

Summary
The tumor microenvironment (TME) is influenced by a “disorganized” extracellular matrix (ECM) that sensitizes cancer cells toward mechanical stress, signaling, and structural alterations. In hepatocellular carcinoma (HCC), lack of knowledge about key ECM proteins driving the TME refractory to targeted therapies poses a barrier to the identification of new therapeutic targets. Herein, we discuss the contributions of various ECM components that impact hepatocytes and their surrounding support network during tumorigenesis. In addition, the underpinnings by which ECM proteins transduce mechanical signals to the liver TME are detailed. Finally, in view of the bidirectional feedback between the ECM, transformed hepatocytes, and immune cells, we highlight the potential role of the ECM disorganization process in shaping responses to immune checkpoint inhibitors and targeted therapies. Our comprehensive characterization of these ECM components may provide a roadmap for innovative therapeutic approaches to restrain HCC.
Keywords: extracellular matrix, hepatocellular carcinoma, fibrosis, chronic liver inflammation, collagen, agrin, glypican-3, immunotherapy, mechanotransduction, YAP/TAZ, targeted therapies
Graphical abstract
Roy et al. review the emerging roles of a disorganized extracellular matrix during the development of hepatocellular carcinoma. They highlight the potential impact of a highly disorganized ECM in shaping up responses toward targeted therapies.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common malignancy and the fourth leading cause of cancer-related mortality globally.1 Multiple etiological factors are associated with the development of HCC, including obesity, alcoholism, viral hepatitis, and non-alcoholic fatty liver disease (NAFLD), which can progress to non-alcoholic steatohepatitis (NASH) that causes chronic inflammation and fibrosis of the liver.1 While viral hepatitis may progress to HCC without the presence of cirrhosis, the majority of etiological factors cause cirrhosis prior to the onset of HCC.2,3 Despite recent advancements in treatments for HCC, such as surgical resection, tumor ablation, chemoembolization, liver transplantation, and the use of oral tyrosine kinase inhibitors (TKIs), anti-vascular endothelial growth factor (VEGF) agents, and immunotherapies, the 5-year survival rate for HCC and intrahepatic bile duct cancer in the United States remains low at only 20.8%.4 In this regard, apart from surgical resection, TKIs were the only approved therapies for HCC until the recent US Food and Drug Administration (FDA) approval of the combination therapy with the immune checkpoint inhibitors (ICIs) atezolizumab (anti-PD-L1), bevacizumab (anti-VEGF), durvalumab (anti-PD-L1), and tremelimumab (anti-CTLA-4) for advanced unresectable or metastatic HCC.5,6,7,8 TKI therapy (sorafenib) has a survival advantage of ∼3 months over placebo as noted in the human dataset from SHARP (Sorafenib Hepatocellular Carcinoma Assessment Randomized Protocol) clinical trial. Atezolizumab plus bevacizumab has a survival advantage of ∼6 months in comparison with that of sorafenib, and the survival advantage for durvalumab plus tremelimumab is ∼2.5 months over sorafenib, collectively improving the prognosis of patients with HCC.5,9 However, the underlying reasons for resistance of HCC to cytotoxic chemotherapy and differential responses to immunotherapy in HCC remain unclear.
The extracellular matrix (ECM) is an essential part of the tumor microenvironment (TME) and is composed of proteins, glycoproteins, and polysaccharides; the ECM acts as a reservoir of cytokines and growth factors, maintains tissue morphology, and regulates cellular behavior.10 The core ECM consists of over 300 macromolecules that can be stratified as collagens, proteoglycans, and glycoproteins. There are several regulatory mechanisms and signaling pathways that can initiate degradation and remodeling of the ECM, and disruption of such mechanisms alters the structure and function of the ECM which ultimately affects organ homeostasis and functions.11 In addition, the interstitial matrix of the ECM serves as a “molecular sink” that binds to several soluble growth factors and facilitates their controlled release with corresponding impacts on cellular signaling and behavior. The ECM is now becoming increasingly recognized as a dynamic communicative layer that actively instructs the underlying tissues to respond and adapt to environmental changes.10 In healthy tissues, the dynamic ECM supports physiological functions and limits tumorigenesis.12 While ECM dynamicity is retained by several conditions including aging, hypoxia, stress, wound injury, inflammatory diseases, and cancer, the protective virtues are gradually lost with the concomitant gain of pathological characteristics. The altered mechanical and topographical parameters represented by the disorganized ECM may also influence cell fate and functions via defined mechanosignaling pathways.13 Intuitively, this highly refurbished ECM impacts amplification of growth factor signaling pathways, inhibition of tumor suppressors, and promotion of angiogenesis. On the other end of this oncogenic corruption loop, cancer cells, stromal cells, including cancer-associated fibroblasts (CAFs), immune cells, epithelial cells, and mesenchymal cells can actively interact with and modify the 3D organization of the ECM (or its components), and these are thought to represent the main effectors of tumor growth and progression.14,15
The activation of stromal cells and the excessive deposition of ECM leads to enhanced integrin signaling and stiffening of the liver tissue in cirrhosis and eventually development of HCC.16 Clinically, increased liver stiffness has been associated with greater HCC risks.17,18,19 However, the molecular contributors to a complex HCC TME remain a topic of debate in the field. Furthermore, the production of ECM components and associated enzymes accelerates when cancer cells preferentially respond to extrinsic growth factor signaling harbored within the ECM, which often results in rapid disease progression. The magnitude of these changes in the liver ECM and details of the corresponding oncogenic regulatory program is still unknown. This represents a major knowledge gap, which must be fulfilled to understand how a disorganized ECM may respond to HCC treatments.
Cancer cells have several strategies for evading immune surveillance, and these have emerged collectively as one of the hallmarks of cancer.20 One strategy effectively exploited by cancer cells is ECM remodeling, which develops immunosuppressive network. The tumor-infiltrating T cells and natural killer (NK) cells in the immunosuppressive TME have a major role in the response to immunotherapy.21 Thus, understanding the extent of remodeling in the TME with new ECM deposition is vital to appreciate the mechanisms of resistance to immunotherapy, to identify reliable biomarkers or response predictors for immunotherapy, and to develop new therapeutic targets for HCC.
By assembling evidence from liver carcinoma cell lines, mouse models, multiregional transcriptomic analyses of human HCC, and concluded human clinical trials, herein, we review the bidirectional feedback between the complex ECM network and its microenvironment as a potential vulnerability to restrain HCC.
Constituents of a disorganized ECM in the HCC microenvironment
Human liver proteomics has identified more than 150 ECM proteins that constitute the healthy liver matrix, and the ECM adds up to 10% of the total volume.22 Collagen-related proteins, transmembrane proteoglycans, ECM regulators such as lysyl oxidases (LOXs), matrix metalloproteinases (MMPs), and cytokines that bind to the ECM such as transforming growth factor β (TGF-β) also contribute to tissue homeostasis and are referred to as the “matrisome.”23 Figure 1A shows the contributions of key ECM components in promoting several cancer hallmark phenotypes. Prominent roles of collagens, agrin, glypican-3, syndecan, osteopontin, and others are associated with sustaining cancer cell proliferation, invasion, and angiogenesis, while ECM-harbored TGF-β, hyaluronic acid, and fibronectin have tumor-inflammatory actions. Though hepatocytes are the prominent parenchymal cell type in the liver (50%–60%), 40% of the liver cells are non-parenchymal (NP) cells of the following types: liver sinusoidal endothelial cells (LSECs), acting as a filtration barrier; Kupffer cells, i.e., liver-resident macrophages; hepatic stellate cells (HSCs), i.e., responsible for hepatic fibrosis; biliary cells; and liver-associated lymphocytes. In view of different regions of healthy livers contributing to subsequent disease conditions, a spatial proteogenomic approach identified localized zones within healthy liver tissues that influence the development of a specific myeloid cell niche, which is subsequently altered in diseased livers.24 Upon tissue injury and inflammation, the inflamed liver is infiltrated by the myeloid cells even before the development of HCC.25 Overall, both parenchymal and NP cell types are responsible for the synthesis, degradation, and remodeling of the ECM proteins and liver homeostasis.
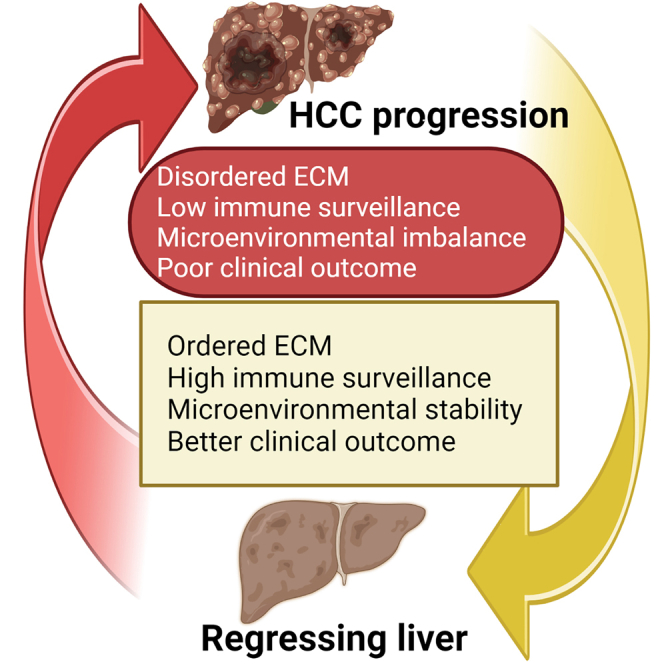
Figure 1.

ECM regulating the “hallmarks of liver cancer” and its status in healthy liver, chronic disease conditions, and hepatocellular carcinoma
(A) The liver ECM network as a molecular sink that harbors structural components, proteoglycans, and several growth factors. The impact of key ECM components (light blue boxes) known to regulate the specific cancer hallmark (reddish brown) are highlighted.
(B) ECM in healthy liver tissues that act to maintain liver homeostasis by controlling the proliferation of hepatocytes and sustaining immune-suppressive functions. When chronic liver diseases develop, the ECM is altered by increases in collagen deposition, which enhance stiffness, elastin-derived peptides (EDPs), and PGs, as well as augments the influx of fibroblasts. This initiates an ECM corruption process, as manifested by a low immune response that impacts the hepatocytes, activates HSCs, and promotes abnormal vasculature leading to liver fibrosis. Increased ECM disorganization, in turn, increases the mechanosignaling and contractility of the hepatocytes, which leads to their neoplastic transformation. Cancerous hepatocytes further increase ECM disorganization, causing a vicious oncogenic feedforward loop accompanied by enhanced tissue rigidity, resistance to targeted therapies, and immune-escape mechanisms.
In healthy liver, ECM components are concentrated in the portal area, around the central vein, and in the space surrounding the liver sinusoids (space of Disse). Collagen, laminins, fibronectin, entactin (nidogens), and perlecan are the major constituents in the healthy liver26 (Figure 1B). Among these, collagen is the most common ECM component and is expressed mainly in the basement membrane (BM) lining of the bile ducts, in the interstitial matrix of the portal tracts, and in the space of Disse. Collagen IV is the most represented collagen family member in healthy liver.26,27 Decorin, biglycan, integrin, and heparan sulfate also commonly inhabit the healthy liver ECM27 (Figure 1B).
