

Summary
HIV-1 persists indefinitely in people living with HIV (PLWH) on antiretroviral therapy (ART). If ART is stopped, the virus rapidly rebounds from long-lived latently infected cells. Using a humanized mouse model of HIV-1 infection and CD4+ T cells from PLWH on ART, we investigate whether antagonizing host pro-survival proteins can prime latent cells to die and facilitate HIV-1 clearance. Venetoclax, a pro-apoptotic inhibitor of Bcl-2, depletes total and intact HIV-1 DNA in CD4+ T cells from PLWH ex vivo. This venetoclax-sensitive population is enriched for cells with transcriptionally higher levels of pro-apoptotic BH3-only proteins. Furthermore, venetoclax delays viral rebound in a mouse model of persistent HIV-1 infection, and the combination of venetoclax with the Mcl-1 inhibitor S63845 achieves a longer delay in rebound compared with either intervention alone. Thus, selective inhibition of pro-survival proteins can induce death of HIV-1-infected cells that persist on ART, extending time to viral rebound.
Graphical abstract
Highlights
-
•
Venetoclax delays viral rebound in a humanized mouse model of HIV infection
-
•
Venetoclax depletes intact HIV DNA ex vivo in cells from people living with HIV on ART
-
•
The HIV reservoir is enriched in cells with higher expression of BH3-only proteins
Arandjelovic et al. report that venetoclax delays viral rebound following ART cessation in a mouse model of HIV infection and reduces intact HIV DNA following ex vivo treatment of cells from people with HIV on ART. Venetoclax can selectively eliminate HIV-infected cells and should be further investigated as a cure intervention.
Introduction
Although antiretroviral therapy (ART) suppresses HIV-1 replication, pro-virus can persist in long-lived and proliferating CD4+ T cells and is the likely source of rapid viral rebound when ART is interrupted.1,2,3 Persistence of HIV-1 on ART necessitates lifelong treatment and represents the key obstacle to cure. Despite early optimism, it is apparent that transcriptional reactivation of latently infected cells is not sufficient to induce cell death and delay viral rebound,4,5,6,7 and recent evidence suggests that antiapoptotic mechanisms are in place that maintain survival of latently infected cells despite the highly apoptotic environment of productive infection.8,9,10 BH3 mimetics are small-molecule therapeutics that lower the threshold for induction of intrinsic apoptosis by antagonizing the function of Bcl-2 family pro-survival proteins.11,12 Venetoclax is a BH3 mimetic that specifically antagonizes Bcl-213,14 and is approved for the treatment of chemotherapy-refractory chronic lymphocytic leukemia (CLL).15,16 While the ability of venetoclax to kill latent and productively infected cells in vitro has been explored by others,9,10,17 we sought to examine the efficacy of venetoclax in a physiologically relevant in vivo model of HIV-1 persistence on ART and in CD4+ T cells from people living with HIV (PLWH) on ART.
Results
Venetoclax does not delay viral rebound after 3 weeks of treatment in vivo
We generated humanized mice (hu-mice) by reconstituting immunodeficient NOD-scid IL2Rγnull (NSG) neonates with human umbilical cord-derived CD34+ hematopoietic stem cells. At 16 weeks of age, hu-mice were infected intraperitoneally with CCR5-tropic HIV-1JR-CSF. A stable level of plasma HIV-1 RNA was established in animals 3 weeks post-infection and was associated with reductions in peripheral CD4+ T cells (Figures S1B and SD). Following the establishment of viral setpoint, hu-mice received daily ART in food, which reliably suppressed viremia within 7–8 weeks (Figures S1A and SB). To investigate the importance of Bcl-2 in HIV-1 persistence on ART in vivo, virally suppressed mice were dosed orally with 100 mg/kg venetoclax13,18,19 every weekday for 3 weeks. This is an allometrically scaled dosage that corresponds to approximately 480 mg/day in an average human adult, a safe dosage range as determined by extensive clinical experience.15,20 At the end of treatment, ART was interrupted and animals were bled weekly to assess viral load in plasma (Figure S1A). The time between ART interruption and plasma viral RNA detection, known as time to viral rebound, is considered a key surrogate marker for the intact, replication-competent reservoir.21,22 Following a 3-week regimen, both control- and venetoclax-treated mice rebounded within 2 weeks of ART interruption (Figure S1B; median time to rebound: 1 week controls vs. 1 week venetoclax). Overall, there was no difference in time to viral rebound between the two groups (Figure S1C). We did not observe any overt toxicities in venetoclax-treated mice compared with controls. There was also no difference in the frequency of total CD4+ T cells in blood between treatment and control groups at any time post-ART interruption (Figure S1D; p > 0.05 corrected for multiple comparisons), suggesting that venetoclax was not causing non-specific collateral lymphopenia in our model.
Extended venetoclax treatment in vivo delays viral rebound by up to 2 weeks
Short courses of an intervention may only elicit small reductions in the size of the replication-competent reservoir. Our ability to detect such small differences may be compromised by heterogeneity in the frequency of and response to an apoptosis-inducing stimulus across the pool of latently infected cells.23,24,25 Detecting these small differences would require daily bleeding and large numbers of animals. We speculated that an extended intervention would eliminate a greater proportion and diversity of infected cells that persist on ART, revealing measurable differences in rebound kinetics. Before proceeding with our HIV latency model, we sought to determine the impact of long-term venetoclax treatment in vivo on different white blood cell (WBC) subsets in blood. To do this, uninfected hu-mice were treated with venetoclax every weekday for 6 weeks, with a week-long drug holiday halfway through the regime (Figure S2). We observed a transient but statistically significant drop in CD19+ B cells following the first 3 weeks of treatment (Figure S2F), an expected result given the well-documented susceptibility of B cells to venetoclax.18 This difference was no longer significant by week 7, and B cell levels were comparable to control by week 10. Other than a small baseline difference in CD8+ T cells—an inadvertent consequence of our strategy to equilibrate groups based on total CD4+ T cell count—for all other T cell subsets, we did not detect any significant changes in the number of cells. Our in vivo hu-mouse results are corroborated by available human data demonstrating limited T cell lymphopenia after extended venetoclax exposure.20,26,27
To test our hypothesis that an extended intervention would eliminate a greater proportion of the latent reservoir, hu-mice were infected with HIV-1JR-CSF and treated with ART before receiving venetoclax every weekday for 6 weeks (Figure 1A). Similar to the prior experiment with 3-week venetoclax treatment, there was no statistically significant difference in overall numbers of peripheral CD4+ T cells in venetoclax-treated mice compared to controls (Figure 1D; p > 0.05 corrected for multiple comparisons). To further assess the effect of venetoclax on different subsets of lymphocytes, we also sacrificed mice at the conclusion of treatment but before ART interruption and processed lymph nodes for analysis. We observed no statistically significant differences in either the total number or percentage of CD4+ T cells, CD8+ T cells, central memory CD4+ T cells, or effector memory CD4+ T cells (Figure S3). As with the uninfected mice, we observed a significant decrease in CD19+ B cells.18 In all mice treated with vehicle control, virus rebounded within 1 week of ART interruption. However, after 6 weeks of venetoclax, in 5 of 8 (62.5%) treated mice, virus did not rebound until 2 weeks post-ART interruption, while in the remaining 2 of 8 animals (25%), virus did not rebound until 3 weeks post-ART (Figure 1B; median time to rebound: 1 week controls vs. 2 weeks venetoclax). No mice remained aviraemic by 4 weeks post-ART discontinuation. Overall, venetoclax significantly delayed viral rebound by up to 3-fold following cessation of ART in this pre-clinical model (Figure 1C; log-rank Mantel-Cox test p = 0.0018). These results highlight the pro-survival role of Bcl-2 in a proportion of HIV-1-infected cells that persist on ART and support Bcl-2 antagonism as a strategy to preferentially eliminate HIV-1-infected cells.
Figure 1.

Viral rebound was delayed after a 6 week course of venetoclax
(A) Schematic timeline depicting HIV-1 infection, suppressive ART period, and the beginning of each venetoclax treatment cycle (red arrows; administered every weekday).
(B) Plasma viral loads of individual hu-mice over the course of the experiment (n = 6–8 mice per group). Limit of detection (LOD) is indicated with a dotted horizontal line (2.3 log10 RNA copies mL−1).
(C) Kaplan-Maier curve representing the time to viral rebound following ART interruption.
(D) Peripheral CD4+ T cell counts over the course of the experiment.
(E) Frequency of CD4+ T cells expressing HIV-1 msRNA constitutively (i.e., unstimulated) (black) and following 24-h maximal stimulation with PMA/ionomycin (red).
For (E), all data points represent one individual hu-mouse except for gray points, which represent pooled CD4+ T cells from 2 animals. For (E), n = 2 independent experiments. p value for Kaplan-Maier curve (C) was calculated using a log-rank Mantel-Cox test. p values for (E) were calculated using Wilcoxon matched-pairs signed-rank test. p value for each time point in (D) was analyzed using an unpaired t test corrected for multiple comparisons with the Holm-Šìdák method.
To quantify the magnitude of the inducible reservoir in our hu-mouse model of HIV-1 persistence, and to quantify the effect of venetoclax on this reservoir, we employed an ex vivo tat/rev induced limiting-dilution assay (TILDA).28 TILDA uses nested real-time PCR and a limiting-dilution format to measure the frequency of cells harboring viral genomes that produce tat/rev HIV multiply spliced RNA (msRNA) upon maximal stimulation. Following 6-week venetoclax treatment under ART cover, spleen and peripheral lymph nodes from hu-mice were harvested, and total CD4+ T cells were used for TILDA analysis. Spontaneous (baseline) production of msRNA as well as production of msRNA upon stimulation was detected in every sample except one mouse who received vehicle control, for which we did not detect spontaneous production of msRNA (Figure 1E). For both treatment and control groups, and across all samples, the frequency of CD4+ T cells spontaneously producing tat/rev msRNA was lower than the frequency of CD4+ T cells producing msRNA upon maximal PMA/ionomycin stimulation (Figure 1E; median UnstimControl = 53.5, median StimControl = 166; median UnstimVNX = 69.5, median StimVNX = 224.5). Regardless of treatment, the frequency of CD4+ T cells expressing HIV-1 msRNA was significantly greater following stimulation with PMA/ionomycin, consistent with the presence of an inducible HIV-1 reservoir in the peripheral lymph nodes and spleen of our ART-suppressed hu-mice (pcontrol = 0.0156; pVNX = 0.0312). However, we were unable to detect a difference in the frequency of CD4+ T cells with inducible HIV-1 msRNA using the TILDA when comparing control and venetoclax conditions (Figure 1E). These results suggest that in our hu-mouse model, we are unable to directly measure small reductions in the number of HIV-1-infected cells that persist on ART, which may contribute to the observed delay in viral rebound. Importantly, there was no reduction in the frequency of CD4+ T cells that constitutively express HIV-1 msRNA following venetoclax treatment, indicating that our observed delays in rebound are unlikely to be the result of killing actively infected cells.
