

Abstract
Nucleotides are the foundational elements of life. Proliferative cells acquire nutrients for energy production and synthesis of macromolecules, including proteins, lipids, and nucleic acids. Nucleotides are continuously replenished through the activation of the nucleotide synthesis pathways. Despite the importance of nucleotides in cell physiology, there is still much to learn about how the purine and pyrimidine synthesis pathways are regulated upon intracellular and exogenous signals. Over the past decade, evidence has emerged that several signaling pathways (Akt, mTORC1, RAS, TP53, and Hippo-YAP signaling) alter nucleotide synthesis activity and influence cell function. Here we examine the mechanisms by which these signaling networks impinge on de novo nucleotide synthesis in mammalian cells. We also discuss how these molecular links can be targeted in diseases such as cancers and immune disorders.
Keywords: de novo purine and pyrimidine synthesis, signaling pathways, cancer metabolism, nucleotide signaling, immune disorders, metabolic vulnerability
Cell cycle, signaling, and nucleotide metabolism
Nucleotides are the fundamental building blocks for the preservation of life. As monomeric units, purine and pyrimidine nucleotides are polymerized into ribonucleic acids (RNA) and deoxyribonucleic acids (DNA). Mammalian cells require extracellular growth factors-initiated signals to leave the quiescent state and enter the cell cycle. Once in the S phase, cells use ribonucleotide reductase (RNR) to promote deoxynucleoside triphosphate (dNTPs) synthesis by converting nucleoside triphosphate (NTPs) into dNTPs [1]. However, further research is needed to comprehensively understand the impact of the cell cycle on de novo nucleotide synthesis (see Glossary).
Cells have evolved signaling networks that enable them to adjust growth and metabolic capacity in response to stress conditions and nutrient availability [2]. For example, the signaling pathways that regulate nucleotide metabolism include the RAS-ERK cascade, the mechanistic target of rapamycin complex I (mTORC1) pathway, the proto-oncogene MYC, and the tumor suppressor TP53. Corruptions in these signaling pathways, caused by gain or loss of function mutations, are responsible for complex diseases such as immune disorders, infectious diseases, metabolic syndrome, and cancers [3–9].
While recent reviews have discussed the importance of metabolic dependencies in diseased cells [10,11], there has been little emphasis on how the signaling pathways regulate cellular metabolism, particularly de novo nucleotide synthesis. Here we review the molecular mechanisms enabling selective control of nucleotide synthesis and the metabolic pathways supporting nucleotide metabolism downstream of critical signaling networks and discuss the relevance of these findings in physiological and pathological states.
Nucleotide metabolism
Cells use two metabolic routes to generate nucleotides: the de novo and salvage pathways. The de novo nucleotide pathways assemble the purine and pyrimidine rings using precursors such as amino acids, activated sugar, ATP, and bicarbonate. On the other hand, the salvage pathway uses intermediate metabolites (nucleobases, nucleosides) derived from nucleotide catabolism or the surrounding environment to maintain cellular nucleotide pools [8,12]. Although several studies have implicated the signaling pathways in the control of de novo nucleotide synthesis, mechanistic avenues remain to be explored to address whether the cell signaling pathways could regulate the purine and pyrimidine salvage pathways. For example, some studies suggest that Akt inhibition or mutation of p53 controls the de novo and salvage pathways [13,14]. This review focuses on how cell signaling networks regulate the metabolic pathways required for the de novo synthesis of pyrimidine and purine nucleotides, building upon previous studies that have elucidated details of metabolic control of nucleotide metabolism [1,8,12,15,16].
Metabolism of pyrimidine nucleotides
De novo biosynthesis of pyrimidine nucleotide is catalyzed by three gene products: CAD ((carbamoyl-phosphate synthetase 2 (CPS2), aspartate transcarbamoylase (ATC), and dihydroorotase (DHO)), DHODH (dihydroorotate dehydrogenase) and UMPS (uridine monophosphate synthetase) [17]. Pyrimidine nucleotides are essential for nucleic acid synthesis in all organisms with different temporal and/or spatial roles in cellular metabolism [18]. Proliferating cells activate the de novo pyrimidine pathway, whereas differentiated cells rely primarily on the pyrimidine salvage pathway to generate their nucleotide pools [19] (Figure 1). Uridine and cytidine can be recycled and phosphorylated by the enzyme uridine-cytidine kinase (UCK) to synthesize UMP and CMP, respectively. Additionally, the recycling activity of cytidine deaminase (CDA) within the pyrimidine salvage pathway plays a critical role in cellular DNA and RNA synthesis [20]. For a comprehensive review of the biological and pathological roles of pyrimidine nucleotides, readers are referred to previous publications [18,21].
Figure 1. The de novo pyrimidine and purine synthesis metabolic network.
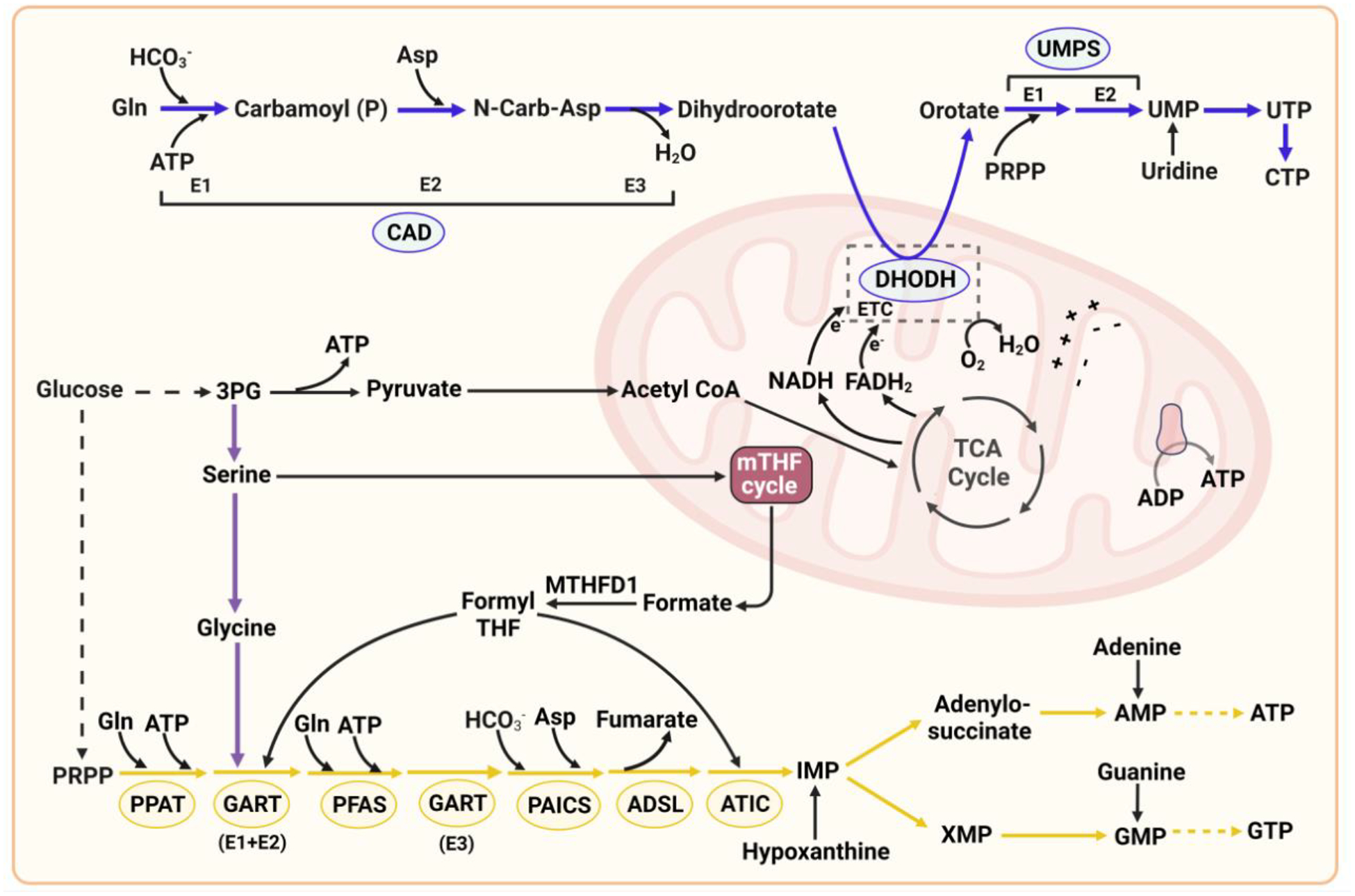
De novo pyrimidine synthesis: CAD, a trifunctional multi-domain enzyme, catalyzes the first three reaction steps of the de novo pyrimidine pathway in the cytosol, which uses atoms from glutamine, bicarbonate, aspartate, and ATP to synthesize dihydroorotate. The dihydroorotate dehydrogenase reaction step occurs in the mitochondria. Phosphoribosylpyrophosphate (PRPP), generated from the glucose-derived pentose phosphate pathway, is incorporated at the UMPS step in the cytosol. Mitochondrial TCA cycle intermediates such as oxaloacetate could also be used to generate cytosolic aspartate for de novo pyrimidine synthesis. Pyrimidine synthesis enzymes: CAD: carbamoyl-phosphate synthetase 2 (E1), aspartate transcarbamoylase (E2), and dihydroorotase (E3); DHODH: dihydroorotate dehydrogenase; UMPS: uridine monophosphate synthetase. UMPS is a bifunctional enzyme and has two domains: orotate phosphoribosyl transferase (OPRT, E1) and orotidine-5’-phosphate decarboxylase (ODC, E2). De novo purine synthesis: Glucose metabolism produces glycolytic intermediates that can be used by auxiliary pathways including the pentose phosphate pathway to generate PRPP, and the mitochondrial tetrahydrofolate (mTHF) cycle to synthesize glycine, 10N-formyl-THF for incorporation into the purine ring and 5N,10N-methylene-THF for thymidylate production. The de novo purine pathway requires the coordinated actions of six enzymes to catalyze ten sequential reactions to synthesize the first purine nucleotide inosine 5’-monophosphate (IMP) from PRPP. IMP is the precursor of AMP and GMP. Purine synthesis enzymes: PPAT: phosphoribosyl pyrophosphate amidotransferase; GART: glycinamide ribonucleotide transformylase; PFAS: phosphoribosylformylglycinamidine synthase; PAICS: phosphoribosylaminoimidazole carboxylase and phosphoribosylamino-imidazolesuccinocarboxamide synthase; ADSL: adenylosuccinate lyase; ATIC: 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase.
