

Summary
Upper tract urothelial carcinoma (UTUC) is often diagnosed late and exhibits poor prognosis. Limited data are available on potential non-invasive biomarkers for disease monitoring. Here, we investigate the proteomic profile of plasma in 362 UTUC patients and 239 healthy controls. We present an integrated tissue-plasma proteomic approach to infer the signature proteins for identifying patients with muscle-invasive UTUC. We discover a protein panel that reflects lymph node metastasis, which is of interest in identifying UTUC patients with high risk and poor prognosis. We also identify a ten-protein classifier and establish a progression clock predicting progression-free survival of UTUC patients. Finally, we further validate the signature proteins by parallel reaction monitoring assay in an independent cohort. Collectively, this study portrays the plasma proteomic landscape of a UTUC cohort and provides a valuable resource for further biological and diagnostic research in UTUC.
Keywords: plasma proteomics, mass spectrometry, DIA, upper tract urothelial carcinoma, prognosis
Graphical abstract
Highlights
-
•
Proteomic profiles of plasma in UTUC patients with a 5-year follow-up
-
•
Discovery of the plasma signature proteins associated with UTUC
-
•
Construction of predictive model distinguishing between MI-UTUCs and NMI-UTUCs
-
•
Identification of progression clock predicting progression-free survival of UTUC patients
Upper tract urothelial cancer (UTUC) is a relatively uncommon but more aggressive form of urothelial cancer. It is often diagnosed late and has poor prognosis. Qu et al. portray a plasma proteomic landscape of UTUC and provide predictive models that could potentially improve the diagnosis and management of UTUC.
Introduction
Upper tract urothelial carcinoma (UTUC), which comprises cancer of the ureter and renal pelvis, is relatively uncommon and accounts for only approximately 5%–10% of urothelial carcinomas (UCs).1 Furthermore, 90%–95% of UCs occur in the bladder (UCB). Because UTUC is relatively rare, clinical decision making for UTUC patients is extrapolated based on treatment data for UCB. In a more recent study of 116 UCB patients, we reported three proteome-based subtypes (U-I, U-II, and U-III) with different clinical outcomes and molecular characteristics.2 However, the molecular pathogenesis of UTUC is poorly understood, and no useful biomarkers are available for accurate diagnosis and classification.
Patients suspected of having UTUC need to undergo invasive procedures such as computed tomography urography, retrograde pyelography, or ureteroscopy for a definitive diagnosis. However, these tools struggle to detect early disease progression. Urine cytology is a non-invasive method for the detection of UC but also fails to detect most UTUCs.3 Plasma samples are easily obtained, and they provide an indication of cancer status including various cellular elements such as proteins.4 However, the available data on plasma-based protein biomarkers in UTUC are limited.5
Approximately 55%–59% of UTUC cases are muscle invasive at the time of diagnosis compared to only 15% of UCBs.6,7 Muscle-invasive UTUC patients (MI-UTUCs) usually have a very poor prognosis, and the 5-year specific survival rate is <50% and <10% for the pT2/pT3 and pT4 stages, respectively.8 Up to 30% of MI-UTUCs have metastasis in the regional lymph nodes,9 which represents a well-established poor prognostic factor.1 A major concern is the difficulty in determining whether a patient has muscle-invasive or more advanced disease because imaging studies, cytology, or biopsy findings cannot reliably diagnose the disease stage and identify patients with lymph node invasion preoperatively.10 Hence, the discovery of reliable plasma biomarkers for identifying MI-UTUCs and lymph node involvement in UTUC is critical to improve clinical management and patient outcomes.
Accurate prediction of postoperative recurrence and progression could help guide patient management of adjuvant therapies and the design of clinical trials. Prognostic models have been developed for various malignancies and are utilized in clinical decision making across different disease stages. For example, Kim et al. reported an eight-gene prognostic model with strong predictive value for determining disease outcome in bladder cancer.11 Wu et al. identified a five-gene classifier that predicts the progression-free survival (PFS) interval for papillary thyroid carcinoma.12 The time of recurrence and progression in UTUC patients is highly dynamic, and some patients do not experience recurrence within 2 years whereas others do within 2 months.7 Therefore, the temporal prediction of progression and recurrence is important in guiding counseling of UTUC patients, follow-up scheduling, and administration of adjuvant therapies.
Plasma proteins are the major functional component of the plasma and play key roles in various biological processes, including signaling, transport, and defense against infections.13 Recently, plasma proteomics technology has been widely used to study various diseases such as prostate cancer,14,15 coronavirus disease 2019 (COVID-19),16,17 and alcohol-related liver disease.18 Therefore, the development of plasma-based proteomics provides enormous possibilities for clinical transformation and application.
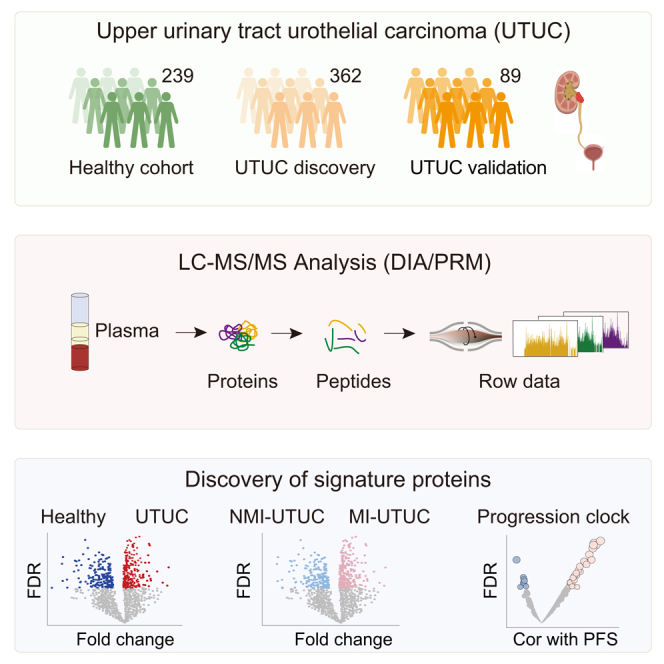
In this study, we analyzed 601 plasma samples (including 362 UTUC samples and 239 healthy control samples) using mass spectrometry-based, data-independent acquisition (DIA) quantitative proteomics. The integrated tissue-plasma proteomics approach identified the signature proteins for identifying MI-UTUCs. Furthermore, we identified a ten-protein classifier and established a progression clock that predicted the PFS of UTUC patients. We then validated the findings in an independent UTUC cohort. Collectively, this study presented the plasma proteomic landscape of a UTUC cohort and provided a valuable resource for further biological, diagnostic, and drug-discovery efforts.
Results
Overall synopsis of the plasma proteome profiling of patients with UTUC
Plasma samples from 451 UTUC patients (discovery [n = 362], validation [n = 89]) and 239 healthy controls were analyzed using quantitative proteomic analysis with DIA strategy (Figure 1A and STAR Methods). UTUC is defined as urothelial carcinoma arising from the ureter (UUC) or renal pelvis (RPUC). We included 163 RPUCs, 170 UUCs, and 29 with a combination of both cancers in the UTUC discovery cohort (Figure 1B). The overall workflow of this study is presented in Figure 1A. The demographic and clinical data of all the study participants are summarized in Table S1. Additionally, the basic features—age, gender, and history of smoking—were similar among patients with RPUC or UUC, or a combination of both (Figure 1C).
Figure 1.

Overall synopsis of the plasma proteome profiling of UTUC patients
(A) The proteomics workflow involved three modules: cohort construction (discovery cohort and validation cohort), proteomic profiling, and data analysis.
(B) Pie chart showing the component proportion of UTUC discovery cohort.
(C) Clinical data of UTUC discovery cohort.
(D) Proteins identified in UTUC and normal plasma samples of UTUC discovery cohort.
(E) Number of proteins identified in UTUC and normal plasma samples of UTUC discovery cohort. Boxplots show median (central line), upper and lower quartiles (box limits), 1.5× interquartile range (whiskers).
(F) Dynamic range of the protein identification of each sample according to the descending sort of protein abundance in UTUC and normal plasma samples of UTUC discovery cohort.
(G) Components identified in plasma proteome.
(H) Cancer-related proteins and proteins associated with drugs approved by the FDA identified in UTUC discovery cohort.
A total of 9,336 protein groups were identified in all plasma samples (Figure S1A), with an average of 1,860 and 1,789 protein groups per UTUC and normal plasma (healthy control) samples, respectively (Figures 1D and 1E). Proteome quantification was conducted using the intensity-based absolute quantification (iBAQ) algorithm,19 followed by the fraction of total (FOT) normalization as reported previously.20 The proteome was highly dynamic, spanning approximately about two orders of magnitude measured by the protein abundance (FOT) (Figure 1F). The distribution of log2-transformed FOT abundance of identified proteins in 601 plasma samples is shown in Figure S1B, and the consistency of the samples indicated the stability of our mass spectrometry platform.
To monitor the liquid chromatography-tandem mass spectrometry (LC-MS/MS) platform instrument stability, the mixture of all plasma samples from UTUC patients was measured every 20 samples, which was adopted in proteomic studies. The quality control (QC) plasma samples were analyzed using the same method and conditions used for our cohort plasma samples.21 The average Pearson’s correlation coefficient, calculated for all quality control runs of QC samples, was 0.95, indicating that the MS data were of high quality (Figures S1C and S1D). In addition, we analyzed the proteome of ten matched UTUC tumor tissue samples with plasma samples and ten normal urothelial epithelial tissues with a data-dependent acquisition method, quantifying 11,372 proteins in total (Figures S1E and S1F; STAR Methods).
Additionally, as shown in Figure 1G, the number of proteins identified in UTUC plasma that were annotated as extracellular matrix or were in the extracellular space was not significantly different from that in normal plasma. Interestingly, a slightly higher number of proteins annotated as cancer-related proteins and associated with drugs approved by the US Food and Drug Administration (FDA) were identified in UTUC than in normal plasma (Figure 1H).
Plasma proteome profiles differed between UTUC and normal samples
We compared the plasma proteome profiles of UTUC patients to those of the healthy controls. This revealed a dramatic shift in the quantitative proteome composition, reflected by 550 significantly differentially expressed proteins (DEPs), of which 307 proteins were upregulated and 243 proteins were downregulated (Figure 2A; Wilcoxon rank-sum test, Benjamini-Hochberg [BH] P < 0.05, UTUC/normal ratio >1.5 or <0.67; Table S2). Pathway enrichment analysis of DEPs showed that normally enriched proteins were involved in the cleavage of the damaged purine, deubiquitination, and sulfur metabolism, whereas proteins enriched in UTUC plasma mainly participated in transfer RNA (tRNA) aminoacylation, fatty acid degradation, and acute-phase response pathway (Figure 2B).
Figure 2.

Plasma proteome profiles differed between UTUC and normal samples
(A) Protein abundance differences between UTUC and normal plasma samples.
(B) Different pathways between UTUC and normal plasma samples.
(C) Spearman correlation between plasma and tissue proteomes.
(D) Fold changes of plasma and tissue proteins in UTUC and normal samples (left), and pathways enriched for respective specifically changed proteins (right).
(E) Heatmap showing the proteins that meet the screening criteria. Their presence in blood was annotated from HPA.
(F) Box plot showing plasma protein abundance of CTSB (BH P = 4.27E-7) and S100A8 (BH P = 8.25E-15). Boxplots show median (central line), upper and lower quartiles (box limits), 1.5× interquartile range (whiskers).
(G) (Top) Strategy for plasma signature proteins to distinguish between UTUC and normal samples. (Bottom) Heatmap of the selected proteins expressed in UTUC and normal plasma samples.
(H) ROC curve of plasma signature proteins in UTUC discovery cohort.
Among the 8,923 proteins identified in UTUC plasma proteome, 234 were kidney-tissue-specific proteins annotated by the Human Protein Atlas (HPA, https://www.proteinatlas.org) (Figure S2A and Table S2). Differential protein analysis between UTUC and normal plasma samples resulted in the identifications of 46 kidney-tissue-specific proteins (Figure S2B; Wilcoxon rank-sum test, BH P < 0.05, UTUC/normal ratio >1.5 or <0.67). Further pathway enrichment analysis using these differential proteins indicated that UTUC-enriched proteins were involved in fatty acid metabolic pathways, whereas those enriched in normal samples mainly participated in cellular sodium ion homeostasis (Figure S2C). These indicated that leaking of these cancer-associated proteins into the plasma might influence plasma composition.
