

Abstract
Mechanoluminescent materials, which emit light in response to mechanical stimuli, have recently been explored as promising candidates for photonic skins, remote optogenetics, and stress sensing. All mechanoluminescent materials reported thus far are bulk solids with microns-sized grains, and their light emission is only produced when fractured or deformed in bulk form. In contrast, mechanoluminescence has never been observed in liquids and colloidal solutions, thus limiting its biological application in living organisms. Here we report the synthesis of mechanoluminescent fluids via a suppressed dissolution approach. We demonstrate that this approach yields stable colloidal solutions comprising mechanoluminescent nanocrystals with bright emission in the range of 470–610 nm and diameters down to 20 nm. These colloidal solutions can be recharged and discharged repeatedly under photoexcitation and hydrodynamically focused ultrasound, respectively, thus yielding rechargeable mechanoluminescent fluids that can store photon energy in a reversible manner. This rechargeable fluid can facilitate a systemically delivered light source gated by tissue-penetrant ultrasound for biological applications that require light in the tissue, such as optogenetic stimulation in the brain.
Graphical Abstract
Introduction
Mechanoluminescent (ML) materials emit light in response to mechanical stimuli, such as compression, stretching, fracture, and friction.1–4 Conventional ML materials comprise trap-controlled inorganic solids with defects, as well as inorganic and organic materials that emit light when their chemical bonds are broken during the fracture of the solid.3–5 The reversibility, color tunability, and bright emission (usually visible to the naked eye) under moderate mechanical stimuli of ML materials make them attractive candidates for sensing and imaging stress distribution, visualizing crack propagation, lighting and display, as well as developing photonic skins.1–3,5,6 In addition, combining the deep tissue penetration of ultrasound7 and ultrasound-responsive ML materials may yield a minimally invasive method for optogenetic neuromodulation and other light-based biomedical applications.1,8
Despite their expanding applications, all ML materials reported thus far only emit light under stress in bulk solid form,3,9 while their large sizes and poor solubility prohibit their stable suspension in water. As a result, the ML phenomenon has never been reported in liquids under hydrostatic and hydrodynamic pressures.10 Instead, an extremely high peak negative pressure is required to produce sonoluminescence in liquid water, a distinct phenomenon from mechanoluminescence, with weak light emission due to cavitation and the implosion of gas bubbles.11 Since mechanoluminescence represents a much more efficient and biologically safer way of converting mechanical energy into light than sonoluminescence, it is desirable to develop liquids that exhibit mechanoluminescence under hydrodynamic pressure. Compared to conventional ML solids, ML liquids can be easily delivered in a flow system or in a living organism to facilitate many biological applications therein.
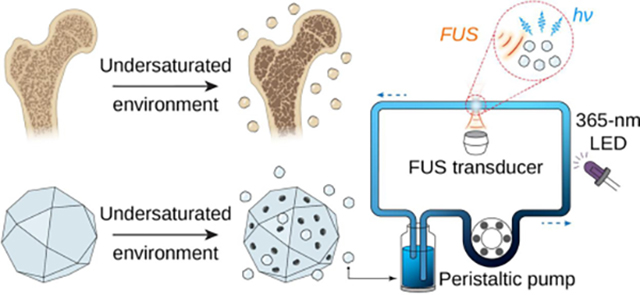
In this paper, we demonstrate the production of ML fluids, implemented by stably suspending colloidal ML nanocrystals in an aqueous solution. These ML colloids are composed of (470 nm), ZnS:Cu,Al (534 nm), ZnS:Mn (578 nm), and CaTiO3:Pr (610 nm) with their ML emissions spanning the entire visible spectrum. Since all these ML materials require the creation of point defects in the host materials with desired phases,12 solid-state reactions at elevated temperatures of >1000 °C constitute the only synthesis approach of these ML materials.13 The elevated temperature of the solid-state reactions necessarily results in bulk particles of >10 μm sizes. To turn these bulk ML materials into colloidal solutions, we leveraged a suppressed dissolution approach by learning from the intricate nanostructures found in natural biominerals (Scheme 1).14 This approach effectively produces water-soluble ML nanocrystals with diameters down to 20 nm while preserving the strong ML emission of their solid-state synthesized counterparts. The suppressed dissolution approach enables the synthesis of ML fluids with a variety of emission wavelengths ranging from 470 nm to 610 nm under hydrodynamically focused ultrasound (FUS). Furthermore, these ML fluids can be recharged and discharged repeatedly under photoexcitation and hydrodynamically focused ultrasound, respectively (Scheme 1). Taken together, these properties of ML fluids, enabled by a suppressed dissolution approach, may facilitate reversible photon energy storage in an optical flow battery and an intravenously delivered light source gated by FUS for deep-tissue imaging and optogenetics.15
Scheme 1.
The suppressed dissolution approach leverages a kinetically preserved dissolution mechanism of natural biominerals to produce colloidal ML materials as a rechargeable light source under FUS.
Results and Discussion
We set out by choosing an array of ML materials with their emissions spanning the visible spectrum. To produce intense ML emission at desirable wavelengths, we chose host materials with a large reported piezoelectric coefficient, a sufficient band gap, lattice atoms facilitating defect formation, and a crystal field enabling tuning of the electron orbital states of emitter centers. Specifically, melilite (, 470 nm), wurtzite (ZnS:Cu,Al, 534 nm and ZnS:Mn, 578 nm), and perovskite (CaTiO3:Pr, 610 nm) are trap-controlled ML materials with their ML emissions arising from point defects in host materials with specific phases. On one hand, point defects, such as substitutional atoms (e.g., DySr•) and vacancies (e.g., vS••), act as electron or hole traps that give rise to mechanoluminescence.3 A high synthesis temperature (usually >1000 °C) is thus necessary to fulfill the thermodynamic and kinetic requirements of point defect formation.12 On the other hand, materials lacking central symmetry are required for the production of mechanoluminescence.5 For example, only the non-centrosymmetric wurtzite phase of ZnS, which is favored at a high temperature (>1020 °C),16 exhibits ML emission, while its centrosymmetric zincblende phase does not. Therefore, these ML materials cannot be synthesized at a low temperature via wet chemical, bottom-up approaches alone, while solid-state reactions or annealing at elevated temperatures of >1000 °C constitute the only synthetic route to satisfy the desirable point defects and polymorphs.13 As a result, the resultant ML materials usually exhibit large grain sizes (>10 μm) after high-temperature annealing and solid-state reactions.3 Their large sizes prevent them from forming a stable solution in water.9 Although mechanical milling represents a plausible route to produce nanoparticles from solid-state products, the plastic deformation process introduces undesired built-in stresses and dislocations into the resulting nanoparticles.17 These built-in stresses and dislocations lead to mechanical and defect-induced quenching of ML emission,18,19 thus making mechanical milling an unfavorable approach for producing colloidal ML solutions from solid-state precursor particles. Other approaches, such as ultrasonic pyrolysis and pulsed laser deposition, also fail to synthesize ML colloids that can be stably suspended in water.20,21 In summary, none of the existing approaches has yet succeeded in producing liquids or colloidal solutions with ML properties.
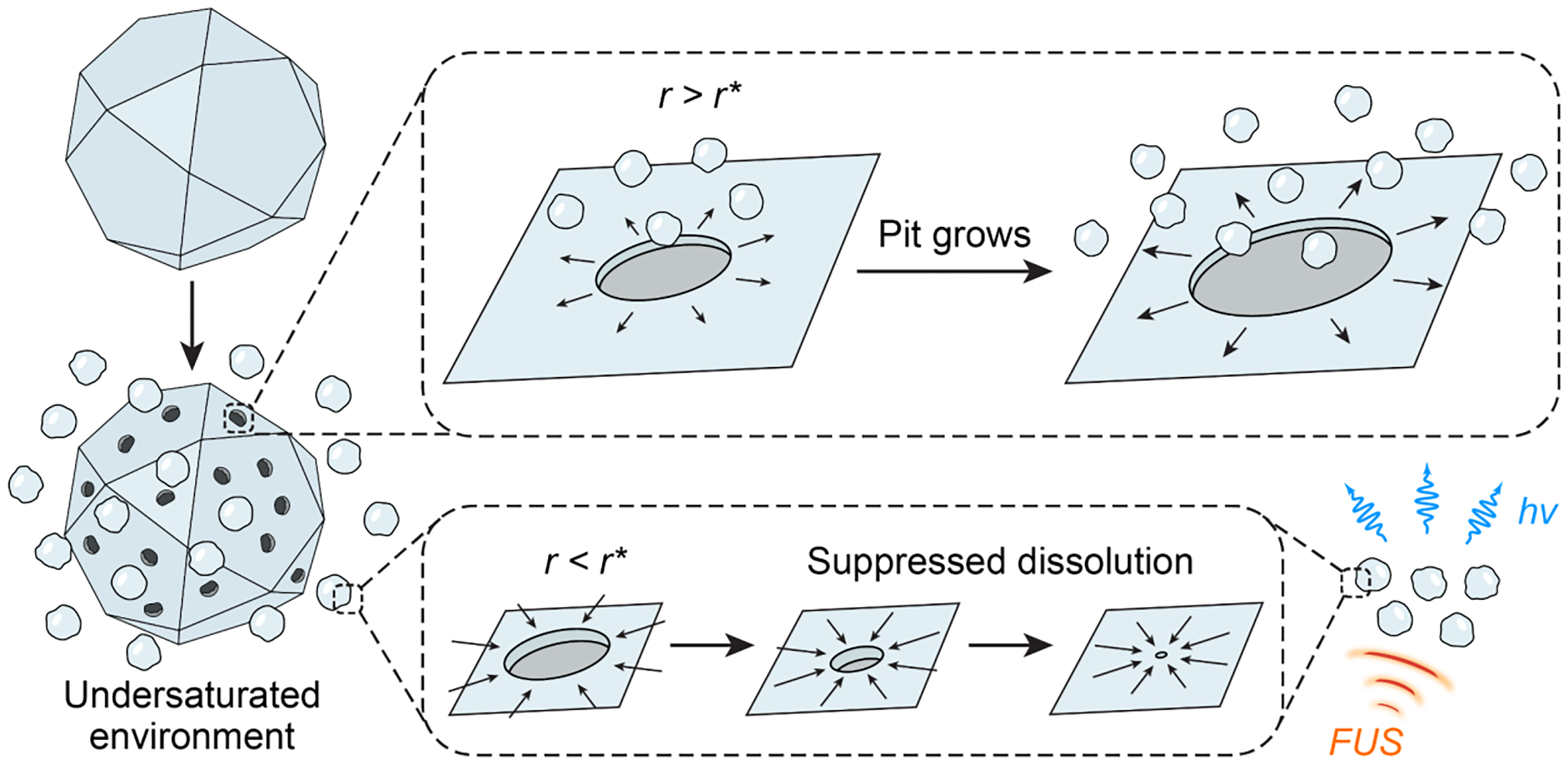
To mitigate this challenge in producing ML fluids, we developed a suppressed dissolution approach by leveraging an unusual finding in the demineralization process of bone and tooth enamel.22 Biominerals, such as bones, teeth, and avian eggshells, usually exhibit intricate nanostructures on their surfaces in a physiological undersaturated aqueous environment, such as during the process of bone resorption (Scheme 1).22–24 Surprisingly, nanostructures on the surface and nanoparticles released into the aqueous solution remain resistant to further dissolution despite a negative Gibbs free energy for the dissolution reaction (i.e., ). It has been found that biomineral nanoparticles are kinetically stabilized in an undersaturated solution despite their thermodynamic instability, thus yielding a self-preservation behavior.
