

Abstract
Asthma exacerbations significantly impact millions of patients worldwide to pose large disease burdens on affected patients, families, and health care systems. Although numerous environmental factors cause asthma exacerbations, viral respiratory infections are the principal triggers. Advances into the pathophysiology of asthma have elucidated dysregulated protective immune responses and upregulated inflammation that create susceptibility and risks for exacerbation. Biologics for treatment of severe asthma reduce rates of exacerbations and also identify specific pathways of inflammation that contribute to altered pathophysiology, novel therapeutic targets and informative biomarkers. Major steps to prevent exacerbations include the identification of molecular pathways whose blockage will prevent asthma attacks safely, predictably, and effectively.
Keywords: Asthma exacerbations, airway inflammation, T2 inflammation, rhinovirus infections, biologics
The Morbidity and Health Care Burden of Asthma Exacerbations (see Glossary)
Asthma is the most common, chronic respiratory disease in the world with an estimated prevalence of 320 million patients [1,2]. Asthma is also a heterogeneous disease with multiple phenotypes that reflect variability in disease severity, responsiveness to treatment, and susceptibility for adverse risks, including exacerbations. Exacerbations (see Glossary), or asthma attacks, are major contributors to burdens of asthma, and, although they may occur in any patient with asthma, are more frequent and profound in those with severe disease (see Glossary).
Asthma exacerbations are characterized by a worsening of symptoms that dramatically increase breathlessness, difficulty breathing, and compromise lung function. When severe, asthma attacks are life threatening and may result in respiratory failure and death. Asthma attacks can occur suddenly, but usually begin with a gradual increase in symptoms 3 to 5 days after an upper respiratory tract infection (URI). Given their burden and associated health care risks, understanding the mechanisms of exacerbations is an essential step to prevent the consequences and improve asthma care.
Despite significant advancements in understanding the mechanisms of exacerbations and the advent of effective and safe biologics now available, current guideline-directed care of severe disease has experienced limitations to prevent these attacks. Furthermore, repetitive exacerbations create a vicious cycle by promoting progressive loss of lung function, creating, in turn, a greater susceptibility of exacerbations [3]. Therefore, asthma exacerbations remain the Achilles heel in asthma management for many patients. To overcome these shortcomings and provide protection not afforded for the mythological Achilles, a greater understanding of the molecular bases of these events is needed to establish more effective treatment (see Highlights).
Highlights.
Asthma affects 10% of the world’s population but is a heterogeneous disease in terms of clinical manifestations and underlying molecular control of airway inflammation. In most asthma patients, a type (T)-2 inflammatory pattern of inflammation drives clinical characteristics, susceptibility to exacerbations, and forms the target for therapy. Asthma causes major health care burdens for many patients and exacerbations are principal contributors to these disease burdens.
The majority of asthma exacerbations are provoked by viral respiratory infections, with the common cold virus, rhinovirus, the major infectious cause. Rhinoviruses infect the airway epithelium to activate inflammation, increase an eosinophilic response, and cause greater airflow obstruction and symptoms.
Although inhaled corticosteroids are the primary treatment in asthma, they do not always prevent exacerbations in patients, particularly, with severe disease. The development of monoclonal biologics directed towards components of T2 inflammation have begun to revolutionize treatment of severe asthma and more effectively prevent exacerbations in asthma and reduce the need for systemic corticosteroids.
Pathophysiology of asthma
Asthma is a heterogeneous clinical syndrome characterized by wheezing, shortness of breath, variable airflow limitation, and airway hyperresponsiveness, which includes a susceptibility for exacerbations [4]. Airway inflammation is central to the pathophysiology of asthma, clinical characteristics, susceptibility for risks, and target of treatment. Airway inflammation in asthma is complex, heterogeneous, multicellular, and interactive, and collectively leads to different patterns of inflammation: eosinophilic, neutrophilic, mixed, and paucigranulocytic and clinical patterns of disease [5]. The resulting inflammation in asthma is caused by the dominant cytokines expressed and categorized as type (T) 2-high (see Glossary) or non-T2 inflammation (see Glossary) (Figure 1). When a respiratory virus infects airway epithelium in susceptible asthma patients, there is an activation and enhancement of T2 inflammation to compromise airway structure and produce greater airflow obstruction. Furthermore, current treatments do not predictably prevent further activation of existing T2 inflammation during an exacerbation.