We also compared the plasma proteome profiles of UTUC patients to those of UTUC tissue proteome. In total, 7,913 proteins were commonly quantified in the UTUC tissue and plasma samples, and we confirmed the significantly positive correlation between the proteome of UTUC tissue and plasma (Figure 2C). Moreover, 165 proteins (Figure 2D; Wilcoxon rank-sum test, BH P < 0.05, UTUC/normal ratio >1.5) that were upregulated in UTUCs in both plasma proteome and tissue proteome were enriched in positive regulation of nuclear factor κB signaling, response to drug, and endocytosis. We also found a divergence between the proteome of UTUC tissue and plasma. The results showed that 234 proteins were significantly upregulated only in UTUC plasma proteome (Figure 2D; Wilcoxon rank-sum test, BH P < 0.05, UTUC/normal ratio >1.5), and they were mainly enriched in proteolysis and cell-adhesion pathways.
To explore plasma enhanced proteins, we performed supervised analysis to filter plasma proteins https://www.proteinatlas.org/, whereby 21 proteins met the criteria, nine of which were cancer-related proteins (Figure 2E). Among the nine proteins, CTSB, S100A8, HP, and CHI3L1 were associated with bad prognosis in urothelial cancer according to the HPA dataset (Figures 2E and S2D). Notably, CTSB and S100A8 upregulated in UTUC plasma were involved in proteolysis and cell-adhesion pathway, respectively (Figure 2F). The overexpression of CTSB could promote cell invasion and metastasis of colorectal cancer, liver cancer, gastric cancer, glioma, and ovarian cancer.22,23,24,25,26
We employed stepwise logistic regression based on the DEPs to identify a subset of proteins that could distinguish between UTUC and normal (termed UTUC/normal-sig) (Figure 2G and STAR Methods). Based on the UTUC/normal-sig proteins, 10-fold cross-validation was applied which generated a mean area under the receiver-operating characteristics curve (ROC-AUC) of 0.942 (Figure 2H). We further evaluated the performance of our model in another independent validation cohort (n = 89) using DIA quantitative proteomics, whereby the AUC was 0.925 (Figure S2E). Taken together, the classifier model could represent a potential predictive model to identify UTUC patients.
Difference between plasma proteome profiles and urine proteome profiles
Urine is a good source for profiling localized diseases, and it serves as an ideal source for discovering biomarkers for diseases in kidney and other tissues of the urogenital system. We collected urine samples from nine UTUC patients (T1 [n = 2], T2 [n = 4], T3 [n = 3]) and five healthy controls and performed proteome analysis. A total of 5,898 protein groups were identified in all urine samples, with an average of 2,042 protein groups per urine samples of UTUC and 2,203 protein groups per urine samples of normal (Figures S2F and S2G; Table S2; STAR Methods). The number of proteins identified in both UTUC and normal samples was 3,488 (Figure S2H).
To identify UTUC-associated changes in the urine proteome, we determined which proteins were differentially present in the urine of UTUC patients compared to the healthy controls. The result showed that 293 proteins were upregulated while 387 proteins were downregulated in urine samples of UTUC patients (Figure S2I; Wilcoxon rank-sum test, BH P < 0.05, UTUC/normal ratio >1.5 or <0.67). Among the DEPs, 56 upregulated proteins and 71 downregulated proteins were urine exosome annotated according to the urinary exosome protein database (https://esbl.nhlbi.nih.gov/UrinaryExosomes/).27
We further compared the urine proteome profiles with UTUC to that of UTUC plasma proteome. Differential protein analysis showed that 307 upregulated proteins and 243 downregulated proteins were observed in UTUC plasma samples, as opposed to 293 upregulated proteins and 387 downregulated proteins in UTUC urine samples (Figure S2J; Wilcoxon rank-sum test, BH P < 0.05, UTUC/normal ratio >1.5 or <0.67). We further found that 29 proteins upregulated in UTUC samples in both plasma proteome and urine proteome compared with normal samples were enriched in tRNA aminoacylation and proteolysis pathway (Figure S2K).
When we combined the UTUC tissue samples, we observed that six proteins were upregulated in UTUC plasma, UTUC urine, and UTUC tissues (Figure S2L and Table S2). We defined the six proteins as the potential UTUC-enhanced proteins, which might be indicators for UTUC. Furthermore, we found that five out of these six proteins were associated with the prognosis in our previously reported UCB cohort2 (Figure S2M). We further investigated these six proteins by assessing immunohistochemistry (IHC) expression in the HPA (Figure S2N). Four of these six proteins showed medium to strong tumor staining in urothelial cancer samples, while two other proteins showed low staining in the HPA and merit further investigation (Figure S2O).
To explore the signatures in urine for NMI-UTUC diagnostics, we compared the proteins identified in the urine of NMI-UTUCs and healthy controls. The result showed 1,254 proteins identified in NMI-UTUC urine samples (100% identification), of which 37 proteins were only identified in urine of NMI-UTUCs compared to healthy controls (Figure S2P). We investigated the 37 proteins in the plasma proteome of NMI-UTUCs, whereby the result showed that 15 proteins were observed in less than 10% of NMI-UTUC plasma samples (Figure S2Q and Table S2). We further found that three out of 15 proteins (RALB, FRY, and WDR45B) were not detected in the MI-UTUC urine samples. These results suggested that three proteins only detected in NMI-UTUC urine samples might be associated with NMI-UTUC disease, which could be further verified in a larger cohort in the future.
Difference between plasma proteome profiles and EV proteome profiles
Exosomes are extracellular vesicles (EVs) secreted by most eukaryotic cells and participate in intercellular communication.28 Tumor-derived exosomes participate in formation and progression of different cancer processes, including tumor microenvironment remodeling, angiogenesis, invasion, metastasis, and drug resistance.29 Plasma-derived EVs share some similar components with plasma; however, EVs also have unique protein composition due to their relatively independent subcellular structure. In this study, we also isolated EVs from 60 plasma samples, comprising 33 UTUCs and 27 healthy controls, to explore the UTUC-associated EV proteins. We used classical approaches,30,31,32,33 including ultracentrifugation and size-exclusion chromatography, for isolation of EVs from plasma samples (STAR Methods). The expression levels of the conventional EV markers CD9, TSG101, and HSP7034,35 were measured using western blotting, and these markers were detectable in plasma-derived EVs (Figure 3A). Label-free MS quantification measurement of all EV samples resulted in a total of 3,628 protein groups, with an average of 1,061 protein groups per normal sample and 1,126 protein groups per UTUC sample (Figure 3B and Table S2). The 3,628 proteins were next compared with the multiple proteomics exosome studies published in Vesiclepedia,36 revealing that 3,401 (93.7%) have previously been identified by human exosome proteomics studies (Figure 3C). These results suggested that our identifications were consistent with data originating from exosomes previously reported.
Figure 3.

Difference between plasma proteome profiles and EV proteome profiles
(A) Western blot of EVs isolated from plasma for conventional identified EV markers.
(B) Box plot showing the proteins identified in normal and UTUC EV samples. Boxplots show median (central line), upper and lower quartiles (box limits), 1.5× interquartile range (whiskers).
(C) Venn diagram showing proteins identified in EVs herein and belonging to human EV proteins in the Vesiclepedia dataset.
(D) Venn diagram showing proteins identified in more than 30% of UTUC plasma observed in EVs.
(E) Venn diagram showing proteins upregulated in UTUC plasma (left) or downregulated in UTUC plasma (right) observed in EVs.
(F) Volcano plot showing the EV protein abundance differences between UTUC and normal plasma samples.
(G) Bubble plot showing pathways enriched in normal and UTUC samples.
(H) Venn diagram of upregulated or downregulated proteins identified in both EVs and plasma.
(I) Heatmap of EV proteins abundance differences between normal and UTUC samples.
(J) Box plot showing EV protein abundance of PSMD2 (BH P = 0.028) and ICAM1 (BH P = 0.040).
To understand the characteristics and composition of UTUC-associated EV proteins, we tried to determine which of these proteins were present in the plasma of UTUC patients. The result showed that 2,022 proteins were identified in more than 30% of plasma samples in the UTUC discovery cohort, 1,207 of which (60%) were also detected in EVs (Figure 3D). We further focused on the 550 DEPs between UTUC plasma samples and normal samples. When compared to the EV proteome, 298 DEPs were identified in EVs, of which 187 proteins were upregulated and 111 proteins were downregulated (Figure 3E). These results showed that the DEPs in plasma proteome could be detected in EV proteome; however, these DEPs may not necessarily have significant differences between normal and UTUC samples in the EV proteome.
To identify EV proteins associated with UTUC patients, we sought to identify the proteins that were upregulated in UTUC EVs by comparing the proteome of UTUC EVs with that of normal EVs. This revealed a dramatic shift in the quantitative proteome composition, reflected by 283 DEPs, of which 142 proteins were upregulated and 141 proteins were downregulated (Figure 3F; Wilcoxon rank-sum test, BH P < 0.05, UTUC/normal ratio >1.5 or <0.67). Pathway analysis showed that UTUC-enriched EV proteins were involved in cell adhesion, cell migration, and complement and coagulation cascades, whereas normal-enriched EV proteins mainly participated in cellular oxidant detoxification and proteasome (Figure 3G). These findings indicated the different biological functions of normal EVs and UTUC EVs.
Furthermore, we compared the UTUC EV proteome to that of UTUC plasma proteome. The number of proteins identified in both UTUC plasma proteome and matched EV proteome was 2,200, accounting for 69.8% of the proteins identified in EV samples. Among these, 299 and 260 DEPs were observed in the plasma and EV samples, respectively (Figure 3H). Further analysis showed that 18 proteins were upregulated, whereas nine proteins were downregulated in both UTUC plasma and UTUC EVs (Figures 3I and 3J; Table S2; Wilcoxon rank-sum test, BH P < 0.05, UTUC/normal ratio >1.5 or <0.67). These 18 proteins upregulated in both UTUC plasma and UTUC EVs might serve as potential signature proteins for UTUC patients and warrant further validation.
Plasma proteome profiles differed between RPUC and UUC samples
UTUC combines RPUC and UUC, which have homogeneous and heterogeneous biology. We investigated the similarities and differences between RPUC and UUC. First, we performed correlation analysis among RPUC, UUC, and normal samples. The results showed that the frequencies of proteins identified in RPUC were more closely correlated with UUC than normal samples (Figure 4A). To investigate altered proteomic features of UTUC, we compared proteins with significantly differential expression (Wilcoxon rank-sum test, BH P < 0.05, UTUC/normal ratio >1.5) between UTUC patients and normal samples in RPUC and UUC, respectively (Figures 4B and 4C). We identified 580 proteins upregulated in RPUC, in which 375 (64.6%) proteins were also upregulated in UUC (66.5% of upregulated proteins in UUC). Pathway analysis showed that the 375 proteins were mainly enriched in fatty acid degradation and cell proliferation pathways (Figure 4D), which was consistent with UTUC being characterized by disorders of cell proliferation and metabolism in a previous study.37 We further found that 24 of 375 commonly upregulated proteins were associated with prognosis, such as ACDAS and PPP1R9B (Figures 4E and 4F). These results indicated that, in comparison to normal plasma samples, the directions of most dysregulations in RPUC and UUC plasma samples were consistent.
Figure 4.

Similarities and differences between RPUC and UUC
(A) Spearman correlation of plasma proteins among RPUC, UUC, and normal samples.
(B) Volcano plot showing protein abundance differences between RPUC and normal plasma samples.
(C) Volcano plot showing protein abundance differences between UUC and normal plasma samples.
(D) Pathway analysis of proteins commonly identified in both renal pelvis and ureter samples.
(E) Heatmap showing protein abundance differences between normal and UTUC samples.
(F) Overall survival (OS) analyses of UTUC patients with high or low levels of ACADS (top) or PPP1R9B (bottom) protein abundance in UTUC discovery cohort.
(G) OS and progression-free survival (PFS) analyses of RPUC versus UUC patients.
(H) The association of RPUC with UUC in terms of clinical information.
(I) Volcano plot showing protein abundances between RPUC and UUC.
(J) Pathway analysis of RPUC (blue) and UUC (red).
(K) (Left) Heatmap showing the protein abundance among normal, RPUC, and UUC. (Right) The hazard ratio of each protein.