Combined Bcl-2 and Mcl-1 inhibition achieves a greater delay in viral rebound compared with either strategy alone
The eventual viral rebound following ART cessation in mice treated with venetoclax suggested that not all infected cells that persist are sensitive to killing through Bcl-2 inhibition over the 6-week course of treatment. Together with our 3-week treatment data, we speculated that HIV-infected cells that persist on ART may vary in their sensitivity to apoptosis over time, reflecting heterogeneity in the viral reservoir. This could be examined by extending the duration of venetoclax to determine if longer courses progressively kill more infected cells. However, such an analysis is precluded in our model, as HIV-1-infected hu-mice eventually develop bone marrow failure.29 Instead of lengthening treatment courses of venetoclax alone, we sought to cause a more profound reduction in the apoptotic threshold by combining complementary BH3 mimetics. As a first step, we investigated whether additional Bcl-2 family members are important in the survival of infected cells on ART, employing a different BH3 mimetic designed to specifically inhibit the pro-survival protein Mcl-1.30 A critical regulator of T cell development and survival, Mcl-1 represents an additional target for priming infected cells to die and delaying viral rebound.18,31,32 Hu-mice undergoing suppressive ART were administered the Mcl-1 inhibitor S63845 intravenously twice weekly for 3 weeks (Figure 2A). Treatment with S63845 alone did not significantly delay viral rebound after cessation of ART compared with vehicle controls (Figures 2B and 2C; median time to rebound: 1 week controls vs. 1 week S63845; p = 0.1255). In control mice, virus rebounded within 1 week of ART interruption, as occurred in 6 of 8 mice (75%) treated with S63845. However, in 2 of 8 (25%) mice treated with S63845, virus did not rebound until 2 weeks post-ART interruption (Figure 2C). Similar to 3 weeks of venetoclax treatment, no difference in blood total CD4+ T cells was observed between S63845-treated mice and vehicle controls (Figure 3D; p > 0.05 corrected for multiple comparisons).
Figure 2.

A delay in viral rebound was not detected after 3 weeks of S63845 treatment
(A) Schematic timeline depicting HIV-1 infection, suppressive ART period, and the dosing regimen of S63845 (blue arrows; twice weekly).
(B) Plasma viral loads of individual hu-mice over the course of the experiment (n = 8 mice per group). LOD is indicated with a dotted horizontal line (2.3 log10 RNA copies mL−1).
(C) Kaplan-Maier curve representing the time to viral rebound following ART interruption.
(D) Peripheral CD4+ T cell counts over the course of the experiment.
p value for Kaplan-Maier curve (C) was calculated using a log-rank Mantel-Cox test. p value for each time point in (D) was analyzed using an unpaired t test corrected for multiple comparisons with the Holm-Šìdák method.
Figure 3.

Viral rebound was delayed after a 3 week combined course of venetoclax and S63845
(A) Schematic timeline depicting HIV-1 infection, suppressive ART, and the dosing regimen of venetoclax (red arrows; administered every weekday) and S63845 (blue arrows; twice weekly).
(B) Plasma viral loads of individual hu-mice over the course of the experiment (n = 4–7 mice per group). LOD is indicated with a dotted horizontal line (2.3 log10 RNA copies mL−1).
(C) Kaplan-Maier curve representing the time to viral rebound following ART interruption.
(D) Peripheral CD4+ T cell counts over the course of the experiment.
p value for Kaplan-Maier curve (C) was calculated using a log-rank Mantel-Cox test. p value for each time point in (D) was analyzed using an unpaired t test corrected for multiple comparisons with the Holm-Šìdák method. “845” = S63845.
We next examined if the combination of a Bcl-2 and Mcl-1 inhibitor over a short window could effectively sensitize infected cells that persist on ART to apoptosis and delay time to viral rebound post-cessation of ART. Hu-mice were suppressed with ART before undergoing 3 weeks of combined venetoclax (anti-Bcl-2) and S63845 (anti-Mcl-1) treatment (Figure 3A). In the cohort of mice receiving both BH3 mimetics, 3 of 7 mice were euthanized due to unexpected illness. This combination regimen was repeated in HIV-1-infected, ART-suppressed (n = 13) as well as uninfected (n = 5) hu-mice, and after 3 weeks of combination therapy, no animals required euthanization due to morbidity. Following treatment and throughout ART interruption follow-up, there was no statistically significant difference in overall numbers of blood total CD4+ T cells in combination-treated mice compared with controls (Figure 3D; p > 0.05 corrected for multiple comparisons). Similar to previous experiments, in the majority of vehicle-treated control mice, virus rebounded within 1 week of ART interruption (5 of 7 mice; 71%) (Figure 3B). Of those receiving combination therapy that underwent analytical treatment interruption, in 2 of 4 mice (50%), virus rebounded 2 weeks post-ART discontinuation, while in a further 2 mice, virus did not rebound until 4 weeks post-ART (Figures 3B and 3C). During the treatment interruption period, there was a trend toward decreased viral loads in mice receiving combination therapy, although this was only observed up until 19 weeks post-infection (Figure 3B). Following only a 3-week therapeutic cycle, the combination of venetoclax and S63845 significantly delayed time to viral rebound by up to 4-fold after ART cessation in our hu-mouse model of HIV-1 infection (Figure 3C; log-rank Mantel-Cox test p = 0.0255; median time to rebound: 1 week controls vs. 3 week combination therapy). Therefore, combined antagonism of Bcl-2 and Mcl-1 sensitizes infected cells to die in a shorter time period, resulting in a longer time to viral rebound, which was not seen with either venetoclax or S63845 alone when the drug was administered for only 3 weeks.
Venetoclax administration reduces total and intact HIV-1 pro-viral DNA in CD4+ T cells from PLWH on ART
To investigate the impact of venetoclax on the latent reservoir ex vivo, we collected peripheral blood mononuclear cells from PLWH on suppressive ART (Table S1). We quantified HIV-1 DNA in the pool of remaining CD4+ T cells following 24-h venetoclax exposure. We first measured total integrated HIV-1 DNA using Alu PCR as previously described4,33,34 and observed a significant dose-dependent reduction in total integrated HIV-1 DNA, with a median fold change of 0.39× at the 100-nM dose relative to DMSO control (Figures 4A and 4B; p100nM = 0.031). Notably, this PCR assay does not distinguish between defective and intact pro-viral DNA. To determine whether venetoclax is also capable of eliminating intact HIV-1 DNA, we performed the intact pro-viral DNA assay (IPDA) on 11 donors.24 We detected intact pro-virus in 10 out of these 11 donors, observing a dose-dependent reduction in the fold change of intact HIV-1 DNA relative to DMSO (Figures 4C and 4D). In one donor (donor 3), intact HIV-1 DNA was eliminated to below the limit of detection at the 100-nM dose. Overall, we observed a 0.58× median fold change in intact HIV-1 DNA at the highest 100 nM venetoclax dose, although this did not achieve statistical significance (p = 0.064) (Figures 4C and 4D). We did not observe comparable dose-dependent reductions in the proportion of 3′ or 5′ defective pro-virus, relative to DMSO-treated samples (Figures S4D and S4E). Additionally, our analysis showed that, at the 10 nM and 100 nM doses, the fold change reduction of HIV-1 DNA was greater for intact compared to 3′ and 5′ defective DNA (Figures 4C, 4D, and S4). This difference was greatest at the 100 nM dose, where we detected median fold increases of 1.06× and 1.15× for 5′ and 3′ defective pro-virus, respectively, compared with a 0.58× fold decrease in intact HIV-1 DNA. These comparisons support the conclusion that venetoclax preferentially depletes a pool of intact HIV-1-infected cells that persist on ART.
Figure 4.

CD4+ T cells from PLWH on ART that survive ex vivo venetoclax co-culture have reduced levels of total and intact HIV-1 pro-viral DNA and exhibit a less apoptotic transcriptional signature
Total CD4+ T cells isolated from peripheral blood of PLWH on ART were co-cultured with venetoclax (VNX) or DMSO control for 24 h before washing and harvesting 24 h later for quantification of HIV-1 DNA. For flow cytometry analysis and RNA sequencing (RNA-seq), cells were harvested immediately following 24 h VNX co-culture.
(A) Fold change in total integrated HIV-1 DNA per million CD4+ T cells relative to DMSO control (n = 6 donors).
(B) Absolute frequency of total integrated HIV-1 DNA per million CD4+ T cells for the 100 nM dose.
(C) Fold change in intact HIV-1 DNA per million CD4+ T cells (n = 11 donors).
(D) Absolute frequency of intact HIV-1 DNA per million CD4+ T cells for the 100 nM dose.
(E) Heatmap visualizing the normalized proportion of dead/dying Violet+ cells within stated CD4+ T cell subsets at different VNX concentrations (n = 5 donors).
(F and G) Normalized expression values by VNX treatment group for (F) pro-death and (G) pro-survival human Bcl-2 family genes. NA, naive; CM, central memory; EM, effector memory; TM, transitional memory; TD, terminally differentiated. For (B) and (D), MFC, median fold change.
p values for (A) and (C) were calculated using a one-sample Wilcoxon signed-rank test. p values for (B) and (D) were calculated using a Wilcoxon matched-pairs signed-rank test. Error bars in (A) and (C) show median ± 95% confidence interval (CI). Boxplots in (F) and (G) show median ± interquartile range. Each symbol represents a different donor.
Naive and effector memory CD4+ T cells display the greatest sensitivity to venetoclax treatment ex vivo
Although venetoclax does not cause significant CD4+ T cell lymphopenia in either our hu-mice (Figure S2) or human clinical trials,20 we sought to examine the effect of venetoclax on different cell subsets ex vivo. Total CD4+ T cells from leukapheresis samples of PLWH on ART were isolated and treated with increasing concentrations of venetoclax up to 100 nM (Figure 4E). We found that naive and effector memory CD4+ T cells had the greatest sensitivity to depletion with venetoclax, with average proportions of 32% and 29.9% Violet+ dead/dying cells, respectively, observed in these cell types at the highest venetoclax concentration (Figure 4E). Conversely, we observed minimal cell death in central and transitional memory CD4+ T cells and only moderate impact in terminally differentiated cells. These data suggest that under ex vivo culture conditions, venetoclax preferentially sensitizes naive and effector memory CD4+ T cells to death while mostly sparing central and transitional memory T cells as well as terminally differentiated cells. Interestingly, at the most efficacious dose of 100 nM, we also noted that the fold change reductions in total integrated DNA (Figures 4A and 4B; median fold change [MFC]100nM = 0.39) and intact HIV-1 DNA (Figure 4B; MFC100nM = 0.58) were greater than the proportion of cell death induced by venetoclax across all subsets analyzed. These instances of depletion of HIV-1 DNA beyond the level of background cell death are consistent with preferential targeting of a pool of infected cells that persist on ART.
