

Abstract
The lipid peroxidation product malondialdehyde and the DNA peroxidation product base-propenal react with dG to generate the exocyclic adduct, M1dG. This mutagenic lesion has been found in human genomic and mitochondrial DNA. M1dG in genomic DNA is enzymatically oxidized to 6-oxo-M1dG, a lesion of currently unknown mutagenic potential. Here, we report the synthesis of an oligonucleotide containing 6-oxo-M1dG and the results of extension experiments aimed at determining the effect of the 6-oxo-M1dG lesion on the activity of human polymerase iota (hPol ι). For this purpose, an LC-MS/MS assay was developed to obtain reliable quantitative data on the utilization of poorly incorporated nucleotides. Results demonstrate that hPol ι primarily incorporates deoxycytidine triphosphate (dCTP) and thymidine triphosphate (dTTP) across from 6-oxo-M1dG with approximately equal efficiency whereas deoxyadenosine triphosphate (dATP) and deoxyguanosine triphosphate (dGTP) are poor substrates. Following incorporation of a single nucleotide opposite the lesion, 6-oxo-M1dG blocks further replication by the enzyme.
Keywords: Translesion synthesis, 6-oxo-M1dG, M1dG, LC-MS/MS, oligonucleotide synthesis, DNA adduct
Graphical Abstract
INTRODUCTION
M1dG is the major DNA adduct formed by the reaction of deoxyguanosine with the DNA peroxidation product base-propenal or the lipid peroxidation product malondialdehyde (Figure 1A)1–3. M1dG is present in mammalian genomic DNA and has been detected in human leukocyte DNA at levels that range from 0.004–9 adducts per 108 bases4, 5. Mitochondrial DNA contains levels of M1dG that are 50–100-fold higher than in genomic DNA and are increased by agents that disrupt mitochondrial electron transport or by mutations in bone morphogenetic peptide receptor-26. Single-adduct mutagenesis experiments indicate that M1dG induces transitions to dA and transversions to dT as well as frameshift mutations7, 8. It is removed from genomic DNA by nucleotide excision repair and oxidation by AlkB7, 9. M1dG that is detectable in urine from healthy humans may reflect whole body production followed by nucleotide excision repair10.
Figure 1.
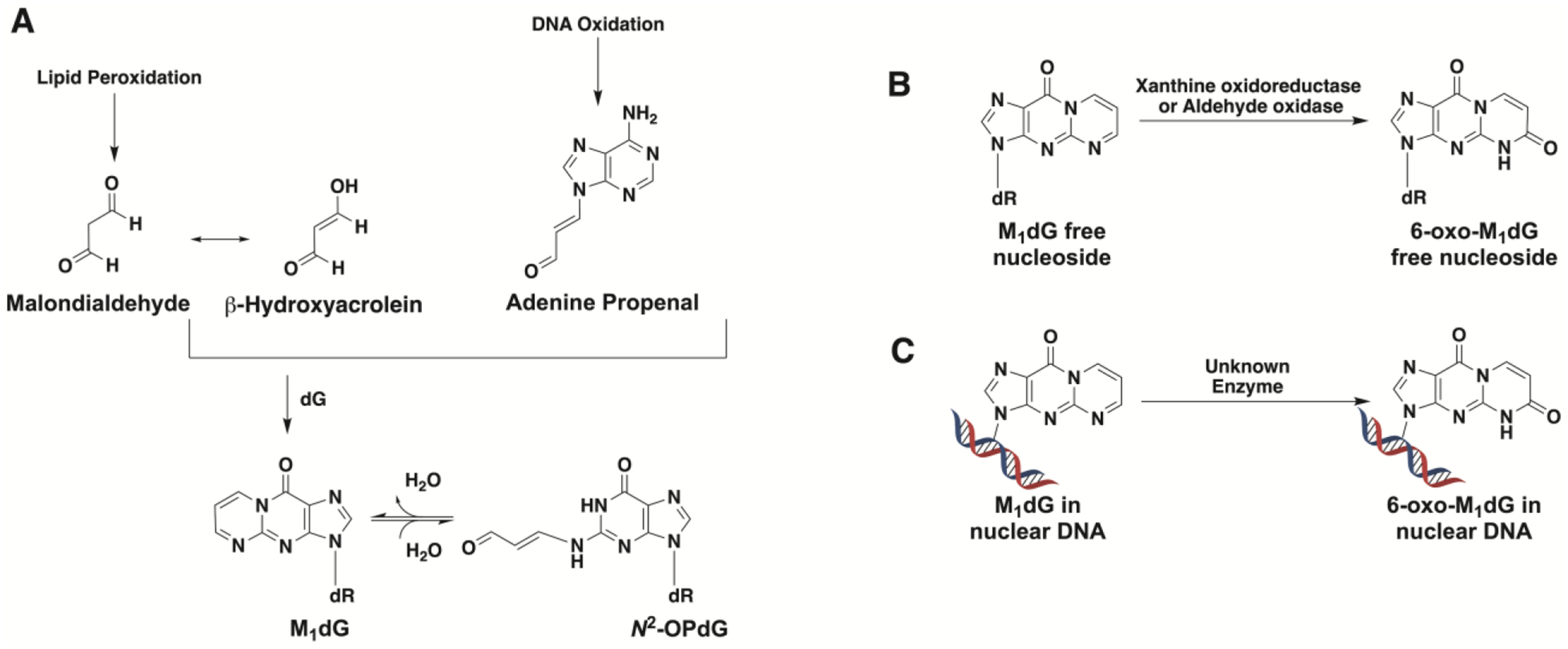
(A) Formation of M1dG from the lipid peroxidation product malondialdehyde via its tautomer β-hydroxyacrolein or from the DNA oxidation product adenine propenal. M1dG is subject to ring-opening to form N2-(3-oxo-1-propenyl)-dG (N2-OP-dG). (B) M1dG generated by base excision repair of damaged DNA is oxidized to 6-oxo-M1dG by xanthine oxidoreductase or aldehyde oxidase. (C) M1dG in nuclear DNA is oxidized to 6-oxo-M1dG.
Injection of M1dG into rats results in rapid metabolism to a single oxidation product, 6-oxo-M1dG (Figure 1B)11–14. Whereas M1dG is excreted in urine, 6-oxo-M1dG is excreted in feces13. Production of 6-oxo-M1dG from M1dG by rat liver cytosol is suppressed by inhibitors of xanthine oxidoreductase or aldehyde oxidase, suggesting a role for these two enzymes in the reaction11. 6-Oxo-M1dG is also formed in genomic DNA (Figure 1C)15. Treatment of several human cell lines with adenine propenal led to a rapid elevation in the levels of genomic M1dG, which was then converted to 6-oxo-M1dG by an unidentified oxidase. The rate of oxidation of M1dG in genomic DNA to 6-oxo-M1dG was faster than that of M1dG removal by nucleotide excision repair in these experiments.
The structural impact, mutagenicity, and repair of 6-oxo-M1dG in duplex DNA have not been determined because of the unavailability of oligonucleotides containing the adduct at defined positions. We report here the first synthesis of DNA oligonucleotides containing 6-oxo-M1dG. Reaction schemes that have been used to incorporate other exocyclic DNA adducts into oligonucleotides proved unsuccessful with 6-oxo-M1dG, requiring a reversal of the routinely employed acylation-cyclization sequence.
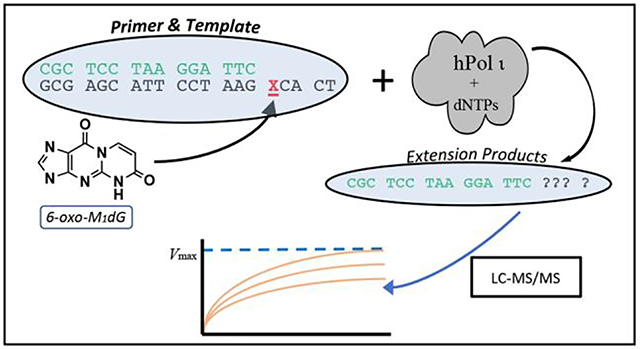
The newly synthesized template containing 6-oxo-M1dG at a defined position afforded the opportunity to conduct in vitro translesion bypass experiments using the Y family DNA polymerases, which are known for their ability to replicate across from sites of DNA damage16, 17. We chose to first examine bypass of the 6-oxo-M1dG lesion by hPol ι using a conventional gel-electrophoresis-based polymerase assay. hPol ι inserts a nucleotide across from a wide variety of adducts, which makes it an attractive starting point for translesion bypass studies. In addition, it often incorporates only a single nucleotide across from the site of damage without further extension of the primer18. This has been shown to occur in the hPol ι-mediated bypass of M1dG19. Thus, the products obtained from the action of hPol ι on the 6-oxo-M1dG-containing template were expected to be primarily or solely single nucleotide incorporations, resulting in a straightforward product profile. Our initial experiments revealed preferential incorporation of dCTP and dTTP opposite the lesion. However, the very low efficiency of dATP and dGTP incorporation generated weak bands on the polyacrylamide gels, thereby limiting our ability to acquire reliable data for kinetic analysis of the reaction. To address this problem, we developed a liquid chromatography/tandem mass spectrometry (LC-MS/MS) assay for the quantification of oligonucleotide primers and extension products. This assay, which detects the intact oligonucleotides directly via selected reaction monitoring (SRM), does not require a restriction digest step common to many MS-based oligonucleotide analyses20, 21. Thus, we describe here the synthesis of a novel 6-oxo-M1dG-containing oligonucleotide, the kinetics of hPol ι-mediated nucleotide insertion opposite the 6-oxo-M1dG lesion, and an LC-MS/MS assay for the direct and accurate quantitation of the products of the hPol ι-mediated extension.
EXPERIMENTAL PROCEDURES
Reagents.
All DNA oligonucleotides with the exception of the 6-oxo-M1dG oligonucleotide were from Integrated DNA Technologies (IDT), Inc. DNase I and dNTPs were from New England Biolabs. Micrococcal nuclease, phosphodiesterase I, and alkaline phosphatase were from Worthington Biochemical. hPol ι was purified as described previously22. Methanol, acetonitrile, and water were HPLC-grade and were purchased from Fisher. Nuclease P1, formic acid, triethylamine, and hexafluoroisopropanol were from Sigma. All chemicals were the best available quality.
Synthesis of the 6-oxo-M1dG-containing oligonucleotide
General methods.
1H NMR spectra were recorded at 400, 500, or 600 MHz on a Brucker AM NMR spectrometer in acetonitrile-d3 (CD3CN) and dimethyl sulfoxide-d6 (DMSO-d6). Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad, app = apparent), coupling constants (Hz), and integration. 13C NMR data were recorded on a Bruker instrument at 125 MHz. Thin-layer chromatography was performed on silica gel glass plates (Silica XHL TLC Plates, w/UV254, glass backed, layer thickness 250 μm, Sorbent Technologies Atlanta, GA). The chromatograms were visualized under UV light (254 nm) or by staining with an anisaldehyde/H2SO4 solution, followed by heating. Purification of the compounds was performed by a Combi-flash Rf (plus-UV) Automated Flash Chromatography System using silica gel (60 Å, 63–200 μm, Sorbent Technologies Atlanta, GA). The silica gel was suspended in dichloromethane, and triethylamine was added. The suspension was stirred occasionally with a glass rod and then placed into a loading cartridge. The cartridge was sealed, attached to the Combi-flash RF, and washed with 300 mL of dichloromethane. The cartridge was then removed from the instrument, loaded with the reaction mixture, and attached back to the instrument for elution.