During liver inflammation/injury, the balance between the synthesis and degradation of ECM proteins is perturbed, which contributes to tissue scarring and fibrosis.28 During cirrhosis, the myofibroblasts generally believed to be derived from activated HSCs act as the major source of altered ECM deposition (mainly collagen) that inaugurates a dysregulated ECM, resulting in subsequent carcinogenesis.29 In contrast, the ECM in some cases may restrain the primary tumor and/or vascular infiltration. For example, in 40%–50% of cases, HCC is surrounded by a fibrous capsule with trabecula, postulated to arise from tumor-host liver interactions. Patients with HCC who have encapsulated tumors have a better prognosis than patients whose tumors lack the capsule.30 Type I, III, and IV pro-collagens, which is deposited by myofibroblast CAFs, are the major components of this capsule.30
We revisit the major ECM components, their roles in chronic liver diseases (CLDs)/precancerous conditions, tumor development, tumor dissemination, and angiogenesis in mouse models and human studies.
Structural architecture of the ECM in HCC
Collagen
Collagens are proteins that form fibers or networks in tissues and are the major structural component of the ECM in most solid cancers. The progression of alcoholic liver disease, NAFLD, and NASH and the development of HCC are all related to fibrosis.31,32 In CLDs and cirrhosis/fibrosis, inflammation activates the HSCs, which undergo transdifferentiation into myofibroblasts that increase collagen secretion, creating a dense matrix (Figure 1B). In a carbon-tetrachloride (CCL4)-induced mouse cirrhotic model, the BM was enriched with type IV collagen α5 that was activated by HSCs, and this led to the discovery of an ECM signature that predicted early stages of cirrhosis.33 BM collagens such as type IVs, VI, and I, III are variably increased in cirrhosis and promote HCC, with type I being the most predominant type associated with progression of cirrhosis.34,35 The expression of integrins α1β1 and α2β1, the main receptors for collagen, is upregulated during liver injury and also promotes hepatic fibrosis.36 The activation of HSCs is modulated by immune cells that interact with the integrins to regulate cell proliferation, migration, and tumor dissemination.37,38 Zone-specific ECM and its association with vasculature may impact collagen production during fibrosis and HCC. Notably, in a NASH-based rodent model, collagen produced by central vein-associated HSCs was pathogenic for fibrosis.39 Collagen activation within specific HSC populations was recently shown to be a decisive factor for liver tumorigenesis.40 While cytokine-enriched (quiescent) HSCs oppose tumorigenesis, the myofibroblast subtypes are enriched in collagen I, which activates the Hippo effector TAZ in pretumorigenic liver tissues, thereby creating a highly stiff, tumor-prone liver microenvironment.40 Once tumors develop, the collagen I-enriched HSC population further engages in a crosstalk with its canonical tyrosine kinase discoid in domain receptor-1 (DDR1) and integrin β1 to sustain carcinogenesis. Hence, we speculate that zone-specific cell types may deposit collagen to generate a localized fibrotic niche that ultimately initiates tumorigenesis (Figure 1B). Collagen acts as a high-affinity ligand for the leukocyte-associated immune receptor (LAIR)-1, promoting an immunosuppressive phenotype.41 On the premise that myofibroblasts predominantly activate HSCs in CLDs, the imbalance between cytokine-enriched versus myofibroblast HSC subtypes may dictate the pro-tumorigenic microenvironment accompanied with tissue stiffness.42 Whether liver zone-specific integration of collagen alters with HCC stages also requires special attention.
Elastin
Elastin aggregates to form elastin fibers in tissues; it is particularly abundant within arteries and lungs, skin, cartilage, and certain ligaments.43 Although elastin is a minor ECM component in healthy liver, tropoelastin is synthesized by hepatic myofibroblasts during fibrosis.44 In CLDs, elastin fiber accumulates in the hepatic artery and portal vein, leading to fibrosis.45 Elastin accumulation forms elastin-derived peptides (EDPs) (Figure 1B, EDP formation is shown in the middle and third panels in yellow). EDPs stimulate inflammation, cytokine expression, and remodeling of the hepatic ECM, thereby creating fibrosis and transition of NAFLD to NASH.46 The expression of elastin fibers during the fibrotic stages may predict the incidence of HCC.47 Supporting the hypothesis that a highly crosslinked ECM may stimulate elastin fiber secretion, CCL4-derived mouse liver fibrosis models displayed a strong correlation between elastin, collagen, and LOX-like 1 (LOXL1), a major ECM crosslinking enzyme.48 Also, a higher association between elastin and collagen fibers was found to be significantly correlated with multinodular macroscopic lesions.49 Elastin-based magnetic resonance imaging probes were implemented to detect ECM deposition in a rabbit HCC model.50 Expectedly, EDPs can modulate tumor angiogenesis.43 Despite these observations, how elastin responds to ECM crosslinking and stiffness are not clearly understood, as mouse models and human HCC studies analyzing the role of elastin are limited.
Fibronectin
Being the most abundant glycoprotein in the ECM of hepatic tissue, fibronectin (FN) accumulates rapidly in response to injury and inflammation.51 Exogenous FN-stimulated liver cells increased the production of pro-inflammatory cytokines such as tumor necrosis factor α (TNF-α), interleukin-6 (IL-6), and pro-fibrogenic factors.52 FN exists in several different forms, namely a plasma-associated FN (PFN), synthesized by hepatocytes and secreted into the ECM. Cellular FN (CFN) is usually observed at low levels in healthy liver but accumulates rapidly during chronic injuries (Figure 1B). CFN potentially serves as a biomarker for early-stage alcoholic liver disease and HCC.33,51,53 Mechanistically, mechanical forces establish FN connections to integrins, collagen, and fibrin for cell adhesion and growth differentiation in tumorigenesis and metastasis.53,54,55 FN and its integrin receptors are abundantly expressed in the tumor vasculature and promote angiogenesis.56
Proteoglycan landscape of a disorganized liver ECM
Proteoglycans (PGs) consist of glycosaminoglycan (GAG) chains that are covalently linked to a protein core and constitute an important portion of the ECM. Several PGs are overexpressed in HCC, at the cell surface (syndecan-1 and glypican-3), in the pericellular space (agrin and collagen XVIII/endostatin), and in extracellular space (versican and decorin). Due to their extensive roles in promoting cancer cell migration, modification of the stromal microenvironment, and angiogenesis, thereby supporting cancer progression, PGs remain attractive as therapeutic targets.57
Syndecan-1
Syndecan-1 (SDC1) is a cell surface PG expressed in the liver. Serum SDC1 levels were high in patients with NAFLD, although no significant correlation between SDC1 serum levels and their corresponding liver biopsy-sourced immunohistochemistry scores was detected.58 SDC1 immunostaining in CLD positively correlated with the severity of fibrosis.59 Soluble SDC1 can be used as a circulating biomarker for the detection and staging of HCC. Serum SDC1 levels are high in patients with HCC with disease progression and recurrence.60,61 SDC1 expressed by human mammary fibroblasts, in association with integrin, promoted the assembly of ECM fibers in parallel arrays, which increased migration and invasion of breast cancer cells.62 The same phenotypic effect of SDC1 is expected to be mirrored in HCC. Interestingly, the expression of pro-angiogenic factors VEGF and fibroblast growth factor 2 (FGF-2) was reduced in thioacetamide-induced rats through the inhibition of SDC1, a finding suggestive of its role in angiogenesis and HCC progression.63
Glypican-3
Glypican-3 (GPC3) is a cell surface PG that increases proliferation of HCC cells, epithelial-to-mesenchymal transition (EMT), and angiogenesis. GPC3 has a diagnostic and prognostic value in HCC.64 While not present in healthy liver, GPC3 is highly expressed in HCC tissues. Circulating levels of GPC3 are elevated in the serum of patients with HCC.65 GPC3 is usually expressed in embryonal tissues, but its reappearance as an oncofetal protein during malignant hepatocyte transformation corresponds to a poor HCC prognosis.66,67 GPC3 alone and combined with α-fetoprotein (AFP) showed increased sensitivity and specificity for the early detection of HCC.68 It stimulates Wnt signaling and the insulin-like growth factor (IGF) and promotes EMT via extracellular signal-regulated kinase (ERK) signaling.69,70 Several preclinical studies and ongoing clinical trials are using GPC3 peptide vaccines, monoclonal antibodies against GPC3, and chimeric antigen receptor (CAR) T cell therapy targeting GPC3 to assess the utility of GPC3 as an attractive immunotherapeutic target in HCC.71,72,73
Agrin
Agrin is a heparan sulfate PG that represents a major secretory component in neuromuscular junctions (NMJs).74 As a key ECM component, agrin is overexpressed in HCC.75 In HCC, both agrin and its receptor repertoire, comprised of lipoprotein-related receptor-4 (Lrp4), muscle-specific tyrosine kinase (MuSK), and integrin β1, are overexpressed.75 Agrin promotes HCC cell proliferation, migration, and invasiveness, and supplementation with a recombinant soluble agrin (sAgrin) readily revives cancer cell proliferation and migration defects caused by the genetic ablation of agrin.75 Mechanistically, agrin sustains the integrity of the focal adhesion kinase (FAK)-induced proliferative and invasive functions of cancer cells.75 In validation of these studies, agrin has been recently shown to promote metastatic pancreatic ductal adenocarcinoma (PDAC) via similar EMT mechanisms.76 Moreover, HSCs activated by platelet-derived growth factors (PDGFs) have been shown to secrete agrin that may contribute to HCC.77 Agrin activates the transcription factors Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), the central effectors of the Hippo pathway78 (Figure 2A). Besides serving as major oncogenic factors that promote liver fibrosis and HCC by engaging transcriptional enhanced associate domain (TEAD) transcriptional factors, YAP/TAZ are widely regarded as mechanosensors that respond to a variety of extrinsic physical stimuli (Figure 2A). Interestingly, studies on agrin have provided the first evidence on how an ECM PG may orchestrate extrinsic mechanical signals and transduce them to cancer cells to engage YAP/TAZ mechanosensing (Figure 2A).78 Agrin in the TME recruits endothelial cells (ECs) and controls their angiogenic properties.79,80 It promotes angiogenesis by increasing VEGF receptor 2 (VEGFR2) stability in a matrix stiffness-sensed fashion (Figure 2B).79,81 Mirroring the liver TME, agrin has also been recently shown to promote skin repair by mechanically stimulating the wounded microenvironment by activating MMP12.82 However, it remains to be seen whether the agrin-MMP12 oncogenic axis can remodel the ECM during HCC.
Figure 2.