Figure 1.
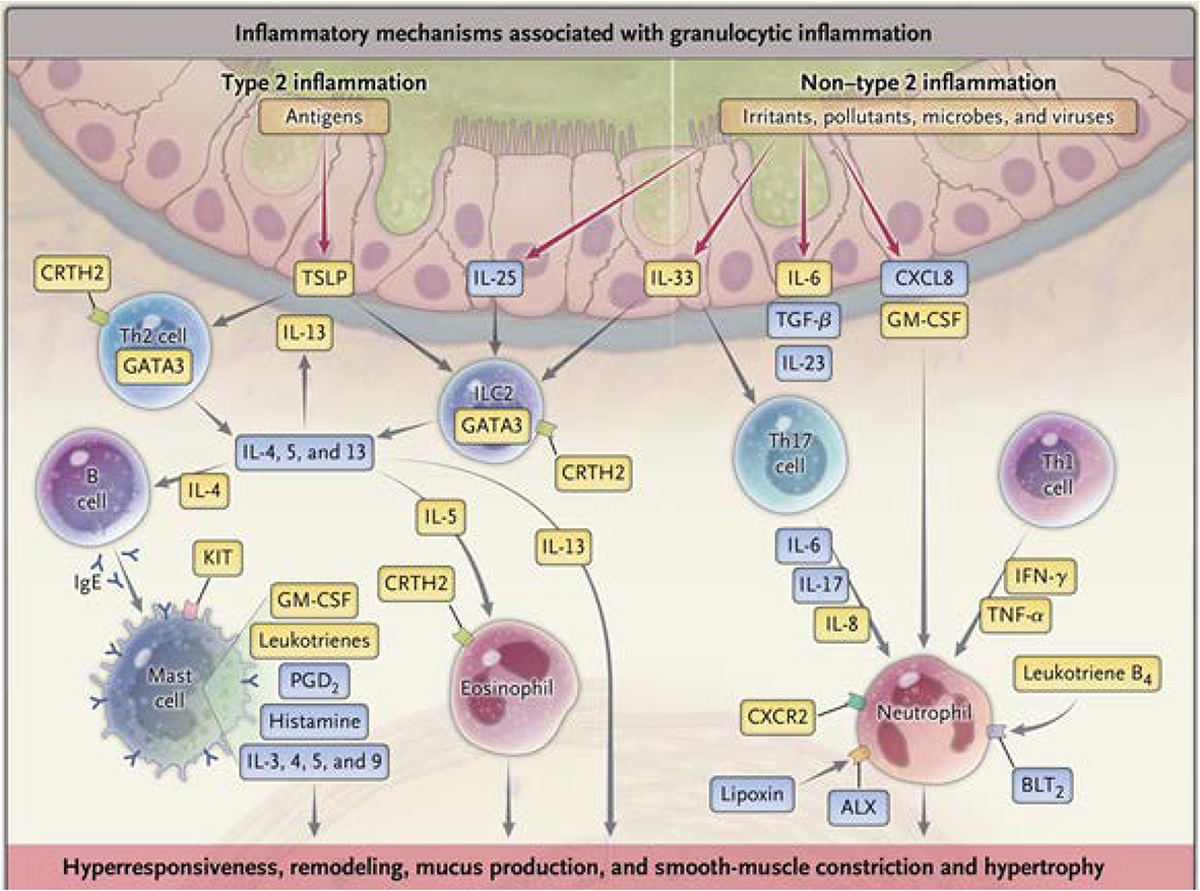
The inflammatory mechanisms that contribute to the pathophysiology of severe asthma are heterogeneous. ALX, lipoxin A4 receptor; BLT2, leukotriene B4 receptor 2; CRTH2, chemoattractant receptor-homologous molecule expressed on Th2; CXCL8, CXC ligand 8; CXCR2. CXC chemokine receptor 2; GATA3, GATA binding protein 3; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, Interferon; IL. interleukin; Ig. immunoglobulin; KIT, tyrosine kinase receptor; PGD2, prostaglandin D2; TGF-β, transforming growth factor β; Th2, Type 2 helper; TNF-α, tumor necrosis factor α; TSLP, thymic stromal lymphopoietin [89]. (Reprinted with permission).