(L) OS analyses of UTUC patients with high or low levels of DBN1 (top) or YAP1 (bottom) protein abundance in UTUC discovery cohort.
Second, UUC showed a worse prognosis than RPUC (Figure 4G; log-rank test, p < 0.05), which indicated that UUC and RPUC had different molecular features and clinical characteristics. In our cohort, clinical information showed the similarity of patients’ basic features between RPUC and UUC, besides the tumor size being significantly bigger in RPUC (Figure 4H and Table S1; Fisher’s exact test, p < 0.05). To further investigate the tumor heterogeneity between UUC and RPUC, we compared the differential expression at the plasma proteome. The result showed that a total of 284 proteins differentially expressed between RPUC and UUC (Figure 4I; Wilcoxon rank-sum test, BH P < 0.05, fold change >1.5 or <0.67). Interestingly, the kidney enhanced proteins (e.g., ACSF2, DDAH1, MSRA) annotated by the HPA dataset were upregulated in the RPUC plasma samples.
Furthermore, pathway enrichment analysis of the proteins upregulated in the RPUC samples were enriched in the regulation of cell growth, fatty acid oxidation, and peroxisome proliferator-activated receptor signaling pathways, while proteins upregulated in the UUC samples were mainly involved in cell-cell adhesion, Hippo signaling pathway, and mitogen-activated protein kinase cascade (Figures 4J and 4K). The proteins participating in cell-cell adhesion, and Hippo signaling pathway, such as CD93, DBN1, and YAP1, were correlated with clinical outcomes (Figures 4K and 4L). Previous studies had reported that YAP1 is essential for cancer initiation or growth of most solid tumors and its activation induces cancer stem cell attributes, proliferation, and metastasis.38 These results indicated that YAP1, and molecules (CSTA, CD93, DBN) of cell adhesion over-represented in UUC, might play a role in UUC, indicating that RPUC and UUC had different pathogenic mechanisms.
Plasma proteomic profiles identify patients with muscle-invasive UTUC
Tumor stage was reported as the most important prognostic factor for UTUC.39 Muscle-invasive UTUC has a very poor prognosis, with 5-year survival less than 50% for stage pT2/pT3, and <10% for pT4,8 which was also observed with our data (Figure 5A). Our discovery cohort contained 351 UTUC patients with T category, comprising 109 non-muscle-invasive UTUCs (NMI-UTUCs) (Ta [n = 34], T1 [n = 75]) and 242 muscle-invasive UTUCs (MI-UTUCs) (T2 [n = 87], T3 [n = 140], T4 [n = 15]) (Figure 5B). Differential plasma protein analysis between NMI-UTUCs and MI-UTUCs resulted in 42 DEPs (Figure 5C and Table S3; Wilcoxon rank-sum test, BH P < 0.05, MI-UTUCs/NMI-UTUCs ratio >1.5 or <0.67). Pathway enrichment analysis of the DEPs showed that MI-UTUC-enriched proteins were involved in acute-phase response, tRNA aminoacylation, and fatty acid degradation, whereas proteins enriched in NMI-UTUCs mainly participated in sulfur metabolism and deubiquitination (Figure 5D). We found that the abundance of acute-phase response proteins, such as SAA1 and CRP, showed the highest positive correlation with the T category (Figure S3A). For further comparison, we surveyed the divergence of the DEPs among the normal, NMI-UTUCs, and MI-UTUCs (Figures 5E and S3B). Interestingly, we found that the expression level of proteins over-represented in MI-UTUCs gradually increased from normal to NMI-UTUCs to MI-UTUCs, whereas that of proteins over-represented in NMI-UTUCs gradually decreased (Figure S3C). Furthermore, we also observed some plasma proteins (e.g., ABLIM3, SCGN, PYGO2) exclusively expressed in NMI-UTUCs or MI-UTUCs (Figure S3D), but the identification frequency of these proteins was low (less than 30%), and more validation is needed to confirm these proteins.
Figure 5.

Plasma proteomic profiles identify patients with muscle-invasive UTUC
(A) Kaplan-Meier curves for OS and PFS of NMI-UTUCs versus MI-UTUCs.
(B) Bar plot for T category in NMI-UTUCs and MI-UTUCs.
(C) Difference in protein abundance between NMI-UTUCs and MI-UTUCs.
(D) Pathways enriched for DEPs in NMI-UTUCs and MI-UTUCs.
(E) Fold changes of DEPs from comparison of NMI-UTUCs and MI-UTUCs in UTUC and normal plasma samples.
(F) Strategy for screen diagnosis plasma signature proteins.
(G) Protein abundance of TST and HPCAL1 among normal, NMI-UTUCs, and MI-UTUCs in tissue (top) and plasma (bottom) cohorts. Boxplots show median (central line), upper and lower quartiles (box limits), 1.5× interquartile range (whiskers).
(H) RNA expression level of TST and HPCAL1 in NMI-UTUC and MI-UTUC tissues in WCM UTUC cohort.
(I) ROC curves of classifier model in predicting NMI-UTUCs and MI-UTUCs in 70% training and 30% test set.
To filter out credible plasma signature proteins associated with NMI-UTUCs and MI-UTUCs, we performed supervised analysis (Figure 5F). We reasoned that ideal signatures should be commonly differentially abundant in the tumor tissue and plasma proteome. Therefore, we included data from our recently published proteomic landscape of 116 UCB patients.2 By performing comparative analysis, a total of 42 and 1,034 DEPs were observed at the plasma and tissue samples, respectively, among which two proteins (TST and HPCAL1) overlapped in both plasma and tissue samples (Figure 5F and Table S3). The protein abundance of TST was significantly reduced in the muscle-invasive samples compared with the normal and non-muscle-invasive samples in both the plasma and tissue cohorts (Figure 6G, Wilcoxon rank-sum test, BH P < 0.05). Notably, we found that patients showing higher expression levels of TST protein in tumors appeared to have better prognostic outcomes (Figures S3E and S3F; log-rank test, p < 0.05). HPCAL1, a member of the visinin-like subfamily of neural calcium sensors, was overexpressed in muscle-invasive samples and associated with poor patient prognosis (Figure 6G). In addition, we validated the TST and HPCAL1 RNA expression levels in MI-UTUC and NMI-UTUC tissue samples in the Weill Cornell Medicine (WCM UTUC) cohort.40 As a result, consistent with our plasma proteomic data, HPCAL1 was confirmed to be over-represented in MI-UTUC tissue samples, whereas TST was over-represented in NMI-UTUC tissue samples (Figure 6H; Wilcoxon rank-sum test, BH P < 0.05).
Figure 6.

Clinical features associated with proteomic profiles
(A) Pairwise Spearman correlation of proteins and clinical variables for UTUC plasma discovery cohort, resulting in matrix of correlation coefficients where each variable is compared to all others. Main clusters are functionally annotated with keywords.
(B) Magnified area highlights fibrinogen (FIB) (red) and 16 proteins, quantified using plasma proteome profiling (black).
(C) Volcano plot shows correlation between plasma FIB and protein abundance.
(D) Pathways enriched for proteins significantly positively or negatively correlated with FIB.
(E) Heatmap of plasma FIB level and protein abundance significantly positively correlated with FIB.
(F) (Left) Heatmap of lymph node involvement (LNI) score and abundance of 14 proteins highly positively associated with LNI. (Right) The prognostic risk scores (hazard ratios) of each protein.
We employed stepwise logistic regression to identify a subset of proteins that discriminated between NMI-UTUCs and MI-UTUCs (termed NMI/MI-sig), which is robust to noise and overfitting (STAR Methods). To train and subsequently test the classifier, samples were partitioned based on sample type (i.e., NMI-UTUCs or MI-UTUCs), and 60% and 40% were used as the training and testing sets, respectively. Based on the NMI/MI-sigs (n = 12) (Figure S3G and Table S3), we applied 10-fold cross-validation to the 60% of training samples, yielding a classifier model with a mean ROC-AUC of 0.865. To further validate the classifier model, we employed the targeted MS approach, the parallel reaction monitoring (PRM) assay, which has been adopted in classifier validation in recent proteomic studies,35,41 to quantify the proteins of the classifier model in the validation cohort with 89 UTUC plasma samples (STAR Methods). We then selected a set of target peptides unique to NMI/MI-sigs using the library search results (Table S3). The fragment total areas of targeted peptides reported by Skyline-daily (4.2.1.19004, University of Washington, USA) were used to quantify these proteins. Based on the PRM quantification, the mean ROC-AUC was 0.75 (Figure S3G). Furthermore, we also identified the model by using more splits of 80/20 or 70/30 in the discovery cohort. As with the modeling method of the 60/40 training/testing sets, we employed stepwise logistic regression in the 80% training set to identify a subset of proteins. We found that the 12 NMI/MI-sigs from 80% training set overlapped highly with that of the 60% training set, with only two proteins not included in the 60% training set of NMI/MI-sigs (Figure S3H). Based on the 12 NMI/MI-sigs from the 80% training set, the predictive model achieved a mean ROC-AUC of 0.876 and 0.73 in training set and testing set, respectively (Figure S3I). The same modeling method was applied to the 70/30 training/testing sets and identified a subset of proteins. We found that 11 out of 12 NMI/MI-sigs from the 70% training set were coincident with the NMI/MI-sigs from the 60% training set (Figure S3J). Based on the 12 NMI/MI-sigs from the 70% training set, the predictive model achieved a mean ROC-AUC of 0.864 and 0.748 in the training set and testing set, respectively (Figure 6I). The predictive model was also confirmed in the validation cohort and achieved a mean ROC-AUC of 0.752 (Figure S3K). In conclusion, the classifier could be a potential predictive model to distinguish between MI-UTUCs and NMI-UTUCs.
Clinical features associated with proteomic profiles
To evaluate the association between each factor and oncological outcomes, we performed a Cox regression (Figure S4A). The univariate Cox regression results showed that plasma fibrinogen (FIB), a routine coagulation parameter, was associated with both poor overall survival (OS) and inferior PFS (Figure S4B and STAR Methods), which was consistent with the previous study.42,43 To evaluate whether the FIB was associated with other plasma proteins, we employed a correlation analysis of the plasma proteome.44,45 Filtering for a quantitative data completeness of at least 70%, pairwise correlation of the FIB and the quantified proteins resulted in a data matrix with 883 proteins and 55 continuous clinical variables. Correlating all variables with each other followed by hierarchical clustering produced a correlation map with four main clusters (clusters 1–4) (Figure 6A). Cluster 1 was chiefly enriched for proteins with the gene ontology (GO) term “acute-phase response and complement and coagulation cascades” as well as other significant terms. Cluster 2 was enriched for proteins with the GO terms “glycometabolism” and “mitotic cell cycle.” Cluster 3 was enriched for proteins with the GO terms “immunoglobulin” and “B cell receptor,” suggesting that these proteins originated from immune cells. Cluster 4 was enriched for proteins with the GO terms “antigen processing and presentation” and “phagosome.” Specifically, cluster 1 contained the FIB associated with poor prognosis (Figure 6B).
We used the same proteins and continuous clinical variables to perform weighted gene correlation network analysis (WGCNA) (STAR Methods), which was an unsupervised manner to identify groups of co-regulated proteins and the association with clinical variables.46 Consistent with the result of hierarchical clustering, four modules were yielded (Figure S4C). In addition, the pathways enriched in the modules presented results similar to those of the hierarchical clustering (Figure S4D). For example, the proteins in MEblue module were mainly participating in immunoglobulin production and B cell receptor, which was consistent with cluster 3. The proteins in MEgrey module were enriched in antigen processing and presentation, consistent with cluster 4. Specifically, the MEbrown module was significantly correlated with FIB (Figure S4C).
We observed that FGG, FGA, and FGB quantified by plasma proteome profiling were the proteins with the strongest correlation with FIB, which demonstrated the reliability of the plasma proteomic data (Figures 6B and S4E; Table S4). In addition, we found that the proteins participating in inflammation (SAA1, CRP, and SAA2) and cell migration (ITGB2) were positively correlated with FIB (Figures 6C and 6E). High expression levels of these proteins were associated with a poor prognosis (Figure S4F). It has been reported that fibrinogen and platelets facilitate each other in protecting tumor cells from natural killer cytotoxicity by forming thrombin, which depends on β-integrins expressed on human cancer cells.47 Further pathway enrichment analysis showed that the proteins significantly positively correlated with FIB (n = 250) were enriched in platelet activation, acute-phase response, and cell migration pathways, whereas the proteins significantly negatively correlated with FIB (n = 139) were enriched in cellular oxidant detoxification pathways (Figures 6D and 6E; Table S4).