We hypothesize that this self-preservation behavior of biomineral nanoparticles can be leveraged to produce stable ML nanocrystals in an aqueous solution via the suppressed dissolution approach. We argue that both biominerals and ML materials are ceramic materials with intrinsically low solubility in water, thus yielding a high interfacial tension, , between these solid materials and liquid water when they are immersed in a solution.25 Such a high interfacial tension is energetically unfavorable for the formation of a dissolution pit with a radius of on the two-dimensional (2D) surface of the solid, which incurs a Gibbs free energy change as follows
| (1) |
Therefore, dissolution can only happen when the change in Gibbs free energy of the dissolution reaction, , overcomes the energy barrier of pit formation, yielding a net negative :
| (2) |
where is the change in Gibbs free energy per dissolution unit, while is the area occupied by each dissolution unit. It is obvious from Eq. (2) that yields a positive value for a very small , which is upper-bounded by the size of the dissolving particle. Therefore, a critical size exists, below which no dissolution can occur:
| (3) |
The critical size of most ML materials lies in the range of 10–100 nm, thus suggesting the possibility of synthesizing stable colloidal solutions of ML materials via the suppressed dissolution approach.
We experimentally validated our hypothesis by applying the suppressed dissolution approach on an array of ML materials with their emission spanning the visible spectrum. Specifically, melilite (, 470 nm), wurtzite (ZnS:Cu,Al, 534 nm and ZnS:Mn, 578 nm), and perovskite (CaTiO3:Pr, 610 nm) ML materials were synthesized via solid-state reactions (see Materials and Methods). Scanning electron microscopy (SEM) images of as-synthesized ML materials revealed large sizes of >10 μm (Fig. 1a), thus resulting in quick sedimentation after these materials are dispersed in water (Fig. 1a, insets). We then applied the suppressed dissolution approach to these ML materials by simply mixing bulk ML particles with an undersaturated solution of citrate until a metastable equilibrium was reached (see Materials and Methods). This simple and generic approach yielded stable colloidal solutions containing their corresponding ML nanocrystals resistant to further dissolution (Fig. 1b, insets), in contrast to the more common, self-accelerating dissolution process (e.g., the dissolution of table salt in water). Transmission electron microscopy (TEM) images revealed a diameter of resulting colloidal nanocrystals between 10 and 110 nm for these materials (Fig. 1b & 1c), in agreement with the size range of kinetically stabilized nanoparticles predicted by Eq. (3). In addition, the size distribution of ML colloids was confirmed with dynamic light scattering (Fig. S1a). X-ray diffraction (XRD) patterns of ML nanocrystals and their corresponding bulk precursors confirmed the preservation of the crystal structure of the host materials after the suppressed dissolution process (Fig. S2).
Fig. 1.
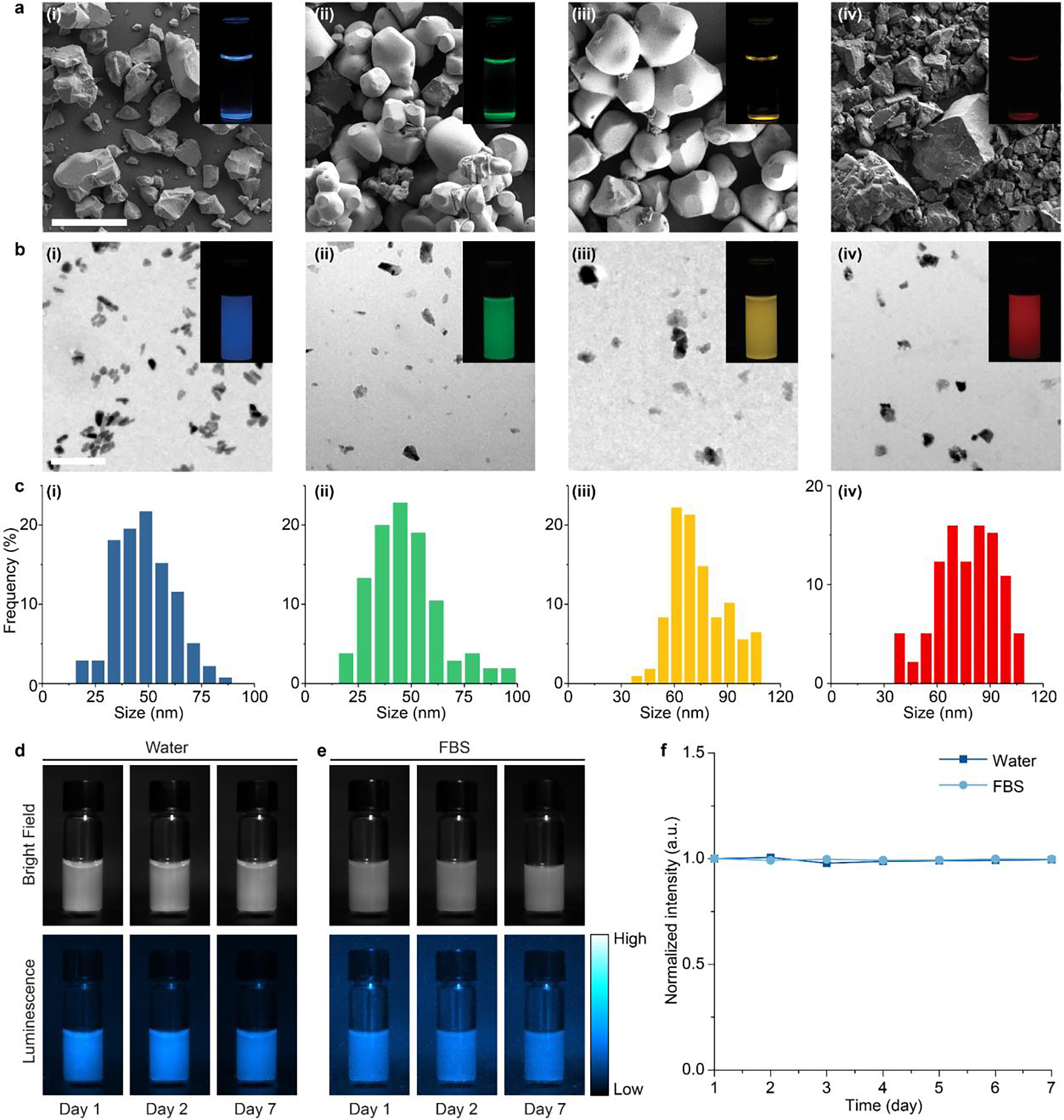
Synthesis of multicolor mechanoluminescent colloids via a bioinspired approach. (a) SEM images of (i), ZnS:Cu,Al (ii), ZnS:Mn (iii), and CaTiO3:Pr (iv) bulk ML materials after solid-state reactions. (b) TEM images of (i), ZnS:Cu,Al (ii), ZnS:Mn (iii), and CaTiO3:Pr (iv) ML colloidals produced by the suppressed dissolution approach. The insets in a&b are luminescence images of the corresponding ML materials in an aqueous solution. The scale bars represent 50 μm in a and 200 nm in b. (c) Histograms showing the size distributions of (i), ZnS:Cu,Al (ii), ZnS:Mn (iii), and CaTiO3:Pr (iv) colloidals. (d&e) Brightfield (top) and luminescence (bottom) images of colloids in water (d) and FBS (e). (f) Luminescence intensity of colloids in water and FBS over the course of 1 week.
The small sizes (down to 10 nm) of as-produced ML nanocrystals impart excellent colloidal stability over extended times (Fig. 1d&e). The abundance of citrate anions in the undersaturated solution during suppressed dissolution offers a convenient means to confer a net negative charge to stabilize ML colloids (Fig. S1b), resulting in negligible precipitation (< 0.4%) over a week (Fig. 1d&f). Similar colloidal stability was also observed for the same ML colloids suspended in fetal bovine serum (FBS), confirming their ability to resist aggregation in a biological environment (Fig. 1e&f). Besides citrate, we have shown that the original surface-capping agent can be replaced by poly(ethylene glycol) (PEG) to yield similar colloidal stability (Fig. S3). This chronic stability of ML colloids contrasts that of bulk ML particles, which exhibited rapid precipitation within merely 1 h (Fig. S4).
We next sought to provide a quantitative theoretical framework of the suppressed dissolution mechanism in the context of the demineralization process of many biominerals. A characteristic hallmark of demineralization is the roughening of crystal surfaces, covered by nonequilibrium, nanostructured morphologies.26 SEM images of all ML particles after the suppressed dissolution process exhibit much rougher surfaces (Fig. S5) than before (Fig. 1a). Similar rough surfaces have been found on enamel surfaces under physiological conditions, while nanoparticles loosely attached to the surface during the roughening process can be released directly into the solution by the diffusion flux.24 Based on the TEM images and concentrations of ML nanocrystals found in the solution after the suppressed dissolution process, we found that each , ZnS:Cu,Al, ZnS:Mn, and CaTiO3:Pr particle releases 106, 105, 105, and 105 nanocrystals on average, respectively.