Metabolism of purine nucleotides
One of the significant differences between de novo pyrimidine and purine synthesis lies in the integration of the activated ribose (PRPP) to the nucleobase ring. The de novo purine synthesis pathway commences with PRPP. It requires the coordinated actions of six enzymes to catalyze ten sequential reactions to synthesize the first purine nucleotide inosine 5’-monophosphate (IMP) from PRPP. On the other hand, the pyrimidine ring is first synthesized before incorporating PRPP at the UMPS step. The purine ring is built on the phosphoribosyl moiety, wherein PRPP is catalyzed by the phosphoribosyl pyrophosphate amidotransferase (PPAT) enzyme to bind with the amide nitrogen from glutamine, causing the formation of 5-phosphoribosylamine along with the release of pyrophosphate [22]. The next steps include 9 metabolic reactions from which 5-phosphoribosylamine is converted into IMP through the purinosome assembly [3,23], producing adenine and guanine nucleotides (Figure 1).
Purines are also obtained through the salvage of preformed purine bases and the recycling of cellular degradation products. In the purine salvage pathway, hypoxanthine-guanine phosphoribosyltransferase 1 (HPRT1) catalyzes the conversion of hypoxanthine and guanine into IMP and GMP, respectively and adenine phosphoribosyltransferase (APRT) catalyzes the conversion of adenine into AMP. The de novo purine synthesis pathway requires 6 moles of ATP to generate each mole of purine nucleotide product, while HPRT1 and APRT require one mole of ATP. Much of the cellular energy is conserved in the purine salvage reactions compared with the de novo purine pathway, and 90% of free purines generated are recycled rather than degraded or excreted [24].
Signaling and transcriptional control of the de novo nucleotide synthesis pathways
The RAS-ERK (extracellular signal-regulated kinase), PI3K (phosphatidylinositol 3-kinase)-mTORC1, and Hippo-Yes-associated protein (YAP) signaling pathways are cell master regulators that control cell growth, proliferation, motility, differentiation, survival, and metabolism in response to extracellular cues. The metabolic effects can be mediated through post-translational, translational, post-transcriptional, or transcriptional mechanisms (Table 1). Herein, we review the current understanding of the connections between these pathways and nucleotide metabolism and how they are altered in disease conditions.
Table 1:
Molecular mechanisms controlling de novo nucleotide synthesis in cancer and immune cells
| Regulators / Signaling proteins / nutrients | Mechanism(s) | References |
|---|---|---|
| Ras/ERK | Phosphorylation of CAD at the T456 site by ERK | [33] |
| ERK2 directly phosphorylates PFAS on T619 and stimulates de novo purine synthesis | [3] | |
| PI3K/Akt/mTORC1 | mTORC1-mediated control of the oxidative branch of PPP through SREBP1 to increase PRPP production for nucleotide synthesis | [34] |
| mTORC1 increases de novo pyrimidine synthesis through S6K1-mediated CAD phosphorylation on S1859 | [26,27] | |
| mTORC1 promotes one carbon formyl unit production and purine synthesis through ATF4-dependent control of MTHFD2 | [35] | |
| mTORC1 stimulates SLC4A7 mRNA translation to increase bicarbonate inflow for de novo nucleotide synthesis | [36] | |
| Akt stimulates the nonoxidative PPP through direct phosphorylation of TKT on Thr382 to promote PRPP production for nucleotide synthesis. | [37] | |
| Akt phosphorylates NADK to stimulate NADP(H) production for reductive metabolism and nucleotide synthesis | [38] | |
| The S6K-dependent CAD phosphorylation on S1859 enhances pyrimidine synthesis to generate effector CD8+ T cells | [39] | |
| Type I interferon | IFN signaling augments nucleotide catabolism through regulation of ataxia telangiectasia and RAD3-related protein (ATR) | [40] |
| SIRT3 | Inactivation of SIRT3 promotes de novo nucleotide synthesis in part through mTORC1 activation | [41] |
| PKM1 | PKM1 expression regulates nucleotide production in certain conditions | [42] |
| K-RAS | K-Ras stimulates nucleotide synthesis through regulation of RPIA expression downstream of c-MYC | [9] |
| c-Myc | MYC binds to the promoters of nucleotide-related genes and regulates their expressions (e.g., during MYC activation, eIF4E controls PRPS2 mRNA translation) | [7,43] |
| The fitness of BTIC necessitates the regulation of nucleotide synthesis | [44] | |
| MYC upregulates IMPDH2 to increase GTP synthesis | [45] | |
| LKB1 and KRAS | Concurrent activation of KRAS and loss of LKB1 increase CPS1 expression and promote nitrogen flow from urea cycle intermediates into pyrimidines | [46] |
| PTEN | PTEN deficiency stimulates de novo pyrimidine synthesis through activation of the mTORC1 pathway | [47] |
| p53 | Expression of mtp53 enhances the expression of several nucleotide metabolism genes | [48] |
| Yap1 | Yap1 stimulates glutamine synthetase (GLUL) expression, thereby increasing cellular glutamine levels and de novo nucleotide synthesis | [49] |
| Through the stimulation of glucose transporter 1 expression (GLUT1), Yap1 regulates nucleotide synthesis | [50] | |
| Yap1 stimulates RRM2 expression and deoxythymidylate kinase (DTYMK) enzymes for producing dNTPs | [51] | |
| ARL13B | Activated ARL13B regulates IMPDH2 to enhance purine synthesis | [52] |
| Glucose | PKM2 controls the glycolytic rate and shunt of glycolytic intermediates into the pentose phosphate and serine/glycine synthesis pathways to control nucleotide synthesis | [42,53] |
| Oxidative and nonoxidative arms of PPP enable the production of ribose 5-phosphate (R5P) and the activated ribose (PRPP) for nucleotide synthesis | [4,8] | |
| Upon glucose starvation, AMPK phosphorylates PRPS1/2 and inhibits PRPP synthesis and nucleotide synthesis | [54] | |
| Upon glucose starvation, high levels of ASS1 render tumor cells dependent on purine synthesis through the reprogramming of central carbon metabolism | [55] | |
| Energy stress | Energy stress regulates purine synthesis, possibly through the AMPK-dependent sequestration of PFAS enzyme from the purinosome metabolon | [56] |
| Urea cycle and CAD | In cancers with decreased ASS1, the reduced usage of aspartate by the urea cycle leads to the accumulation of cytosolic aspartate pools that can be consumed by CAD to boost pyrimidine synthesis | [57] |
Abbreviations: ERK, extracellular signal–regulated kinase; SIRT3, NAD-dependent deacetylase sirtuin-3, mitochondrial; RPIA, ribose 5-phosphate isomerase A; PPP, pentose phosphate pathway; CPS1, carbamoyl phosphate synthetase-1; SLC4A7, solute carrier family 4 member 7; MTHFD2, methylene tetrahydrofolate dehydrogenase 2; BTIC, brain tumor initiating cells; IMPDH, inosine monophosphate dehydrogenase; RRM2, ribonucleotide reductase member 2; GLUL, glutamine synthetase, PRPP, 5-phosphoribosyl pyrophosphate; PTEN, phosphatase and tensin homolog; AMPK, AMP-activated protein kinase; SREBP1, sterol regulatory element-binding transcription factor 1; ARL13B, ADP-ribosylation factor-like protein 13B; ASS1, argininosuccinate synthase.
Regulation of de novo pyrimidine synthesis by the cell signaling pathways
mTORC1 and pyrimidine synthesis
mTORC1 signaling is regulated by hormones, growth factors, amino acids, purines, cytokines, oncogenes, and tumor suppressors [25]. It promotes the production of new nucleotides to facilitate an increased demand for RNA and DNA synthesis [26,27]. The PI3K/AKT/mTORC1 pathway is physiologically stimulated by growth factors and hormonal signals such as insulin and insulin-like growth factor 1 (IGF1). In proliferating cells, activation of this pathway promotes an increase in pyrimidine synthesis flux through S6K1-mediated phosphorylation of CAD on S1859 [26,27] (Figure 2A). Although S6K1-mediated CAD phosphorylation is not required to maintain CAD basal activity, its phosphorylation increases the cellular pyrimidine pool size for macromolecular synthesis and cell growth. Rheb, a lysosomal GTPase, required for mTORC1 activation and mLST8, a component of the mTORC1 complex, have been shown to interact with CAD and positively regulate its activity [28,29]. Furthermore, loss of mitochondrial sirtuin 3 (SIRT3) leads to mitochondrial stress increasing mTORC1 activity, as evidenced by the induction of CAD phosphorylation and increased de novo pyrimidine synthesis [30]. Further research is needed to determine the precise mechanisms driving mTORC1 activation downstream of SIRT3 loss. Hyperactivation of the PI3K pathway, a common feature in many cancers, results in metabolic reprogramming to support anabolic metabolism and biomass production [31]. Such PI3K-mTORC1-dependent activation of nucleotide synthesis renders PI3K-dependent cancers sensitive to PI3K inhibitors, as nucleotide depletion and DNA damage occur [32].
Figure 2. Regulation of de novo purine and pyrimidine synthesis by the cell signaling networks.
