

Abstract
The clinical significance of the tumor‐immune interaction in breast cancer is now established, and tumor‐infiltrating lymphocytes (TILs) have emerged as predictive and prognostic biomarkers for patients with triple‐negative (estrogen receptor, progesterone receptor, and HER2‐negative) breast cancer and HER2‐positive breast cancer. How computational assessments of TILs might complement manual TIL assessment in trial and daily practices is currently debated. Recent efforts to use machine learning (ML) to automatically evaluate TILs have shown promising results. We review state‐of‐the‐art approaches and identify pitfalls and challenges of automated TIL evaluation by studying the root cause of ML discordances in comparison to manual TIL quantification. We categorize our findings into four main topics: (1) technical slide issues, (2) ML and image analysis aspects, (3) data challenges, and (4) validation issues. The main reason for discordant assessments is the inclusion of false‐positive areas or cells identified by performance on certain tissue patterns or design choices in the computational implementation. To aid the adoption of ML for TIL assessment, we provide an in‐depth discussion of ML and image analysis, including validation issues that need to be considered before reliable computational reporting of TILs can be incorporated into the trial and routine clinical management of patients with triple‐negative breast cancer. © 2023 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: deep learning, machine learning, digital pathology, guidelines, image analysis, pitfalls, prognostic biomarker, triple‐negative breast cancer, tumor‐infiltrating lymphocytes
Introduction
The prognostic and predictive significance of the tumor‐immune interaction in breast cancer (BC) has been investigated intensively in recent years [1, 2], and tumor‐infiltrating lymphocytes (TILs) have emerged as a robust biomarker with reasonable reproducibility [3, 4, 5]. Within BC, triple‐negative BC (TNBC) (estrogen receptor, progesterone receptor, and HER2‐negative) and HER2‐positive BC exhibit a more pronounced tumor‐associated immune cell infiltrate. There is good evidence to suggest both a prognostic and predictive potential for TILs in TNBC, even in the absence of systemic chemotherapy [6]. In TNBC, each 10% increment in TIL is associates with a 17% relative increase in overall survival (OS) [7], and TILs can predict chemotherapy response [8, 9]. Therefore, routine evaluation of TILs during diagnostic workup of TNBC patients was recommended in the 2019 St Gallen International Breast Cancer Consensus [10], and TIL assessment is now incorporated into several national guidelines as a biomarker for TNBC and HER2‐positive BC and is used prognostically until predictiveness is validated in new trials [11, 12].
To move TIL evaluation from research and single‐center clinical use to routine cancer care, the clinical evaluation must be accurate and reproducible. The International Immuno‐Oncology Biomarker Working Group on Breast Cancer (also called the TILs‐WG: www.tilsinbreastcancer.org) has formulated a set of guidelines for visual TIL assessment (VTA) on hematoxylin and eosin (H&E)‐stained slides [13]. Although this method of analyzing TILs is reproducible among trained pathologists [14, 15], there remains a need for additional training, particularly because tumor heterogeneity affects reproducibility [16]. For this reason, the US Food and Drug Administration (FDA) recently provided an online publicly available continuing medical education (CME) accredited TIL training course for pathologists (https://ceportal.fda.gov/).
Recent developments in machine learning (ML) have had a major impact on computational pathology [17], including automated evaluation of TILs using ML and image analysis – also referred to as computational TIL assessment (CTA). CTA is a promising solution for many of the issues of VTA and may lead to a standardized and more reliable evaluation of TILs to complement local TIL assesment when needed.
Using ML and digital image analysis to analyze immune cell infiltration is not a new idea, having been studied sporadically for the last decade or so, mainly employing immunohistochemistry (IHC) [18, 19, 20, 21]. Several novel approaches have demonstrated the promise of deep neural network‐based algorithms for this task on H&E stains [22, 23, 24]. However, important issues must be taken into consideration during the development of algorithms to evaluate TILs in BC, and new research, development, and validation are required before ML tools can be incorporated into the routine clinical management of BC.
In this review, we provide a perspective of the current state of CTA and focus on how pitfalls with manual assessment [14] also impact ML‐based methods. This is achieved by categorizing the inconsistent cases reported in recent studies [22, 23, 24], and we extend the analysis to include the unique challenges involved in solving pitfalls with automated TIL evaluation. We group our findings into four main areas: (1) general pathology pitfalls, (2) ML and image analysis, (3) data challenges, and (4) validation.
Background
In the TIL‐WG VTA guidelines [13], TILs are defined as mononuclear immune cells, lymphocytes, and plasma cells. Intratumoral TILs (iTILs) that are in direct contact with tumor cells are distinguished from stromal TILs (sTILs) located in the stromal tissue between tumor cells islands. The guidelines recommend focusing on sTILs because their evaluation is more reproducible [7]. sTILs are assessed as the ratio of the area occupied by sTILs divided by the total tumor‐associated stromal area, with the final score reported as a percentage value. It is imperative that areas of necrosis, ductal, and lobular carcinoma in situ (DCIS/LCIS), and normal breast tissue are excluded from the analysis.
The TIL‐WG has reported on how computational assessment of TILs could be designed, with the recommendation that ‘computational TILs assessment (CTA) algorithms need to account for the complexity involved in TIL‐scoring procedures, and to closely follow guidelines for visual assessment where appropriate’ [25]. Several approaches to CTA can be considered, from more granular approaches, closely mimicking the guidelines recommended by TIL‐WG, to coarser strategies, with methods also varying in their level of automation. In this article, we focus predominantly on recently created CTA algorithms that adhere to the guidelines.
Bai et al [23] produced an algorithm using the open‐source software QuPath [26], in which inclusion of tumor regions and exclusion of noninvasive epithelium (DCIS/LCIS and normal ducts and lobules) are manually annotated by a specialist pathologist. Therefore, the algorithm relies on an experienced pathologist since it cannot identify the correct areas for analysis, nor can it eliminate common artifacts [23]. This algorithm applies color normalization before cells are segmented using a traditional image analysis algorithm (background subtraction, thresholding, and watershed) to compensate for H&E variability. Finally, a model trained on extracted handcrafted cellular features classifies all cells as tumor cells, TILs, fibroblasts, or others. The algorithm then outputs five quantitative variables with prognostic significance, including the TIL‐WG definition: (1) total area of TILs within the stroma (percentage); (2) number of TILs in the annotated region; (3) amount of stromal cells in the annotated region; (4) the total number of cells in the annotated region; and (5) the proportional number of TILs relative to the tumor [13].
Sun et al [24] presented a more comprehensive approach, although still relying on manually annotated regions by a pathologist. After identifying tumor regions and excluding noninvasive regions, a tissue‐level model automatically detects and excludes necrosis to ensure that necrotic cells are not misclassified as lymphocytes. Cells are subsequently detected and classified as malignant epithelial cells, TILs, or others using a cell‐level model. The classified cell algorithms are then used to identify the tumor‐, stroma‐, and lymphocyte‐dense regions using a rule‐based system, which produces a regional‐based quantitative variable of the area coverage of sTILs.
Thagaard et al [22] reported a fully automatic system using commercial software (Visiopharm A/S, Hørsholm, Denmark), in which a tissue‐level model identifies the tissue types and then, with no manual interaction, automatically identifies the invasive tumor, noninvasive breast structures, and stromal and necrotic regions. A cell‐level model then identifies TILs and reports sTIL density as a quantitative variable. Other studies have proposed alternative metrics [21, 27, 28] or used stains other than H&E [29, 30, 31]. We have found that these methods show inconsistency with the TIL‐WG VTA guidelines (see [25]).
The common findings from these studies are that CTA has good to excellent agreement with VTA and, more importantly, is independently associated with clinical outcome, confirming that patients with TNBC and a high CTA score have improved survival [22, 24]. In addition, the studies indicate that current CTA is not a panacea for the limitations of VTA, and more research is needed to address handling pitfalls, along with further development and clinical validation of CTA.
Common pitfalls between visual and computational assessments
On behalf of the TIL‐WG, Kos et al [14] identified and reported the most common pitfalls of evaluating TILs by eye. Some are also relevant when developing ML approaches and will be discussed here, as will those unique to CTA.