As surface roughening happens, the dissolution rate is expected to decrease over time when the feature sizes of the roughened surface and released nanoparticles approach the critical size . This size-dependent dissolution represents another hallmark of the kinetically preserved demineralization process in biology. Eq. (3) predicts that the dissolution rate should approach zero when , with an analytical formula of the dissolution rate described in previous reports as follows:
| (4) |
Using a reported constant composition (CC) technique,27 we measured the overall dissolution flux of ML crystals, , which is proportional to the dissolution rate , as a function of dissolution time, based on the corresponding titration curves (Fig. 2a&b). The dissolution flux exhibits a characteristic decay for all added ML particles (Fig. 2b), thus confirming the prediction by Eq. (4). Importantly, only 44%, 9%, 9%, and 34% of added , ZnS:Cu,Al, ZnS:Mn, and CaTiO3:Pr bulk seeds underwent dissolution, respectively, as evidenced by a plateau in the titration curve lower than complete dissolution (Fig. 2a). In addition, we measured the titrant volume and dissolution flux of the same amount of under different undersaturations, which were in turn determined by the of the solution. Specifically, a lower results in greater undersaturation, according to the following chemical equation at equilibrium:
| (5) |
The undersaturation, σ, can be defined by28
| (6) |
where is the change in chemical potential per molecule in the reaction of eq. (5), is the Boltzmann constant, is absolute temperature, and concentrations with the subscript “0” indicate those in equilibrium with bulk particles. The CC condition in our experiments (see Materials and Methods) ensures that and are constant and equal to and at all times. Therefore, is dependent only on the of the solution, becoming more positive as the decreases and reaching zero at the saturation of 9.8 for the selected and . Notably, Fig. 2c&d reveal markedly decreasing dissolution rates tending to zero at all undersaturations, with smaller overall dissolution rates and more rapid suppression of dissolution at a lower undersaturation (i.e., a higher closer to the of 9.8). These observations agree with the dissolution process found in many biominerals,22,23,27,29 thus confirming a similar suppressed dissolution mechanism at play during the formation of ML colloids.
Fig. 2.
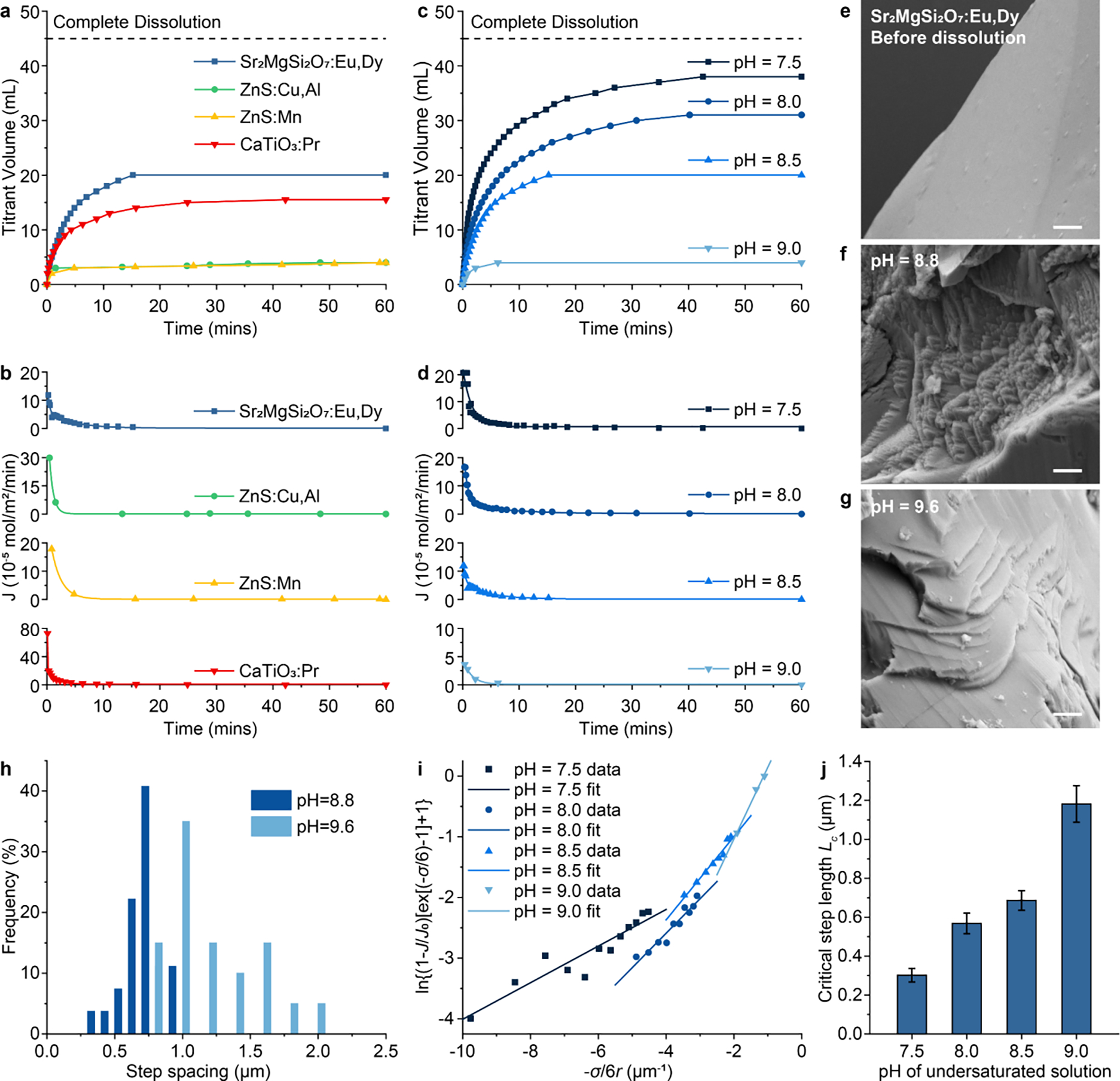
Mechanistic study of the suppressed dissolution approach in ML materials. (a&b) Titration curves (a) and dissolution flux (b) of , ZnS:Cu,Al, ZnS:Mn, and CaTiO3:Pr under constant composition (CC) conditions. (c&d) Titration curves (c) and dissolution flux (d) of at different undersaturations as determined by the of the CC solution. (e-g) SEM images of surface before dissolution (e), and after suppressed dissolution at (f) and 9.6 (g), respectively. Dissolution steps can be seen in (f&g) after suppressed dissolution. All scale bars represent 1 μm. (h) Histograms showing the step spacing distribution on the surface after suppressed solution at and 9.6 in f&g. (i) Experimental data and linear fitting of vs. under different undersaturations. (j) The critical lengths of receding steps on the surface of under different undersaturations, extracted from the slopes of linear fitting curves in (i). The error bars represent the uncertainty in fitting parameters.
Besides these macroscopic kinetic measurements of the suppressed dissolution process, we also sought to quantitatively describe the dissolution process from a microscopic perspective. Previous studies leveraging in situ atomic force microscopy (AFM) and TEM liquid cells have experimentally observed the advancement and recession of stepped terraces during the growth and dissolution of biominerals, respectively.28,30 The validation of the same demineralization mechanism underlying our method (Fig. 2a–d and Fig. S5) justifies applying this microscopic step-recession model to the suppressed dissolution approach. Specifically, the Gibbs free energy change, , for the recession of a single, straight step due to the dissolution of its constituent atoms is
| (7) |
where is the length of the receding step, is the intermolecular distance along this step, and is the distance between the neighboring steps. Eq. (7) is thus the microscopic presentation of eq. (2) above. Setting eq. (7) to zero shows that the steps recede only if exceeds a critical length, , given by
| (8) |
Combining eq. (8) and eq. (6) yields
| (9) |
Alternatively, for any given length of a step, its equilibrium concentration of hydroxide ions, , is related to as follows:
| (10) |
Solving for yields
| (11) |
where , by definition above, is the concentration of in equilibrium with bulk particles. Inserting eq. (9) into eq. (11) yields
| (12) |
The receding velocity of the step has been reported as28
| (13) |
Combining eq. (6), (12), and (13) yields
| (14) |
where is the receding velocity for an infinitely long step:
| (15) |
This microscopic dissolution model allows us to validate the mechanism in the dissolution of ML materials in two ways. First, eq. (9) reveals an inverse relationship between the critical length and undersaturation during the recession of steps. Since the spacing between steps has been reported to be proportional to and a lower yields a higher (eq. (6)), one should expect a smaller spacing between steps at a lower . To verify this expectation, we performed SEM imaging of the surface of bulk ML particles in the middle of the dissolution process at the of 8.8 and 9.6. Fig. 2e–h confirms the presence of stepped terraces28,30 and reveals much smaller spacings between the neighboring steps at the of 8.8 than those at the of 9.6, thus confirming the prediction by eq. (9). Second, it has been reported that the step receding velocity, , is proportional to the dissolution flux, .22 Eq. (14) can thus be simplified as
| (16) |
Since the average receding step length is related to the average particle radius of the resulting colloid,27 eq. (16) can be further simplified and rearranged to yield
| (17) |
Plotting vs. yields a linear relationship at different undersaturations, with the slope of linear fitting equal to (Fig. 2i). The extracted from Fig. 2i increases with an increasing (thus a decreasing ; Fig. 2j), agreeing with the observed step spacings in Fig. 2e–h and further confirming the validity of the step-recession model underlying the suppressed dissolution approach.
Having confirmed the mechanism of the suppressed dissolution approach, we then characterized the excitation and emission spectra of bulk ML materials (Fig. 3a) and resulting colloidal solutions (Fig. 3b). The identical spectral properties between bulk ML materials and their corresponding colloids suggest effective preservation of dopant ions (which act as color centers) in their corresponding host materials. Owing to the presence of identical luminescence centers after dissolution, we hypothesized that the resulting colloidal solutions should have the same ML properties as their bulk counterparts, owing to the mechanism of mechanoluminescence in trap-controlled ML materials (Fig. S6). To experimentally verify this hypothesis (Fig. 3c), we measured the mechanoluminescence spectra of bulk ML materials (Fig. 3d) and colloidal nanoparticles in a polydimethylsiloxane (PDMS) phantom (Fig. 3e) and found identical peak locations between bulk and colloidal ML materials. Importantly, the ML spectra of colloids span the entire visible spectrum, thus yielding a palette of ML colloids with different colors of emission under mechanical stress.
Fig. 3.