UTUC mainly spreads by direct invasion/extension and via lymphatics. Elevated FIB was significantly associated with lymph node involvement (LNI) but showed a weak correlation of 0.12 (Figure 6E). To identify proteins that were more predictive of LNI, we correlated all quantified plasma proteins directly to LNI. We generated an LNI score for our samples based on 14 proteins that were highly positively associated with LNI (Figure 6F and Table S4; STAR Methods). As expected, the LNI score showed a correlation of 0.41, which was better than FIB’s weak correlation of 0.12 (Figure 6F; Spearman’s r = 0.41, p = 3.5 × 10−15). Taken together, the protein panel was associated with lymph node metastasis, which is of interest to identify high-risk and poor-prognosis UTUC patients.
Progression clock of patients with UTUC identified by machine learning
The prediction of the UTUC progression clock is essential for disease treatment planning and medication management but remains one of the challenging tasks in the medical field. We sought to build a progression clock to predict the progression time based on individual plasma samples. To screen survival-related proteins, we determined the correlation between PFS and the protein expression level. The result showed that 461 and 365 proteins exhibited a significantly positive and significantly negative correlation with PFS (Figure 7A and Table S5). Proteins showing a significantly positive correlation with PFS were mainly associated with the acute-phase response, cell adhesion, and protein transport pathways (Figure 7B).
Figure 7.

Progression-related protein classifier of UTUC patients
(A) Volcano plot of proteins correlated with PFS.
(B) Pathways enriched for proteins significantly positively or negatively correlated with PFS.
(C) Forest plots of the univariate Cox hazard model for PFS.
(D) AUC was 0.742, 0.816, and 0.877 at 1, 3, and 5 years, respectively, in UTUC discovery cohort.
(E) Calibration curves of nomograms between predicted and observed 1-, 3-, and 5-year PFS in discovery UTUC cohort.
(F) AUC was 0.812, 0.88, and 0.905 at 1, 3, and 5 years, respectively, in validation cohort using PRM assays.
To explore the prognostic value of proteins, we constructed a univariate Cox hazards model to filter the proteins significantly related to PFS. The subsequent lasso Cox regression successfully selected ten highly relevant proteins (variables) from among 93 proteins (univariate Cox regression: p < 0.05; Table S5), obtaining the results of ten proteins (Figures S5A and S5B). We then identified the following ten proteins as predictors of progression in UTUC patients: ARIH1, EIF4H, PPP5C, NDUFAF3, LRG1, NXF1, SNRPB, AK2, TMPO, and IFITM3 (Figure S5B). Subsequently, we established the PFS score by multivariate Cox proportional hazards regression model including the ten proteins screened by lasso regression (STAR Methods). Additionally, the forest plot of the associations between each protein and PFS is shown in Figure 7C.
To determine the potential clinical usefulness of the model, we next tested whether plasma sample data could be used in the predicting protein model to predict the progression time in UTUC patients. For the progression clock operating characteristic analysis of the PFS score, the AUC was 0.742, 0.816, and 0.877 at 1, 3, and 5 years, respectively (Figure 7D). Subsequently, we integrated the ten proteins to construct a nomogram (Figure S5C). Based on the calibrated plots, the predicted 1-, 3-, and 5-year survival probabilities of the nomogram performed well in our cohort (Figure 7E), and the concordance index (C index) of the nomogram was 0.7387. Additionally, decision curve analysis (DCA) was used to assess the predicting nomogram’s differential advantage (Figure S5D), and DCA displayed a net benefit of the nomogram, showing that it had good clinical utility.
We further evaluated the accuracy of the progression clock model in the independent cohort (n = 89) using PRM assays. We selected a set of target peptides that were unique to these proteins using the library search results (Table S5). Based on the PRM quantification, the AUC was 0.812, 0.880, and 0.905 at 1, 3, and 5 years, respectively (Figure 7F). Interestingly, the accuracy of predictions substantially improves for more distant time points into the future, which is worthy of further analysis. This indicates progression-related plasma protein features as a potential predictive model to predict the progression time on the basis of blood samples from UTUC patients.
Discussion
Although diagnosis of UTUC has been substantially improved with minimally invasive procedures using advanced endoscopic devices, there remain risks for adverse events such as ureter injury, infection, and difficultly detecting early progression of diseases.48 Urine cytology also fails to detect the majority of UTUCs.3 In this regard, easily available plasma proteins would be a plausible target of diagnostics, and only a few reports have investigated its potential for the monitoring and diagnosis of UTUC.49 In this study, we investigated the plasma proteomic landscape of UTUC patients and healthy controls and provided predictive models that could potentially enhance the diagnosis and management of UTUC.
Proteins that are present in plasma originate from a variety of sources. Solid tissues, especially from intestine and liver, secrete a large number of proteins that execute their functions in the plasma.50 In this study, the plasma proteome captured more than 50% of kidney-associated proteins, and these intracellular proteins could serve as signature proteins reflecting tissue damage actually induced by tumor formation.51 Furthermore, to further explore the dynamic changes in response to major changes in tumor burden, we collected post-surgery and post-chemotherapy plasma samples matched with their paired pre-surgery and pre-chemotherapy plasma samples. Functional enrichment analysis showed that the proteins downregulated in post-surgery samples were involved in fatty acid degradation and tRNA aminoacylation, indicating that these pathways decreased alongside the diminishing tumor burden. Conversely, the proteins upregulated in post-surgery samples were mainly enriched in leukocyte transendothelial migration and platelet activation, delineating the stress response to surgery (Figures S6A–S6H). Similar trends were also observed in post-chemotherapy plasma samples (Figures S6I–S6N). The plasma partially returned to normal after the surgery or chemotherapy, and the changes linked to the tumor burden were captured.
We also performed differential expression analysis of UTUC and normal samples and constructed a classifier to identify UTUC patients. Because the UTUC samples contained RPUC, UUC, and a combination of both cancers, we investigated whether a different distribution of cohort assignments with possibly a different mixture has an effect on the performance of a proteomics-based prediction model. First, we divided the UTUC samples into RPUC (n = 163), UUC (n = 165), and combined (n = 34), and evaluated the performance of the model. The results showed that the AUC of RPUC (1:0:0), UUC (0:1:0), and combined samples (0:0:1) was 0.938, 0.935, and 0.943, respectively (Figures S7A–S7C). Furthermore, we remixed the UTUC samples in a different ratio of 1:1:1 and 2:4:1 (UUC/RPUC/combined) in the discovery cohort, where the AUC was 0.932 and 0.942 (Figures S7D and S7E), respectively. Additionally, we remixed the UTUC samples in a ratio of 1:1:1 and 2:4:1 (UUC/RPUC/combined) in the validation cohort, where the AUC was 0.898 and 0.906, respectively (Figures S7F and S7G). The data showed that the different distribution of cohort assignments with possibly a different mixture would not have an effect on the final results.
Exosomes, since being discovered as signaling carriers and communication media, have positioned themselves at the forefront of cancer research in the last decade.28 In this study, we found that 18 proteins upregulated in both UTUC plasma and UTUC EVs might serve as potential signature proteins for UTUC patients and warranted further validation. Additionally, urine plays a crucial role in the early diagnosis and forecast status of clinical diseases. Our findings supported the presence of three proteins that were only detected in urine samples from NMI-UTUCs, suggesting a potential association with NMI-UTUCs. Taken together, we performed an integrated analysis, including the proteome of tissue, plasma, EVs, and urine. The dysregulated proteins identified in different compartments partially overlapped, and each compartment yielded a different UTUC-associated protein expression signature.
The key prognostic factor at the time of diagnosis of UTUC is whether the tumor is in the muscle-invasive or non-muscle-invasive stage.8 However, urine cytology has been found to perform poorly in the prediction of muscle-invasive or high-grade disease in UTUC (sensitivity 56% for high-grade tumors, sensitivity 62% for muscle-invasive UTUC).52 Hurel et al. combined gender, locally advanced stage on pre-operative workup, and positive cytology to predict MI-UTUCs with an accuracy of 65.3%.53 We constructed a plasma protein model to predict MI-UTUCs with >80% sensitivity/specificity. The model was further validated in an independent cohort. In addition, we found some proteins (e.g., ABLIM3, SCGN, PYGO2) exclusively expressed in MI-UTUC or NMI-UTUC samples. Nevertheless, the identification frequency of these proteins was low owing to the sample itself or instrument sensitivity, and more validation is needed to confirm these proteins.
Through correlation analysis, we deciphered the relationship between clinical indicators and tumors. For example, we observed that FIB was associated with poor prognosis and lymph node metastasis, which was proved in a previous study,43 but the mechanism was unclear. By the identification of plasma proteins, we added nearly 2,000 protein features to each sample, which allowed us to associate clinical indicators with plasma proteins in networks. We identified a cluster of four proteins of special interest, SAA1, CRP, SAA2, and ITGB2, which was significantly correlated with FIB. The proteins participating in inflammation and cell migration were linked to the pathogenesis of urologic cancers or identified as a risk factor for the disease.54,55,56 These proteins in the network provided a basis for investigating the potential mechanism of FIB and tumor progression.
Predicting the progression time for individual patients could improve their clinical outcomes.57 Recently, several prognostic models have been developed to predict oncological outcomes in UTUC patients.58,59,60 However, these accurate prognostic models were based on clinical features. In this study, we built a progression-related protein model using the lasso-Cox method to predict the progression clock based on the plasma proteome. The protein panel underlying the progression-related protein model was selected in an unbiased manner, driven by the proteins’ diagnostic performance. We further proved that the progression clock model was an indicator for the 1-, 3-, and 5-year survival of UTUC patients, and the model was also verified in an independent cohort by PRM assays. These results indicated that progression-related plasma protein features could be a potential predictive model to predict the progression time on the basis of plasma samples from UTUC patients.
In conclusion, in this study we captured the changes in the plasma proteome of UTUC patients that were linked to the pathological features and clinical manifestations of underlying disease. We demonstrated that plasma proteome profiling enables the discovery of molecular signatures related to disease, which could have a major impact on important aspects of disease management: (1) the diagnostic classifier might help diagnose UTUC, (2) the potential predicted model could distinguish between MI-UTUCs and NMI-UTUCs, and (3) the progression clock could provide information about disease progression.
