 2022). Furthermore, it is not uncommon for bacteria to produce more than one β-lactamase, making certain pathogens completely resistant to all β-lactams, as seen for example in Stenotrophomonas maltophilia that encodes the co-evolved β-lactamases L1 and L2 with entirely complementary hydrolytic spectra (Brooke, 2012; Nakamura, Oota, Matsumoto, Sato, & Yamano, 2021).
6.1.1. Classical β-lactamase inhibitors.
The search for solutions against β-lactam-resistant infections has led to another success story for modern antibacterial drug development, the generation of β-lactamase inhibitors. These compounds were designed to be co-administered with existing β-lactam antibiotics and to bind, often irreversibly, into the active site of β-lactamases, abrogating their activity. The function of many class A enzymes can be inhibited through the use of clavulanic acid, tazobactam or sulbactam, otherwise known as classical β-lactamases inhibitors (Papp-Wallace, 2019). Unfortunately, evolved representatives of class A enzymes, such as members of the KPC and CTX-M families, along with most enzymes of classes B, C, and D, evade inhibition by classical inhibitors and have necessitated the development of broader and more potent adjuvant compounds (Bush & Bradford, 2019; Tooke et al., 2019).
6.1.2. New-generation β-lactamase inhibitors.
Over the last decade, new β-lactam-adjuvant combinations have been brough to the clinic, including ceftazidime, aztreonam/avibactam, meropenem/vaborbactam, and imipenem/cilastatin/relebactam (Papp-Wallace, 2019). Jointly, these adjuvants target the majority of class A and C enzymes, with avibactam also exhibiting activity against the slightly divergent class D species (Papp-Wallace, 2019).
Because of their unique structure and mechanism, class B metallo-β-lactamases continue to evade inhibition and pose a major challenge for multidrug-resistant infections (Tooke et al., 2019). Research is underway to identify new compounds capable of targeting class B enzymes, as well as enzymes from other classes that have evolved resistance to previously introduced inhibitor molecules. An interesting approach is based on the use of bismuth-containing compounds that force metal exchange in the metallo-β-lactamase active site, thus disrupting the activity of these enzymes in vitro and in vivo (R. Wang et al., 2018). In 2022, a highly anticipated cefepime-taniborbactam combination passed Phase III clinical trials and is intended for the treatment of complicated urinary tract infections; FDA approval is expected in 2023 (Pharmaceuticals, 2022). Taniborbactam exhibits efficacy against metallo-β-lactamases, including the broadly disseminated NDM and VIM families (Liu et al., 2020). Other compounds currently under development, such as the boronic acid inhibitor VNRX-7145 (Trout et al., 2021) and the sulbactam-durlobactam combination for treatment of A. baumannii, are in Phase I and Phase III trials, respectively (Entasis, 2022; Granata, Taglietti, Schiavone, & Petrosillo, 2022). Most recently, a siderophore-conjugated thiol-based compound was also shown to inhibit class B1 metallo-β-lactamases (Rotter et al., 2023).
6.2. Countering resistance to colistin
Until recently, colistin resistance was predominantly associated with mutations in chromosomal two-component pathways, like PmrA/PrmB and PhoP/PhoQ, or their regulators, such as MgrB, which result in the upregulation of native ArnT glycosyltranferases or EptA phosphoethanolamine transferases (H. Zhang, Srinivas, Xu, Wei, & Feng, 2019). More recently, zinc-dependent mobile colistin resistance (MCR) proteins, similar to EptA enzymes, emerged in clinical isolates, with ten distinct families identified in pathogens like E. coli, S. enterica, K. pneumoniae and E. cloacae (H. Zhang et al., 2019). Both ArnT glycosyltransferases and phosphoethanolamine enzymes (EptA or MCR) modify the phosphate groups of the lipid A component of the LPS through the addition of 4-amino-4-deoxy-L-arabinose or ethanolamine moieties, respectively. When modified LPS is integrated into the outer membrane, the overall surface charge of the bacterium decreases. This alters the interaction of colistin with the cell surface, protecting the cell from lysis and causing antibiotic resistance (H. Zhang et al., 2019). Unlike chromosomally harbored arnT and eptA genes, genes encoding MCR enzymes are exclusively found on mobilizable genetic elements (predominantly plasmids) that allow their rapid spread among Gram-negative bacteria through horizonal gene transfer. Therefore, the emergence of these resistance proteins threatens the longevity of colistin, which is an invaluable last-resort drug against carbapenem-resistant pathogens (Du, Chen, Tang, & Kreiswirth, 2016).
Several inhibitors have been described against MCR proteins to date. A previously explored strategy, whereby catalytically critical metal ions are exchanged by silver, was shown to inhibit MCR-1 activity in several Gram-negative pathogens, and combination treatment with colistin succeeded in clearing K. pneumoniae in vivo.(Q. Zhang et al., 2022). Although use of free metals is usually considered unsuitable for therapy, the low concentration of silver required for its use as an adjuvant might allow topical applications (Q. Zhang et al., 2022). Synergistic effects with colistin against resistant Enterobacteriaceae were also observed for several natural products, such as osthole (Y. Zhou et al., 2019), pterostilbene (Y. Zhou et al., 2018), honokiol (Guo et al., 2020), and cajanin stilbene acid (Jia et al., 2023). More recently, pyrazolone (Hanpaibool et al., 2023) and phenyl-2-(phenylamino)-ethanone (Lan et al., 2019) scaffolds were identified as potential inhibitors through a combination of microbiological assays and molecular simulations. Together the above studies have helped determine appropriate MCR binding sites, demonstrated the possibility of targeting these resistance proteins, and identified scaffolds for future inhibitor development. Development of MCR protein inhibitors represents a critical step towards prolonging the efficacy colistin, as well as achieving colistin potentiation against bacterial pathogens that are intrinsically resistant to this antibiotic like Serratia marcescens or N. meningitidis (Gogry, Siddiqui, Sultan, & Haq, 2021).
6.3. Countering efflux pumps
Efflux pumps are essential for the upkeep of cytoplasmic and periplasmic homeostasis. Six families of efflux pumps have been described to date (ABC, MFS, MATE, SMR, RND, and PACE pumps (Hassan, Elbourne, et al., 2015; Hassan, Liu, Henderson, & Paulsen, 2015)), with the majority being single-component plasma-membrane apparatuses that remove metabolic side products from the cytoplasm (Aeschlimann, 2003; Bazzini et al., 2011; Nikaido, 2016; Poole, 2001; J. Sun, Deng, & Yan, 2014). Several complex tripartite efflux systems, comprising a cytoplasmic pump, an outer-membrane channel, and a periplasmic adaptor protein, span the cell envelope and promote extracellular extrusion. These include the ATP-binding-cassette (ABC) transporters and the more notable Resistance-Nodulation-Division (RND) pumps, like AcrAB-TolC found in E. coli and Salmonella typhimurium, or the MexAB-OprM pump of P. aeruginosa, that remove toxic molecules and antibiotics, such as fluoroquinolones, aminoglycosides, or β-lactams from the periplasm (Nikaido, 2016; Opperman & Nguyen, 2015; Paulsen, 2003; Poole, 2007). Copper and silver ion extrusion can also be driven by dedicated efflux systems, like the CusCFBA pump in E. coli (Rademacher & Masepohl, 2012). Many bacterial species encode more than one efflux pump (Dawson & Locher, 2006), and while generally pump genes are chromosomally encoded, specialized pumps are sometimes found on resistance plasmids (Laws, Shaaban, & Rahman, 2019) where cooperation between these pumps and other resistance determinants results in extensive antibiotic resistance. These characteristics make efflux pumps a long-standing target for antibiotic and adjuvant drug development (Poole, 2001). For a more thorough overview on the expansive landscape of efflux pumps, their structures and mechanisms of action, we refer the reader to several excellent reviews (Anes, McCusker, Fanning, & Martins, 2015; Laws et al., 2019; Poole, 2007; Thakur et al., 2021).
Despite years of extensive research, only a few successful efflux pump inhibitors have been discovered. Development of such compounds is hampered by toxicity issues arising from similarities between eukaryotic and prokaryotic pumps, functional redundancy between pumps expressed by the same organisms, and lack of suitable and efficient activity assays that can help guide rational drug design (Lv et al., 2021; Roy et al., 2021; J. Sun et al., 2014). The most successful pump inhibitors rely on decoupling the pump from its energy source, steric inhibition of the substrate binding sites, or suppression of pump expression, and are discussed briefly below. For a more comprehensive overview of efflux pump inhibitors we refer the reader to the review by Sharma, Gupta & Pathania (A. Sharma et al., 2019).
6.3.1. Energy decoupling.
The ABC transporters derive their energy from the hydrolysis of ATP (Lubelski, Konings, & Driessen, 2007), the RND, SMR, MFS and likely also the PACE pumps rely on the PMF, while the MATE pumps depend either on the PMF or on a sodium ion antiporter system (Piddock, 2006). As such, disruption of energy production or dissipation of the plasma membrane potential as discussed previously (see section 2.2.2) is promising avenue for targeting these apparatuses.
6.3.2. Steric inhibition.
Small-molecule inhibitors binding directly to efflux pump components, generally known as EPIs (efflux pump inhibitors), are mostly alkaloid, flavonoid, polyphenol, quinolone, aryl and heterocyclic derivatives, and can be categorized based on whether they have competitive or non-competitive mechanisms of action (Laws et al., 2019; A. Sharma et al., 2019).
The competitive inhibitor Phe-Arg β-naphthylamide (PaβN), potentiates erythromycin and chloramphenicol against P. aeruginosa and E. coli (Lomovskaya et al., 2001), likely through interaction with the AcrB cytoplasmic pump component (Jamshidi, Sutton, & Rahman, 2017; Kinana, Vargiu, May, & Nikaido, 2016; Vargiu & Nikaido, 2012). Controversially, PaβN has also been linked to increased membrane permeability and transient membrane depolarization, raising doubts about whether it is an actual direct efflux pump inhibitor (Lamers, Cavallari, & Burrows, 2013). This is a commonly encountered issue with inhibitors of bacterial efflux because distinguishing inhibitors that impair pumps by directly binding to their components from compounds that cause efflux defects through energy decoupling is challenging. For example, CCCP, verapamil, or IITR08027 are commonly labelled as EPIs, but have all recently been reclassified as molecules that cause indirect efflux effects through PMF disruption.
The non-competitive inhibitor 1-(1-Naphthylmethyl)-piperazine (NMP), which potentiates multiple effluxed antibiotic classes against E. coli (Bohnert & Kern, 2005) is more likely to be a true pump inhibitor, given that error prone PCR on acrB produced NMP-resistant mutants and helped identify the binding site on this pump component (Anes, Sivasankaran, Muthappa, Fanning, & Srikumar, 2019; A. Sharma et al., 2019; Vargiu & Nikaido, 2012). The FDA-approved drug chlorpromazine, initially developed for treating schizophrenia, also inhibits AcrB, but is unsuitable for use due to toxicity issues (Grimsey et al., 2020). More recently, a novel set of pyanopyridine compounds was crystalized in the E. coli AcrB substrate-binding domain (Sjuts et al., 2016), and a ramA:gfp reporter fusion combined with high-throughput screening identified potentiators that sensitized E. coli and Salmonella to chloramphenicol (Marshall et al., 2020). These results, along with previously identified inhibitors suggest that AcrB is a promising target for counteracting the activity of RND tripartite pumps. Finally, the cationic AMP plantaricin A, which is thought to increase membrane permeability, was reported to synergize with a variety of antibiotics and to inhibit ciprofloxacin efflux by disrupting the folding of an S. aureus efflux pump (Meng et al., 2022). Notably, this peptide also exhibits low toxicity against eukaryotic cells highlighting its potential for further development (Meng et al., 2022).
6.3.3. Gene regulation.
Targeting bacterial efflux through controlling gene expression has been proposed as an alternative strategy for abrogating pump function. Although this is not a straightforward task, primarily because of the large number of pump genes that are scattered across bacterial genomes, it can be achieved by targeting efflux pump signaling pathways and regulatory systems, by inhibiting pump gene transcription, or through binding of relevant transcription factors.
Research into terpenes, natural components of essential oils, identified derivatives capable of blocking the expression of specific efflux systems in several pathogens (Dias et al., 2022). For example, clerodane diterpene dampens the expression of the key S. aureus pump NorA, which is involved in fluoroquinolone resistance (V. K. Gupta et al., 2016). The FDA-approved hypertension medication reserpine can also stop the production of the same pump, but is highly toxic at the concentrations required for inhibition (V. K. Gupta et al., 2016; Neyfakh, Borsch, & Kaatz, 1993; Schmitz et al., 1998). Similarly, andrographolide is associated with downregulation of mexB, a component of the MexAB-OprM pump from P. aeruginosa (C. M. Wu et al., 2007), while celastrol inhibits smeYZ, which is part of an RND pump found in S. maltophilia (H.-R. Kim, Lee, & Eom, 2018). A slightly different approach, based on the use of a synthetic DNA mimic (a peptide nucleic acid) decreased the transcription of the specific efflux gene cmeA in Campylobacter jejuni, resulting in sensitization of this organism to ciprofloxacin and erythromycin (Jeon & Zhang, 2009). Together these studies show the potential of approaches that target efflux gene expression. Nonetheless, since thorough understanding of the complex transcriptional and translational regulation of efflux systems remains elusive, such strategies will likely require significant time investment in the future.
6.3.4. Biofilm formation.
Bacterial efflux is strongly associated with biofilm formation, making resistant infections even harder to treat. For a more comprehensive overview of this relationship we refer the reader to the review by Alav, Sutton & Rahamn (Alav, Sutton, & Rahman, 2018). Based on this link, impairment of efflux through the use of thioridazine, PaβN, or NMP decreases the formation of biofilm in E. coli and K. pneumoniae (Kvist, Hancock, & Klemm, 2008), while a similar effect was reported for the application of the triterpenoid celastrol against S. malthophilia (H.-R. Kim et al., 2018).
Overall, development of efflux pump inhibitors requires thorough knowledge of the diversity and regulation of these systems in the context of bacterial infections. That said, the efforts to identify molecules with activity against efflux pumps inadvertently highlighted avenues for antimicrobial therapies that extend beyond the inhibition of these apparatuses and led to the discovery of compounds capable of disrupting energy production, cellular homeostasis, and biofilm formation. Jointly, these effects are likely to have a much greater impact in the treatment of persistent and multidrug-resistant infections compared to the inhibition of bacterial efflux.
6.4. Double-arrow strategies
Novel adjuvant strategies that simultaneously disable at least two unrelated antibiotic resistance determinants, thus potentiating multiple antibiotic classes, have recently been proposed (Furniss et al., 2022; Oliveira-Tintino et al., 2023; H. Sun et al., 2020). With traditional adjuvant compounds having activity spectra that are limited within a specific resistance protein family, these double-arrow approaches are unprecedented and full of promise.
In 2020, the FDA-approved antirheumatic drug auranofin was shown to inhibit the function of both metallo-β-lactamases and MCR enzymes (H. Sun et al., 2020). This gold(I)-based compound interacts with the active-site cysteine residue in VIM and NDM β-lactamases and displaces their catalytically active zinc ion (H. Sun et al., 2020). In a similar fashion, it coordinates with the zinc-binding residues of the MCR active site and disrupts the phosphoethanolamine cleavage step, ultimately preventing lipid A modification and subsequent colistin resistance (H. Sun et al., 2020). It was demonstrated that auranofin can sensitize Enterobacteriaceae to the carbapenem antibiotic meropenem, and to colistin (H. Sun et al., 2020). Moreover, combination of this molecule with colistin decreases the K. pneumoniae load and completely eradicates E. coli in a murine model of infection (H. Sun et al., 2020). More recently, it was found that auranofin has activity against other critical Gram-negative pathogens, including Neisseria gonorrhoeae and A. baumannii (Elhassanny, Abutaleb, & Seleem, 2022; H. R. Kim & Eom, 2022). Several clinical trials are now underway, so as to evaluate the pharmacokinetic profile of this compound and its potential to be repurposed as a novel dual-action adjuvant for the treatment of multidrug-resistant infections (Y. Liu et al., 2022).