Synthesis of 2-amino-9-((2R,4S,5R)-5-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-4-((triethylsilyl)oxy)tetrahydrofuran-2-yl)-1,9-dihydro-6H-purin-6-one (Scheme 3, compound 9).
2-Amino-9-((2R,4S,5R)-5-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-4-hydroxytetrahydrofuran-2-yl)-1,9-dihydro-6H-purin-6-one (0.5 g, 0.87 mmol) was dissolved in anhydrous dimethyl formamide (5 mL). The solution was cooled to 0 °C, and imidazole (0.177 g, 2.6 mmol) was added followed by the dropwise addition of chlorotriethylsilane (0.190 mL, 1.13 mmol). The reaction mixture was stirred for 1 h at 0 °C and at room temperature for 1 h. The reaction mixture was cooled to 0 °C, quenched with water (25 mL), and extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with brine and dried with anhydrous sodium sulfate. The drying agent was filtered off, and the filtrate was concentrated. The residue was purified by Combi-flash Rf, (0–10% methanol in dichloromethane over 30 min). The procedure yielded 505 mg (85%) of product. 1H NMR (DMSO-d6) δ 7.83 (s, 1H), 7.35–7.33 (m, 2H), 7.29–7.20 (m, 7H), 6.85–6.83 (m, 4H), 6.44 (s, 2H), 6.12 (t, 1H, J = 8 Hz), 4.49–4.47 (m, 1H), 3.85–3.80 (m, 1H), 3.73 (s, 6H), 3.22–3.19 (m, 1H), 3.11–3.08 (m, 1H), 2.69–2.63 (m, 1H), 2.30–2.25 (m, 1H), 0.84 (t, 9H, J = 8 Hz), 0.54–0.50 (m, 6H) (Supplementary Figure S1); 13C NMR (DMSO-d6) 158.47, 157.25, 154.19, 151.45, 145.15, 135.82, 135.19, 130.04, 128.13, 128.03, 127.05, 117.21, 113.49, 86.06, 85.86, 82.39, 72.01, 63.74, 55.39, 55.29, 6.94, 4.74, 4.45, 4.15 (Supplementary Figure S2). High resolution MS: m/z calculated for C37H46N5O6Si [M + H]+ 684.3212, found 684.3202.
Synthesis of 3-((2R,4S,5R)-5-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-4-hydroxytetrahydrofuran-2-yl)-3,5-dihydropyrimido[1,2-a]purine-6,10-dione (Scheme 3, Compound 11).
2-Amino-9-((2R,4S,5R)-5-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-4-((triethylsilyl)oxy)tetrahydrofuran-2-yl)-1,9-dihydro-6H-purin-6-one (0.4 g, 0.58 mmol) was dissolved in anhydrous dimethyl formamide (8 mL). The solution was cooled to 0 °C, and triethylamine (0.41 mL, 2.92 mmol) was added followed by the dropwise addition of (E)-3-ethoxyacryloyl chloride (0.09 mL, 0.75 mmol). The reaction mixture was stirred for 1 h at 0 °C. Thin-layer chromatography showed a complete reaction. Water (20 mL) was added to quench the reaction, and the product was extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with brine and dried with anhydrous sodium sulfate. The drying agent was filtered off, and the filtrate was concentrated to give an oily product. The product was placed under high vacuum for 1 h, then dissolved in anhydrous tetrahydrofuran (5 mL). The reaction mixture was cooled to 0 °C, and triethylamine (0.49 mL, 3.51 mmol) was added followed by dropwise addition of triethylamine trihydrofluoride (0.19 mL, 1.17 mmol). The reaction mixture was then stirred at 0 °C for 30 min. Thin-layer chromatography showed a complete reaction. Water (20 mL) was added to quench the reaction, and the product was extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with brine and dried with anhydrous sodium sulfate. The drying agent was filtered off, and the filtrate was concentrated to give an oily product. The product was placed under high vacuum for 1 h, then dissolved in anhydrous methanol (5 mL). Anhydrous potassium carbonate (0.16 g, 1.17 mmol) was added, and the mixture was heated at 37 °C for 1 h. The methanol was removed in vacuo, and the residue was dissolved in dichloromethane, transferred into a separatory funnel, and washed with 5% acetic acid. The organic layer was separated and dried with anhydrous sodium sulfate. The drying agent was filtered off, and the filtrate was concentrated down to give a solid reside that was purified by Combi-flash Rf, (0–10% methanol in dichloromethane over 30 min). Obtained 300 mg (83%) of product. 1H NMR (DMSO-d6) δ 8.62 (d, J = 8.4 Hz, 1H), 8.08 (s, 1H), 7.34–7.32 (m, 2H), 7.25–7.17 (m, 7H), 6.83–6.77 (m, 4H), 6.27 (t, 1H, J = 8 Hz), 6.18 (d, J = 8.4 Hz, 1H), 5.34 (br s, 1H), 4.44–4.42 (m, 1H), 3.99–3.95 (m, 1H), 3.71 (s, 6H), 3.27–3.23 (m, 1H), 3.15–3.12 (m, 1H), 2.75–2.70 (m, 1H), 2.36–2.30 (m, 1H) (Supplementary Figures S3 and S4). 13C NMR (DMSO-d6), 163.05, 158.38, 158.33, 125.62, 149.68, 148.17, 145.28, 138.39, 135.95, 135.90, 134.41, 130.10, 130.04, 128.07, 126.94, 116.84, 113.43, 113.39, 109.52, 86.50, 85.80, 83.17, 70.97, 64.73 (Supplementary Figure S5); High resolution MS: m/z calculated for C34H32N5O7 [M + H]+ 622.2296, found 622.2280.
Synthesis of (2R,3S,5R)-2-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-5-(6,10-dioxo-5,10-dihydropyrimido[1,2-a]purin-3(6H)-yl)tetrahydrofuran-3-yl-(2-cyanoethyl) diisopropylphosphoramidite (Scheme 3, Compound 12).
3-((2R,4S,5R)-5-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-4-hydroxytetrahydrofuran-2-yl)-3,5-dihydropyrimido[1,2-a]purine-6,10-dione (0.05 g, 0.08 mmol) was dried by co-evaporation with anhydrous pyridine and dried overnight under high vacuum. The gummy residue was dissolved in dry dichloromethane (15 mL), and N,N-diisopropylethylamine (0.056 mL, 0.32 mmol) was added. 2-Cyanoethyl-N,N-diisopropylchlorophosphoramidite (0.022 mL, 0.09 mmol) was added dropwise, and the reaction mixture was stirred at room temperature for 2 h. The reaction was quenched by the addition of saturated sodium bicarbonate. The organic layer was collected, and the solvent was removed in vacuo with a rotary evaporator. The crude product was purified by Combi-flash Rf, (0–3% methanol in dichloromethane over 60 min) to give the corresponding phosphoramidite (39 mg, 60%). 1H NMR (CD3CN) 8.66 (d, 1H, J = 8Hz), 7.80 (s, 1H), 7.43–7.40 (m, 2H), 7.29–7.21 (m, 7H), 6.30 (t, 1H, J = 8 Hz), 6.14 (d, J = 8.4 Hz, 1H), 4.73–4.69 (m, 1H), 4.28–4.18 (m, 1H), 3.85–3.58 (m,2H), 3.75 (s, 6H), 3.34–3.28 (m, 2H), 3.18–3.15 (m, 2H), 2.84–2.81 (m, 1H), 2.68–2.55 (m, 3H), 1.13–1.06 (m, 12H) (Supplementary Figure S6); 31P NMR (CD3CN, 121 MHz) δ 148.25, 148.03 (Supplementary Figure S7). 13C NMR (CD3CN) 166.51, 159.61, 159.58, 154.04, 151.33, 151.24, 150.83, 150.73, 146.04, 138.06, 138.00, 136.79, 136.73, 134.78, 131.03, 130.99, 130.95, 130.93, 128.98, 128.92, 128.73, 127.76, 113.95, 113.93, 110.17, 87.04, 86.38, 84.41, 84.29, 74.84, 74.66, 74.37, 64.80, 59.42, 59.23, 55.84, 49.90, 44.10, 43.97, 39.91, 39.49, 24.89, 24.84, 24.77, 21.01, 20.59, 16.56 (Supplementary Figure S8); High resolution MS (fast atom bombardment+) m/z calculated for C43H49N7O8P [M + H]+ 822.3374, found 822.3368.
Oligonucleotide synthesis.
The DNA oligonucleotides were synthesized on a Perseptive Biosystems Model 8909 DNA synthesizer at a 1-μmol scale using their Expedite reagents with the standard synthetic protocol for the coupling of the unmodified bases. The coupling of the 6-oxo-M1dG phosphoramidite (12) was performed off-line manually for 30 min as previously described23.
The remainder of the synthesis was performed on-line using standard protocols. Unless otherwise noted, the modified oligonucleotides were cleaved from the solid support, and the exocyclic amino groups were deprotected in a single step using K2CO3 (0.5 M, methanol) at room temperature overnight.
HPLC purification and analysis.
HPLC purification and analysis were carried out on a gradient HPLC (Beckman Instruments; System Gold Software) equipped with pump module 125 and photodiode array detector module 168. A Phenomenex Gemini-C18 column (250 × 4.6 mm, flow rate 1.5 mL/min, 250 × 4.6 mm, flow rate 5 mL/min) was used to monitor reactions and for oligonucleotide purification. Oligonucleotides were detected by their UV absorbance at 254 nm. The HPLC elution solvents consisted of acetonitrile and 100 mM aqueous ammonium formate. The following gradient was used: initial conditions were 1% acetonitrile, followed by a 15 min linear gradient to 15% acetonitrile, then a 5 min linear gradient to 20% acetonitrile, 5 min of isocratic elution at 20% acetonitrile, then a 2.5 min linear gradient to 80% acetonitrile, 3 min of isocratic elution at 80% acetonitrile, then a 3 min linear gradient to the initial conditions. A chromatogram of the purified oligonucleotide indicated >98% purity, and the mass spectrum revealed an m/z value of 1537.3 (z = 4, m/z theoretical 1536.8) (Supplementary Figure S9).
Enzymatic digestion of oligonucleotides.
The 6-oxo-M1dG oligonucleotide was heated at 95 °C for 3 min. For LC-MS/MS analysis, the oligonucleotide (40 pmol) was combined with nuclease P1 (0.8 U), ZnCl2 (5 mM), MgCl2 (15 mM), 3-(N-morpholino)propanesulfonic acid (MOPS, 40 mM, pH 7.9), [15N]-labeled deoxyguanosine (100 pmol), and [15N]-labeled 6-oxo-M1dG (50 pmol). This enzyme mixture was incubated for 1 h at 60 °C. Next, phosphodiesterase I (0.4 U) and alkaline phosphatase (0.75 U) were added in a final reaction volume of 59 μL, and the mixture was incubated at 37 °C overnight. For HPLC analysis, the digestion procedure was the same except 400 pmol of oligonucleotide was digested, and [15N]-labeled dG and 6-oxo-M1dG were not added.
LC-MS/MS analysis of digested oligonucleotides.