Disorganized ECM modulate mechanical checkpoints in the liver TME
(A) A stiff ECM crosstalks with hepatocytes and HSCs, leading to increases in their rigidity, contractility, and geometrical confinement. Members of the disorganized ECM such as agrin, tenascin (TNCN), or osteopontin (OPN) activate their respective mechanosensory receptor integrins-lipoprotein related receptor-4 (Lrp4)-muscle-specific tyrosine kinase (MuSK), frizzled receptors (Fzds), or receptor tyrosine kinases (RTKs), respectively, to transmit biophysical signals. In response to stiff ECM, agrin stimulates integrin-FAK mechanosignaling and actomyosin-dependent RhoA activation, which potentiate the YAP/TAZ transcriptional program. TNCN induces nuclear β-catenin, and OPN via the integrin-EGFR pathway activates the MAPK-dependent transcription of genes that enhance tumorigenesis.
(B) Secreted proteins from the disorganized ECM also impact other cell types in the liver TME. For instance, agrin, perlecan, and OPN stimulate the VEGF-VEGFR2 pathway in liver endothelial cells, which increases shear stress and angiogenesis that favors tumor growth. These impacts transformed hepatocytes that further remodel the ECM. Therefore, a bidirectional network establishes a physical continuum between the extrinsic “disorganized ECM” and intrinsic “oncogenic behavior.”
Tenascin
Tenascin (TNCN) is a glycoprotein that binds to several ECM proteins, including epidermal growth factor receptor (EGFR) and integrins, and with several growth factors such as TGF-β, VEGF, PDGF, and IGF. The TNCN family consists of four glycoproteins: TNCN-C, TNCN-X, TNCN-R, and TNCN-W. TNCN is not usually detected in healthy tissues but is expressed upon tissue injury and inflammation and in multiple cancers including HCC.83 The overexpression of TNCN in HCC is associated with a poor prognosis.84 Further, TNF-α derived from the inflammatory TME upregulates TNCN expression in HCC cells and in turn enhances migration through the WNT/β-catenin pathway (Figure 2A).84,85 However, the precise interactions and mechanisms by which TNCN mediates liver disease and HCC remain unclear.
Endostatin
Endostatin is a C-terminal fragment of type XVIII collagen that binds to cell surface proteins and receptors.57 It binds to VEGF, GPC1, and GPC4 and elicits anti-angiogenic effects. Additionally, it interacts with integrin α5β1 and disrupts EC migration and proliferation.86 Hence, its ability to inhibit neovascularization is of prime importance to HCC prevention. Paradoxically, the increased expression of endostatin correlates with elevated VEGF levels and tumor stages, and this is associated with a poor prognosis in HCC.87 Owing to its influence on tumor angiogenesis, we speculate that endostatin may play an important role in predicting responses to targeted therapies and ICIs.
GAGs (hyaluronic acid)
Hyaluronic acid (HA) is a GAG composed of repeating disaccharides that form chains of variable length.88 The HSCs secrete HA in the liver and act through its native ligand transmembrane receptor, CD44.89 Mechanical forces, oxidative stress, injury, and inflammation that occurs in the CLDs produce HA breakdown products. This activates TGF-β signaling, which increases cell proliferation, migration, and invasion and aids in the development of HCC (Figure 1B).90,91,92 HA promotes immune responses through the maturation of antigen-presenting cells and helps in cell migration and angiogenesis.93 HA promotes EC proliferation and migration and neo-angiogenesis through endogenous release of VEGF by its binding to the receptor CD44, a receptor for hyaluronan-mediated motility (RHAMM), and to Toll-like receptor-4 (TLR-4) in the development and progression of tumors.94 Indeed, RHAMM is upregulated in HCC and is positively correlated with the stage, degree of vascular invasion, and tumor recurrence and is associated with poor overall survival (OS) and disease-free survival (DFS).95
Osteopontin (OPN)
OPN is a phosphorylated glycoprotein that is widely expressed in many cells such as T cells, B cells, myeloid cells, NK cells, and osteoblasts. It is found in excess in chronic inflammatory stages like alcoholic liver disease, NAFLD, cirrhosis, liver fibrosis from hepatitis, and HCC.96,97 Secreted OPN is markedly elevated in the plasma of patients with HCC and has been proposed as a diagnostic marker.98 OPN is associated with tumor grade, tumor size, and recurrence of HCC.99,100 It binds to CD44 and integrin receptor αvβ3 and induces proliferation, infiltration, metastasis, and tumor progression through mitogen-activated protein kinases (MAPKs), nuclear factor κB (NF-κB), and the PIK3/AKT pathway (Figure 2A).98,99,101 OPN promotes HCC by increasing reactive oxygen species (ROS) by activating the JAK2/STAT3/NADPH oxidase 1 (NOX1) pathway.97 OPN levels can be used to predict the prognosis of HCC and are associated with poor OS and DFS.100 OPN also induces angiogenesis via VEGF-PI3K/AKT/ERK1/2 stimulation in ECs.102 OPN inhibits cytotoxic T-lymphocytes; thus, tumors likely modulate OPN expression as a measure to control the balance between immune tolerance versus escape.103 Furthermore, deletion of OPN in mouse HCC models decreased the infiltration of tumor-associated macrophages (TAMs) and T cells, which helped to establish an anti-PD-L1 blockade in these tumors.104
Other ECM proteins involved in the development and progression of HCC
The expression of versican, a chondroitin sulfate PG, was found to be increased in advanced liver fibrosis caused by viral hepatitis, fatty liver disease, and liver cirrhosis.105 As essential components of the BM, agrin, versican, and perlecan are upregulated in HCC.106 Cystine-rich protein 61, or CCN1, is a matricellular protein associated with the ECM, and it is involved in the proliferation, migration, and apoptosis of cells. It is also secreted in high amounts during liver injury/inflammation and promotes proliferation of HCC cells.107,108 CCN1 may be associated with the progression of the hepatic cirrhosis-HCC axis through activation of HSCs.108 CCN1 is also involved in suppressing hepatic carcinogenesis. It inhibits EGFR-dependent hepatocyte proliferation through integrin α6β1 and subsequent ROS production.109 Another ECM matricellular protein that assembles collagen fiber is osteonectin, a secreted protein acidic and rich in cystine (SPARC). Its increased expression in cirrhosis and HCC induces collagen deposition, recruitment of inflammatory cells, production of TGF-β1, and mesenchymal stem cell proliferation.110 Thrombospondin-1 (TSP-1) is a glycoprotein that regulates cell-matrix interactions and is usually expressed more in pathological conditions.111 In patients with congenital liver fibrosis, alcoholic cirrhosis, and NASH-related cirrhosis, TSP-1 is expressed in higher amounts.112
The disorganized ECM orchestrates mechanotransduction in the liver TME
Cells experience a variety of physical forces including tension or compression, shear stress of circulating blood, and static mechanical forces.113 These inputs are processed as defined signaling responses within cells, a process termed mechanotransduction. A well-regarded concept is that CLDs marked by excessive ECM deposition may confer physical stress to underlying hepatocytes, portal fibroblasts, LSECs, and HSCs. The forces experienced by these cells and reinforced by the ECM are measured as elastic modulus I and shear modulus (G), which largely can be used to define rigidity parameters. Real-time shear-wave elastography has revealed that liver stiffness drastically increases from 1–2 to 5–6 kPa during cirrhosis and/or fibrotic stages. In HCC, stiffness values of tumor-laden livers could range in excess of 20 kPa.18,19 The ability of tissue stiffness to mediate broadscale changes to the HCC TME requires further analysis. Despite observations suggestive of increased stiffness in the presence of liver tumors, the overall consistency of HCC nodules remains heterogeneous, especially when compared with non-tumor liver areas. As such, the degree of local stiffness may vary based on nodule size, region of interest within tumors, stage of disease (early versus metastatic), and clinical interventions used on patients. Similarly, HCC cells cultured on mechanically tunable “stiff gels” that conferred mitogenic signaling-dependent growth versus those on “soft gels” simulated stem cell properties.114 Therefore, we propose that aberrant and selective deposition of ECM components needs to be considered in addition to overall tumor stiffness to comprehend HCC progression. The abnormal tissue stiffness is likely an outcome of anomalous ECM deposition primarily by HSCs and hepatocytes. Substrate stiffness is sufficient to activate FAK, proto-oncogene-protein tyrosine kinase Src, Ras homolog gene family member (RhoA), and YAP/TAZ mechanosensors in HSCs.115,116 FAK-YAP mechanosignaling activates gene transcription for HSC activation and mediates durotaxis/migration of HSCs toward a stiffer environment, which promotes increased production of ECM proteins, further increasing matrix rigidities (Figure 2A). The mechanical force from the increased stiffness in the ECM induces activation of integrins and the formation of complexes on cell membranes comprised of actin-binding proteins and signaling molecules such as FAK, Src, and PI3K. The biochemical signaling from these complexes through RhoA and direct force transfer through actin filaments, i.e., external forces that can cause gene transcription, actomyosin contractility, and cytoskeleton remodeling, results in the dedifferentiation, proliferation, adhesion, and migration of hepatocytes (Figure 2A). The mechanosignaling induced from sheer stress also leads to the release of hepatocyte growth factor in a VEGFR3-integrin β1-dependent fashion, which supports the proliferation and survival of hepatocytes and angiogenesis.117 PGs such as agrin and SDC1 are effectors of “outside-in” mechanotransduction that reside at the interface of a stiff ECM and cancer cells78,81 (Figures 2A and 2B). In addition to its impact on cancer cells, the stiffened microfibrotic niche modulates the function of LSECs and angiogenesis, leading toward liver fibrosis as precursor events toward HCC.118
Impact of a disordered liver ECM on the immune surveillance machinery
Intrinsic immune surveillance forms an important barrier to cancer progression.119 However, cancer cells override this immune-surveillance repertoire by generating an immunosuppressive TME. The reduced recognition of tumor antigens; the accumulation of cells with negative regulatory immune activity such as regulatory T cells (Tregs); the presence of B regulatory cells, myeloid-derived suppressor cells (MDSCs), or M2-polarized TAMs; and the upregulation of coinhibitory lymphocyte signals including immune checkpoint ligands and receptors cumulatively reflect traits of an immunosuppressive TME.120 Interestingly, the ECM directly modulates immune responses and influences immune responses through the modification of growth factors, cytokines, and other components in the TME (Figure 3). TGF-β1, which is secreted by the ECM during chronic inflammation, increases the rate of hepatocyte death and transdifferentiates HSCs to myofibroblasts that enhance the deposition of ECM proteins and fibrosis.121,122 It can also drive both pro-inflammatory as well as inhibitory immune responses that are pro-tumorigenic (Figure 3). TGF-β1 causes impairment of NK cell activity and polarizes macrophages toward the M2 phenotype associated with immunosuppressive functions.123,124 At the same time, it causes terminal differentiation of Tregs, which negatively regulates liver inflammation.125 Importantly, TGF-β1 is a driver of matrix stiffness that modulates the invasiveness of HCC cells.126 However, little is currently known about the impact of TGF-β1-induced mechanotransduction on specific immune compartments in the liver tissue. Moreover, distinct macrophage and Kupffer cell populations exist within specialized zones in healthy versus cancerous liver tissues. A specialized group of macrophages termed as LAMs (lipid-associated macrophages) have been shown to be induced by lipids within steatosis regions of liver tissues.24
Figure 3.