Limitations of the study
The single-center cohort study conducted at the Fudan University Shanghai Cancer Center was retrospective and included only Chinese patients, which may lead to potential selection bias. Additional prospective studies are needed to validate our findings in multiple centers and cohorts of other ethnicities. The signature proteins may not be tumor specific but could have stemmed from other affected organs or may be indirectly induced by the effects of the tumors on their microenvironment or even systemically. More solid methods are needed to validate the signature proteins from this study. The progression clock contained a ten-protein classifier that needs to be further investigated in future confirmatory trials to advance the mechanistic understanding of UTUC progression. Follow-up research on the biological mechanisms is still needed in future studies to guide clinical management and develop drug targets.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Plasma/Urine/Tissue/EVs samples | The Department of Urology of Fudan University Shanghai Cancer Center (FUSCC) | This paper |
| Antibodies | ||
| Calnexin Rabbit mAb | ABclonal | Cat# A4846; RRID: AB_2863359 |
| CD9 Rabbit mAb | ABclonal | Cat# A19027; RRID: AB_2862519 |
| HSP70 Rabbit mAb | ABclonal | Cat# A21180; RRID: AB_2943041 |
| TSG101/VPS23 Rabbit mAb | ABclonal | Cat# A5789; RRID: AB_2863517 |
| Chemicals, peptides, and recombinant proteins | ||
| HPLC-grade water | J.T. Baker | Catalog: 4218-03 |
| Phenylmethylsulfonyl fluoride | Amresco | Catalog: M145 |
| Dithiothreitol | Sigma | Catalog: 43815 |
| Iodoacetamide | Sigma | Catalog: V900335 |
| Formic acid | Sigma | Catalog: 507 |
| Trypsin | Promega | Catalog: V528A |
| Ammonium bicarbonate | J.T. Baker | Catalog: 3003-01 |
| Acetonitrile | J.T. Baker | Catalog: 9829-03 |
| Iodoacetamide | Sigma | Catalog: V900335 |
| Dithiothreitol | Sigma | Catalog: 43815 |
| qEVoriginal | IZON | Catalog: ICO-70 |
| Instrumentation | N/A | |
| Q Exactive HF-X Hybrid | N/A | |
| Quadrupole-Orbitrap | ThermoFisher | Catalog: 726042 |
| Mass Spectrometer | N/A | |
| EASY-nLC 1200 system | ThermoFisher | Catalog: LC140 |
| Vacuum centrifuge | Eppendorf | Catalog: 07-748-15 |
| Deposited data | ||
| Proteomic data | This paper | iProx: IPX0005194000 |
| Fudan bladder cancer cohort | Xu et al.2 | PMID: 35659036 |
| TCGA cohort | Robertson et al., 201761 | PMID: 30096301 |
| The human protein atlas database (version: 21.1, released 2022-05-31) | Human protein atlas (HPA) | https://www.proteinatlas.org/ |
| Software and algorithms | ||
| R version v 3.5.1 | The R Project for Statistical Computing |
https://www.r-project.org/ |
| RStudio (v1.2.5019) | R Studio Team, 2015 | https://rstudio.com/products/rstudio/download/ |
| GraphPad Prism (version 8.0.1) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Firmiana | Feng et al.62 | https://phenomics.fudan.edu.cn/firmiana/gardener/ |
| Mascot (version 2.4) | Matrix Science | http://www.matrixscience.com/ |
| UniProt human protein database (released on 09-06-2021) | The UniProt Consortium |
https://www.uniprot.org/ |
| ConsensusPathDB | Herwig et al., 201663 | http://cpdb.molgen.mpg.de/CPDB/ |
Resource availability
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Chen Ding (chend@fudan.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Participant recruitment and clinical data collection
In this study, plasma samples from all participants in two independent cohorts (Cohort 1 and 2) were recruited from the Department of Urology, Fudan University Shanghai Cancer Center (FUSCC). Cohort 1 was the discovery cohort consisting of 239 healthy controls and 362 UTUC patients. Cohort 2 was the validation cohort consisting of 89 UTUC patients. We also collected 10 plasma corresponding UTUC tumor tissue samples and 10 normal tissue samples from the Department of FUSCC. The normal tissue samples were normal urothelial epithelia obtained from 10 patients with kidney cancer undergoing nephrectomy at the FUSCC. The healthy controls matched for age, gender and BMI were peripheral blood samples for physical examination from healthy persons. We excluded control samples in case of any medication or any cancer among other exclusion criteria (Table S1). Patients with UTUC who did not undergo any anti-cancer treatments prior to surgery were randomly selected from April 2013 to September 2022 on their first visit. Clinical information of patients, including tumor location, gender, age, tumor node metastasis (TNM) staging, and routine blood test results, is listed in Table S1. All the samples were stored at −80°C until sample processing. The study was compliant with the ethical standards of Helsinki Declaration II and was approved by the institutional review board of FUSCC (050432-4-1911D). Written informed consent was obtained from each patient before any study-specific investigation was conducted.
Method details
Pathology review
The tissue samples were systematically evaluated by three expert genitourinary pathologists to confirm the histopathologic diagnosis according to the World Health Organization (WHO) classification. Additionally, the tumor tissues samples were assessed for tumor content, the presence and extent of tumor necrosis, and signs of invasion into the muscularis propria. Tumor sections were required to contain an average of 70% tumor cell nuclei with equal to or less than 20% necrosis for inclusion in the study. Any non-concordant diagnoses among the three pathologists were re-reviewed, and a resolution was reached following discussion. Each sample was assigned a new research ID, and the patient’s name or medical record number used during hospitalization was de-identified.
Plasma protein extraction and trypsin digestion
Plasma samples were mixed with 100 μL 50 mM ammonium bicarbonate (ABC) buffer, and the proteins were inactivated at 95°C for 5 min. The samples were cooled to room temperature, digested using trypsin at an enzyme to protein mass ratio of 1:25 for 17 h in a 37°C incubator. Then, 5 μL aqueous ammonia was added to each tube, vortexed to quench the digestion reaction, and the supernatant was subsequently dried using a 60°C vacuum drier (SpeedVac, Eppendorf). Then, the peptides were dissolved in 100 μL 0.1% formic acid (FA), followed by vortexing for 3 min, and then sedimentation for 3 min (12,000 ×g). The supernatant was picked into new tube and then desalinated. Before desalination, the activation of pillars with 2 slides of 3M C18 disk is required, and the lipid is as follows: 90 μL 100% acetonitrile (ACN) twice, 90 μL 50% and 80% ACN once in turn, and then 90 μL 50% ACN once. After pillar balance with 90 μL 0.1% FA twice, the supernatant of the tubes was loading into the pillar twice, and decontamination with 90 μL 0.1% FA twice. Lastly, 90 μL elution buffer (0.1% FA in 50% ACN) was added into the pillar fir elution twice and only the effluent was collected for MS. Finally, the collected peptides were dried using a 60°C vacuum drier.
Tissue protein extraction and trypsin digestion
The tissue samples were minced and lysed in lysis buffer (8 M urea, 100 mM Tris-HCl, pH 8.0) containing protease and phosphatase inhibitors (Thermo Scientific) and then sonicated for 1 min (3s on and 3 s off, amplitude 25%). The lysates were centrifuged at 14,000×g for 10 min and supernatants were collected as whole-tissue extracts. Protein concentrations were determined by the Bradford protein assay (TaKaRa, T9310A). Extracts were reduced with 10 mM DTT (dithiothreitol) at 56°C for 30 min and alkylated with 10 mM iodoacetamide at room temperature in the dark for 30 min. Protein samples were then digested with trypsin using a filter-aided sample preparation method64 at 37°C overnight with an enzyme to protein mass ratio of 1:25. Peptides were dried using a 60°C vacuum drier.
Urine protein extraction and trypsin digestion
The 200 μL urine supernatant samples were heated at 99°C for 10 min to inactivate the proteins. The samples were cooled to room temperature and was loaded into a 10 kD Microcon filtration device (Millipore). Adsorbed proteins were washed two times with 100 μL of 50 mm ABC buffer (12,000 × g for 20 min). The samples were digested using trypsin at an enzyme to protein mass ratio of 1:25 for 17 h in a 37°C incubator. Peptides were dried using a 60°C vacuum drier.
EVs purification from plasma samples
The ultracentrifugation and size exclusion chromatography (SEC) was used to isolate EVs from plasma samples, which is a widely adopted method for the isolation of EVs.31,32,33 For ultracentrifugation, the centrifugal force used typically ranges from ∼100,000 to 120,000 × g. Before the start of isolation, a cleaning step is usually carried out for samples to rid of large bioparticles in a sample and the sample is spiked with protease inhibitors to prevent the degradation of proteins.65 After rinsing the columns with phosphate buffered saline (PBS), samples were applied on top of a qEVoriginal column (Izon Science).30 After removal of buffer volume (Fractions 1–6, 3.0 mL), the EVs-rich Fractions 7–9 (purified collection volume, 1.5 mL) were pooled. Then purified EVs samples (50 μL) were obtained by concentrating using an Amicon Utra-4-10k centrifugal filter device (Merck Millipore). The EVs protein concentration was measured by BCA (bicinchoninic acid) (Pierce, Thermo Fisher Scientific). The EVs size and particle number were analyzed using the LM10 nanoparticle characterization system (NanoSight, Malvern, Instruments) equipped with a violet laser (405 nm). Isolated EVs were fixed with 2% paraformaldehyde and then spread onto glow discharged Formvar-coated copper mesh grids and stained with 2% uranyl acetate for 5 min, post drying, grids were photographed under a JEOL 100 CX electron microscope operated at 80 kV. The presence of EV-enriched proteins determined by western blotting in 10 μg of lysates using the following antibodies: Calnexin (A4846, ABclonal, dilution 1: 1000), CD9 (A19027, ABclonal, dilution 1: 1000), HSP70 (A21180, ABclonal, dilution 1: 1000), TSG101 (A5789, ABclonal, dilution 1: 1000).
EVs protein extraction and trypsin digestion
The EVs samples were lysed in TCEP buffer (2% deoxycholic acid sodium salt, 40 mM 2-chloroacetamide, 100 mM Tris-HCl, 10 mM Tris(2-chloroethyl) phosphate, and 1 mM PMSF mixed with MS water, pH 8.5), and heated in a metal bath at 99°C for 30 min. After cooling to room temperature, trypsin was added and digested for 17 h at 37°C. 10% FA was added to each tube, vortexed for 3 min, and then centrifuging 12, 000g for 5 min. The supernatant was collected in a new 1.5 mL tube and dried using a 60°C vacuum drier. Then, the peptides were dissolved in 100 μL 0.1% FA, followed by vortexing for 3 min, and then sedimentation for 5 min (12,000 ×g). The supernatant was picked into new tube and then desalinated. Before desalination, the activation of pillars with 2 slides of 3M C18 disk is required, and the lipid is as follows: 90 μL 100% ACN twice, 90 μL 50% and 80% ACN once in turn, and then 90 μL 50% ACN once. After pillar balance with 90 μL 0.1% FA twice, the supernatant of the tubes was loading into the pillar twice, and decontamination with 90 μL 0.1% FA twice. Lastly, 90 μL elution buffer (0.1% FA in 50% ACN) was added into the pillar fir elution twice and only the effluent was collected for MS. Finally, the collected peptides were dried using a 60°C vacuum drier.
LC-MS/MS analysis
The acquisition of samples was randomized to avoid bias. The plasma samples and urine samples were measured using LC-MS instrumentation consisting of an EASY- nLC 1200 ultra-high-pressure system (Thermo Fisher Scientific) coupled via a nano-electrospray ion source to a Q Exactive HF-X Hybrid Quadrupole-Orbitrap mass spectrometer (all Thermo Fisher Scientific). Eluted peptides were separated at 60°C on 150 μm I.D. × 8 cm column (C18, 1.9μm, 120 Å, Dr. Maisch GmbH). Mobile phases A and B were 99.9/0.1% water/FA (v/v) and 80/20/0.1% ACN/water/FA (v/v/v). The MS analysis was performed in a data-independent acquisition (DIA) mode. The DIA method consisted of MS1 scan from 300 to 1,400 m/z at 30,000 resolution and the automatic gain control (AGC) target 3e6 or a maximum of 20 ms. Then, 30 DIA segments were acquired at 15,000 resolution with an AGC target 1e6 or 22 ms for maximal injection time. The setting “inject ions for all available parallelizable time” was enabled. Higher energy collision dissociation (HCD) fragmentation was set to normalized collision energy of 27%. The spectra were recorded in profile mode. The default charge state for the MS2 was set to 3.
The tissue samples and EVs samples were analyzed on Easy-nLC 1200 liquid chromatography system (Thermo Fisher Scientific) coupled to an Orbitrap Exploris 480 via a nano-electrospray ion source (Thermo Fisher Scientific). The dried peptides were redissolved in 20 μL loading buffer (0.1% FA), and 10 μL of the sample was loaded onto a trap column (100 μm × 2 cm, home-made; particle size, 3 μm; pore size, 120 Å; SunChrom) with a maximum pressure of 280 bar using solvent A, then separated on home-made 150 μm × 12 cm silica microcolumn (particle size, 1.9 μm; pore size, 120 Å; SunChrom) with a gradient of 5–35% mobile phase B (ACN and 0.1% FA) at a flow rate of 600 nL/min for 150 min. The eluted peptides were ionized and detected using high-field asymmetric waveform ion mobility spectrometry coupled with OE 480 MS (Thermo Fisher Scientific). The DV was set to −45 V and −65 V. Mass spectrometry was performed in data-dependent acquisition (DDA) mode. For the MS1 Spectra full scan, ions with m/z ranging from 300 to 1,400 were acquired by an Orbitrap mass analyzer at a high resolution of 120,000. The AGC target value was set to 3E+06. The maximal ion injection time was 80 ms. MS2 spectral acquisition was performed in the ion trap in a rapid speed mode. Precursor ions were selected and fragmented with HCD with a normalized collision energy of 27%. Fragment ions were analyzed by an ion trap mass analyzer with an AGC target at 5E+04. The maximal ion injection time of MS2 was 20 ms. Peptides that triggered MS/MS scans were dynamically excluded from further MS/MS scans for 12 s.