LC-MS/MS analyses were performed on a Shimadzu Nexera X2 system in-line with a SCIEX 6500 QTrap mass spectrometer, and the SCIEX Analyst software package was used to collect and process data. The oligonucleotide digestion mixture (10 μL) was added to water (90 μL), and 20 μL was injected. An Acquity C18 column (10 × 0.2 cm, 1.7 μm) at 32 °C was used for analyses. The mobile phase consisted of methanol:acetonitrile (3:1, 0.5% formic acid, B) and water (0.5% formic acid, A), and the flow rate was 0.3 mL/min. The following gradient was used: initial conditions were 2% B for 0.5 min, followed by a 6 min linear gradient to 80% B, then 1 min of isocratic elution at 80% acetonitrile, then a 0.5 min linear gradient to 2% B. The column was equilibrated for 2 min at the initial conditions prior to each injection. 6-Oxo-M1dG and dG were detected by SRM in positive ion mode with the following transitions: m/z 268.1 to 152.1 (dG) and m/z 320.1 to 204.1 (6-oxo-M1dG). The analytes were quantitated against their [15N]-labeled analogs, and the SRM reactions for these internal standards were m/z 273.1 to 157.2 (15N5-dG) and m/z 325.1 to 209.1 (15N5-6-oxo-M1dG).
HPLC of digested and intact oligonucleotides.
HPLC analyses of undigested and digested oligonucleotides were carried out on a Waters 1525 binary pump in-line with a Waters 2996 photodiode array detector using a Phenomenex Jupiter Proteo 90 Å column (15 cm × 0.46 cm). Waters Empower software was used for instrument control, data acquisition, and data processing. UV data were collected from 215 to 400 nm, and quantitative data were generated for all samples by extracting and processing chromatograms at 258 nm. The mobile phase consisted of methanol:50 mM triethylammonium acetate, pH = 7.2 (1:1) (B), and 50 mM triethylammonium acetate, pH = 7.2 (A). The following gradient was used: initial conditions were 1% B for 1.25 min, followed by a 15 min linear gradient to 40% B, then 1 min of isocratic elution at 40% acetonitrile. The column was equilibrated for 8 min at the initial conditions prior to each injection.
MS analysis of the 6-oxo-M1dG-containing oligonucleotide.
MS analysis of the 6-oxo-M1dG oligonucleotide was performed at the Mass Spectrometry Research Center at Vanderbilt University on an Acquity UPLC system using an Acquity UPLC BEH C18 column (1 μM, 1.0 mm × 100 mm) (Waters, Milford, MA) connected to a Finnigan LTQ mass spectrometer (ThermoElectron) equipped with an Ion Max API source and a standard electrospray probe. LC conditions were as follows: buffer A contained 10 mM ammonium acetate plus 2% acetonitrile (v/v), and buffer B contained 10 mM ammonium acetate plus 95% acetonitrile (v/v). The following gradient program was used with a flow rate of 0.15 mL/min: initial conditions were 0% B, followed by a 3 min linear gradient to 3% B, then a 1.5 min linear gradient to 20% B, then a 0.5 min linear gradient to 100% B, then 0.5 min of isocratic elution at 100% B, then a 1 min linear gradient to 0% B, then 3 min of isocratic elution at 0% B. The temperature of the column was maintained at 50 °C, and the samples (10 μL) were injected with an auto-sampler. The electrospray conditions were as follows: source voltage 4 kV, source current 100 μA, auxiliary gas N2, flow rate setting 20, sweep gas flow rate setting 5, sheath gas flow setting 34, capillary voltage −49 V, capillary temperature 350 °C, and tube lens voltage −90 V. No collision-induced dissociation offset was employed. MS/MS conditions were as follows: normalized collision energy 35%, activation Q 0.250, and activation time 30 ms. The isolation width in MS/MS was 2. The automatic gain control settings in full MS were 10000. The maximum injection time in full MS was 10 ms. The MS data were acquired in negative ion mode. Helium was used as the collision damping gas in the ion trap and was set at a pressure of 1 mTorr. The number of μscans used for data acquisition in full MS was 2. Product ion spectra were acquired over the range m/z 345 to 2000.
Low and high-resolution fast atom bombardment mass spectra were obtained at the Mass Spectrometry Facility at the University of Notre Dame, Notre Dame, IN.
Single nucleotide insertion and primer extension assays
Generation of primer-template DNA duplexes for in vitro assays.
Control oligonucleotide (5′-TC AGC GAA TCC TTA CGA GCG-3′) or 6-oxo-M1dG oligonucleotide (5′-TC ACX GAA TCC TTA CGA GCG-3′, X = site of 6-oxo-M1dG incorporation) was mixed with an equimolar amount of 5′-fluorescein- (FAM)-labeled complementary primer (5′-FAM-CGC TCG TAA GGA TTC-3′). The mixture was heated at 95 °C for 4 min and allowed to cool slowly overnight.
Primer extension and single nucleotide insertion assays.
Primer extension assays were conducted at 37 °C with 5 mM dithiothreitol, 0.05% bovine serum albumin, 5% glycerol, 5 mM MgCl2, 50 mM NaCl, 40 mM Tris-HCl (pH 7.5), 5 μM primer-template complex, and 500 μM dNTP mix (all four dNTPs) or 500 μM individual dNTPs (dATP, dCTP, dGTP, or dTTP). The concentration of hPol ι varied from 50 to100 nM. The total reaction volume was 10 μL. The reactions were quenched at time points from 0 to 90 min by adding 2 μL of the reaction mixture to 8 μL stop solution [10 mM ethylenediaminetetraacetic acid, 95% formamide (v/v), 0.03% bromophenol blue (w/v), and 0.03% xylene cyanol (w/v)]. Quenched products (2 μL aliquots) were separated by electrophoresis on a 20% (w/v) denaturing polyacrylamide gel with 7 M urea. The gels were then imaged on a Typhoon Trio Mode Imager in fluorescence mode with the green (532 nm) laser in conjunction with the 526-nm short-pass filter.
Steady-state kinetic analysis.
Steady-state kinetic reactions were performed using a duplex of control oligonucleotide annealed to the complementary FAM-labeled primer and a duplex of 6-oxo-M1dG annealed to the same complementary FAM-labeled primer. The reaction time was optimized for initial velocities, and reactions were carried out at 37 °C for 5 to 12 min. The reaction mixture was the same as listed for primer extension and single nucleotide insertion assays except dNTP concentrations varied from 0 to 1000 μM. Reactions were terminated, and products were electrophoresed and visualized as described for primer extension and single nucleotide insertion assays. Bands were quantitated using Image J software (downloaded from nih.gov), and kinetic data were analyzed as described previously24.
MS analysis of oligonucleotides.
The remainder of the quenched reaction volume (8 μL for dATP and dGTP; 7.3 μL for dCTP and dTTP) from the steady-state kinetics experiments and from the primer extension and single nucleotide insertion assays (8 μL) was analysed by LC-MS/MS. All LC-MS/MS analyses were done on a Shimadzu Nexera X2 system in-line with a SCIEX 6500 QTrap mass spectrometer. Oligonucleotides were chromatographed on a reverse-phase system using an Acquity BEH C8 column (10 cm × 0.2 cm, 1.7 μm) held at 60 °C under a gradient elution scheme. Mobile phase component A was water with 15 mM triethylamine and 100 mM hexfluoroisopropanol plus 1% (v/v) methanol. Component B was 1:1 (v:v) methanol:acetonitrile with 15 mM triethylamine and 100 mM hexfluoroisopropanol. A typical gradient was 4% B for 0 to 0.5 min, followed by a linear increase to 19% B over 6 min followed by a 2 min hold at 19% B. The column was equilibrated for 2 min before each injection. The SCIEX Analyst software package was used to collect and process data. The predicted mass of each oligonucleotide and its w- and y- fragments were calculated using the Mongo Oligo Mass Calculator from Prof. Jef Rozenski (ORCID 0000–0001-9624–5536) (http://mass.rega.kuleuven.be/mass/mongo.htm).
Statistical Analyses.
From the LC-MS/MS data, the ratio product/(product + substrate) was calculated (Rp). The turnover value (ν in min−1) was calculated according to the equation below with Di as the initial oligonucleotide duplex concentration, E as the enzyme concentration, and t as time in min:
Using GraphPad Prism, the turnover values were plotted for each nucleotide concentration. The graphs were analyzed by nonlinear regression and the Michaelis-Menten entry to determine kcat and Km. Tabular results of kinetic parameters are from an experiment done in triplicate for each dNTP. GraphPad Prism was used to calculate the SEM for each parameter.
RESULTS
Site-specific synthesis of oligonucleotides containing 6-oxo-M1dG.
We attempted to incorporate 6-oxo-M1dG into DNA oligonucleotides by the standard approach of synthesizing a DMT-protected mononucleoside activated at the 3′ position as a phosphoramidite. Unfortunately, repeated attempts to protect the 5′-hydroxyl group of the 6-oxo-M1dG nucleoside with 4,4′-dimethoxytrityl (DMT) chloride were unsuccessful, presumably because of derivatization of the 6-oxo-M1dG adduct. Therefore, we next attempted a post-oligomerization strategy in which we synthesized phenoxyacetyl-protected oligonucleotides containing an uncyclized precursor that could later be cyclized to 6-oxo-M1dG upon base deprotection (Scheme 1). Even though the synthesis of the phosphoramidite 7 turned out to be successful, there were complications during the deprotection of the oligonucleotide and subsequent formation of the 6-oxo-M1dG adduct. When the deprotection of the oligonucleotide was performed with K2CO3 in methanol, a mixture of two oligonucleotides was observed, the 6-oxo-M1dG-containing desired oligonucleotide (minor) and an N1(-acrylic acid)-N2-phenoxyacetyl-dG-containing oligonucleotide (major) (Scheme 2). HPLC did not completely separate the product oligonucleotides. If the deprotection of the oligonucleotide was carried out with 0.1 M NaOH at 37 °C, a mixture of two oligonucleotides was also observed, one that contained the 6-oxo-M1dG adduct (minor) and another containing N1(-acrylic acid)-dG (major) (Scheme 2). The chromatographic separation of the two product oligonucleotides was challenging, and the yield of the 6-oxo-M1dG oligonucleotide was not adequate for chemical or biological studies.
Scheme 1.