A disorganized liver ECM impacts immune surveillance machinery
A low ECM complexity state within the matrix triggers tumor necrosis factor (TNF) and interferon γ (IFN-γ) to facilitate Kupffer cell and natural killer (NK) cell migration and recruitment within tumors for efficient phagocytosis and killing of cancer cells. Moreover, Kupffer cells stimulate regulatory T cells (Tregs) to recruit cytotoxic CD8+ T cells that are induced by increased TNF and IFN harbored within a compliant ECM. CD8+ T cells release perforin and granzyme to mediate tumor killing. With increased ECM disorganization, high collagen and elastin fiber deposition create a stiff matrix for trapping NK cells. Increased pro-angiogenic growth factors (VEGF, IL-8, and PDGFβ) and matrix metalloproteinases MMP9 and MMP14 generate abnormal vasculature and an EMT that favors tumor growth via TAM recruitment. Highly contractile CAFs also stimulate an EMT under the guidance of TGF-β and IL-6. Independently, a stiff ECM also induces PD-L1 via dendritic cells (DCs), TGF-β, and cyclooxygenase-2 (COX2), all of which suppress CD8+ T cell activity to generate a pro-tumorigenic immune response.
The bioactive domains in the ECM proteins act as ligands for several immune cell receptors and potentially participate in heterotypic “lock and key” ligand-receptor interactions in the liver TME. For example, most of the immune cells including T cells, B cells, NK cells, macrophages, and monocytes express LAIR-1, and the interaction of collagen with LAIR-1 creates an immunosuppressive TME.127 CCN1 induces macrophage adhesion and activation through integrin αMβ2,107 allowing the infiltration of macrophages in the liver. Further, CCN1 induces the expansion of MDSCs that suppress T cell proliferation.107,128
Another limitation in cancer immunotherapy is that TAMs can suppress the activity of tumor-associated T cells and support tumor growth and the notion that TAM subtypes may also vary across the spectrum of solid cancers.129,130 Macrophages cultured in high-density collagen (representative of increased matrix rigidity) were capable of inhibiting T cell proliferation and attracting cytotoxic T cells131 (Figure 3). ECM-sequestered TGF-β1, besides providing matrix stiffness, also promotes the M2 polarization of TAMs.132 When cancer cells are presented as tumor antigens, effector T cells are activated and migrate toward the TME, where they selectively kill cancer cells by releasing granzyme, perforin, and interferon-γ (IFN-γ).133 This cancer-suppressive immune response is inhibited by immune checkpoints and VEGFs.134 The abnormal deposition of ECM components may pose an inhibitory barrier for normal immune surveillance and may prevent cancer cell death by decreasing tumor-derived antigens (Figure 3). There are multiple scenarios by which matrix stiffness caused by aberrant ECM proteins can regulate immune responses in the liver TME, such as through NK cell trapping, TAM recruitment, and PD-L1 expression (Figure 3). During the inflammatory stage of hepatocytes in NASH and HCC, the number of NK cells and cytotoxic CD8+ T cells increases. The NK cells prevent liver fibrosis by destroying the HSCs that initiate fibrosis. The cytolytic CD8+ T cells target the cancer cells via perforin and granzyme B secretion (Figure 3).133 However, the stiff ECM in HCC serves as a physical barrier to the migration and infiltration of NK cells and CD8+ T cells into the tumor, which disrupts immune recognition and tumor cell destruction processes.135 Indeed, the stiffness of the TME can upregulate the expression of PD-L1 and subsequently lead to rapid tumor growth.136 Interestingly, the “lock and key” simulated functional interaction has been recently validated between HCC tumors and their immune cell counterparts via multiregional single-cell RNA sequencing.137 The interaction between OPN with prostaglandin E receptor-4 (PTGER4) in TAMs is predictive of poor survival among patients with HCC.137 Hence, the dysregulated ECM may affect ICI responses in the following ways: (1) formation of a dense matrix barrier that interferes with ICI diffusion and access to cancer cells, (2) increased ECM disorganization and its altered stiffness may act as an immune-cell trap, thereby preventing the immune-surveillance machinery from accessing certain regions of tumors, and (3) creation of a hypoxic liver environment that alters angiogenesis and impacts ICI activity.
Current therapeutic interventions targeting ECM in HCC: What have we learned from other cancer models?
Anti-PD-L1 atezolizumab in combination with anti-VEGF agent bevacizumab has been approved as the first-line treatment in HCC.6 Several other ICIs, such as nivolumab and pembrolizumab, have also been used as single-agent treatments in HCC with good response.120 However, some patients do not respond to ICIs. Despite the extensive usage of different ICIs in different combinations, there is a lack of suitable biomarkers that can predict which ICIs and their combinations would be appropriate to use in HCC. It could be speculated that one of the reasons for the decreased response to ICIs could be the immunosuppressive TME that is generated by the disorganization of the ECM. Through the above-mentioned mechanisms, the disorganized ECM in HCC helps tumor cells to evade the immune system and promotes rapid tumor cell proliferation, angiogenesis, decreased apoptosis, and metastasis (Figure 3). The utilization of renin-angiotensin system (RAS) inhibitors (RASis) such as angiotensin-converting enzyme inhibitors (ACEis) and angiotensin receptor blockers (ARBs) have shown to improve survival in patients with HCC.138 Moreover, patients treated with sorafenib and RASis had better overall outcomes.138 Though the mechanistic involvement of how RASis may act in HCC remains to be resolved, insights derived from pancreatic cancers should pave the way forward. The main target of RASi acts on AngII/AT1R signaling, which shapes the TME by supporting an immunosuppressive milieu.139,140 Preclinical studies confirmed that the angiotensin inhibitor losartan impacts stromal collagen and hyaluronan production associated with decreased expression of profibrotic signals TGF-β1, CCN2, and ET-1. Losartan reduced bulk stress in tumors, resulting in increased vascular perfusion that increased intratumoral delivery of RASis.141 Similar to liver diseases, obesity also increased desmoplasia and impaired drug delivery, both of which were shown to be reversed by genetic and pharmacological inhibition of angiotensin-II type-1 receptor.139 Patients with locally advanced PDAC who received neoadjuvant FOLFIRINOX and losartan followed by chemoradiotherapy were associated with improved survival.142 Thus, the anti-fibrotic effect and the RAS signaling blockade could be beneficial in halting tumor progression.138,140,141 The targeted therapies in HCC with their targets and possible interacting ECM proteins are illustrated in Table 1.
Table 1.
Potential impact of targeted therapies and their combinations on ECM components in HCC
| Targeted therapies | Pharmacological target | Affected ECM protein |
|---|---|---|
| Single-agent ICI | ||
| Pembrolizumab | PD-1 | TGF-β1143 |
| Nivolumab | PD-1 | collagen144,145 |
| Dostarlimaba | PD-1 | collagen144,145 |
| Durvalumaba | PD-L1 | collagen144,145 |
| Combination ICI therapies | ||
| Atezolizumab + bevacizumab | PD-L1 + VEGF | VEGF, fibronectin,146 collagen144,145 |
| Ipilimumab + nivolumab | CTLA-4 + PD-1 | pro-collagen type-III147 |
| Durvalumab + tremelimumab | PD-L1 + CTLA-4 | pro-collagen type-III147 |
| Pembrolizumab + lenvatinib | PD-1 + TKI | collagen144,145 |
| Atezolizumab + cabozantinib | PD-L1 + TKI | collagen144,145 |
| Other targeted therapies | ||
| Sorafenib | multiple receptor tyrosine kinases (RTKs) | agrin, VEGF5,77 |
| CAR T cell therapy | immunotherapy | GPC371,73 |
| GC33 | monoclonal antibodies | GPC371,73 |
| GPC3 peptide vaccine | vaccines | GPC371,73 |
| Recombinant endostatin | peptides | VEGF (ClinicalTrials.gov: NCT03208335) |
| ACEi and ARBs | RASis | collagen, hyaluronic acid140 |
In view of the lack of evidence for HCC-specific studies, the role of collagen as a key mediator of anti-PD-1 (dostarlimab) and anti-PD-L1 (durvalumab) resistance remains speculative.
As collagen deposition and stiffness of the ECM is the main underlying reason for the progression of fibrosis and HCC, different collagen isoforms may be useful as potential targets for treatments. In other solid tumors such as in lung, increased collagen deposition is resistant to PD-1/PD-L1 inhibitors.144 Therefore, therapies directed toward the reduction of collagen may increase T cell infiltration to the tumor and reverse the resistance to ICIs. Cetuximab, an anti-EGFR antibody, can achieve longer periods of retention in collagen-abundant tumors without loss of efficacy and has shown better therapeutic effects.148 Likewise, the retention of anti-inflammatory antibodies at the inflammatory site can be enhanced by conferring a collagen-binding affinity and increasing their therapeutic efficacy.149 By restoring normalcy within the liver vasculature, the combination of anti-PD1 and VEGFR2 has been shown to sensitize HCC cells toward targeted therapy and improve survival.150
Targeting of mechanotransduction at the interface of ECM and liver cancer cells may also represent a new paradigm in HCC therapies. For instance, antibodies targeting agrin reduced oncogenic signaling and liver tumorigenesis by reducing mechanosignaling and tumor angiogenesis.75 Agrin can be secreted by HSCs upon activation by PDGF.77 Sorafenib reduced inflammation, fibrosis, and liver carcinogenesis through PDGF receptor inhibition, which decreased agrin secretion.5,77 Thus, agrin can be considered an emerging therapeutic target in HCC.
Conclusions and future directions
We propose a three-pronged strategy to better understand ECM complexity in driving HCC. First, identify the liver cell types that cause differential deposition of ECM proteins across different stages of cancer initiation and cause heterogeneity in local stiffness of liver tissues and signaling responses. Second, ECM components may serve as biomarkers of HCC and predict responses toward ICI therapies. Therapy-induced changes in the ECM of liver tissues that create a matrix for disease recurrence need special attention. Third, owing to their prominent role in bidirectional feedback in the liver TME, ECM proteins can be potentially targeted to decrease inflammation, fibrosis, and HCC progression based on their relative spatial and temporal distributions. Here, we anticipate that anti-ECM targeting strategies will “loosen up” ECM density, thereby allowing therapies access to cancer cells. Monoclonal antibodies to ECM proteins, engineered fusion constructs containing bioactive domains of ECM moieties, and their associated growth factors, as well as PD-L1 antibodies (Fab portion), could serve as novel recombinant decoy protein traps that provide specificity for targeting aggressive tumor regions. These may antagonize an immunosuppressive liver TME and improve the delivery of standard-of-care therapeutic agents. In summary, we are hopeful that future clinical trials focusing on ECM targeting strategies will pave the way forward for the development of new and effective HCC-restraining therapies.