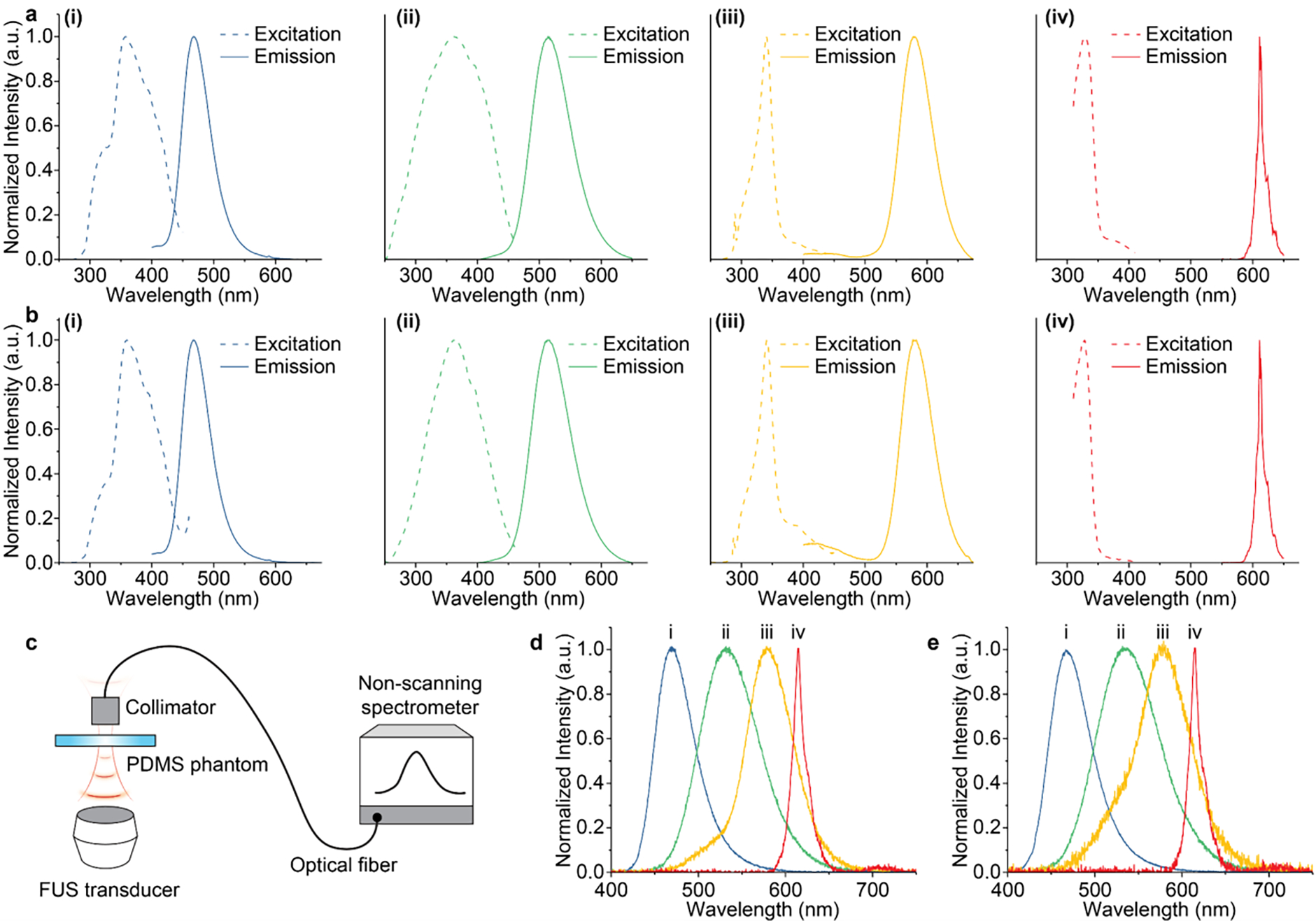
Optical properties of multicolor mechanoluminescent colloids. (a&b) Excitation (dashed) and emission (solid) spectra of (i), ZnS:Cu,Al (ii), ZnS:Mn (iii), and CaTiO3:Pr (iv) bulk ML materials (a) and their corresponding colloids (b). (c) Schematic showing the setup for mechanoluminescence spectroscopy. (d&e) Mechanoluminescence spectra of (i), ZnS:Cu,Al (ii), ZnS:Mn (iii), and CaTiO3:Pr (iv) bulk materials (d) and their corresponding colloids (e). Data are plotted in colors representative of the emission of corresponding materials.
We next sought to demonstrate the rechargeability of produced ML colloids. Specifically, we used a 365-nm light source to recharge ML colloids suspended in a PDMS phantom, before the phantom is subject to incident FUS pulses (see Materials and Methods). The same process was repeated 500 cycles and the intensity was measured in real time (Fig. 4a–d, i). Within each cycle, ML intensity reached maximum at 0.1 s after the application of the FUS pulse, followed by a decay corresponding to gradual depletion of energy traps triggered by the FUS pulse (Fig. 4a–d, ii). Despite repeated recharging and FUS-triggered depletion of energy traps, we found the stability of ML intensity over at least 500 cycles, suggesting excellent rechargeability for all materials (Fig. 4a–d, iii).
Fig. 4.
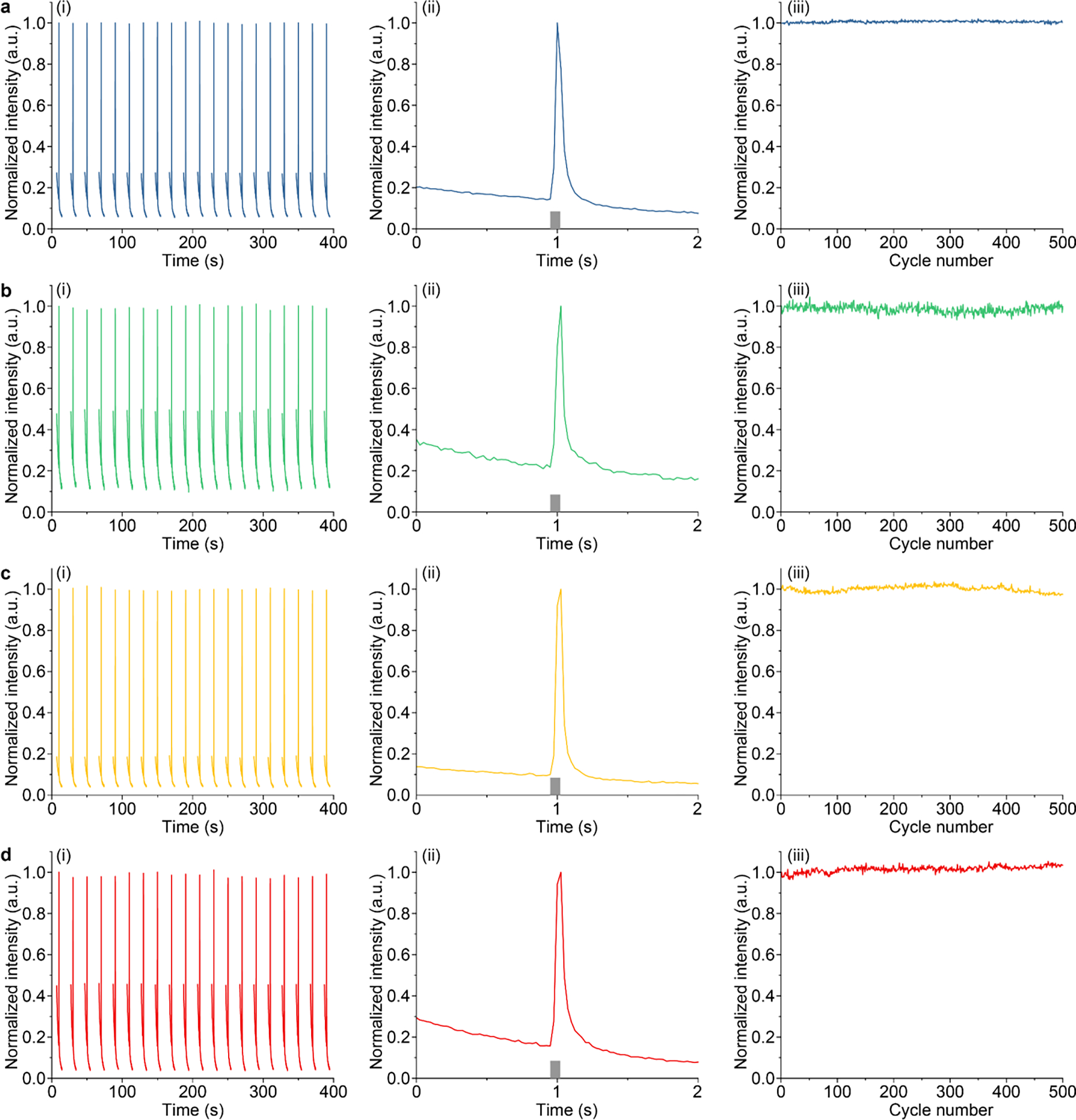
Rechargeability of (a), ZnS:Cu,Al (b), ZnS: Mn (c), and CaTiO3:Pr (d) colloids. Each panel includes (i) temporal dynamics of luminescence intensity over 20 cycles of recharging and FUS-mediated emission; (ii) temporal dynamics of a representative cycle; and (iii) normalized peak intensity of ML emission as a function of cycle numbers.
Having demonstrated the rechargeability of ML colloids in PDMS, we then studied the rechargeability of their corresponding ML fluids by suspending the colloids in an aqueous solution. All ML nanoparticles exhibit excellent colloidal stability in water owing to their small sizes of <100 nm (insets, Fig. 1b; Fig. 1d–f). Therefore, the resulting ML fluid should be able to flow in a circulatory system to be recharged and depleted at two separate locations, rather than at the same location as shown in PDMS above. To test this hypothesis, we constructed an artificial circulatory system as a bioinspired device to mimic the endogenous circulatory system in the mammalian body. Specifically, the artificial circulatory system is composed of a closed loop of Tygon tubing to mimic the blood vessel and a peristaltic pump to mimic the heart. In addition, one segment of the tubing is exposed to the 365-nm recharging light, while another segment is stimulated by 1.5-MHz FUS to produce localized ML emission (Fig. 5a). We found obvious emission of different colors at the focus of ultrasound in the circulating ML fluid (Fig. 5b–e). Furthermore, real-time intensity measurements of light emission at the focus of ultrasound revealed reproducible peaks of identical intensity, thus validating the rechargeability of ML fluids (Fig. 5f–i). Lastly, we confirmed the stability of ML intensity over at least 600 cycles when these ML fluids are recharged and discharged repeatedly over at least 10 min (Fig. S7). Therefore, the circulating ML fluids can be used as an FUS-activated light source with recharging stability over a time period sufficient to optogenetically probe biological processes occurring at a similar timescale.31
Fig. 5.