In 2022, the oxidative protein folding pathway (see section 5.2) was demonstrated to be a promising target for disabling antibiotic resistance caused both by β-lactamases and MCR proteins (Furniss et al., 2022). Loss of dsbA resulted in inactivation of numerous clinically important, broad-spectrum class A, B, and D β-lactamases, including carbapenemases from the KPC family and the metallo-β-lactamase L1 from S. maltophilia (Furniss et al., 2022). This effect was shown to be due to the fact that β-lactamases lacking their disulfide bonds were either degraded or misfolded. Similarly, absence of DsbA destabilized MCR proteins and abrogated colistin resistance (Furniss et al., 2022). Chemical inhibition of DsbB (see section 5.2.1), the partner protein of DsbA, sensitized clinical strains of Enterobacteriaceae to imipenem and colistin, while dsbA1 deletion in multidrug-resistant P. aeruginosa isolates led to potentiation of first-line anti-pseudomonal β-lactams and increased survival in a Galleria mellonella infection model (Furniss et al., 2022). Interestingly, impairment of cell envelope proteostasis through disruption of oxidative protein folding also indirectly affected bacteria efflux, leading to sensitization of E. coli to chloramphenicol (Furniss et al., 2022). Together, these findings highlight the central role of systems that safeguard cell envelope protein homeostasis (see section 5) for resistance and highlight their inhibition as a promising strategy for the deployment of much-needed broad-acting resistance breakers.
Finally, in 2023, a 1,8-naphthyridine scaffold was shown to inhibit an S. aureus β-lactamase along with the species-specific QacABC efflux pump, although its mechanism of action has not yet been elucidated (Oliveira-Tintino et al., 2023). Together, approaches that target conserved features among resistance proteins, such as metal-binding pockets or disulfide bonds, hold promise for the development of next-generation adjuvants with the capacity to evade the pitfalls of traditional inhibitors, where high target specificity can give rise to resistance evolution. Given the prevalence of antimicrobial plasmids carrying numerous resistance genes, the possibility of simultaneously disabling multiple mechanisms of antimicrobial resistance is exciting.
7. CONCLUDING REMARKS
Some of the most important antibiotics in clinical use target the Gram-negative cell envelope. Nonetheless, while the antibiotic discovery pipeline has been running dry, deliberate efforts to inhibit the constituents of this compartment or impair its biogenesis have been limited. The adherence to traditional antibiotic discovery strategies established in the 20th century, along with the pursuit of the “ideal antibiotic target” and the search for antibacterial “silver bullets”, has become obsolete in the current era of resistance emergence and dwindling antimicrobial resources (Hurdle et al., 2011; Quadri, 2007; Silver, 2016). It is only the unprecedented success of β-lactamase inhibitors, which highlighted the promise of strategic development of therapeutics that do not impede essential bacterial processes, that enabled a critical shift in the antibacterial drug discovery paradigm, and that is allowing us to, somewhat, keep pace with antibiotic-resistant bacteria.
Alternative strategies that can complement the conventional antibiotic discovery pipeline need to be investigated. To this effect, development of combination therapy approaches should be pursued more actively, while generation of conjugate or chimeric therapeutics, essentially antibiotic-adjuvant combinations condensed into a single molecule, should be at the forefront of drug discovery (Luther et al., 2019). The “mix-and-match” nature of the latter allows the amalgamation of desired pharmacokinetic and pharmacodynamic properties, resulting in increased efficacy, decreased toxicity, and limited resistance evolution. For example, a murepavidin β-hairpin peptide derivative combined with a polymyxin B tail fragment is effective against a range of clinically important Gram-negative pathogens, with the advantage of significantly lower kidney toxicity compared to its parent compounds (Luther et al., 2019). Unrealized potential also lies in the use of tailored or species-specific compounds, especially ones aimed at highly resistant bacteria like A. baumannii (Brochado et al., 2018; Shim, 2023), or at the development of broader-acting alternative approaches to target the outer membrane, like light-activatable membrane-disrupting nano molecules (Ornes, 2023; Santos et al., 2022), phage therapy (Broncano-Lavado, Santamaría-Corral, Esteban, & García-Quintanilla, 2021), or antibody-based drugs (Mullard, 2021). The success of these therapies also depends on the simultaneous development of rapid and accurate diagnostic methods that will allow timely identification of the organism or target of interest, therefore decreasing the risk of treatment failure and resistance evolution.
The continuous emergence of antibiotic resistance will always necessitate the deployment of new antibiotic strategies or chemical scaffolds against traditional targets. Nonetheless, exploitation of unusual and underexplored pathways, with the aim of abrogating resistance, attenuating virulence (Dickey, Cheung, & Otto, 2017; Sionov & Steinberg, 2022), or eradicating untreatable intrinsically-resistant pathogens should also be pursued. The recently discovered double-arrow adjuvant approaches, that can be used to simultaneously impair multiple resistance mechanisms, are an example of such unexpected breakthroughs (Furniss et al., 2022; H. Sun et al., 2020). Ultimately, a diversified portfolio of antibiotic and adjuvant options, supported by appropriate and efficient diagnostic technologies is likely to stand us in a better stead towards safeguarding current and future antibiotic repertoires in the race against antibiotic-resistant bacterial pathogens.
FUNDING INFORMATION:
Publication of this review was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI158753 (to D.A.I.M.); the content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- ABC
ATP-binding-cassette
- AMPs
Antimicrobial peptides
- ATP
Adenosine triphosphate
- Bam
β-barrel assembly machinery
- CCCP
Carbonyl cyanide-m-chlorophenylhydrazone
- CL
Cardiolipin
- DCAP
2-((3-(3,6-dichloro-9H-carbazol-9-yl)-2-hydroxypropyl)amino)-2-(hydroxymethyl)propane-1,3-diol
- DCCD
N,N′-Dicyclohexylcarbodiimide
- DSB
Disulfide bond formation
- EPI-I
Epinecidin I
- EPIs
Efflux pump inhibitors
- ES1
Eyarestatin 1
- FDA
Food and Drug Administration
- GTP
Guanosine-5’-triphosphate
- Lol
Localization of lipoproteins
- Lpp
Braun’s lipoprotein
- LPS
Lipopolysaccharide
- Lpt
Lipopolysaccharide transport
- MATE
Multidrug and toxic compound extrusion
- MCR
Mobile colistin resistance
- MFS
Major facilitator superfamily
- NADH
Nicotinamide adenine dinucleotide (NAD) + hydrogen (H)
- NMP
1-(1-Naphthylmethyl)-piperazine
- OM
Outer membrane
- OMP
Outer membrane protein
- PACE
Proteobacterial antimicrobial compound efflux
- PaβN
Phe-Arg β-naphthylamide
- PBPs
Penicillin-binding proteins
- PE
Phosphatidylethanolamine
- PG
Phosphatidylglycerol
- PG
Peptidoglycan
- PM
Plasma membrane
- PMF
Proton motive force
- RND
Resistance-Nodulation-Division
- Sec
General secretory pathway
- SMR
Small multidrug resistance
- Tat
Twin-arginine translocation
- ΔpH
Transmembrane chemical proton gradient
- ΔΨ
Membrane electric potential
Footnotes
CONFLICTS OF INTEREST: The authors declare that there are no conflicts of interest.
REFERENCES
- Achaogen I (2013). A study to assess the safety, tolerability, and pharmacokinetics of ACHN-975 in healthy volunteers. from https://clinicaltrials.gov/ct2/show/NCT01597947, urldate = 2023-03-04
- Achouak W, Heulin T, & Pagès JM (2001). Multiple facets of bacterial porins. FEMS Microbiology Letters, 199(1), 1–7. [DOI] [PubMed] [Google Scholar]
- Aeschlimann JR (2003). The role of multidrug efflux pumps in the antibiotic resistance of Pseudomonas aeruginosa and other Gram-negative bacteria. Pharmacotherapy, 23(7), 916–924. [DOI] [PubMed] [Google Scholar]
- Agar SL, Sha J, Baze WB, Erova TE, Foltz SM, Suarez G, et al. (2009). Deletion of Braun lipoprotein gene (lpp) and curing of plasmid pPCP1 dramatically alter the virulence of Yersinia pestis CO92 in a mouse model of pneumonic plague. Microbiology, 155(10), 3247–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alav I, Sutton JM, & Rahman KM (2018). Role of bacterial efflux pumps in biofilm formation. Journal of Antimicrobial Chemotherapy, 73(8), 2003–2020. [DOI] [PubMed] [Google Scholar]
- Alba BM, & Gross CA (2004). Regulation of the Escherichia coli σE-dependent envelope stress response. Molecular Microbiology, 52(3), 613–619. [DOI] [PubMed] [Google Scholar]
- Alexander MK, Miu A, Oh A, Reichelt M, Ho H, Chalouni C, et al. (2018). Disrupting Gram-negative bacterial outer membrane biosynthesis through inhibition of the lipopolysaccharide transporter MsbA. Antimicrobial Agents and Chemotherapy, 62(11), e01142–01118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MS, & Raetz CR (1987). Biosynthesis of lipid A precursors in Escherichia coli. A cytoplasmic acyltransferase that converts UDP-N-acetylglucosamine to UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine. Journal of Biological Chemistry, 262(11), 5159–5169. [PubMed] [Google Scholar]
- Andrade FF, Silva D, Rodrigues A, & Pina-Vaz C (2020). Colistin update on its mechanism of action and resistance, present and future challenges. Microorganisms, 5(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anes J, McCusker MP, Fanning S, & Martins M (2015). The ins and outs of RND efflux pumps in Escherichia coli. Frontiers in Microbiology, 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anes J, Sivasankaran SK, Muthappa DM, Fanning S, & Srikumar S (2019). Exposure to Sub-inhibitory Concentrations of the Chemosensitizer 1-(1-Naphthylmethyl)-Piperazine Creates Membrane Destabilization in Multi-Drug Resistant Klebsiella pneumoniae. Frontiers in Microbiology, 10, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaout TE, & Soulimane T (2019). Targeting lipoprotein biogenesis: considerations towards antimicrobials. Trends in Biochemical Sciences, 44(8), 701–715. [DOI] [PubMed] [Google Scholar]
- Babinski KJ, Kanjilal SJ, & Raetz CRH (2002). Accumulation of the lipid A precursor UDP-2,3-diacylglucosamine in an Escherichia coli mutant lacking the lpxH gene. Journal of Biological Chemistry, 277(29), 25947–25956. [DOI] [PubMed] [Google Scholar]
- Babinski KJ, Ribeiro AA, & Raetz CRH (2002). The Escherichia coli gene encoding the UDP-2,3-diacylglucosamine pyrophosphatase of lipid A biosynthesis. Journal of Biological Chemistry, 277(29), 25937–25946. [DOI] [PubMed] [Google Scholar]
- Bageshwar UK, VerPlank L, Baker D, Dong W, Hamsanathan S, Whitaker N, et al. (2016). High throughput screen for Escherichia coli twin arginine translocation (Tat) inhibitors. PloS One, 11(2), 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker EP, & Mangerich WE (1981). Interconversion of components of the bacterial proton motive force by electrogenic potassium transport. Journal of Bacteriology, 147(3), 820–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Huryn DM, & Smith AB (2013). Carboxylic acid (bio)isosteres in drug design. ChemMedChem, 8(3), 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleza D, & Gómez-Lagunas F (2008). Conserved motifs in mechanosensitive channels MscL and MscS. European Biophysics Journal, 38(7), 1013–1027. [DOI] [PubMed] [Google Scholar]
- Barh D, Jain N, Tiwari S, Parida BP, D’Afonseca V, Li L, et al. (2011). A novel comparative genomics analysis for common drug and vaccine targets in Corynebacterium pseudotuberculosis and other CMN group of human pathogens. Chemical Biology & Drug Design, 78(1), 73–84. [DOI] [PubMed] [Google Scholar]
- Barreto-Santamaría A, Arévalo-Pinzón G, Patarroyo MA, & Patarroyo ME (2021). How to combat Gram-negative bacteria using antimicrobial peptides: a challenge or an unattainable goal? Antibiotics, 10(12), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartling CM, & Raetz CRH (2008). Steady-state kinetics and mechanism of LpxD, the N-acyltransferase of lipid A biosynthesis. Biochemistry, 47(19), 5290–5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini S, Udine C, Sass A, Pasca MR, Longo F, Emiliani G, et al. (2011). Deciphering the role of RND efflux transporters in Burkholderia cenocepacia. PloS One, 6(4), e18902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechinger B, & Gorr SU (2017). Antimicrobial peptides: mechanisms of action and resistance. Journal of Dental Research, 96(3), 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe KD, & Pei D (1998). A continuous fluorimetric assay for tail-specific protease. Analytical Biochemistry, 263(1), 51–56. [DOI] [PubMed] [Google Scholar]
- Bell EW, Zheng EJ, & Ryno LM (2018). Identification of inhibitors of the E. coli chaperone SurA using in silico and in vitro techniques. Bioorganic and Medicinal Chemistry Letters, 28(22), 3540–3548. [DOI] [PubMed] [Google Scholar]
- Bertani B, & Ruiz N (2018). Function and biogenesis of lipopolysaccharides. EcoSal Plus, 8(1), 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertsche U, Breukink E, Kast T, & Vollmer W (2005). In vitro murein (peptidoglycan) synthesis by dimers of the bifunctional transglycosylase-transpeptidase PBP1B from Escherichia coli. Journal of Biological Chemistry, 280(45), 38096–38101. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya T, Sharma A, Akhter J, & Pathania R (2017). The small molecule IITR08027 restores the antibacterial activity of fluoroquinolones against multidrug-resistant Acinetobacter baumannii by efflux inhibition. International Journal of Antimicrobial Agents, 50(2), 219–226. [DOI] [PubMed] [Google Scholar]