Outstanding questions
-
•
How can advanced transcriptomics and proteomic approaches comprehend ECM heterogeneity in the liver TME?
-
•
Is the connection between aberrant ECM deposition and matrix stiffness merely a correlative phenotype in HCC or a reflection of initiatory steps in HCC that contribute to a CLD-prone microenvironment?
-
•
Are there any correlative and reliable ECM biomarkers for HCC tumor burden that predict responses to therapies? If so, does prolonged TKI/ICI therapy induce changes in these ECM proteins that may promote HCC recurrence?
-
•
What are potential strategies to target key ECM components in HCC? Can a single agent or combination with present standard-of care treatments be utilized?
-
•
How do we estimate the crosstalk of immune surveillance machinery in rendering changes to the ECM architecture with tumor progression? Would multiregional sequencing of tumors identify local differences in immune responses and the heterogeneity of immune cells?
Acknowledgments
We thank Deanna Conners, Jamie Brooks, and Dr. Sandra Gollnick for critically reading our manuscript. Editorial assistance for this publication was provided by Roswell Park’s Scientific Editing and Research Communications Core (SERCC) Resource supported by a National Cancer Institute (NCI) Cancer Center Support Grant (grant no.NCI P30CA016056). This review was funded in part from start-up funds and the “Developmental Therapeutics” grant from Cancer Center Support Grant – Developmental Funds (5P30CA016056-45) Department of Pharmacology and Therapeutics, RPCC to S.C..
Author contributions
Conceptualization, S.C. and R.I.; writing – original draft preparation, A.M.R. and S.C.; writing – review & editing, S.C., R.I., and A.M.R.; figure preparation, A.M.R. and S.C.; supervision, S.C.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Renuka Iyer, Email: renuka.iyer@roswellpark.org.
Sayan Chakraborty, Email: sayan.chakraborty@roswellpark.org.
References
- 1.Waller L.P., Deshpande V., Pyrsopoulos N. Hepatocellular carcinoma: A comprehensive review. World J. Hepatol. 2015;7:2648–2663. doi: 10.4254/wjh.v7.i26.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 2004;11:97–107. doi: 10.1046/j.1365-2893.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- 3.Tarao K., Nozaki A., Ikeda T., Sato A., Komatsu H., Komatsu T., Taguri M., Tanaka K. Real impact of liver cirrhosis on the development of hepatocellular carcinoma in various liver diseases-meta-analytic assessment. Cancer Med. 2019;8:1054–1065. doi: 10.1002/cam4.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 5.Llovet J.M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J.F., de Oliveira A.C., Santoro A., Raoul J.L., Forner A., et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 6.Finn R.S., Qin S., Ikeda M., Galle P.R., Ducreux M., Kim T.Y., Kudo M., Breder V., Merle P., Kaseb A.O., et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020;382:1894–1905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 7.Casak S.J., Donoghue M., Fashoyin-Aje L., Jiang X., Rodriguez L., Shen Y.L., Xu Y., Jiang X., Liu J., Zhao H., et al. FDA Approval Summary: Atezolizumab Plus Bevacizumab for the Treatment of Patients with Advanced Unresectable or Metastatic Hepatocellular Carcinoma. Clin. Cancer Res. 2021;27:1836–1841. doi: 10.1158/1078-0432.CCR-20-3407. [DOI] [PubMed] [Google Scholar]
- 8.Abou-Alfa G.K., Lau G., Kudo M., Chan S.L., Kelley R.K., Furuse J., Sukeepaisarnjaroen W., Kang Y.-K., Van Dao T., De Toni E.N., et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evidence. 2022;1 doi: 10.1056/EVIDoa2100070. EVIDoa2100070. [DOI] [PubMed] [Google Scholar]
- 9.Cheng A.L., Qin S., Ikeda M., Galle P.R., Ducreux M., Kim T.Y., Lim H.Y., Kudo M., Breder V., Merle P., et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022;76:862–873. doi: 10.1016/j.jhep.2021.11.030. [DOI] [PubMed] [Google Scholar]
- 10.Lu P., Weaver V.M., Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu P., Takai K., Weaver V.M., Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harbor Perspect. Biol. 2011;3:a005058. doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bissell M.J., Hines W.C. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Humphrey J.D., Dufresne E.R., Schwartz M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014;15:802–812. doi: 10.1038/nrm3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhowmick N.A., Neilson E.G., Moses H.L. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singer N.G., Caplan A.I. Mesenchymal stem cells: mechanisms of inflammation. Annu. Rev. Pathol. 2011;6:457–478. doi: 10.1146/annurev-pathol-011110-130230. [DOI] [PubMed] [Google Scholar]
- 16.Zhang D.Y., Friedman S.L. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology. 2012;56:769–775. doi: 10.1002/hep.25670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim M.N., Kim S.U., Kim B.K., Park J.Y., Kim D.Y., Ahn S.H., Song K.J., Park Y.N., Han K.H. Increased risk of hepatocellular carcinoma in chronic hepatitis B patients with transient elastography-defined subclinical cirrhosis. Hepatology. 2015;61:1851–1859. doi: 10.1002/hep.27735. [DOI] [PubMed] [Google Scholar]
- 18.Akima T., Tamano M., Hiraishi H. Liver stiffness measured by transient elastography is a predictor of hepatocellular carcinoma development in viral hepatitis. Hepatol. Res. 2011;41:965–970. doi: 10.1111/j.1872-034X.2011.00846.x. [DOI] [PubMed] [Google Scholar]
- 19.Wang H.M., Hung C.H., Lu S.N., Chen C.H., Lee C.M., Hu T.H., Wang J.H. Liver stiffness measurement as an alternative to fibrotic stage in risk assessment of hepatocellular carcinoma incidence for chronic hepatitis C patients. Liver Int. 2013;33:756–761. doi: 10.1111/liv.12118. [DOI] [PubMed] [Google Scholar]
- 20.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 21.Melero I., Rouzaut A., Motz G.T., Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014;4:522–526. doi: 10.1158/2159-8290.CD-13-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naba A., Clauser K.R., Whittaker C.A., Carr S.A., Tanabe K.K., Hynes R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer. 2014;14:518. doi: 10.1186/1471-2407-14-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naba A., Clauser K.R., Hoersch S., Liu H., Carr S.A., Hynes R.O. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guilliams M., Bonnardel J., Haest B., Vanderborght B., Wagner C., Remmerie A., Bujko A., Martens L., Thoné T., Browaeys R., et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. 2022;185:379–396.e38. doi: 10.1016/j.cell.2021.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weston C.J., Zimmermann H.W., Adams D.H. The Role of Myeloid-Derived Cells in the Progression of Liver Disease. Front. Immunol. 2019;10:893. doi: 10.3389/fimmu.2019.00893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mak K.M., Mei R. Basement Membrane Type IV Collagen and Laminin: An Overview of Their Biology and Value as Fibrosis Biomarkers of Liver Disease. Anat. Rec. 2017;300:1371–1390. doi: 10.1002/ar.23567. [DOI] [PubMed] [Google Scholar]
- 27.Klaas M., Kangur T., Viil J., Mäemets-Allas K., Minajeva A., Vadi K., Antsov M., Lapidus N., Järvekülg M., Jaks V. The alterations in the extracellular matrix composition guide the repair of damaged liver tissue. Sci. Rep. 2016;6:27398. doi: 10.1038/srep27398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wells R.G. Cellular sources of extracellular matrix in hepatic fibrosis. Clin. Liver Dis. 2008;12:759–768. doi: 10.1016/j.cld.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yavuz B.G., Pestana R.C., Abugabal Y.I., Krishnan S., Chen J., Hassan M.M., Wolff R.A., Rashid A., Amin H.M., Kaseb A.O. Origin and role of hepatic myofibroblasts in hepatocellular carcinoma. Oncotarget. 2020;11:1186–1201. doi: 10.18632/oncotarget.27532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishizaki M., Ashida K., Higashi T., Nakatsukasa H., Kaneyoshi T., Fujiwara K., Nouso K., Kobayashi Y., Uemura M., Nakamura S., Tsuji T. The formation of capsule and septum in human hepatocellular carcinoma. Virchows Arch. 2001;438:574–580. doi: 10.1007/s004280000391. [DOI] [PubMed] [Google Scholar]
- 31.Tsuchida T., Friedman S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017;14:397–411. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 32.He Y., Liu T., Dai S., Xu Z., Wang L., Luo F. Tumor-Associated Extracellular Matrix: How to Be a Potential Aide to Anti-tumor Immunotherapy? Front. Cell Dev. Biol. 2021;9:739161. doi: 10.3389/fcell.2021.739161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Y., Cao Y., Xu K., Zhu Y., Qiao Y., Wu Y., Chen J., Li C., Zeng R., Ge G. Dynamically remodeled hepatic extracellular matrix predicts prognosis of early-stage cirrhosis. Cell Death Dis. 2021;12:163. doi: 10.1038/s41419-021-03443-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rojkind M., Giambrone M.A., Biempica L. Collagen types in normal and cirrhotic liver. Gastroenterology. 1979;76:710–719. [PubMed] [Google Scholar]
- 35.Bedossa P., Paradis V. Liver extracellular matrix in health and disease. J. Pathol. 2003;200:504–515. doi: 10.1002/path.1397. [DOI] [PubMed] [Google Scholar]
- 36.Zhou X., Murphy F.R., Gehdu N., Zhang J., Iredale J.P., Benyon R.C. Engagement of alphavbeta3 integrin regulates proliferation and apoptosis of hepatic stellate cells. J. Biol. Chem. 2004;279:23996–24006. doi: 10.1074/jbc.M311668200. [DOI] [PubMed] [Google Scholar]