Including wrong areas or cells
The most frequent cause of inconsistent CTA results compared to manual scoring is the inclusion of incorrect areas for evaluation. These tissue‐level pitfalls include (1) TILs around noninvasive structures (DCIS/LCIS, benign lesions, and normal ducts and lobules) (Figure 1) [22, 24]; (2) lymphocytes associated with other structures (such as lymphovascular invasion and vessels in general; Figure 1) [24]; (3) necrotic areas [24]; and (4) tertiary lymphoid structures (TLSs), possibly as an aggregate because H&E staining does not allow differentiation between B‐ and T‐cells [23]. In addition, training algorithms are commonly developed on ductal tumors, making potential pitfalls for lobular histology and less common histologic subtypes such as mucinous, metaplastic, apocrine, and papillary cancers [22, 24]. Both Sun et al and Bai et al suggested excluding these confounding regions manually, which is therefore subject to the same pitfalls as full VTA and reduces time efficiency due to pathologist involvement [23, 24]. In Thagaard et al this step was performed automatically, with issues for complex pattern equivocal DCIS but not benign regions or uniform DCIS. Overall, manual and automatic approaches have the same pitfalls regarding regions of equivocal DCIS. The extent to which this impacts the accuracy of computational tools for TILs has yet to be fully resolved [22].
Figure 1.
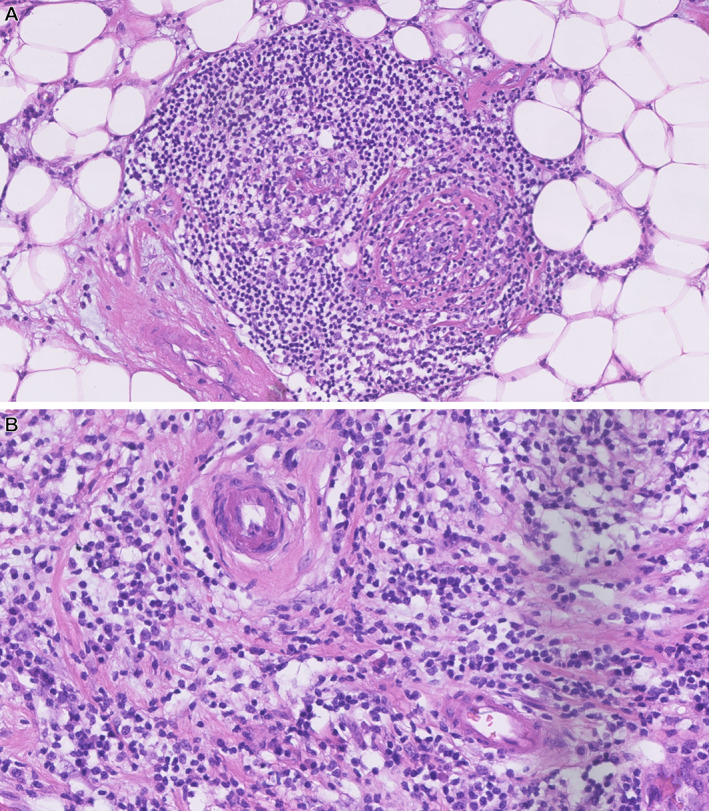
Lymphocyte‐dense regions associated with other structures should be excluded as the inflammation is not necessarily an immune response to the tumor. (A) TLS. (B) Lymphocytes surrounding vessels. These areas are reported [24] as possible false‐positive areas in CTA at much higher levels than VTA. Images by Elisabeth Specht Stovgaard from Herlev cohort used in [22].
Cell‐level problems where incorrect cell detections are included are less prevalent in CTA. Bai et al reported substantial segmentation failure in 1–2%, i.e. where the segmentation model is the major cause of discordant cases. The main cause was an inability to distinguish iTILs from sTILs, causing tumors with a high proportion of iTILs to be excluded from the study. Apoptotic bodies, neutrophils, and low‐grade or neuroendocrine tumors can also lead to false‐positive TIL detection [23].
The aforementioned performance pitfalls can be addressed using approaches described in the subsequent section ‘Image analysis challenges when adhering to a clinical guideline’ and by using variable data for model training described in ‘Training data challenges to create robust and generalizable algorithms’.
Technical factors that impact ML algorithms
Slide‐related issues are a common challenge for VTA [14] and also impact CTA. Variables in the preanalytic workflow include cautery artifacts, as well as tissue dyes used to mark resection margins during macroscopic examination. Artifacts of histological preparations include those derived from tissue processing, microtomy, staining, and mounting (zonal fixation, blade lines, tissue disruption, microchatters, air bubbles, floaters). Out‐of‐focus areas, pen markings, tissue folds, blurring, air bubbles, thick sections, and crush artifacts can each confuse tissue‐ or cell‐level models and consequently lead to inaccurate quantification. For example, poor sectioning can cause false‐negative TILs, thereby producing an underestimation of the true TIL density [22].
Scanning variability among different manufacturers is also a problem when comparing cohorts of multi‐institutional studies because of the lack of standardized acquisition parameters. The extent of this issue in CTA has yet to be properly investigated [22], but for applications such as detection of prostate cancer, variation influences the uniform interpretation of CTA. Similarly, inter‐ and intrasite variation in slide preparation and/or staining may contribute to differences in CTA between cohorts [23, 24], similar to other applications [32].
There are two main approaches to combating these problems. First, they can be handled manually or by employing a separate model, e.g. excluding out‐of‐focus cells [33] or fields [34] in a preanalysis phase. Second, more variability in scanning and staining quality metrics can be incorporated into the dataset used to develop CTA, and we cover key aspects of these issues in what follows (‘Training data challenges to create robust and generalizable algorithms’).
Heterogeneity in sTIL distribution and tumor‐compartment definitions
One pitfall that causes the highest manual interobserver variation is the presence of increased sTILs at the leading edge of a tumor compared to the central tumor area [14] (Figure 2A). This aspect of CTA compared to VTA was highlighted in recent studies [22, 24]. The increased density of sTILs at a tumor's leading edge can cause a lower CTA score, because the immune‐deserted stromal region in the central tumor region will contribute to a larger stromal area quantification than would be estimated by manual assessment. In contrast, if stroma is scarce in the central tumor, the high‐density margin will contribute most to the overall score, resulting in a higher CTA score.
Figure 2.
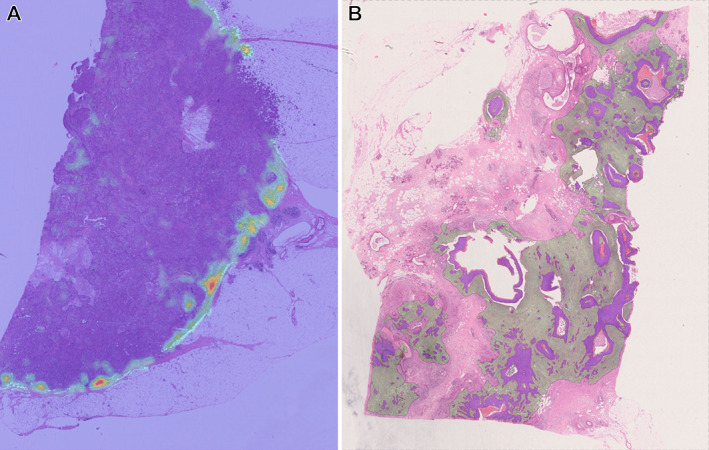
Examples of discrepant cases from Herlev cohort used in [22]; purple areas: tumor nests, heatmap areas: sTIL regions. (A) A case of high sTIL density at tumor margin compared to central area. As the stroma is scarce inside the tumor, sTIL density is reported to be very high in CTA as mostly the margin contributes to the score. (B) The tumor grows irregularly with small tumor nests between larger invasive tumor areas. In these cases, the CTA includes more stroma than VTA, resulting in a lower sTIL density score (larger denominator) than the manual score.
In general, the identification of tumor‐associated stromal regions in which sTILs should be scored is not strictly defined in manual guidelines. Sometimes there are larger stromal areas within the tumor core, but the allowable distance between stromal TILs and tumor nests for quantitation remains unclear (Figure 2B). Similarly, there is no quantitative definition of outlier TIL density hotspots that should be excluded. This can lead to discrepancies between VTA and CTA [22], depending on the CTA integration details method and the validation approach employed (discussed further in the sections ‘Image analysis challenges when adhering to a clinical guideline’ and ‘Validation challenges when comparing CTA with VTA’).