T2-high inflammation is seen in the majority of patients with asthma and is associated with an over expression of interleukin (IL)-4, IL-5, and IL-13 and eosinophilic inflammation. Th2 and innate lymphoid cells (ILC) 2 cells are the primary sources of IL-5 and IL-13 with Th2 cells contributing IL-4. Activation of mast cells through IgE releases bronchoconstricting mediators along with IL-4, IL-5, and IL-13. IL-5 produces eosinophils whose lipid mediators, and granule-associated proteins stimulate smooth muscle contraction, damage airway epithelium, and express cytokines, including IL-4, IL-5, GM-CSF and TGF-β. Collectively, activation of inflammation contributes to airway injury and remodeling, fibrosis, and mucus secretion through activation of the gene, MUC5AC [5].
Airway epithelium is the interface between the environment and the lung and can be activated by respiratory viruses and aeroallergens to generate a family of innate mediators, alarmins, including IL-33, IL-25, and thymic stromal lymphopoietin (TSLP). These cytokines, in turn, activate innate responses from ILC2 to further promote T2 inflammation through generation of IL-5 and IL-13 [6–8]. IL-33 augments T2 inflammation in asthma exacerbations [9]. T2-low inflammation is less well characterized and includes Th17 cells to generate IL-17, which is increased in severe asthma and associated with neutrophilic inflammation [5].
Asthma is characterized by an imbalance between protective and injurious factors in the airway. With a respiratory infection provoking an exacerbation, there is a further shift in the balance towards greater airway injury and airflow obstruction (Figure 2). Respiratory viral infections that trigger exacerbations begin in the upper airway. Within 2 to 3 days, the effects of a respiratory virus infects the lower airway to cause a rapid accentuation of existing inflammation and tip the existing imbalance towards an exacerbation. Although anti-viral factors, principally interferons, are generated with the infection, there may be an existing deficiency of interferon-associated anti-viral activity making asthma patients more susceptible to the adverse events of an infection. Within 4 to 7 days, interferons are generated and may serve to eliminate the viral infection. Despite this anti-viral activity, the increase in inflammation has already occurred and caused an exacerbation. Collectively, the progression to an exacerbation is an interaction between environmental triggers (e.g., viral infection) and host susceptibility reflecting the abnormal gene-by-environment processes.