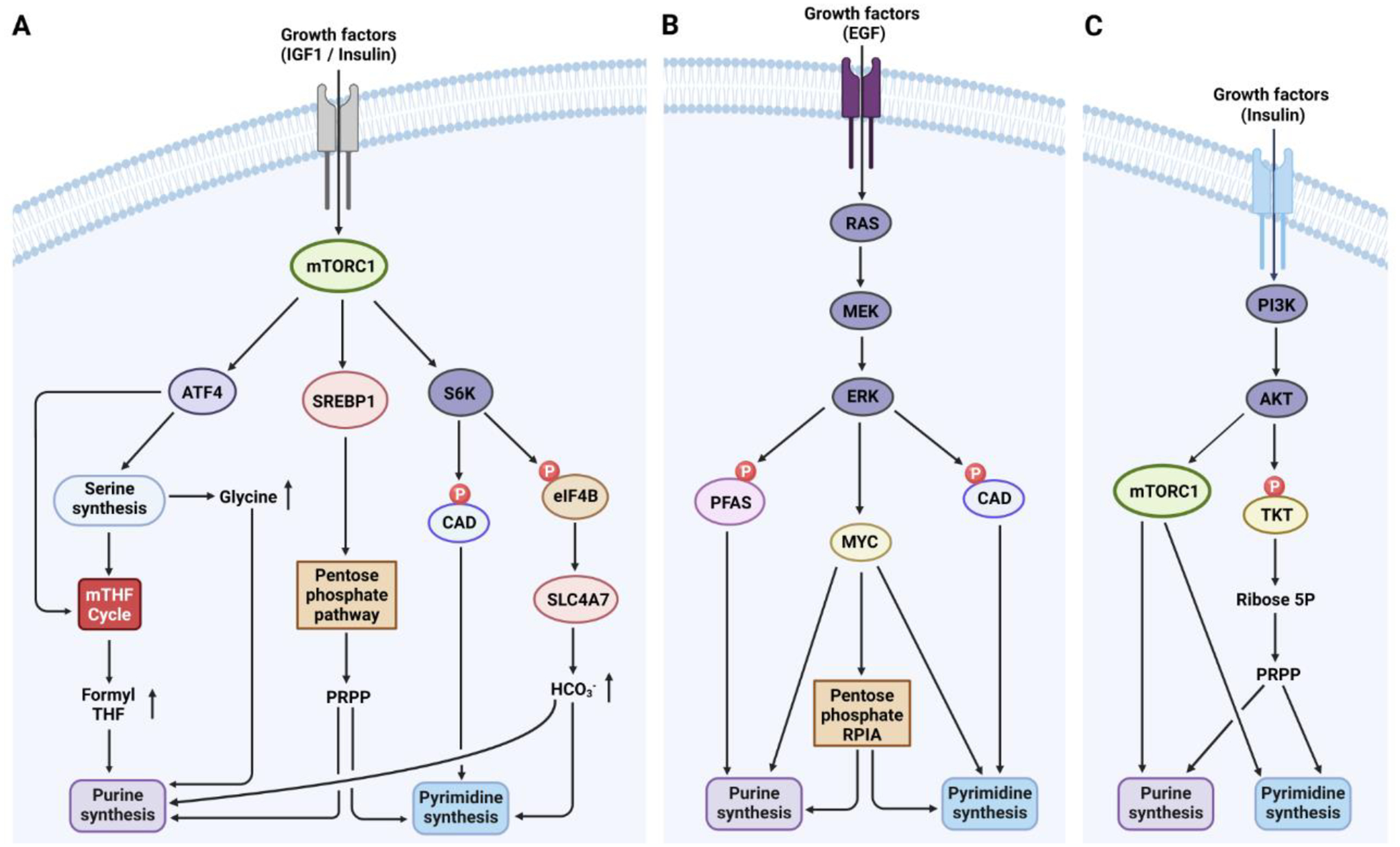
A. In response to growth factors, mTORC1 is activated downstream of the PI3K/Akt pathway and promotes S6K1-dependent phosphorylation of CAD, thereby increasing de novo pyrimidine synthesis. Moreover, mTORC1 activation fuels de novo purine and pyrimidine synthesis by increasing cellular bicarbonate abundance through the stimulation of SLC4A7 mRNA translation via the S6K-dependent phosphorylation of eIF4B. Downstream of mTORC1, the transcription factor SREBP1 stimulates the oxidative pentose phosphate pathway (PPP) to enhance de novo purine and pyrimidine synthesis via increased availability of PRPP. Additionally, mTORC1 activation enhances ATF4 expression, promoting serine/glycine synthesis and 10N-formyl-THF production, which increases de novo purine synthesis. B. In response to growth factors, ERK phosphorylates CAD and PFAS, enhancing de novo pyrimidine or purine synthesis, respectively. MYC, a downstream transcription factor of RAS-ERK, controls the expression of genes involved in de novo purine and pyrimidine synthesis. C. Insulin activates the PI3K Akt pathway, which leads to mTORC1 activation. Besides, Akt activates the nonoxidative PPP through TKT phosphorylation, boosting PRPP synthesis for nucleotide synthesis. EGF, epidermal growth factor; eIF4B, eukaryotic translation initiation factor 4B; IGF1, insulin-like growth factor 1; PRPP, 5’-phosphoribosyl-pyrophosphate; PFAS, phosphoribosylformylglycinamidine synthase; RPIA, ribose 5-phosphate isomerase A; SLC4A7, solute carrier family 4 member 7; SREBP1, sterol regulatory element-binding protein 1; TKT, transketolase.
As the tumor suppressor PTEN negatively regulates the PI3K-Akt pathway, mutation or loss of PTEN activates Akt and mTORC1 signaling, thus promoting cellular growth [58]. PTEN loss significantly stimulated the growth of mouse embryonic fibroblasts (MEFs) through increased de novo pyrimidine synthesis [47]. In cells with PTEN loss, the de novo pyrimidine synthesis rate was also enhanced through the mTORC1-dependent phosphorylation of CAD [27]. Moreover, the observation that DHODH inhibition considerably decreases the growth of PTEN-null cells [47] further supports the notion that cells with a hyperactive PI3K-AKT-mTORC1 axis depend on the continual supply of metabolites to fuel nucleotide synthesis.
A recent study has revealed the functional implications of mTORC1-induced CAD stimulation on memory CD8+ T cells [39]. The authors found that CAD plays a critical role in promoting the rapid recall response of previously activated CD8+ T cells. The ability of mTORC1 to generate effector CD8+ T cells depends on CAD-dependent induction of pyrimidine synthesis and/or CAD-mediated pre-rRNA synthesis [39]. Moreover, the authors demonstrated in vivo that CAD remains phosphorylated in memory T cells long after antigen treatment, despite mTORC1 returning to baseline [39]. The mechanism underlying the persistence of CAD phosphorylation and the continued synthesis of de novo pyrimidines in resting T cells beyond the cessation of faster cell division following activation remain unclear. This raises outstanding questions, such as the involvement of other kinases in maintaining this continual phosphorylation, or the presence of mechanisms that inhibit phosphatases that mediate CAD dephosphorylation in memory T cells.
RAS-ERK signaling and pyrimidine synthesis
ERK is part of the mitogen-activated protein kinase (MAPK) family, which plays a role in cascade signaling and transmits extracellular signals to intracellular targets. Hence, MAPK cascades are central signaling elements that regulate basic processes, including cell proliferation, differentiation, and stress responses [59]. The over-activation of upstream proteins and kinases in the ERK pathway induces various diseases, including cancer, developmental disorders, inflammation, and neurodegenerative diseases [60]. In fact, ERK activation stimulates the transcriptional activity of MYC [61], which was dubbed the master regulator of nucleotide metabolism because of its direct control of pyrimidine and purine synthesis gene expression [7]. While an increased nucleotide synthesis rate is required for DNA replication in normal proliferating cells, it is tempting to speculate that the amplification of MYC in cancers could create an enhanced expression of nucleotide metabolism genes, which would, in turn, abnormally elevates the cellular nucleotide levels that could contribute to DNA instability and cell growth. Moreover, ERK1/2 activation was also shown to positively regulate de novo pyrimidine synthesis through the direct phosphorylation of CAD at the T456 site on the CPS2 domain, diminishing the ability of UTP to allosterically inhibit CAD [33,62] (Figure 2B).
Regulation of de novo purine synthesis by the cell signaling pathways
mTORC1 and purine synthesis
Compared to the regulation of de novo pyrimidine synthesis, mTORC1 signaling stimulates de novo purine synthesis through long-term mediating mechanisms via the induction of the transcription factors ATF4 (activating transcription factor 4) (Figure 2A) and MYC, which both control the expression of specific metabolic enzymes contributing to or directly required for de novo purine synthesis. For example, in response to mTORC1 activation, ATF4 enhances the production of enzymes that belong to the serine/glycine synthesis pathway and the mitochondrial tetrahydrofolate cycle, which produce glycine and one-carbon formyl units, respectively, for de novo purine synthesis [35]. Moreover, mTORC1 activation stimulates cellular glutathione synthesis partly through the ATF4-dependent induction of the cystine transporter SLC7A11, a key provider of cellular cysteine for protein and glutathione synthesis [63]. These findings support a logic of coordinated metabolic programs downstream of mTORC1, enhancing glutathione synthesis to help mitigate high levels of oxidative stress that may occur in highly proliferating cells with increased nucleotide and protein synthesis rates [63]. mTORC1 signaling controls the assembly of the purinosome, a metabolon located at proximity to mitochondria that enables metabolic channeling through de novo purine synthesis. Studies using cell culture systems have demonstrated that rapamycin, a mTORC1 inhibitor, blocks the colocalization of purinosomes with mitochondria [64,65]. In the context of cellular demand, mTORC1 increases the association of purinosomes with mitochondria, possibly facilitating the shuttling of mitochondria-derived metabolic intermediates (such as formate) to the purinosome complex to enhance de novo nucleotide synthesis [66]. With this colocalization, the purinosome perhaps also takes advantage of the high levels of mitochondrially generated ATP and substrates to promote flux through the de novo purine pathway [67]. However, additional studies are warranted to identify how the purinosome-mitochondria localization is regulated by mTORC1 signaling. Of equal importance, the mTORC1-mediated control of the oxidative branch of the pentose phosphate pathway through the transcription factor SREBP1 also results in enhanced production of activated ribose required for nucleotide synthesis [34]. Downstream of mTORC1, MYC activation stimulates purine biosynthesis by upregulating the levels of de novo purine enzymes [68,69]. Questions remain as to how mTORC1 activation, under physiological conditions, integrates all the distinct inputs to coherently enable metabolic output stimulation and preserve cellular homeostasis.
ERK and purine synthesis
While the metabolic effects of MYC downstream of ERK signaling are well-recognized, studies on the acute metabolic changes directly controlled by ERK are still emerging. In fact, ERK was shown to enhance the de novo purine synthesis rate through direct phosphorylation of the purine synthesis enzyme PFAS on T619 upon physiological stimuli such as the epidermal growth factor and in ERK-driven cancer cells [3] (Figure 2B). The ERK-mediated PFAS phosphorylation participates in cell proliferation and growth of RAS-driven tumors, indicating that this posttranslational mechanism could be a metabolic vulnerability in cancers with hyperactivated ERK signaling. However, further investigations are necessary to confirm this hypothesis. It remains critical to understand whether the ERK-dependent regulation of PFAS could participate in resistance to chemotherapy. Developing small molecules that specifically inhibit PFAS activity in cancer cells is the next step in understanding whether targeting PFAS in RAS/ERK-driven cancers could be an effective therapeutic strategy against cancer growth and progression.
Indirect regulation of de novo nucleotide synthesis by the Hippo-Yap pathway
Besides the mTORC1 and ERK signaling, the Hippo-Yap signaling is an evolutionarily conserved pathway that significantly controls tissue growth and organ size in response to different stimuli [70]. Through the induction of glutamine synthetase/glutamate-ammonia ligase (GLUL), Yap1 was recently shown to increase cellular glutamine levels and subsequently enhance de novo nucleotide synthesis in zebrafish [49]. Furthermore, pharmacological inhibition of GLUL sensitized Yap1-driven liver tumors to nucleotide synthesis inhibition. Further work expanded these findings by demonstrating that Yap1, through the induction of the glucose transporter GLUT1, increases uptake and anabolic utilization of glucose to stimulate de novo nucleotide synthesis [50]. Besides regulating nucleotide synthesis by reprogramming glutamine or glucose metabolism, YAP also regulates dNTP synthesis by promoting the expression of enzymes necessary for producing dNTPs, including RRM2 and deoxythymidylate kinase (DTYMK) [51]. Moreover, YAP/TAZ can bypass environmental cues-induced senescence by adjusting the rate of nucleotide synthesis. The regulation of dNTP synthesis by YAP can contribute to resistance to conventional chemotherapies that target dNTP synthesis or, possibly, inhibition of oncogene-induced senescence. Depletion of dNTP metabolic enzymes DTYMK and RRM2 impaired YAP-induced cancer cell growth in vitro models; however, this therapeutic opportunity needs to be tested further in pre-clinical models [51].