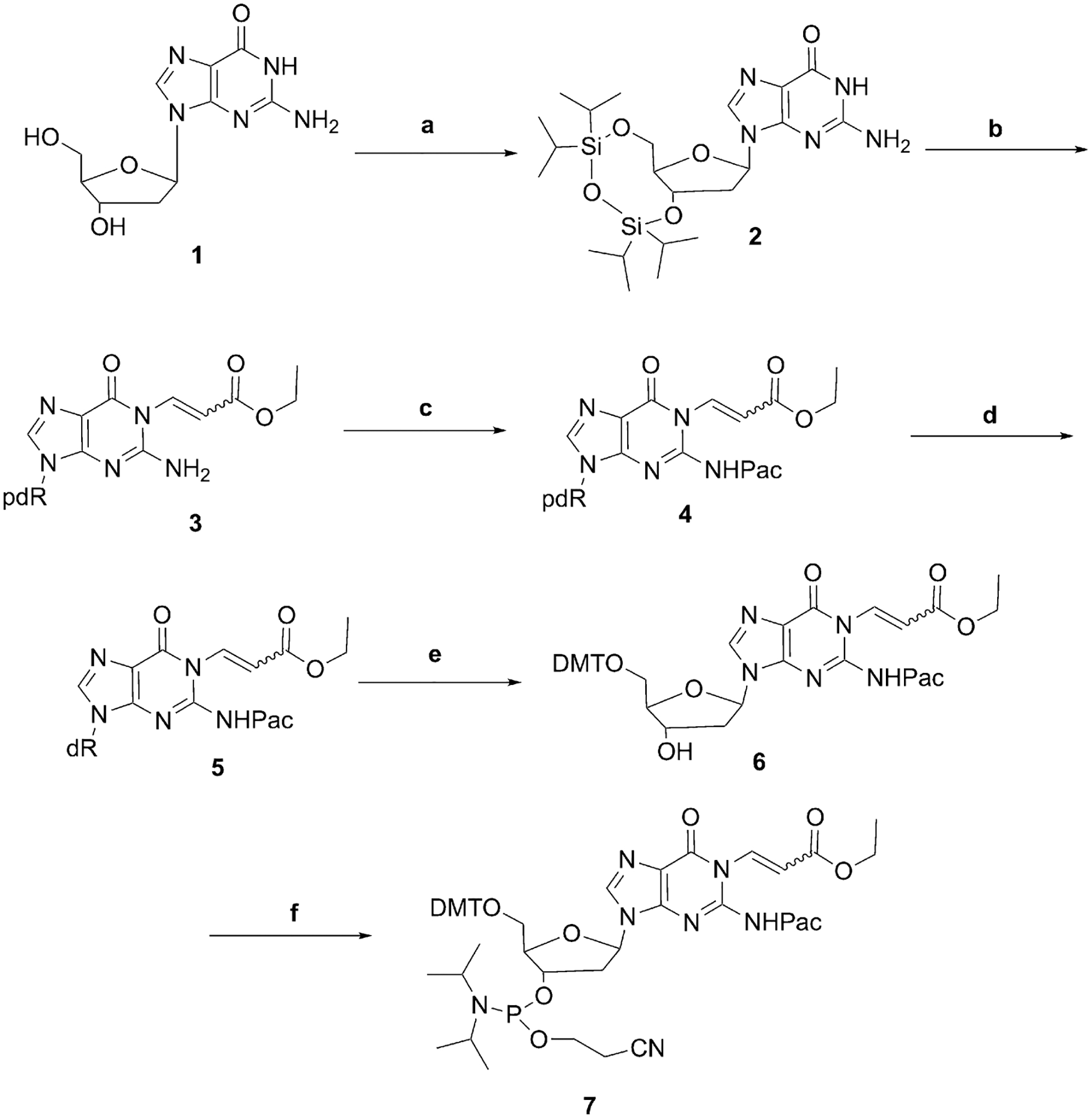
Initial proposed synthesis using a post-oligomerization strategy.
Reagents and Conditions: (a) 1,3-Dichloro-1,1,3,3-tetraisopropyldisiloxane, imidazole, dimethylformamide, 1 h, 0 °C, overnight, room temperature, 85%; (b) ethyl (Z)-3-bromoacrylate, K2CO3, dimethylformamide, 1 h, 0 °C, 50%; (c) phenoxyacetyl chloride, triethyl amine, dichloromethane, 30 min, 0 °C, 80%; (d) tetrabutyl ammonium fluoride, 2 h, room temperature, 80%; (e) 4,4′-dimethoxytriphenylmethyl chloride, N,N-diisopropylethylamine, pyridine, overnight, room temperature, 85%; (f) 2-cyanoethyl N,N-diisopropylchlorophosphoramidite, N,N-diisopropylethylamine, dichloromethane, 2 h, room temperature, 70%.
Scheme 2.
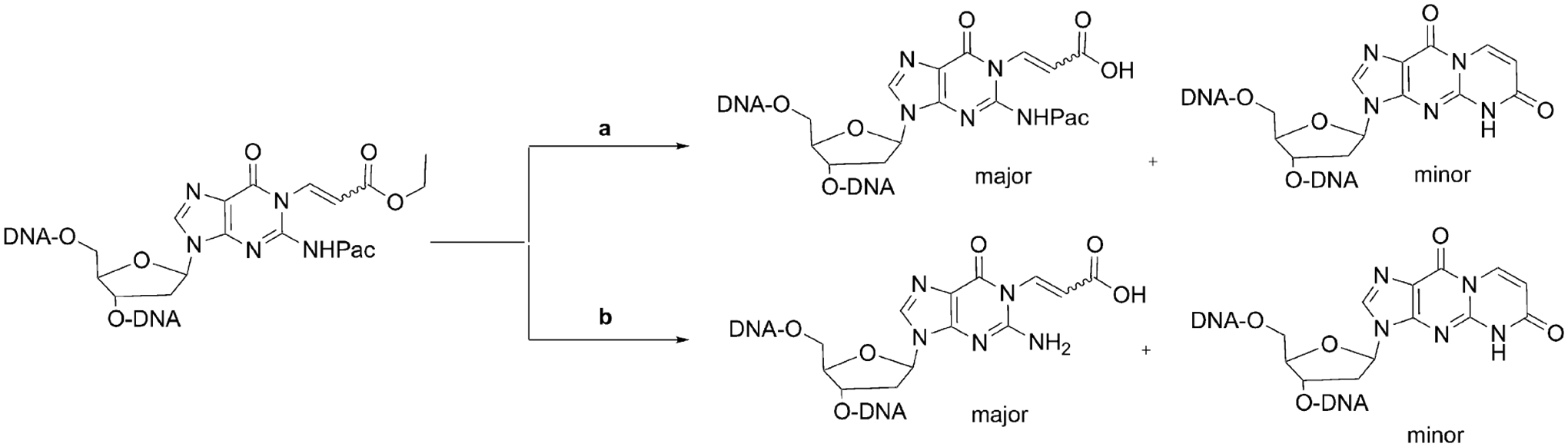
Products of deprotection of the oligonucleotide synthesized using Scheme 1.
Reagents and Conditions: (a) K2CO3 (50 mM, methanol); (b) NaOH (0.1 M).
Although the post-oligomerization approach failed, preliminary results obtained with the mononucleoside indicated that it would be possible to synthesize the originally desired DMT-phosphoramidite of 6-oxo-M1dG if the sequence of acylation-cyclization normally employed for exocyclic DNA adduct synthesis was reversed (Scheme 3). Specifically, the 3′-hydroxyl group of 5′-DMT-dG (8) was protected with a triethylsilyl group. Then, compound 9 was treated with (E)-3-ethoxyacryloyl chloride to produce the amide 10, which was isolated and characterized (Supplementary Figures S10 and S11). Amide 10 is a product of initial amide formation at N2 followed by N1 Michael addition to the double bond. Interestingly, during purification, amide 10 underwent elimination to form minor amounts of 6-oxo-M1dG, which complicated the chromatographic purification. The formation of 6-oxo-M1dG was due to the use of triethylamine as a mobile phase modifier. We decided not to isolate amide 10 at the larger scale of synthesis to simplify the purification. Instead, amide 10 was treated sequentially with triethylamine hydrofluoride to remove the silyl protecting group followed by K2CO3 in methanol to facilitate the formation of the double bond-containing compound, 11. Compound 11 was converted to the phosphoroamidite 12 using Hunig’s base and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite. Phosphoramidite 12 was used for the preparation of oligonucleotides containing the 6-oxo-M1dG lesion at defined locations. Incorporation of the modified nucleotide was performed off-line, using a manual coupling protocol23.
Scheme 3.
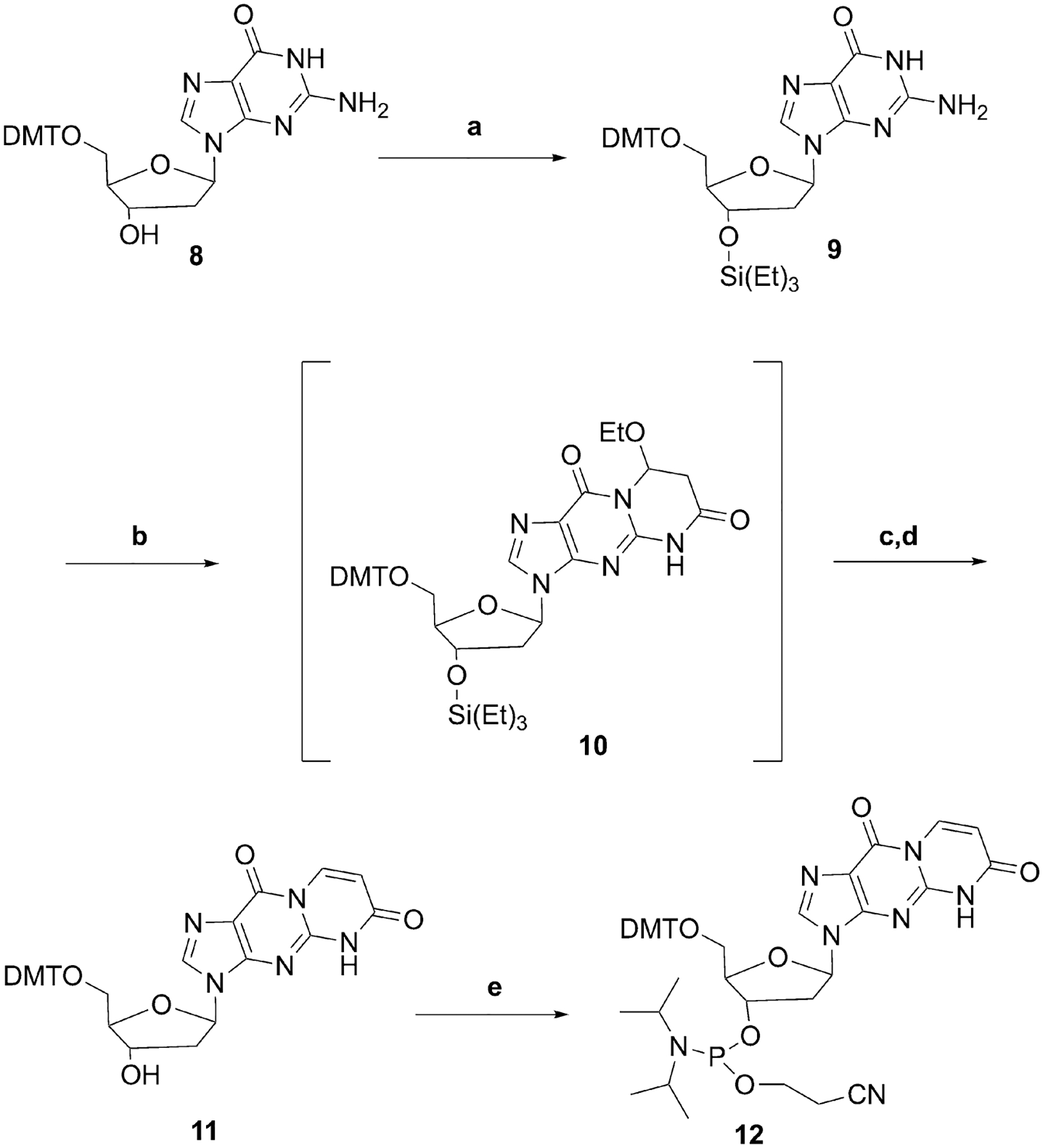
Successful route for phosphoramidite synthesis.
Reagents and Conditions: (a) Triethyl silyl chloride, imidazole, dimethylformamide, 1 h, 0 °C, 85%; (b) (E)-3-ethoxyacryloyl chloride, triethylamine, dimethylformamide, 0 °C, 1 h; (c) triethylamine trihydrofluoride, triethylamine, tetrahydrofuran, 30 min, 0 °C; (d) K2CO3, methanol, 1 h, 37 °C, 83%; (e) 2-cyanoethyl N,N-diisopropylchlorophosphoramidite, N,N-diisopropylethylamine, dichloromethane, 2 h, room temperature, 65%.