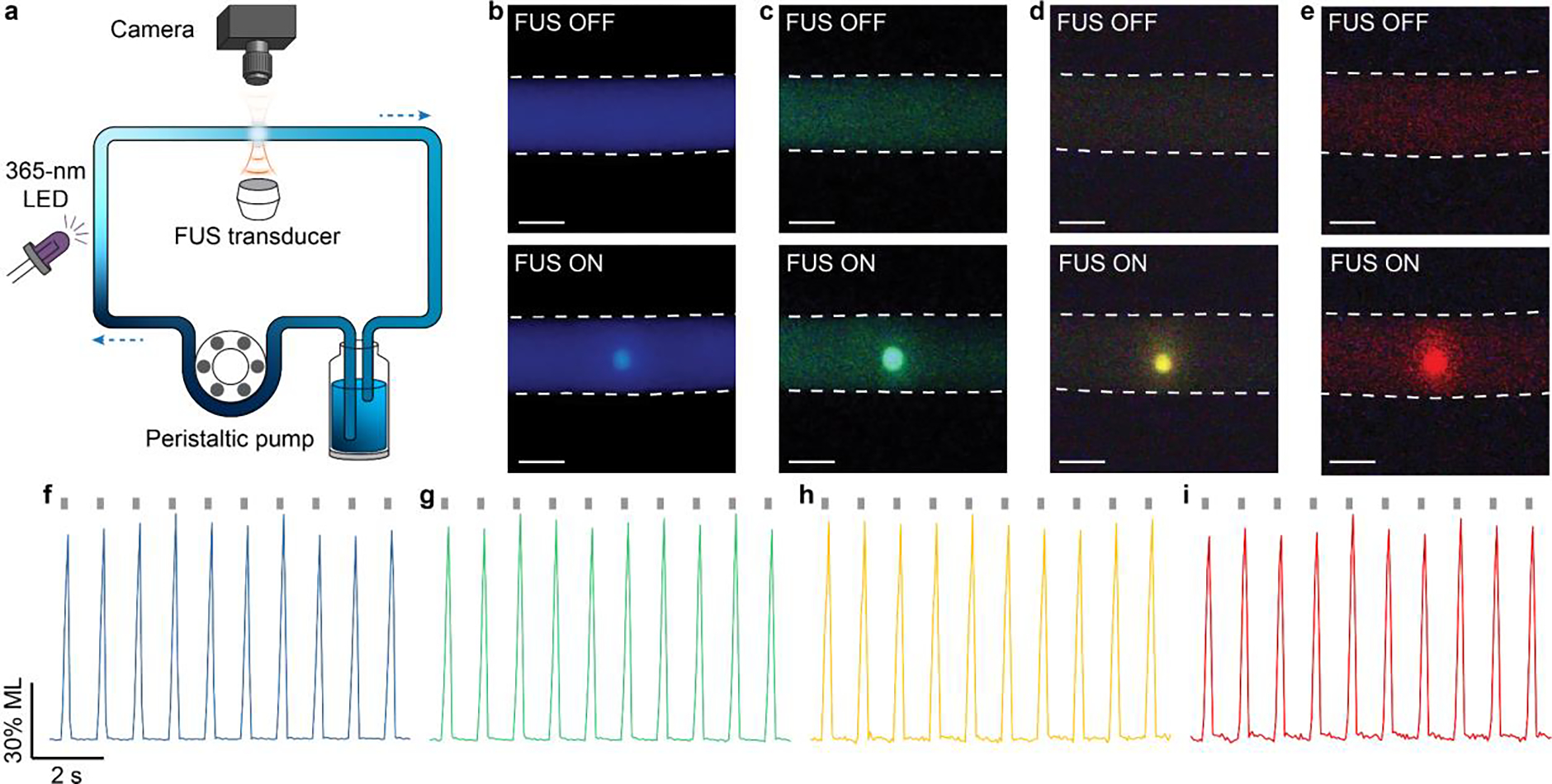
Rechargeable emission of multicolor ML fluids in an artificial circulatory system. (a) Schematic showing the setup of the artificial circulatory system. (b-e) True-color images of the tubing filled with ML fluids composed of (b), ZnS:Cu,Al (c), ZnS:Mn (d), and CaTiO3:Pr (e) when FUS was on (top) and off (bottom). All scale bars represent 1 mm. (f-g) Representative dynamics of light emission intensity from ML fluids composed of (f), ZnS:Cu,Al (g), ZnS:Mn (h), and CaTiO3:Pr (i) in the artificial circulatory system. The gray ticks on the top represent FUS pulses.
The results in Fig. 5 confirmed mechanoluminescence produced by hydrodynamic pressure in a solution, unlike the conventional mechanoluminescence produced by bulk stress in a solid. To understand how FUS-induced hydrodynamic pressure correlates with emission intensity, we varied the pressure of FUS and measured the ML intensity from the focus of ultrasound in the solution. Our measurements revealed a thresholded behavior between the FUS peak pressure and ML intensity. Specifically, ML intensity increases slowly with an increase in FUS peak pressure below the threshold, beyond which ML emission significantly intensifies with pressure (Fig. 6a–d). This thresholded behavior of ML fluids resembles that of bulk ML materials,32,33 thus confirming a similar origin of mechanoluminescence despite the difference in pressure exertion. All materials exhibited a similar threshold of 1 MPa, suggesting an energy barrier to overcome before sufficient ML emission can be produced. The thermoluminescence spectra of all ML colloids reveal a similar emission peak near 350 K (Fig. S8), thus confirming a comparable energy barrier for ML emission. Importantly, the ability to produce intense light emission at a peak pressure as low as 1 MPa disproves sonoluminescence as a plausible mechanism to account for the observed emission in ML fluids. Specifically, we observed no light emission in the carrier medium (i.e., water) without ML colloids under the same pressure (Fig. S9). In addition, this threshold of 1 MPa corresponds to a mechanical index of 0.8 MPa/MHz1/2, well below the FDA-recommended threshold of 1.9 MPa/MHz1/2 to produce cavitation.34 Therefore, the ML fluids synthesized in this work represent a new mechanism for light generation in liquids under acoustic waves besides sonoluminescence.
Fig. 6.
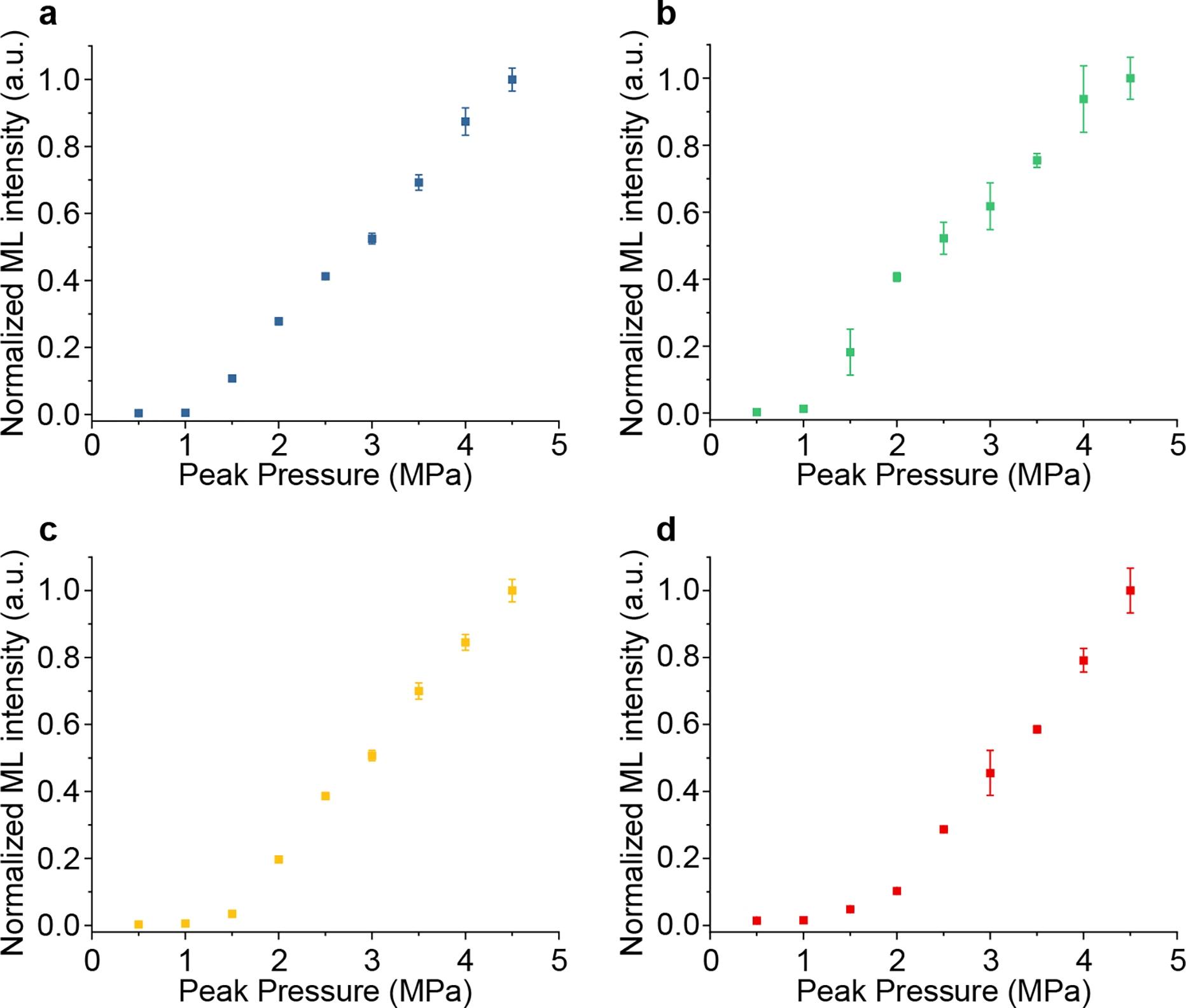
The dependence of light emission intensity of ML fluids on the hydrodynamic pressure induced by FUS. Normalized ML emission intensity is plotted as a function of FUS peak pressure for (a), ZnS:Cu,Al (b), ZnS:Mn (c), and CaTiO3:Pr (d) colloidal fluids in the artificial circulatory system. All data are presented as mean ± SD of 10 independent measurements.