- 37.Seki E., Schwabe R.F. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066–1079. doi: 10.1002/hep.27332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hastings J.F., Skhinas J.N., Fey D., Croucher D.R., Cox T.R. The extracellular matrix as a key regulator of intracellular signalling networks. Br. J. Pharmacol. 2019;176:82–92. doi: 10.1111/bph.14195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobie R., Wilson-Kanamori J.R., Henderson B.E.P., Smith J.R., Matchett K.P., Portman J.R., Wallenborg K., Picelli S., Zagorska A., Pendem S.V., et al. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep. 2019;29:1832–1847.e8. doi: 10.1016/j.celrep.2019.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Filliol A., Saito Y., Nair A., Dapito D.H., Yu L.X., Ravichandra A., Bhattacharjee S., Affo S., Fujiwara N., Su H., et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature. 2022;610:356–365. doi: 10.1038/s41586-022-05289-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lebbink R.J., Raynal N., de Ruiter T., Bihan D.G., Farndale R.W., Meyaard L. Identification of multiple potent binding sites for human leukocyte associated Ig-like receptor LAIR on collagens II and III. Matrix Biol. 2009;28:202–210. doi: 10.1016/j.matbio.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 42.Mederacke I., Hsu C.C., Troeger J.S., Huebener P., Mu X., Dapito D.H., Pradere J.P., Schwabe R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinet A., Fahem A., Cauchard J.H., Huet E., Vincent L., Lorimier S., Antonicelli F., Soria C., Crepin M., Hornebeck W., Bellon G. Elastin-derived peptides enhance angiogenesis by promoting endothelial cell migration and tubulogenesis through upregulation of MT1-MMP. J. Cell Sci. 2005;118:343–356. doi: 10.1242/jcs.01613. [DOI] [PubMed] [Google Scholar]
- 44.Kanta J., Velebný V., Mergancová J., Ettlerová E., Chlumská A. Elastin content in human fibrotic and cirrhotic liver. Sb. Ved. Pr. Lek. Fak. Karlovy Univerzity. Hradci Kralove. 1990;33:489–494. [PubMed] [Google Scholar]
- 45.Kanta J. Elastin in the Liver. Front. Physiol. 2016;7:491. doi: 10.3389/fphys.2016.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Romier B., Ivaldi C., Sartelet H., Heinz A., Schmelzer C.E.H., Garnotel R., Guillot A., Jonquet J., Bertin E., Guéant J.L., et al. Production of Elastin-Derived Peptides Contributes to the Development of Nonalcoholic Steatohepatitis. Diabetes. 2018;67:1604–1615. doi: 10.2337/db17-0490. [DOI] [PubMed] [Google Scholar]
- 47.Yasui Y., Abe T., Kurosaki M., Higuchi M., Komiyama Y., Yoshida T., Hayashi T., Kuwabara K., Takaura K., Nakakuki N., et al. Elastin Fiber Accumulation in Liver Correlates with the Development of Hepatocellular Carcinoma. PLoS One. 2016;11:e0154558. doi: 10.1371/journal.pone.0154558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao W., Yang A., Chen W., Wang P., Liu T., Cong M., Xu A., Yan X., Jia J., You H. Inhibition of lysyl oxidase-like 1 (LOXL1) expression arrests liver fibrosis progression in cirrhosis by reducing elastin crosslinking. Biochim. Biophys. Acta, Mol. Basis Dis. 2018;1864:1129–1137. doi: 10.1016/j.bbadis.2018.01.019. [DOI] [PubMed] [Google Scholar]
- 49.Maehara J., Masugi Y., Abe T., Tsujikawa H., Kurebayashi Y., Ueno A., Ojima H., Okuda S., Jinzaki M., Shinoda M., et al. Quantification of intratumoral collagen and elastin fibers within hepatocellular carcinoma tissues finds correlations with clinico-patho-radiological features. Hepatol. Res. 2020;50:607–619. doi: 10.1111/hepr.13484. [DOI] [PubMed] [Google Scholar]
- 50.Keller S., Borde T., Brangsch J., Reimann C., Kader A., Schulze D., Buchholz R., Kaufmann J.O., Karst U., Schellenberger E., et al. Assessment of the hepatic tumor extracellular matrix using elastin-specific molecular magnetic resonance imaging in an experimental rabbit cancer model. Sci. Rep. 2020;10:20785. doi: 10.1038/s41598-020-77624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kelsh-Lasher R.M., Ambesi A., Bertram C., McKeown-Longo P.J. Integrin alpha4beta1 and TLR4 Cooperate to Induce Fibrotic Gene Expression in Response to Fibronectin's EDA Domain. J. Invest. Dermatol. 2017;137:2505–2512. doi: 10.1016/j.jid.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aziz-Seible R.S., Lee S.M., Kharbanda K.K., McVicker B.L., Casey C.A. Ethanol feeding potentiates the pro-inflammatory response of Kupffer cells to cellular fibronectin. Alcohol Clin. Exp. Res. 2011;35:717–725. doi: 10.1111/j.1530-0277.2010.01389.x. [DOI] [PubMed] [Google Scholar]
- 53.Kim H., Park J., Kim Y., Sohn A., Yeo I., Jong Yu S., Yoon J.H., Park T., Kim Y. Serum fibronectin distinguishes the early stages of hepatocellular carcinoma. Sci. Rep. 2017;7:9449. doi: 10.1038/s41598-017-09691-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rick J.W., Chandra A., Dalle Ore C., Nguyen A.T., Yagnik G., Aghi M.K. Fibronectin in malignancy: Cancer-specific alterations, protumoral effects, and therapeutic implications. Semin. Oncol. 2019;46:284–290. doi: 10.1053/j.seminoncol.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin T.C., Yang C.H., Cheng L.H., Chang W.T., Lin Y.R., Cheng H.C. Fibronectin in Cancer: Friend or Foe. Cells. 2019;9 doi: 10.3390/cells9010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murphy P.A., Begum S., Hynes R.O. Tumor angiogenesis in the absence of fibronectin or its cognate integrin receptors. PLoS One. 2015;10:e0120872. doi: 10.1371/journal.pone.0120872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dituri F., Gigante G., Scialpi R., Mancarella S., Fabregat I., Giannelli G. Proteoglycans in Cancer: Friends or Enemies? A Special Focus on Hepatocellular Carcinoma. Cancers. 2022;14:1902. doi: 10.3390/cancers14081902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yilmaz Y., Eren F., Colak Y., Senates E., Celikel C.A., Imeryuz N. Hepatic expression and serum levels of syndecan 1 (CD138) in patients with nonalcoholic fatty liver disease. Scand. J. Gastroenterol. 2012;47:1488–1493. doi: 10.3109/00365521.2012.725093. [DOI] [PubMed] [Google Scholar]
- 59.Charchanti A., Kanavaros P., Koniaris E., Kataki A., Glantzounis G., Agnantis N.J., Goussia A.C. Expression of Syndecan-1 in Chronic Liver Diseases: Correlation With Hepatic Fibrosis. Vivo. 2021;35:333–339. doi: 10.21873/invivo.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Metwaly H.A., Al-Gayyar M.M.H., Eletreby S., Ebrahim M.A., El-Shishtawy M.M. Relevance of serum levels of interleukin-6 and syndecan-1 in patients with hepatocellular carcinoma. Sci. Pharm. 2012;80:179–188. doi: 10.3797/scipharm.1110-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nault J.C., Guyot E., Laguillier C., Chevret S., Ganne-Carrie N., N'Kontchou G., Beaugrand M., Seror O., Trinchet J.C., Coelho J., et al. Serum proteoglycans as prognostic biomarkers of hepatocellular carcinoma in patients with alcoholic cirrhosis. Cancer Epidemiol. Biomarkers Prev. 2013;22:1343–1352. doi: 10.1158/1055-9965.EPI-13-0179. [DOI] [PubMed] [Google Scholar]
- 62.Yang N., Mosher R., Seo S., Beebe D., Friedl A. Syndecan-1 in breast cancer stroma fibroblasts regulates extracellular matrix fiber organization and carcinoma cell motility. Am. J. Pathol. 2011;178:325–335. doi: 10.1016/j.ajpath.2010.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Metwaly H.A., El-Gayar A.M., El-Shishtawy M.M. Inhibition of the signaling pathway of syndecan-1 by synstatin: A promising anti-integrin inhibitor of angiogenesis and proliferation in HCC in rats. Arch. Biochem. Biophys. 2018;652:50–58. doi: 10.1016/j.abb.2018.06.007. [DOI] [PubMed] [Google Scholar]
- 64.Allegretta M., Filmus J. Therapeutic potential of targeting glypican-3 in hepatocellular carcinoma. Anti-Cancer Agents Med. Chem. 2011;11:543–548. doi: 10.2174/187152011796011109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Capurro M., Wanless I.R., Sherman M., Deboer G., Shi W., Miyoshi E., Filmus J. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. 2003;125:89–97. doi: 10.1016/s0016-5085(03)00689-9. [DOI] [PubMed] [Google Scholar]
- 66.Ota S., Hishinuma M., Yamauchi N., Goto A., Morikawa T., Fujimura T., Kitamura T., Kodama T., Aburatani H., Fukayama M. Oncofetal protein glypican-3 in testicular germ-cell tumor. Virchows Arch. 2006;449:308–314. doi: 10.1007/s00428-006-0238-x. [DOI] [PubMed] [Google Scholar]
- 67.Shirakawa H., Suzuki H., Shimomura M., Kojima M., Gotohda N., Takahashi S., Nakagohri T., Konishi M., Kobayashi N., Kinoshita T., Nakatsura T. Glypican-3 expression is correlated with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2009;100:1403–1407. doi: 10.1111/j.1349-7006.2009.01206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jia X., Liu J., Gao Y., Huang Y., Du Z. Diagnosis accuracy of serum glypican-3 in patients with hepatocellular carcinoma: a systematic review with meta-analysis. Arch. Med. Res. 2014;45:580–588. doi: 10.1016/j.arcmed.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 69.Wu Y., Liu H., Weng H., Zhang X., Li P., Fan C.L., Li B., Dong P.L., Li L., Dooley S., Ding H.G. Glypican-3 promotes epithelial-mesenchymal transition of hepatocellular carcinoma cells through ERK signaling pathway. Int. J. Oncol. 2015;46:1275–1285. doi: 10.3892/ijo.2015.2827. [DOI] [PubMed] [Google Scholar]
- 70.Cheng W., Tseng C.J., Lin T.T.C., Cheng I., Pan H.W., Hsu H.C., Lee Y.M. Glypican-3-mediated oncogenesis involves the Insulin-like growth factor-signaling pathway. Carcinogenesis. 2008;29:1319–1326. doi: 10.1093/carcin/bgn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shimizu Y., Suzuki T., Yoshikawa T., Endo I., Nakatsura T. Next-Generation Cancer Immunotherapy Targeting Glypican-3. Front. Oncol. 2019;9:248. doi: 10.3389/fonc.2019.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li D., Li N., Zhang Y.F., Fu H., Feng M., Schneider D., Su L., Wu X., Zhou J., Mackay S., et al. Persistent Polyfunctional Chimeric Antigen Receptor T Cells That Target Glypican 3 Eliminate Orthotopic Hepatocellular Carcinomas in Mice. Gastroenterology. 2020;158:2250–2265.e20. doi: 10.1053/j.gastro.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li D., Lin S., Hong J., Ho M. Immunotherapy for hepatobiliary cancers: Emerging targets and translational advances. Adv. Cancer Res. 2022;156:415–449. doi: 10.1016/bs.acr.2022.01.013. [DOI] [PubMed] [Google Scholar]