The presence of 6-oxo-M1dG was confirmed by enzymatic digestion of the oligonucleotide followed by LC-MS/MS and HPLC-photodiode array analysis. Supplementary Figure S12A shows an LC-MS/MS chromatogram of the digested oligonucleotide. The top chromatogram shows the 6-oxo-M1dG nucleoside (black trace) and its internal standard (red trace). The retention time of the liberated 6-oxo-M1dG nucleoside matches that of the 15N5-labeled internal standard and that of an authentic standard (data not shown). Relative quantitation of the 5 nucleosides in the 6-oxo-M1dG oligonucleotide was performed by HPLC-photodiode array (Supplementary Figure S12B). Again, the liberated 6-oxo-M1dG nucleoside eluted at 10.0 min, which matched the retention time of an authentic standard, and the UV spectrum associated with this peak (Supplementary Figure S12C) matched that of the authentic standard. The measured relative ratios of the nucleosides agreed with the theoretical ratio (Table 1). Together, these results confirm the composition of the 6-oxo-M1dG oligonucleotide.
Table 1.
Comparison of the theoretical ratio of bases to those obtained from analysis of an enzymatic digest of the 6-oxo-M1dG oligonucleotide. Values are normalized to 6-oxo-M1dG.
| dC | dG | dT | dA | 6-oxo-M1dG | |
|---|---|---|---|---|---|
| 6-oxo Oligo | 6.20 | 4.01 | 4.15 | 4.90 | 1 |
| Theoretical | 6 | 4 | 4 | 5 | 1 |
Primer extension opposite dG and 6-oxo-M1dG by hPol ι.
Experiments to investigate the impact of the 6-oxo-M1dG adduct on replication by a DNA polymerase were conducted with hPol ι. The control (unmodified) or 6-oxo-M1dG-containing oligonucleotide was annealed to a 5′-FAM-labeled 15-mer complementary primer strand (Figure 2A). With this primer-template, the first nucleotide should be incorporated immediately opposite dG in the control template or the site of adduction in the 6-oxo-M1dG-containing template. A time course of extension by hPol ι in the presence of all four dNTPs is shown in Figure 2B. With the control oligonucleotide template, hPol ι extended the primer by four nucleotides to a 19-mer. A slight amount of 20-mer product was also present upon overexposure of the gel (data not shown). However, hPol ι was able to incorporate only a single nucleotide directly across from the 6-oxo-M1dG when the modified template-primer substrate was used. We observed no evidence for further extension, even upon overexposure of the gel. Two bands were observed on the gel at the position corresponding to a 16-mer, suggesting that two or more different nucleotides were incorporated in the site directly across from the adduct. The time course of extension in the presence of single dNTPs was also evaluated. Incorporation of dATP and dGTP is shown in Figure 3A. In both cases, a single nucleotide was inserted, and there was no significant additional nucleotide incorporation after 45 min, regardless of the template employed. Using the control oligonucleotide, only 11% of the FAM-labeled primer was extended to the 16-mer with dATP and only 4.2% with dGTP. Similar results were obtained with the 6-oxo-M1dG oligonucleotide duplex. Only 10% of the FAM-labeled primer was extended to the 16-mer with dATP, and 7.1% was extended with dGTP as the nucleotide substrate (Figure 3A). The time course of incorporation for dCTP and dTTP is shown in Figure 3B. Extension to the 16-mer product increased with each time point measured for both templates.
Figure 2.
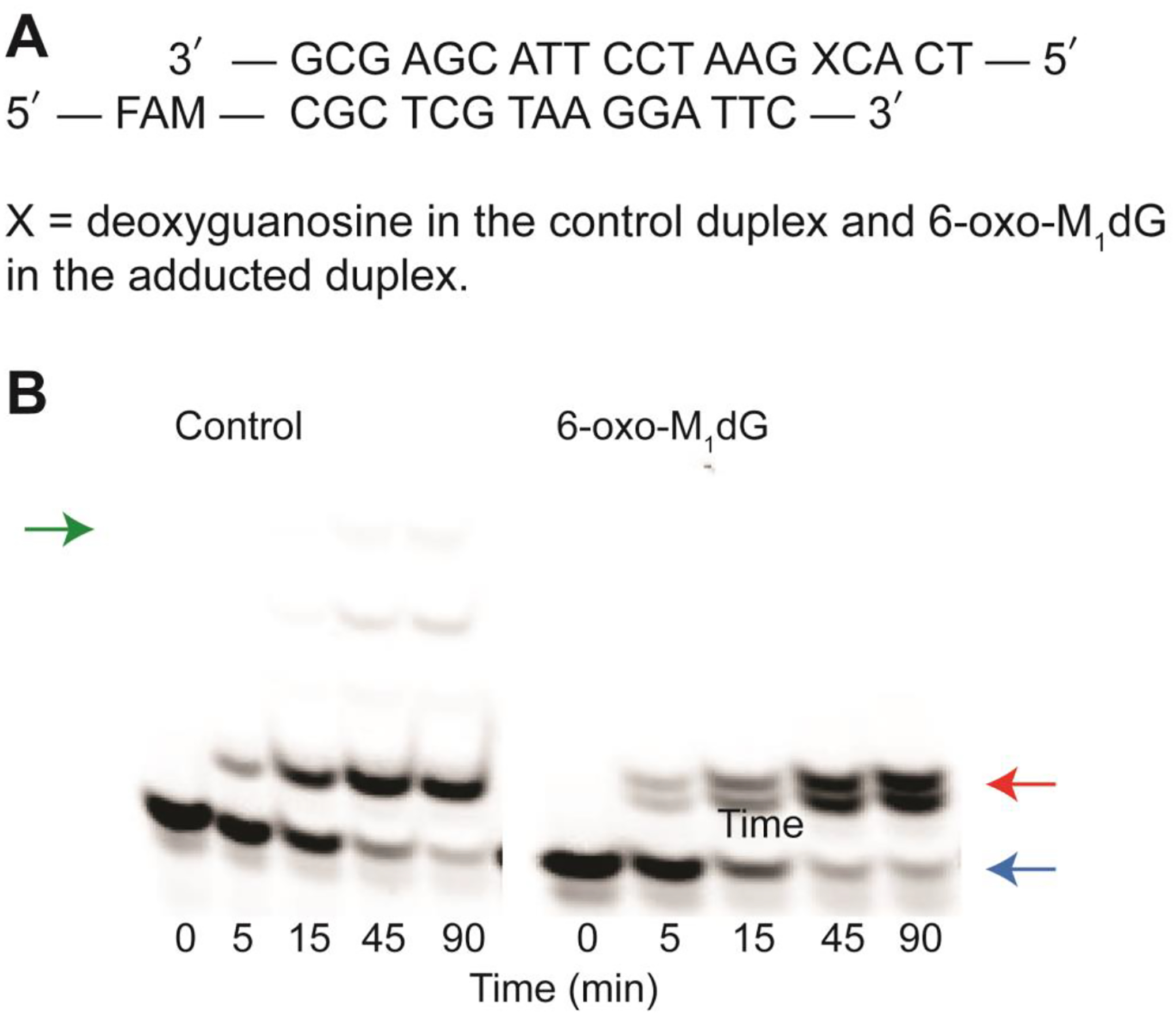
Incorporation of nucleotides across from the 6-oxo-M1dG adduct. (A) sequence of the DNA duplex. (B) time course of nucleotide incorporation. hPol ι (60–85 nM) was incubated with 5 μM DNA duplex and a 500 μM mix of all four dNTPs. The reaction was stopped at 5, 15, 45, and 90 min. Green, red, and blue arrows indicate the locations of the 19-mer and 16-mer products and the 15-mer primer, respectively.
Figure 3.
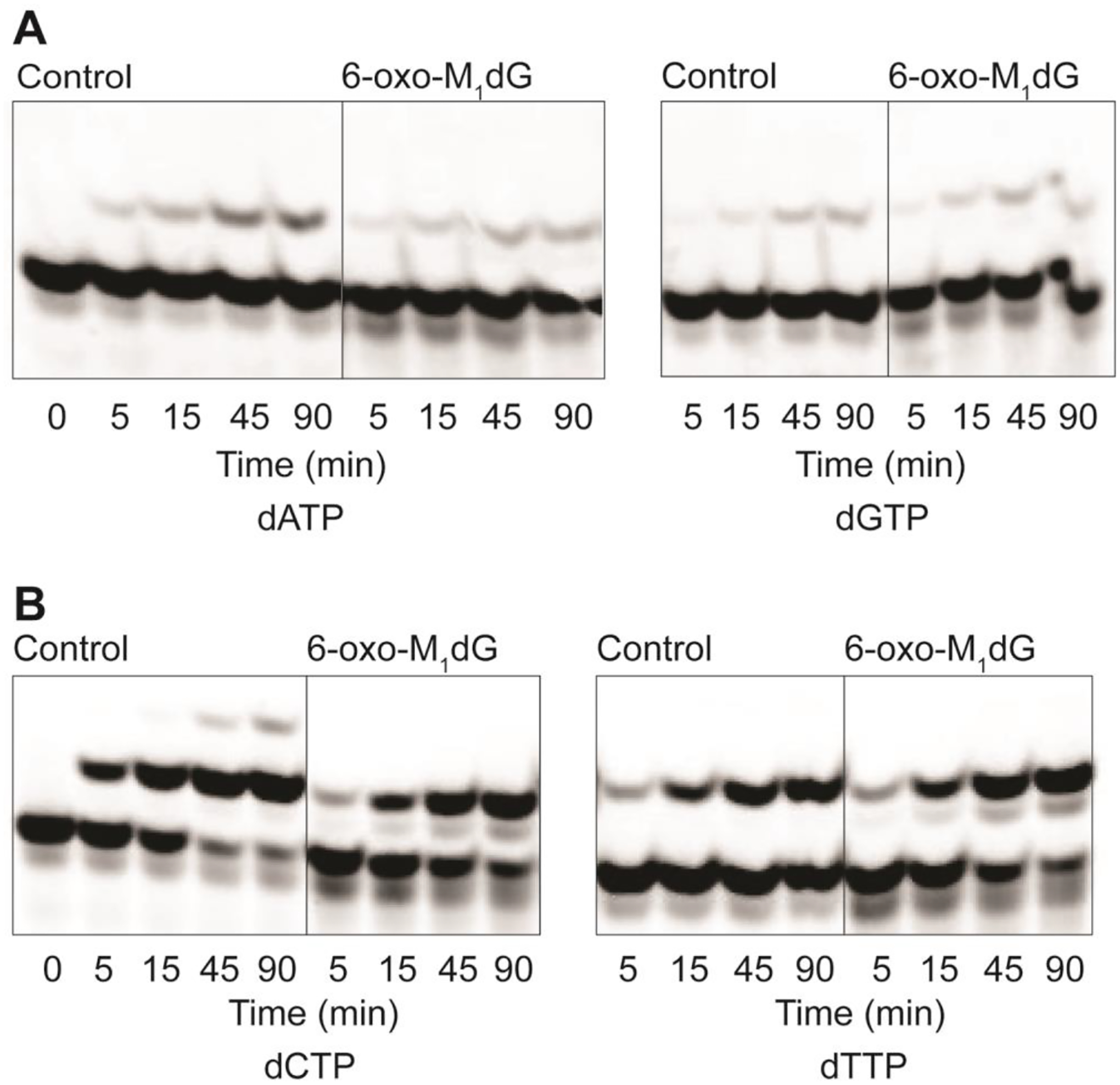
Time course of incorporation of single dNTPs across from dG and the 6-oxo-M1dG adduct. (A) Incorporation of dATP and dGTP. (B) Incorporation of dCTP and dTTP. For both A and B, 60–85 nM Pol ι was incubated with 5 μM DNA duplex and 500 μM of each individual dNTP. The reaction was stopped at 5, 15, 45, and 90 min.