It is important to note, however, that venetoclax appeared to be causing apoptosis more broadly in our ex vivo biospecimens compared with our in vivo observations (Figures 1, 2, 3, and S2) as well as human trial data.20,26 It is likely that confounding stressors associated with ex vivo culturing, processing, and freeze/thaw cycles may sensitize a broader array of cells to venetoclax-induced apoptosis. Collectively, our in vivo and ex vivo results align to support the notion that venetoclax is clearing a subset of HIV-1-infected cells that persist on ART, including cells with intact HIV-1 DNA, an outcome that is not associated with overt toxicity in our animal model and that would not be predicted to cause toxicity in humans based on the mean concentration of venetoclax achieved in vivo at the licensed dose of 400 mg/day.15,31
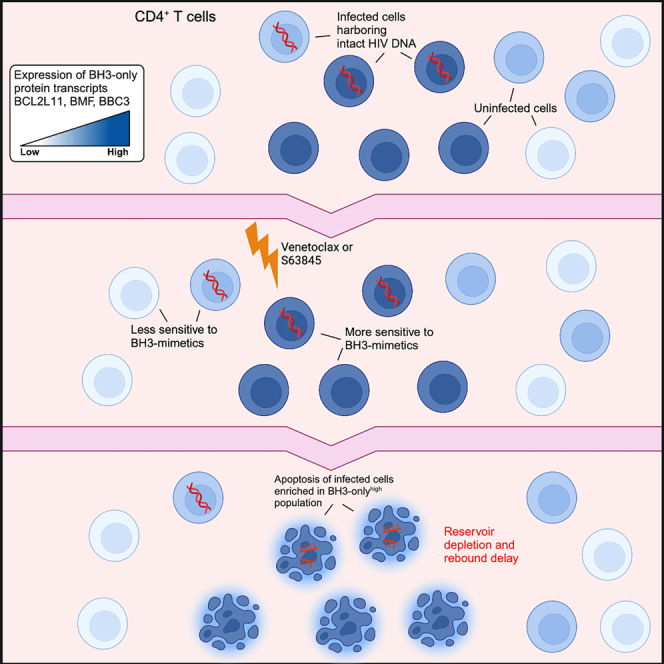
The pro-apoptotic proteins BIM, BMF, and BBC3 are expressed at higher baseline levels in CD4+ T cells sensitive to venetoclax killing
We also performed bulk RNA sequencing on DMSO- and venetoclax-treated total CD4+ T cells. Specifically, we wanted to document transcriptional changes within the network of intrinsic apoptosis proteins, which we hypothesized would help us better understand the profile of cells being sensitized to death by venetoclax. Transcriptomic analysis revealed a dose-dependent downregulation of the pro-apoptotic BH3-only proteins BIM (BCL2L11; false discovery rate [FDR] = 1.23 × 10−5, log fold change [logFC] = −0.84), BMF (FDR = 0.046, logFC = −0.42), and BBC3 (FDR = 0.006, logFC = −0.54) relative to DMSO, suggesting enrichment for a population with cell-intrinsic decreases in apoptotic potential within the surviving CD4+ T cell pool following venetoclax co-culture (Figures 4F and 4G). We did not detect changes in relative expression for any pro-survival Bcl-2 family members (Figure 4G). Overall, this sequencing result suggests that venetoclax eliminates a CD4+ T cell population ex vivo that is sensitive to pro-apoptotic intervention by virtue of increased baseline BIM, BMF, and BBC3 transcriptional expression. We propose that this population is enriched for HIV-1-infected cells that are primed for apoptosis but that survive and persist on ART, thus representing an attractive target for apoptosis-inducing compounds.
Discussion
We proposed that HIV-1-infected cells that persist on ART will have diversity in their apoptotic thresholds due to cell-intrinsic variability in levels of pro- and antiapoptotic factors. Those CD4+ T cells that survive productive infection or that were latent initially—persisting on ART, escaping cell death, and contributing to a pool of cells harboring replication-competent pro-virus that rebounds upon ART interruption—may retain a survival capacity that tolerates higher levels of apoptotic stress and higher levels of pro-apoptotic proteins. This may be analogous to virus-specific CD8+ T cells that survive the contraction phase of an immune response after acute viral infection in mice, which express, but tolerate, higher levels of pro-apoptotic proteins.35 Due to this apoptotic legacy, we suggest that infected cells that persist on ART are distinct from other infected or healthy cells such that their pro-survival phenotype allows them to persist in a way that renders them susceptible to pro-apoptotic therapeutics, including the BH3 mimetic venetoclax. This phenomenon is also observed in clinical studies, in which malignancies tolerating and expressing the highest level of pro-apoptotic proteins are the most susceptible to venetoclax treatment.36 The present study used samples from PLWH on ART to interrogate the transcriptional profile of intrinsic apoptosis mediators in CD4+ T cells that are resistant to venetoclax killing ex vivo. From our results, we were able to infer that the pro-apoptotic BH3-only proteins BIM, BMF, and BBC3 are transcriptionally expressed at higher baseline levels in the venetoclax-sensitive population of CD4+ cells. Furthermore, the accompanying decrease in intact HIV-1 DNA suggests that this eliminated cell subset is enriched for infected cells that persist on ART in the majority of ex vivo samples tested. Indeed, the in vivo and ex vivo findings presented herein are supported by two recent reports37,38 describing antiapoptotic compensatory mechanisms, including increased BCL2 expression, in HIV-DNA+ cells from PLWH on ART, thus reinforcing our strategy for reservoir elimination. Additionally, Ren et al. have shown that BMF is overexpressed in productively HIV-1-infected total and central memory CD4+ T cells, further supporting our hypothesis that infected cells that persist on ART may harbor intrinsic tolerance mechanisms that counter the pro-apoptotic milieu of productive infection.39 BIM and BMF are critical regulators of lymphocyte homeostasis,40 and both can antagonize Bcl-2, while BIM also mediates apoptotic priming through a dynamic interaction with Mcl-1.41,42 Of note, binding of released BIM to Mcl-1 has been suggested as a mechanism of venetoclax resistance,41,43 and this is consistent with our in vivo pre-clinical data demonstrating a longer delay to viral rebound following combined Bcl-2 and Mcl-1 antagonism. Together, these findings support a model where compensatory antiapoptotic mechanisms allow infected CD4+ T cells to tolerate higher levels of BH3-only proteins, thereby favoring persistence on ART while simultaneously priming them for BH3-mimetic-mediated killing.
It remains unclear in our pre-clinical model, as well as in PLWH, whether virus persists on ART entirely in a latent form as productively infected cells in distinct anatomical sites or whether there is a spectrum of transcriptional activity in infected cells (reviewed in Zerbato et al.44). Although the initial descriptions of HIV-1 latency highlighted the long half-life of latently infected resting CD4+ central memory cells,1 we now know that virus can persist in all T cell subsets,45,46,47,48,49,50,51,52,53 in long-lived and proliferating cells,54 as non-inducible pro-virus,55 with intact and defective virus,24,56,57 and in cells that are transcriptionally and translationally active.58,59 We are unable to distinguish between these possibilities in our pre-clinical mouse model. However, we argue that the effects we describe are not due to apoptosis of productively infected cells or residual virus replication that persists on ART. This conclusion is supported by our in vivo TILDA results, which do not show a venetoclax-mediated decrease in the frequency of cells that constitutively express HIV-1 msRNA. In addition, if it was the case that venetoclax was killing productively infected cells, then we would expect pro-apoptotic therapies to delay rebound even with very short courses of treatment. Instead, we found that pro-apoptotic therapies needed a longer therapeutic interval for efficacy to manifest, perhaps reflecting variable sensitivity of the latent reservoir over time and/or stochastic release of virus from latently infected cells. Spatiotemporal, extrinsic, and intrinsic physiological perturbations, including T cell activation, may variably sensitize latently infected cells over time, making them susceptible to pro-apoptotic drugs.60
Interestingly, our ex vivo flow cytometry analysis of CD4+ T cells treated with venetoclax revealed a marked sensitivity among the naive and effector memory populations, both of which have been shown to harbor a large and inducible HIV-1 latent reservoir.61,62 While this may partially explain the decrease in intact HIV-1 DNA observed in our ex vivo donor samples, we notably did not observe significant depletion of naive or effector memory CD4+ T cells in our hu-mice following 6 weeks of venetoclax. Indeed, our in vivo experiments did not reveal meaningful changes in any major CD4+ lymphocyte subsets of interest, despite a significant delay in time to viral rebound upon analytical treatment interruption. This population analysis aligns with available studies characterizing human T cell subsets post-venetoclax administration20,26 and may help to address concerns surrounding non-specific lymphopenia in vivo. Using a pre-clinical model of HIV-1 latency, our work provides in vivo evidence that Bcl-2 inhibition depletes HIV-1-infected cells that persist on ART. The hu-mouse model recapitulates key aspects of viral rebound following ART-mediated suppression to support the translational merit of our data.63,64 Nevertheless, our hematopoietic stem cell (HSC)-derived hu-mice are unable to elicit a strong and consistent HIV-1-specific CTL response, and reconstitution with human macrophages and natural killer (NK) cells is almost non-existent.65,66 While innate and adaptive immune responses are ineffective in clearing HIV-infected cells that persist on ART, HIV-1-specific T cells enhance clearance of latently infected cells ex vivo,67 their frequency is inversely correlated to the size of the reservoir in PLWH on ART,68,69,70 and HIV-1-specific T cells could potentially augment sensitivity to pro-apoptotic therapies, as recently described.10,71 In addition, Chandrasekar and colleagues used hu-mice to investigate the efficacy of venetoclax in limiting HIV-1 viral load during uncontrolled acute infection.71 Although this mouse model differs substantially from our latency model, the authors demonstrate reduced HIV RNA copies relative to CD4+ T cell count and propose that venetoclax augments the death of productively HIV-infected cells via innate and adaptive immune mediators. It is therefore conceivable that we have underestimated the efficacy of venetoclax in clearing latently infected cells and that we may observe greater efficacy in immunocompetent PLWH on ART.
Our data show that venetoclax depletes HIV-1-infected cells in blood from PLWH on ART ex vivo and prolongs time to viral rebound following cessation of ART in HIV-infected hu-mice. We did not observe a significant loss of peripheral or lymph node (LN) CD4+ T cells in venetoclax-treated hu-mice, consistent with observations in human clinical trials.15,20,27 However, it is also important to note that our mice lack cytochrome P450 3A4 (CYP3A4), a major metabolism pathway for venetoclax,72,73 and further study is warranted to determine how this may affect HIV-1-infected cell clearance or lymphocyte depletion. Nonetheless, the pharmacological and toxicity profile of venetoclax is well documented in humans, with few major side effects relative to other chemotherapeutics,15 thus positioning it as a prime candidate for further clinical investigation in the setting of HIV-1 cure research. The optimal strategy for eliminating the HIV-1 reservoir remains unclear and may well require a combinatorial regimen involving transcriptional reactivators,74 death sensitizers,17,75 or neutralizing antibodies.76,77 Importantly, our results demonstrate that BH3 mimetics alone induce a significant delay in viral rebound post-cessation of ART and are a potentially feasible and effective clinical approach toward achieving a cure for HIV-1.