Moving beyond human capabilities
Some of the aforementioned discrepancies may be eliminated by algorithm improvements, such as more accurate delineation of stroma [24]. However, other pitfalls may arise when tissue‐level outlining becomes too precise [22]. Specifically, CTA detects very small areas of stroma within tumor nests, which a pathologist might not consider due to their size, but no rules exist to define how small a stromal area can be to be included. This problem can lead to higher or lower measurements of TILs than a manual score if these areas include many TILs (larger TIL count) or do not include TILs (larger stromal area). The highly accurate quantification allowed by CTA will therefore lead to discrepancies with VTA pathologic evaluation; the gold standard will be the method that provides the highest clinical benefit, measured by its predictive or prognostic accuracy [22]. The choice between standard VTA and CTA‐derived guidelines will be settled by discrepancy aspects, which is discussed subsequently.
Image analysis challenges when adhering to a clinical guideline
Many of the pitfalls mentioned can be attributed to the image analysis approach that is used to implement the rules of the VTA guideline. However, the gold reference for scoring most existing histology‐based biomarkers is currently the pathologists' assessment, for instance, HER2 [34] and the VTA guideline [13]. Hence, the computational pathology community always needs to answer the same question first: What strategy do we want to use to translate the rules of manual guidelines into something a computer can execute? There are many valid answers to this question and we review the pros and cons of CTA in the following sections.
Different approaches to the computer vision problems relevant for building interpretable TIL algorithms
Here, we focus on three main categories of computational approaches for quantifying tissues and cells: (1) patch classification, (2) object detection, and (3) image segmentation. Although several other methods exist, we focus on the most commonly used approaches (see [35] for a more extensive review).
First, the simplest approach is classification, sometimes referred to as patch‐based approaches in digital pathology, especially for methods employing deep learning models. A supervised learning algorithm incorporates an image patch/field of view (FOV) for classification into one discrete label/class from a predefined set of labels. Second, object detection extends classification by producing one class per object of an image along with its spatial location. The location of objects of interest is marked with a box around the detected object, or the object is marked at its center. Finally, segmentation goes beyond detection by assigning a label/class to every pixel in the image to create a semantic map of the types of objects and their location. In contrast to object detection, segmentation can outline the border of the objects at very high resolution and accuracy. The general consideration when selecting a category for a computer vision problem is to determine the level of precision/resolution needed (i.e. how coarse the output can be to still adhere to the guideline). There are also differences in terms of training data and validation requirements that we will cover in later sections.
According to the TIL‐WG guideline [24], a CTA algorithm should be able to (1) detect and compartmentalize tissue into tumor structures, tumor‐associated stroma, and stroma outside tumor borders and (2) quantify TILs in the compartment. Different considerations apply to these two topics.
Considerations for tissue‐level models
Object detection is not suited for subdividing complex tissue structures into distinct areas (e.g. highly infiltrating tumor nests) and is therefore often excluded for the recognition and compartmentalization of tissue. Most CTA algorithms use a classification approach [21, 27] or full segmentation [20, 36]. The main difference is the granularity of the annotated maps produced, with segmentation being finer than classification, although resolution varies depending on the overlap and tile size of the input image patches and the size of the sliding window.
When using a direct classification of image patches as tumor, stromal, or lymphocyte regions, an individual patch may contain different tissue components, making classification not only difficult to train because of the inherent noise in the annotations but also imprecise for prediction due to the presence of multiple classes in a single image for only one output class. Moreover, a patch‐based approach may not provide detailed, quantitative information on TIL density; for instance, an accurate patch‐based lymphocyte classifier would produce the same output whether only one or many lymphocytes are within an input image. Bai et al [23] used manual outlining by a pathologist and did not discriminate between tumor and stromal areas, which was problematic for high‐iTIL cases, as mentioned previously. Sun et al [24] also used manual outlining in combination with a patch‐based model to identify and exclude necrosis. They then used the cell‐level output (see the next section for details) and empirically defined a tumor area as a patch containing more than two tumor cells. This sliding window approach produced relatively coarse boundaries compared to a full segmentation model [20, 22].
Providing models that segment the tissue allows for the construction of more detailed and quantitative information at the cellular level. Even though segmentation seems the obvious choice to carry out the tissue‐level task of detecting the stromal areas necessary for CTA, the approach also has disadvantages. First are potential segmentation artifacts from a sliding window analysis, which is preferred due to the gigapixel size of whole‐slide images (WSIs). This can also lead to tiling‐induced issues that result in incorrect labeling/misleading categorization (e.g. one glandular structure is divided in two, and these are analyzed independently, with one part being segmented as invasive tumor and the other part as DCIS). In the naïve setup, the ML model only takes into account one part at a time in what is called the receptive field (i.e. the tissue structure that the model sees at each prediction). Such inconsistencies along the edges of each FOV need to be handled, and in this postprocessing strategies can be helpful. If two segments of DCIS and invasive tumor regions touch as a single object, the size and shape of the DCIS segment can be considered in a logical postprocessing step to determine whether both should be segmented as DCIS or invasive tumor [22]. The important point is that these events are handled consistently, and with relevance to the clinical guideline. For example, one should rather exclude DCIS because there is often a high density of stromal TILs around these preinvasive lesions, and including false‐positive regions around DCIS structures would heavily influence the overall TIL score. Systematic inclusion of clinical guidelines in the digital framework is needed at either the data preprocessing or postprocessing stage.
Considerations for cell‐level models
The objective of TIL quantitation is to output the percentage of TILs in a given tissue region. This step is usually performed at the same or higher magnification than is used for tissue‐level analysis to include sufficient cell‐level image features for accurate model predictions. The main goal is to distinguish mononuclear immune cells from other cells. Previous studies performed this task by classification, detection, and segmentation. Janowczyk and Madabushi [37] used a classification model with a small sliding window to obtain the most likely location of each lymphocyte. A potential drawback of this method is computational inefficiency, as its high precision requires highly overlapping predictions. More recently, several studies [22, 38, 39] employed segmentation models to directly predict the center of all TILs in a FOV, avoiding the latter inefficiency issue. Others [24, 40] used a combination of both object detection and segmentation [41] to obtain the location and outline of TILs simultaneously. There are minor differences between the methods; the main challenges for cell‐level models relate to the requirements of the training data required for their development, which we discuss in the following section. Future work should also investigate improving the accuracy of cell classification to guide the model with information about the location of cells derived from compartment classification/segmentation. The idea is that the probability that cells will belong to a particular category (e.g. lymphocytes, fibroblasts) depends on their location in tissue compartments (like epithelium or stroma).
Another consideration here is the definition of the final sTIL score as a quantitative output variable, and recent methods have used different definitions. The VTA guideline uses an area coverage approach, which is the most accessible for humans to estimate. However, this introduces a slight size bias toward larger TIL nuclei. Does this mean that a CTA should do the same? We argue that as long as the CTA quantifies the degree of immune cell infiltration and is interpretable by pathologists (either by heatmaps or a score), then it is a valid score, and validation methods will then identify the most appropriate scoring system. That recent papers [22, 23, 24] found seven output variables associated with survival is evidence for this. Interestingly, although the VTA guideline explicitly states that sTILs should not be scored as a fraction of TILs compared to other cell populations, two variables of this assessment type consistently provide better results [23]. Thus, there might be other ways of creating a CTA, but it could also just be a derivative of the model design proposed in that paper.
Training data challenges to create robust and generalizable algorithms
The described models are exclusively built using deep learning, a powerful form of ML that, given sufficient training examples, learns to unravel and identify complex patterns. We will not review all aspects of this field but instead refer the reader to other excellent review articles [35, 42]. However, since the most promising CTA algorithms use deep learning, we will cover one of the main challenges of creating such algorithms: obtaining the training data required.
Data variation considerations
The general rule for creating a development/training dataset (i.e. the data used to develop the algorithm) is to include as much interclinical variation as the algorithm can be expected to encounter. Therefore, the requirements depend on the scope of the CTA algorithm, meaning the level of generalization required. For instance, single‐center research studies are deployed in only one laboratory. In a multicenter study, different laboratories participating in the training relate to internal validation, or a laboratory outside of model development is related to external validation. The answer to these questions indicates what boundaries of variation the CTA algorithm is expected to handle. The main sources of variation originate from the significant challenges in standardization within pathology. As such, before beginning image analysis, quality control of tissue, histology slide, stain, and WSI should be confirmed to ensure that a standard is met that will allow the collection of reliable data. Variability across pathology laboratories in preanalytical (e.g. fixation, sectioning) and analytical (e.g. staining protocol, scanner model) variables causes distributional shifts in the image data. Studies have investigated the impact of such variables, and methods to normalize and/or decrease variability from scanners [43, 44, 45] and staining [32, 46] have been developed.