- 74.Uhm C.S., Neuhuber B., Lowe B., Crocker V., Daniels M.P. Synapse-forming axons and recombinant agrin induce microprocess formation on myotubes. J. Neurosci. 2001;21:9678–9689. doi: 10.1523/JNEUROSCI.21-24-09678.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chakraborty S., Lakshmanan M., Swa H.L.F., Chen J., Zhang X., Ong Y.S., Loo L.S., Akıncılar S.C., Gunaratne J., Tergaonkar V., et al. An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat. Commun. 2015;6:6184. doi: 10.1038/ncomms7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tian C., Öhlund D., Rickelt S., Lidström T., Huang Y., Hao L., Zhao R.T., Franklin O., Bhatia S.N., Tuveson D.A., Hynes R.O. Cancer Cell-Derived Matrisome Proteins Promote Metastasis in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2020;80:1461–1474. doi: 10.1158/0008-5472.CAN-19-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lv X., Fang C., Yin R., Qiao B., Shang R., Wang J., Song W., He Y., Chen Y. Agrin para-secreted by PDGF-activated human hepatic stellate cells promotes hepatocarcinogenesis in vitro and in vivo. Oncotarget. 2017;8:105340–105355. doi: 10.18632/oncotarget.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chakraborty S., Njah K., Pobbati A.V., Lim Y.B., Raju A., Lakshmanan M., Tergaonkar V., Lim C.T., Hong W. Agrin as a Mechanotransduction Signal Regulating YAP through the Hippo Pathway. Cell Rep. 2017;18:2464–2479. doi: 10.1016/j.celrep.2017.02.041. [DOI] [PubMed] [Google Scholar]
- 79.Njah K., Chakraborty S., Qiu B., Arumugam S., Raju A., Pobbati A.V., Lakshmanan M., Tergaonkar V., Thibault G., Wang X., Hong W. A Role of Agrin in Maintaining the Stability of Vascular Endothelial Growth Factor Receptor-2 during Tumor Angiogenesis. Cell Rep. 2019;28:949–965.e7. doi: 10.1016/j.celrep.2019.06.036. [DOI] [PubMed] [Google Scholar]
- 80.Chakraborty S., Njah K., Hong W. Agrin Mediates Angiogenesis in the Tumor Microenvironment. Trends Cancer. 2020;6:81–85. doi: 10.1016/j.trecan.2019.12.002. [DOI] [PubMed] [Google Scholar]
- 81.Chakraborty S., Hong W. Linking Extracellular Matrix Agrin to the Hippo Pathway in Liver Cancer and Beyond. Cancers. 2018;10:45. doi: 10.3390/cancers10020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chakraborty S., Sampath D., Yu Lin M.O., Bilton M., Huang C.K., Nai M.H., Njah K., Goy P.A., Wang C.C., Guccione E., et al. Agrin-Matrix Metalloproteinase-12 axis confers a mechanically competent microenvironment in skin wound healing. Nat. Commun. 2021;12:6349. doi: 10.1038/s41467-021-26717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.El-Karef A., Kaito M., Tanaka H., Ikeda K., Nishioka T., Fujita N., Inada H., Adachi Y., Kawada N., Nakajima Y., et al. Expression of large tenascin-C splice variants by hepatic stellate cells/myofibroblasts in chronic hepatitis C. J. Hepatol. 2007;46:664–673. doi: 10.1016/j.jhep.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 84.Nong Y., Wu D., Lin Y., Zhang Y., Bai L., Tang H. Tenascin-C expression is associated with poor prognosis in hepatocellular carcinoma (HCC) patients and the inflammatory cytokine TNF-alpha-induced TNC expression promotes migration in HCC cells. Am. J. Cancer Res. 2015;5:782–791. [PMC free article] [PubMed] [Google Scholar]
- 85.Fabris L., Cadamuro M., Cagnin S., Strazzabosco M., Gores G.J. Liver Matrix in Benign and Malignant Biliary Tract Disease. Semin. Liver Dis. 2020;40:282–297. doi: 10.1055/s-0040-1705109. [DOI] [PubMed] [Google Scholar]
- 86.Walia A., Yang J.F., Huang Y.H., Rosenblatt M.I., Chang J.H., Azar D.T. Endostatin's emerging roles in angiogenesis, lymphangiogenesis, disease, and clinical applications. Biochim. Biophys. Acta. 2015;1850:2422–2438. doi: 10.1016/j.bbagen.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hu T.H., Huang C.C., Wu C.L., Lin P.R., Liu S.Y., Lin J.W., Chuang J.H., Tai M.H. Increased endostatin/collagen XVIII expression correlates with elevated VEGF level and poor prognosis in hepatocellular carcinoma. Mod. Pathol. 2005;18:663–672. doi: 10.1038/modpathol.3800336. [DOI] [PubMed] [Google Scholar]
- 88.Bollyky P.L., Falk B.A., Wu R.P., Buckner J.H., Wight T.N., Nepom G.T. Intact extracellular matrix and the maintenance of immune tolerance: high molecular weight hyaluronan promotes persistence of induced CD4+CD25+ regulatory T cells. J. Leukoc. Biol. 2009;86:567–572. doi: 10.1189/jlb.0109001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bhattacharya D., Svechkarev D., Souchek J.J., Hill T.K., Taylor M.A., Natarajan A., Mohs A.M. Impact of structurally modifying hyaluronic acid on CD44 interaction. J. Mater. Chem. B. 2017;5:8183–8192. doi: 10.1039/C7TB01895A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.West D.C., Hampson I.N., Arnold F., Kumar S. Angiogenesis induced by degradation products of hyaluronic acid. Science. 1985;228:1324–1326. doi: 10.1126/science.2408340. [DOI] [PubMed] [Google Scholar]
- 91.Sugahara K.N., Murai T., Nishinakamura H., Kawashima H., Saya H., Miyasaka M. Hyaluronan oligosaccharides induce CD44 cleavage and promote cell migration in CD44-expressing tumor cells. J. Biol. Chem. 2003;278:32259–32265. doi: 10.1074/jbc.M300347200. [DOI] [PubMed] [Google Scholar]
- 92.Nikitovic D., Zafiropoulos A., Katonis P., Tsatsakis A., Theocharis A.D., Karamanos N.K., Tzanakakis G.N. Transforming growth factor-beta as a key molecule triggering the expression of versican isoforms v0 and v1, hyaluronan synthase-2 and synthesis of hyaluronan in malignant osteosarcoma cells. IUBMB Life. 2006;58:47–53. doi: 10.1080/15216540500531713. [DOI] [PubMed] [Google Scholar]
- 93.McQuitty C.E., Williams R., Chokshi S., Urbani L. Immunomodulatory Role of the Extracellular Matrix Within the Liver Disease Microenvironment. Front. Immunol. 2020;11:574276. doi: 10.3389/fimmu.2020.574276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pardue E.L., Ibrahim S., Ramamurthi A. Role of hyaluronan in angiogenesis and its utility to angiogenic tissue engineering. Organogenesis. 2008;4:203–214. doi: 10.4161/org.4.4.6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.He X., Liao W., Li Y., Wang Y., Chen Q., Jin J., He S. Upregulation of hyaluronan-mediated motility receptor in hepatocellular carcinoma predicts poor survival. Oncol. Lett. 2015;10:3639–3646. doi: 10.3892/ol.2015.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wen Y., Jeong S., Xia Q., Kong X. Role of Osteopontin in Liver Diseases. Int. J. Biol. Sci. 2016;12:1121–1128. doi: 10.7150/ijbs.16445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu Q., Li L., Miao C., Hasnat M., Sun L., Jiang Z., Zhang L. Osteopontin promotes hepatocellular carcinoma progression through inducing JAK2/STAT3/NOX1-mediated ROS production. Cell Death Dis. 2022;13:341. doi: 10.1038/s41419-022-04806-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shang S., Plymoth A., Ge S., Feng Z., Rosen H.R., Sangrajrang S., Hainaut P., Marrero J.A., Beretta L. Identification of osteopontin as a novel marker for early hepatocellular carcinoma. Hepatology. 2012;55:483–490. doi: 10.1002/hep.24703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sun J., Xu H.M., Zhou H.J., Dong Q.Z., Zhao Y., Fu L.Y., Hei Z.Y., Ye Q.H., Ren N., Jia H.L., Qin L.X. The prognostic significance of preoperative plasma levels of osteopontin in patients with TNM stage-I of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2010;136:1–7. doi: 10.1007/s00432-009-0629-x. [DOI] [PubMed] [Google Scholar]
- 100.Sun T., Li P., Sun D., Bu Q., Li G. Prognostic value of osteopontin in patients with hepatocellular carcinoma: A systematic review and meta-analysis. Medicine (Baltim.) 2018;97:e12954. doi: 10.1097/MD.0000000000012954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Phillips R.J., Helbig K.J., Van der Hoek K.H., Seth D., Beard M.R. Osteopontin increases hepatocellular carcinoma cell growth in a CD44 dependant manner. World J. Gastroenterol. 2012;18:3389–3399. doi: 10.3748/wjg.v18.i26.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dai J., Peng L., Fan K., Wang H., Wei R., Ji G., Cai J., Lu B., Li B., Zhang D., et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene. 2009;28:3412–3422. doi: 10.1038/onc.2009.189. [DOI] [PubMed] [Google Scholar]
- 103.Klement J.D., Paschall A.V., Redd P.S., Ibrahim M.L., Lu C., Yang D., Celis E., Abrams S.I., Ozato K., Liu K. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Invest. 2018;128:5549–5560. doi: 10.1172/JCI123360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhu Y., Yang J., Xu D., Gao X.M., Zhang Z., Hsu J.L., Li C.W., Lim S.O., Sheng Y.Y., Zhang Y., et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68:1653–1666. doi: 10.1136/gutjnl-2019-318419. [DOI] [PubMed] [Google Scholar]
- 105.Ramnath D., Irvine K.M., Lukowski S.W., Horsfall L.U., Loh Z., Clouston A.D., Patel P.J., Fagan K.J., Iyer A., Lampe G., et al. Hepatic expression profiling identifies steatosis-independent and steatosis-driven advanced fibrosis genes. JCI Insight. 2018;3:e120274. doi: 10.1172/jci.insight.120274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Duncan M.B. Extracellular matrix transcriptome dynamics in hepatocellular carcinoma. Matrix Biol. 2013;32:393–398. doi: 10.1016/j.matbio.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 107.Bian Z., Peng Y., You Z., Wang Q., Miao Q., Liu Y., Han X., Qiu D., Li Z., Ma X. CCN1 expression in hepatocytes contributes to macrophage infiltration in nonalcoholic fatty liver disease in mice. J. Lipid Res. 2013;54:44–54. doi: 10.1194/jlr.M026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li Z.Q., Wu W.R., Zhao C., Zhao C., Zhang X.L., Yang Z., Pan J., Si W.K. CCN1/Cyr61 enhances the function of hepatic stellate cells in promoting the progression of hepatocellular carcinoma. Int. J. Mol. Med. 2018;41:1518–1528. doi: 10.3892/ijmm.2017.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen C.C., Kim K.H., Lau L.F. The matricellular protein CCN1 suppresses hepatocarcinogenesis by inhibiting compensatory proliferation. Oncogene. 2016;35:1314–1323. doi: 10.1038/onc.2015.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Francki A., Bradshaw A.D., Bassuk J.A., Howe C.C., Couser W.G., Sage E.H. SPARC regulates the expression of collagen type I and transforming growth factor-beta1 in mesangial cells. J. Biol. Chem. 1999;274:32145–32152. doi: 10.1074/jbc.274.45.32145. [DOI] [PubMed] [Google Scholar]
- 111.Smalling R.V., Delker D.A., Zhang Y., Nieto N., McGuiness M.S., Liu S., Friedman S.L., Hagedorn C.H., Wang L. Genome-wide transcriptome analysis identifies novel gene signatures implicated in human chronic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2013;305:G364–G374. doi: 10.1152/ajpgi.00077.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.El-Youssef M., Mu Y., Huang L., Stellmach V., Crawford S.E. Increased expression of transforming growth factor-beta1 and thrombospondin-1 in congenital hepatic fibrosis: possible role of the hepatic stellate cell. J. Pediatr. Gastroenterol. Nutr. 1999;28:386–392. doi: 10.1097/00005176-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 113.Wells R.G. The role of matrix stiffness in regulating cell behavior. Hepatology. 2008;47:1394–1400. doi: 10.1002/hep.22193. [DOI] [PubMed] [Google Scholar]