A salient advantage of ML fluids, in comparison to conventional bulk ML solids, arises from their ability to absorb and store photon energy in a flow medium. Specifically, ML fluids carry the stored photon energy in the flow until they are discharged by FUS pulses to release the energy in the form of light emission. Therefore, using ML fluids as a medium for energy transport and light delivery requires understanding the temporal kinetics of FUS-triggered light emission. To this end, we performed real-time ML intensity measurements in response to FUS stimuli of different pulse widths. Specifically, we varied the FUS pulse widths between 10 ms and 500 ms while maintaining their repetition rate at 1 Hz. ML fluids were allowed to circulate in the same system as Fig. 5a while being continuously recharged to eliminate the interference of insufficient recharging power on temporal kinetics measurements. Fig. 7 reveals fast onset and offset kinetics of ML emission when FUS is applied and removed, respectively. We plotted the rise and fall times of ML emission at different pulse widths for all ML fluids, and found a short onset time of ≤5 ms for all ML fluids regardless of the FUS duration. This result suggests that all ML fluids emit light within ca. 5 ms after receiving the FUS pulse, thus satisfying the fast temporal requirements of many light-based biomedical applications such as optogenetics and fluorescence imaging.15,35,36 Although the offset times (10–50 ms) are generally longer than onset times (≤5 ms) for all materials, they do not impose a significant challenge for applying ML fluids for aforementioned applications. As a comparison, the intrinsic deactivation time of opsins and the intrinsic decay time of fluorescent calcium indicators are at a similar level or longer (tens of milliseconds for opsins and hundreds of milliseconds for jCaMP7f).35,37,38
Fig. 7.
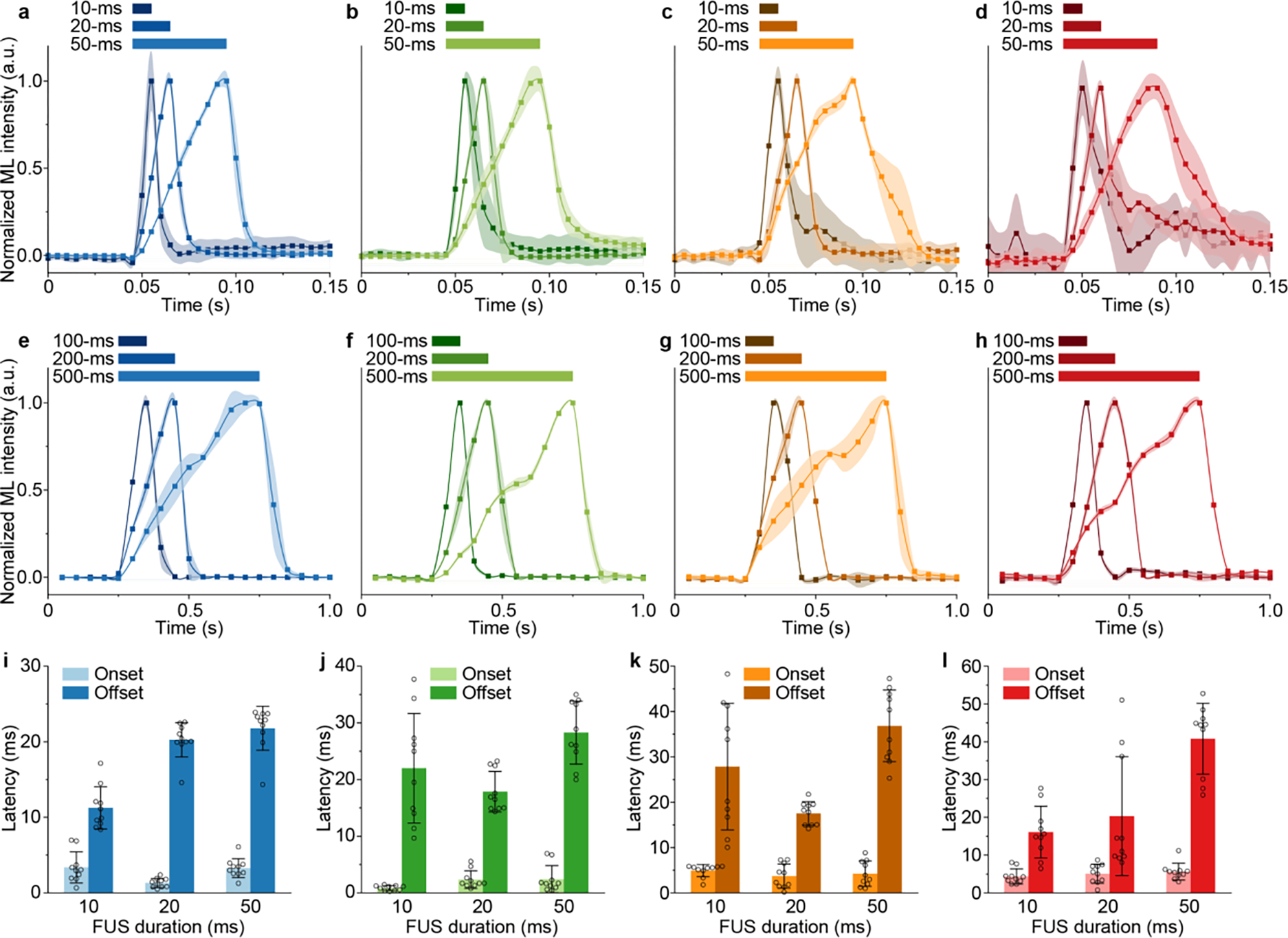
Temporal kinetics of FUS-triggered light emission from ML fluids. (a-d) ML dynamics of (a), ZnS:Cu,Al (b), ZnS:Mn (c), and CaTiO3:Pr (d) colloidal fluids upon 10-ms, 20-ms, and 50-ms FUS pulses in the artificial circulatory system. The camera frame rate was 200 Hz. (e-h) ML dynamics of (e), ZnS:Cu,Al (f), ZnS:Mn (g), and CaTiO3:Pr (h) colloidal fluids upon 100-ms, 200-ms, and 500-ms FUS pulses in the artificial circulatory system. The camera frame rate was 20 Hz. (i-l) Bar charts summarizing the ML onset and offset times of (i), ZnS:Cu,Al (j), ZnS:Mn (k), and CaTiO3:Pr (l) colloidal fluids with 10-ms, 20-ms, and 50-ms FUS pulses in the artificial circulatory system. Onset and offset times were only analyzed from a-d due to the sufficient frame rate used therein (200 Hz), while e-h have insufficient frame rates (20 Hz) for accurate measurements of onset and offset times. All data are presented as mean ± SD of 10 independent measurements.
The utility of the ML fluids lies in their ability to be delivered into blood vessels to produce local light emission in vascularized biological tissues under FUS. To prove this utility, we demonstrated light emission in different mouse organs with their corresponding vasculature perfused with ML fluids (see Materials and Methods). Specifically, with the ML fluid composed of colloids delivered into the vasculature in the liver, kidney, and brain of a mouse, light emission can be triggered reproducibly at the focus of ultrasound that penetrates through the entire organ (Fig. 8a–c). We found a full width at half maximum (FWHM) of 150–500 μm for light emission spots from these organs (Fig. 8d–f). This sub-millimeter range of FWHM sets an achievable spatial resolution on par with that of an implanted optical fiber while eliminating the invasiveness associated with fiber insertion in the tissue.39 Lastly, we also demonstrated reproducible light emission in response to FUS pulses in all organs by measuring the temporal kinetics of light intensity during repeated ultrasound stimulation (Fig. 8g–i).
Fig. 8.
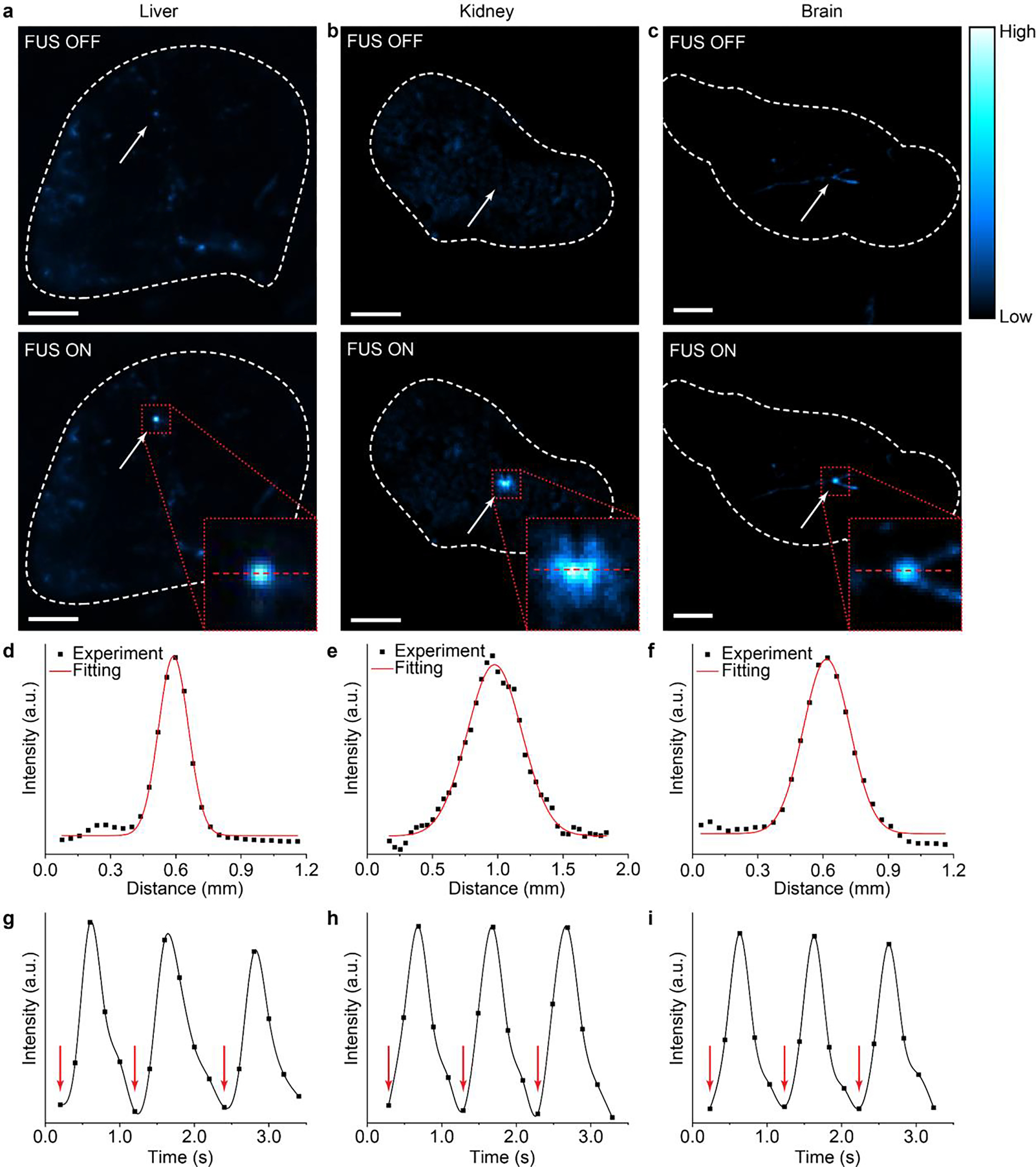
ML emission from different mouse organs. (a-c) Luminescence images of the mouse liver (a), kidney (b), and brain (c) with their vessels filled with the ML fluid of during the off (top) and on (bottom) phases of FUS pulses. All scale bars represent 2 mm. (d-f) Intensity profiles of ML emission from the liver (d), kidney (e), and brain (f) along the red dashed lines in the insets of a, b, and c, respectively. (g-i) Temporal kinetics of ML emission from the liver (g), kidney (h), and brain (i) upon repetitive FUS pulse stimulation (red arrows).