Figure 2.
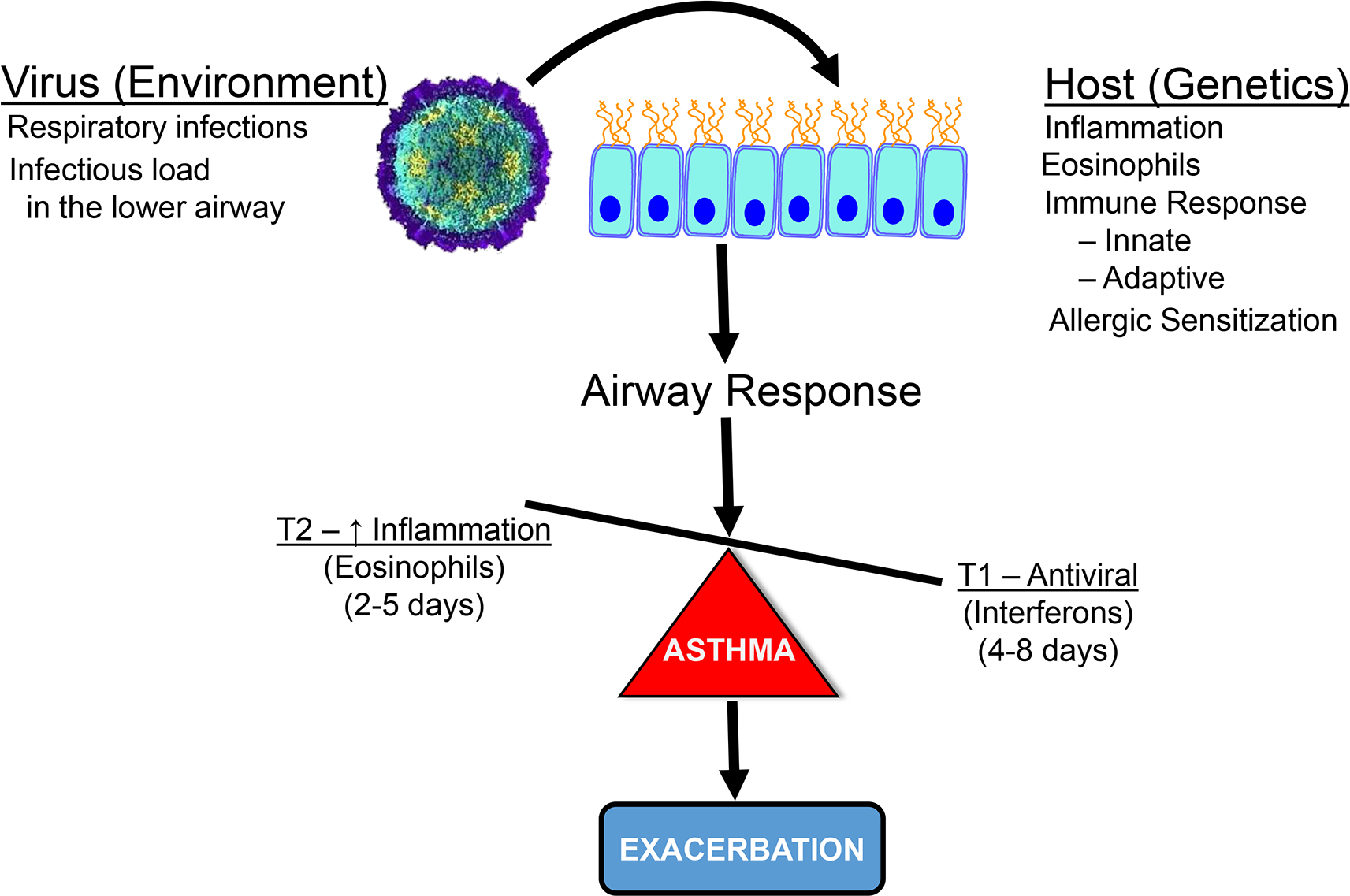
Factors in the development of an exacerbation involve interactions with an environmental agent, respiratory virus, and host (genetics) factors to promote airway inflammation and lead to an exacerbation. In the susceptible asthma patient, a respiratory virus infects airway epithelium to activate an inflammatory response, which is characterized by eosinophils. This occurs early in the infection. This shifts the imbalance in asthma between inflammation to protection towards greater inflammation and a progression to an exacerbation. There is a later onset counterbalance with the production of antiviral interferons, which may gradually control the viral infection.
How are asthma exacerbations provoked?
Numerous environmental factors participate in exacerbations: microbes, allergens, and pollutants. Although the mechanisms associated with responses to these provocations are likely distinct, the clinical outcomes are similar: airflow obstruction and increased symptoms of asthma[10].
Respiratory viruses
Respiratory viral infections are the triggers most commonly associated with exacerbations. Although any viral respiratory infection can provoke an exacerbation, rhinoviruses (RV) are the most common cause [11,12] and account for about 80% of exacerbations in children and 50% in adults.[13] Respiratory syncytial virus [14], human parainfluenza virus [15], and influenza [16] are also linked to exacerbations but their contributions are less frequent.
RV are members of the picornavirus family and cause the “common cold.” Although RVs were felt to infect only the upper airway, they infect the lower airways in asthma and precipitate a T2 eosinophilic inflammatory response [11,17]. One hundred and sixty strains of RV exist and fall into three species (A, B, and C) [12]. RV-A and -C are the predominant species provoking asthma exacerbations. RV-A binds to ICAM-1 and, in adolescents and adults, is most likely to cause exacerbations [18,19]. RV-C is newly described and binds to cadherin-related protein-3 (CDHR3), a corresponding gene linked to risks for childhood asthma [20]. In young children, RV-C infections are more frequent causes of asthma exacerbations, and can be severe, requiring intensive care [21–23].