Limitations of the study
Our study has several limitations. The in vivo hu-mouse model does not recapitulate key elements of the human immune response, such as HIV-1-specific CD8+ T cells or complete reconstitution with human innate immune cells. Our ability to characterize reservoir and/or rebound dynamics over a longer period of time is hampered by the fact that our hu-mice display eventual bone marrow failure. Furthermore, using the TILDA, we were unable to detect a decrease in the frequency of cells with inducible HIV-1 msRNA following venetoclax administration. Alternative quantitative assays for measuring the in vivo reservoir in mice may be needed in future experiments.
While we were able to demonstrate a significant reduction in HIV-1 DNA following venetoclax treatment ex vivo, the specific reservoir being depleted remains unclear. Whether this depletion is primarily attributable to a decline of truly latent pro-virus, or the active reservoir, is unknown. In future studies, employment of more complex reservoir measurements, such as parallel HIV RNA, integration site, and proviral sequencing (PRIP-seq),78 may help to distinguish these two populations. Moreover, we utilized the IPDA to enumerate intact HIV-1 DNA; however, this assay has been shown to overestimate the quantity of intact pro-virus.79,80
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BV786 Mouse anti-Human CD45 | BD Horizon | Cat# 563716; RRID:AB_2716864 |
| APC-H7 Mouse anti-Human CD4 | BD Pharmingen | Cat# 560158; RRID:AB_1645478 |
| PerCP-Cy5.5 Mouse anti-Human CD8 | BD Pharmingen | Cat# 560662; RRID:AB_1727513 |
| APC Mouse anti-Human CD8 | BD Pharmingen | Cat# 555369; RRID:AB_398595 |
| PE-Cy7 Mouse anti-Human CD3 | BD Pharmingen | Cat# 563423; RRID:AB_2738196 |
| BV510 Mouse anti-Human CD19 | BD Pharmingen | Cat# 562947; RRID:AB_2737912 |
| PE-CF594 Mouse Anti-human CD45RO BD | BD Biosciences | Cat# 562299; RRID:AB_11154398 |
| BV421 Mouse Anti-human CD14 | BD Biosciences | Cat# 558121; RRID:AB_397041 |
| PE-Cy7 Mouse Anti-human CD45RA | BD Biosciences | Cat# 337167; RRID:AB_647424 |
| PE Mouse anti-Mouse CD45.1 | BD Pharmingen | Cat# 561872; RRID:AB_395044 |
| PE-Cy5 Mouse anti-Human CD62L | BD Pharmingen | Cat# 555545; RRID:AB_395929 |
| BV421 Mouse anti-Human CD44 | BD Horizon | Cat# 562890; RRID:AB_2737869 |
| PE-Cy5 Mouse anti-Human CD45 | BD Pharmingen | Cat# 555484; RRID:AB_395876 |
| PE Rat anti-Mouse LY6C + LY6G (Gr-1) | BD Pharmingen | Cat# 553128; RRID:AB_394644 |
| Alexa Fluor 488 Mouse anti-Human CD197 (CCR7) | BioLegend | Cat# 353205; RRID:AB_10916389 |
| BV711 Mouse anti-Human CD27 | BD Horizon | Cat# 563167; RRID:AB_2738042 |
| PE Mouse anti-Human CD3 | BD Pharmingen | Cat# 555340; RRID:AB_395746 |
| APC Mouse anti-Human CD4 | BD Pharmingen | Cat# 555349; RRID:AB_398593 |
| Bacterial and virus strains | ||
| OneShot™ Stbl3™ chemically-competent E.coli | ThermoFisher Scientific | Cat# C737303 |
| Biological samples | ||
| Cord Blood CD34+ Stem/Progenitor cells | Lonza | Cat# 2C-101 |
| PBMC samples from people living with HIV on ART | The Alfred Hospital and The Avenue Hospital, Melbourne, AUS | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Venetoclax | Active Biochem or SelleckChem | Cat# A-1231 or S8048, respectively |
| S63845 | MedChemExpress | Cat# HY-100741 |
| Tenofovir disoproxil fumarate | Gilead Sciences | Eviplera |
| Emtricitabine | Gilead Sciences | Eviplera |
| Rilpivirine | Gilead Sciences | Eviplera |
| Raltegravir (for in vivo use) | Merck | Isentress |
| Efavirenz | NIH HIV Reagent Program | Cat# HRP-4624, https://www.hivreagentprogram.org/Catalog/HRPAntimicrobialCompoundsandOtherChemicals/HRP-4624.aspx |
| Zidovudine | NIH HIV Reagent Program | Cat# ARP-3485, https://www.hivreagentprogram.org/Catalog/HRPAntimicrobialCompoundsandOtherChemicals/ARP-3485.aspx |
| Raltegravir (for ex vivo use) | NIH HIV Reagent Program | Cat# HRP-11680, https://www.hivreagentprogram.org/Catalog/HRPAntimicrobialCompoundsandOtherChemicals/HRP-11680.aspx |
| Phorbol-12-myristate-13-acetate (PMA) | Sigma | Cat# P8139 |
| Ionomycin calcium salt | Sigma | Cat# I0634 |
| Critical commercial assays | ||
| Invisorb Virus RNA HTS 96 Kit/C | Stratec | Cat# 7043300300 |
| SYBR™ Green RNA-to-CT™ 1-step Kit | ThermoFisher Scientific | Cat# 4389986 |
| HIV-1 p24 ELISA kit | Jomar Life Research | Cat# XB-1000 |
| AllPrep DNA/RNA Mini kit | Qiagen | Cat# 80204 |
| TruSeq RNA Library Prep kit v2 | Illumina | Cat# RS-122-2001 |
| Qubit™ DNA BR assay kit | ThermoFisher Scientific | Cat# Q32850 |
| LIVE/DEAD™Fixable Violet Dead Cell Stain kit | ThermoFisher Scientific | Cat# L34955 |
| QIAamp DNA Mini Kit (250) | Qiagen | Cat# 51306 |
| EasySep™ Human CD4+ T cell Isolation kit | StemCell Technologies | Cat# 17952 |
| Deposited data | ||
| RNA-Seq data at Gene Expression Omnibus | Gene Expression Omnimus (GEO) | GEO: GSE222783 |
| Original R script used in the processing and analysis of the RNA-Seq data | GitHub | https://github.com/sheerind-wehi/venetoclax_RNA_seq/blob/main/QC%20%26%20DGE%20analysis.Rmd |
| Experimental models: Cell lines | ||
| HEK293T cells | Pellegrini Lab, The Walter and Eliza Hall Institute of Medical Research, Melbourne, AUS | N/A |
| Experimental models: Organisms/strains | ||
| NOD.Cg-PrkdcscidIl2rgtm1Wjl/Sz mice (NSG) | The Jackson Laboratory | Strain# 005557 |
| Oligonucleotides | ||
| HIV-1 Gag forward primer: AAC ACC ATG CTA AAC ACA GTG G | Integrated DNA Technologies | N/A |
| HIV-1 Gag reverse primer: GCT TCC TCA TTG ATG GTC TCT T | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| HIV-1, strain JR-CSF infectious molecular clone (pYK-JRCSF) | NIH HIV Reagent Program | Cat# ARP-2708 |
| Software and algorithms | ||
| BD FACSDiva v9.0 | BD Biosciences | https://www.bdbiosciences.com/enau/products/software |
| FlowJo v10 | BD | https://www.flowjo.com/solutions/flowjo/downloads |
| Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Limma | Ref. 71 | v3.47.9 |
| EdgeR | Ref. 72 | v3.33.2 |
| Rsubread | Ref. 69 | v2.4.2 |
| Featurecounts | Ref. 70 | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marc Pellegrini (pellegrini@wehi.edu.au).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human participants
All participants provided informed consent to undergoing leukapharesis and the protocol was approved by the Human Research and Ethics Committees at Alfred Health and University of Melbourne, Melbourne, Australia. People living with HIV on ART were recruited from The Alfred Hospital and The Avenue Hospital in Melbourne, Australia. Collection of leukapheresis samples was approved by the relevant Institutional Review Boards. Clinical and demographic characteristics of study donors are summarized in Table S1.
Animal studies
NOD.Cg-Prkdcscid Il2rgtm1Wjl/Sz (NSG) mice were purchased from The Jackson Laboratory (USA) and bred and housed at the Walter and Eliza Hall Institute animal facility in individually-ventilated cages (IVC; Techniplast) at a temperature of 18°C–22°C, humidity of 50–70%, 50 air exchanges per hour in the cages, and a 12/12-h light/dark cycle. The maximum cage density was six mice from the same litter and sex beginning at weaning. As bedding, corn cob granulate was provided (SAFE-lab). Mice were fed a standardised pellet diet (unless ART was administered as per method details) and provided sterilized water ad libitum. All materials were autoclaved before use. Animals were monitored daily by visual health-check. Adult male and female mice were rationally assigned to experimental groups in order to ensure comparable baseline levels of human CD4+ T cells as well as plasma viral setpoint. All animal experiments using live HIV-1 were performed in a Physical Containment 3 (PC3) facility approved by the Office of the Gene Technology Regulator at the Walter and Elisa Hall Institute.
Cell lines
HEK293T cell line used in this study was maintained in DMEM-GlutaMAX medium (ThermoFisher Scientific) + 10% fetal bovine serum (FBS; Sigma) + 1% penicillin/streptomycin (ThermoFisher) at 37°C, 5% CO2 conditions. All cell lines used in this study were confirmed mycoplasma-free by PCR. Isolated CD4+ T cells from PLWH on ART were cultured at 37°C, 5% CO2 in RPMI-1640 medium (ThermoFisher Scientific) supplemented with 10% FBS, 100 U/ml penicillin, 100 mg/mL streptomycin, 29.2 mg/mL L-glutamine plus 1 U/ml recombinant human IL-2 (Merck) and 1 mM raltegravir (NIH HIV Reagent Program).
Method details
Human CD4+ T cell isolation
Upon receiving informed consent, large numbers of peripheral blood mononuclear cells (PBMCs) were collected from PLWH on suppressive ART for at least 3 years by leukapheresis with approval from the relevant Institutional Review Boards (Alfred Hospital, Melbourne, Australia, HREC 214/15 & 48/16; The Avenue Hospital, Windsor, Australia, HREC 00242, protocol 202; The University of Melbourne, Australia, HREC 15452271 & 1443071). PBMCs were isolated using Ficoll Histopaque-1077 (Sigma). Total CD4+ T cells from PBMC were isolated using a EasySep Human CD4+ T cell Isolation kit (StemCell Technologies). Isolated CD4+ T cells were cultured at 37°C, 5% CO2 in RF10 (RPMI-1640 medium; ThermoFisher Scientific) supplemented with 10% FBS, 100 U/ml penicillin, 100 mg/mL streptomycin and 29.2 mg/mL L-glutamine) plus 1 U/ml recombinant human IL-2 (Merck) and 1 μM raltegravir (NIH HIV Reagent Program).