- Blount P, & Iscla I (2020). Life with Bacterial Mechanosensitive Channels, from Discovery to Physiology to Pharmacological Target. Microbiol Mol Biol Rev, 84(1), e00055–00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blümmel A-S, Drepper F, Knapp B, Eimer E, Warscheid B, Müller M, et al. (2017). Structural features of the TatC membrane protein that determine docking and insertion of a twin-arginine signal peptide. Journal of Biological Chemistry, 292(52), 21320–21329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Pyrshev K, Yesylevskyy S, Ryabichko S, Boiko V, Ivanchenko P, et al. (2020). Phospholipid distribution in the cytoplasmic membrane of Gram-negative bacteria is highly asymmetric, dynamic, and cell shape-dependent. Science Advances, 6(23), eaaz6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert J. r. A., & Kern WV (2005). Selected arylpiperazines are capable of reversing multidrug resistance in Escherichia coli overexpressing RND efflux pumps. Antimicrobial Agents and Chemotherapy, 49(2), 849–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhringer N, Green R, Liu Y, Mettal U, Marner M, Modaresi SM, et al. (2021). Mutasynthetic production and antimicrobial characterization of darobactin analogs. Microbiology Spectrum, 9(3), e01535–01521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolhuis A, Venema G, Quax WJ, Bron S, & Dijl J. M. v. (1999). Functional analysis of paralogous thiol-disulfide oxidoreductases in Bacillus subtilis. Journal of Biological Chemistry, 274(35), 24531–24538. [DOI] [PubMed] [Google Scholar]
- Bongard J, Schmitz AL, Wolf A, Zischinsky G, Pieren M, Schellhorn B, et al. (2019). Chemical validation of DegS a a target for the development of antibiotics with a novel mode of action. ChemMedChem, 14(11), 1074–1078. [DOI] [PubMed] [Google Scholar]
- Bonnemain C, Raynaud C, Réglier-Poupet H, Dubail I, Frehel C, Lety MA, et al. (2004). Differential roles of multiple signal peptidases in the virulence of Listeria monocytogenes. Molecular Microbiology, 51(5), 1251–1266. [DOI] [PubMed] [Google Scholar]
- Booth IR, & Blount P (2012). The MscS and MscL families of mechanosensitive channels act as microbial emergency release valves. Journal of Bacteriology, 194(18), 4802–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born P, Breukink E, & Vollmer W (2006). In vitro synthesis of cross-linked murein and its attachment to sacculi by PBP1A from Escherichia coli. Journal of Biological Chemistry, 281(37), 26985–26993. [DOI] [PubMed] [Google Scholar]
- Botos I, Majdalani N, Mayclin Stephen J., McCarthy Jennifer G., Lundquist K, Wojtowicz D, et al. (2016). Structural and functional characterization of the LPS transporter LptDE from Gram-negative pathogens. Structure, 24(6), 965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce JH, Dang B, Ary B, Edmondson Q, Craik CS, DeGrado WF, et al. (2020). Platform to discover protease-activated antibiotics and application to siderophore–antibiotic conjugates. Journal of the American Chemical Society, 142(51), 21310–21321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd SE, Livermore DM, Hooper DC, & Hope WW (2020). Metallo-β-Lactamases: structure, function, epidemiology, treatment options, and the development pipeline. Antimicrobial Agents and Chemotherapy, 64(10), e00397–00320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochado AR, Telzerow A, Bobonis J, Banzhaf M, Mateus A, Selkrig J, et al. (2018). Species-specific activity of antibacterial drug combinations. Nature, 559(7713), 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogden KA (2005). Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nature Reviews Microbiology, 3(3), 238–250. [DOI] [PubMed] [Google Scholar]
- Broncano-Lavado A, Santamaría-Corral G, Esteban J, & García-Quintanilla M (2021). Advances in bacteriophage therapy against relevant multidrug-resistant pathogens. Antibiotics, 10(6), 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke JS (2012). Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clinical Microbiology Reviews, 25(1), 2–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruni GN, & Kralj JM (2020). Membrane voltage dysregulation driven by metabolic dysfunction underlies bactericidal activity of aminoglycosides. eLife, 9, e58706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck ED, Lammertyn E, & Anné J (2008). The importance of the twin-arginine translocation pathway for bacterial virulence. Trends in Microbiology, 16(9), 442–453. [DOI] [PubMed] [Google Scholar]
- Budd A, Blandin S, Levashina EA, & Gibson TJ (2004). Bacterial α2-macroglobulins: colonization factors acquired by horizontal gene transfer from the metazoan genome? Genome Biology, 5(6), R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K, & Bradford PA (2019). Interplay between β-lactamases and new β-lactamase inhibitors. Nature Reviews Microbiology, 17(5), 295–306. [DOI] [PubMed] [Google Scholar]
- Bush K, & Jacoby GA (2010). Updated functional classification of β-lactamases. Antimicrobial Agents and Chemotherapy, 54(3), 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzder-Lantos P, Bockstael K, Anné J, & Herdewijn P (2009). Substrate based peptide aldehyde inhibits bacterial type I signal peptidase. Bioorganic and Medicinal Chemistry Letters, 19(10), 2880–2883. [DOI] [PubMed] [Google Scholar]
- C.V SR, Waelheyns ED, Economou A, & Anné J (2014). Antibiotic targeting of the bacterial secretory pathway. Biochimica et Biophysica Acta, 1843(8), 1762–1783. [DOI] [PubMed] [Google Scholar]
- Callewaert L, Walmagh M, Michiels CW, & Lavigne R (2011). Food applications of bacterial cell wall hydrolases. Current Opinion in Biotechnology, 22(2), 164–171. [DOI] [PubMed] [Google Scholar]
- Campbell J, Singh AK, Maria JPS, Kim Y, Brown S, Swoboda JG, et al. (2011). Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chemical Biology, 6(1), 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Yi F, Tian Q, Dang G, Si W, Liu S, et al. (2017). Targeting the Gram-negative bacteria peptidoglycan synthase MraY as a new approach for monoclonal antibody anti-bacterial activity. Human Vaccines & Immunotherapeutics, 13(9), 2086–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanié-Cornet M-P, Bruel N, & Genevaux P (2014). Chaperone networking facilitates protein targeting to the bacterial cytoplasmic membrane. Biochimica et Biophysica Acta, 1843(8), 1442–1456. [DOI] [PubMed] [Google Scholar]
- Cesare GBD, Cristy SA, Garsin DA, & Lorenz MC (2020). Antimicrobial peptides: a new frontier in antifungal therapy. mBio, 11(6), e02123–02120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee C, Paul M, Xie L, & Donk W. A. v. d. (2005). Biosynthesis and mode of action of lantibiotics. Chemical Reviews, 105(2), 633–684. [DOI] [PubMed] [Google Scholar]
- Chaturvedi D, & Mahalakshmi R (2017). Transmembrane β-barrels: evolution, folding and energetics. Biochimica et Biophysica Acta, 1859(12), 2467–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee PY, Mang M, Lau ES, Tan LT-H, He Y-W, Lee W-L, et al. (2019). Epinecidin-1, an antimicrobial peptide derived from grouper (Epinephelus coioides): pharmacological activities and applications. Frontiers in Microbiology, 10, 2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Gardete S, Jansen RS, Shetty A, Dick T, Rhee KY, et al. (2018). Verapamil targets membrane energetics in Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy, 62(5), e02107–02117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, & Lu TK (2020). Development and challenges of antimicrobial peptides for therapeutic applications. Antibiotics, 9(1), 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimalakonda G, Ruiz N, Chng S-S, Garner RA, Kahne D, & Silhavy TJ (2011). Lipoprotein LptE is required for the assembly of LptD by the β-barrel assembly machine in the outer membrane of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America, 108(6), 2492–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Choi Y, Min K, Son JB, Park H, Lee HH, et al. (2020). Over-activation of a nonessential bacterial protease DegP as an antibiotic strategy. Communications Biology, 3(1), 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Lee M, Cochrane CS, Webster CG, Fenton BA, Zhao J, et al. (2020). Structural basis of the UDP-diacylglucosamine pyrophosphohydrolase LpxH inhibition by sulfonyl piperazine antibiotics. Proceedings of the National Academy of Sciences of the United States of America, 117(8), 4109–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi U, & Lee C-R (2019). Distinct roles of outer membrane porins in antibiotic resistance and membrane integrity in Escherichia coli. Frontiers in Microbiology, 10, 953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SR, & Foster SJ (2006). Surface adhesins of Staphylococcus aureus. Advances in Microbial Physiology, 51, 187–224. [DOI] [PubMed] [Google Scholar]
- Coleman WG, & Leive L (1979). Two mutations which affect the barrier function of the Escherichia coli K-12 outer membrane. Journal of Bacteriology, 139(3), 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello SM, Plummer AM, Fleming PJ, & Fleming KG (2016). Dynamic periplasmic chaperone reservoir facilitates biogenesis of outer membrane proteins. Proceedings of the National Academy of Sciences of the United States of America, 113(33), E4794–E4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote JM, & Taylor EA (2017). The glycosyltransferases of LPS Core: a review of four heptosyltransferase enzymes in context. International Journal of Molecular Sciences, 18(11), 2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther GJ, Weller SM, Jones JC, Weaver T, Fan E, Voorhis WCV, et al. (2015). The bacterial Sec pathway of protein export. Journal of Biomolecular Screening, 20(7), 921–926. [DOI] [PubMed] [Google Scholar]
- Cui P, Li X, Zhu M, Wang B, Liu J, & Chen H (2017). Design, synthesis and antimicrobial activities of thiouracil derivatives containing triazolo-thiadiazole as SecA inhibitors. European Journal of Medicinal Chemistry, 127, 159–165. [DOI] [PubMed] [Google Scholar]
- Dash R, & Bhattacharjya S (2021). Thanatin: an emerging host defense antimicrobial peptide with multiple modes of action. International Journal of Molecular Sciences, 22(4), 1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson RJP, & Locher KP (2006). Structure of a bacterial multidrug ABC transporter. Nature, 443(7108), 180–185. [DOI] [PubMed] [Google Scholar]
- DeDent A, Bae T, Missiakas DM, & Schneewind O (2008). Signal peptides direct surface proteins to two distinct envelope locations of Staphylococcus aureus. EMBO Journal, 27(20), 2656–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- delaVega AL, & Delcour AH (1995). Cadaverine induces closing of E. coli porins. EMBO Journal, 14(23), 6058–6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennehy R, Romano M, Ruggiero A, Mohamed YF, Dignam SL, Troncoso CM, et al. (2017). The Burkholderia cenocepacia peptidoglycan-associated lipoprotein is involved in epithelial cell attachment and elicitation of inflammation. Cellular Microbiology, 19(5), e12691. [DOI] [PubMed] [Google Scholar]
- Depuydt M, Leonard SE, Vertommen D, Denoncin K, Morsomme P, Wahni K, et al. (2009). A periplasmic reducing system protects single cysteine residues from oxidation. Science, 326(5956), 1109–1111. [DOI] [PubMed] [Google Scholar]
- Dhiman RK, Mahapatra S, Slayden RA, Boyne ME, Lenaerts A, Hinshaw JC, et al. (2009). Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Molecular Microbiology, 72(1), 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Komura R, Sano T, Pantua H, Storek KM, Inaba H, et al. (2021). Inhibition of Escherichia coli lipoprotein diacylglyceryl transferase is insensitive to resistance caused by deletion of Braun’s lipoprotein. Journal of Bacteriology, 203(13), e00149–00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias KJSDO, Miranda GM, Bessa JR, Araùjo ACJD, Freitas PR, Almeida RSD, et al. (2022). Terpenes as bacterial efflux pump inhibitors: A systematic review. Frontiers in Pharmacology, 13, 953982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey SW, Cheung GYC, & Otto M (2017). Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nature Reviews Drug Discovery, 16(7), 457–471. [DOI] [PubMed] [Google Scholar]
- Dong H, Xiang Q, Gu Y, Wang Z, Paterson NG, Stansfeld PJ, et al. (2014). Structural basis for outer membrane lipopolysaccharide insertion. Nature, 511(7507), 52–56. [DOI] [PubMed] [Google Scholar]
- Dramsi S, Magnet S, Davison S, & Arthur M (2008). Covalent attachment of proteins to peptidoglycan. FEMS Microbiology Reviews, 32(2), 307–320. [DOI] [PubMed] [Google Scholar]
- Du H, Chen L, Tang Y-W, & Kreiswirth BN (2016). Emergence of the mcr-1 colistin resistance gene in carbapenem-resistant Enterobacteriaceae. Lancet Infectious Diseases, 16(3), 287–288. [DOI] [PubMed] [Google Scholar]
- Dubin DT, Hancock R, & Davis BD (1963). The sequence of some effects of streptomycin in Escherichia coli. Biochimica et Biophysica Acta, 74, 476–489. [DOI] [PubMed] [Google Scholar]
- Duprez W, Bachu P, Stoermer MJ, Tay S, McMahon RM, Fairlie DP, et al. (2015). Virtual screening of peptide and peptidomimetic fragments targeted to inhibit bacterial dithiol oxidase DsbA. PloS One, 10(7), e0133805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprez W, Premkumar L, Halili MA, Lindahl F, Reid RC, Fairlie DP, et al. (2015). Peptide inhibitors of the Escherichia coli DsbA oxidative machinery essential for bacterial virulence. Journal of Medicinal Chemistry, 58(2), 577–587. [DOI] [PubMed] [Google Scholar]
- Dutton RJ, Boyd D, Berkmen M, & Beckwith J (2008). Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proceedings of the National Academy of Sciences of the United States of America, 105(33), 11933–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan AJF, Errington J, & Vollmer W (2020). Regulation of peptidoglycan synthesis and remodelling. Nature Reviews Microbiology, 18(8), 446–460. [DOI] [PubMed] [Google Scholar]
- Elhassanny AEM, Abutaleb NS, & Seleem MN (2022). Auranofin exerts antibacterial activity against Neisseria gonorrhoeae in a female mouse model of genital tract infection. PloS One, 17(4), e0266764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entasis THI (2022). Entasis therapeutics presents efficacy and safety data from landmark phase 3 ATTACK trial at ECCMID 2022 conference.