The demonstrated ability of ML fluids to produce on-demand, focused illumination in vascularized biological tissues facilitates any applications that require light deep inside the body. One of such applications is optogenetic stimulation, which usually requires an implanted optical fiber to locally deliver light in the live mouse brain.37 However, the implanted optical fiber requires an invasive procedure that induces acute and chronic tissue responses, which may alter the endogenous activity and connectivity of neurons and non-neuronal cells.40 In contrast, ML fluids can be delivered via a minimally invasive systemic route, thereby enabling optogenetic stimulation with tissue-penetrant ultrasound. To validate this utility of ML fluids, we injected colloids into the mouse circulatory system, recharged them in superficial vessels during their circulation in vivo, and discharged them with FUS to emit light in the motor cortex of the brain (Fig. 9a; see Materials and Methods). We verified the efficacy of FUS-mediated optogenetic stimulation via systemically-delivered ML fluids by carrying out immunohistochemical staining of c-Fos, an immediate early gene that labels recent neuronal activity (Fig. 9b).41 We found a significant increase in the number of c-Fos+ cells in Thy1-ChR2-YFP transgenic mice that express channelrhodopsin-2 (ChR2) in neurons, compared to that in wildtype C57BL/6J mice (Fig. 9c). This result also rules out the possibility of nonspecific, direct neural activation with FUS in the mouse brain since the WT mouse brain shows minimal baseline activity. In conclusion, ML fluids, which are recharged during blood circulation, can produce sufficient light intensity to activate ChR2 for wireless optogenetics in vivo.
Fig. 9.
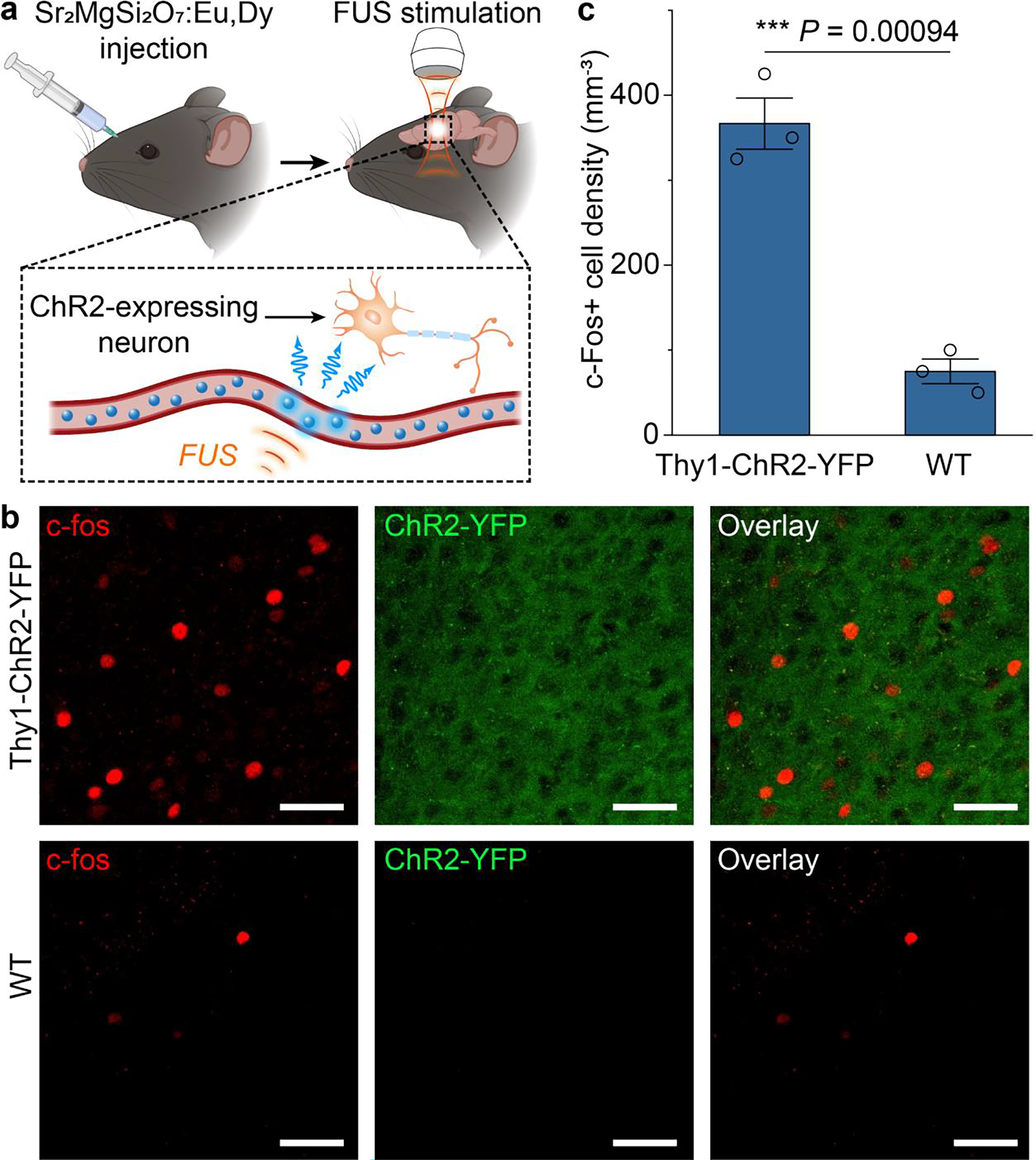
In vivo ultrasound-mediated optogenetic stimulation with circulating colloids. (a) Schematics showing the experimental procedure of ultrasound-mediated optogenetic stimulation with ML colloids composed of . (b) Representative confocal images of c-fos (left), ChR2-YFP (middle) and the overlay (right) of the same motor cortex region in Thy1-ChR2-YFP (top) and WT (bottom) mice after ML colloids composed of were injected, recharged during circulation, and discharged by FUS to emit light. All scale bars represent 40 μm. (c) Statistical analysis of the density of c-fos+ cells for the Thy1-ChR2-YFP and WT mice in b. All data are presented as mean ± SD. per group. One way analysis of variance (ANOVA), *** P < 0.0001.
Conclusions
In summary, we leveraged a bioinspired strategy (“the suppressed dissolution approach”) to synthesize a palette of ML fluids with distinct colors of emission under FUS. These ML fluids exhibit similar spectral characteristics as their bulk, solid counterparts and represent the example of a stable aqueous solution with mechanoluminescence. Unlike conventional mechanoluminescence produced by bulk stress in a solid, ML fluids demonstrate the feasibility of generating light emission from a different origin of hydrodynamic pressure. Importantly, intense light is emitted from ML fluids under hydrodynamic pressures well below the cavitation threshold, thus representing a new mechanism with lower power requirements for light generation in liquids under acoustic waves (the other mechanism being sonoluminescence). We characterized the rechargeability, FUS power dependence, and temporal kinetics of these ML fluids in an artificial circulatory system. Furthermore, we demonstrated that these ML fluids can be delivered into the mouse body via the endogenous circulatory system to produce light emission in different organs, thus enabling minimally-invasive optogenetic stimulation of the mouse brain via tissue-penetrant FUS. The ML fluids produced in this work and the method to synthesize them open up a new avenue for understanding the interaction between mechanical waves and liquids. Moreover, the straightforward implementation of the suppressed dissolution approach highlights a general mechanism, originally found in biominerals and heretofore unexploited with ML materials, for producing stable colloids of potentially any solid-state ceramic materials. Furthermore, ML fluids enable a minimally invasive approach to deliver photon energy via the endogenous circulatory system for in vivo applications that require a light source beyond the typical tissue penetration depth of visible photons. Lastly, we envision that ML liquids may act as an energy storage medium to implement an optical flow battery and as a working medium in a microfluidic photonic circuit.
Supplementary Material
Acknowledgement
Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-2026822. Confocal imaging was performed at the Stanford Wu Tsai Neuroscience Microscopy Service. G.H. acknowledges three awards by NIH (5R00AG056636-04, 1R34NS127103-01, and R01NS126076-01), a National Science Foundation (NSF) CAREER award (2045120), an NSF EAGER award (2217582), a Rita Allen Foundation Scholars Award, a Beckman Technology Development Grant, a grant from the focused ultrasound (FUS) Foundation, a gift from the Spinal Muscular Atrophy (SMA) Foundation, a gift from the Pinetops Foundation, two seed grants from the Wu Tsai Neurosciences Institute, and two seed grants from the Bio-X Initiative of Stanford University. X.W. acknowledges the support by a Stanford Graduate Fellowship. Z.O. acknowledges the support by a Wu Tsai Neurosciences Institute Interdisciplinary Scholar Award. Some schematics were created with BioRender.com.