HIV-1 DNA quantification and CD4+ T cell cytotoxicity studies
Total CD4+ T cells from PLWH were cultured overnight in RF10 plus 1 U/ml recombinant human IL-2 (Merck) and 1 μM raltegravir (NIH HIV Reagent Program) to rest, and 3 × 106 cells treated with 5–100 nM venetoclax (SelleckChem) or a matched percentage of DMSO diluent for 24 h. Cells were then washed to remove the drugs and cultured for a further 24 h before being harvested for HIV-1 DNA quantification. For donors 1–8, cell-associated DNA and/or RNA was extracted using AllPrep DNA/RNA Mini kit (Qiagen), according to manufacturer’s instructions. For donors 9–13, cell-associated DNA was extracted using QIAamp DNA Mini kit (Qiagen), all according to manufacturer’s protocol except with the following change: the first elution was performed using 40 μL, followed by a second elution using 30 μL, for a final eluant volume of 70 μL. Total integrated HIV-1 DNA was quantified as previously described4,33 and intact proviral DNA assay (IPDA) was performed according to established protocols,24 with the following minor amendment to the IPDA analysis: venetoclax is a known pro-apoptotic drug which we speculate has capacity to preferentially kill infected cells that contribute to the HIV-1 reservoir. Therefore, to avoid biasing our data, we did not mathematically compensate for DNA Shearing Index (DSI) associated with dead/dying cells. For IPDA, we used the same primer sequences as previously published.24 For cytotoxicity and RNA-Seq studies, total CD4+ T cells were treated with venetoclax for 24 h as above, but without the additional 24 h wash step preceding harvest and DNA/RNA extraction. For subset cytotoxicity study, cells were harvested and stained with the following surface antibodies: anti-human CD3-PE, CD4-APC, CD197 (CCR7)-AF488, CD27-BV711, CD45RA-PE-Cy. Viability was assessed using LIVE/DEAD Fixable Violet Dead Cell Stain kit (ThermoFisher Scientific) according to manufacturer’s instructions. Samples were acquired by flow cytometry on a BD LSRFortessa (BD Biosciences) followed by analysis using FlowJo v10 software (BD) for the percentage of Violet+ cells.
Mice
Human cord blood CD34+ haematopoietic stem cells (HSC) (Lonza) were thawed, counted and resuspended according to manufacturer’s protocols. NOD.Cg-Prkdcscid Il2rgtm1Wjl/Sz mice (NSG; The Jackson Laboratory) were sub-lethally irradiated (100 cGy) between 24 and 48 h after birth. Irradiation was immediately (within ∼2 h) followed by intravenous injection of 0.5–1 × 105 CD34+ HSCs into the retro-orbital vein. Mandibular bleeds were performed at sixteen weeks of age to assess haematopoietic reconstitution by flow cytometry. Mice with comparable levels of human hematopoietic reconstitution were injected intraperitoneally with HIV-1JR-CSF (200ng p24 equivalent). Plasma viral load was determined three weeks post-infection and mice with viral loads > 103 copies ml−1 were used for experiments. A sample size analysis was performed to determine animal numbers based on 80% power, a two-sided confidence level of 95%, and assuming 90% of control animals rebound in 1 week and that interventions would reduce this to 10%. All animal experiments performed were approved by Walter and Eliza Hall Institute Animal Ethics Committee and Human Research and Ethics Committee.
Transformation of bacteria with HIV-1JR-CSF
10 ng of HIV-1JR-CSF plasmid DNA (pYK-JRCSF, NIH HIV Reagent Program) was added to 50 mL of One Shot Stbl3 chemically competent E. coli (ThermoFisher Scientific) and incubated for 30 min on ice. Bacteria were heat-shocked for 90 s at 42°C, placed on ice for a further 2 min, before the addition of 500 mL of S.O.C Medium (ThermoFisher Scientific) followed by incubation at 37°C for 30 min under shaking. The sample was centrifuged at 2000 xg for 2 min and all but 200 mL of supernatant was removed. The remaining supernatant was used to resuspend the bacterial pellet before 100 mL of culture was plated onto Lysogeny Broth (LB) agar plates containing carbenicillin (1 L LB broth – 10 g tryptone, 5 g yeast extract, 10 g NaCl, 15 g agar in 1 L deionized H2O, supplemented with 100 mg/mL carbenicillin, pH 7.0) and incubated overnight at 37°C. The next day, HIV-1 plasmid DNA was amplified in 300 mL LB broth overnight at 37°C in a shaking incubator. DNA was extracted using an endotoxin-free EndoFree Plasmid Maxi kit (Qiagen), following manufacturer’s protocol. Purified plasmid DNA was resuspended in TE buffer at 1 mg/mL and stored at −20°C until needed.
Transfection of HEK293T cells
HEK293T cells were seeded into 12x T150 tissue culture flasks under the following conditions per flask: 5 × 106 cells per 36 mL Dulbecco’s Modified Eagle Medium-GlutaMAX (DMEM-GlutaMAX) (ThermoFisher Scientific) supplemented with 10% FBS (ThermoFisher Scientific), 1% penicillin/streptomycin (ThermoFisher Scientific). Cells were allowed to adhere for 24 h before undergoing transfection the following afternoon. Stock HIV-1JR-CSF plasmid DNA was diluted 1:100 in 50 mL room temperature (RT) serum-free OptiMEM (ThermoFisher Scientific) to make a final concentration of 10 mg/mL. X-tremeGene DNA Transfection Mix (Merck) was warmed to room temperature before 750 μL was added dropwise to the plasmid/OptiMEM solution (this represents a ratio of 1 mg DNA:1.5 mL X-tremeGene). The transfection mix was inverted gently before incubating at RT for 15 min. Next, 4 mL of DNA/X-tremeGene transfection mix was added dropwise to each flask containing HEK293T cells, giving a final volume of 40 mL per flask. Cells were incubated at 37°C, 5% CO2 for 36 h. Supernatant was harvested and filtered through 0.22 mm Nalgene vacuum filters before proceeding to viral concentration.
Centrifugation and concentration of HIV-1JR-CSF
Filtered, virus-infected media was transferred to ultracentrifuge tubes (Nalgene PPCO with sealing closure; 24 mL per tube) before being underlaid with a 6 mL cushion of 20% sucrose (Merck) in PBS, using a serological pipette. Virus was concentrated by centrifuging the tubes at 40,000 xg for 1–2 h at 4°C (Sigma 3-30KS high-speed centrifuge) to pellet the virus. Supernatant was carefully decanted before the viral pellet was resuspended in a 100–200x concentrated volume of sterile PBS. Viral aliquots were stored at −80°C.
Quantifying HIV-1 viral stocks
Quantification of frozen HIV-1JR-CSF aliquots was performed using an HIV-1 p24 ELISA kit (XB-1000, Jomar Life Research), according to the manufacturer’s protocol.
Measuring HIV-1 viral load
Total viral RNA was extracted from plasma-EDTA using Invisorb Virus RNA HTS 96 Kit/C (Stratec), and samples were analyzed for HIV-1 RNA by qRT-PCR. The following forward and reverse Gag-specific primers were used: 5′-AAC ACC ATG CTA AAC ACA GTG G-3′ and 5′-GCT TCC TCA TTG ATG GTC TCT T-3′, respectively (Integrated DNA Technologies). The reaction mix was prepared using the Power SYBR Green RNA-to-CT 1-Step Kit (ThermoFisher Scientific). Cycle threshold (CT) values were correlated to standard samples of known HIV-1 RNA copy number. The lower limit of detection was 2.3 log10 HIV-1 RNA copies ml−1 blood.
Viral suppression
Three weeks post-infection, mice were administered a cocktail of four antiretroviral drugs: tenofovir disoproxil fumarate (TDF; 90 mg/kg), emtricitabine (FTC; 60 mg/kg), rilpivirine (7 mg/kg), and raltegravir (80 mg/kg). TDF, emtricitabine and rilpivirine were obtained under the commercial formulation “Eviplera” (Gilead), while raltegravir was purchased as “Isentress” (Merck). Tablets were crushed into powder and delivered to mice in mash. Pelleted feed was removed during this period. ART mash was administered daily for seven weeks before mice were analyzed for viral suppression.
BH3-mimetic treatment
Venetoclax (Active Biochem) was delivered on weekdays by oral gavage (100 mg/kg) in a formulation of 60% Phosal50PG (Lipoid), 30% PEG400 (Sigma), and 10% ethanol (Chem-Supply). The Mcl-1 inhibitor S63845 (MedChemExpress) was formulated in a vehicle of 2% Vitamin E-TPGS (Sigma) in normal saline, and was injected intravenously into the tail vein (30 mg/kg) twice weekly.
Analytical treatment interruption
Mice were returned to standard, ART-free pelleted feed at the conclusion of a given dosing regimen. Bleeds were taken once a week post-ART discontinuation to assess for viral rebound.
Immunophenotyping
Flow cytometry was used to immunophenotype humanised mice. Briefly, mice were bled and 30–50 mL of blood was transferred into a 96-well U-bottom plate. Erythrocytes were removed by resuspending cells in ice-cold ACK Red Cell Lysis buffer (155 mM NH4Cl, 10 mM KHCO3 and 100 mM EDTA, pH 7.3) for 1 min. All centrifuge spins were performed at 450 xg for 5 min at 4°C. ACK lysis was repeated twice before cells were washed once and stained with extracellular antibodies. The following antibodies were used for blood immunophenotyping: anti-mouse CD45.1-PE (BD Pharmingen), and anti-human CD45-BV786 (BD Horizon), CD4-APC-H7 (BD Pharmingen), CD8-PerCP-Cy5.5 (BD Pharmingen), CD19-BV510 (BD Horizon) and CD3-PeCy7 (BD Pharmingen). For immune subset analysis, mice were sacrificed at the conclusion of BH3-mimetic treatment but before ART interruption, and lymph nodes were harvested and pooled. Lymph nodes (LN) were processed into single cell suspension by mashing through a 40 mM cell strainer into a solution of 2% FBS in PBS (wash buffer). This single cell suspension was centrifuged at 450 xg for 5 min and resuspended in 1-2mL wash buffer. Cells were counted and 100 mL transferred into a 96-well U-bottom plate for antibody staining. The following antibodies were used for LN immunophenotyping: anti-mouse CD45.1-PE (BD Pharmingen), and anti-human CD45-BV786 (BD Horizon), CD4-APC-H7 (BD Pharmingen), CD8-APC (BD Pharmingen), CD19-BV510 (BD Horizon), CD3-PeCy7 (BD Pharmingen), CD45RO-PeCF594 (BD Biosciences), CD44-BV421 (BD Biosciences), and CD62L-PeCy5 (BD Biosciences). All stains were performed for 1 h at 4°C. After staining, cells were washed before being fixed with 2% paraformaldehyde (diluted in PBS) for 10 min at room temp. To determine absolute cell numbers in blood, 5 × 103 CountBright absolute counting beads (Life Technologies) were added to each sample prior to transfer into FACS cluster tubes (Corning) for analysis. Samples were analyzed using a LSRFortessa X20 (BD Biosciences) and data was analyzed using FlowJo v10 software (BD).