Additionally, in the case of immune cells, YAP plays a critical role in the fine-tuning process of the adaptive immune response through the regulation of amino acid levels, TCA cycle intermediates, and in part, through nucleotide synthesis [71]. However, since the specific mechanisms by which YAP or TAZ differentially regulate either pyrimidine or purine metabolism remain unknown, the role of the Hippo-YAP/TAZ pathway in the control of nucleotide synthesis necessitates further investigations. While signaling networks can regulate the nucleotide synthesis pathways, cellular nucleotide levels profoundly impact signaling activity and cellular metabolism (see Box 1 for more information).
Box 1. Nucleotide signaling and control of cell function.
Purine nucleotides such as 3′,5′-cyclic AMP (cAMP) and cyclic GMP (cGMP) are well-established intracellular signaling molecules that convey the effects of glucagon and nitric oxide (NO) signals, respectively. cAMP activates protein kinase A (PKA), inducing the phosphorylation cascade of proteins that regulate glycogen metabolism [113–115]. cGMP binds to and activates the protein kinase G (PKG), enabling the phosphorylation of several substrates mediating the cGMP-dependent vasodilatation of various vessel types [116]. AMP-activated protein kinase (AMPK) is the primary cellular energy sensor, which is highly conserved across eukaryotic species. While AMPK represents the perfect example of a purine kinase sensor since it binds directly to AMP, the mechanisms by which cells sense intracellular pyrimidine levels remains unknown. Thus, it is possible that molecular mechanisms exist in cells that can mediate such sensing and signal needed to alter cellular processes accordingly. Addressing this scientific conundrum will improve our knowledge and eventually enable the generation of targeted therapeutics for treating diseases associated with malfunctions in nucleotide sensing. Intracellular purine levels, but not pyrimidine levels, also maintain mTORC1 activity (mTORC1) [117,118]. Moreover, formate through purine synthesis was shown to activate mTORC1 and induce pyrimidine synthesis via the mTORC1-dependent phosphorylation of CAD [119]. However, the identity of the purine sensor upstream of mTORC1 remains to be determined. An imbalance of purine nucleotides, which is not sensed by mTORC1 signaling, disrupts the coordination between cell growth and division and renders proliferating cells dependent on replication stress signaling for survival [120]. Intracellular purine levels have been shown to control of the ability of the glycolytic intermediate 3-phosphoglycerate to shunt into the de novo serine/glycine synthesis pathway [121]
Small G-proteins, such as Ras, Rags, Rheb, and Rabs, use GDP and GTP as interchangeable substrates for controlling signaling systems, cellular trafficking, and cytoskeletal dynamics [122–124]. While the role of pyrimidine nucleotides as signaling molecules has yet to be established, their role as allosteric regulators and substrates for key cellular metabolic processes is well recognized. Specially, UTP facilitates the production of substrates for glycosylation processes [125]. Cytidine triphosphate (CTP) maintains phospholipid synthesis via its consumption by the CDP-choline and CDP-diacylglycerol pathways [126]. At the tissue, organ, or system level, pyrimidine nucleotides are also crucial for liver glucuronidation to enable the clearance of exogenous drugs and hormones [127].
The metabolic interplay between nucleotide metabolism in tumor cells and immune cells is recently emerging [11,128]. Abnormal nucleotide secretion in the tumor microenvironment (TME) also modifies the normal immune response and tumor function, suggesting that targeting nucleotide metabolism could be a promising approach to enhance immunotherapy [81,129,130]. Moreover, an increased level of purine nucleotides is sufficient to induce the expression of MICA (glycoprotein MHC class IA), a cell-surface glycoprotein which allows for the detection and elimination of cancer cells or virus-infected cells by natural killer (NK) cells [131,132]. Additionally, nucleoside analogs targeting nucleotide synthesis can exhibit antiviral activity by activating the host’s innate immunity [133], while depletion of cellular pyrimidine nucleotides has been found to induce mitochondrial DNA (mtDNA) release and stimulate immune responses [134]. However, further research is needed to elucidate the exact mechanisms by which nucleotide depletion can activate innate immunity.
Nutrient abundance and de novo nucleotide synthesis
Glucose metabolism
The glycolytic enzyme pyruvate kinase M2 (PKM2), which phosphorylates ADP to ATP and mediates pyruvate synthesis from phosphoenolpyruvate, controls glycolysis rate and enables the shunt of glycolytic intermediates upstream of phosphoenolpyruvate into the pentose phosphate and serine/glycine synthesis pathways. This shunt enhances the production of substrates (ribose 5-phosphate, NADPH (from PPP), glycine (from the serine biosynthesis pathway) and one-carbon units (from the tetrahydrofolate pathway) for de novo purine and pyrimidine synthesis [42,53].
Upstream of PKM2, the PPP (pentose phosphate pathway) enables the production of activated ribose (PRPP) for nucleotide synthesis and NAD metabolism [4,8]. PRPP synthesis is regulated by adenylate/guanylate levels and is decreased upon AMPK activation during glucose starvation [54,72]. Energy stress also regulates purine synthesis through AMPK-dependent sequestration of the PFAS enzyme [56]. Growth signals, via Akt signaling, activate TKT, promoting PRPP production [37] (Figure 2C). Additionally, Akt and PKC stimulate NADP(H) production, essential for nucleotide synthesis and redox homeostasis [38,73–75].
Argininosuccinate synthase 1 and de novo pyrimidine synthesis
The metabolic connection between the urea cycle and nucleotide synthesis was discovered in a subset of cancer cells with high or low expression of the argininosuccinate synthase (ASS1) enzyme. ASS1 is involved in the urea cycle, which synthesizes argininosuccinate from citrulline and aspartate. In cancers with reduced ASS1 expression, the decreased consumption of aspartate by ASS1 results in the accumulation of cytosolic aspartate pools. These pools are then utilized by CAD to produce N-carbamoyl-L-aspartate, which fuels pyrimidine synthesis and cancer growth [57]. However, excess pyrimidine in tumor cells with a dysfunctional urea cycle promotes genomic instability and mutations, which may enhance the response to immune checkpoint inhibitors [76]. On the other hand, cancers with upregulated ASS1 expression exhibit increased gluconeogenesis and metabolic flux towards the serine/glycine pathway, which is required to generate one-carbon units and glycine precursors for purine synthesis. The increased rate of purine synthesis in cancer cells with a hyperactive urea cycle may lead to resistance mechanisms against immune checkpoint inhibitors. In fact, inhibiting purine synthesis was shown to increase the pyrimidine-to-purine ratio, promote immunoproteasome expression and improve the response of CD8+ T cells to anti-PD-1 therapy [55]. The molecular mechanisms that underpin the “Jekyll and Hyde” behavior of ASS1 in different types of cancer remain an open question. Specific oncogenes may have differential effects on urea cycle activity in different cancer types, which may underlie the complexity behind the pro-oncogenic or tumor-suppressive role of ASS1.
mTORC1 signaling controls bicarbonate abundance to support nucleotide synthesis
Sodium bicarbonate (NaHCO3), also known as baking soda, maintains cellular pH homeostasis in eukaryotic cells. However, its contribution to anabolic metabolism, cell biomass, and proliferation as an anabolic substrate for nucleotide metabolism is largely understudied. Ali and colleagues demonstrated that intracellular bicarbonate availability limits de novo purine and pyrimidine synthesis in growing cells [36]. Among the ten bicarbonate transporters of the SLC4 family, the NaHCO3 co-transporter SLC4A7 is specifically upregulated by growth factor stimuli through the activation of the PI3K-Akt-mTORC1 signaling pathway. Stimulation of SLC4A7 mRNA translation was found to be mediated by S6K-dependent phosphorylation of eIF4B downstream of mTORC1 activation. Notably, SLC4A7 upregulation is observed in breast cancers, and its inhibition has been shown to delay breast cancer development and slow tumor growth [77].
Moreover, SLC4A7 loss sensitized PI3K-driven breast cancer tumors to mTORC1 inhibition in xenograft mouse models [36]. Consistent with its pro-oncogenic role, SLC4A7 expression supports the growth of Ras-driven tumors [78], suggesting that SLC4A7 is a metabolic vulnerability in various types of cancer. While the pro-proliferative effects of SLC4A7 were mainly observed in cancer cell settings, it remains to define if SLC4A7 expression is required to enable proper physiological function through the regulation of de novo nucleotide synthesis.
As our comprehension of cellular metabolism regulation progresses, we are gaining a better understanding of the short- and long-term mechanisms that govern nucleotide synthesis. This involves the connections between cell signaling, metabolic pathways, and nucleotide synthesis in both normal proliferating cells and cancer cells.
Targeting nucleotide synthesis as a therapeutic avenue to fight cancer and immune diseases
Cancer cells and cells infected with viruses undergo significant changes in nucleotide metabolism, which enables them to survive and multiply. Nucleotide synthesis provides cancer cells and viruses with building blocks to proliferate and evade the immune system, making it a target for therapeutic strategies in both diseases [6,11]. While previous studies have discussed various therapeutic approaches for targeting nucleotide metabolism in different disorders [1,6,8,11,21,79–81], this review article will solely present the most recent advancements in using nucleotide synthesis inhibitors as therapeutic agents.
Antimetabolites for targeting metabolic dependencies in cancer and viral-infected cells
Antimetabolites are small molecules that competitively inhibit the enzymes involved in nucleotide synthesis, and their effects have been investigated for decades in cancer cells. Aminopterin, an analog of folic acid, was the first antimetabolite discovered in 1948 and was shown to be active against a subset of childhood leukemias [82]. Subsequently, several analogs of aminopterin were identified, and methotrexate (MTX), a folate analog that inhibits DHFR, emerged as a promising chemotherapeutic agent. Attempts to reduce the broad spectrum of toxicity and enhance the efficacy of MTX and the events of intrinsic and acquired resistance to MTX have prompted the search for more effective antifolates. For instance, pemetrexed, a derivative of MTX, inhibits multiple folate-dependent enzymes, including DHFR, GART, and thymidylate synthase (TYMS) [83]. The purine analogs 6-mercaptopurine (6-MP), the folate analog methotrexate, and the pyrimidine analog 5-fluorouracil (5-FU) and gemcitabine are examples of widely used chemotherapeutic antimetabolites [8,84] (Figure 3). Their clinical success against cancer progression is attributed to the increased metabolic demand of tumor cells for nucleotide synthesis and DNA replication. For example, the proper activation of gemcitabine relies on the nucleoside salvage pathways, but the overexpression of cytidine deaminase can deaminate gemcitabine, making it less effective in suppressing cell proliferation and tumor growth [20,85]. Despite their effectiveness against some cancer types, antimetabolites have limitations, suggesting that further research is needed to better understand their metabolic effects. A detailed understanding of their mechanisms of action and chemoresistance might result in more effective treatments, especially in solid tumors.
Figure 3. Mechanisms of action of antimetabolites used in cancer therapy.