- Epand RM, & Epand RF (2011). Bacterial membrane lipids in the action of antimicrobial agents. Journal of Peptide Science, 17(5), 298–305. [DOI] [PubMed] [Google Scholar]
- Epand RM, Walker C, Epand RF, & Magarvey NA (2016). Molecular mechanisms of membrane targeting antibiotics. Biochimica et Biophysica Acta, 1858(5), 980–987. [DOI] [PubMed] [Google Scholar]
- Erickson HP, Anderson DE, & Osawa M (2010). FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiology and Molecular Biology Reviews, 74(4), 504–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin AL (2016). Antibacterial drug discovery targeting the lipopolysaccharide biosynthetic enzyme LpxC. Cold Spring Harbor Perspectives in Medicine, 6(7), a025304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun Y-J, Foss MH, Kiekebusch D, Pauw DA, Westler WM, Thanbichler M, et al. (2012). DCAP: a broad-spectrum antibiotic that targets the cytoplasmic membrane of bacteria. Journal of the American Chemical Society, 134(28), 11322–11325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun Y-J, Zhou M, Kiekebusch D, Schlimpert S, Trivedi RR, Bakshi S, et al. (2013). Divin: a small molecule inhibitor of bacterial divisome assembly. Journal of the American Chemical Society, 135(26), 9768–9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman JW, Noinaj N, & Buchanan SK (2011). The structural biology of β-barrel membrane proteins: a summary of recent reports. Current Opinion in Structural Biology, 21(4), 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farha Maya A., Verschoor Chris P., Bowdish D, & Brown Eric D. (2013). Collapsing the proton motive force to identify synergistic combinations against Staphylococcus aureus. Chemistry and Biology, 20(9), 1168–1178. [DOI] [PubMed] [Google Scholar]
- Feng X, Zhu W, Schurig-Briccio LA, Lindert S, Shoen C, Hitchings R, et al. (2015). Antiinfectives targeting enzymes and the proton motive force. Proceedings of the National Academy of Sciences of the United States of America, 112(51), e7073–e7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fereshteh S, Kalhor H, Sepehr A, Rahimi H, Zafari M, Cohan RA, et al. (2022). Rational design of inhibitors against LpxA protein of Acinetobacter baumannii using a virtual screening method. Journal of the Indian Chemical Society, 99(2), 100319. [Google Scholar]
- Fiorentino F, Sauer JB, Qiu X, Corey RA, Cassidy CK, Mynors-Wallis B, et al. (2021). Dynamics of an LPS translocon induced by substrate and an antimicrobial peptide. Nature Chemical Biology, 17(2), 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredenhagen A, Fendrich G, MÄRki F, MÄRki W, Gruner J, Raschdorf F, et al. (1990). Duramycins B and C, two new lanthionine containing antibiotics as inhibitors of phospholipase A2. Structural revision of duramycin and cinnamycin. The Journal of Antibiotics, 43(11), 1403–1412. [DOI] [PubMed] [Google Scholar]
- Früh V, Zhou Y, Chen D, Loch C, Ab E, Grinkova YN, et al. (2010). Application of fragment-based drug discovery to membrane proteins: Identification of ligands of the integral membrane enzyme DsbB. Chemistry and Biology, 17(8), 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Wang Y, Shao H, Ma J, Song X, Zhang M, et al. (2018). DegP functions as a critical protease for bacterial acid resistance. The FEBS Journal, 285(18), 3525–3538. [DOI] [PubMed] [Google Scholar]
- Furniss RCD, Kaderabkova N, Barker D, Bernal P, Maslova E, Antwi AAA, et al. (2022). Breaking antimicrobial resistance by disrupting extracytoplasmic protein folding. eLife, 11, e57974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürst M, Zhou Y, Merfort J, & Müller M (2018). Involvement of PpiD in Sec-dependent protein translocation. Biochimica et Biophysica Acta, 1865(2), 273–280. [DOI] [PubMed] [Google Scholar]
- Gai Z, Samodelov SL, Kullak-Ublick GA, & Visentin M (2019). Molecular mechanisms of colistin-induced nephrotoxicity. Molecules, 24(3), 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdiero S, Falanga A, Cantisani M, Tarallo R, Pepa MED, D’Oriano V, et al. (2012). Microbe-host interactions: structure and role of Gram-negative bacterial porins. Current Protein & Peptide Science, 13(8), 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamayun I, O’Keefe S, Pick T, Klein M-C, Nguyen D, McKibbin C, et al. (2019). Eeyarestatin compounds selectively enhance Sec61-mediated Ca2+ leakage from the endoplasmic reticulum. Cell Chemical Biology, 26(4), 571–583.e576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garde S, Chodisetti PK, & Reddy M (2021). Peptidoglycan: Structure, Synthesis, and Regulation. EcoSal Plus, 9(2), 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett TA, Kadrmas JL, & Raetz CRH (1997). Identification of the gene encoding the Escherichia coli lipid A 4′-kinase. Facile phosphorylation of endotoxin analogs with recombinant LpxK. Journal of Biological Chemistry, 272(35), 21855–21864. [DOI] [PubMed] [Google Scholar]
- Garrett TA, Que NLS, & Raetz CRH (1998). Accumulation of a lipid A precursor lacking the 4′-phosphate following inactivation of the Escherichia coli lpxK gene. Journal of Biological Chemistry, 273(20), 12457–12465. [DOI] [PubMed] [Google Scholar]
- Ge X, Lyu Z-X, Liu Y, Wang R, Zhao XS, Fu X, et al. (2014). Identification of FkpA as a key quality control factor for the biogenesis of outer membrane proteins under heat shock conditions. Journal of Bacteriology, 196(3), 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genentech. (2023). Genentech: our Pipeline. from https://www.gene.com/medical-professionals/pipeline
- Gentry DR, Wilding I, Johnson JM, Chen D, Remlinger K, Richards C, et al. (2010). A rapid microtiter plate assay for measuring the effect of compounds on Staphylococcus aureus membrane potential. Journal of Microbiological Methods, 83(2), 254–256. [DOI] [PubMed] [Google Scholar]
- Gessmann D, Chung YH, Danoff EJ, Plummer AM, Sandlin CW, Zaccai NR, et al. (2014). Outer membrane β-barrel protein folding is physically controlled by periplasmic lipid head groups and BamA. Proceedings of the National Academy of Sciences of the United States of America, 111(16), 5878–5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M, Miller PA, Möllmann U, Claypool WD, Schroeder VA, Wolter WR, et al. (2017). Targeted antibiotic delivery: selective siderophore conjugation with daptomycin confers potent activity against multidrug resistant Acinetobacter baumannii both in vitro and in vivo. Journal of Medicinal Chemistry, 60(11), 4577–4583. [DOI] [PubMed] [Google Scholar]
- Goemans C, Denoncin K, & Collet J-F (2014). Folding mechanisms of periplasmic proteins. Biochimica et Biophysica Acta, 1843(8), 1517–1528. [DOI] [PubMed] [Google Scholar]
- Gogry FA, Siddiqui MT, Sultan I, & Haq QMR (2021). Current update on intrinsic and acquired colistin resistance mechanisms in bacteria. Frontiers in Medicine, 8, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottler LM, & Ramamoorthy A (2009). Structure, membrane orientation, mechanism, and function of pexiganan - a highly potent antimicrobial peptide designed from magainin. Biochimica et Biophysica Acta, 1788(8), 1680–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götzke H, Palombo I, Muheim C, Perrody E, Genevaux P, Kudva R, et al. (2014). YfgM Is an ancillary subunit of the SecYEG translocon in Escherichia coli. Journal of Biological Chemistry, 289(27), 19089–19097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowicz M (2019). Lipoproteins and their trafficking to the outer membrane. EcoSal Plus, 8(2), 1–8. [DOI] [PubMed] [Google Scholar]
- Grabowicz M, & Silhavy TJ (2017). Redefining the essential trafficking pathway for outer membrane lipoproteins. Proceedings of the National Academy of Sciences of the United States of America, 114(18), 4769–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granata G, Taglietti F, Schiavone F, & Petrosillo N (2022). Durlobactam in the treatment of multidrug-resistant Acinetobacter baumannii infections: a systematic review. Journal of Clinical Medicine, 11(12), 3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grein F, Müller A, Scherer KM, Liu X, Ludwig KC, Klöckner A, et al. (2020). Ca2+-Daptomycin targets cell wall biosynthesis by forming a tripartite complex with undecaprenyl-coupled intermediates and membrane lipids. Nature Communications, 11(1), 1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsey EM, Fais C, Marshall RL, Ricci V, Ciusa ML, Stone JW, et al. (2020). Chlorpromazine and amitriptyline are substrates and inhibitors of the AcrB multidrug efflux pump. mBio, 11(3), e00465–00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grizot S, Salem M, Vongsouthi V, Durand L, Moreau F, Dohi H, et al. (2006). Structure of the Escherichia coli heptosyltransferase WaaC: binary complexes with ADP and ADP-2-deoxy-2-fluoro heptose. Journal of Molecular Biology, 363(2), 383–394. [DOI] [PubMed] [Google Scholar]
- Gronenberg LS, & Kahne D (2010). Development of an activity assay for discovery of inhibitors of lipopolysaccharide transport. Journal of the American Chemical Society, 132(8), 2518–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasinghe SD, Shiota T, Stubenrauch CJ, Schulze KE, Webb CT, Fulcher AJ, et al. (2018). The WD40 protein BamB mediates coupling of BAM complexes into assembly precincts in the bacterial outer membrane. Cell Reports, 23(9), 2782–2794. [DOI] [PubMed] [Google Scholar]
- Guo Y, Lv X, Wang Y, Zhou Y, Lu N, Deng X, et al. (2020). Honokiol restores polymyxin susceptibility to MCR-1-positive pathogens both in vitro and in vivo. Applied and Environmental Microbiology, 86(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Cohen KA, Winglee K, Maiga M, Diarra B, & Bishai WR (2014). Efflux inhibition with verapamil potentiates bedaquiline in Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy, 58(1), 574–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta VK, Tiwari N, Gupta P, Verma S, Pal A, Srivastava SK, et al. (2016). A clerodane diterpene from Polyalthia longifolia as a modifying agent of the resistance of methicillin resistant Staphylococcus aureus. Phytomedicine, 23(6), 654–661. [DOI] [PubMed] [Google Scholar]
- Halili MA, Bachu P, Lindahl F, Bechara C. r., Mohanty B, Reid RC, et al. (2015). Small molecule inhibitors of disulfide bond formation by the bacterial DsbA-DsbB dual enzyme system. ACS Chemical Biology, 10(4), 957–964. [DOI] [PubMed] [Google Scholar]
- Hall BS, Hill K, McKenna M, Ogbechi J, High S, Willis AE, et al. (2014). The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathogens, 10(4), e1004061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto H, Urai M, Ishii K, Yasukawa J, Paudel A, Murai M, et al. (2015). Lysocin E is a new antibiotic that targets menaquinone in the bacterial membrane. Nature Chemical Biology, 11(2), 127–133. [DOI] [PubMed] [Google Scholar]
- Han S, Zaniewski RP, Marr ES, Lacey BM, Tomaras AP, Evdokimov A, et al. (2010). Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences of the United States of America, 107(51), 22002–22007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Ma X, Balibar CJ, Rath CMB, Benton B, Bermingham A, et al. (2020). Two distinct mechanisms of inhibition of LpxA acyltransferase essential for lipopolysaccharide biosynthesis. Journal of the American Chemical Society, 142(9), 4445–4455. [DOI] [PubMed] [Google Scholar]
- Hanpaibool C, Ngamwongsatit N, Ounjai P, Yotphan S, Wolschann P, Mulholland AJ, et al. (2023). Pyrazolones potentiate colistin activity against MCR-1-producing resistant bacteria: computational and microbiological study. ACS Omega, 8(9), 8366–8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DA, Powers ME, & Romesberg FE (2009). Synthesis and biological evaluation of penem inhibitors of bacterial signal peptidase. Bioorganic and Medicinal Chemistry Letters, 19(14), 3787–3790. [DOI] [PubMed] [Google Scholar]
- Hart EM, Mitchell AM, Konovalova A, Grabowicz M, Sheng J, Han X, et al. (2019). A small-molecule inhibitor of BamA impervious to efflux and the outer membrane permeability barrier. Proceedings of the National Academy of Sciences of the United States of America, 116(43), 21748–21757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan KA, Elbourne LDH, Li L, Gamage HKAH, Liu Q, Jackson SM, et al. (2015). An ace up their sleeve: a transcriptomic approach exposes the AceI efflux protein of Acinetobacter baumannii and reveals the drug efflux potential hidden in many microbial pathogens. Frontiers in Microbiology, 6, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan KA, Liu Q, Henderson PJF, & Paulsen IT (2015). Homologs of the Acinetobacter baumannii acei transporter represent a new family of bacterial multidrug efflux systems. mBio, 6(1), e01982–01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselblatt H, Kurzbauer R, Wilken C, Krojer T, Sawa J, Kurt J, et al. (2007). Regulation of the σE stress response by DegS: how the PDZ domain keeps the protease inactive in the resting state and allows integration of different OMP-derived stress signals upon folding stress. Genes and Development, 21(20), 2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, & Wu HC (1985). Accumulation of prolipoprotein in Escherichia coli mutants defective in protein secretion. Journal of Bacteriology, 161(3), 949–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, et al. (2008). An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science, 321(5896), 1673–1675. [DOI] [PubMed] [Google Scholar]
- He D, Zhang M, Liu S, Xie X, & Chen PR (2019). Protease-mediated protein quality control for bacterial acid resistance. Cell Chemical Biology, 26(1), 144–150.e143. [DOI] [PubMed] [Google Scholar]
- Henderson JC, Zimmerman SM, Crofts AA, Boll JM, Kuhns LG, Herrera CM, et al. (2015). The power of asymmetry: architecture and assembly of the Gram-negative outer membrane lipid bilayer. Annual Review of Microbiology, 70(1), 1–24. [DOI] [PubMed] [Google Scholar]
- Heras B, Kurz M, Shouldice SR, & Martin JL (2007). The name’s bond…… disulfide bond. Current Opinion in Structural Biology, 17(6), 691–698. [DOI] [PubMed] [Google Scholar]
- Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, & Martin JL (2009). DSB proteins and bacterial pathogenicity. Nature Reviews Microbiology, 7(3), 215–225. [DOI] [PubMed] [Google Scholar]
- Hernéndez SB, Cava F, Pucciarelli MG, Portillo F. G. d., Pedro MA, & Casadesús J (2015). Bile-induced peptidoglycan remodelling. Environmental Microbiology, 17(4), 1081–1089. [DOI] [PubMed] [Google Scholar]
- Higgins DL, Chang R, Debabov DV, Leung J, Wu T, Krause KM, et al. (2005). Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrobial Agents and Chemotherapy, 49(3), 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiniker A, Collet J-F, & Bardwell JCA (2005). Copper stress causes an in vivo requirement for the Escherichia coli disulfide isomerase DsbC. Journal of Biological Chemistry, 280(40), 33785–33791. [DOI] [PubMed] [Google Scholar]
- Hirakawa H, Suzue K, & Tomita H (2022). Roles of the Tol/Pal System in bacterial pathogenesis and its application to antibacterial therapy. Vaccines, 10(3), 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DJ, Bae IK, Jang I-H, Jeong SH, Kang H-K, & Lee K (2015). Epidemiology and characteristics of metallo-β-lactamase-producing Pseudomonas aeruginosa. Infection & Chemotherapy, 47(2), 81–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W-C, Lin C-Y, Hashimoto M, Wu J-J, Wang M-C, Lin W-H, et al. (2020). The role of the bacterial protease Prc in the uropathogenesis of extraintestinal pathogenic Escherichia coli. Journal of Biomedical Science, 27(1), 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YJ, Wang H, Gao FB, Li M, Yang H, Wang B, et al. (2012). Fluorescein analogues inhibit SecA ATPase: the first sub-micromolar inhibitor of bacterial protein translocation. ChemMedChem, 7(4), 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber G, & Nesemann G (1968). Moenomycin, an inhibitor of cell wall synthesis. Biochemical and Biophysical Research Communications, 30(1), 7–13. [DOI] [PubMed] [Google Scholar]
- Hurdle JG, O’Neill AJ, Chopra I, & Lee RE (2011). Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nature Reviews Microbiology, 9(1), 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Meyer KJ, Iinishi A, Favre-Godal Q, Green R, Manuse S, et al. (2019). A new antibiotic selectively kills Gram-negative pathogens. Nature, 576(7787), 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Murakami S, Nakagawa A, Iida H, Kinjo M, Ito K, et al. (2009). Dynamic nature of disulphide bond formation catalysts revealed by crystal structures of DsbB. EMBO Journal, 28(6), 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, et al. (2006). Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell, 127(4), 789–801. [DOI] [PubMed] [Google Scholar]
- Inouye S, Franceschini T, Sato M, Itakura K, & Inouye M (1983). Prolipoprotein signal peptidase of Escherichia coli requires a cysteine residue at the cleavage site. EMBO Journal, 2(1), 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iscla I, Levin G, Wray R, & Blount P (2007). Disulfide trapping the mechanosensitive channel MscL into a gating-transition state. Biophysical Journal, 92(4), 1224–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iscla I, Wray R, & Blount P (2008). On the structure of the N-terminal domain of the MscL channel: helical bundle or membrane interface. Biophysical Journal, 95(5), 2283–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iscla I, Wray R, Wei S, Posner B, & Blount P (2014). Streptomycin potency is dependent on MscL channel expression. Nature Communications, 5(1), 4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Kanamaru K, Taniguchi N, Miyamoto S, & Tokuda H (2006). A novel ligand bound ABC transporter, LolCDE, provides insights into the molecular mechanisms underlying membrane detachment of bacterial lipoproteins. Molecular Microbiology, 62(4), 1064–1075. [DOI] [PubMed] [Google Scholar]