Tat/Rev Induced Limiting Dilution Assay (TILDA)
At the conclusion of dosing, spleen and peripheral lymph nodes were harvested and pooled for each mouse. Spleen and lymph nodes were processed into single-cell suspension before treatment with ACK Red Cell Lysis buffer for 15 s. PBMCs were counted and stained with the following surface panel: anti-mouse CD45.1-PE (BD Pharmingen), anti-mouse LY6C + LY6G (Gr-1)-PE (BD Pharmingen), anti-human CD45-PeCy5 (BD Pharmingen), anti-human CD14-BV421 (BD Horizon), anti-human CD8-APC (BD Pharmingen), and anti-human CD19-BV510 (BD Horizon). Human CD4+ T cells (hCD45+ hCD19- hCD8- hCD14-) were isolated via 2-way purity sorting through a 70 μm nozzle on a BD FACSAria Fusion (BD Biosciences). After sorting, purified CD4+ T cells were divided into ‘stimulated’ and ‘unstimulated’ conditions. ‘Stimulated’ cells were resuspended in RPMI-1640-GlutaMAX medium (ThermoFisher Scientific) supplemented with 10% FBS, 1% penicillin/streptomycin, 1x antiretroviral (ARV) cocktail (180 nM Zidovudine, 300 nM Efavirenz, 200 nM Raltegravir), 100 ng/mL Phorbol-12-myristate-13-acetate (PMA; Sigma), and 1 μg/mL Ionomycin (Sigma). All ARVs are available through the NIH HIV Reagent Program (https://www.hivreagentprogram.org/). ‘Unstimulated’ cells were resuspended in the same media as above except without PMA/Ionomycin. Cells were transferred to appropriate tissue culture plates and incubated for 18 h at 37°C, 5% CO2. Following overnight stimulation, cells were harvested and counted before resuspension with 10% FBS to 9,000 live cells per mL. Using 1x 96-well PCR plate (ThermoFisher Scientific) per mouse, the stimulated and unstimulated cells were serially diluted before processing for TILDA real-time PCR as previously described.23
RNA-seq analysis
Total CD4+ T cells from PLWH were cultured overnight in RF10 plus 1 U/ml IL-2 and 1 mM raltegravir to rest. Following this, 3 × 106 cells were treated with 5–100 nM venetoclax (Selleck Chemicals) or a matched concentration of DMSO diluent for 24 h. Cell-associated DNA/RNA was extracted using AllPrep DNA/RNA Mini kit (Qiagen) according to manufacturer’s protocol. An input of 10–100 ng of total RNA was prepared and indexed for Illumina sequencing using the TruSeq RNA Sample prep kit (Illumina) with RiboGlobin depletion as per manufacturer’s instruction. Each library was quantified using the Agilent Tapestation (using RNA ScreenTape) on a 2200 Tapestation system (Agilent Technologies) and the Qubit DNA BR assay kit for Qubit 3.0 Fluorometer (ThermoFisher Scientific). Libraries were sequenced on an Illumina NextSeq 500 instrument (Illumina) to produce 80 bp paired-end reads. Sequence reads were aligned to the human hg38 genome using Rsubread 2.4.2.81 Read counts were obtained for Entrez Gene IDs using featureCounts82 and Rsubread’s built-in RefSeq annotation. An average of 20 million reads were assigned per sample. Gene annotation was downloaded from ftp.ncbi.nlm.nih.gov/gene/on 21 January 2021. Protein-coding and long ncRNA genes on the reference chromosomes were retained for downstream analysis. Statistical analysis used the limma 3.47.8 and edgeR 3.33.2 software packages.83,84 Genes expressed in fewer than 8 samples were filtered using edgeR’s filterByExpr function. Library sizes were normalized by the TMMwsp method. Linear models were fitted using edgeR’s voomLmFit function adjusting for baseline differences between the human donors. Differential expression following venetoclax treatment was assessed using empirical Bayes t-statistics with robust estimation of hyperparameters.85 The false discovery rate (FDR) was controlled below 0.05 using the Benjamini and Hochberg method.
Statistical analyses
Analysis of data was performed using GraphPad Prism v9.0 (GraphPad Software). Kaplan-Maier curves showing viral rebound were analyzed using a log rank Mantel-Cox test. Humanised mouse peripheral CD4+ T cell counts as well as cell subsets in uninfected hu-mice were analyzed with an unpaired t test corrected for multiple comparisons with Holm-Šìdák method. Integrated HIV-1 DNA and IPDA fold-change results were analyzed using one-sample Wilcoxon signed rank test (theoretical median = 1.0). Hu-mouse TILDA results were analyzed using Wilcoxon matched-pairs signed rank test. Integrated HIV-1 DNA and IPDA absolute frequencies were analyzed using Wilcoxon matched-pairs signed rank test. FDR-adjusted p values for RNA-Seq data were analyzed using the limma-zoom method described above.
Acknowledgments
We thank Merle Dayton for assistance with facial vein injections, the Australian Red Cross Blood Service for supplying buffy coats, and Melissa Hobbs for technical support. We also thank PLWH for generously providing blood samples for this study. We are grateful to Ajantha Solomon, Ashanti Dantanarayana, Surekha Tennakoon, Socheata Chea, Judy Chang, Thomas Rasmussen, Barbara Scher, and Jared Stern at The University of Melbourne for providing clinical information, samples from PLWH, and technical support, plus Tina Luke from the Doherty Institute Flow Cytometry Facility for support. This work was supported by the National Health and Medical Research Council Australia (grants 1006592, 1045549, and 1065626 to M.P.; 1052979 and 118864 to S.R.L.; and 1154970 to G.K.S.); the Sylvia & Charles Viertel Senior Medical Research Fellowship (M.P.); an Australian Centre for HIV and Hepatitis Virology Research 2018 grant (J.L.A. and S.R.L.); the Victorian State Government Operational Infrastructure Support; and the Independent Research Institutes Infrastructure Support Scheme of the Australian Government National Health and Medical Research Council.
Author contributions
P.A., C.C.A., and M.P. designed research in the mouse model. Y.K., J.L.A., and S.R.L. designed research using primary human peripheral blood mononuclear cells (PBMCs). J.H.M. provided samples from PLWH on ART. P.A., C.C.A., S.P.P., Y.K., J.L.A., S.E.G., J.M.Z., C.T., J.O., and J.P.C. performed experiments. P.A., C.C.A., Y.K., M.R., D.S., J.L.A., G.K.S., S.R.L., and M.P. analyzed and interpreted the data. P.A. and M.P. wrote the paper. All authors reviewed and contributed to the manuscript.
Declaration of interests
The Walter and Eliza Hall Institute receives milestone and royalty payments related to venetoclax and has a commercial collaboration with Servier with respect to Mcl-1 inhibitors under which it may receive future payments. M.P. is eligible for financial benefits related to these payments. S.R.L. has received investigator-initiated, industry-funded research support from Merck Sciences, Gilead Sciences, and ViiV and provision of reagents from Infinity Pharmaceuticals, Merck Sciences, and BMS for investigator-initiated research. S.R.L. and J.L.A. have research collaborations with Merck Sciences unrelated to this work.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: August 30, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101178.
Supplemental information
Data and code availability
This paper does not report original code.
RNA-Seq data have been deposited at NCBI GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Original R script used in the processing and analysis of the RNA-Seq data can be found at GitHub (https://github.com/sheerind-wehi/venetoclax_RNA_seq/blob/main/QC%20%26%20DGE%20analysis.Rmd).
Any additional information/data from this paper is available from the lead contact upon request.
References
- 1.Finzi D., Blankson J., Siliciano J.D., Margolick J.B., Chadwick K., Pierson T., Smith K., Lisziewicz J., Lori F., Flexner C., et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 2.Wong J.K., Hezareh M., Günthard H.F., Havlir D.V., Ignacio C.C., Spina C.A., Richman D.D. Recovery of Replication-Competent HIV Despite Prolonged Suppression of Plasma Viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 3.Colby D.J., Trautmann L., Pinyakorn S., Leyre L., Pagliuzza A., Kroon E., Rolland M., Takata H., Buranapraditkun S., Intasan J., et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat. Med. 2018;24:923–926. doi: 10.1038/s41591-018-0026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasmussen T.A., Tolstrup M., Brinkmann C.R., Olesen R., Erikstrup C., Solomon A., Winckelmann A., Palmer S., Dinarello C., Buzon M., et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet. HIV. 2014;1:e13–e21. doi: 10.1016/s2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- 5.Ke R., Lewin S.R., Elliott J.H., Perelson A.S. Modeling the Effects of Vorinostat In Vivo Reveals both Transient and Delayed HIV Transcriptional Activation and Minimal Killing of Latently Infected Cells. PLoS Pathog. 2015;11 doi: 10.1371/journal.ppat.1005237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elliott J.H., Wightman F., Solomon A., Ghneim K., Ahlers J., Cameron M.J., Smith M.Z., Spelman T., McMahon J., Velayudham P., et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elliott J.H., McMahon J.H., Chang C.C., Lee S.A., Hartogensis W., Bumpus N., Savic R., Roney J., Hoh R., Solomon A., et al. Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study. Lancet. HIV. 2015;2:e520–e529. doi: 10.1016/s2352-3018(15)00226-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sainski A.M., Dai H., Natesampillai S., Pang Y.-P., Bren G.D., Cummins N.W., Correia C., Meng X.W., Tarara J.E., Ramirez-Alvarado M., et al. Casp8p41 generated by HIV protease kills CD4 T cells through direct Bak activation. J. Cell Biol. 2014;206:867–876. doi: 10.1083/jcb.201405051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cummins N.W., Sainski A.M., Dai H., Natesampillai S., Pang Y.-P., Bren G.D., de Araujo Correia M.C.M., Sampath R., Rizza S.A., O’Brien D., et al. Prime, Shock, and Kill: Priming CD4 T Cells from HIV Patients with a BCL-2 Antagonist before HIV Reactivation Reduces HIV Reservoir Size. J. Virol. 2016;90:4032–4048. doi: 10.1128/jvi.03179-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren Y., Huang S.-H., Patel S., Alberto W.D.C., Magat D., Ahimovic D., Macedo A.B., Durga R., Chan D., Zale E., et al. BCL-2 antagonism sensitizes cytotoxic t cell-resistant hiv reservoirs to elimination ex vivo. J. Clin. Invest. 2020;130:2542–2559. doi: 10.1172/jci132374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willis S.N., Fletcher J.I., Kaufmann T., van Delft M.F., Chen L., Czabotar P.E., Ierino H., Lee E.F., Fairlie W.D., Bouillet P., et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science (New York, N.Y.) 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 12.Carrington E.M., Vikstrom I.B., Light A., Sutherland R.M., Londrigan S.L., Mason K.D., Huang D.C.S., Lew A.M., Tarlinton D.M. BH3 mimetics antagonizing restricted prosurvival Bcl-2 proteins represent another class of selective immune modulatory drugs. Proc. Natl. Acad. Sci. USA. 2010;107:10967–10971. doi: 10.1073/pnas.1005256107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Souers A.J., Leverson J.D., Boghaert E.R., Ackler S.L., Catron N.D., Chen J., Dayton B.D., Ding H., Enschede S.H., Fairbrother W.J., et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 14.Leverson J.D., Phillips D.C., Mitten M.J., Boghaert E.R., Diaz D., Tahir S.K., Belmont L.D., Nimmer P., Xiao Y., Ma X.M., et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015;7:279ra40. doi: 10.1126/scitranslmed.aaa4642. [DOI] [PubMed] [Google Scholar]