Footnotes
Supporting Information
Materials and methods, supplementary figures and supplementary references.
References
- (1).Hong G Seeing the Sound. Science 2020, 369 (6504), 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ghanem MA; Basu A; Behrou R; Boechler N; Boydston AJ; Craig SL; Lin Y; Lynde BE; Nelson A; Shen H; Storti DW The Role of Polymer Mechanochemistry in Responsive Materials and Additive Manufacturing. Nature Reviews Materials 2020, 6 (1), 84–98. [Google Scholar]
- (3).Zhang J-C; Wang X; Marriott G; Xu C-N Trap-Controlled Mechanoluminescent Materials. Prog. Mater Sci. 2019, 103, 678–742. [Google Scholar]
- (4).Zhang Y; Wang Z; Kouznetsova TB; Sha Y; Xu E; Shannahan L; Fermen-Coker M; Lin Y; Tang C; Craig SL Distal Conformational Locks on Ferrocene Mechanophores Guide Reaction Pathways for Increased Mechanochemical Reactivity. Nat. Chem. 2021, 13 (1), 56–62. [DOI] [PubMed] [Google Scholar]
- (5).Feng A; Smet APF A Review of Mechanoluminescence in Inorganic Solids: Compounds, Mechanisms, Models and Applications. Materials 2018, 11 (4), 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Qian X; Cai Z; Su M; Li F; Fang W; Li Y; Zhou X; Li Q; Feng X; Li W; Hu X; Wang X; Pan C; Song Y Printable Skin-driven Mechanoluminescence Devices via Nanodoped Matrix Modification. Adv. Mater. 2018, 30 (25), 1800291. [DOI] [PubMed] [Google Scholar]
- (7).Yu K; Niu X; Krook-Magnuson E; He B Intrinsic Functional Neuron-Type Selectivity of Transcranial Focused Ultrasound Neuromodulation. Nat. Commun. 2021, 12 (1), 2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhang Y; Zhang X; Wang H; Tian Y; Pan H; Zhang L; Wang F; Chang J Remote Regulation of Optogenetic Proteins by a Magneto-luminescence Microdevice. Adv. Funct. Mater. 2021, 31 (4), 2006357. [Google Scholar]
- (9).Chen B; Zhang X; Wang F Expanding the Toolbox of Inorganic Mechanoluminescence Materials. Acc. Mater. Res. 2021, 2 (5), 364–373. [Google Scholar]
- (10).King LV On the Acoustic Radiation Pressure on Spheres. Proceedings of the Royal Society of London. Series A - Mathematical and Physical Sciences 1934, 147 (861), 212–240. [Google Scholar]
- (11).Flannigan DJ; Suslick KS Inertially Confined Plasma in an Imploding Bubble. Nat. Phys. 2010, 6 (8), 598–601. [Google Scholar]
- (12).Cai W; Nix WD Imperfections in Crystalline Solids; Cambridge University Press, 2016. [Google Scholar]
- (13).Smet PF; Van den Eeckhout K; De Clercq OQ; Poelman D Persistent Phosphors. Including Actinides. 2015, 48, 1–108. [Google Scholar]
- (14).Li C; Qi L Bioinspired Fabrication of 3D Ordered Macroporous Single Crystals of Calcite from a Transient Amorphous Phase. Angew. Chem. Int. Ed Engl. 2008, 47 (13), 2388–2393. [DOI] [PubMed] [Google Scholar]
- (15).Chen R; Canales A; Anikeeva P Neural Recording and Modulation Technologies. Nature Reviews Materials. 2017, 2 (2), 16093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Baars J; Brandt G Structural Phase Transitions in ZnS. J. Phys. Chem. Solids 1973, 34 (5), 905–909. [Google Scholar]
- (17).Suryanarayana C Mechanical Alloying and Milling. Prog. Mater Sci. 2001, 46 (1), 1–184. [Google Scholar]
- (18).Tu D; Xu C-N; Fujio Y; Kamimura S; Sakata Y; Ueno N Phosphorescence Quenching by Mechanical Stimulus in CaZnOS:Cu. Applied Physics Letters. 2014, 105 (1), 011908. [Google Scholar]
- (19).Zhang P; Zheng Z; Wu L; Kong Y; Zhang Y; Xu J Self-Reduction-Related Defects, Long Afterglow, and Mechanoluminescence in Centrosymmetric Li2ZnGeO4:Mn2+. Inorg. Chem. 2021, 60 (23), 18432–18441. [DOI] [PubMed] [Google Scholar]
- (20).Li C; Imai Y; Adachi Y; Yamada H; Nishikubo K; Xu C-N One-Step Synthesis of Luminescent Nanoparticles of Complex Oxide, Strontium Aluminate. J. Am. Ceram. Soc. 2007, 90 (7), 2273–2275. [Google Scholar]
- (21).Sohn K-S; Park DH; Kim JS Luminescence of Pulsed-Laser-Deposited SrAl2O4 : Eu, Dy Thin Film and Its Role as a Stress Indicator. J. Electrochem. Soc. 2005, 152 (10), H161. [Google Scholar]
- (22).Wang L; Nancollas GH Calcium Orthophosphates: Crystallization and Dissolution. Chem. Rev. 2008, 108 (11), 4628–4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Athanasiadou D; Jiang W; Goldbaum D; Saleem A; Basu K; Pacella MS; Böhm CF; Chromik RR; Hincke MT; Rodríguez-Navarro AB; Vali H; Wolf SE; Gray JJ; Bui KH; McKee MD Nanostructure, Osteopontin, and Mechanical Properties of Calcitic Avian Eggshell. Sci Adv 2018, 4 (3), eaar3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tang R; Wang L; Orme CA; Bonstein T; Bush PJ; Nancollas GH Dissolution at the Nanoscale: Self-Preservation of Biominerals. Angewandte Chemie. 2004, 116, 2751–2755. [DOI] [PubMed] [Google Scholar]
- (25).Wu W; Nancollas GH Determination of Interfacial Tension from Crystallization and Dissolution Data: A Comparison with Other Methods. Adv. Colloid Interface Sci. 1999, 79 (2–3), 229–279. [DOI] [PubMed] [Google Scholar]
- (26).Meldrum FC; Cölfen H Controlling Mineral Morphologies and Structures in Biological and Synthetic Systems. Chem. Rev. 2008, 108 (11), 4332–4432. [DOI] [PubMed] [Google Scholar]
- (27).Tang R; Nancollas GH; Orme CA Mechanism of Dissolution of Sparingly Soluble Electrolytes. J. Am. Chem. Soc. 2001, 123 (23), 5437–5443. [DOI] [PubMed] [Google Scholar]
- (28).Teng HH; Dove PM; Orme CA; De Yoreo JJ. Thermodynamics of Calcite Growth: Baseline for Understanding Biomineral Formation. Science 1998, 282 (5389), 724–727. [DOI] [PubMed] [Google Scholar]
- (29).Athanasiadou D; Carneiro KMM. DNA Nanostructures as Templates for Biomineralization. Nature Reviews Chemistry. 2021, 5 (2), 93–108. [DOI] [PubMed] [Google Scholar]
- (30).Ye X; Jones MR; Frechette LB; Chen Q; Powers AS; Ercius P; Dunn G; Rotskoff GM; Nguyen SC; Adiga VP; Zettl A; Rabani E; Geissler PL; Alivisatos AP Single-Particle Mapping of Nonequilibrium Nanocrystal Transformations. Science 2016, 354 (6314), 874–877. [DOI] [PubMed] [Google Scholar]
- (31).Liu X; Ramirez S; Pang PT; Puryear CB; Govindarajan A; Deisseroth K; Tonegawa S Optogenetic Stimulation of a Hippocampal Engram Activates Fear Memory Recall. Nature 2012, 484 (7394), 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chandra VK; Chandra BP; Jha P Strong Luminescence Induced by Elastic Deformation of Piezoelectric Crystals. Appl. Phys. Lett. 2013, 102 (24), 241105. [Google Scholar]
- (33).Petit RR; Michels SE; Feng A; Smet PF Adding Memory to Pressure-Sensitive Phosphors. Light Sci Appl 2019, 8, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Stride E; Coussios C Nucleation, Mapping and Control of Cavitation for Drug Delivery. Nature Reviews Physics 2019, 1 (8), 495–509. [Google Scholar]
- (35).Boyden ES; Zhang F; Bamberg E; Nagel G; Deisseroth K Millisecond-Timescale, Genetically Targeted Optical Control of Neural Activity. Nat. Neurosci. 2005, 8 (9), 1263–1268. [DOI] [PubMed] [Google Scholar]
- (36).Winter PW; Shroff H Faster Fluorescence Microscopy: Advances in High Speed Biological Imaging. Curr. Opin. Chem. Biol. 2014, 20, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Fenno L; Yizhar O; Deisseroth K The Development and Application of Optogenetics. Annu. Rev. Neurosci. 2011, 34, 389–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Dana H; Sun Y; Mohar B; Hulse BK; Kerlin AM; Hasseman JP; Tsegaye G; Tsang A; Wong A; Patel R; Macklin JJ; Chen Y; Konnerth A; Jayaraman V; Looger LL; Schreiter ER; Svoboda K; Kim DS High-Performance Calcium Sensors for Imaging Activity in Neuronal Populations and Microcompartments. Nat. Methods 2019, 16 (7), 649–657. [DOI] [PubMed] [Google Scholar]
- (39).Stujenske JM; Spellman T; Gordon JA Modeling the Spatiotemporal Dynamics of Light and Heat Propagation for In Vivo Optogenetics. Cell Rep. 2015, 12 (3), 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Salatino JW; Ludwig KA; Kozai TDY; Purcell EK Glial Responses to Implanted Electrodes in the Brain. Nat Biomed Eng 2017, 1 (11), 862–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sheng M; Greenberg ME The Regulation and Function of c-Fos and Other Immediate Early Genes in the Nervous System. Neuron 1990, 4 (4), 477–485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.