Another important factor when curating a dataset is the impact of histological subtype variability (invasive ductal, invasive lobular, mucinous) on the underlying data distribution. Even the most powerful computational models, such as deep learning, may not generalize outside the subtype seen during training [47, 48] – one should not expect to successfully implement a model on lobular carcinoma if the data used for model development include only ductal carcinomas. This aspect sets some requirements on how to source and sample the patient cohort as part of relevant inclusion and exclusion criteria in the study design and should yield a balanced and realistic dataset. It is important to remember that for any digital model to work in a generalizable manner, interclass (between‐group) variation must be higher than the intraclass (within‐group) variance.
Generally, the solution to these issues is straightforward. Simply including relevant and sufficient variation in the development dataset aids in making the algorithm robust and generalizable. But even with increased sample numbers, the training set will only partially represent the full data distribution, and the trained algorithm will therefore be confronted with some previously unseen situations during application. Methods to identify, monitor, and flag additional novel classes [47], dataset shifts [48, 49], and normalization schemes [32, 43, 46] should help to reduce this problem.
Data labeling considerations
Acquiring an adequate number of manual labels is a critical step in computational pathology, given the time and effort required from pathologists and others with the specific expertise required. Several approaches have been proposed to address the need for manual labels in large‐scale datasets. The requirements for time, expertise, and methods depend on the model type being trained. The magnitude of investment correlates with the precision of the output required. In general, classification labels are the simplest to obtain (only one value is needed per image), then object detection labels (one click and one label per object), and then segmentation labels (many clicks and one label per object). For the new approaches being proposed, the major objective is to limit pathologist involvement to avoid the high cost, the time constraints of clinical practice, and the repetitive nature of annotating multiple examples.
The most straightforward strategy is manual annotations by a large number of experts. This approach generates high‐quality labels because ambiguous labels are identified and corrected, but it remains expensive and suffers from interlabeler variability and the subjectiveness inherent in histopathology. One solution is to ask multiple annotators to annotate the same data and produce a consensus label or model label variability [50]. A crowdsourcing framework for both tissue‐level segmentation and cell‐level classification, object detection, and segmentation was proposed to reduce pathologist effort and to model the interlabel variability of multiple labelers [40, 51]. Multiple nonpathologists (up to six) were required to match the performance of a senior pathologist. However, the benefit is restricted to annotating predominant and visually distinctive patterns, implying that pathologist involvement, and possibly full‐scale labeling effort, will be needed to supplement uncommon and difficult classes that require expertise. Of note, training and test sets must include borderline cases that are encountered in real life but might be hard to annotate. Otherwise, when trained and tested exclusively on ‘clean’ data, the algorithm may have difficulties with data for which the decision is harder to establish.
One of the most important aspects of developing a labeled dataset for CTA is the consistency of labels and annotations, i.e. minimization of ambiguous samples in the dataset. This consistency is difficult to adhere to when relying on manual labels. Compared to other fields, such as radiology, histopathology is unique in terms of creating a ground‐truth definition. For many applications, we rely on experts for ground truth, but we can also use the antibody–antigen specificity of immunohistochemical stains. Recently, multiple labeling schemes were proposed to obtain tissue‐ and cell‐level labels [22]. The idea is to use IHC to guide semiautomatic labels that can be transferred to primary H&E slides, and models can then be trained and deployed on H&E only. The obvious pitfall is the need to prepare new serial sections, which means using more tissue. Also of relevance for TILs, cellular information might be compromised between consecutive sections. Alternatively, the H&E section can be restained if the expertise is available, thereby ensuring that IHC‐stained lymphocytes can be found in the previously H&E‐stained slide. Even though this approach requires additional developmental effort, the quality and consistency of the labels were reported to be higher than those of manual labels, and only one pathologist was needed to review the labels, decreasing the time and effort required for model development [22].
Because WSIs are gigapixel files, it is intractable to manually label entire WSIs. Therefore, one needs to sample training regions, where it is important to use the same principles of including data (label) variation, e.g. regions with low‐, medium‐, and high‐density TILs should be included, also with varying proximity to invasive cells. One solution is to build weakly supervised image segmentation models that do not require detailed cell‐level labels.
Although many schemes can be employed to optimize the time and necessity for pathologist involvement, such procedures have their own pitfalls, as discussed earlier. We always advise developing an annotation protocol and labeling strategy in collaboration with a pathologist and treat it as an iterative process to identify errors and inconsistencies that will enhance the quality and scale of the training labels [52].
Data access and sharing considerations
It is obvious that access to raw data is a prerequisite for developing CTA algorithms. However, there are substantial challenges in collecting and/or accessing appropriate sets of data. Not all laboratories systematically scan all slides on modern scanners into an image management system (IMS) or picture archiving and communication system (PACS). Even fewer departments have digitized their archived slides or, indeed, have enough computer storage for such archiving. Scanning large retrospective datasets is, on the one hand, time‐consuming since most scanners need to be manually checked for quality, although on the other hand this may simplify future research.
A key aspect of developing successful CTA algorithms is collaboration between partners, among academic centers or academia and industry. The development of such algorithms on WSIs will be reviewed by a pathologist to communicate with the data scientists to improve algorithm adjustment and ML training. Another important aspect is analysis of the pathologist's notes. These notes and the complete clinical‐morphological data include information regarding stage, molecular profile, previous biopsies, and post‐treatment changes. Such analysis might be performed using a natural language processing pipeline to extract data for further standardized reporting. Getting the legal terms and conditions into place to share data can be a lengthy process. Sharing is recommended because it substantially eases this process for patient information protection regulations and the included requirement on information technology infrastructure and security. Another important consideration is the size of the datasets and how local or cloud platforms can be used to store, access, and share data.
There are successful studies sharing high‐quality histology datasets publicly under Creative Commons (CC) licenses [53, 54], either fully public [51, 55] or restricted for noncommercial use [56]. The latter can hinder academia–industry collaborations. The most commonly used platforms for public sharing of datasets are the Grand Challenges website [57] and The Cancer Genome Atlas (TCGA) [58]. Historically, there has been a shortage of publicly available datasets for developing CTA systems, with TCGA a notable exception and providing the foundation for many CTA studies [21, 22, 24, 40]. Nonetheless, care must be taken to avoid bias and batch effect implications from public datasets, which were not necessarily created for TIL evaluation [59]. There are recent joint efforts from the FDA and TIL‐WG to create datasets for algorithm validation [50, 60], to fill the critical need for the availability of development datasets. Collecting a large number of WSIs is time‐consuming and is subject to approval by institutional review boards and a data protection officer to comply with privacy and patient laws. Conversely, curating remains a barrier to the scaling of CTA algorithms.
Validation challenges when comparing CTA with VTA
Quantitative metrics on the performance of the different parts of a CTA algorithm need to be evaluated during development and, especially, during validation of the image analysis model [61]. As previously reviewed by the TIL‐WG [24], there are different levels of performance measurement. Briefly, analytical validation (AV) refers to low‐level metrics such as accuracy and reproducibility; clinical validation (CV) describes the discrimination of patients into clinical subgroups; and clinical utility measures the overall benefit in a clinical setting. In the following subsections, we discuss potential pitfalls in model validation.
Subcomponents of modular systems need different evaluation metrics
It is clear that to adhere to the TILS‐WG guideline, an accurate CTA algorithm must consist of multiple models aimed at solving different parts of the guideline. Hence, AV applies to the subcomponents as well as to the entire system. As the subcomponents can be different model approaches, the AV metric needs to capture aspects of each approach while providing information in situations where failure of a subcomponent will cause failure of CTA. Metrics such as accuracy, precision, recall, F‐scores, and Matthew's correlation coefficient are some of the other measures used to evaluate model performance.