- 15.Roberts A.W., Davids M.S., Pagel J.M., Kahl B.S., Puvvada S.D., Gerecitano J.F., Kipps T.J., Anderson M.A., Brown J.R., Gressick L., et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016;374:311–322. doi: 10.1056/nejmoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stilgenbauer S., Eichhorst B., Schetelig J., Coutre S., Seymour J.F., Munir T., Puvvada S.D., Wendtner C.-M., Roberts A.W., Jurczak W., et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17:768–778. doi: 10.1016/s1470-2045(16)30019-5. [DOI] [PubMed] [Google Scholar]
- 17.Cummins N.W., Sainski-Nguyen A.M., Natesampillai S., Aboulnasr F., Kaufmann S., Badley A.D. Maintenance of the HIV Reservoir Is Antagonized by Selective BCL2 Inhibition. J. Virol. 2017;91 doi: 10.1128/jvi.00012-17. e00012-17–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrington E.M., Zhan Y., Brady J.L., Zhang J.-G., Sutherland R.M., Anstee N.S., Schenk R.L., Vikstrom I.B., Delconte R.B., Segal D., et al. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ. 2017;24:878–888. doi: 10.1038/cdd.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teh T.-C., Nguyen N.-Y., Moujalled D.M., Segal D., Pomilio G., Rijal S., Jabbour A., Cummins K., Lackovic K., Blombery P., et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia. 2018;32:303–312. doi: 10.1038/leu.2017.243. [DOI] [PubMed] [Google Scholar]
- 20.Lok S.W., Whittle J.R., Vaillant F., Teh C.E., Lo L.L., Policheni A.N., Bergin A.R.T., Desai J., Ftouni S., Gandolfo L.C., et al. A phase 1b dose-escalation and expansion study of the BCL-2 inhibitor venetoclax combined with tamoxifen in ER and BCL-2-positive metastatic breast cancer. Cancer Discov. 2019;9 doi: 10.1158/2159-8290.CD-18-1151. [DOI] [PubMed] [Google Scholar]
- 21.Bar K.J., Sneller M.C., Harrison L.J., Justement J.S., Overton E.T., Petrone M.E., Salantes D.B., Seamon C.A., Scheinfeld B., Kwan R.W., et al. Effect of HIV Antibody VRC01 on Viral Rebound after Treatment Interruption. N. Engl. J. Med. 2016;375:2037–2050. doi: 10.1056/nejmoa1608243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scheid J.F., Horwitz J.A., Bar-On Y., Kreider E.F., Lu C.-L., Lorenzi J.C.C., Feldmann A., Braunschweig M., Nogueira L., Oliveira T., et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature. 2016;535:556–560. doi: 10.1038/nature18929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hill A.L., Rosenbloom D.I.S., Goldstein E., Hanhauser E., Kuritzkes D.R., Siliciano R.F., Henrich T.J. Real-Time Predictions of Reservoir Size and Rebound Time during Antiretroviral Therapy Interruption Trials for HIV. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruner K.M., Wang Z., Simonetti F.R., Bender A.M., Kwon K.J., Sengupta S., Fray E.J., Beg S.A., Antar A.A.R., Jenike K.M., et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature. 2019;566:120–125. doi: 10.1038/s41586-019-0898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosmane N.N., Kwon K.J., Bruner K.M., Capoferri A.A., Beg S., Rosenbloom D.I.S., Keele B.F., Ho Y.-C., Siliciano J.D., Siliciano R.F. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J. Exp. Med. 2017;214:959–972. doi: 10.1084/jem.20170193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nader A., Minocha M., Othman A.A. Exposure-Response Analyses of the Effects of Venetoclax, a Selective BCL-2 Inhibitor, on B-Lymphocyte and Total Lymphocyte Counts in Women with Systemic Lupus Erythematosus. Clin. Pharmacokinet. 2020;59:335–347. doi: 10.1007/s40262-019-00818-5. [DOI] [PubMed] [Google Scholar]
- 27.Teh C.E., Peng H., Luo M.X., Tan T., Trussart M., Howson L.J., Chua C.C., Muttiah C., Brown F., Ritchie M.E., et al. Venetoclax treatment in cancer patients has limited impact on circulating T and NK cells. Blood Adv. 2023;7:2733–2745. doi: 10.1182/bloodadvances.2022008221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Procopio F.A., Fromentin R., Kulpa D.A., Brehm J.H., Bebin A.-G., Strain M.C., Richman D.D., O’Doherty U., Palmer S., Hecht F.M., et al. A Novel Assay to Measure the Magnitude of the Inducible Viral Reservoir in HIV-infected Individuals. EBioMedicine. 2015;2:874–883. doi: 10.1016/j.ebiom.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li G., Zhao J., Cheng L., Jiang Q., Kan S., Qin E., Tu B., Zhang X., Zhang L., Su L., Zhang Z. HIV-1 infection depletes human CD34+CD38- hematopoietic progenitor cells via pDC-dependent mechanisms. PLoS Pathog. 2017;13 doi: 10.1371/journal.ppat.1006505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kotschy A., Szlavik Z., Murray J., Davidson J., Maragno A.L., Le Toumelin-Braizat G., Chanrion M., Kelly G.L., Gong J.-N., Moujalled D.M., et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–482. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 31.Opferman J.T., Letai A., Beard C., Sorcinelli M.D., Ong C.C., Korsmeyer S.J. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 32.Tripathi P., Koss B., Opferman J.T., Hildeman D.A. Mcl-1 antagonizes Bax/Bak to promote effector CD4(+) and CD8(+) T-cell responses. Cell Death Differ. 2013;20:998–1007. doi: 10.1038/cdd.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vandergeeten C., Fromentin R., Merlini E., Lawani M.B., DaFonseca S., Bakeman W., McNulty A., Ramgopal M., Michael N., Kim J.H., et al. Cross-clade ultrasensitive PCR-based assays to measure HIV persistence in large-cohort studies. J. Virol. 2014;88:12385–12396. doi: 10.1128/jvi.00609-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cameron P.U., Saleh S., Sallmann G., Solomon A., Wightman F., Evans V.A., Boucher G., Haddad E.K., Sekaly R.-P., Harman A.N., et al. Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc. Natl. Acad. Sci. USA. 2010;107:16934–16939. doi: 10.1073/pnas.1002894107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurtulus S., Tripathi P., Moreno-Fernandez M.E., Sholl A., Katz J.D., Grimes H.L., Hildeman D.A. Bcl-2 Allows Effector and Memory CD8+ T Cells To Tolerate Higher Expression of Bim. J. Immunol. 2011;186:5729–5737. doi: 10.4049/jimmunol.1100102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts A.W., Seymour J.F., Brown J.R., Wierda W.G., Kipps T.J., Khaw S.L., Carney D.A., He S.Z., Huang D.C.S., Xiong H., et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012;30:488–496. doi: 10.1200/jco.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark I.C., Mudvari P., Thaploo S., Smith S., Abu-Laban M., Hamouda M., Theberge M., Shah S., Ko S.H., Pérez L., et al. HIV silencing and cell survival signatures in infected T cell reservoirs. Nature. 2023;614:318–325. doi: 10.1038/s41586-022-05556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun W., Gao C., Hartana C.A., Osborn M.R., Einkauf K.B., Lian X., Bone B., Bonheur N., Chun T.-W., Rosenberg E.S., et al. Phenotypic signatures of immune selection in HIV-1 reservoir cells. Nature. 2023;614:309–317. doi: 10.1038/s41586-022-05538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ren Y., Huang S.H., Macedo A.B., Ward A.R., Alberto W.D.C., Klevorn T., Leyre L., Copertino D.C., Mota T.M., Chan D., et al. Selective BCL-XL Antagonists Eliminate Infected Cells from a Primary-Cell Model of HIV Latency but Not from Ex Vivo Reservoirs. J. Virol. 2021;95 doi: 10.1128/jvi.02425-20. e0242520–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hübner A., Cavanagh-Kyros J., Rincon M., Flavell R.A., Davis R.J. Functional Cooperation of the Proapoptotic Bcl2 Family Proteins Bmf and Bim In Vivo. Mol. Cell Biol. 2010;30:98–105. doi: 10.1128/mcb.01155-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niu X., Zhao J., Ma J., Xie C., Edwards H., Wang G., Caldwell J.T., Xiang S., Zhang X., Chu R., et al. Binding of Released Bim to Mcl-1 is a Mechanism of Intrinsic Resistance to ABT-199 which can be Overcome by Combination with Daunorubicin or Cytarabine in AML Cells. Clin. Cancer Res. 2016;22:4440–4451. doi: 10.1158/1078-0432.ccr-15-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conage-Pough J.E., Boise L.H. Phosphorylation alters Bim-mediated Mcl-1 stabilization and priming. FEBS J. 2018;285:2626–2640. doi: 10.1111/febs.14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luedtke D.A., Niu X., Pan Y., Zhao J., Liu S., Edwards H., Chen K., Lin H., Taub J.W., Ge Y. Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal Transduct. Targeted Ther. 2017;2 doi: 10.1038/sigtrans.2017.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zerbato J.M., Purves H.V., Lewin S.R., Rasmussen T.A. Between a shock and a hard place: challenges and developments in HIV latency reversal. Curr. Opin. Virol. 2019;38:1–9. doi: 10.1016/j.coviro.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neidleman J., Luo X., Frouard J., Xie G., Hsiao F., Ma T., Morcilla V., Lee A., Telwatte S., Thomas R., et al. Phenotypic analysis of the unstimulated in vivo HIV CD4 T cell reservoir. Elife. 2020;9:e60933. doi: 10.7554/elife.60933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Banga R., Procopio F.A., Noto A., Pollakis G., Cavassini M., Ohmiti K., Corpataux J.-M., de Leval L., Pantaleo G., Perreau M. PD-1+ and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat. Med. 2016;22:754–761. doi: 10.1038/nm.4113. [DOI] [PubMed] [Google Scholar]