- Iyer R, & Delcour AH (1997). Complex inhibition of OmpF and OmpC bacterial porins by polyamines. Journal of Biological Chemistry, 272(30), 18595–18601. [DOI] [PubMed] [Google Scholar]
- Jackman JE, Raetz CRH, & Fierke CA (1999). UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase of Escherichia coli is a zinc metalloenzyme. Biochemistry, 38(6), 1902–1911. [DOI] [PubMed] [Google Scholar]
- Jamshidi S, Sutton JM, & Rahman KM (2017). Computational study reveals the molecular mechanism of the interaction between the efflux inhibitor PAβN and the AdeB transporter from Acinetobacter baumannii. ACS Omega, 2(6), 3002–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaudzems K, Kurbatska V, Jēkabsons A, Bobrovs R, Rudevica Z, & Leonchiks A (2020). Targeting bacterial sortase A with covalent inhibitors: 27 new starting points for structure-based hit-to-lead optimization. ACS Infectious Diseases 6(2), 186–194. [DOI] [PubMed] [Google Scholar]
- Jauss B, Petriman N-A, Drepper F, Franz L, Sachelaru I, Welte T, et al. (2019). Noncompetitive binding of PpiD and YidC to the SecYEG translocon expands the global view on the SecYEG interactome in Escherichia coli. Journal of Biological Chemistry, 294(50), 19167–19183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RJ, & Dotson GD (2012). Dual targeting antibacterial peptide inhibitor of early lipid A biosynthesis. ACS Chemical Biology, 7(7), 1170–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon B, & Zhang Q (2009). Sensitization of Campylobacter jejuni to fluoroquinolone and macrolide antibiotics by antisense inhibition of the CmeABC multidrug efflux transporter. Journal of Antimicrobial Chemotherapy, 63(5), 946–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeżowska-Bojczuk M, & Stokowa-Soltys K (2018). Peptides having antimicrobial activity and their complexes with transition metal ions. European Journal of Medicinal Chemistry, 143, 997–1009. [DOI] [PubMed] [Google Scholar]
- Jia Y, Liu J, Yang Q, Zhang W, Efferth T, Liu S, et al. (2023). Cajanin stilbene acid: a direct inhibitor of colistin resistance protein MCR-1 that restores the efficacy of polymyxin B against resistant Gram-negative bacteria. Phytomedicine, 114, 154803. [DOI] [PubMed] [Google Scholar]
- Jin J, Hsieh Y-H, Chaudhary AS, Cui J, Houghton JE, Sui S. f., et al. (2018). SecA inhibitors as potential antimicrobial agents: differential actions on SecA-only and SecA-SecYEG protein-conducting channels. FEMS Microbiology Letters, 365(15), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CH, Bolken TC, Jones KF, Zeller GO, & Hruby DE (2001). Conserved DegP protease in Gram-positive bacteria is essential for thermal and oxidative tolerance and full virulence in Streptococcus pyogenes. Infection and Immunity, 69(9), 5538–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junkes C, Wessolowski A, Farnaud S, Evans RW, Good L, Bienert M, et al. (2008). The interaction of arginine- and tryptophan-rich cyclic hexapeptides with Escherichia coli membranes. Journal of Peptide Science, 14(4), 535–543. [DOI] [PubMed] [Google Scholar]
- Junne T, Wong J, Studer C, Aust T, Bauer BW, Beibel M, et al. (2015). Decatransin, a new natural product inhibiting protein translocation at the Sec61/SecYEG translocon. Journal of Cell Science, 128(6), 1217–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice SS, Hunstad DA, Harper JR, Duguay AR, Pinkner JS, Bann J, et al. (2005). Periplasmic peptidyl prolyl cis-trans isomerases are not essential for viability, but SurA is required for pilus biogenesis in Escherichia coli. Journal of Bacteriology, 187(22), 7680–7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaderbhai NN, Khan T, & Kaderbhai MA (2008). An anti-microbial peptide derivative of flesh fruit fly mimics secretory signal sequence and inhibits signal peptidase-I in the export pathway. International Journal of Peptide Research and Therapeutics, 14(2), 173–181. [Google Scholar]
- Kalinin DV, & Holl R (2017). LpxC inhibitors: a patent review (2010-2016). Expert Opinion on Therapeutic Patents, 27(11), 1227–1250. [DOI] [PubMed] [Google Scholar]
- Kaplan E, Greene NP, Jepson AE, & Koronakis V (2022). Structural basis of lipoprotein recognition by the bacterial Lol trafficking chaperone LolA. Proceedings of the National Academy of Sciences of the United States of America, 119(36), e2208662119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakonstantis S, Kritsotakis EI, & Gikas A (2019). Pandrug-resistant Gram-negative bacteria: a systematic review of current epidemiology, prognosis and treatment options. Journal of Antimicrobial Chemotherapy, 75(2), 271–282. [DOI] [PubMed] [Google Scholar]
- Kaur H, Hartmann J-B, Jakob RP, Zahn M, Zimmermann I, Maier T, et al. (2019). Identification of conformation-selective nanobodies against the membrane protein insertase BamA by an integrated structural biology approach. Journal of Biomolecular NMR, 73(6-7), 375–384. [DOI] [PubMed] [Google Scholar]
- Kaur H, Jakob RP, Marzinek JK, Green R, Imai Y, Bolla JR, et al. (2021). The antibiotic darobactin mimics a β-strand to inhibit outer membrane insertase. Nature, 593(7857), 125–129. [DOI] [PubMed] [Google Scholar]
- Khoury ME, Swain J, Sautrey G, Zimmermann L, Smissen PVD, Décout J-L, et al. (2017). Targeting bacterial cardiolipin enriched microdomains: an antimicrobial strategy used by amphiphilic aminoglycoside antibiotics. Scientific Reports, 7(1), 10697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-R, Lee D, & Eom Y-B (2018). Anti-biofilm and anti-virulence efficacy of celastrol against Stenotrophomonas maltophilia. International Journal of Medical Sciences, 15(6), 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, & Eom YB (2022). Auranofin promotes antibacterial effect of doripenem against carbapenem-resistant Acinetobacter baumannii. Journal of Applied Microbiology, 133(3), 1422–1433. [DOI] [PubMed] [Google Scholar]
- Kinana AD, Vargiu AV, May T, & Nikaido H (2016). Aminoacyl β-naphthylamides as substrates and modulators of AcrB multidrug efflux pump. Proceedings of the National Academy of Sciences of the United States of America, 113(5), 1405–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura S, Owensby A, Wall D, & Wolan DW (2018). Lipoprotein signal peptidase inhibitors with antibiotic properties identified through design of a robust in vitro HT platform. Cell Chemical Biology, 25(3), 301–308.e312. [DOI] [PubMed] [Google Scholar]
- Konovalova A, Grabowicz M, Balibar CJ, Malinverni JC, Painter RE, Riley D, et al. (2018). Inhibitor of intramembrane protease RseP blocks the σE response causing lethal accumulation of unfolded outer membrane proteins. Proceedings of the National Academy of Sciences of the United States of America, 115(28), E6614–E6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouwen TRHM, Trip EN, Denham EL, Sibbald MJJB, Dubois J-YF, & Dijl J. M. v. (2009). The large mechanosensitive channel MscL determines bacterial susceptibility to the bacteriocin sublancin 168. Antimicrobial Agents and Chemotherapy, 53(11), 4702–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeck KG, Sacco MD, Smith EW, Zhang X, Shoun D, Akhtar A, et al. (2019). Discovery of dual-activity small-molecule ligands of Pseudomonas aeruginosa LpxA and LpxD using SPR and X-ray crystallography. Scientific Reports, 9(1), 15450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kureisaite-Ciziene D, Varadajan A, McLaughlin SH, Glas M, Silva AM, Luirink R, et al. (2018). Structural analysis of the interaction between the bacterial cell division proteins FtsQ and FtsB. mBio, 9(5), e01346–01318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurittu P, Khakipoor B, Aarnio M, Nykasenoja S, Brouwer M, Myllyniemi AL, et al. (2021). Plasmid-borne and chromosomal ESBL/AmpC genes in Escherichia coli and Klebsiella pneumoniae in global food products. Frontiers in Microbiology, 12, 592291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvist M, Hancock V, & Klemm P (2008). Inactivation of efflux pumps abolishes bacterial biofilm formation. Applied and Environmental Microbiology, 74(23), 7376–7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai GC, Cho H, & Bernhardt TG (2017). The mecillinam resistome reveals a role for peptidoglycan endopeptidases in stimulating cell wall synthesis in Escherichia coli. PLoS Genetics, 13(7), e1006934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JS, Philbrick WM, Hayashi S, Inukai M, Arai M, Hirota Y, et al. (1981). Globomycin sensitivity of Escherichia coli and Salmonella typhimurium: effects of mutations affecting structures of murein lipoprotein. Journal of Bacteriology, 145(1), 657–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers RP, Cavallari JF, & Burrows LL (2013). The efflux inhibitor phenylalanine-arginine β-naphthylamide (PAβN) permeabilizes the outer membrane of Gram-negative bacteria. PloS One, 8(3), e60666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan X.-j., Yan H.-t., Lin F, Hou S, Li C.-c., Wang G.-s., et al. (2019). Design, synthesis and biological evaluation of 1-phenyl-2-(phenylamino) ethanone derivatives as novel MCR-1 inhibitors. Molecules, 24(15), 2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeta C, Blazyk JL, Hatahet F, Meehan BM, Eser M, Myrick A, et al. (2015). Compounds targeting disulfide bond forming enzyme DsbB of Gram-negative bacteria. Nature Chemical Biology, 11(4), 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeta C, Boyd D, & Beckwith J (2018). Disulfide bond formation in prokaryotes. Nature Microbiology, 3(3), 270–280. [DOI] [PubMed] [Google Scholar]
- Landeta C, McPartland L, Tran NQ, Meehan BM, Zhang Y, Tanweer Z, et al. (2019). Inhibition of Pseudomonas aeruginosa and Mycobacterium tuberculosis disulfide bond forming enzymes. Molecular Microbiology, 111(4), 918–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley KE, Hawrot E, & Kennedy EP (1982). Membrane assembly: movement of phosphatidylserine between the cytoplasmic and outer membranes of Escherichia coli. Journal of Bacteriology, 152(3), 1033–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langsrud S, Steinhauer K, Lüthje S, Weber K, Goroncy-Bermes P, & Holck AL (2016). Ethylhexylglycerin impairs membrane integrity and enhances the lethal effect of phenoxyethanol. PloS One, 11(10), e0165228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laws M, Shaaban A, & Rahman KM (2019). Antibiotic resistance breakers: current approaches and future directions. FEMS Microbiology Reviews, 43(5), 490–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BS, Hards K, Engelhart CA, Hasenoehrl EJ, Kalia NP, Mackenzie JS, et al. (2021). Dual inhibition of the terminal oxidases eradicates antibiotic-tolerant Mycobacterium tuberculosis. EMBO Molecular Medicine, 13(1), e13207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Sutterlin HA, Wzorek JS, Mandler MD, Hagan CL, Grabowicz M, et al. (2018). Substrate binding to BamD triggers a conformational change in BamA to control membrane insertion. Proceedings of the National Academy of Sciences of the United States of America, 115(10), 2359–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legood S, Seng D, Boneca IG, & Buddelmeijer N (2022). A defect in lipoprotein modification by Lgt leads to abnormal morphology and cell death in Escherichia coli that is independent of major lipoprotein Lpp. Journal of Bacteriology, 204(9), e00164–00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman KM, Smith HC, & Grabowicz M (2022). A biological signature for the inhibition of outer membrane lipoprotein biogenesis. mBio, 13(3), e00757–00722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levina N, Tötemeyer S, Stokes NR, Louis P, Jones MA, & Booth IR (1999). Protection of Escherichia coli cells against extreme turgor by activation of MscS and MscL mechanosensitive channels: identification of genes required for MscS activity. EMBO Journal, 18(7), 1730–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LA, Rice PA, & Ram S (2019). Role of gonococcal neisserial surface protein A (NspA) in serum resistance and comparison of its factor H binding properties with those of its meningococcal counterpart. Infection and Immunity, 87(2), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GW, Burkhardt D, Gross C, & Weissman JS (2014). Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell, 157(3), 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhu X, Zhang J, Lin Y, You X, Chen M, et al. (2020). Identification of a compound that inhibits the growth of Gram-negative bacteria by blocking BamA–BamD interaction. Frontiers in Microbiology, 11, 1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B-R, Hsieh Y-H, Jiang C, & Tai PC (2012). Escherichia coli membranes depleted of SecYEG elicit SecA-dependent ion-channel activity but lose signal peptide specificity. Journal of Membrane Biology, 245(11), 747–757. [DOI] [PubMed] [Google Scholar]
- Lin T-Y, & Weibel DB (2016). Organization and function of anionic phospholipids in bacteria. Applied Microbiology and Biotechnology, 100(10), 4255–4267. [DOI] [PubMed] [Google Scholar]
- Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, et al. (2015). A new antibiotic kills pathogens without detectable resistance. Nature, 517(7535), 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Trout REL, Chu G-H, McGarry D, Jackson RW, Hamrick JC, et al. (2020). Discovery of taniborbactam (VNRX-5133): a broad-spectrum serine- and metallo-β-lactamase inhibitor for carbapenem-resistant bacterial infections. Journal of Medicinal Chemistry, 63(6), 2789–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Tong J, Wu Q, Liu J, Yuan M, Tian C, et al. (2022). Natural inhibitors targeting the localization of lipoprotein system in Vibrio parahaemolyticus. International Journal of Molecular Sciences, 23(22), 14352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lu Y, Xu Z, Ma X, Chen X, & Liu W (2022). Repurposing of the gold drug auranofin and a review of its derivatives as antibacterial therapeutics. Drug Discovery Today, 27(7), 1961–1973. [DOI] [PubMed] [Google Scholar]
- Lock RL, & Harry EJ (2008). Cell-division inhibitors: new insights for future antibiotics. Nature Reviews Drug Discovery, 7(4), 324–338. [DOI] [PubMed] [Google Scholar]
- Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, et al. (2001). Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrobial Agents and Chemotherapy, 45(1), 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz C, Dougherty TJ, & Lory S (2016). Transcriptional responses of Escherichia coli to a small-molecule inhibitor of LolCDE, an essential component of the lipoprotein transport pathway. Journal of Bacteriology, 198(23), 3162–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering AL, Castro L. H. d., Lim D, & Strynadka NCJ (2007). Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science, 315(5817), 1402–1405. [DOI] [PubMed] [Google Scholar]
- Lubelski J, Konings WN, & Driessen AJM (2007). Distribution and physiology of ABC-type transporters contributing to multidrug resistance in bacteria. Microbiology and Molecular Biology Reviews, 71(3), 463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist K, Billings E, Bi M, Wellnitz J, & Noinaj N (2021). The assembly of β-barrel membrane proteins by BAM and SAM. Molecular Microbiology, 115(3), 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther A, Urfer M, Zahn M, Müller M, Wang S-Y, Mondal M, et al. (2019). Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature, 576(7787), 452–458. [DOI] [PubMed] [Google Scholar]
- Lv F, Cai J, He Q, Wang W, Luo Y, Wang X, et al. (2021). Overexpression of efflux pumps mediate pan resistance of Klebsiella pneumoniae sequence type 11. Microbial Drug Resistance, 27(10), 1405–1411. [DOI] [PubMed] [Google Scholar]
- Ma B, Niu C, Zhou Y, Xue X, Meng J, Luo X, et al. (2016). The disulfide bond of the peptide thanatin is dispensible for its antimicrobial activity in vivo and in vitro. Antimicrobial Agents and Chemotherapy, 60(7), 4283–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Prathapam R, Wartchow C, Chie-Leon B, Ho C-M, Vicente JD, et al. (2020). Structural and biological basis of small molecule inhibition of Escherichia coli LpxD acyltransferase essential for lipopolysaccharide biosynthesis. ACS Infectious Diseases 6(6), 1480–1489. [DOI] [PubMed] [Google Scholar]
- Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, & Dessen A (2006). Penicillin binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiology Reviews, 30(5), 673–691. [DOI] [PubMed] [Google Scholar]
- Mahalakshmi S, Sunayana MR, SaiSree L, & Reddy M (2014). Regulation of lipid A synthesis by yciM. Molecular Microbiology, 91(1), 145–157. [DOI] [PubMed] [Google Scholar]
- Mahmud B, Wallace MA, Reske KA, Alvarado K, Muenks CE, Rasmussen DA, et al. (2022). Epidemiology of plasmid lineages mediating the spread of extended-spectrum β-lactamases among clinical Escherichia coli. mSystems, 7(5), e0051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden MM, & Waters CM (2020). Triclosan depletes the membrane potential in Pseudomonas aeruginosa biofilms inhibiting aminoglycoside induced adaptive resistance. PLoS Pathogens, 16(10), e1008529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinverni JC, Werner J, Kim S, Sklar JG, Kahne D, Misra R, et al. (2006). YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Molecular Microbiology, 61(1), 151–164. [DOI] [PubMed] [Google Scholar]