If a subcomponent is a segmentation model (e.g. the tumor, necrosis, and noninvasive tissue‐level model), standardized metrics such as the F1 score can be used to evaluate AV. The F1 score can be interpreted as the weighted average of the precision and recall/sensitivity. However, it is important to consider that the F1 score on a FOV with no true‐positive segments of any given class will be evaluated as zero for that class, implying that potential false positives will not be captured as false positives, invalidating the overall F1 score. Another challenge for subcomponent AV is the impact of the exact test score of the model. A benchmark for the exact model selection does not always exist; hence, it is difficult to know if an exact score is sufficient or if a better (or worse) model would impact the AV and/or CV of the CTA.
Dudgeon et al [50] proposed both a metric and a dataset that might qualify as a FDA Medical Device Development Tool [60]. The metric is a multireader, multicase version of the mean squared error. Similar metrics such as Spearman rank‐based correlation are often used for the algorithm‐to‐pathologist comparison [20, 22]. One of the pitfalls of such count‐based metrics is that they do not capture whether the pathologist and algorithm are counting the same or different TILs because they compare only the sum of TILs. However, the metrics are easy to use and interpret, and they capture the most clinically relevant aspect of the algorithm – the extent of TILs in a defined region.
Considerations regarding clinical validation and utility
For the AV of the full algorithm, the same metrics can be used for the algorithm‐to‐pathologist comparison. However, as recently commented [62], the best method to evaluate digitally assessed biomarkers, such as CTA for both AV and CV, remains an open question. This point to the paradox of selecting the ground truth for digital pathology in TILs as either concordance between the pathologist and computational score or patient outcome, or a combination of both. This also raises the question of clinical cut‐off value for sTILs, since there are no formal recommendations at this time. The lack of manual VTA‐based cut‐off for patient stratification into clinically meaningful subgroups makes the process of CV more challenging for CTA because any cut‐off comparison between VTA and CTA might be arbitrary. Current CTA studies [22, 23, 24] use other cut‐off points than those used for VTA [3, 62, 63, 64] to identify two patient groups (TILs‐high versus TILs‐low) and find different levels of agreement between manual and automated methods at different cut‐offs. Sun et al [24] found moderate to substantial agreement depending on the exact cut‐offs, but only moderate agreement at a 10% cut‐off. In contrast, a different cohort [22] showed substantial agreement at 10% cut‐off. Interestingly, the former findings might imply different TIL cut‐off values are important, depending on the cohort and patient ethnicities, although no significant difference in TIL distribution was found between Asians and Caucasians [24]. This highlights the general difficulties of finding a cut‐off for biomarkers, which still involves a high degree of uncertainty [62]. In contrast, both studies found that CTA score as a continuous variable was associated with disease‐free survival (DFS) and OS. Hence, TILs could be better integrated into prognostic modeling containing existing clinical variables such as age, lymph node status, tumor size, tumor grade, and tumor type, removing the need to determine a cut‐off even for different ethnicities. The optimal method of TIL assessment – threshold versus continuous may be different for VTA versus CTA and remains an area of active research, e.g. against alternative endpoints such as BC progression [65].
Discussion
Current state‐of‐art CTA algorithms suggest that sTILs can be assessed computationally and represent a crucial prognostic and predictive factor for TNBC [7]. This review highlights different methodological approaches to designing algorithms. Beyond methodological design, many of the same pitfalls exist for VTA [14]. Whether these influence the clinical validation of CTA is to be determined, given that it depends on the future approaches taken to validate these algorithms. The TIL‐WG is currently organizing a grand challenge using phase 3 clinical trial data, which is a crucial step in validating any CTA algorithm [62]. This may answer many of the questions related to the clinical importance of CTA precision that are currently difficult to evaluate. However, similar collaborative community‐driven initiatives are needed to create robust and generalizable CTA algorithms. Many technological and procedural standardizations and harmonizations are necessary to counteract model‐decay and interinstitutional differences in workflow, especially in difficult tissues (e.g. small, deformed morphology or poor tissue integrity). Currently, there is no public framework or infrastructure to work collaboratively on different labeling strategies ensuring that CTA algorithms can identify and handle all histological components, including DCIS, fibrosis, hyalinization, and a larger number of granulocytes. There is currently no easy and practical way of building combined versioned datasets of standardized WSI and label formats, largely due to institutional data‐sharing restrictions and privacy requirements.
Another important unresolved aspect is the human–algorithm interaction, i.e. when and how the algorithm should be introduced into the workflow. Should the pathologist be required to open a case and manually annotate or edit regions, send the case for analysis, and wait for the result? Or should the algorithm be automated so that a case is analyzed based on slide metadata readily available after scanning, meaning that the case will have already been analyzed when the pathologist opens it for the first time? We deem the former unrealistic due to time and workload constraints. Different implementations will need to be optimized to augment and not disrupt the current workflow. Similarly, uncertainties remain on the best way to present the quantitative results of CTA, e.g. a precise count of TILs per square millimeter or a relative area. A dichotomous score of both computational and manual measurement may predict outcomes better than either variable alone [24]. This might affect whether the CTA should provide the primary score or work as a secondary reader on difficult cases.
It is clear that CTA is a powerful tool, but it is beneficial only when in the hands of expert pathologists. Work is in progress on many of these challenges as we look to an exciting future. Aware of the responsibility of the pathologist's decision‐making, we hold as our ultimate goal the development of robust tools for pathologists that assist with personalized precision care in a standardized and time‐efficient manner. We hope that by highlighting the specific pitfalls in using ML for sTIL assessment during both the model development and the clinical translation stages, future developments and collaborations will be positioned/forged to find the solutions needed to ensure reliable computational reporting of sTILs, with the end goal of using this tool in the routine clinical management of BC.
Author contributions statement
JT, GB and ES conceptualized, developed methodology and wrote the original draft. JT and ES were responsible for visualization. All authors were involved in reviewing and editing the original draft. SH, AD, TE, JD, EB and RS supervised. ZK, GA, NB, FC, EH, MK, RM, FP, JMR and ES were involved in writing, reviewing and editing the original draft. All authors have read and agreed to publish the final version of the manuscript.