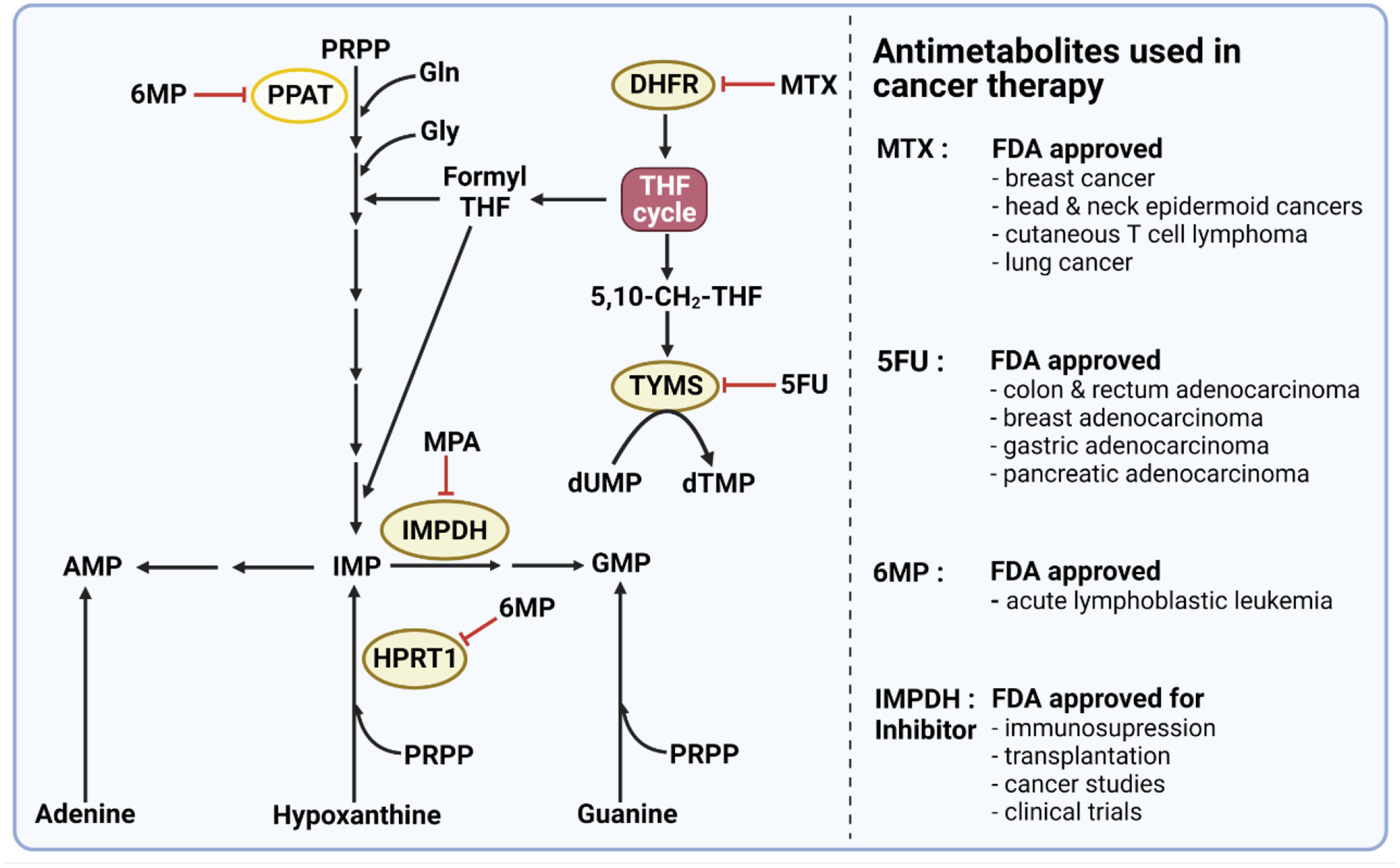
Methotrexate (MTX) is a folate analog that inhibits dihydrofolate reductase (DHFR) required for de novo nucleotide synthesis and several other metabolic processes. DHFR catalyzes the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF). THF is required for the action of folate-dependent enzymes and is therefore vital for DNA synthesis and methylation reactions. The purine analog 6-mercaptopurine (6MP) inhibits 5-phosphoribosyl-1-pyrophosphate amidotransferase (PPAT), the first enzyme in de novo purine biosynthesis as well as hypoxanthine-guanine phosphoribosyltransferase 1 (HPRT1), a key enzyme in the purine salvage pathway. The pyrimidine analog 5-fluorouracil (5FU) is a synthetic analog of uracil that inhibits thymidylate synthase, limiting the availability of deoxythymidine nucleotides required for cellular DNA synthesis. Moreover, FDA has recently approved IMPDH inhibitor (MPA) for organ transplants and several clinical trials of cancer studies. PRPP, phosphoribosylpyrophosphate; MTX, methotrexate; HPRT1: hypoxanthine phosphoribosyltransferase 1; IMPDH, inosine 5’-monophosphate dehydrogenase. 6MP, 6-mercaptopurine; 5FU, 5-fluorouracil; MPA, mycophenolic acid. dUMP, deoxyuridine monophosphate; dTMP, deoxythymidine monophosphate.
Strategies for targeting pyrimidine metabolism in cancer and immune cells
Cancer cells rewire key metabolic pathways to increase substrate availability for nucleotide synthesis and escape DNA damage induced by chemotherapeutic agents. Brown and colleagues observed that doxorubicin induced an increase in pyrimidine synthesis in triple-negative breast cancer (TNBC) cells, associated with the ERK-dependent phosphorylation of CAD on T456, without any marked changes in phosphorylation of S6K1-dependent site on CAD [86]. Inhibition of de novo pyrimidine synthesis with leflunomide (DHODH inhibitor) could enhance the sensitivity of TNBC cells to doxorubicin. Since then, several DHODH inhibitors, including brequinar, BAY2402234, and PTC299, are currently tested against various cancers and different models of viral infections in humans [87–90] (Figure 4). Additionally, DHODH-dependency was widely reported in PTEN-deficient tumors [47], K-Ras mutant pancreatic tumor cells [91], isocitrate dehydrogenase (IDH) mutant gliomas [92], diffuse midline gliomas [89], and MYC-driven medulloblastomas [44]. Thus, these preclinical studies strongly suggest that targeting de novo pyrimidine synthesis by inhibiting DHODH may be an effective strategy for treating certain cancers, particularly undruggable brain cancers [44,89,92]. In addition to disrupting the mitochondrial electron transport chain and energy production [93], DHODH inhibition creates a metabolic imbalance between intracellular purine and pyrimidine levels, resulting in replicative stress, DNA damage, or MYC instability, all of which may trigger cancer cell death.
Figure 4. Cellular and phenotypic alterations during therapy with DHODH Inhibitors.
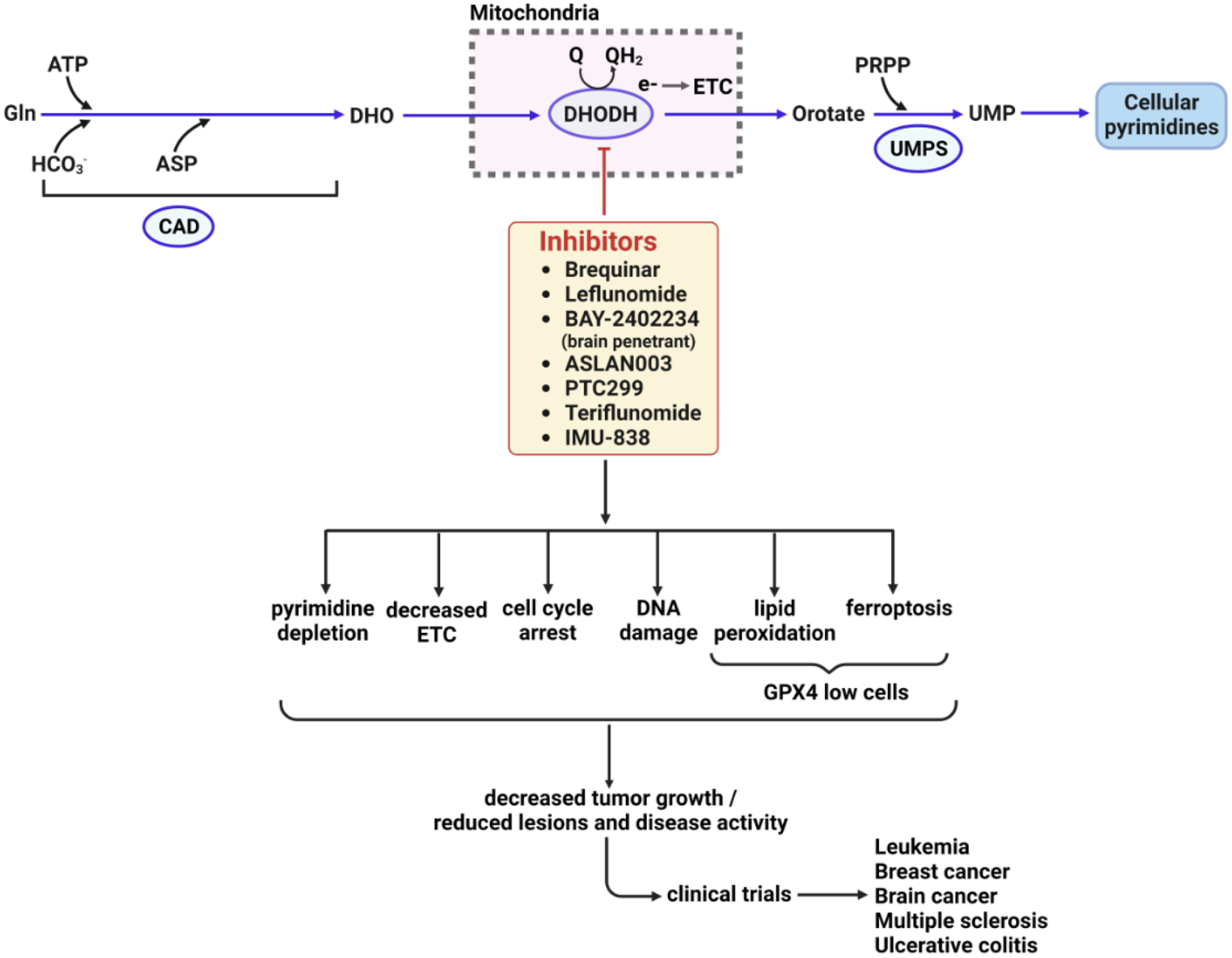
DHODH is necessary for the generation of cellular pyrimidine levels, and as such, its inhibitors have been long used to treat various autoimmune diseases, and are in clinical trials for cancer and immune disorders. A DHODH inhibitor quickly depletes cellular pyrimidine levels, forcing cells to limit the synthesis of RNA and DNA. Additionally, functional imbalance in ETC, cell cycle arrest, DNA damage, DNA replication stress could play critical roles in reducing tumor growth or inflammatory lesions. Furthermore, inhibition of DHODH might also increase mitochondrial lipid peroxidation and ferroptosis in GPX4low cancer cells and suppress GPX4low tumor growth [90]. DHO, dihydroorotate; UMP, uridine monophosphate; ETC, electron transport chain. GPX4, glutathione peroxidase 4.