MS database searching
Peptide identification and protein quantification
All data were processed using “Firmiana” (a one-stop proteomic cloud platform, https://phenomics.fudan.edu.cn/firmiana/).62 The data were search against UniProt human protein database (updated on 2019.12.17, 20406 entries) using FragPipe (v12.1) with MSFragger (2.2) (DIA data)66 and Mascot search engine (DDA data). The mass tolerances were 20 ppm for precursor and 50 mmu for product ions. Up to two missed cleavages were allowed. The search engine set cysteine carbamidomethylation as a fixed modification and N-acetylation and oxidation of methionine as variable modifications. Precursor ion score charges were limited to +2, +3, and +4. The data were also searched against a decoy database so that protein identifications were accepted at a false discovery rate (FDR) of 1%. The results of DDA data were combined into spectra libraries using SpectraST software. A total of 327 libraries were used as reference spectra libraries.
DIA data was analyzed using DIA-NN (v1.7.0).67 The default settings were used for DIA-NN (Precursor FDR: 5%, Log lev: 1, Mass accuracy: 20 ppm, MS1 accuracy: 10 ppm, Scan window: 30, Implicit protein group: genes, Quantification strategy: robust LC (high accuracy)). Quantification of identified peptides was calculated as the average of chromatographic fragment ion peak areas across all reference spectra libraries. Label-free protein quantifications were calculated using a label-free, intensity-based absolute quantification (iBAQ) approach.68 We calculated the peak area values as parts of corresponding proteins. The fraction of total (FOT) was used to represent the normalized abundance of a particular protein across samples. The FOT values was defined as a protein’s iBAQ divided by the total iBAQ of all identified proteins within a sample. The FOT values were multiplied by 105 for the ease of presentation.
Quality control of the mass spectrometry data
For quality control of performance of mass spectrometry, the mixture of all plasma samples from UTUC patients were measured every 20 samples, which was adopted in proteomic studies.21,69 The quality control (QC) plasma samples were analyzed using the same method and conditions as our plasma cohort. A Pearson’s correlation coefficient was calculated for all QC runs. The average Pearson’s correlation coefficient of QC plasma samples in this study was 0.96 (range, 0.95–1.00) (Figure S1D; Table S1), demonstrating consistent stability of the mass spectrometry platform. Assessed on the quality control samples, median workflow coefficient of variation was 22% (Figure S1C).
Targeted PRM analysis
To evaluate the accuracy of the classifiers, we designed parallel reaction monitoring (PRM) strategy to quantify the classifier proteins in plasma samples from the independent cohort composed of 89 UTUC patients. Using the library search results, a set of target peptides that unique to the classifier proteins were selected, including distinguishing between muscle-invasive and non-muscle invasive UTUC classifier proteins, and predicting progression-free survival classifier proteins. Besides, house-keeping proteins, such as VCP, RPLP0, PSMB4, were also included for the reference. Equal amount of plasma from each sample (the validation cohort with 89 UTUC patients) was digested as described in the part of profiling preparation. Peptide samples were injected into the Q Exactive HF-X Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific) operating in PRM mode with quadrupole isolation and HCD fragmentation. The mobile phase buffers were the same as the buffers mentioned in the method of DIA section. The full MS mode was measured at resolution 60,000 with AGC target value of 3E6 and maximum IT of 20 ms, with scanning range of 300–1400 m/z. Target precursors were then isolated through an m/z window of 1.2 Th, followed by fragmentation at 27% normalized collision energy. The product ions were scanned with a resolution of 15,000, AGC target value of 1E6 or a maximum injection time of 25 ms.
Raw data was searched by Skyline-daily (4.2.1.19004, University of Washington, USA). The proteins were quantified with the fragment total area reported by Skyline-daily. We selected peptides and tested their stability of signal and shape of peaks in the pool sample for final quantification, and referred to the ranking offered by skyline.
Quantification and statistical analysis
Missing value imputation
For the proteomic data, FOTs multiplied by 1E5 were used for quantification, and missing values were imputed with 1E-5 and finally, log2 transformed, if necessary.
Differential protein analysis
Proteins expressed in more than 30% of the samples were selected for differential expression analysis. The Wilcoxon rank-sum test was used to examine whether proteins were differentially expressed between UTUC and normal, or NMI-UTUCs and MI-UTUCs. Upregulated or downregulated proteins are defined as proteins differentially expressed in one group compared with the other group (Wilcoxon rank-sum test, BH p < 0.05, Fold change >1.5 or <0.67).
Pathway enrichment analysis
Differentially expressed proteins were subjected to Gene Ontology and KEGG pathway enrichment analysis in DAVID (https://david.ncifcrf.gov/)70 and ConsensusPathwayDB (http://cpdb.molgen.mpg.de/)63 with an FDR <0.05. We used gene sets of molecular pathways from the KEGG71/Reactome72/GO73 databases to compute pathways.
Construction and validation of predictive models to distinguish between UTUC and normal or MI-UTUCs and NMI-UTUCs
Binomial logistic regression analysis was used to construct the distinguishing between UTUC and normal or MI-UTUCs and NMI-UTUCs prediction model based on the significantly differentially expressed proteins in UTUC and Normal or MI-UTUCs and NMI-UTUCs plasma samples using in the R software v3.5.1. The backward stepwise method was utilized to feature selection. Samples were randomly divided into the training set and the testing set.74 Moreover, the diagnostic value of this model was verified using ROC analysis (pROC R package version 1.16.2 and Caret R package version 6.0–86). Sensitivity, specificity, accuracy, and AUC were used to determine predictive values. The predictive model was validated in validation cohort.
WGCNA analysis
Weighted gene correlation network analysis (WGCNA)46 was used to identify groups of co-regulated genes in an unsupervised manner. A sample network was constructed to identify outlying samples with a standardized connectivity score of less than −2.5. A signed gene co-expression network was constructed with a soft threshold power of 10. Groups of co-regulated genes (modules) correlated with each other with a Pearson correlation coefficient of 0.9, or better, were merged. Pathway enrichment analysis was used for the functional annotation of the identified modules. The eigengenes of each module were used to measure the association between modules and clinical information.
Lymph node involvement (LNI) score
Single-sample gene set enrichment analysis (ssGSEA)75 was utilized to obtain score for each sample based on proteomic data using the R package GSVA.76 Correlations between the plasma proteins and lymph node involvement were determined using Spearman’s correlation. The inferred lymph node involvement (LNI) score was performed using ssGSEA implemented in the R package GSVA.
Risk score
The risk scores (hazard ratios) of each protein were calculated based on the overall survival or progression-free survival. The hazard ratios and their 95% confidence intervals (CIs) for each protein were based on Cox proportional hazards models by the R package “survival”.
Survival analysis
Kaplan-Meier survival curves (log rank test) were used to determine the overall survival (OS) and progression-free survival (PFS) of patients with muscle-invasive versus non-muscle invasive and patients with high plasma fibrinogen versus low plasma fibrinogen. The coefficient value, which is equal to ln (HR), was calculated using Cox proportional hazards regression analysis. P-values less than 0.05, were considered significantly different and selected for Cox regression multivariate analysis. Prior to the log rank test of a given protein, survminer (version 0.2.4, R package) with maxstat (maximally selected rank statistics; http://r-addict.com/2016/11/21/Optimal-Cutpoint-maxstat.html) was used to determine the optimal cut-off point for the selected samples according to a previous study.77 OS and PFS curves were then calculated based on the optimal cut-off point. The 95% pointwise CIs for the two groups of subjects were produced by the function ggsurvplot () [in Survminer R package].
Multivariate Cox proportional hazards regression analysis
Multivariate Cox proportional hazards regression analysis was used to establish the progression-free survival-predicting model for UTUC. The results were plotted using a forest map with the R package “ggplot2”. The PFS score for each UTUC patient was calculated using the formula: PFS score = Σβi∗Xi (Xi represents the screened protein expression, and βi represents the corresponding coefficient).
Statistical analysis
Standard statistical tests were used to analyze the clinical data, including but not limited to Student’s t test, Wilcoxon rank-sum test, Chi-square test, Fisher’s exact test, Kruskal-Wallis test, Log rank test. The Wilcoxon rank-sum test was used to examine whether proteins were differentially expressed between UTUC and normal, or NMI-UTUCs and MI-UTUCs. The Kruskal-Wallis test was used to test whether proteins were differentially expressed among the three groups (Normal, NMI-UTUC, and MI-UTUC). For correlation analysis, Spearman or Pearson correlation was used. All statistical tests were two-sided, and statistical significance was considered when p value <0.05. To account for multiple-testing, the p values were adjusted using the Benjamini-Hochberg FDR correction. Kaplan–Meier plots (Log rank test) were used to describe OS and PFS probabilities. Variables associated with overall survival and progression-free survival were identified using univariate Cox proportional hazards regression models. Significant factors in univariate analysis were further subjected to a multivariate Cox regression analysis in a forward LR manner. All the analyses of clinical data were performed in R (version 3.5.1). The p values less than 0.05, 0.01, 0.001, 0.0001 were marked with ∗, ∗∗, ∗∗∗, ∗∗∗∗, respectively. All the statistical analysis had been checked by two statisticians.
Acknowledgments
This work is supported by National Key R&D Program of China (2019YFC1316005, 2022YFA1303200, 2022YFA1303201, 2020YFE0201600, 2018YFE0201600, 2018YFE0201603, 2018YFA0507500, 2018YFA0507501, 2017YFA0505100, 2017YFA0505102, 2017YFA0505101, 2017YFC0908404, 2016YFA0502500), sponsored by Program of Shanghai Academic/Technology Research Leader (22XD1420100), Shuguang Program of Shanghai Education Development Foundation and Shanghai Municipal Education Commission (19SG02), National Natural Science Foundation of China (31972933, 31770886, 31700682, 82172817, 82172741), the Major Project of Special Development Funds of Zhangjiang National Independent innovation Demonstration Zone (ZJ2019-ZD-004), Shanghai Municipal Science and Technology Major Project (2017SHZDZX01), Natural Science Foundation of Shanghai (20ZR1413100), Shanghai "Science and Technology Innovation Action Plan" medical innovation research project (22Y11905100), Shanghai Rising-Star Program (23QA1408900), Shanghai Municipal Health Bureau Project (2020CXJQ03), Beijing Xisike Clinical Oncology Research Foundation (Y-HR2020MS-0948), Shanghai Anti-Cancer Association Eyas Project (SACA-CY21A06 and SACA-CY21B01), and the Fudan Original Research Personalized Support Project. Ethics approval and consent to participate: the study was compliant with the ethical standards of Helsinki Declaration II and was approved by the institutional review board of the Department of Urology of Fudan University Shanghai Cancer Center (FUSCC) (050432-4-1911D).
Author contributions
Conceptualization, C.D., D.Y., H.Z., and J. Zhao; experimental work and data collection, Y.Q., Z.Y., N.X., G.S., J.S., Y.C., and K.L.; data curation, J.F., Y.W., N.X., Z.Y., G.S., Y.Q., K.L., and C.D.; data analysis, Y.Q., Z.Y., N.X., G.S., D.C., M.S., H.G., Y.W., S. Tang, and J.S.; visualization, Y.Q., Z.Y., N.X., J. Zhu, F.M., S. Tang, Wenhao Xu, X.T., S. Tian, Wenbo Xu, S.Z., X.W., and C.D.; patient sample management and QC, Y.Q., Z.Y., N.X., G.S., J.S., S.Y., K.C., K.L., J.L., X.P., Y.W., and S. Tang; supervision, C.D., D.Y., H.Z., and J. Zhao; writing, Z.Y., N.X., Y.Q., G.S., J.S., and C.D.
Declaration of interests
The authors declare no competing interests.
Published: August 25, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101166.
Contributor Information
Jianyuan Zhao, Email: zhaojy@fudan.edu.cn.
Hailiang Zhang, Email: zhanghl918@163.com.
Dingwei Ye, Email: dwyeli@163.com.
Chen Ding, Email: chend@fudan.edu.cn.
Supplemental information
A. Inclusion and exclusion criteria. B. Clinical characteristics of the UTUC discovery cohort and healthy cohort. C. Clinical characteristics of the UTUC validation cohort. D. Correlation of quality control (QC) plasma samples. E. Raw expression data of identified plasma protein in the UTUC discovery and healthy control samples (identification frequency >30%).