PCR testing has dramatically increased the sensitivity to detect respiratory viruses and also solidified the importance of RV infections for 80% of exacerbations in children [24]. Not all RV infections lead to exacerbation, nor does every asthma patient wheeze with a RV infection. The mechanisms determining variability and susceptibility between patients and viral infections have yet to be fully identified. There are also differences with RV infections in asthma compared to healthy subjects. Experimental RV16 infections cause more respiratory symptoms and an acute decline in lung function in asthma vs. healthy controls [9]. Furthermore, provocation of pulmonary function changes with RV are greater in the presence of uncontrolled asthma [25].
RV have a seasonal pattern of infections and their relationship to asthma exacerbations with a greater frequency of both events in the spring and fall. These seasonal associations are related to children returning to school or co-existing allergy symptoms, and referred to as the “September Epidemic” of asthma, with highly predictable rates of hospitalization for asthma exacerbations in both children and adults [24,26].
Major concerns arose that COVID-19 infections would be a potent asthma exacerbation trigger. Consequently, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) pandemic raised alarms for asthmatic patients. This has not borne out. Angiotensin-converting enzyme-2 (ACE2) is the major receptor for SARS-CoV-2 via its structural spike glycoprotein. ACE2 expression is decreased in the presence of allergic sensitization and T2 cytokines possibly explaining an apparent protection and molecular regulation of virus receptors to explain a decrease in infection susceptibility in asthma [27]. Multiple studies have not demonstrated an increased risk of severe COVID-19 disease or asthma exacerbations in children and adults [28–35]. However, more severe COVID-19 outcomes have been reported in adults who had evidence of recent uncontrolled disease [36].
Respiratory bacteria
The role and contribution of respiratory bacteria to asthma exacerbations are more complex. Airway colonization with S. pneumonia, H. influenza, and M. catarrhalis colonization increases the risk for wheezing and a later development of asthma [37]. However, the scope of bacteria as direct causes of wheezing has shifted to include consequences of interactions between respiratory viruses and the airway microbiome. For example, patients with T2-high asthma had a lower bacterial burden and increased responsiveness to corticosteroids [38,39]. Moreover, when RV was found in the presence of either S. pneumonia or M. catarrhalis, the likelihood of experiencing asthma symptoms increased [40]. These data suggest that RV contributes to a pathogenic role of some airway bacteria and leads to a loss of asthma control. The molecular mechanisms underscoring virus-bacteria in relationships to exacerbation risks are a focus of current investigation.
Allergic sensitization
Allergic sensitization (see Glossary) is common in asthma, especially in children [10]. Allergic sensitization also appears to precede RV-induced wheezing in children suggesting early life allergic sensitization is a risk factor for wheezing with RV to occur and the later development of asthma.[41] Allergic sensitization also increases susceptibility for exacerbations in the presence of respiratory viruses [42]. In a study of children presenting to an emergency room in Costa Rica with acute wheezing, the presence of RV and IgE sensitization to house dust mite was evaluated to determine their relationships to wheezing. Although allergy to house dust mite by itself was a risk for wheezing, the presence of a RV infection plus allergic sensitization significantly increased probabilities for wheezing. Thus, allergic sensitization influences whether a co-existing RV infection provokes wheezing and vice-versa. [42]
Defective anti-viral activity in asthma
Multiple studies have found diminished airway epithelial cell interferon (β and γ) generation when cultured with RV in asthma. [43, 44] In cultured dendritic cells from asthma patients with RV, interferon-α generation was diminished and related to the presence of cell-bound IgE [45]. An apparent acquired deficiency of interferon antiviral activity in patients with asthma may lead to respiratory infections that are more severe, persistent, and likely to trigger exacerbations [10].
Both T2-high and T2-low asthma patients have deficient induction of type I and III interferon following RV infection, with the level of interferon (IFN) production related to the severity of the existing infection [46]. Type I interferon inhibits ILC2 function; reduced interferon generation may explain increased T2 inflammation during exacerbations from activation of non-regulated ILC2 cells [8]. Additionally, it has been demonstrated that high T2 inflammation and low type 1 interferon gene expression in nasal samples at baseline predict short-term exacerbation risks with a viral respiratory infection [47].