Several clinical trials are currently underway to explore the potential benefits of de novo nucleotide synthesis inhibitors for developing safer and more effective cancer therapies. For instance, leflunomide, which is effective in treating rheumatoid arthritis, may be used as an alternative to methotrexate in cases where the latter is contraindicated or not well-tolerated [94]. Furthermore, a phase I study of leflunomide in patients with recalcitrant myeloma showed manageable side effects, and 82% of patients achieved stable conditions [95]. Teriflunomide, another inhibitor of the DHODH enzyme, has shown promise in improving T cell subsets and T cell receptor groups in patients with relapsing multiple sclerosis, suggesting that it could be used to treat other immune disorders [96]. More potent and selective inhibitors of DHODH are currently being developed for treating autoimmune diseases and SARS-CoV-2 [97,98], including P1788, which has been found to enhance cellular antitumor immunity via interferon signaling [99]. Co-targeting CAD and EGFR-tyrosine kinase receptor enhances the efficacy of immune checkpoint blockade (ICB) therapies [100]. These studies indicate that inhibiting de novo pyrimidine synthesis may be promising or boosting innate immunity. Additionally, CAD’s ability to promote ribosomal biogenesis in memory T cells may be essential to enhance the efficacy of chimeric antigen receptors (CAR) T-cell therapy [39]. Nevertheless, the mechanisms by which pyrimidine synthesis inhibition activates the innate immune system remain unclear. It is unsettled why some antimetabolites, such as 5-fluorouracil and raltitrexed, which inhibit thymidylate synthesis, promote immunosuppressive effects while others, such as DHODH inhibitors, activate the immune system.
Strategies for targeting purine metabolism in cancer and immune cells
Recent years have seen substantial progress in targeting purine metabolism and purinosome to reprogram cancer or immune metabolism in various model systems. In particular, it has been demonstrated that elevated IMPDH activity facilitates the synthesis of GTP, which supplies the necessary components for rapid proliferation. Inhibition of IMPDH was shown to selectively trigger apoptosis of activated mTORC1-driven proliferating cells [101]. Moreover, combined inhibition of PI3K signaling and IMPDH activity proved most effective in reducing tumor growth in a PDX mouse model of hepatocellular carcinoma [102] and in different ovarian cancer models [103]. Notably, purine dependency was also detected in glioblastoma and acute lymphoblastic leukemia [104], as well as in a subset of small-cell lung cancers defined by low expression of the transcription factor ASCL1 and high expression of oncogenic MYC [68]. Inhibition of IMPDH by mycophenolic acid (MPA) suppressed the growth of these lung cancer cells. Furthermore, inhibiting GTP synthesis, or the RNR enzyme radio-sensitized glioblastoma cells and patient-derived neurospheres by impairing DNA repair [105,106], thereby increasing the therapeutic efficacy of temozolomide [52]. Furthermore, the upregulation of IMPDH2 in glioblastoma cells has been associated with aberrant nucleolar function and augmented anabolic processes to support gliomagenesis [45].
It is noted that both MPA and mizoribine (a guanosine analog that inhibits IMPDH) are already used in humans as immunosuppressants in organ transplantation and autoimmune diseases, with mizoribine being well tolerated [107]. However, the application of IMPDH inhibitor has been restricted by adverse effects at high doses and differential distributions of IMPDH levels in different tumors [103].
Targeting purine metabolism is further known to enhance the efficacy of cancer immunotherapy in experimental and clinical settings. For example, mizoribine has been shown to promote the release of the immunoproteasome, which induced an enhanced response of autologous CD8+ T cells to anti-PD1 therapies in specific ASS1-expressing cancers [55]. In addition, it is recently believed that in some particular cancers, disrupting the balance of nucleotide pools by inhibitor(s) might produce mutations that affect antigen presentation and, thus, the immune response against the disease [76]. Some studies also demonstrated that purine synthesis inhibitors can improve immune suppression, as some secreted purines might bind the inhibitory receptors on immune cells [55,108,109].
The Cyclic dinucleotide cGAMP was shown to activate innate immune responses via the stimulator of interferon genes (STING), and eliciting a protective anti-microbial immunity response in mammalian cells [110,111]. Future studies are needed to investigate how to effectively activate or maintain this immune sensor cGAMP in the body to face microbial pandemics. Moreover, in several inflammatory disease models, the deficiency of MTHFD2, a gene supporting purine synthesis, reduced disease severity [112]. This finding suggests that purine nucleotides can play a critical role in immune disorders.
Concluding remarks
Cancer, viral-infected, or immune cells exploit the metabolism of nucleotides to overcome the challenges they face during abnormal growth and proliferation. Several signaling pathways regulate nucleotide synthesis in cells, but our understanding of the intricate regulatory network impinging on the nucleotide synthesis pathways under normal and pathological states is still emerging. Inhibiting the catalytic activity of the nucleotide metabolic enzymes with small molecules is a potential therapeutic strategy. As a result, we are likely to see an increasing number of antimetabolite-based therapies in the future. However, many questions still need to be answered due to the complicated and metabolically interconnected nature of nucleotide metabolism (see Outstanding Questions).
Outstanding Questions.
How do de novo nucleotide synthesis pathways respond to different signals in both normal and disease states?
Does the sodium bicarbonate co-transporter SLC4A7 regulate nucleotide synthesis in vivo and enable physiological or pathological functions?
What molecular mechanisms are responsible for the differential behavior of argininosuccinate synthase 1 (ASS1) in various types of cancer?
What are the mechanisms of nucleotide sensing in human cells?
What are the mechanisms through which the loss of SIRT3 leads to the activation of
mTORC1?
How does DHODH inhibition trigger the activation of the innate immune system?
The distinctive nucleotide-associated phenotype of cancer or immune disorders induced by specific oncogenes or pathogens may enable us to differentiate pathological proliferation from normal cellular growth providing a potential therapeutic window. High-resolution single-cell metabolomics and next-generation mass spectrometric metabolite imaging to evaluate nucleotide metabolism in tumors at a high resolution may facilitate our understanding of nucleotide metabolism’s heterogeneity and spatial regulation. This may lead to the identification of signaling and metabolic liabilities that can be exploited to develop effective cancer treatments. With improved experimental designs and the ability to quantitatively measure metabolic pathway rates becoming more accessible and sophisticated, there is potential for a promising future.
Highlights.
Activation of several signaling pathways, including RAS and mTORC1, tunes nucleotide synthesis through a variety of mechanisms.
Like glutamine or aspartate, bicarbonate abundance is regulated and limiting for nucleotide synthesis.
Targeting nucleotide metabolism may enhance the effectiveness of cancer immunotherapy by disrupting nucleotide pools and promoting immunogenicity.
CAD-induced ribosomal biogenesis in memory T cells might improve chimeric antigen receptors (CAR) T-cell therapy.
De novo pyrimidine synthesis emerges as a key metabolic vulnerability in certain cancers for which drug therapies are limited.
Acknowledgements
We apologize to our colleagues whose work we were not able to cover in this review due to space constraints. We thank Hina Anjum for contributing to figure preparation. This work was supported by grants from the National Institutes of Health, United States, R01GM135587, R01GM143334 (I.B.-S.), and by the LAM Foundation, United States, Established Investigator Award LAM0151E01–22 (I.B.-S.).
Glossary
- De novo nucleotide synthesis pathways
Metabolic pathways that assemble purine or pyrimidine nucleotide rings from simple precursors such as amino acids, activated ribose, ATP, and bicarbonate. Rapidly dividing cells activate the de novo nucleotide synthesis pathways for their growth and proliferation.
- Nucleotide salvage pathways
Metabolic pathways in which purines or pyrimidine nucleotides are recycled after partial degradation. Broadly, the salvage pathways use intermediate metabolites (nucleobases, nucleosides) derived from nucleotide catabolism or the circulation to maintain cellular nucleotide pools. Most differentiated cells produce nucleotides via the salvage pathways from nucleosides or nucleobases.
- Glycolysis (glycolytic pathway)
A catabolic pathway with a ten-step reaction that converts glucose (a hexose) into two three-carbon compounds (pyruvate) and generates energy (ATP).
- Pentose phosphate pathway
It is a catabolic pathway that generates NADPH and pentoses (5-carbon sugars) from glycolytic intermediates for reductive and anabolic metabolism.
- Gluconeogenesis
A metabolic pathway that generates glucose from non-carbohydrate substrates such as pyruvate, lactate, gluconeogenic amino acids, or glycerol.
- TCA (Tricarboxylic acid) cycle
A metabolon responsible for the generation of redox cofactors, anabolic intermediates, epigenetic regulators, and mitochondrial ATP.
- TNBC (triple-negative breast cancer)
In this type of breast cancer, cancer cells have little or no HER2 protein and do not express estrogen or progesterone receptors.
- PDX (Patient derived xenografts) model
A study model of cancer where the cells or tissue from a patient’s tumor are implanted into an immunodeficient or humanized rodent.
- Anti-PD1 therapy
A novel immunotherapy aims not to kill cancer cells directly but to block a pathway that shields cancer cells from the immune system.
- Memory T Cells
A subset of T lymphocytes that is antigen-specific and is known to protect hosts against previously encountered pathogens.
- Tumor microenvironment
A local environment of a tumor consisting of proliferating tumor cells, stromal cells, blood vessels, infiltrating inflammatory cells, immune cells, any metabolites, proteins or a variety of other molecules.