Using dCTP as the substrate with the control oligonucleotide duplex, 89% of the total added primer had one or two dCTPs added by 90 min, yielding either a 16-mer or 17-mer product. A significant amount of product formation would be expected with the control oligonucleotide and dCTP since canonical Watson-Crick base pairing would occur between the dG and incoming dCTP. The small amount of 17-mer product may have resulted from an additional dCTP incorporated opposite cytosine or slippage of the template strand to bring a G to the insertion site in the control oligonucleotide (see sequence in Figure 2A). This additional nucleotide incorporation was only seen with the control oligonucleotide and dCTP. When the 6-oxo-M1dG oligonucleotide primer-template was incubated with hPol ι and dCTP, formation of the 16-mer product accounted for 78% of the total starting primer. Finally, with dTTP as the substrate, the percent 16-mer product formation was 43% with control oligonucleotide duplex and 82% with the 6-oxo-M1dG oligonucleotide duplex.
Considering the much higher efficiency of incorporation of dCTP and dTTP as opposed to dGTP and dATP across from 6-oxo-M1dG (Figure 3), one would expect that when all four dNTPs are present, the predominant products obtained from the action of hPol ι on the 6-oxo-M1dG-containing duplex would result from incorporation of dCTP or dTTP. This is consistent with our observation of two 16-mer product bands following incubation of the 6-oxo-M1dG-containing template/primer with all four dNTPs (Figure 2B). Notably, the different extension products varied in their mobility on denaturing polyacrylamide gel electrophoresis, with dCTP- and dATP-containing products migrating further on the gel than dGTP- and dTTP-containing products. Indeed, the faster migrating band observed in the reactions with dNTP co-migrated with the product of dCTP incorporation whereas the slower migrating band co-migrated with the product of dTTP incorporation. This assignment was confirmed by mass spectrometric analysis of the products as described below.
Steady state kinetic analysis of dNTP insertion across from dG and 6-oxo-M1dG by hPol ι.
Steady state kinetic analysis was performed to quantify the single nucleotide insertion preference of hPol ι across from 6-oxo-M1dG. hPol ι was incubated with the control oligonucleotide primer-template or the 6-oxo-M1dG oligonucleotide primer-template and increasing amounts of each individual dNTP. Initial experiments were performed using gel electrophoresis to quantify nucleotide incorporation. As anticipated, robust incorporation was observed for dCTP and dTTP but not dATP or dGTP (Supplementary Figure S13). The slight amount of incorporation of dATP and dGTP led to errors in quantification because of the large amount of remaining 15-mer primer that saturated the response of the fluorescence detector (Supplementary Figures S13A and C). Thus, these data could not be used to derive kinetic constants for incorporation of dATP and dGTP.
Mass spectrometry of intact oligonucleotides.
The poor quality of the gel-based analyses of primer-template extension products resulting from low efficiency incorporation prompted us to develop an LC-MS/MS assay for quantification of the oligonucleotides in order to take advantage of the high sensitivity and specificity afforded by MS analysis. FAM-labeled oligonucleotides corresponding to all potential products of single nucleotide incorporation (Table 2) were infused into the 6500 QTrap in negative ion/high mass mode. For each oligonucleotide, the Q1 spectrum revealed an array of peaks corresponding to various charge states. Typically, peaks corresponding to z = 4 to 10 were observed, with those corresponding to z = 7 to 9 predominating. Figure 4A shows the Q1 spectrum of oligonucleotide 16C. The dominant ion, m/z 673.1, represents the z = 8 state of 16C.
Table 2.
Sequences of oligonucleotides used for gel electrophoresis- and LC-MS/MS-based kinetics assay and structure of the FAM fluorescent tag and 5′ terminus of the FAM-labeled oligonucleotides.
| Oligo Name | Sequence |
|---|---|
| Control (template) | 5′- TCA CXG AAT CCT TAC GAG CG* |
| FAM | 5′-/56FAM/ CGC TCG TAA GGA TTC |
| FAM+1 (16C) | 5′-/56FAM/ CGC TCG TAA GGA TTC C |
| 16G | 5′-/56FAM/ CGC TCG TAA GGA TTC G |
| 16A | 5′-/56FAM/ CGC TCG TAA GGA TTC A |
| 16T | 5′-/56FAM/ CGC TCG TAA GGA TTC T |
| FAM+2 | 5′-/56FAM/ CGC TCG TAA GGA TTC CG |
| FAM+3 | 5′-/56FAM/ CGC TCG TAA GGA TTC CGT |
| FAM+4 | 5′-/56FAM/ CGC TCG TAA GGA TTC CGT G |
X is either G in the control oligonucleotide or 6-oxo-M1dG in the 6-oxo-M1dG oligonucleotide
Figure 4.
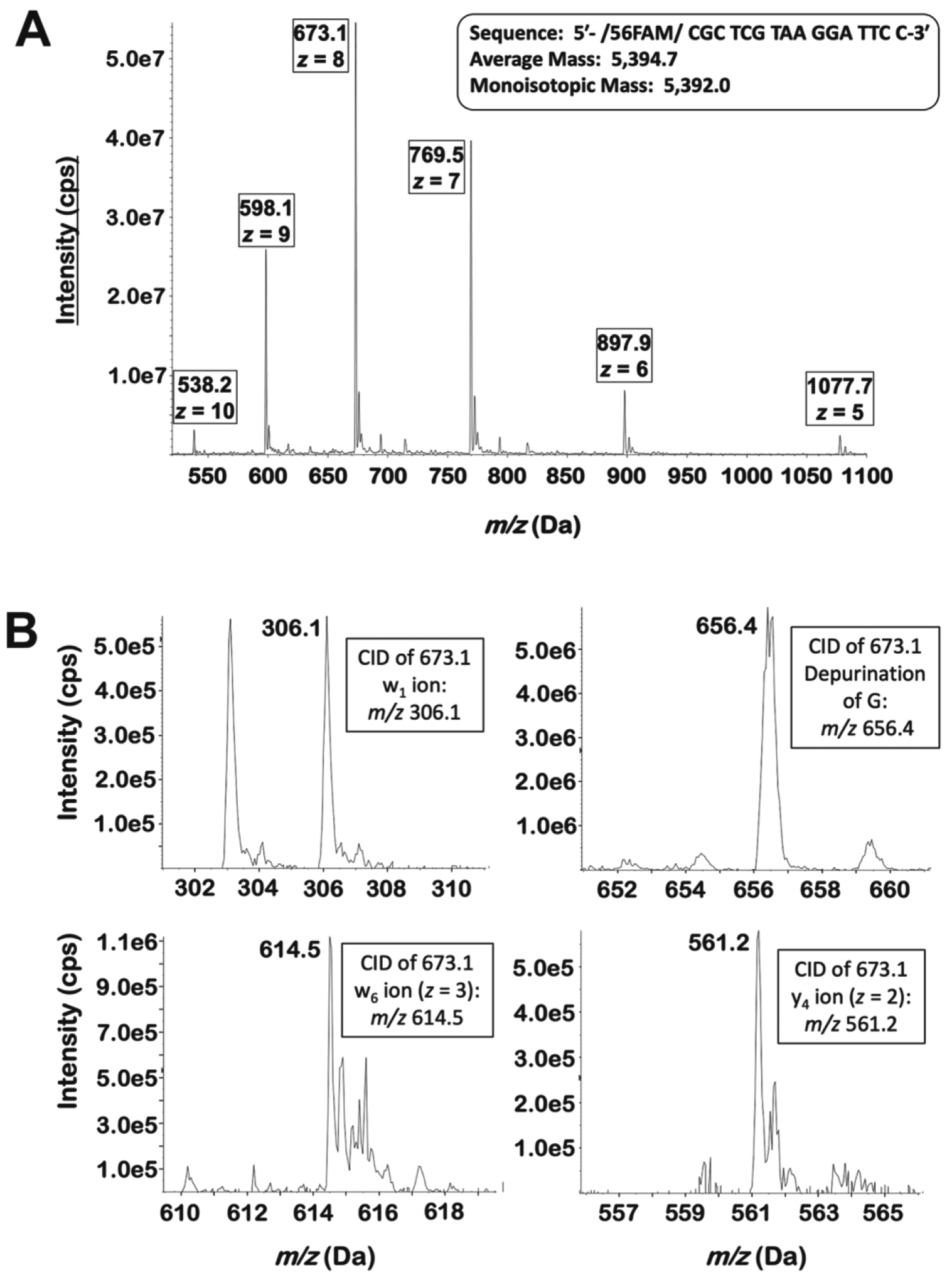
Mass spectra of the 16C oligonucleotide. (A) The Q1 spectrum of oligonucleotide 16C when infused directly. Multiple charge states are observed, with the z = 8 peak predominating. (B) Upon collision-induced dissociation of the z = 8 peak (m/z 673.1), many of the predicted w and y ions are observed. Additionally, a peak corresponding to the depurination of G is seen.
For all oligonucleotides, the most abundant Q1 peak was then subjected to collision-induced dissociation. Under the conditions used in these experiments, oligonucleotides primarily undergo fragmentation at four locations around the phosphodiester bridge. McLuckey et al. proposed a nomenclature scheme for the various ions generated by this fragmentation pattern25. A simplified version of their scheme is shown in Supplementary Figure S14. Fragments are designated w, x, y, and z based on the exact location of the fragmentation around the phosphodiester bond and numbered according to their position relative to the 3′ end of the oligonucleotide. Because all nucleotide additions resulting from the action of hPol ι occur at the 3′ end, we are considering only the fragment ions generated from that region of the oligonucleotide. These fragments are unique to each single extension product (and, indeed, to all other extension products considered here). Thus, a unique and specific SRM transition can be obtained for each oligonucleotide by specifying a high-abundance charge state as the Q1 value and as the Q3 value, a unique w-, y- or dG-depurination fragment of that ion.
On the 6500 QTrap system described here, fragments containing bases 1–6 were the most abundant, whereas those containing bases above 6 were observed at very low abundance or not at all. Practically speaking, the w- and y- ions were the predominant fragment ions. This is seen in Figure 4B, which shows several product ions of oligonucleotide 16C (m/z 673.1). The 6500 QTrap was tuned on many pairs of parent/fragment ions (w-, y-, or depurination) for the relevant oligonucleotides. Those pairs providing the greatest signal were chosen for use in SRM detection. Supplementary Table S1 gives the Q1 and Q3 values of the SRM transitions used for detection and quantitation of each oligonucleotide. However, many potential Q1/Q3 pairs are available for each oligonucleotide.
As a first test of the utility of LC-MS/MS, we examined the products of hPol ι-mediated primer extension using the 6-oxo-M1dG-containing duplex in the presence of all four dNTPs. Incubations were carried out as described in Materials and Methods. LC-MS/MS of the resulting oligonucleotides generated after a 90 min incubation enabled definitive identification of the nucleotides inserted opposite 6-oxo-M1dG by the enzyme. Consistent with the gel electrophoresis data (Figure 2B), the LC-MS/MS assay demonstrated that oligonucleotides containing dC or dT in the position opposite 6-oxo-M1dG in the template comprised 40% and 53% of the total products, respectively. Thus, products containing dC or dT accounted for 93% of the total, with dT slightly favored over dC.