- 47.Tsunetsugu-Yokota Y., Kobayahi-Ishihara M., Wada Y., Terahara K., Takeyama H., Kawana-Tachikawa A., Tokunaga K., Yamagishi M., Martinez J.P., Meyerhans A. Homeostatically Maintained Resting Naive CD4+ T Cells Resist Latent HIV Reactivation. Front. Microbiol. 2016;7:1944. doi: 10.3389/fmicb.2016.01944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsiao F., Frouard J., Gramatica A., Xie G., Telwatte S., Lee G.Q., Roychoudhury P., Schwarzer R., Luo X., Yukl S.A., et al. Tissue memory CD4+ T cells expressing IL-7 receptor-alpha (CD127) preferentially support latent HIV-1 infection. PLoS Pathog. 2020;16 doi: 10.1371/journal.ppat.1008450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grau-Expósito J., Luque-Ballesteros L., Navarro J., Curran A., Burgos J., Ribera E., Torrella A., Planas B., Badía R., Martin-Castillo M., et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLoS Pathog. 2019;15 doi: 10.1371/journal.ppat.1007991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Venanzi Rullo E., Cannon L., Pinzone M.R., Ceccarelli M., Nunnari G., O’Doherty U. Genetic Evidence That Naive T Cells Can Contribute Significantly to the Human Immunodeficiency Virus Intact Reservoir: Time to Re-evaluate Their Role. Clin. Infect. Dis. 2019;69:2236–2237. doi: 10.1093/cid/ciz378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pinzone M.R., Weissman S., Pasternak A.O., Zurakowski R., Migueles S., O’Doherty U. Naive infection predicts reservoir diversity and is a formidable hurdle to HIV eradication. JCI Insight. 2021;6 doi: 10.1172/jci.insight.150794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venanzi Rullo E., Pinzone M.R., Cannon L., Weissman S., Ceccarelli M., Zurakowski R., Nunnari G., O’Doherty U. Persistence of an intact HIV reservoir in phenotypically naive T cells. JCI Insight. 2020;5 doi: 10.1172/jci.insight.133157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chomont N., El-Far M., Ancuta P., Trautmann L., Procopio F.A., Yassine-Diab B., Boucher G., Boulassel M.-R., Ghattas G., Brenchley J.M., et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z., Gurule E.E., Brennan T.P., Gerold J.M., Kwon K.J., Hosmane N.N., Kumar M.R., Beg S.A., Capoferri A.A., Ray S.C., et al. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc. Natl. Acad. Sci. USA. 2018;115:E2575–E2584. doi: 10.1073/pnas.1720665115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ho Y.-C., Shan L., Hosmane N.N., Wang J., Laskey S.B., Rosenbloom D.I.S., Lai J., Blankson J.N., Siliciano J.D., Siliciano R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bruner K.M., Murray A.J., Pollack R.A., Soliman M.G., Laskey S.B., Capoferri A.A., Lai J., Strain M.C., Lada S.M., Hoh R., et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016;22:1043–1049. doi: 10.1038/nm.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peluso M.J., Bacchetti P., Ritter K.D., Beg S., Lai J., Martin J.N., Hunt P.W., Henrich T.J., Siliciano J.D., Siliciano R.F., et al. Differential decay of intact and defective proviral DNA in HIV-1-infected individuals on suppressive antiretroviral therapy. Jci Insight. 2020;5 doi: 10.1172/jci.insight.132997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu G., Zuck P., Goh S.L., Milush J.M., Vohra P., Wong J.K., Somsouk M., Yukl S.A., Shacklett B.L., Chomont N., et al. Gag p24 is a Marker of HIV Expression in Tissues and Correlates with Immune Response. J. Infect. Dis. 2021;224:1593–1598. doi: 10.1093/infdis/jiab121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Estes J.D., Kityo C., Ssali F., Swainson L., Makamdop K.N., Del Prete G.Q., Deeks S.G., Luciw P.A., Chipman J.G., Beilman G.J., et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat. Med. 2017;23:1271–1276. doi: 10.1038/nm.4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hill A.L., Rosenbloom D.I.S., Fu F., Nowak M.A., Siliciano R.F. Predicting the outcomes of treatment to eradicate the latent reservoir for HIV-1. Proc. Natl. Acad. Sci. USA. 2014;111:13475–13480. doi: 10.1073/pnas.1406663111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zerbato J.M., McMahon D.K., Sobolewski M.D., Mellors J.W., Sluis-Cremer N. Naive CD4+ T Cells Harbor a Large Inducible Reservoir of Latent, Replication-competent Human Immunodeficiency Virus Type 1. Clin. Infect. Dis. 2019;69:1919–1925. doi: 10.1093/cid/ciz108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hiener B., Horsburgh B.A., Eden J.-S., Barton K., Schlub T.E., Lee E., von Stockenstrom S., Odevall L., Milush J.M., Liegler T., et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4(+) T Cells from Effectively Treated Participants. Cell Rep. 2017;21:813–822. doi: 10.1016/j.celrep.2017.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watanabe S., Terashima K., Ohta S., Horibata S., Yajima M., Shiozawa Y., Dewan M.Z., Yu Z., Ito M., Morio T., et al. Hematopoietic stem cell-engrafted NOD/SCID/IL2Rgamma null mice develop human lymphoid systems and induce long-lasting HIV-1 infection with specific humoral immune responses. Blood. 2007;109:212–218. doi: 10.1182/blood-2006-04-017681. [DOI] [PubMed] [Google Scholar]
- 64.Satheesan S., Li H., Burnett J.C., Takahashi M., Li S., Wu S.X., Synold T.W., Rossi J.J., Zhou J. HIV replication and latency in a humanized NSG mouse model during suppressive oral combinational ART. J. Virol. 2018;92 doi: 10.1128/jvi.02118-17. 021188-e2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marsden M.D. Benefits and limitations of humanized mice in HIV persistence studies. Retrovirology. 2020;17 doi: 10.1186/s12977-020-00516-2. 7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Denton P.W., Garcia J.V. Novel humanized murine models for HIV research. Curr. HIV AIDS Rep. 2009;6:13–19. doi: 10.1007/s11904-009-0003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deng K., Pertea M., Rongvaux A., Wang L., Durand C.M., Ghiaur G., Lai J., McHugh H.L., Hao H., Zhang H., et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. 2015;517:381–385. doi: 10.1038/nature14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pollack R.A., Jones R.B., Pertea M., Bruner K.M., Martin A.R., Thomas A.S., Capoferri A.A., Beg S.A., Huang S.-H., Karandish S., et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe. 2017;21:494–506.e4. doi: 10.1016/j.chom.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hatano H., Jain V., Hunt P.W., Lee T.-H., Sinclair E., Do T.D., Hoh R., Martin J.N., McCune J.M., Hecht F., et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J. Infect. Dis. 2013;208:50–56. doi: 10.1093/infdis/jis630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Graf E.H., Pace M.J., Peterson B.A., Lynch L.J., Chukwulebe S.B., Mexas A.M., Shaheen F., Martin J.N., Deeks S.G., Connors M., et al. Gag-Positive Reservoir Cells Are Susceptible to HIV-Specific Cytotoxic T Lymphocyte Mediated Clearance. PLoS One. 2013;8 doi: 10.1371/journal.pone.0071879.t001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chandrasekar A.P., Cummins N.W., Natesampillai S., Misra A., Alto A., Laird G., Badley A.D. The BCL-2 Inhibitor Venetoclax Augments Immune Effector Function Mediated by Fas Ligand, TRAIL, and Perforin/Granzyme B, Resulting in Reduced Plasma Viremia and Decreased HIV Reservoir Size during Acute HIV Infection in a Humanized Mouse Model. J. Virol. 2022;96 doi: 10.1128/jvi.01730-22. e0173022–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu H., Michmerhuizen M.J., Lao Y., Wan K., Salem A.H., Sawicki J., Serby M., Vaidyanathan S., Wong S.L., Agarwal S., et al. Absorption, Metabolism, and Excretion of a Novel Bcl-2 Inhibitor Venetoclax in Humans. Drug Metab. Dispos. 2017;45:294–305. doi: 10.1124/dmd.116.071613. [DOI] [PubMed] [Google Scholar]
- 73.Gonzalez F.J. Cytochrome P450 humanised mice. Hum. Genom. 2004;1:300–306. doi: 10.1186/1479-7364-1-4-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leth S., Schleimann M.H., Nissen S.K., Højen J.F., Olesen R., Graversen M.E., Jørgensen S., Kjær A.S., Denton P.W., Mørk A., et al. Combined effect of Vacc-4x, recombinant human granulocyte macrophage colony-stimulating factor vaccination, and romidepsin on the HIV-1 reservoir (REDUC): a single-arm, phase 1B/2A trial. Lancet. HIV. 2016;3:e463–e472. doi: 10.1016/s2352-3018(16)30055-8. [DOI] [PubMed] [Google Scholar]
- 75.Campbell G.R., Bruckman R.S., Chu Y.-L., Trout R.N., Spector S.A. SMAC Mimetics Induce Autophagy-Dependent Apoptosis of HIV-1-Infected Resting Memory CD4+ T Cells. Cell Host Microbe. 2018;24:689–702.e7. doi: 10.1016/j.chom.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Borducchi E.N., Liu J., Nkolola J.P., Cadena A.M., Yu W.-H., Fischinger S., Broge T., Abbink P., Mercado N.B., Chandrashekar A., et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature. 2018;563:360–364. doi: 10.1038/s41586-018-0600-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gunst J.D., Pahus M.H., Rosás-Umbert M., Lu I.-N., Benfield T., Nielsen H., Johansen I.S., Mohey R., Østergaard L., Klastrup V., et al. Early intervention with 3BNC117 and romidepsin at antiretroviral treatment initiation in people with HIV-1: a phase 1b/2a, randomized trial. Nat. Med. 2022:1–12. doi: 10.1038/s41591-022-02023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Einkauf K.B., Osborn M.R., Gao C., Sun W., Sun X., Lian X., Parsons E.M., Gladkov G.T., Seiger K.W., Blackmer J.E., et al. Parallel analysis of transcription, integration, and sequence of single HIV-1 proviruses. Cell. 2022;185:266–282.e15. doi: 10.1016/j.cell.2021.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reeves D.B., Gaebler C., Oliveira T.Y., Peluso M.J., Schiffer J.T., Cohn L.B., Deeks S.G., Nussenzweig M.C. Impact of misclassified defective proviruses on HIV reservoir measurements. Nat. Commun. 2023;14:4186. doi: 10.1038/s41467-023-39837-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gaebler C., Lorenzi J.C.C., Oliveira T.Y., Nogueira L., Ramos V., Lu C.-L., Pai J.A., Mendoza P., Jankovic M., Caskey M., Nussenzweig M.C. Combination of quadruplex qPCR and next-generation sequencing for qualitative and quantitative analysis of the HIV-1 latent reservoir. J. Exp. Med. 2019;216:2253–2264. doi: 10.1084/jem.20190896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 83.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCarthy D.J., Chen Y., Smyth G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Phipson B., Lee S., Majewski I.J., Alexander W.S., Smyth G.K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 2016;10:946–963. doi: 10.1214/16-aoas920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper does not report original code.
RNA-Seq data have been deposited at NCBI GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Original R script used in the processing and analysis of the RNA-Seq data can be found at GitHub (https://github.com/sheerind-wehi/venetoclax_RNA_seq/blob/main/QC%20%26%20DGE%20analysis.Rmd).
Any additional information/data from this paper is available from the lead contact upon request.