- Single-cell metabolomics
An evolving mass spectrometry-based technique that enables a large variety of metabolites to be concurrently detected from individual cells, providing the phenotypes on the single-cell level.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- 1.Buj R and Aird KM (2018) Deoxyribonucleotide Triphosphate Metabolism in Cancer and Metabolic Disease. Front Endocrinol (Lausanne) 9, 177. 10.3389/fendo.2018.00177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward PS and Thompson CB (2012) Signaling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol 4, a006783. 10.1101/cshperspect.a006783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali ES et al. (2020) ERK2 Phosphorylates PFAS to Mediate Posttranslational Control of De Novo Purine Synthesis. Mol Cell 78, 1178–1191.e1176. 10.1016/j.molcel.2020.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Sahra I and Manning BD (2017) mTORC1 signaling and the metabolic control of cell growth. Current Opinion in Cell Biology 45, 72–82. 10.1016/j.ceb.2017.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franklin DA et al. (2016) p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Scientific Reports 6, 38067. 10.1038/srep38067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ariav Y et al. (2021) Targeting nucleotide metabolism as the nexus of viral infections, cancer, and the immune response. Sci Adv 7. 10.1126/sciadv.abg6165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu YC et al. (2008) Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 3, e2722. 10.1371/journal.pone.0002722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villa E et al. (2019) Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers 11, 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santana-Codina N et al. (2018) Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun 9, 4945. 10.1038/s41467-018-07472-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bi J et al. (2018) Targeting cancer’s metabolic co-dependencies: A landscape shaped by genotype and tissue context. Biochim Biophys Acta Rev Cancer 1870, 76–87. 10.1016/j.bbcan.2018.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu H. l. et al. (2022) Targeting nucleotide metabolism: a promising approach to enhance cancer immunotherapy. Journal of Hematology & Oncology 15, 45. 10.1186/s13045-022-01263-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puigserver P (2018) Chapter 7 - Signaling Transduction and Metabolomics. In Hematology (Seventh Edition) (Hoffman R et al., eds), pp. 68–78, Elsevier [Google Scholar]
- 13.Wang W et al. (2009) The phosphatidylinositol 3-kinase/akt cassette regulates purine nucleotide synthesis. Journal of Biological Chemistry 284, 3521–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt V et al. (2017) Control of Nucleotide Metabolism Enables Mutant p53’s Oncogenic Gain-of-Function Activity. Int J Mol Sci 18. 10.3390/ijms18122759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J et al. (2022) De novo nucleotide biosynthetic pathway and cancer. Genes & Diseases. 10.1016/j.gendis.2022.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armenta-Medina D et al. (2014) Comparative genomics of nucleotide metabolism: a tour to the past of the three cellular domains of life. BMC Genomics 15, 800. 10.1186/1471-2164-15-800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lane AN and Fan TW (2015) Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 43, 2466–2485. 10.1093/nar/gkv047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siddiqui A and Ceppi P (2020) A non-proliferative role of pyrimidine metabolism in cancer. Mol Metab 35, 100962. 10.1016/j.molmet.2020.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walter M and Herr P (2022) Re-Discovery of Pyrimidine Salvage as Target in Cancer Therapy. Cells 11. 10.3390/cells11040739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frances A and Cordelier P (2020) The Emerging Role of Cytidine Deaminase in Human Diseases: A New Opportunity for Therapy? Mol Ther 28, 357–366. 10.1016/j.ymthe.2019.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang W et al. (2021) Targeting Pyrimidine Metabolism in the Era of Precision Cancer Medicine. Frontiers in Oncology 11. 10.3389/fonc.2021.684961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pareek V et al. (2021) Human de novo purine biosynthesis. Crit Rev Biochem Mol Biol 56, 1–16. 10.1080/10409238.2020.1832438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson AD et al. (2020) Dysregulation of de novo nucleotide biosynthetic pathway enzymes in cancer and targeting opportunities. Cancer letters 470, 134–140 [DOI] [PubMed] [Google Scholar]
- 24.Moffatt BA and Ashihara H (2002) Purine and pyrimidine nucleotide synthesis and metabolism. Arabidopsis Book 1, e0018. 10.1199/tab.0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu GY and Sabatini DM (2020) mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 21, 183–203. 10.1038/s41580-019-0199-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robitaille AM et al. (2013) Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339, 1320–1323. 10.1126/science.1228771 [DOI] [PubMed] [Google Scholar]
- 27.Ben-Sahra I et al. (2013) Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakashima A et al. (2013) Association of CAD, a multifunctional protein involved in pyrimidine synthesis, with mLST8, a component of the mTOR complexes. Journal of Biomedical Science 20, 24. 10.1186/1423-0127-20-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato T et al. (2015) Rheb protein binds CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, and dihydroorotase) protein in a GTP- and effector domain-dependent manner and influences its cellular localization and carbamoyl-phosphate synthetase (CPSase) activity. J Biol Chem 290, 1096–1105. 10.1074/jbc.M114.592402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herrera KNG et al. (2018) Small-molecule screen identifies de novo nucleotide synthesis as a vulnerability of cells lacking SIRT3. Cell reports 22, 1945–1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He Y et al. (2021) Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduction and Targeted Therapy 6, 425. 10.1038/s41392-021-00828-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juvekar A et al. (2016) Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc Natl Acad Sci U S A 113, E4338–4347. 10.1073/pnas.1522223113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graves LM et al. (2000) Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature 403, 328–332. 10.1038/35002111 [DOI] [PubMed] [Google Scholar]
- 34.Düvel K et al. (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular cell 39, 171–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ben-Sahra I et al. (2016) mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ali ES et al. (2022) The mTORC1-SLC4A7 axis stimulates bicarbonate import to enhance de novo nucleotide synthesis. Molecular Cell 82, 3284–3298.e3287. 10.1016/j.molcel.2022.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saha A et al. (2014) Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Molecular cell 55, 264–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoxhaj G et al. (2019) Direct stimulation of NADP+ synthesis through Akt-mediated phosphorylation of NAD kinase. Science 363, 1088–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Claiborne MD et al. (2022) Persistent CAD activity in memory CD8(+) T cells supports rRNA synthesis and ribosomal biogenesis required at rechallenge. Sci Immunol 7, eabh4271. 10.1126/sciimmunol.abh4271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abt ER et al. (2022) Reprogramming of nucleotide metabolism by interferon confers dependence on the replication stress response pathway in pancreatic cancer cells. Cell Reports 38, 110236. 10.1016/j.celrep.2021.110236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez Herrera KN et al. (2018) Small-Molecule Screen Identifies De Novo Nucleotide Synthesis as a Vulnerability of Cells Lacking SIRT3. Cell reports 22, 1945–1955. 10.1016/j.celrep.2018.01.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lunt SY et al. (2015) Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Molecular cell 57, 95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tong X et al. (2009) The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Current opinion in genetics & development 19, 32–37. 10.1016/j.gde.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gwynne WD et al. (2022) Cancer-selective metabolic vulnerabilities in MYC-amplified medulloblastoma. Cancer Cell. 10.1016/j.ccell.2022.10.009 [DOI] [PubMed] [Google Scholar]
- 45.Kofuji S et al. (2019) IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nature Cell Biology 21, 1003–1014. 10.1038/s41556-019-0363-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J et al. (2017) CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172. 10.1038/nature22359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mathur D et al. (2017) PTEN Regulates Glutamine Flux to Pyrimidine Synthesis and Sensitivity to Dihydroorotate Dehydrogenase Inhibition. Cancer discovery 7, 380–390. 10.1158/2159-8290.cd-16-0612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kollareddy M et al. (2015) Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun 6, 7389. 10.1038/ncomms8389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cox AG et al. (2016) Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nature cell biology 18, 886–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cox AG et al. (2018) Yap regulates glucose utilization and sustains nucleotide synthesis to enable organ growth. The EMBO journal 37. 10.15252/embj.2018100294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santinon G et al. (2018) d NTP metabolism links mechanical cues and YAP/TAZ to cell growth and oncogene-induced senescence. The EMBO journal 37, e97780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shireman JM et al. (2021) De novo purine biosynthesis is a major driver of chemoresistance in glioblastoma. Brain 144, 1230–1246. 10.1093/brain/awab020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye J et al. (2012) Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proceedings of the National Academy of Sciences 109, 6904–6909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qian X et al. (2018) Conversion of PRPS Hexamer to Monomer by AMPK-Mediated Phosphorylation Inhibits Nucleotide Synthesis in Response to Energy StressAMPK Phosphorylates PRPS to Reduce Nucleotide Synthesis. Cancer discovery 8, 94–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keshet R et al. (2020) Targeting purine synthesis in ASS1-expressing tumors enhances the response to immune checkpoint inhibitors. Nature Cancer 1, 894–908. 10.1038/s43018-020-0106-7 [DOI] [PubMed] [Google Scholar]
- 56.Schmitt DL et al. (2016) Sequestration-mediated downregulation of de novo purine biosynthesis by AMPK. ACS chemical biology 11, 1917–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rabinovich S et al. (2015) Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 527, 379–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stambolic V et al. (1998) Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39 [DOI] [PubMed] [Google Scholar]
- 59.Brunet A et al. (1994) Growth factor-stimulated MAP kinase induces rapid retrophosphorylation and inhibition of MAP kinase kinase (MEK1). FEBS Lett 346, 299–303. 10.1016/0014-5793(94)00475-7 [DOI] [PubMed] [Google Scholar]
- 60.Kim EK and Choi EJ (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 1802, 396–405. 10.1016/j.bbadis.2009.12.009 [DOI] [PubMed] [Google Scholar]
- 61.Murphy LO et al. (2004) A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol Cell Biol 24, 144–153. 10.1128/mcb.24.1.144-153.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shin J et al. (2023) Allosteric regulation of CAD modulates de novo pyrimidine synthesis during the cell cycle. Nature Metabolism 5, 277–293. 10.1038/s42255-023-00735-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Torrence ME et al. (2021) The mTORC1-mediated activation of ATF4 promotes protein and glutathione synthesis downstream of growth signals. Elife 10. 10.7554/eLife.63326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.French JB et al. (2016) Spatial colocalization and functional link of purinosomes with mitochondria. Science 351, 733–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pareek V et al. (2020) Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 368, 283–290. 10.1126/science.aaz6465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ma EH and Jones RG (2016) (TORC)ing up purine biosynthesis. Science 351, 670–671. doi: 10.1126/science.aaf1929 [DOI] [PubMed] [Google Scholar]
- 67.De Vitto H et al. (2021) The Intersection of Purine and Mitochondrial Metabolism in Cancer. Cells 10, 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang F et al. (2018) Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell metabolism 28, 369–382.e365. 10.1016/j.cmet.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mannava S et al. (2008) Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 7, 2392–2400. 10.4161/cc.6390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piccolo S et al. (2014) The biology of YAP/TAZ: hippo signaling and beyond. Physiological reviews 94, 1287–1312. 10.1152/physrev.00005.2014 [DOI] [PubMed] [Google Scholar]