- Mamou G, Corona F, Cohen-Khait R, Housden NG, Yeung V, Sun D, et al. (2022). Peptidoglycan maturation controls outer membrane protein assembly. Nature, 606(7916), 953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manioglu S, Modaresi SM, Ritzmann N, Thoma J, Overall SA, Harms A, et al. (2022). Antibiotic polymyxin arranges lipopolysaccharide into crystalline structures to solidify the bacterial membrane. Nature Communications, 13(1), 6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoor UF, Vitharana D, Reddy PA, Daubaras DL, McNicholas P, Orth P, et al. (2011). Design and synthesis of potent Gram-negative specific LpxC inhibitors. Bioorganic and Medicinal Chemistry Letters, 21(4), 1155–1161. [DOI] [PubMed] [Google Scholar]
- Mao G, Zhao Y, Kang X, Li Z, Zhang Y, Wang X, et al. (2016). Crystal structure of E. coli lipoprotein diacylglyceryl transferase. Nature Communications, 7(1), 10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RL, Lloyd GS, Lawler AJ, Element SJ, Kaur J, Ciusa ML, et al. (2020). New multidrug efflux inhibitors for Gram-negative bacteria. mBio, 11(4), e01340–01320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JK, Sheehan JP, Bratton BP, Moore GM, Mateus A, Li SH-J, et al. (2020). A dual-mechanism antibiotic kills Gram-negative bacteria and avoids drug resistance. Cell, 181(7), 1518–1532.e1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Guitián M, Vázquez-Ucha JC, Álvarez-Fraga L, Conde-Pérez K, Bou G, Poza M, et al. (2019). Antisense inhibition of lpxB gene expression in Acinetobacter baumannii by peptide-PNA conjugates and synergy with colistin. Journal of Antimicrobial Chemotherapy, 75(1), 51–59. [DOI] [PubMed] [Google Scholar]
- Martorana AM, Moura ECCM, Sperandeo P, Vincenzo FD, Liang X, Toone E, et al. (2021). Degradation of components of the Lpt transenvelope machinery reveals LPS-dependent Lpt complex stability in Escherichia coli. Frontiers in Molecular Biosciences, 8, 758228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matagne A, Lamotte-Brasseur J, & FrÉRe J-M (1998). Catalytic properties of class A β-lactamases: efficiency and diversity. Biochemical Journal, 330(2), 581–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelié-Guinlet M, Asmar AT, Collet J-F, & Dufrêne YF (2020). Lipoprotein Lpp regulates the mechanical properties of the E. coli cell envelope. Nature Communications, 11(1), 1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S. i., Yokota N, & Tokuda H (1997). A novel outer membrane lipoprotein, LolB (HemM), involved in the LolA (p20)-dependent localization of lipoproteins to the outer membrane of Escherichia coli. EMBO Journal, 16(23), 6947–6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteelli A, Carvalho ACC, Dooley KE, & Kritski A (2010). TMC207: the first compound of a new class of potent anti-tuberculosis drugs. Future Microbiology, 5(6), 849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod SM, Fleming PR, MacCormack K, McLaughlin RE, Whiteaker JD, Narita S.-i., et al. (2015). Small-molecule inhibitors of Gram-negative lipoprotein trafficking discovered by phenotypic screening. Journal of Bacteriology, 197(6), 1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mdluli KE, Witte PR, Kline T, Barb AW, Erwin AL, Mansfield BE, et al. (2006). Molecular validation of LpxC as an antibacterial drug target in Pseudomonas aeruginosa. Antimicrobial Agents and Chemotherapy, 50(6), 2178–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier M, Blatter XL, Seelig A, & Seelig J (2006). Interaction of verapamil with lipid membranes and P-glycoprotein: connecting thermodynamics and membrane structure with functional activity. Biophysical Journal, 91(8), 2943–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister B, Gömüç B, Suarez E, Ishii Y, Dürr K, & Gillberg L (2006). Hypothalamic proopiomelanocortin (POMC) neurons have a cholinergic phenotype. European Journal of Neuroscience, 24(10), 2731–2740. [DOI] [PubMed] [Google Scholar]
- Meng F, Nie T, Lyu Y, Lyu F, Bie X, Lu Y, et al. (2022). Plantaricin A reverses resistance to ciprofloxacin of multidrug-resistant Staphylococcus aureus by inhibiting efflux pumps. Environmental Microbiology, 24(10), 4818–4833. [DOI] [PubMed] [Google Scholar]
- Messens J, & Collet J-F (2006). Pathways of disulfide bond formation in Escherichia coli. International Journal of Biochemistry and Cell Biology, 38(7), 1050–1062. [DOI] [PubMed] [Google Scholar]
- Milicaj J, Hassan BA, Cote JM, Ramirez-Mondragon CA, Jaunbocus N, Rafalowski A, et al. (2022). Discovery of first-in-class nanomolar inhibitors of heptosyltransferase I reveals a new aminoglycoside target and potential alternative mechanism of action. Scientific Reports, 12(1), 7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani M, Mukaiyama K, Xiao J, Mori M, Satou R, Narita S.-i., et al. (2013). Functional differentiation of structurally similar membrane subunits of the ABC transporter LolCDE complex. FEBS Letters, 587(1), 23–29. [DOI] [PubMed] [Google Scholar]
- Moffatt JH, Harper M, Harrison P, Hale JDF, Vinogradov E, Seemann T, et al. (2010). Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrobial Agents and Chemotherapy, 54(12), 4971–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretta A, Scieuzo C, Petrone AM, Salvia R, Manniello MD, Franco A, et al. (2021). Antimicrobial peptides: a new hope in biomedical and pharmaceutical fields. Frontiers in Cellular and Infection Microbiology, 11, 668632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muheim C, Götzke H, Eriksson AU, Lindberg S, Lauritsen I, Nørholm MHH, et al. (2017). Increasing the permeability of Escherichia coli using MAC13243. Scientific Reports, 7(1), 17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A (2021). FDA approves 100th monoclonal antibody product. Nature Reviews Drug Discovery, 20(7), 491–495. [DOI] [PubMed] [Google Scholar]
- Murray CJL, Ikuta KS, Sharara F, Swetschinski L, Aguilar GR, Gray A, et al. (2022). Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet, 399(10325), 629–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na S-H, Jeon H, Oh M-H, Kim Y-J, Chu M, Lee I-Y, et al. (2021). Therapeutic effects of inhibitor of ompA expression against carbapenem-resistant Acinetobacter baumannii strains. International Journal of Molecular Sciences, 22(22), 12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura R, Oota M, Matsumoto S, Sato T, & Yamano Y (2021). In vitro activity and in vivo efficacy of cefiderocol against Stenotrophomonas maltophilia. Antimicrobial Agents and Chemotherapy, 65(4), e01436–01420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale P, Bröser T, & Driessen AJM (2008). Sec- and Tat-mediated protein secretion across the bacterial cytoplasmic membrane - distinct translocases and mechanisms. Biochimica et Biophysica Acta, 1778(9), 1735–1756. [DOI] [PubMed] [Google Scholar]
- Nayar AS, Dougherty TJ, Ferguson KE, Granger BA, McWilliams L, Stacey C, et al. (2015). Novel antibacterial targets and compounds revealed by a high-throughput cell wall reporter assay. Journal of Bacteriology, 197(10), 1726–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus FC, & Baddiley J (2003). A continuum of anionic charge: structures and functions of d -alanyl-teichoic acids in Gram-positive bacteria. Microbiology and Molecular Biology Reviews, 67(4), 686–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyfakh AA, Borsch CM, & Kaatz GW (1993). Fluoroquinolone resistance protein NorA of Staphylococcus aureus is a multidrug efflux transporter. Antimicrob Agents Chemother 37(1), 128–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson NN, Jao CC, Xu Y, Quinn J, Skippington E, Alexander MK, et al. (2018). A novel inhibitor of the LolCDE ABC transporter essential for lipoprotein trafficking in Gram-negative bacteria. Antimicrobial Agents and Chemotherapy, 62(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijeholt J. A. L. a., & Driessen AJM (2012). The bacterial Sec-translocase: structure and mechanism. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences, 367(1592), 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H (2016). Structure and mechanism of RND-type multidrug efflux pumps. Advances in Enzymology and Related Areas of Molecular Biology, 77, 1–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen T, Nes IF, & Holo H (2003). Enterolysin A, a cell wall-degrading bacteriocin from Enterococcus faecalis LMG 2333. Applied and Environmental Microbiology, 69(5), 2975–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E, Downing KH, Amos LA, & Löwe J (1998). Tubulin and FtsZ form a distinct family of GTPases. Nature Structural Biology, 5(6), 451–458. [DOI] [PubMed] [Google Scholar]
- Novo DJ, Perlmutter NG, Hunt RH, & Shapiro HM (2000). Multiparameter flow cytometric analysis of antibiotic effects on membrane potential, membrane permeability, and bacterial counts of Staphylococcus aureus and Micrococcus luteus. Antimicrobial Agents and Chemotherapy, 44(4), 827–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara JA, Ambe LA, Casella LG, Townsend BM, Pelletier MR, Ernst RK, et al. (2013). Activities of vancomycin-containing regimens against colistin-resistant Acinetobacter baumannii clinical strains. Antimicrobial Agents and Chemotherapy, 57(5), 2103–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs MM, McCusker MP, Bains M, & Hancock REW (1999). Negative regulation of the Pseudomonas aeruginosa outer membrane porin OprD selective for imipenem and basic amino acids. Antimicrobial Agents and Chemotherapy, 43(5), 1085–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Freinkman E, & Kahne D (2012). Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. coli. Science, 338(6111), 1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Sherman DJ, Silhavy TJ, Ruiz N, & Kahne D (2016). Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nature Reviews Microbiology, 14(6), 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, & Tokuda H (2009). Model of mouth-to-mouth transfer of bacterial lipoproteins through inner membrane LolC, periplasmic LolA, and outer membrane LolB. Proceedings of the National Academy of Sciences of the United States of America, 106(14), 5877–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, & Tokuda H (2011). Lipoprotein sorting in bacteria. Annual Review of Microbiology, 65(1), 239–259. [DOI] [PubMed] [Google Scholar]
- Okuda S, Watanabe S, & Tokuda H (2008). A short helix in the C-terminal region of LolA is important for the specific membrane localization of lipoproteins. FEBS Letters, 582(15), 2247–2251. [DOI] [PubMed] [Google Scholar]
- Oliveira-Tintino C. D. d. M., Tintino SR, Araujo A. C. J. d., Barbosa C. R. d. S., Freitas PR, Neto J. B. d. A., et al. (2023). Efflux pump (QacA, QacB, and QacC) and β-lactamase inhibitors? An evaluation of 1,8-naphthyridines against Staphylococcus aureus strains. Molecules, 28(4), 1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoto S, Suzuki H, & Inouye S (2006). Isolation and structure of Sf-1902 A5 a new globomycin analogue. The Journal of Antibiotics, 32(1), 83–86. [DOI] [PubMed] [Google Scholar]
- Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen M-H, et al. (1996). Antibacterial agents that inhibit lipid A biosynthesis. Science, 274(5289), 980–982. [DOI] [PubMed] [Google Scholar]
- Opperman TJ, & Nguyen ST (2015). Recent advances toward a molecular mechanism of efflux pump inhibition. Frontiers in Microbiology, 6, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornes S (2023). Researchers turn to tiny robots to fight antibiotic resistance. Proceedings of the National Academy of Sciences of the United States of America, 120(7), e2300515120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn M, Stachulski N, Sun H, Blais J, Venishetty V, Raccuglia M, et al. (2019). A first-in-human study to assess the safety and pharmacokinetics of LYS228, a novel intravenous monobactam antibiotic in healthy volunteers. Antimicrobial Agents and Chemotherapy, 63(7), e02592–02518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paetzel M, Dalbey RE, & Strynadka NCJ (2000). The structure and mechanism of bacterial type I signal peptidases A novel antibiotic target. Pharmacology and Therapeutics, 87(1), 27–49. [DOI] [PubMed] [Google Scholar]
- Paetzel M, Karla A, Strynadka NCJ, & Dalbey RE (2002). Signal peptidases. Chemical Reviews, 102(12), 4549–4580. [DOI] [PubMed] [Google Scholar]
- Palmer T, & Berks BC (2012). The twin-arginine translocation (Tat) protein export pathway. Nature Reviews Microbiology, 10(7), 483–496. [DOI] [PubMed] [Google Scholar]
- Palmer T, & Stansfeld PJ (2020). Targeting of proteins to the twin-arginine translocation pathway. Molecular Microbiology, 113(5), 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panahandeh S, Maurer C, Moser M, DeLisa MP, & Müller M (2008). Following the path of a twin-arginine precursor along the TatABC translocase of Escherichia coli. Journal of Biological Chemistry, 283(48), 33267–33275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Sharma A, Tripathi D, Kumar A, Khubaib M, Bhuwan M, et al. (2016). Mycobacterium tuberculosis peptidyl-prolyl isomerases also exhibit chaperone like activity in vitro and in vivo. PloS One, 11(3), e0150288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandeya A, Yang L, Alegun O, Karunasena C, Risko C, Li Z, et al. (2021). Biotinylation as a tool to enhance the uptake of small molecules in Gram-negative bacteria. PloS One, 16(11), e0260023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp-Wallace KM (2019). The latest advances in β-lactam/β-lactamase inhibitor combinations for the treatment of Gram-negative bacterial infections. Expert Opinion on Pharmacotherapy, 20(17), 2169–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis-Bleau C, Markovski M, Uehara T, Lupoli TJ, Walker S, Kahne DE, et al. (2010). Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell, 143(7), 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JT, & Uehara T (2008). How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiology and Molecular Biology Reviews, 72(2), 211–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquina LW, Maria JPS, & Walker S (2013). Teichoic acid biosynthesis as an antibiotic target. Current Opinion in Microbiology, 16(5), 531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania R, Zlitni S, Barker C, Das R, Gerritsma DA, Lebert J, et al. (2009). Chemical genomics in Escherichia coli identifies an inhibitor of bacterial lipoprotein targeting. Nature Chemical Biology, 5(11), 849–856. [DOI] [PubMed] [Google Scholar]
- Patil SD, Sharma R, Srivastava S, Navani NK, & Pathania R (2013). Downregulation of yidC in Escherichia coli by antisense RNA expression results in sensitization to antibacterial essential oils eugenol and carvacrol. PloS One, 8(3), e57370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen IT (2003). Multidrug efflux pumps and resistance: regulation and evolution. Current Opinion in Microbiology, 6(5), 446–451. [DOI] [PubMed] [Google Scholar]
- Pharmaceuticals, V. (2022). Press Releases
- Piddock LJV (2006). Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clinical Microbiology Reviews, 19(2), 382–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl EE, Krylov AV, Block M, & Pohl P (1998). Changes of the membrane potential profile induced by verapamil and propranolol. Biochimica et Biophysica Acta, 1373(1), 170–178. [DOI] [PubMed] [Google Scholar]
- Poole K (2001). Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms. Journal of Molecular Microbiology and Biotechnology, 3(2), 255–264. [PubMed] [Google Scholar]
- Poole K (2007). Efflux pumps as antimicrobial resistance mechanisms. Annals of Medicine, 39(3), 162–176. [DOI] [PubMed] [Google Scholar]
- Potluri L, Karczmarek A, Verheul J, Piette A, Wilkin JM, Werth N, et al. (2010). Septal and lateral wall localization of PBP5, the major D,D-carboxypeptidase of Escherichia coli, requires substrate recognition and membrane attachment. Molecular Microbiology, 77(2), 300–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad NK, Seiple IB, Cirz RT, & Rosenberg OS (2022). Leaks in the pipeline: a failure analysis of Gram-negative antibiotic development from 2010 to 2020. Antimicrobial Agents and Chemotherapy, 66(5), e00054–00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujari V, Rozman K, Dhiman RK, Aldrich CC, & Crick DC (2022). Mycobacterial MenG: partial purification, characterization, and inhibition. ACS Infectious Diseases 8(12), 2430–2440. [DOI] [PubMed] [Google Scholar]
- Puyenbroeck VV, & Vermeire K (2018). Inhibitors of protein translocation across membranes of the secretory pathway: novel antimicrobial and anticancer agents. Cellular and Molecular Life Sciences, 75(9), 1541–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri L (2007). Strategic paradigm shifts in the antimicrobial drug discovery process of the 21st century. Infectious Disorders - Drug Targets, 7(3), 230–237. [DOI] [PubMed] [Google Scholar]
- Radchenko M, Symersky J, Nie R, & Lu M (2015). Structural basis for the blockade of MATE multidrug efflux pumps. Nature Communications, 6(1), 7995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademacher C, & Masepohl B (2012). Copper-responsive gene regulation in bacteria. Microbiology, 158(10), 2451–2464. [DOI] [PubMed] [Google Scholar]
- Radika K, & Raetz CR (1988). Purification and properties of lipid A disaccharide synthase of Escherichia coli. Journal of Biological Chemistry, 263(29), 14859–14867. [PubMed] [Google Scholar]