Acknowledgements
The authors would like to thank Jeannette Parrodi, PA assistant to Professor Sherene Loi, for het extensive help and administrative support for the International Immuno‐Oncology Biomarker Working Group (TIL working group). Without her, this working group would not even exist. Furthermore, the authors make the following acknowledgments regarding support and funding. GB: Funded by Gilead Breast Cancer Research Grant 2023. SV: Supported by Interne Fondsen KU Leuven/Internal Funds KU Leuven. BA: supported by the Swedish Society for Medical Research (Svenska Sällskapet för Medicinsk Forskning) postdoctoral grant, Swedish Breast Cancer Association (Bröstcancerförbundet) Research grant 2021. GC: Peer Reviewed Cancer Research Program (Award W81XWH‐21‐1‐0160) from the US Department of Defense and the Mayo Clinic Breast Cancer SPORE grant P50 CA116201 from the National Institutes of Health (NIH). CF‐M: Funded by the Horizon 2020 European Union Research and Innovation Programme under the Marie Sklodowska Curie Grant agreement No. 860627 (CLARIFY Project). SBF: NHMRC GNT1193630. WMG: Support by the Higher Education Authority, Department of Further and Higher Education, Research, Innovation and Science, and the Shared Island Fund [AICRIstart: A Foundation Stone for the All‐Island Cancer Research Institute (AICRI): Building Critical Mass in Precision Cancer Medicine, https://www.aicri.org/aicristart]: Irish Cancer Society (Collaborative Cancer Research Centre BREAST‐PREDICT; CCRC13GAL; https://www.breastpredict.com), the Science Foundation Ireland Investigator Programme (OPTi‐PREDICT; 15/IA/3104), the Science Foundation Ireland Strategic Partnership Programme (Precision Oncology Ireland; 18/SPP/3522; https://www.precisiononcology.ie). SG: Partially supported by NIH grants CA224319, DK124165, CA263705, and CA196521. AG: Supported by Breast Cancer Now (and their legacy charity Breakthrough Breast Cancer) and Cancer Research UK (CRUK/07/012, KCL‐BCN‐Q3). TRK: Japan Society for the Promotion of Science (JSPS) KAKENHI (21K06909). UK: Funded by Horizon 2020 European Union Research and Innovation Programme under the Marie Sklodowska Curie Grant agreement 860627 (CLARIFY Project). JKL: This work is in part supported by NIH R37 CA225655 to JKL. AM: Research reported in this publication was supported by the National Cancer Institute under award numbers R01CA268287A1, U01CA269181, R01CA26820701A1, R01CA249992‐01A1, R01CA202752‐01A1, R01CA208236‐01A1, R01CA216579‐01A1, R01CA220581‐01A1, R01CA257612‐01A1, 1U01CA239055‐01, 1U01CA248226‐01, and 1U54CA254566‐01, National Heart, Lung and Blood Institute 1R01HL15127701A1, R01HL15807101A1, National Institute of Biomedical Imaging and Bioengineering 1R43EB028736‐01, VA Merit Review Award IBX004121A from the US Department of Veterans Affairs Biomedical Laboratory Research and Development Service the Office of the Assistant Secretary of Defense for Health Affairs, through the Breast Cancer Research Program (W81XWH‐19‐1‐0668), the Prostate Cancer Research Program (W81XWH‐20‐1‐0851), the Lung Cancer Research Program (W81XWH‐18‐1‐0440, W81XWH‐20‐1‐0595), the Peer Reviewed Cancer Research Program (W81XWH‐18‐1‐0404, W81XWH‐21‐1‐0345, W81XWH‐21‐1‐0160), the Kidney Precision Medicine Project (KPMP) Glue Grant, and sponsored research agreements from Bristol Myers‐Squibb, Boehringer‐Ingelheim, Eli‐Lilly, and Astrazeneca. SKM: Kay Pogue‐Geile, Director of Molecular Profiling at NSABP for her constant support and encouragement, Roberto Salgado, for initiating me into the wonderful subject of Immuno‐Oncology and its possibilities. FuAAM: Funding from EPSRC EP/W02909X/1 and PathLAKE consortium. FP‐L: Research grants from Fondation ARC, La Ligue contre le Cancer. RDP: The Melbourne Research Scholarship and a scholarship from the Peter MacCallum Cancer Centre. JSR‐F: Funded in part by the Breast Cancer Research Foundation, by a Susan G. Komen Leadership grant, and by the NIH/NCI grant P50 CA247749 01. JS: NIH/NCI grants UH3CA225021 and U24CA215109. ST: Supported by Interne Fondsen KU Leuven/Internal Funds KU Leuven. JT: Supported by institutional grants of the Dutch Cancer Society and the Dutch Ministry of Health, Welfare and Sport. EAT: Breast Cancer Research Foundation grant 22‐161. GEV: Supported by Breast Cancer Now (and their legacy charity Breakthrough Breast Cancer) and Cancer Research UK (CRUK/07/012, KCL‐BCN‐Q3). TW: Support by the French government under management of Agence Nationale de la Recherche as part of the Investissements d'avenir’ program, reference ANR‐19‐P3IA‐0001 (PRAIRIE 3IA Institute), and by Q‐Life (ANR‐17‐CONV‐0005). HYW: Funded in part by the NIH/NCI grant P50 CA247749 01. YY: Funding from Cancer Research UK Career Establishment Award (CRUK C45982/A21808). PS: Funding support from the National Health and Medical Research Council, Australia. SL: Supported by the National Breast Cancer Foundation of Australia (NBCF) (APP ID: EC‐17‐001), the Breast Cancer Research Foundation, New York [BCRF (APP ID: BCRF‐21‐102)], and a National Health and Medical Council of Australia (NHMRC) Investigator Grant (APP ID: 1162318). RS: Supported by the Breast Cancer Research Foundation (BCRF, grant 17‐194).
Conflict of interest statement: JT: Employee of Visiopharm A/S. GB: Speaker's fee received from MSD, Novartis, advisory boards for Roche and MSD, Consultant for MSD, Novartis and Roche, travel and conference support from Roche, MSD, and Gilead. ZK: Paid advisory role for Eli Lilly and AstraZeneca Canada. GA: Employee of Merck. KRB: Scientific advisory board for CDI Labs, Research funding from Carevive. FC: Chair of the Scientific and Medical Advisory Board of TRIBVN Healthcare, France, and received advisory board fees from TRIBVN Healthcare, France in the last 5 years. He is shareholder of Aiosyn BV, the Netherlands. LAC: Participation in Tempus Algorithm Advisors program. AC: Contracted researcher for Oncoinvent AS and Novocure and a consultant for Sotio a.s. and Epics Therapeutics SA. JDH: Cofounder of Visiopharm A/S. TE: Employee of Visiopharm A/S. ME: Egyptian missions sector. WMG: Cofounder, shareholder and part‐time Chief Scientific Officer of OncoAssure Limited, shareholder in Deciphex, and member of scientific advisory board of Carrick Therapeutics. JMG: Employee and stockholder of Roche/Genentech. SG: Research funding from Regeneron Pharmaceuticals, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Genentech, EMD Serono, Pfizer, and Takeda, unrelated to the current work; named co‐inventor on an issued patent for multiplex immunohistochemistry to characterize tumors and treatment responses. The technology is filed through Icahn School of Medicine at Mount Sinai (ISMMS) and is currently unlicensed. NH: Patent on a technology to measure immune infiltration in cancer to predict treatment outcome (WO2012038068A2). MGH: Consultant for PaigeAI, VolastraTx, and advisor for PathPresenter. JH: Speaker's honoraria or advisory board remunerations from Roche, Novartis, AstraZeneca, Eli Lilly, and MSD. Cofounder and shareholder of Stratipath AB. AIH: Research fund received from Visiopharm A/S. KK: Employee and stockholder of Roche. AK: Honorarium from Roche, MSD, and Pfizer and is a member of the advisory board of Pfizer. A‐VL: Institutional grants from AstraZeneca and personal grants from AstraZeneca (travel and honorarium from advisory board), MSD (honorarium from advisory board), and Daiichi Sankyo (travel). XL: Eli Lilly Company, Advisor, Cancer Expert Now, Advisor, Champions Oncology, Research fund. AM: Equity holder in Picture Health, Elucid Bioimaging, and Inspirata Inc., advisory board of Picture Health, Aiforia Inc., and SimBioSys, Consultant for SimBioSys, sponsored research agreements with AstraZeneca, Boehringer‐Ingelheim, Eli‐Lilly, and Bristol Myers‐Squibb, technology licensed to Picture Health and Elucid Bioimaging, involovement in three different R01 grants with Inspirata Inc. DKM: Consulting: Astrazeneca, Lilly USA LLC, Hologic. Sponsored research: Merck, Agendia. SM: Scientific Committee Study member: Roche, data and safety monitoring member of clinical trials: Sensorion, Biophytis, Servier, IQVIA, Yuhan, Kedrion. FuAAM: Research studentship funding from GSK. DAM: Speaker fees from AstraZeneca, Eli Lilly, and Takeda, consultancy fees from AstraZeneca, Thermo Fisher, Takeda, Amgen, Janssen, MIM Software, Bristol‐Myers Squibb, and Eli Lilly and has received educational support from Takeda and Amgen. FP‐L: Personal financial interests: AbbVie, Agendia, Amgen, Astellas, AstraZeneca, Bayer, BMS, Daiichi‐Sankyo, Eisai, Exact Science, GSK, Illumina, Incyte, Janssen, Lilly, MERCK Lifa, Merck‐MSD, Myriad, Novartis, Pfizer, Pierre‐Fabre, Roche, Sanofi, Seagen, Takeda, Veracyte, Servier. Institutional financial interests: AstraZeneca, Bayer, BMS, MSD, Myriad, Roche, Veracyte. Congress invitations: AbbVie, Amgen, AstraZeneca, Bayer, BMS, Gilead, MSD, Novartis, Roche, Lilly, Pfizer. NMR: CoFounder, director and CSO of Histofy Ltd, UK. JSR‐F: JSR‐F is an Associate Editor of The Journal of Pathology; he reports receiving personal/consultancy fees from Goldman Sachs, Bain Capital, REPARE Therapeutics, Saga Diagnostics, and Paige.AI, membership in scientific advisory boards of VolitionRx, REPARE Therapeutics, and Paige.AI, membership on the board of directors of Grupo Oncoclinicas, and ad hoc membership on the scientific advisory boards of Astrazeneca, Merck, Daiichi Sankyo, Roche Tissue Diagnostics, and Personalis, outside the scope of this study. ES: Employee of BMS. AS: AS: Advisory board/speaker's bureau: Aignostics, Astra Zeneca, Bayer, BMS, Eli Lilly, Illumina, Incyte, Janssen, MSD, Novartis, Pfizer, Roche, Seagen, Takeda, and Thermo Fisher, as well as grants from Bayer, BMS, Chugai, and Incyte. FS: Expert advisory panel for AXDEV Group. TT: Employee of Tempus Labs. JT: Shareholder of Ellogon.AI BV. TT: Speaker's fee received from Pfizer. JvdL: Member of advisory boards of Philips, the Netherlands, and ContextVision, Sweden, and received research funding from Philips, the Netherlands, ContextVision, Sweden, and Sectra, Sweden, in the last 5 years. He is chief scientific officer (CSO) and shareholder of Aiosyn BV, the Netherlands. TW: Collaboration with the company TRIBUN Health on automatic grading of biopsies for head and neck cancer and a patent on the prediction of homologous recombination deficiency (HRD) in breast cancer. YW: Employee of CellCarta. HYW: Advisory faculty of AstraZeneca. YY: Speaker/consultant for Roche and Merck. PS: Consultant (uncompensated) to Roche‐Genentech. SL: Research funding to her institution from Novartis, Bristol‐Meyers Squibb, Merck, Puma Biotechnology, Eli Lilly, Nektar Therapeutics, Astra Zeneca, Roche‐Genentech, and Seattle Genetics. SLoi has acted as consultant (not compensated) to Seattle Genetics, Novartis, Bristol‐Meyers Squibb, Merck, AstraZeneca, Eli Lilly, Pfizer, and Roche‐Genentech. SLoi has acted as consultant (paid to her institution) to Aduro Biotech, Novartis, GlaxoSmithKline, Roche‐Genentech, Astra Zeneca, Silverback Therapeutics, G1 Therapeutics, PUMA Biotechnologies, Pfizer, Gilead Therapeutics, Seattle Genetics, Daiichi‐Sankyo, Amunix, Tallac therapeutics, Eli Lilly, and Bristol‐Meyers Squibb. RS: Nonfinancial support from Merck and Bristol Myers Squibb (BMS), research support from Merck, Puma Biotechnology and Roche, and personal fees from Roche, BMS, and Exact Sciences for advisory boards.