Using LC-MS/MS analysis, the amounts of the 15-mer FAM primer and 16-mer FAM products were measured to determine the kinetics of incorporation of each nucleotide by hPol ι. Michaelis-Menten plots of the data are provided in Supplementary Figure S15, and the derived kinetic constants are summarized in Table 3. The specificity constant (kcat/KM) provides a measure of the catalytic efficiency of each dNTP insertion reaction and allows comparisons to be made between dNTP insertion events. Across from 6-oxo-M1dG, hPol ι incorporated both dCTP and dTTP equally well. Although the kcat value is lower for dCTP compared to dTTP, the Km value is also smaller for dCTP compared to dTTP. Thus, the specificity constant values for incorporation of these two nucleotides, 0.017 μM−1min−1 are the same within the error of the experiment. The specificity constants were approximately 4-fold lower for both dATP and dGTP. This suggests that in vitro, hPol ι would preferentially incorporate either dCTP or dTTP over dATP and dGTP across from the 6-oxo-M1dG adduct. This is consistent with the LC- MS/MS analysis of the extension products of the 6-oxo-M1dG duplex with hPol ι and all four dNTPs. The doublet seen on the gel (Figure 2B) has been shown to be a 16-mer with 93% of the products containing either dCTP or dTTP. With the control oligonucleotide, insertion of dCTP was the favored nucleotide to be placed across from the guanine (Table 3). dTTP had the second highest specificity constant and was incorporated approximately 11-fold less efficiently than dCTP. dATP and dGTP were both poorly incorporated, with kcat/KM values 45- and 160-fold lower than for dCTP, respectively. In comparing the incorporation of nucleotides between the control duplex and the 6-oxo-M1dG duplex, the greatest difference in specificity constants was for incorporation of dCTP with an approximately 10-fold lower value for the 6-oxo-M1dG duplex. In contrast, the efficiency of incorporation for TTP differed by less than 2-fold between the control and 6-oxo-M1dG oligonucleotides. dATP and dGTP exhibited the lowest specificity constants for both substrates. There was not a significant difference in incorporation of dATP between the control duplex and the 6-oxo-M1dG duplex. With dGTP incorporation, the specificity constant for the 6-oxo-M1dG duplex was 5-fold higher than that for the control duplex.
Table 3.
Steady state kinetic parameters for insertion of single nucleotides opposite dG and 6-oxo-M1dG by hPol ι.
| Nucleotide | kcat (min−1) | Km (μM) | kcat/Km (μM−1min−1) |
|---|---|---|---|
| dATP | 0.31 ± 0.03 | 95 ± 33 | 0.0033 |
| dCTP | 4.1 ± 0.4 | 27 ± 10 | 0.15 |
| dGTP | 0.12 ± 0.01 | 130 ± 40 | 0.00092 |
| dTTP | 2.5 ± 0.3 | 200 ± 50 | 0.013 |
| Nucleotide | kcat (min−1) | Km (μM) | kcat/Km (μM−1min−1) |
| dATP | 0.36 ± 0.011 | 81 ± 9 | 0.0044 |
| dCTP | 1.2 ± 0.1 | 72 ± 18 | 0.017 |
| dGTP | 0.14 ± 0.02 | 31 ± 13 | 0.0045 |
| dTTP | 2.9 ± 0.1 | 170 ± 20 | 0.017 |
DISCUSSION
We describe for the first time the synthesis of oligonucleotides containing 6-oxo-M1dG. Traditional routes employed for the synthesis of exocyclic DNA adducts, either as a mononucleoside or oligonucleotide precursor, proved unsuccessful so a new approach was developed. DMT- and triethylsilyl-protected dG was acylated on the exocyclic nitrogen and, following removal of the triethylsilyl group, cyclization was promoted with K2CO3. The DMT-protected 6-oxo-M1dG was converted to the phosphoramidite, which was utilized for oligonucleotide synthesis. The 6-oxo-M1dG was stable to all steps in the synthesis and deprotection of the oligonucletides. The 6-oxo-M1dG-containing oligonucleotide was produced in high purity, and the content of 6-oxo-M1dG was in line with expectations from the oligonucleotide sequence. Thus, the synthetic route described enables the facile synthesis of a variety of sequences for in vitro and in vivo experiments.
We initiated studies of hPol ι-mediated replication of the 6-oxo-M1dG-containing oligonucleotide using a traditional gel electrophoresis-based polymerase assay. This approach yielded good quantitative data for the incorporation of dCTP and dTTP, which were favored substrates for the reaction. However, it lacked the sensitivity required to quantitate the products of incorporation of dATP and dGTP, which were generated in low amounts. It is notable that kinetic data for the hPol ι-dependent insertion of dATP or dGTP across from M1dG have not been reported, presumably because they are poorly incorporated across from this adduct as well. To address this deficiency, we developed the described LC-MS/MS method, which proved capable of identifying unambiguously the extension products generated during hPol ι experiments and was superior to the polyacrylamide gel-based system for the quantitation of the low-level products. By utilizing LC-MS/MS to quantitate products, reliable kinetic values are achievable, even for nucleotides that were not abundantly incorporated. In addition, LC-MS/MS was useful for characterizing the products of hPol ι using the 6-oxo-M1dG-containing template and all dNTPs. The method confirmed that dCTP and dTTP were the major nucleotides incorporated by hPol ι when all dNTPs were present. This was consistent with the results from the steady state kinetics experiments. Values generated by the LC-MS/MS assay for incorporation of dTTP and dCTP were similar to those obtained using the gel-based assay (Table 3 and Supplementary Table S2). Discrepancies between the two methods were greater for values of Km than for kcat, which is consistent with the higher statistical variability in the Km results.
Our LC-MS/MS approach is generally applicable as a polymerase assay with both advantages and disadvantages over prior methods. In the present setting, the number of potential products was limited, which made it feasible to obtain authentic standards and sufficiently characterize all of them. An advantage of the assay is the elimination of the need to incorporate a FAM (or indeed any other) fluorescent moiety onto the primer oligonucleotide, although FAM-tagged oligonucleotides were used in these experiments to enable direct comparison with gel-based assay results through analysis of the same samples. An additional advantage afforded by the LC-MS/MS assay is ease of sample preparation, which in this case was simply a dilution of the quenched enzyme reaction. This is quite simple in comparison to other reported LC-MS/MS analyses of polymerase extension products, as we do not use a restriction digest step. However, the method described here is unlikely to be applicable in situations where the products of polymerase experiments are very large molecules, such as plasmids.
The results from our kinetics studies show that hPol ι incorporates dCTP and dTTP across from 6-oxo-M1dG with similar efficiency, as indicated by their essentially identical specificity constants of 0.017 μM−1min−1 (Table 3). In contrast, when using an oligonucleotide containing M1dG, dCTP is hPol ι’s preferred substrate for insertion across from the adduct; the specificity constant for dCTP is 14-fold greater than that for dTTP (Supplementary Table S2)19. A possible explanation for this difference is that M1dG is subject to hydrolysis, producing the ring-opened adduct N2-(3-oxo-1-propenyl)-dG (Figure 1A)26, 27. Structural studies of M1dG opposite dC in duplex DNA indicate that the ring-opened form predominates, and that it is less disruptive of DNA structure than the parent exocyclic adduct. Thus, the ring-opened adduct would be expected to be less mutagenic than either the parent M1dG or 6-oxo-M1dG28. Consistent with this explanation, the specificity constants for hPol ι-mediated insertion of dCTP and dTTP across from the structurally related 1,N2-etheno-dG adduct were reported to be identical29. In addition, the ring opened form of γ-hydroxy-1,N2-propano-2′-deoxyguanosine (γ-HOPdG) is bypassed by human Pols η, ι, and κ; however, only hPol ι is able to incorporate a nucleotide across from the structural analog propano-2′-deoxyguanosine (PdG), which remains in the ring-closed form. hPol ι incorporated a single dC or dT across from this adduct and was not able to extend further than the single nucleotide incorporation, as seen in our results with 6-oxo-M1dG30. As noted above, hPol ι’s ability to insert a single nucleotide across from 6-oxo-M1dG and M1dG but then to extend no further has been seen with other adducts18, 31–36. In some investigations, further extension has been achieved with simultaneous treatment with hPol ι and hPol κ; hPol ι incorporates a nucleotide across from the adduct, and hPol κ then extends past this insertion point37–40.
The ability of other human Y-family polymerases, including η, κ, and Rev1 to bypass M1dG has been reported19, 41. hPol κ preferentially incorporates dCTP across from M1dG and can extend past the site of adduction, but incorporation of dCTP opposite M1dG is 12.5-fold less efficient than incorporation across from dG. Rev1 behaved similarly to hPol ι in that it was only able to incorporate a single nucleotide across from M1dG and failed to extend past this point. The specificity constant for dCTP with Rev1 was only 2-fold lower for M1dG than for dG. In contrast to the other Y-family polymerases, hPol η has the highest specificity constant for dATP incorporation across from M1dG. The specificity constant for dCTP is approximately 100-fold lower. However, hPol η is the most efficient of all the Y-family polymerases at extending primers that have already incorporated a dCTP across from M1dG. This suggests that bypass of M1dG in vivo may occur by the action of more than one Y-family polymerase, and the same may be true for 6-oxo-M1dG. We have shown that hPol ι adds only a single nucleotide (predominantly dCTP or dTTP) across from the lesion; the activity of a different Y-family polymerase may be required to extend the primer further. Additional work will investigate the activity of the other Y-family polymerases towards 6-oxo-M1dG.
Our findings suggest that hPol ι has an approximately 50% chance of introducing a mutation across from 6-oxo-M1dG through the incorrect insertion of dTTP as opposed to dCTP. After an additional round of replication, this would produce a G→A transition. In various cell lines treated with adenine propenal, M1dG is first produced and then converted to 6-oxo-M1dG. The latter adduct begins appearing about 3 h post-treatment and is still present 24 h after treatment (28). Thus, if DNA replication occurs after M1dG has been converted to 6-oxo-M1dG, there is a much higher chance that a mutation will be introduced. Further studies are required to fully test the hypothesis that 6-oxo-M1dG conveys a higher mutagenic potential than M1dG in vivo.
In summary, we have outlined a novel synthetic scheme for a unique adduct-containing oligonucleotide (6-oxo-M1dG) and a highly sensitive and specific method for identification and quantification of replication products opposite this adduct using a LC-MS/MS assay. Although we have used this assay to characterize products generated by hPol ι-mediated nucleotide insertion opposite the 6-oxo-M1dG adduct, it could be extrapolated to a variety of DNA adducts and the activity of other Y-family DNA polymerases. The LC-MS/MS method offers a reliable quantification of kinetics for nucleotides that are either abundantly incorporated or poorly incorporated, and it provides indisputable confirmation of which nucleotides are incorporated opposite adducts when all four dNTPs are present. Thus, we demonstrate the utility of LC-MS/MS for the study of translesion DNA synthesis while also opening the door to the understanding of the biological impact of the heretofore uncharacterized DNA adduct, 6-oxo-M1dG.
Supplementary Material
ACKNOWLEDGMENT
LC-MS/MS analyses were carried out in part at the Vanderbilt Mass Spectrometry Core Facility.
Funding Sources
This work was supported by a research grant from the National Institutes of Health [CA87819 to L.J.M.].
ABBREVIATIONS
- M1dG
3-(2′-deoxy-β-D-erythro-pentofuranosyl)pyrimido[1,2-α]purin-10(3H)-one
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- SRM
selected reaction monitoring
- FAM
single isomer fluorescein label
- DMT
dimethoxytrityl
- dCTP
deoxycytidine triphosphate
- dTTP
thymidine triphosphate
- dATP
deoxyadenosine triphosphate
- dGTP
deoxyguanosine triphosphate
- MOPS
3-(N-morpholino)propanesufonic acid
Footnotes
Supporting Information. Supplementary Figures S1–S15, Supplementary Tables S1 and S2.