A. List of differentially expressed proteins (Wilcoxon rank-sum test, BH P < 0.05, UTUC/normal ratio >1.5 or <0.67) in UTUC and normal plasma samples. B. Kidney-specific proteins observed in UTUC plasma. C. List of candidate proteins of classifier model in plasma samples. D. List of proteins identified in urine samples. E. List of proteins identified in EV samples. F. List of the upregulated protein (Wilcoxon rank-sum test, BH P < 0.05, UTUC/Normal ratio >1.5) overlap among UTUC tumor tissue, UTUC plasma, and UTUC urine. G. List of candidate proteins of NMI-UTUC urine samples. H. List of candidate proteins of UTUC EV samples.
A. List of differentially expressed proteins in NMI-UTUC and MI-UTUC plasma samples. B. List of differentially expressed proteins in NMI-UC and MI-UC tissue samples. C. List of targeted peptides unique to the proteins of the classifier model.
A. List of the plasma proteins correlated with plasma fibrinogen. B. List of the plasma protein panel associated with lymph node metastasis.
A. Proteins correlated with PFS. B. Hazard ratio of selected proteins. C. The correlation coefficients of ten proteins. D. List of targeted peptides unique to the proteins of model.
Data and code availability
-
•
The raw mass spectrometry (MS) proteomics data generated in this study have been deposited in the ProteomeXchange Consortium via the iProX partner repository (http://www.iprox.cn/; dataset identifier: PXD043774) under Project ID IPX0005194000. Human Protein Atlas (HPA) IHC Staining Data could be accessed at https://www.proteinatlas.org/. The remaining data are available within the Article and Supplementary Information.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Rouprêt M., Babjuk M., Burger M., Capoun O., Cohen D., Compérat E.M., Cowan N.C., Dominguez-Escrig J.L., Gontero P., Hugh Mostafid A., et al. European association of Urology guidelines on upper urinary tract urothelial carcinoma: 2020 update. Eur. Urol. 2021;79:62–79. doi: 10.1016/j.eururo.2020.05.042. [DOI] [PubMed] [Google Scholar]
- 2.Xu N., Yao Z., Shang G., Ye D., Wang H., Zhang H., Qu Y., Xu F., Wang Y., Qin Z., et al. Integrated proteogenomic characterization of urothelial carcinoma of the bladder. J. Hematol. Oncol. 2022;15:76. doi: 10.1186/s13045-022-01291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baard J., de Bruin D.M., Zondervan P.J., Kamphuis G., de la Rosette J., Laguna M.P. Diagnostic dilemmas in patients with upper tract urothelial carcinoma. Nat. Rev. Urol. 2017;14:181–191. doi: 10.1038/nrurol.2016.252. [DOI] [PubMed] [Google Scholar]
- 4.Hanash S.M., Pitteri S.J., Faca V.M. Mining the plasma proteome for cancer biomarkers. Nature. 2008;452:571–579. doi: 10.1038/nature06916. [DOI] [PubMed] [Google Scholar]
- 5.Yates D.R., Catto J.W.F. Distinct patterns and behaviour of urothelial carcinoma with respect to anatomical location: how molecular biomarkers can augment clinico-pathological predictors in upper urinary tract tumours. World J. Urol. 2013;31:21–29. doi: 10.1007/s00345-012-0946-6. [DOI] [PubMed] [Google Scholar]
- 6.Fujita K., Uemura M., Yamamoto Y., Tanigawa G., Nakata W., Sato M., Nagahara A., Kiuchi H., Nakai Y., Matsumiya K., et al. Preoperative risk stratification for cancer-specific survival of patients with upper urinary tract urothelial carcinoma treated by nephroureterectomy. Int. J. Clin. Oncol. 2015;20:156–163. doi: 10.1007/s10147-014-0695-1. [DOI] [PubMed] [Google Scholar]
- 7.Margulis V., Shariat S.F., Matin S.F., Kamat A.M., Zigeuner R., Kikuchi E., Lotan Y., Weizer A., Raman J.D., Wood C.G., Upper Tract Urothelial Carcinoma CollaborationThe Upper Tract Urothelial Carcinoma Collaboration Outcomes of radical nephroureterectomy: a series from the upper tract urothelial carcinoma collaboration. Cancer. 2009;115:1224–1233. doi: 10.1002/cncr.24135. [DOI] [PubMed] [Google Scholar]
- 8.Lughezzani G., Burger M., Margulis V., Matin S.F., Novara G., Roupret M., Shariat S.F., Wood C.G., Zigeuner R. Prognostic factors in upper urinary tract urothelial carcinomas: a comprehensive review of the current literature. Eur. Urol. 2012;62:100–114. doi: 10.1016/j.eururo.2012.02.030. [DOI] [PubMed] [Google Scholar]
- 9.Zigeuner R., Pummer K. Urothelial carcinoma of the upper urinary tract: surgical approach and prognostic factors. Eur. Urol. 2008;53:720–731. doi: 10.1016/j.eururo.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Secin F.P., Koppie T.M., Salamanca J.I.M., Bokhari S., Raj G.V., Olgac S., Serio A., Vickers A., Bochner B.H. Evaluation of regional lymph node dissection in patients with upper urinary tract urothelial cancer. Int. J. Urol. 2007;14:26–32. doi: 10.1111/j.1442-2042.2006.01664.x. [DOI] [PubMed] [Google Scholar]
- 11.Kim W.J., Kim E.J., Kim S.K., Kim Y.J., Ha Y.S., Jeong P., Kim M.J., Yun S.J., Lee K.M., Moon S.K., et al. Predictive value of progression-related gene classifier in primary non-muscle invasive bladder cancer. Mol. Cancer. 2010;9:3. doi: 10.1186/1476-4598-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu M., Yuan H., Li X., Liao Q., Liu Z. Identification of a five-gene signature and establishment of a prognostic nomogram to predict progression-free interval of papillary thyroid carcinoma. Front. Endocrinol. 2019;10:790. doi: 10.3389/fendo.2019.00790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uhlén M., Karlsson M.J., Hober A., Svensson A.S., Scheffel J., Kotol D., Zhong W., Tebani A., Strandberg L., Edfors F., et al. The human secretome. Sci. Signal. 2019;12 doi: 10.1126/scisignal.aaz0274. [DOI] [PubMed] [Google Scholar]
- 14.Anwaier A., Zhu S.X., Tian X., Xu W.H., Wang Y., Palihati M., Wang W.Y., Shi G.H., Qu Y.Y., Zhang H.L., Ye D.W. Large-Scale proteomics data reveal integrated prognosis-related protein signatures and role of SMAD4 and RAD50 in prognosis and immune infiltrations of prostate cancer microenvironment. Phenomics. 2022;2:404–418. doi: 10.1007/s43657-022-00070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kälin M., Cima I., Schiess R., Fankhauser N., Powles T., Wild P., Templeton A., Cerny T., Aebersold R., Krek W., Gillessen S. Novel prognostic markers in the serum of patients with castration-resistant prostate cancer derived from quantitative analysis of the pten conditional knockout mouse proteome. Eur. Urol. 2011;60:1235–1243. doi: 10.1016/j.eururo.2011.06.038. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y.M., Zheng Y., Yu Y., Wang Y., Huang Q., Qian F., Sun L., Song Z.G., Chen Z., Feng J., et al. Blood molecular markers associated with COVID-19 immunopathology and multi-organ damage. EMBO J. 2020;39 doi: 10.15252/embj.2020105896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shu T., Ning W., Wu D., Xu J., Han Q., Huang M., Zou X., Yang Q., Yuan Y., Bie Y., et al. Plasma proteomics identify biomarkers and pathogenesis of COVID-19. Immunity. 2020;53:1108–1122.e5. doi: 10.1016/j.immuni.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niu L., Thiele M., Geyer P.E., Rasmussen D.N., Webel H.E., Santos A., Gupta R., Meier F., Strauss M., Kjaergaard M., et al. Noninvasive proteomic biomarkers for alcohol-related liver disease. Nat. Med. 2022;28:1277–1287. doi: 10.1038/s41591-022-01850-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 20.Ge S., Xia X., Ding C., Zhen B., Zhou Q., Feng J., Yuan J., Chen R., Li Y., Ge Z., et al. A proteomic landscape of diffuse-type gastric cancer. Nat. Commun. 2018;9:1012. doi: 10.1038/s41467-018-03121-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bittremieux W., Tabb D.L., Impens F., Staes A., Timmerman E., Martens L., Laukens K. Quality control in mass spectrometry-based proteomics. Mass Spectrom. Rev. 2018;37:697–711. doi: 10.1002/mas.21544. [DOI] [PubMed] [Google Scholar]
- 22.Rempel S.A., Rosenblum M.L., Mikkelsen T., Yan P.S., Ellis K.D., Golembieski W.A., Sameni M., Rozhin J., Ziegler G., Sloane B.F. Cathepsin B expression and localization in glioma progression and invasion. Cancer Res. 1994;54:6027–6031. [PubMed] [Google Scholar]
- 23.Liu Y., Xiao S., Shi Y., Wang L., Ren W., Sloane B.F. Cathepsin B on invasion and metastasis of gastric carcinoma. Chin. Med. J. 1998;111:784–788. [PubMed] [Google Scholar]
- 24.Nishikawa H., Ozaki Y., Nakanishi T., Blomgren K., Tada T., Arakawa A., Suzumori K. The role of cathepsin B and cystatin C in the mechanisms of invasion by ovarian cancer. Gynecol. Oncol. 2004;92:881–886. doi: 10.1016/j.ygyno.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 25.Chen W.N., Chen J.Y., Jiao B.Y., Lin W.S., Wu Y.L., Liu L.L., Lin X. Interaction of the hepatitis B spliced protein with cathepsin B promotes hepatoma cell migration and invasion. J. Virol. 2012;86:13533–13541. doi: 10.1128/JVI.02095-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bian B., Mongrain S., Cagnol S., Langlois M.J., Boulanger J., Bernatchez G., Carrier J.C., Boudreau F., Rivard N. Cathepsin B promotes colorectal tumorigenesis, cell invasion, and metastasis. Mol. Carcinog. 2016;55:671–687. doi: 10.1002/mc.22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzales P.A., Pisitkun T., Hoffert J.D., Tchapyjnikov D., Star R.A., Kleta R., Wang N.S., Knepper M.A. Large-scale proteomics and phosphoproteomics of urinary exosomes. J. Am. Soc. Nephrol. 2009;20:363–379. doi: 10.1681/asn.2008040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai J., Su Y., Zhong S., Cong L., Liu B., Yang J., Tao Y., He Z., Chen C., Jiang Y. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct. Targeted Ther. 2020;5:145. doi: 10.1038/s41392-020-00261-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mashouri L., Yousefi H., Aref A.R., Ahadi A.M., Molaei F., Alahari S.K. Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer. 2019;18:75. doi: 10.1186/s12943-019-0991-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Böing A.N., van der Pol E., Grootemaat A.E., Coumans F.A.W., Sturk A., Nieuwland R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles. 2014;3:23430. doi: 10.3402/jev.v3.23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li P., Kaslan M., Lee S.H., Yao J., Gao Z. Progress in exosome isolation techniques. Theranostics. 2017;7:789–804. doi: 10.7150/thno.18133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lobb R.J., Becker M., Wen S.W., Wong C.S.F., Wiegmans A.P., Leimgruber A., Möller A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles. 2015;4 doi: 10.3402/jev.v4.27031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo J., Wu C., Lin X., Zhou J., Zhang J., Zheng W., Wang T., Cui Y. Establishment of a simplified dichotomic size-exclusion chromatography for isolating extracellular vesicles toward clinical applications. J. Extracell. Vesicles. 2021;10 doi: 10.1002/jev2.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kowal J., Arras G., Colombo M., Jouve M., Morath J.P., Primdal-Bengtson B., Dingli F., Loew D., Tkach M., Théry C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA. 2016;113:E968–E977. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoshino A., Kim H.S., Bojmar L., Gyan K.E., Cioffi M., Hernandez J., Zambirinis C.P., Rodrigues G., Molina H., Heissel S., et al. Extracellular vesicle and particle biomarkers define multiple human cancers. Cell. 2020;182:1044–1061.e18. doi: 10.1016/j.cell.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pathan M., Fonseka P., Chitti S.V., Kang T., Sanwlani R., Van Deun J., Hendrix A., Mathivanan S. Vesiclepedia 2019: a compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019;47:D516–D519. doi: 10.1093/nar/gky1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu S., Chen J., Dong P., Zhang S., He Y., Sun L., Zhu J., Cheng Y., Li X., Tang A., et al. Global gene expression profiling identifies ALDH2, CCNE1 and SMAD3 as potential prognostic markers in upper tract urothelial carcinoma. BMC Cancer. 2014;14:836. doi: 10.1186/1471-2407-14-836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zanconato F., Cordenonsi M., Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29:783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L.J., Chou W.C., Pang S.T., Yang C.W., Chuang C.K., Chang Y.H., Hsieh M.L., Wong Y.C. Risk stratification of upper urinary tract urothelial carcinoma patients for survival prediction: a simple summation scoring method. J. Cancer. 2018;9:2284–2294. doi: 10.7150/jca.24815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson B.D., Vlachostergios P.J., Bhinder B., Liu W., Li K., Moss T.J., Bareja R., Park K., Tavassoli P., Cyrta J., et al. Upper tract urothelial carcinoma has a luminal-papillary T-cell depleted contexture and activated FGFR3 signaling. Nat. Commun. 2019;10:2977. doi: 10.1038/s41467-019-10873-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng X., Xu K., Zhou B., Chen T., Huang Y., Li Q., Wen F., Ge W., Wang J., Yu S., et al. A circulating extracellular vesicles-based novel screening tool for colorectal cancer revealed by shotgun and data-independent acquisition mass spectrometry. J. Extracell. Vesicles. 2020;9 doi: 10.1080/20013078.2020.1750202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pichler M., Dalpiaz O., Ehrlich G.C., Stojakovic T., Martín Hernández J.M., Mannweiler S., Pummer K., Zigeuner R., Hutterer G.C. Validation of the preoperative plasma fibrinogen level as a prognostic factor in a European cohort of patients with localized upper tract urothelial carcinoma. J. Urol. 2014;191:920–925. doi: 10.1016/j.juro.2013.10.073. [DOI] [PubMed] [Google Scholar]