- 114.Schrader J., Gordon-Walker T.T., Aucott R.L., van Deemter M., Quaas A., Walsh S., Benten D., Forbes S.J., Wells R.G., Iredale J.P. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192–1205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Olsen A.L., Bloomer S.A., Chan E.P., Gaça M.D.A., Georges P.C., Sackey B., Uemura M., Janmey P.A., Wells R.G. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 2011;301:G110–G118. doi: 10.1152/ajpgi.00412.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lachowski D., Cortes E., Robinson B., Rice A., Rombouts K., Del Río Hernández A.E. FAK controls the mechanical activation of YAP, a transcriptional regulator required for durotaxis. Faseb. J. 2018;32:1099–1107. doi: 10.1096/fj.201700721R. [DOI] [PubMed] [Google Scholar]
- 117.Lorenz L., Axnick J., Buschmann T., Henning C., Urner S., Fang S., Nurmi H., Eichhorst N., Holtmeier R., Bódis K., et al. Mechanosensing by beta1 integrin induces angiocrine signals for liver growth and survival. Nature. 2018;562:128–132. doi: 10.1038/s41586-018-0522-3. [DOI] [PubMed] [Google Scholar]
- 118.Liu L., You Z., Yu H., Zhou L., Zhao H., Yan X., Li D., Wang B., Zhu L., Xu Y., et al. Mechanotransduction-modulated fibrotic microniches reveal the contribution of angiogenesis in liver fibrosis. Nat. Mater. 2017;16:1252–1261. doi: 10.1038/nmat5024. [DOI] [PubMed] [Google Scholar]
- 119.Schreiber R.D., Old L.J., Smyth M.J. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 120.Sangro B., Sarobe P., Hervás-Stubbs S., Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021;18:525–543. doi: 10.1038/s41575-021-00438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oberhammer F.A., Pavelka M., Sharma S., Tiefenbacher R., Purchio A.F., Bursch W., Schulte-Hermann R. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc. Natl. Acad. Sci. USA. 1992;89:5408–5412. doi: 10.1073/pnas.89.12.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dooley S., ten Dijke P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012;347:245–256. doi: 10.1007/s00441-011-1246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gong D., Shi W., Yi S.J., Chen H., Groffen J., Heisterkamp N. TGFbeta signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012;13:31. doi: 10.1186/1471-2172-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Marcoe J.P., Lim J.R., Schaubert K.L., Fodil-Cornu N., Matka M., McCubbrey A.L., Farr A.R., Vidal S.M., Laouar Y. TGF-beta is responsible for NK cell immaturity during ontogeny and increased susceptibility to infection during mouse infancy. Nat. Immunol. 2012;13:843–850. doi: 10.1038/ni.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang L., Pang Y., Moses H.L. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–227. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tang R.Z., Gu S.S., Chen X.T., He L.J., Wang K.P., Liu X.Q. Immobilized Transforming Growth Factor-Beta 1 in a Stiffness-Tunable Artificial Extracellular Matrix Enhances Mechanotransduction in the Epithelial Mesenchymal Transition of Hepatocellular Carcinoma. ACS Appl. Mater. Interfaces. 2019;11:14660–14671. doi: 10.1021/acsami.9b03572. [DOI] [PubMed] [Google Scholar]
- 127.Lebbink R.J., de Ruiter T., Adelmeijer J., Brenkman A.B., van Helvoort J.M., Koch M., Farndale R.W., Lisman T., Sonnenberg A., Lenting P.J., Meyaard L. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J. Exp. Med. 2006;203:1419–1425. doi: 10.1084/jem.20052554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang H., Lian M., Zhang J., Bian Z., Tang R., Miao Q., Peng Y., Fang J., You Z., Invernizzi P., et al. A functional characteristic of cysteine-rich protein 61: Modulation of myeloid-derived suppressor cells in liver inflammation. Hepatology. 2018;67:232–246. doi: 10.1002/hep.29418. [DOI] [PubMed] [Google Scholar]
- 129.Ngambenjawong C., Gustafson H.H., Pun S.H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017;114:206–221. doi: 10.1016/j.addr.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jayasingam S.D., Citartan M., Thang T.H., Mat Zin A.A., Ang K.C., Ch'ng E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019;9:1512. doi: 10.3389/fonc.2019.01512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Larsen A.M.H., Kuczek D.E., Kalvisa A., Siersbæk M.S., Thorseth M.L., Johansen A.Z., Carretta M., Grøntved L., Vang O., Madsen D.H. Collagen Density Modulates the Immunosuppressive Functions of Macrophages. J. Immunol. 2020;205:1461–1472. doi: 10.4049/jimmunol.1900789. [DOI] [PubMed] [Google Scholar]
- 132.Zhang F., Wang H., Wang X., Jiang G., Liu H., Zhang G., Wang H., Fang R., Bu X., Cai S., Du J. TGF-beta induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget. 2016;7:52294–52306. doi: 10.18632/oncotarget.10561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen D.S., Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 134.Fukumura D., Kloepper J., Amoozgar Z., Duda D.G., Jain R.K. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat. Rev. Clin. Oncol. 2018;15:325–340. doi: 10.1038/nrclinonc.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Salmon H., Franciszkiewicz K., Damotte D., Dieu-Nosjean M.C., Validire P., Trautmann A., Mami-Chouaib F., Donnadieu E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest. 2012;122:899–910. doi: 10.1172/JCI45817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Miyazawa A., Ito S., Asano S., Tanaka I., Sato M., Kondo M., Hasegawa Y. Regulation of PD-L1 expression by matrix stiffness in lung cancer cells. Biochem. Biophys. Res. Commun. 2018;495:2344–2349. doi: 10.1016/j.bbrc.2017.12.115. [DOI] [PubMed] [Google Scholar]
- 137.Ma L., Heinrich S., Wang L., Keggenhoff F.L., Khatib S., Forgues M., Kelly M., Hewitt S.M., Saif A., Hernandez J.M., et al. Multiregional single-cell dissection of tumor and immune cells reveals stable lock-and-key features in liver cancer. Nat. Commun. 2022;13:7533. doi: 10.1038/s41467-022-35291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pinter M., Weinmann A., Wörns M.A., Hucke F., Bota S., Marquardt J.U., Duda D.G., Jain R.K., Galle P.R., Trauner M., et al. Use of inhibitors of the renin-angiotensin system is associated with longer survival in patients with hepatocellular carcinoma. United European Gastroenterol. J. 2017;5:987–996. doi: 10.1177/2050640617695698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Incio J., Liu H., Suboj P., Chin S.M., Chen I.X., Pinter M., Ng M.R., Nia H.T., Grahovac J., Kao S., et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016;6:852–869. doi: 10.1158/2159-8290.CD-15-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pinter M., Jain R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017;9:eaan5616. doi: 10.1126/scitranslmed.aan5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chauhan V.P., Martin J.D., Liu H., Lacorre D.A., Jain S.R., Kozin S.V., Stylianopoulos T., Mousa A.S., Han X., Adstamongkonkul P., et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013;4:2516. doi: 10.1038/ncomms3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Murphy J.E., Wo J.Y., Ryan D.P., Clark J.W., Jiang W., Yeap B.Y., Drapek L.C., Ly L., Baglini C.V., Blaszkowsky L.S., et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019;5:1020–1027. doi: 10.1001/jamaoncol.2019.0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Feun L.G., Li Y.Y., Wu C., Wangpaichitr M., Jones P.D., Richman S.P., Madrazo B., Kwon D., Garcia-Buitrago M., Martin P., et al. Phase 2 study of pembrolizumab and circulating biomarkers to predict anticancer response in advanced, unresectable hepatocellular carcinoma. Cancer. 2019;125:3603–3614. doi: 10.1002/cncr.32339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Peng D.H., Rodriguez B.L., Diao L., Chen L., Wang J., Byers L.A., Wei Y., Chapman H.A., Yamauchi M., Behrens C., et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8(+) T cell exhaustion. Nat. Commun. 2020;11:4520. doi: 10.1038/s41467-020-18298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jensen C., Nissen N.I., Von Arenstorff C.S., Karsdal M.A., Willumsen N. Serological assessment of collagen fragments and tumor fibrosis may guide immune checkpoint inhibitor therapy. J. Exp. Clin. Cancer Res. 2021;40:326. doi: 10.1186/s13046-021-02133-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lu L.G., Zhou Z.L., Wang X.Y., Liu B.Y., Lu J.Y., Liu S., Zhang G.B., Zhan M.X., Chen Y. PD-L1 blockade liberates intrinsic antitumourigenic properties of glycolytic macrophages in hepatocellular carcinoma. Gut. 2022;71:2551–2560. doi: 10.1136/gutjnl-2021-326350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jensen C., Madsen D.H., Hansen M., Schmidt H., Svane I.M., Karsdal M.A., Willumsen N. Non-invasive biomarkers derived from the extracellular matrix associate with response to immune checkpoint blockade (anti-CTLA-4) in metastatic melanoma patients. J. Immunother. Cancer. 2018;6:152. doi: 10.1186/s40425-018-0474-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liang H., Li X., Wang B., Chen B., Zhao Y., Sun J., Zhuang Y., Shi J., Shen H., Zhang Z., Dai J. A collagen-binding EGFR antibody fragment targeting tumors with a collagen-rich extracellular matrix. Sci. Rep. 2016;6:18205. doi: 10.1038/srep18205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Katsumata K., Ishihara J., Mansurov A., Ishihara A., Raczy M.M., Yuba E., Hubbell J.A. Targeting inflammatory sites through collagen affinity enhances the therapeutic efficacy of anti-inflammatory antibodies. Sci. Adv. 2019;5:eaay1971. doi: 10.1126/sciadv.aay1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shigeta K., Datta M., Hato T., Kitahara S., Chen I.X., Matsui A., Kikuchi H., Mamessier E., Aoki S., Ramjiawan R.R., et al. Dual Programmed Death Receptor-1 and Vascular Endothelial Growth Factor Receptor-2 Blockade Promotes Vascular Normalization and Enhances Antitumor Immune Responses in Hepatocellular Carcinoma. Hepatology. 2020;71:1247–1261. doi: 10.1002/hep.30889. [DOI] [PMC free article] [PubMed] [Google Scholar]