References
- 1. Bates GJ, Fox SB, Han C, et al. Quantification of regulatory T cells enables the identification of high‐risk breast cancer patients and those at risk of late relapse. J Clin Oncol 2006; 24: 5373–5380. [DOI] [PubMed] [Google Scholar]
- 2. Wang M, Zhang C, Song Y, et al. Mechanism of immune evasion in breast cancer. Onco Targets Ther 2017; 10: 1561–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Savas P, Salgado R, Denkert C, et al. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol 2016; 13: 228–241. [DOI] [PubMed] [Google Scholar]
- 4. Hammerl D, Smid M, Timmermans AM, et al. Breast cancer genomics and immuno‐oncological markers to guide immune therapies. Semin Cancer Biol 2018; 52: 178–188. [DOI] [PubMed] [Google Scholar]
- 5. Hudeček J, Voorwerk L, van Seijen M, et al. Application of a risk‐management framework for integration of stromal tumor‐infiltrating lymphocytes in clinical trials. NPJ Breast Cancer 2020; 6: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leon‐Ferre RA, Jonas SF, Salgado R, et al. Abstract PD9‐05: stromal tumor‐infiltrating lymphocytes identify early‐stage triple‐negative breast cancer patients with favorable outcomes at 10‐year follow‐up in the absence of systemic therapy: a pooled analysis of 1835 patients. Cancer Res 2023; 83: PD9‐05. [Google Scholar]
- 7. Loi S, Drubay D, Adams S, et al. Tumor‐infiltrating lymphocytes and prognosis: a pooled individual patient analysis of early‐stage triple‐negative breast cancers. J Clin Oncol 2019; 37: 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liang H, Li H, Xie Z, et al. Quantitative multiplex immunofluorescence analysis identifies infiltrating pd1 + cd8 + and cd8 + T cells as predictive of response to neoadjuvant chemotherapy in breast cancer. Thorac Cancer 2020; 11: 2941–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Russo L, Maltese A, Betancourt L, et al. Locally advanced breast cancer: tumor‐infiltrating lymphocytes as a predictive factor of response to neoadjuvant chemotherapy. Eur J Surg Oncol 2019; 45: 963–968. [DOI] [PubMed] [Google Scholar]
- 10. Morigi C. Highlights of the 16th St Gallen international breast cancer conference, Vienna, Austria, 20–23 March 2019: personalised treatments for patients with early breast cancer. Ecancermedicalscience 2019; 13: 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Danske Multidisciplinære Cancer Grupper . Patologiprocedurer og molekylærpatologiske analyser ved brystkræft. Danske Multidisciplinære Cancer Grupper: Copenhagen, Denmark. [Accessed 31 August 2021]. Available from: https://dmcg.dk. [Google Scholar]
- 12. Regionala Cancercentrum I Samverkan . Kvalitetsbilaga för bröstpatologi (KVAST‐bilaga). Kunskapsbanken. Regionala Cancercentrum I Samverkan: Stockholm, Sweden. [Accessed 31 August 2021]. Available from: https://kunskapsbanken.cancercentrum.se.
- 13. Salgado R, Denkert C, Demaria S, et al. The evaluation of tumor‐infiltrating lymphocytes (TILs) in breast cancer: recommendations by an international TILs working group 2014. Ann Oncol 2015; 26: 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kos Z, Roblin E, Kim RS, et al. Pitfalls in assessing stromal tumor infiltrating lymphocytes (sTILs) in breast cancer. NPJ Breast Cancer 2020; 6: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O'Loughlin M, Andreu X, Bianchi S, et al. Reproducibility and predictive value of scoring stromal tumour infiltrating lymphocytes in triple‐negative breast cancer: a multi‐institutional study. Breast Cancer Res Treat 2018; 171: 1–9. [DOI] [PubMed] [Google Scholar]
- 16. Kilmartin D, O'Loughlin M, Andreu X, et al. Intra‐tumour heterogeneity is one of the main sources of inter‐observer variation in scoring stromal tumour infiltrating lymphocytes in triple negative breast cancer. Cancer 2021; 13: 4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van der Laak J, Litjens G, Ciompi F. Deep learning in histopathology: the path to the clinic. Nat Med 2021; 27: 775–784. [DOI] [PubMed] [Google Scholar]
- 18. Basavanhally AN, Ganesan S, Agner S, et al. Computerized image‐based detection and grading of lymphocytic infiltration in HER2+ breast cancer histopathology. IEEE Trans Biomed Eng 2010; 57: 642–653. [DOI] [PubMed] [Google Scholar]
- 19. Yuan Y, Failmezger H, Rueda OM, et al. Quantitative image analysis of cellular heterogeneity in breast tumors complements genomic profiling. Sci Transl Med 2012; 4: 157ra143. [DOI] [PubMed] [Google Scholar]
- 20. Amgad M, Sarkar A, Srinivas C, et al. Joint region and nucleus segmentation for characterization of tumor infiltrating lymphocytes in breast cancer. In Medical Imaging 2019: Digital Pathology, Tomaszewski JE, Ward AD (eds). SPIE: San Diego, 2019; 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saltz J, Gupta R, Hou L, et al. Spatial organization and molecular correlation of tumor‐infiltrating lymphocytes using deep learning on pathology images. Cell Rep 2018; 23: 181–193.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thagaard J, Stovgaard ES, Vognsen LG, et al. Automated quantification of sTIL density with H&E‐based digital image analysis has prognostic potential in triple‐negative breast cancers. Cancers 2021; 13: 3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bai Y, Cole K, Martinez‐Morilla S, et al. An open‐source, automated tumor‐infiltrating lymphocyte algorithm for prognosis in triple‐negative breast cancer. Clin Cancer Res 2021; 27: 5557–5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun P, He J, Chao X, et al. A computational tumor‐infiltrating lymphocyte assessment method comparable with visual reporting guidelines for triple‐negative breast cancer. EBioMedicine 2021; 70: 103492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Amgad M, Stovgaard ES, Balslev E, et al. Report on computational assessment of tumor infiltrating lymphocytes from the international immuno‐oncology biomarker working group. NPJ Breast Cancer 2020; 6: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep 2017; 7: 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Le H, Gupta R, Hou L, et al. Utilizing automated breast cancer detection to identify spatial distributions of tumor‐infiltrating lymphocytes in invasive breast cancer. Am J Pathol 2020; 190: 1491–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abousamra S, Gupta R, Hou L, et al. Deep learning‐based mapping of tumor infiltrating lymphocytes in whole slide images of 23 types of cancer. Front Oncol 2022; 11: 806603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. He T‐F, Yost SE, Frankel PH, et al. Multi‐panel immunofluorescence analysis of tumor infiltrating lymphocytes in triple negative breast cancer: evolution of tumor immune profiles and patient prognosis. PLoS One 2020; 15: e0229955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Swiderska‐Chadaj Z, Pinckaers H, van Rijthoven M, et al. Learning to detect lymphocytes in immunohistochemistry with deep learning. Med Image Anal 2019; 58: 101547. [DOI] [PubMed] [Google Scholar]