- 43.Zhang B., Song Y., Jin J., Zhou L.Q., He Z.S., Shen C., He Q., Li J., Liu L.B., Wang C., et al. Preoperative plasma fibrinogen level represents an independent prognostic factor in a Chinese cohort of patients with upper tract urothelial carcinoma. PLoS One. 2016;11 doi: 10.1371/journal.pone.0150193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghorpade D.S., Ozcan L., Zheng Z., Nicoloro S.M., Shen Y., Chen E., Blüher M., Czech M.P., Tabas I. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature. 2018;555:673–677. doi: 10.1038/nature26138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu L., Zhu S. Computational methods for prediction of human protein-phenotype associations: a review. Phenomics. 2021;1:171–185. doi: 10.1007/s43657-021-00019-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng S., Shen J., Jiao Y., Liu Y., Zhang C., Wei M., Hao S., Zeng X. Platelets and fibrinogen facilitate each other in protecting tumor cells from natural killer cytotoxicity. Cancer Sci. 2009;100:859–865. doi: 10.1111/j.1349-7006.2009.01115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Coninck V., Keller E.X., Somani B., Giusti G., Proietti S., Rodriguez-Socarras M., Rodríguez-Monsalve M., Doizi S., Ventimiglia E., Traxer O. Complications of ureteroscopy: a complete overview. World J. Urol. 2020;38:2147–2166. doi: 10.1007/s00345-019-03012-1. [DOI] [PubMed] [Google Scholar]
- 49.Li Y., He S., He A., Guan B., Ge G., Zhan Y., Wu Y., Gong Y., Peng D., Bao Z., et al. Identification of plasma secreted phosphoprotein 1 as a novel biomarker for upper tract urothelial carcinomas. Biomed. Pharmacother. 2019;113 doi: 10.1016/j.biopha.2019.108744. [DOI] [PubMed] [Google Scholar]
- 50.Inal J.M., Kosgodage U., Azam S., Stratton D., Antwi-Baffour S., Lange S. Blood/plasma secretome and microvesicles. Biochim. Biophys. Acta. 2013;1834:2317–2325. doi: 10.1016/j.bbapap.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 51.Geyer P.E., Kulak N.A., Pichler G., Holdt L.M., Teupser D., Mann M. Plasma proteome profiling to assess human Health and disease. Cell Syst. 2016;2:185–195. doi: 10.1016/j.cels.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 52.Messer J., Shariat S.F., Brien J.C., Herman M.P., Ng C.K., Scherr D.S., Scoll B., Uzzo R.G., Wille M., Eggener S.E., et al. Urinary cytology has a poor performance for predicting invasive or high-grade upper-tract urothelial carcinoma. BJU Int. 2011;108:701–705. doi: 10.1111/j.1464-410X.2010.09899.x. [DOI] [PubMed] [Google Scholar]
- 53.Hurel S., Rouprêt M., Seisen T., Comperat E., Phé V., Droupy S., Audenet F., Pignot G., Cathelineau X., Guy L., et al. Influence of preoperative factors on the oncologic outcome for upper urinary tract urothelial carcinoma after radical nephroureterectomy. World J. Urol. 2015;33:335–341. doi: 10.1007/s00345-014-1311-8. [DOI] [PubMed] [Google Scholar]
- 54.Huang J., Baum Y., Alemozaffar M., Ogan K., Harris W., Kucuk O., Master V.A. C-reactive protein in urologic cancers. Mol. Aspect. Med. 2015;45:28–36. doi: 10.1016/j.mam.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 55.Malle E., Sodin-Semrl S., Kovacevic A. Serum amyloid A: an acute-phase protein involved in tumour pathogenesis. Cell. Mol. Life Sci. 2009;66:9–26. doi: 10.1007/s00018-008-8321-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boguslawska J., Kedzierska H., Poplawski P., Rybicka B., Tanski Z., Piekielko-Witkowska A. Expression of genes involved in cellular adhesion and extracellular matrix remodeling correlates with poor survival of patients with renal cancer. J. Urol. 2016;195:1892–1902. doi: 10.1016/j.juro.2015.11.050. [DOI] [PubMed] [Google Scholar]
- 57.Zhou J., Liu J., Narayan V.A., Ye J. Modeling disease progression via fused sparse group Lasso. Kdd. 2012;2012:1095–1103. doi: 10.1145/2339530.2339702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cha E.K., Shariat S.F., Kormaksson M., Novara G., Chromecki T.F., Scherr D.S., Lotan Y., Raman J.D., Kassouf W., Zigeuner R., et al. Predicting clinical outcomes after radical nephroureterectomy for upper tract urothelial carcinoma. Eur. Urol. 2012;61:818–825. doi: 10.1016/j.eururo.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 59.Rouprêt M., Hupertan V., Seisen T., Colin P., Xylinas E., Yates D.R., Fajkovic H., Lotan Y., Raman J.D., Zigeuner R., et al. Prediction of cancer specific survival after radical nephroureterectomy for upper tract urothelial carcinoma: development of an optimized postoperative nomogram using decision curve analysis. J. Urol. 2013;189:1662–1669. doi: 10.1016/j.juro.2012.10.057. [DOI] [PubMed] [Google Scholar]
- 60.Xylinas E., Kluth L., Mangal S., Roupret M., Karakiewicz P.I., Shariat S.F. Predictive tools for clinical decision-making and counseling of patients with upper tract urothelial carcinoma. World J. Urol. 2013;31:31–36. doi: 10.1007/s00345-012-0947-5. [DOI] [PubMed] [Google Scholar]
- 61.Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R., et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. 2017;171:540–556. doi: 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feng J., Ding C., Qiu N., Ni X., Zhan D., Liu W., Xia X., Li P., Lu B., Zhao Q., et al. Firmiana: towards a one-stop proteomic cloud platform for data processing and analysis. Nat. Biotechnol. 2017;35:409–412. doi: 10.1038/nbt.3825. [DOI] [PubMed] [Google Scholar]
- 63.Herwig R., Hardt C., Lienhard M., Kamburov A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc. 2016;11:1889–1907. doi: 10.1038/nprot.2016.117. [DOI] [PubMed] [Google Scholar]
- 64.Wiśniewski J.R., Zougman A., Nagaraj N., Mann M. Universal sample preparation method for proteome analysis. Nat. Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 65.Rechavi O., Erlich Y., Amram H., Flomenblit L., Karginov F.V., Goldstein I., Hannon G.J., Kloog Y. Cell contact-dependent acquisition of cellular and viral nonautonomously encoded small RNAs. Genes Dev. 2009;23:1971–1979. doi: 10.1101/gad.1789609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kong A.T., Leprevost F.V., Avtonomov D.M., Mellacheruvu D., Nesvizhskii A.I. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods. 2017;14:513–520. doi: 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Demichev V., Messner C.B., Vernardis S.I., Lilley K.S., Ralser M. Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods. 2020;17:41–44. doi: 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang W., Zhang J., Xu C., Li N., Liu H., Ma J., Zhu Y., Xie H. LFQuant: a label-free fast quantitative analysis tool for high-resolution LC-MS/MS proteomics data. Proteomics. 2012;12:3475–3484. doi: 10.1002/pmic.201200017. [DOI] [PubMed] [Google Scholar]
- 69.Ku X., Wang J., Li H., Meng C., Yu F., Yu W., Li Z., Zhou Z., Zhang C., Hua Y., et al. Proteomic portrait of human lymphoma reveals protein molecular fingerprint of disease specific subtypes and progression. Phenomics. 2023;3:148–166. doi: 10.1007/s43657-022-00075-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 71.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R., et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48:D498–D503. doi: 10.1093/nar/gkz1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zheng G., Du L., Yang X., Zhang X., Wang L., Yang Y., Li J., Wang C. Serum microRNA panel as biomarkers for early diagnosis of colorectal adenocarcinoma. Br. J. Cancer. 2014;111:1985–1992. doi: 10.1038/bjc.2014.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barbie D.A., Tamayo P., Boehm J.S., Kim S.Y., Moody S.E., Dunn I.F., Schinzel A.C., Sandy P., Meylan E., Scholl C., et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hänzelmann S., Castelo R., Guinney J. Gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Seckinger A., Meissner T., Moreaux J., Depeweg D., Hillengass J., Hose K., Rème T., Rösen-Wolff A., Jauch A., Schnettler R., et al. Clinical and prognostic role of annexin A2 in multiple myeloma. Blood. 2012;120:1087–1094. doi: 10.1182/blood-2012-03-415588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Inclusion and exclusion criteria. B. Clinical characteristics of the UTUC discovery cohort and healthy cohort. C. Clinical characteristics of the UTUC validation cohort. D. Correlation of quality control (QC) plasma samples. E. Raw expression data of identified plasma protein in the UTUC discovery and healthy control samples (identification frequency >30%).
A. List of differentially expressed proteins (Wilcoxon rank-sum test, BH P < 0.05, UTUC/normal ratio >1.5 or <0.67) in UTUC and normal plasma samples. B. Kidney-specific proteins observed in UTUC plasma. C. List of candidate proteins of classifier model in plasma samples. D. List of proteins identified in urine samples. E. List of proteins identified in EV samples. F. List of the upregulated protein (Wilcoxon rank-sum test, BH P < 0.05, UTUC/Normal ratio >1.5) overlap among UTUC tumor tissue, UTUC plasma, and UTUC urine. G. List of candidate proteins of NMI-UTUC urine samples. H. List of candidate proteins of UTUC EV samples.
A. List of differentially expressed proteins in NMI-UTUC and MI-UTUC plasma samples. B. List of differentially expressed proteins in NMI-UC and MI-UC tissue samples. C. List of targeted peptides unique to the proteins of the classifier model.
A. List of the plasma proteins correlated with plasma fibrinogen. B. List of the plasma protein panel associated with lymph node metastasis.
A. Proteins correlated with PFS. B. Hazard ratio of selected proteins. C. The correlation coefficients of ten proteins. D. List of targeted peptides unique to the proteins of model.
Data Availability Statement
-
•
The raw mass spectrometry (MS) proteomics data generated in this study have been deposited in the ProteomeXchange Consortium via the iProX partner repository (http://www.iprox.cn/; dataset identifier: PXD043774) under Project ID IPX0005194000. Human Protein Atlas (HPA) IHC Staining Data could be accessed at https://www.proteinatlas.org/. The remaining data are available within the Article and Supplementary Information.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.