- Raetz CRH, Reynolds CM, Trent MS, & Bishop RE (2007). Lipid A modification systems in Gram-negative bacteria. Annual Review of Biochemistry, 76(1), 295–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz CRH, & Whitfield C (2002). Lipopolysaccharide endotoxins. Biochemistry, 71(1), 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramm K, & Pluckthun A (2001). High enzymatic activity and chaperone function are mechanistically related features of the dimeric E. coli peptidyl-prolyl-isomerase FkpA. Journal of Molecular Biology, 310(2), 485–498. [DOI] [PubMed] [Google Scholar]
- Rao S, Prestidge CA, Miesel L, Sweeney D, Shinabarger DL, & Boulos RA (2016). Preclinical development of Ramizol, an antibiotic belonging to a new class, for the treatment of Clostridium difficile colitis. The Journal of Antibiotics, 69(12), 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayes JE, Rodríguez-Alonso R, & Collet J-F (2021). Lipoproteins in Gram-negative bacteria: new insights into their biogenesis, subcellular targeting and functional roles. Current Opinion in Microbiology, 61, 25–34. [DOI] [PubMed] [Google Scholar]
- Reig S, Gouellec AL, & Bleves S (2022). What is new in the anti-Pseudomonas aeruginosa clinical development pipeline since the 2017 WHO alert? Frontiers in Cellular and Infection Microbiology, 12, 909731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RePORT, N. IND-enabling studies of the potent LpxC inhibitor LPC-233 as a novel antibiotic against Gram-negative pathogens. from https://reporter.nih.gov/search/8TVNFY8ZZ0ikQeLCIb6zsw/project-details/10011609
- Robert V, Volokhina EB, Senf F, Bos MP, Gelder PV, & Tommassen J (2006). Assembly factor Omp85 recognizes its outer membrane protein substrates by a species-specific c-terminal motif. PLoS Biology, 4(11), e377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Rojas A, Moreno-Morales J, Mason AJ, & Rolff J (2018). Cationic antimicrobial peptides do not change recombination frequency in Escherichia coli. Biology Letters, 14(3), 20180006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas ER, & Huang KC (2018). Regulation of microbial growth by turgor pressure. Current Opinion in Microbiology, 42, 62–70. [DOI] [PubMed] [Google Scholar]
- Rosado-Lugo JD, Sun Y, Banerjee A, Cao Y, Datta P, Zhang Y, et al. (2022). Evaluation of 2,6-difluoro-3-(oxazol-2-ylmethoxy)benzamide chemotypes as Gram-negative FtsZ inhibitors. The Journal of Antibiotics, 75(7), 385–395. [DOI] [PubMed] [Google Scholar]
- Rotter MJ, Zentgraf S, Weizel L, Frank D, Burgers LD, Brunst S, et al. (2023). Integrating siderophore substructures in thiol-based metallo-β-lactamase inhibitors. Molecules, 28(4), 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Chatterjee S, Bhattacharjee A, Chattopadhyay P, Saha B, Dutta S, et al. (2021). Overexpression of efflux pumps, mutations in the pumps’ regulators, chromosomal mutations, and AAC(6’)-Ib-cr are associated with fluoroquinolone resistance in diverse sequence types of neonatal septicaemic Acinetobacter baumannii: a 7-year single center study. Frontiers in Microbiology, 12, 602724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinchik E, Schneider T, Elliott M, Scott WRP, Pan J, Anklin C, et al. (2011). Mechanism of action and limited cross-resistance of new lipopeptide MX-2401. Antimicrobial Agents and Chemotherapy, 55(6), 2743–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KR, Taylor JA, & Bowers LM (2010). The BAM complex subunit BamE (SmpA) is required for membrane integrity, stalk growth and normal levels of outer membrane β-barrel proteins in Caulobacter crescentus. Microbiology, 156(3), 742–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MD, Parkes AL, Corbett D, Dickie AP, Southey M, Andersen OA, et al. (2021). Discovery of novel UDP-N-acetylglucosamine acyltransferase (LpxA) inhibitors with activity against Pseudomonas aeruginosa. Journal of Medicinal Chemistry, 64(19), 14377–14425. [DOI] [PubMed] [Google Scholar]
- Sabnis A, Hagart KLH, Klöckner A, Becce M, Evans LE, Furniss RCD, et al. (2021). Colistin kills bacteria by targeting lipopolysaccharide in the cytoplasmic membrane. eLife, 10, e65836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahl HG, Jack RW, & Bierbaum G (1995). Biosynthesis and biological activities of lantibiotics with unique post-translational modifications. European Journal of Biochemistry, 230(3), 827–853. [DOI] [PubMed] [Google Scholar]
- Samsudin F, Ortiz-Suarez Maite L., Piggot Thomas J., Bond Peter J., & Khalid S (2016). OmpA: A flexible clamp for bacterial cell wall attachment. Structure, 24(12), 2227–2235. [DOI] [PubMed] [Google Scholar]
- Samuel G, & Reeves P (2003). Biosynthesis of O-antigens: genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydrate Research, 338(23), 2503–2519. [DOI] [PubMed] [Google Scholar]
- Sankaran K, & Wu HC (1994). Lipid modification of bacterial prolipoprotein. Transfer of diacylglyceryl moiety from phosphatidylglycerol. Journal of Biological Chemistry, 269(31), 19701–19706. [PubMed] [Google Scholar]
- Santos AL, Liu D, Reed AK, Wyderka AM, Venrooy A. v., Li JT, et al. (2022). Light-activated molecular machines are fast-acting broad-spectrum antibacterials that target the membrane. Science Advances, 8(22), eabm2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent F, Bogsch EG, Stanley NR, Wexler M, Robinson C, Berks BC, et al. (1998). Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO Journal, 17(13), 3640–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar P, Yarlagadda V, Ghosh C, & Haldar J (2017). A review on cell wall synthesis inhibitors with an emphasis on glycopeptide antibiotics. MedChemComm, 8(3), 516–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass P, Josten M, Famulla K, Schiffer G, Sahl H-G, Hamoen L, et al. (2011). Antibiotic acyldepsipeptides activate ClpP peptidase to degrade the cell division protein FtsZ. Proceedings of the National Academy of Sciences of the United States of America, 108(42), 17474–17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinpflug K, Krylova O, Nikolenko H, Thurm C, & Dathe M (2015). Evidence for a novel mechanism of antimicrobial action of a cyclic R-,W-rich hexapeptide. PloS One, 10(4), e0125056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler CA, & Schuhardt VT (1964). Lysostaphin: A new bacteriolytic agent for the Staphylococcus. Proceedings of the National Academy of Sciences of the United States of America, 51(3), 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz FJ, Fluit AC, Lückefahr M, Engler B, Hofmann B, Verhoef J, et al. (1998). The effect of reserpine, an inhibitor of multidrug efflux pumps, on the in-vitro activities of ciprofloxacin, sparfloxacin and moxifloxacin against clinical isolates of Staphylococcus aureus. Journal of Antimicrobial Chemotherapy, 42(6), 807–810. [DOI] [PubMed] [Google Scholar]
- Schnapp D, & Harris A (1998). Antibacterial peptides in bronchoalveolar lavage fluid. American Journal of Respiratory Cell and Molecular Biology, 19(3), 352–356. [DOI] [PubMed] [Google Scholar]
- Schwalm J, Mahoney TF, Soltes GR, & Silhavy TJ (2013). Role for Skp in LptD assembly in Escherichia coli. Journal of Bacteriology, 195(16), 3734–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekyere JO, & Amoako DG (2017). Carbonyl cyanide m-chlorophenylhydrazine (CCCP) reverses resistance to colistin, but not to carbapenems and tigecycline in multidrug-resistant Enterobacteriaceae. Frontiers in Microbiology, 8, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane A, Sabbaj A, McMurry LM, & Levy SB (1992). Multiple antibiotic susceptibility associated with inactivation of the prc gene. Journal of Bacteriology, 174(23), 7844–7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha J, Fadl AA, Klimpel GR, Niesel DW, Popov VL, & Chopra AK (2004). The two murein lipoproteins of Salmonella enterica serovar Typhimurium contribute to the virulence of the organism. Infection and Immunity, 72(7), 3987–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao F, Bader MW, Jakob U, & Bardwell JCA (2000). DsbG, a protein disulfide isomerase with chaperone activity. Journal of Biological Chemistry, 275(18), 13349–13352. [DOI] [PubMed] [Google Scholar]
- Sharma A, Gupta VK, & Pathania R (2019). Efflux pump inhibitors for bacterial pathogens: from bench to bedside. Indian Journal of Medical Research, 149(2), 129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AK, Kumar S, H. K, Dhakan DB, & Sharma VK (2016). Prediction of peptidoglycan hydrolases - a new class of antibacterial proteins. BMC Genomics, 17(1), 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Zhou R, Wan L, Feng S, Song K, Xu C, et al. (2021). Mechanism of LolCDE as a molecular extruder of bacterial triacylated lipoproteins. Nature Communications, 12(1), 4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd M, Heras B, Achard ME, King GJ, Argente MP, Kurth F, et al. (2013). Structural and functional characterization of ScsC, a periplasmic thioredoxin-like protein from Salmonella enterica serovar Typhimurium. Antioxidants & Redox Signaling, 19(13), 1494–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DJ, Okuda S, Denny WA, & Kahne D (2013). Validation of inhibitors of an ABC transporter required to transport lipopolysaccharide to the cell surface in Escherichia coli. Bioorganic and Medicinal Chemistry, 21(16), 4846–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DJ, Xie R, Taylor RJ, George AH, Okuda S, Foster PJ, et al. (2018). Lipopolysaccharide is transported to the cell surface by a membrane-to-membrane protein bridge. Science, 359(6377), 798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H (2023). Three innovations of next-generation antibiotics: evolvability, specificity, and non-immunogenicity. Antibiotics, 12(2), 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouldice SR, Cho SH, Boyd D, Heras B, Eser M, Beckwith J, et al. (2010). In vivo oxidative protein folding can be facilitated by oxidation-reduction cycling. Molecular Microbiology, 75(1), 13–28. [DOI] [PubMed] [Google Scholar]
- Sikora AE, Wierzbicki IH, Zielke RA, Ryner RF, Korotkov KV, Buchanan SK, et al. (2018). Structural and functional insights into the role of BamD and BamE within the β-barrel assembly machinery in Neisseria gonorrhoeae. Journal of Biological Chemistry, 293(4), 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy TJ, Kahne D, & Walker S (2010). The bacterial cell envelope. Cold Spring Harbor Perspectives in Biology, 2(5), a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LL (2016). Appropriate targets for antibacterial drugs. Cold Spring Harbor Perspectives in Medicine, 6(12), a030239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LL (2017). Fosfomycin: mechanism and resistance. Cold Spring Harbor Perspectives in Medicine, 7(2), a025262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JA, Perlmutter NG, & Shapiro HM (2003). Correlation of daptomycin bactericidal activity and membrane depolarization in Staphyloccocus aureus. Antimicrobial Agents and Chemotherapy, 47(8), 2538–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson BW, & Trent MS (2019). Pushing the envelope: LPS modifications and their consequences. Nature Reviews Microbiology, 17(7), 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh MP (2006). Rapid test for distinguishing membrane-active antibacterial agents. Journal of Microbiological Methods, 67(1), 125–130. [DOI] [PubMed] [Google Scholar]
- Sionov RV, & Steinberg D (2022). Targeting the holy triangle of quorum sensing, biofilm formation, and antibiotic resistance in pathogenic bacteria. Microorganisms, 10(6), 1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjuts H, Vargiu AV, Kwasny SM, Nguyen ST, Kim H-S, Ding X, et al. (2016). Molecular basis for inhibition of AcrB multidrug efflux pump by novel and powerful pyranopyridine derivatives. Proceedings of the National Academy of Sciences of the United States of America, 113(13), 3509–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skagia A, Vezyri E, Grados K, Venieraki A, Karpusas M, Katinakis P, et al. (2017). Structure-function analysis of the periplasmic Escherichia coli cyclophilin PpiA in relation to biofilm formation. Journal of Molecular Microbiology and Biotechnology, 27(4), 228–236. [DOI] [PubMed] [Google Scholar]
- Sklar JG, Wu T, Gronenberg LS, Malinverni JC, Kahne D, & Silhavy TJ (2007). Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America, 104(15), 6400–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PA, Koehler MFT, Girgis HS, Yan D, Chen Y, Chen Y, et al. (2018). Optimized arylomycins are a new class of Gram-negative antibiotics. Nature, 561(7722), 189–194. [DOI] [PubMed] [Google Scholar]
- Sohlenkamp C, & Geiger O (2016). Bacterial membrane lipids: diversity in structures and pathways. FEMS Microbiology Reviews, 40(1), 133–159. [DOI] [PubMed] [Google Scholar]
- Spero T SPR206 candidate for treating MDR Gram-negative infections. from https://www.sperotherapeutics.com/patients/spr-206 ,urldate = 2023-03-05
- Spiess C, Beil A, & Ehrmann M (1999). A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell, 97(3), 339–347. [DOI] [PubMed] [Google Scholar]
- Spirig T, Weiner EM, & Clubb RT (2011). Sortase enzymes in Gram-positive bacteria. Molecular Microbiology, 82(5), 1044–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, et al. (2010). Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science, 327(5968), 1010–1013. [DOI] [PubMed] [Google Scholar]
- Steeghs L, Cock H. d., Evers E, Zomer B, Tommassen J, & Ley P. v. d. (2001). Outer membrane composition of a lipopolysaccharide-deficient Neisseria meningitidis mutant. EMBO Journal, 20(24), 6937–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenhuis M, Koningstein GM, Oswald J, Pick T, O’Keefe S, Koch HG, et al. (2021). Eeyarestatin 24 impairs SecYEG-dependent protein trafficking and inhibits growth of clinically relevant pathogens. Molecular Microbiology, 115(1), 28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenhuis M, van Ulsen P, Martin NI, & Luirink J (2021). A ban on BAM: an update on inhibitors of the β-barrel assembly machinery. FEMS Microbiology Letters, 368(11). [DOI] [PubMed] [Google Scholar]
- Stokes JM, Yang K, Swanson K, Jin W, Cubillos-Ruiz A, Donghia NM, et al. (2020). A deep learning approach to antibiotic discovery. Cell, 180(4), 688–702.e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone KJ, & Strominger JL (1971). Mechanism of action of bacitracin: complexation with metal ion and C55-isoprenyl pyrophosphate. Proceedings of the National Academy of Sciences of the United States of America, 68(12), 3223–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storek KM, Auerbach MR, Shi H, Garcia NK, Sun D, Nickerson NN, et al. (2018). Monoclonal antibody targeting the β-barrel assembly machine of Escherichia coli is bactericidal. Proceedings of the National Academy of Sciences of the United States of America, 115(14), 3692–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl H, & Hamoen LW (2010). Membrane potential is important for bacterial cell division. Proceedings of the National Academy of Sciences of the United States of America, 107(27), 12281–12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull F, Betton J-M, & Bardwell JCA (2018). Periplasmic chaperones and prolyl isomerases. EcoSal Plus, 8(1), 1–16. [DOI] [PubMed] [Google Scholar]
- Su M-Y, Som N, Wu C-Y, Su S-C, Kuo Y-T, Ke L-C, et al. (2017). Structural basis of adaptor-mediated protein degradation by the tail-specific PDZ-protease Prc. Nature Communications, 8(1), 1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subedi P, Paxman JJ, Wang G, Ukuwela AA, Xiao Z, & Heras B (2019). The Scs disulfide reductase system cooperates with the metallochaperone CueP in Salmonella copper resistance. Journal of Biological Chemistry, 294(44), 15876–15888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugai M, & Wu HC (1992). Export of the outer membrane lipoprotein is defective in secD, secE, and secF mutants of Escherichia coli. Journal of Bacteriology, 174(8), 2511–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugie Y, Inagaki SAE, Kato Y, Nishida H, Pang C-H, Saito T, et al. (2002). CJ-21,058, a new SecA inhibitor isolated from a fungus. The Journal of Antibiotics, 55(1), 25–29. [DOI] [PubMed] [Google Scholar]
- Sun H, Zhang Q, Wang R, Wang H, Wong Y-T, Wang M, et al. (2020). Resensitizing carbapenem- and colistin-resistant bacteria to antibiotics using auranofin. Nature Communications, 11(1), 5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Deng Z, & Yan A (2014). Bacterial multidrug efflux pumps: mechanisms, physiology and pharmacological exploitations. Biochemical and Biophysical Research Communications, 453(2), 254–267. [DOI] [PubMed] [Google Scholar]
- Sun Y, & Shang D (2015). Inhibitory effects of antimicrobial peptides on lipopolysaccharide-induced inflammation. Mediators of Inflammation, 2015, 167572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvorov M, Vakulenko SB, & Mobashery S (2007). Cytoplasmic-membrane anchoring of a class A β-lactamase and its capacity in manifesting antibiotic resistance. Antimicrobial Agents and Chemotherapy, 51(8), 2937–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain J, Khoury ME, Kempf J, Briée F, Smissen PVD, Décout J-L, et al. (2018). Effect of cardiolipin on the antimicrobial activity of a new amphiphilic aminoglycoside derivative on Pseudomonas aeruginosa. PloS One, 13(8), e0201752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri Y, Joković N, Vitorović J, Grundmann O, Maroyi A, & Calina D (2021). The burden of the serious and difficult-to-treat infections and a new antibiotic available: cefiderocol. Frontiers in Pharmacology, 11, 578823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxis Pharmaceuticals. (2023). Our Pipeline – TAXIS Pharmaceutical.