- 31. Balkenhol MCA, Ciompi F, Świderska‐Chadaj Ż, et al. Optimized tumour infiltrating lymphocyte assessment for triple negative breast cancer prognostics. Breast 2021; 56: 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tellez D, Litjens G, Bándi P, et al. Quantifying the effects of data augmentation and stain color normalization in convolutional neural networks for computational pathology. Med Image Anal 2019; 58: 101544. [DOI] [PubMed] [Google Scholar]
- 33. Kohlberger T, Liu Y, Moran M, et al. Whole‐slide image focus quality: automatic assessment and impact on AI cancer detection. J Pathol Inform 2019; 10: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smit G, Ciompi F, Cigéhn M, et al. Quality control of whole‐slide images through multi‐class semantic segmentation of artifacts. In MIDL 2021 Conference Short. Open Review: Amherst, MA, 2021. [Google Scholar]
- 35. Srinidhi CL, Ciga O, Martel AL. Deep neural network models for computational histopathology: a survey. Med Image Anal 2021; 67: 101813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abe N, Matsumoto H, Takamatsu R, et al. Quantitative digital image analysis of tumor‐infiltrating lymphocytes in HER2‐positive breast cancer. Virchows Arch 2020; 476: 701–709. [DOI] [PubMed] [Google Scholar]
- 37. Janowczyk A, Madabhushi A. Deep learning for digital pathology image analysis: a comprehensive tutorial with selected use cases. J Pathol Inform 2016; 7: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu Z, Xu S, Shao W, et al. Deep‐learning‐based characterization of tumor‐infiltrating lymphocytes in breast cancers from histopathology images and multiomics data. JCO Clin Cancer Inform 2020; 4: 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen J, Srinivas C. Automatic lymphocyte detection in H&E images with deep neural networks. ArXiv preprint 2016; 1612.03217. [Not peer reviewed].
- 40. Amgad M, Atteya LA, Hussein H, et al. NuCLS: A scalable crowdsourcing approach and dataset for nucleus classification and segmentation in breast cancer. GigaScience 2022; 11: giac037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He K, Gkioxari G, Dollar P, et al. Mask R‐CNN. IEEE Trans Pattern Anal Mach Intell 2020; 42: 386–397. [DOI] [PubMed] [Google Scholar]
- 42. Litjens G, Kooi T, Bejnordi BE, et al. A survey on deep learning in medical image analysis. Med Image Anal 2017; 42: 60–88. [DOI] [PubMed] [Google Scholar]
- 43. Swiderska‐Chadaj Z, de Bel T, Blanchet L, et al. Impact of rescanning and normalization on convolutional neural network performance in multi‐center, whole‐slide classification of prostate cancer. Sci Rep 2020; 10: 14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zarella MD, Bowman D, Aeffner F, et al. A practical guide to whole slide imaging: a white paper from the digital pathology association. Arch Pathol Lab Med 2019; 143: 222–234. [DOI] [PubMed] [Google Scholar]
- 45. Abels E, Pantanowitz L, Aeffner F, et al. Computational pathology definitions, best practices, and recommendations for regulatory guidance: a white paper from the digital pathology association. J Pathol 2019; 249: 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. de Bel T, Bokhorst J‐M, van der Laak J, et al. Residual cyclegan for robust domain transformation of histopathological tissue slides. Med Image Anal 2021; 70: 102004. [DOI] [PubMed] [Google Scholar]
- 47. Linmans J, van der Laak J, Litjens G. Efficient out‐of‐distribution detection in digital pathology using multi‐head convolutional neural networks. Proc Mach Learn Res 2020; 121: 465–478. [Google Scholar]
- 48. Thagaard J, Hauberg S, van der Vegt B, et al. Can you trust predictive uncertainty under real dataset shifts in digital pathology? In Medical Image Computing and Computer Assisted Intervention – MICCAI 2020 (Vol. 12261. Lecture Notes in Computer Science.), Martel AL, Abolmaesumi P, Stoyanov D, et al. (eds). Springer International Publishing: Cham, 2020; 824–833. [Google Scholar]
- 49. Stacke K, Eilertsen G, Unger J, et al. A closer look at domain shift for deep learning in histopathology. arXiv 2019; 1909.11575. [Not peer reviewed]. [DOI] [PubMed]
- 50. Dudgeon SN, Wen S, Hanna MG, et al. A pathologist‐annotated dataset for validating artificial intelligence: a project description and pilot study. J Pathol Inform 2021; 12: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Amgad M, Elfandy H, Hussein H, et al. Structured crowdsourcing enables convolutional segmentation of histology images. Bioinformatics 2019; 35: 3461–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wahab N, Miligy IM, Dodd K, et al. Semantic annotation for computational pathology: multidisciplinary experience and best practice recommendations. J Pathol Clin Res 2022; 8: 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Creative Commons — CC0 1.0 Universal . Creative Commons, [Accessed 31 August 2021]. Available from: https://creativecommons.org/publicdomain/zero/1.0/.
- 54. Creative Commons — Attribution‐NonCommercial‐ShareAlike 4.0 International — CC BY‐NC‐SA 4.0. Creative Commons, [Accessed 31 August 2021]. Available from: https://creativecommons.org/licenses/by-nc-sa/4.0/.
- 55. Litjens G, Bandi P, Ehteshami Bejnordi B, et al. 1399 H&E‐stained sentinel lymph node sections of breast cancer patients: the CAMELYON dataset. GigaScience 2018; 7: giy065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Prostate cANcer graDe Assessment (PANDA) Challenge, Kaggle. [Accessed 31 August 2021]. Available from: https://www.kagle.com.
- 57. Grand Challenge . Grand Challenge, [Accessed 31 August 2021]. Available from: https://grand-challenge.org/.
- 58. NIH . The Cancer Genome Atlas Program (TCGA). NIH National Cancer Institute: Center for Cancer Genomics. [Accessed 31 August 2021]. Available from: https://www.cancer.gov/tcga.
- 59. Howard FM, Dolezal J, Kochanny S, et al. The impact of site‐specific digital histology signatures on deep learning model accuracy and bias. Nat Commun 2021; 12: 4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. U.S. Food & Drug Administration . Qualification of Medical Device Development Tools, November 2013. [Accessed 31 August 2021]. Available from: https://www.fda.gov.
- 61. Kleppe A, Skrede O‐J, De Raedt S, et al. Designing deep learning studies in cancer diagnostics. Nat Rev Cancer 2021; 21: 199–211. [DOI] [PubMed] [Google Scholar]
- 62. Acs B, Salgado R, Hartman J. What do we still need to learn on digitally assessed biomarkers? EBioMedicine 2021; 70: 103520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stanton SE, Disis ML. Clinical significance of tumor‐infiltrating lymphocytes in breast cancer. J Immunother Cancer 2016; 4: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stanton SE, Adams S, Disis ML. Variation in the incidence and magnitude of tumor‐infiltrating lymphocytes in breast cancer subtypes: a systematic review. JAMA Oncol 2016; 2: 1354. [DOI] [PubMed] [Google Scholar]
- 65. Fassler DJ, Torre‐Healy LA, Gupta R, et al. Spatial characterization of tumor‐infiltrating lymphocytes and breast cancer progression. Cancer 2022; 14: 2148. [DOI] [PMC free article] [PubMed] [Google Scholar]