The authors declare no competing financial interest
REFERENCES
- 1.Dedon PC; Plastaras JP; Rouzer CA; Marnett LJ, Indirect mutagenesis by oxidative DNA damage: formation of the pyrimidopurinone adduct of deoxyguanosine by base propenal. Proc. Natl. Acad. Sci. U. S. A 1998, 95 (19), 11113–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marnett LJ; Basu AK; O’Hara SM; Weller PE; Rahman AFMM; Oliver JP, Reaction of malondialdehyde with guanine nucleosides: formation of adducts containing oxadiazabicyclononene residues in the base-pairing region. J. Am. Chem. Soc 1986, 108 (6), 13480–50. [Google Scholar]
- 3.Plastaras JP; Dedon PC; Marnett LJ, Effects of DNA structure on oxopropenylation by the endogenous mutagens malondialdehyde and base propenal. Biochemistry 2002, 41 (15), 5033–42. [DOI] [PubMed] [Google Scholar]
- 4.Chaudhary AK; Nokubo M; Reddy GR; Yeola SN; Morrow JD; Blair IA; Marnett LJ, Detection of endogenous malondialdehyde-deoxyguanosine adducts in human liver. Science 1994, 265 (5178), 1580–2. [DOI] [PubMed] [Google Scholar]
- 5.Ma B; Villalta PW; Balbo S; Stepanov I, Analysis of a malondialdehyde-deoxyguanosine adduct in human leukocyte DNA by liquid chromatography nanoelectrospray-high-resolution tandem mass spectrometry. Chem. Res. Toxicol 2014, 27 (10), 1829–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wauchope OR; Mitchener MM; Beavers WN; Galligan JJ; Camarillo JM; Sanders WD; Kingsley PJ; Shim HN; Blackwell T; Luong T; deCaestecker M; Fessel JP; Marnett LJ, Oxidative stress increases M1dG, a major peroxidation-derived DNA adduct, in mitochondrial DNA. Nucleic Acids Res. 2018, 46 (7), 3458–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fink SP; Reddy GR; Marnett LJ, Mutagenicity in Escherichia coli of the major DNA adduct derived from the endogenous mutagen malondialdehyde. Proc. Natl. Acad. Sci. U. S. A 1997, 94 (16), 8652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.VanderVeen LA; Hashim MF; Shyr Y; Marnett LJ, Induction of frameshift and base pair substitution mutations by the major DNA adduct of the endogenous carcinogen malondialdehyde. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (24), 14247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson KA; Fink SP; Marnett LJ, Repair of propanodeoxyguanosine by nucleotide excision repair in vivo and in vitro. J. Biol. Chem 1997, 272 (17), 11434–8. [DOI] [PubMed] [Google Scholar]
- 10.Hoberg AM; Otteneder M; Marnett LJ; Poulsen HE, Measurement of the malondialdehyde-2’-deoxyguanosine adduct in human urine by immuno-extraction and liquid chromatography/atmospheric pressure chemical ionization tandem mass spectrometry. J. Mass Spectrom 2004, 39 (1), 38–42. [DOI] [PubMed] [Google Scholar]
- 11.Knutson CG; Akingbade D; Crews BC; Voehler M; Stec DF; Marnett LJ, Metabolism in vitro and in vivo of the DNA base adduct, M1G. Chem. Res. Toxicol 2007, 20 (3), 550–7. [DOI] [PubMed] [Google Scholar]
- 12.Knutson CG; Skipper PL; Liberman RG; Tannenbaum SR; Marnett LJ, Monitoring in vivo metabolism and elimination of the endogenous DNA adduct, M1dG {3-(2-deoxy-beta-D-erythro-pentofuranosyl)pyrimido[1,2-alpha]purin-10(3H)-one}, by accelerator mass spectrometry. Chem. Res. Toxicol 2008, 21 (6), 1290–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knutson CG; Wang H; Rizzo CJ; Marnett LJ, Metabolism and elimination of the endogenous DNA adduct, 3-(2-deoxy-beta-D-erythropentofuranosyl)-pyrimido[1,2-alpha]purine-10(3H)-one, in the rat. J. Biol. Chem 2007, 282 (50), 36257–64. [DOI] [PubMed] [Google Scholar]
- 14.Otteneder MB; Knutson CG; Daniels JS; Hashim M; Crews BC; Remmel RP; Wang H; Rizzo C; Marnett LJ, In vivo oxidative metabolism of a major peroxidation-derived DNA adduct, M1dG. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (17), 6665–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wauchope OR; Beavers WN; Galligan JJ; Mitchener MM; Kingsley PJ; Marnett LJ, Nuclear Oxidation of a Major Peroxidation DNA Adduct, M1dG, in the Genome. Chem. Res. Toxicol 2015, 28 (12), 2334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prakash S; Johnson RE; Prakash L, Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem 2005, 74, 317–53. [DOI] [PubMed] [Google Scholar]
- 17.Yang W; Gao Y, Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu Rev Biochem 2018, 87, 239–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McIntyre J, Polymerase iota - an odd sibling among Y family polymerases. DNA Repair (Amst) 2020, 86, 102753. [DOI] [PubMed] [Google Scholar]
- 19.Maddukuri L; Eoff RL; Choi JY; Rizzo CJ; Guengerich FP; Marnett LJ, In vitro bypass of the major malondialdehyde- and base propenal-derived DNA adduct by human Y-family DNA polymerases kappa, iota, and Rev1. Biochemistry 2010, 49 (38), 8415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chowdhury G; Guengerich FP, Liquid chromatography-mass spectrometry analysis of DNA polymerase reaction products. Curr Protoc Nucleic Acid Chem 2011, Chapter 7, Unit 7 16 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du H; Wang P; Wu J; He X; Wang Y, The roles of polymerases nu and theta in replicative bypass of O (6)- and N (2)-alkyl-2’-deoxyguanosine lesions in human cells. J Biol Chem 2020, 295 (14), 4556–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coggins GE; Maddukuri L; Penthala NR; Hartman JH; Eddy S; Ketkar A; Crooks PA; Eoff RL, N-Aroyl indole thiobarbituric acids as inhibitors of DNA repair and replication stress response polymerases. ACS Chem Biol 2013, 8 (8), 1722–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elmquist CE; Stover JS; Wang Z; Rizzo CJ, Site-specific synthesis and properties of oligonucleotides containing C8-deoxyguanosine adducts of the dietary mutagen IQ. J Am Chem Soc 2004, 126 (36), 11189–201. [DOI] [PubMed] [Google Scholar]
- 24.O’Flaherty DK; Guengerich FP, Steady-state kinetic analysis of DNA polymerase single-nucleotide incorporation products. Curr Protoc Nucleic Acid Chem 2014, 59, 7 21 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLuckey SA; Van Berkel GJ; Glish GL, Tandem mass spectrometry of small, multiply charged oligonucleotides. J Am Soc Mass Spectrom 1992, 3 (1), 60–70. [DOI] [PubMed] [Google Scholar]
- 26.Riggins JN; Daniels JS; Rouzer CA; Marnett LJ, Kinetic and thermodynamic analysis of the hydrolytic ring-opening of the malondialdehyde-deoxyguanosine adduct, 3-(2’-deoxy-beta-D-erythro-pentofuranosyl)- pyrimido[1,2-alpha]purin-10(3H)-one. J Am Chem Soc 2004, 126 (26), 8237–43. [DOI] [PubMed] [Google Scholar]
- 27.Riggins JN; Pratt DA; Voehler M; Daniels JS; Marnett LJ, Kinetics and mechanism of the general-acid-catalyzed ring-closure of the malondialdehyde-DNA adduct, N2-(3-oxo-1-propenyl)deoxyguanosine (N2OPdG-), to 3-(2’-Deoxy-beta-D-erythro-pentofuranosyl)pyrimido[1,2-alpha]purin- 10(3H)-one (M1dG). J Am Chem Soc 2004, 126 (34), 10571–81. [DOI] [PubMed] [Google Scholar]
- 28.Mao H; Schnetz-Boutaud NC; Weisenseel JP; Marnett LJ; Stone MP, Duplex DNA catalyzes the chemical rearrangement of a malondialdehyde deoxyguanosine adduct. Proc Natl Acad Sci U S A 1999, 96 (12), 6615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi JY; Zang H; Angel KC; Kozekov ID; Goodenough AK; Rizzo CJ; Guengerich FP, Translesion synthesis across 1,N2-ethenoguanine by human DNA polymerases. Chem Res Toxicol 2006, 19 (6), 879–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolfle WT; Johnson RE; Minko IG; Lloyd RS; Prakash S; Prakash L, Human DNA polymerase iota promotes replication through a ring-closed minor-groove adduct that adopts a syn conformation in DNA. Mol Cell Biol 2005, 25 (19), 8748–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furrer A; van Loon B, Handling the 3-methylcytosine lesion by six human DNA polymerases members of the B-, X- and Y-families. Nucleic Acids Res 2014, 42 (1), 553–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klug AR; Harbut MB; Lloyd RS; Minko IG, Replication bypass of N2-deoxyguanosine interstrand cross-links by human DNA polymerases eta and iota. Chem Res Toxicol 2012, 25 (3), 755–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki M; Kino K; Kawada T; Morikawa M; Kobayashi T; Miyazawa H, Analysis of nucleotide insertion opposite 2,2,4-triamino-5(2H)-oxazolone by eukaryotic B- and Y-family DNA polymerases. Chem Res Toxicol 2015, 28 (6), 1307–16. [DOI] [PubMed] [Google Scholar]
- 34.Williams NL; Wang P; Wang Y, Replicative Bypass of O(2)-Alkylthymidine Lesions in Vitro. Chem Res Toxicol 2016, 29 (10), 1755–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu W; Ouellette A; Ghosh S; O’Neill TC; Greenberg MM; Zhao L, Mutagenic Bypass of an Oxidized Abasic Lesion-Induced DNA Interstrand Cross-Link Analogue by Human Translesion Synthesis DNA Polymerases. Biochemistry 2015, 54 (50), 7409–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao L; Pence MG; Christov PP; Wawrzak Z; Choi JY; Rizzo CJ; Egli M; Guengerich FP, Basis of miscoding of the DNA adduct N2,3-ethenoguanine by human Y-family DNA polymerases. J Biol Chem 2012, 287 (42), 35516–35526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andersen N; Wang P; Wang Y, Replication across regioisomeric ethylated thymidine lesions by purified DNA polymerases. Chem Res Toxicol 2013, 26 (11), 1730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frank EG; Sayer JM; Kroth H; Ohashi E; Ohmori H; Jerina DM; Woodgate R, Translesion replication of benzo[a]pyrene and benzo[c]phenanthrene diol epoxide adducts of deoxyadenosine and deoxyguanosine by human DNA polymerase iota. Nucleic Acids Res 2002, 30 (23), 5284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith LA; Makarova AV; Samson L; Thiesen KE; Dhar A; Bessho T, Bypass of a psoralen DNA interstrand cross-link by DNA polymerases beta, iota, and kappa in vitro. Biochemistry 2012, 51 (44), 8931–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Washington MT; Minko IG; Johnson RE; Wolfle WT; Harris TM; Lloyd RS; Prakash S; Prakash L, Efficient and error-free replication past a minor-groove DNA adduct by the sequential action of human DNA polymerases iota and kappa. Mol Cell Biol 2004, 24 (13), 5687–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stafford JB; Eoff RL; Kozekova A; Rizzo CJ; Guengerich FP; Marnett LJ, Translesion DNA synthesis by human DNA polymerase eta on templates containing a pyrimidopurinone deoxyguanosine adduct, 3-(2’-deoxy-beta-d-erythro-pentofuranosyl)pyrimido-[1,2-a]purin-10(3H)-one. Biochemistry 2009, 48 (2), 471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.