- 71.Meng KP et al. (2020) Mechanosensing through YAP controls T cell activation and metabolism. Journal of Experimental Medicine 217. 10.1084/jem.20200053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hove-Jensen B et al. (2017) Phosphoribosyl diphosphate (PRPP): biosynthesis, enzymology, utilization, and metabolic significance. Microbiology and Molecular Biology Reviews 81, e00040–00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martínez-Reyes I and Chandel NS (2021) Cancer metabolism: looking forward. Nat Rev Cancer 21, 669–680. 10.1038/s41568-021-00378-6 [DOI] [PubMed] [Google Scholar]
- 74.Rabani R et al. (2020) Protein kinase C activates NAD kinase in human neutrophils. Free Radic Biol Med 161, 50–59. 10.1016/j.freeradbiomed.2020.09.022 [DOI] [PubMed] [Google Scholar]
- 75.Schild T et al. (2021) NADK is activated by oncogenic signaling to sustain pancreatic ductal adenocarcinoma. Cell Rep 35, 109238. 10.1016/j.celrep.2021.109238 [DOI] [PubMed] [Google Scholar]
- 76.Lee JS et al. (2018) Urea cycle dysregulation generates clinically relevant genomic and biochemical signatures. Cell 174, 1559–1570. e1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee S et al. (2016) Disrupting Na+, HCO₃−-cotransporter NBCn1 (Slc4a7) delays murine breast cancer development. Oncogene 35, 2112–2122. 10.1038/onc.2015.273 [DOI] [PubMed] [Google Scholar]
- 78.Ramirez C et al. (2019) Plasma membrane V-ATPase controls oncogenic RAS-induced macropinocytosis. Nature 576, 477–481. 10.1038/s41586-019-1831-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aird KM and Zhang R (2015) Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett 356, 204–210. 10.1016/j.canlet.2014.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luengo A et al. (2017) Targeting Metabolism for Cancer Therapy. Cell Chem Biol 24, 1161–1180. 10.1016/j.chembiol.2017.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ma J et al. (2021) Emerging roles of nucleotide metabolism in cancer development: progress and prospect. Aging 13, 13349–13358. 10.18632/aging.202962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ribatti D (2012) Sidney Farber and the Treatment of Childhood Acute Lymphoblastic Leukemia with a Chemotherapeutic Agent. Pediatric Hematology and Oncology 29, 299–302. 10.3109/08880018.2012.678969 [DOI] [PubMed] [Google Scholar]
- 83.Aravind P et al. (2022) [18F]Fluorothymidine(FLT)-PET imaging of thymidine kinase 1 pharmacodynamics in non-small cell lung cancer treated with pemetrexed. Journal of Clinical Oncology 40, 3070–3070. 10.1200/JCO.2022.40.16_suppl.3070 [DOI] [Google Scholar]
- 84.Kanarek N et al. (2018) Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 559, 632–636. 10.1038/s41586-018-0316-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shukla SK et al. (2017) MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 32, 71–87.e77. 10.1016/j.ccell.2017.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brown KK et al. (2017) Adaptive Reprogramming of De Novo Pyrimidine Synthesis Is a Metabolic Vulnerability in Triple-Negative Breast Cancer. Cancer Discov 7, 391–399. 10.1158/2159-8290.CD-16-0611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Madak JT et al. (2019) Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacology & Therapeutics 195, 111–131. 10.1016/j.pharmthera.2018.10.012 [DOI] [PubMed] [Google Scholar]
- 88.Kaur H et al. (2021) Efficacy and safety of dihydroorotate dehydrogenase (DHODH) inhibitors “leflunomide” and “teriflunomide” in Covid-19: A narrative review. Eur J Pharmacol 906, 174233. 10.1016/j.ejphar.2021.174233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pal S et al. (2022) A druggable addiction to de novo pyrimidine biosynthesis in diffuse midline glioma. Cancer Cell 40, 957–972.e910. 10.1016/j.ccell.2022.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mao C et al. (2021) DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590. 10.1038/s41586-021-03539-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koundinya M et al. (2018) Dependence on the Pyrimidine Biosynthetic Enzyme DHODH Is a Synthetic Lethal Vulnerability in Mutant KRAS-Driven Cancers. Cell chemical biology 25, 705–717.e711. 10.1016/j.chembiol.2018.03.005 [DOI] [PubMed] [Google Scholar]
- 92.Shi DD et al. (2022) De novo pyrimidine synthesis is a targetable vulnerability in IDH mutant glioma. Cancer Cell 40, 939–956.e916. 10.1016/j.ccell.2022.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhou Y et al. (2021) DHODH and cancer: promising prospects to be explored. Cancer Metab 9, 22. 10.1186/s40170-021-00250-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Narváez J et al. (2015) Comparative Effectiveness of Tocilizumab with either Methotrexate or Leflunomide in the Treatment of Rheumatoid Arthritis. PLOS ONE 10, e0123392. 10.1371/journal.pone.0123392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rosenzweig M et al. (2020) Repurposing leflunomide for relapsed/refractory multiple myeloma: a phase 1 study. Leukemia & lymphoma 61, 1669–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.O’Connor P et al. (2011) Randomized trial of oral teriflunomide for relapsing multiple sclerosis. New England Journal of Medicine 365, 1293–1303 [DOI] [PubMed] [Google Scholar]
- 97.Hahn F et al. (2020) IMU-838, a developmental DHODH inhibitor in phase II for autoimmune disease, shows anti-SARS-CoV-2 and broad-spectrum antiviral efficacy in vitro. Viruses 12, 1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stine ZE et al. (2022) Targeting cancer metabolism in the era of precision oncology. Nature Reviews Drug Discovery 21, 141–162. 10.1038/s41573-021-00339-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hayek S et al. (2020) Cerpegin-derived furo [3, 4-c] pyridine-3, 4 (1H, 5H)-diones enhance cellular response to interferons by de novo pyrimidine biosynthesis inhibition. European Journal of Medicinal Chemistry 186, 111855. [DOI] [PubMed] [Google Scholar]
- 100.Tu H-F et al. (2021) Afatinib Exerts Immunomodulatory Effects by Targeting the Pyrimidine Biosynthesis Enzyme CADImmune Modulation of T Cell by Afatinib. Cancer Research 81, 3270–3282 [DOI] [PubMed] [Google Scholar]
- 101.Valvezan AJ et al. (2017) mTORC1 Couples Nucleotide Synthesis to Nucleotide Demand Resulting in a Targetable Metabolic Vulnerability. Cancer cell 32, 624–638.e625. 10.1016/j.ccell.2017.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chong YC et al. (2020) Targeted Inhibition of Purine Metabolism Is Effective in Suppressing Hepatocellular Carcinoma Progression. Hepatology Communications 4, 1362–1381. 10.1002/hep4.1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu J et al. (2022) Targeting purine metabolism in ovarian cancer. Journal of Ovarian Research 15, 93. 10.1186/s13048-022-01022-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang X et al. (2017) Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nature neuroscience 20, 661–673. 10.1038/nn.4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou W et al. (2020) Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nature Communications 11, 3811. 10.1038/s41467-020-17512-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Perrault EN et al. (2021) Ribonucleotide Reductase Regulatory Subunit M2 as a Driver of Glioblastoma TMZ-Resistance through Modulation of dNTP Production. bioRxiv, 2021.2011.2023.469785. 10.1101/2021.11.23.469785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bergan S et al. (2016) Drug target molecules to guide immunosuppression. Clinical biochemistry 49, 411–418. 10.1016/j.clinbiochem.2015.10.001 [DOI] [PubMed] [Google Scholar]
- 108.Passos DF et al. (2018) Adenosine signaling and adenosine deaminase regulation of immune responses: impact on the immunopathogenesis of HIV infection. Purinergic Signalling 14, 309–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mastelic-Gavillet B et al. (2019) Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. Journal for immunotherapy of cancer 7, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chang D et al. (2020) Extracellular cyclic dinucleotides induce polarized responses in barrier epithelial cells by adenosine signaling. Proceedings of the National Academy of Sciences 117, 27502–27508. doi: 10.1073/pnas.2015919117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Whiteley AT et al. (2019) Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature 567, 194–199. 10.1038/s41586-019-0953-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sugiura A et al. (2022) MTHFD2 is a metabolic checkpoint controlling effector and regulatory T cell fate and function. Immunity 55, 65–81.e69. 10.1016/j.immuni.2021.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nygaard P and Saxild HH (2009) Nucleotide Metabolism. In Encyclopedia of Microbiology (Third Edition) (Schaechter M, ed), pp. 296–307, Academic Press [Google Scholar]
- 114.Berthet J et al. (1957) The relationship of epinephrine and glucagon to liver phosphorylase. IV. Effect of epinephrine and glucagon on the reactivation of phosphorylase in liver homogenates. J Biol Chem 224, 463–475 [PubMed] [Google Scholar]
- 115.Suwanichkul A et al. (1993) Identification of a promoter element which participates in cAMP-stimulated expression of human insulin-like growth factor-binding protein-1. J Biol Chem 268, 9730–9736 [PubMed] [Google Scholar]
- 116.Francis SH et al. (2010) cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev 62, 525–563. 10.1124/pr.110.002907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hoxhaj G et al. (2017) The mTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell Rep 21, 1331–1346. 10.1016/j.celrep.2017.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Emmanuel N et al. (2017) Purine Nucleotide Availability Regulates mTORC1 Activity through the Rheb GTPase. Cell Rep 19, 2665–2680. 10.1016/j.celrep.2017.05.043 [DOI] [PubMed] [Google Scholar]
- 119.Tait-Mulder J et al. (2020) The conversion of formate into purines stimulates mTORC1 leading to CAD-dependent activation of pyrimidine synthesis. Cancer Metab 8, 20. 10.1186/s40170-020-00228-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Diehl FF et al. (2022) Nucleotide imbalance decouples cell growth from cell proliferation. Nat Cell Biol 24, 1252–1264. 10.1038/s41556-022-00965-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Soflaee MH et al. (2022) Purine nucleotide depletion prompts cell migration by stimulating the serine synthesis pathway. Nat Commun 13, 2698. 10.1038/s41467-022-30362-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sancak Y et al. (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501. 10.1126/science.1157535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Menon S et al. (2014) Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771–785. 10.1016/j.cell.2013.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gilleron J and Zeigerer A (2022) Endosomal trafficking in metabolic homeostasis and diseases. Nat Rev Endocrinol. 10.1038/s41574-022-00737-9 [DOI] [PubMed] [Google Scholar]
- 125.Butkinaree C et al. (2010) O-linked beta-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta 1800, 96–106. 10.1016/j.bbagen.2009.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gibellini F and Smith TK (2010) The Kennedy pathway--De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 62, 414–428. 10.1002/iub.337 [DOI] [PubMed] [Google Scholar]
- 127.Jarrar Y and Lee SJ (2021) The Functionality of UDP-Glucuronosyltransferase Genetic Variants and their Association with Drug Responses and Human Diseases. J Pers Med 11. 10.3390/jpm11060554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brunner JS and Finley LWS (2022) Metabolic determinants of tumour initiation. Nature Reviews Endocrinology. 10.1038/s41574-022-00773-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mager LF et al. (2020) Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369, 1481–1489 [DOI] [PubMed] [Google Scholar]
- 130.Halbrook CJ et al. (2019) Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell metabolism 29, 1390–1399. e1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lanier LL (2015) NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunology Research 3, 575–582. 10.1158/2326-6066.Cir-15-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.McCarthy MT et al. (2018) Purine nucleotide metabolism regulates expression of the human immune ligand MICA. J Biol Chem 293, 3913–3924. 10.1074/jbc.M117.809459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shin HJ et al. (2018) Gemcitabine and Nucleos(t)ide Synthesis Inhibitors Are Broad-Spectrum Antiviral Drugs that Activate Innate Immunity. Viruses 10, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sprenger H-G et al. (2021) Cellular pyrimidine imbalance triggers mitochondrial DNA–dependent innate immunity. Nature Metabolism 3, 636–650. 10.1038/s42255-021-00385-9 [DOI] [PMC free article] [PubMed] [Google Scholar]