- Thakur V, Uniyal A, & Tiwari V (2021). A comprehensive review on pharmacology of efflux pumps and their inhibitors in antibiotic resistance. European Journal of Pharmacology, 903, 1–12. [DOI] [PubMed] [Google Scholar]
- Thoma J, Burmann BM, Hiller S, & Müller DJ (2015). Impact of holdase chaperones Skp and SurA on the folding of β-barrel outer-membrane proteins. Nature Structural & Molecular Biology, 22(10), 795–802. [DOI] [PubMed] [Google Scholar]
- Tipper DJ, & Strominger JL (1965). Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proceedings of the National Academy of Sciences of the United States of America, 54(4), 1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooke CL, Hinchliffe P, Bragginton EC, Colenso CK, Hirvonen VHA, Takebayashi Y, et al. (2019). β-Lactamases and β-lactamase inhibitors in the 21st century. Journal of Molecular Biology, 431(18), 3472–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrens G, Sánchez-Diener I, Jordana-Lluch E, Barceló IM, Zamorano L, Juan C, et al. (2019). In vivo validation of peptidoglycan recycling as a target to disable AmpC-mediated resistance and reduce virulence enhancing the cell-wall–targeting immunity. Journal of Infectious Diseases, 220(11), 1729–1737. [DOI] [PubMed] [Google Scholar]
- Totsika M, Vagenas D, Paxman JJ, Wang G, Dhouib R, Sharma P, et al. (2018). Inhibition of diverse DsbA enzymes in multi-DsbA encoding pathogens. Antioxidants & Redox Signaling, 29(7), 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trout RE, Zulli A, Mesaros E, Jackson RW, Boyd S, Liu B, et al. (2021). Discovery of VNRX-7145 (VNRX-5236 etzadroxil): an orally bioavailable β-lactamase inhibitor for Enterobacterales expressing Ambler class A, C, and D enzymes. Journal of Medicinal Chemistry, 64(14), 10155–10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, Banzhaf M, Gross CA, & Vollmer W (2012). From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nature Reviews Microbiology, 10(2), 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ude J, Tripathi V, Buyck JM, Soderholm S, Cunrath O, Fanous J, et al. (2021). Outer membrane permeability: antimicrobials and diverse nutrients bypass porins in Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences of the United States of America, 118(31), e2107644118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanus ML, Scotti PA, Fröderberg L, Sääf A, Gier J. W. L. d., Brunner J, et al. (2001). Sec-dependent membrane protein insertion: sequential interaction of nascent FtsQ with SecY and YidC. EMBO Reports, 2(6), 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urfer M, Bogdanovic J, Monte FL, Moehle K, Zerbe K, Omasits U, et al. (2016). A peptidomimetic antibiotic targets outer membrane proteins and disrupts selectively the outer membrane in Escherichia coli. Journal of Biological Chemistry, 291(4), 1921–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urrutia ÍM, Sabag A, Valenzuela C, Labra B, Álvarez SA, & Santiviago CA (2018). Contribution of the twin-arginine translocation system to the intracellular survival of Salmonella Typhimurium in Dictyostelium discoideum. Frontiers in Microbiology, 9, 3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargiu AV, & Nikaido H (2012). Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proceedings of the National Academy of Sciences of the United States of America, 109(50), 20637–20642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasil ML, Tomaras AP, & Pritchard AE (2012). Identification and evaluation of twin-arginine translocase inhibitors. Antimicrobial Agents and Chemotherapy, 56(12), 6223–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaegh R, Becker KA, Edwards MJ, & Gulbins E (2020). Sphingosine kills bacteria by binding to cardiolipin. Journal of Biological Chemistry, 295(22), 7686–7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma VA, Wang L, Labadie SS, Liang J, Sellers BD, Wang J, et al. (2022). Discovery of inhibitors of the lipopolysaccharide transporter MsbA: from a screening hit to potent wild-type Gram-negative activity. Journal of Medicinal Chemistry, 65(5), 4085–4120. [DOI] [PubMed] [Google Scholar]
- Versporten A, Zarb P, Caniaux I, Gros MF, Drapier N, Miller M, et al. (2018). Antimicrobial consumption and resistance in adult hospital inpatients in 53 countries: results of an internet-based global point prevalence survey. Lancet Global Health, 6(6), e619–e629. [DOI] [PubMed] [Google Scholar]
- Vertommen D, Depuydt M, Pan J, Leverrier P, Knoops L, Szikora JP, et al. (2008). The disulphide isomerase DsbC cooperates with the oxidase DsbA in a DsbD-independent manner. Molecular Microbiology, 67(2), 336–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetterli SU, Zerbe K, Muller M, Urfer M, Mondal M, Wang S-Y, et al. (2018). Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli. Science Advances, 4(11), eaau2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Farrés X, Parra-Millán R, Sánchez-Encinales V, Varese M, Ayerbe-Algaba R, Bayó N, et al. (2017). Combating virulence of Gram-negative bacilli by OmpA inhibition. Scientific Reports, 7(1), 14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogeley L, Arnaout TE, Bailey J, Stansfeld PJ, Boland C, & Caffrey M (2016). Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science, 351(6275), 876–880. [DOI] [PubMed] [Google Scholar]
- Vollmer W, Blanot D, & Pedro MAD (2008). Peptidoglycan structure and architecture. FEMS Microbiology Reviews, 32(2), 149–167. [DOI] [PubMed] [Google Scholar]
- Vorachek-Warren MK, Ramirez S, Cotter RJ, & Raetz CRH (2002). A triple mutant of Escherichia coli lacking secondary acyl chains on lipid A. Journal of Biological Chemistry, 277(16), 14194–14205. [DOI] [PubMed] [Google Scholar]
- Voss BJ, & Trent MS (2018). LPS transport: flipping out over MsbA. Current Biology, 28(1), R30–R33. [DOI] [PubMed] [Google Scholar]
- Waelheyns ED, Segers K, Sardis MF, Anné J, Nicolaes GAF, & Economou A (2015). Identification of small-molecule inhibitors against SecA by structure-based virtual ligand screening. The Journal of Antibiotics, 68(11), 666–673. [DOI] [PubMed] [Google Scholar]
- Waite RD, Rose RS, Rangarajan M, Aduse-Opoku J, Hashim A, & Curtis MA (2012). Pseudomonas aeruginosa possesses two putative type I signal peptidases, LepB and PA1303, each with distinct roles in physiology and virulence. Journal of Bacteriology, 194(17), 4521–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller PR, & Sauer RT (1996). Characterization of degQ and degS, Escherichia coli genes encoding homologs of the DegP protease. Journal of Bacteriology, 178(4), 1146–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton TA, Idigo CA, Herrera N, & Rees DC (2015). MscL: channeling membrane tension. Pflügers Archiv. European Journal of Physiology, 467(1), 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C-Y, Wang S-W, Huang W-C, Kim KS, Chang N-S, Wang Y-H, et al. (2012). Prc contributes to Escherichia coli evasion of classical complement-mediated serum killing. Infection and Immunity, 80(10), 3399–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ansari MF, & Zhou C-H (2021). Identification of unique quinazolone thiazoles as novel structural scaffolds for potential Gram-negative bacterial conquerors. Journal of Medicinal Chemistry, 64(11), 7630–7645. [DOI] [PubMed] [Google Scholar]
- Wang M, Chan EWC, Wan Y, Wong M. H.-y., & Chen S (2021). Active maintenance of proton motive force mediates starvation-induced bacterial antibiotic tolerance in Escherichia coli. Communications Biology, 4(1), 1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Lai T-P, Gao P, Zhang H, Ho P-L, Woo PC-Y, et al. (2018). Bismuth antimicrobial drugs serve as broad-spectrum metallo-β-lactamase inhibitors. Nature Communications, 9(1), 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb CT, Selkrig J, Perry AJ, Noinaj N, Buchanan SK, & Lithgow T (2012). Dynamic association of BAM complex modules includes surface exposure of the lipoprotein BamC. Journal of Molecular Biology, 422(4), 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein EA, Yano T, Li L-S, Avarbock D, Avarbock A, Helm D, et al. (2005). Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proceedings of the National Academy of Sciences of the United States of America, 102(12), 4548–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessler S, Schneider G, & Backert S (2017). Bacterial serine protease HtrA as a promising new target for antimicrobial therapy? Cell Communication & Signaling, 15(1), 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield C, & Trent MS (2014). Biosynthesis and export of bacterial lipopolysaccharides. Annual Review of Biochemistry, 83(1), 99–128. [DOI] [PubMed] [Google Scholar]
- Whitfield C, Williams DM, & Kelly SD (2020). Lipopolysaccharide O-antigens - bacterial glycans made to measure. Journal of Biological Chemistry, 295(31), 10593–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiktor M, Weichert D, Howe N, Huang C-Y, Olieric V, Boland C, et al. (2017). Structural insights into the mechanism of the membrane integral N-acyltransferase step in bacterial lipoprotein synthesis. Nature Communications, 8(1), 15952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmaerts D, Dewachter L, Loose P-JD, Bollen C, Verstraeten N, & Michiels J (2019). HokB monomerization and membrane repolarization control persister awakening. Molecular Cell, 75(5), 1031–1042.e1034. [DOI] [PubMed] [Google Scholar]
- Winkle M, Hernández-Rocamora VM, Pullela K, Goodall ECA, Martorana AM, Gray J, et al. (2021). DpaA detaches Braun’s lipoprotein from peptidoglycan. mBio, 12(3), e00836–00821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojdyla JA, Cutts E, Kaminska R, Papadakos G, Hopper JTS, Stansfeld PJ, et al. (2015). Structure and function of the Escherichia coli Tol-Pal stator protein TolR. Journal of Biological Chemistry, 290(44), 26675–26687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JLC, Romano M, Kerry LE, Kwong HS, Low WW, Brett SJ, et al. (2019). OmpK36-mediated carbapenem resistance attenuates ST258 Klebsiella pneumoniae in vivo. Nature Communications, 10(1), 3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley MV, & Estrada SJ (2014). Bedaquiline: a novel antitubercular agent for the treatment of multidrug-resistant tuberculosis. Pharmacotherapy, 34(11), 1187–1197. [DOI] [PubMed] [Google Scholar]
- Wray R, Herrera N, Iscla I, Wang J, & Blount P (2019). An agonist of the MscL channel affects multiple bacterial species and increases membrane permeability and potency of common antibiotics. Mol Microbiol, 112(3), 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray R, Iscla I, & Blount P (2021). Curcumin activation of a bacterial mechanosensitive channel underlies its membrane permeability and adjuvant properties. PLoS Pathogens, 17(12), e1010198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray R, Iscla I, Gao Y, Li H, Wang J, & Blount P (2015). Dihydrostreptomycin directly binds to, modulates, and passes through the MscL channel pore. PLoS Biology, 14(6), e1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray R, Iscla I, Kovacs Z, Wang J, & Blount P (2019). Novel compounds that specifically bind and modulate MscL: insights into channel gating mechanisms. FASEB Journal, 33(3), 3180–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray R, Wang J, Blount P, & Iscla I (2022). Activation of a bacterial mechanosensitive channel, MscL, underlies the membrane permeabilization of dual-targeting antibacterial compounds. Antibiotics, 11(7), 970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray R, Wang J, Iscla I, & Blount P (2020). Novel MscL agonists that allow multiple antibiotics cytoplasmic access activate the channel through a common binding site. PloS One, 15(1), e0228153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CM, Cao JL, Zheng MH, Ou Y, Zhang L, Zhu XQ, et al. (2007). Effect and mechanism of andrographolide on the recovery of Pseudomonas aeruginosa susceptibility to several antibiotics. Journal of International Medical Research, 36(1), 178–186. [DOI] [PubMed] [Google Scholar]
- Wu JY, Srinivas P, & Pogue JM (2020). Cefiderocol: a novel agent for the management of multidrug-resistant Gram-negative organisms. Infectious Diseases and Therapy, 9(1), 17–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, & Kahne D (2005). Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell, 121(2), 235–245. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Gerth K, Müller R, & Wall D (2012). Myxobacterium-produced antibiotic TA (myxovirescin) inhibits type II signal peptidase. Antimicrobial Agents and Chemotherapy, 56(4), 2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue R-Y, Liu C, Xiao Q-T, Sun S, Zou Q-M, & Li H-B (2021). HtrA family proteases of bacterial pathogens: pros and cons for their therapeutic use. Clinical Microbiology and Infection, 27(4), 559–564. [DOI] [PubMed] [Google Scholar]
- Yi K, Liu S, Liu P, Luo X, Zhao J, Yan F, et al. (2022). Synergistic antibacterial activity of tetrandrine combined with colistin against MCR-mediated colistin-resistant Salmonella. Biomedicine and Pharmacotherapy, 149, 112873. [DOI] [PubMed] [Google Scholar]
- Yoshida I, Takata I, Fujita K, Takashima H, & Sugiyama H (2022). TP0586532, a novel non-hydroxamate LpxC inhibitor: potentiating effect on in vitro activity of meropenem against carbapenem-resistant Enterobacteriaceae. Microbiology Spectrum, 10(3), e00828–00822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young HE, Zhao J, Barker JR, Guan Z, Valdivia RH, & Zhou P (2016). Discovery of the elusive UDP-diacylglucosamine hydrolase in the lipid A biosynthetic pathway in Chlamydia trachomatis. mBio, 7(2), e00090–00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J (1998). Inactivation of DsbA, but not DsbC and DsbD, affects the intracellular survival and virulence of Shigella flexneri. Infection and Immunity, 66(8), 3909–3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdybicka-Barabas A, Mak P, Klys A, Skrzypiec K, Mendyk E, Fiołka MJ, et al. (2012). Synergistic action of Galleria mellonella anionic peptide 2 and lysozyme against Gram-negative bacteria. Biochimica et Biophysica Acta, 1818(11), 2623–2635. [DOI] [PubMed] [Google Scholar]
- Zgurskaya HI, & Rybenkov VV (2020). Permeability barriers of Gram-negative pathogens. Annals of the New York Academy of Sciences, 1459(1), 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Baidin V, Pahil KS, Moison E, Tomasek D, Ramadoss NS, et al. (2018). Cell-based screen for discovering lipopolysaccharide biogenesis inhibitors. Proceedings of the National Academy of Sciences of the United States of America, 115(26), 6834–6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Srinivas S, Xu Y, Wei W, & Feng Y (2019). Genetic and biochemical mechanisms for bacterial lipid A modifiers associated with polymyxin resistance. Trends in Biochemical Sciences, 44(11), 973–988. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Wang R, Wang M, Liu C, Koohi-Moghadam M, Wang H, et al. (2022). Re-sensitization of mcr carrying multidrug resistant bacteria to colistin by silver. Proceedings of the National Academy of Sciences of the United States of America, 119(11), e2119417119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, He D, Yang Y, Lin S, Zhang M, Dai S, et al. (2016). Comparative proteomics reveal distinct chaperone-client interactions in supporting bacterial acid resistance. Proceedings of the National Academy of Sciences of the United States of America, 113(39), 10872–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li Y, Wang W, Zhang J, Lin Y, Hong B, et al. (2019). Identification of an anti-Gram-negative bacteria agent disrupting the interaction between lipopolysaccharide transporters LptA and LptC. International Journal of Antimicrobial Agents, 53(4), 442–448. [DOI] [PubMed] [Google Scholar]
- Zhou P, & Zhao J (2017). Structure, inhibition, and regulation of essential lipid A enzymes. Biochimica et Biophysica Acta, 1862(11), 1424–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu S, Wang T, Li H, Tang S, Wang J, et al. (2018). Pterostilbene, a potential MCR-1 inhibitor that enhances the efficacy of polymyxin B. Antimicrobial Agents and Chemotherapy, 62(4), 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang J, Guo Y, Liu X, Liu S, Niu X, et al. (2019). Discovery of a potential MCR-1 inhibitor that reverses polymyxin activity against clinical mcr-1-positive Enterobacteriaceae. Journal of Infection, 78(5), 364–372. [DOI] [PubMed] [Google Scholar]