Pollution.
According to the WHO, 90% of people breathe polluted air and more than 80% of people living in urban areas are exposed to air pollutant levels that exceed WHO guideline limits. Traffic related air pollution, nitric dioxide, and secondhand smoke exposure have all been linked to development of asthma in children. Exposure to pollutants can trigger oxidative stress in the airway and provoke downstream inflammation and remodeling even in the absence of respiratory infections [47].
Host risk factors for exacerbations
Genetic risk factors
The relationship between genetic variants and asthma exacerbations is of interest and importance. Single nucleotide polymorphisms (SNPs) in IL-13 and IL-4Ralpha were associated with risks for asthma exacerbations in adults and children [48,49]. Genome-wide association studies (GWAS) found several loci associated with severe asthma in children including CDHR3, which is highly expressed in airway epithelium and the receptor for RV-C [20,50]. Several loci have been identified to affect risks of exacerbation in children and are modified by environmental exposure, including TGFB1 and IL-9, CH1T1A, ADAM-33, and CD14 and LY96, and CRTAM with modification from dust mite exposure [51,52], household mold exposure [53], tobacco smoke exposure [54], endotoxin exposure [55], and Vitamin D levels [56] respectively. While intriguing, these studies yield a small risk effect and fail to identify risks at an individual level [57]. A meta-analysis of GWAS with over 23, 948 patients with asthma and 118,538 controls found 18 novel loci associated with asthma but only explained ~3.5% of variance in asthma [58]. Polygenic risk studies, which combine thousands of polymorphisms into a single score, have demonstrated association with increased asthma risk and are of emerging importance [59].
Characteristics of asthma vary over the span of a patient’s lifetime lending to the significance of environmental influences on disease expression. Thus, epigenetic and transcriptomic (see Glossary) studies may provide further insight to reflect the environmental influences on gene expression [58]. Epigenetic gene regulation occurs through DNA methylation and histone modification and is expressed in the RNA transcript, called the transcriptome. In a RNA sequencing study of nasal samples from 190 adults (66 with asthma and 124 controls), 90 genes were found to have a differential expression in patients with asthma with high accuracy (AUC=0.99) [60].
Children with RV-induced wheezing have reduced CDHR3 expression in peripheral blood leukocytes [61], a gene previously identified in GWAS. When nasal epithelial cells from children with and without asthma were cultured with RV, RV altered DNA methylation and gene expression, including genes involved in natural killer-cell activation (BAT3 and MICB) and Th2-mediated airway inflammation (NEU1) [62]. These studies have the potential to identify new biomarkers for asthma and uncover previously unknown risk factors [57]. Additionally, gene expression data predicted responses to 12 weeks of anti-IL-13 therapy for asthma. CCL23, SIGLEC8, PTGDR2, CACNG6, IDO1, and HSD3B7 expression correlated with peripheral eosinophil count and forced expiratory volume in one second (FEV1) improvement, but were not superior to the peripheral eosinophil counts or periostin levels as disease biomarkers [63].
History of exacerbations
One of the most predictable risks for future exacerbations is the history of previous exacerbations [69]. The underlying mechanisms are not established for this relationship but may relate to a post-infection persistence of increased airway inflammation, altered antiviral activity, or a greater progressive loss of lung function in high-risk patients.
Characteristics of asthma exacerbation.
The use of biomarkers to identify exacerbation risk.
Biomarkers for asthma exacerbations are of potential benefit to identify risks for these events and patient selection for biologics. The biomarkers for T2 inflammation in asthma include eosinophils, FeNO, and total IgE. Blood eosinophils directly correlate with risks for exacerbation [64]. Fraction of exhaled nitric oxide (FeNO) is also a biomarker of T2 inflammation and exacerbation risks [65]. In a post-hoc analysis of LIBERTY ASTHMA QUEST (NCT02414852), higher baseline FeNO levels correlated with increased risks of severe asthma exacerbation, but greater in combination with eosinophilia or a history of exacerbations [66]. Total IgE variability is also associated with risk of asthma exacerbation but less predictable than either eosinophils or FeNO [67].
Inflammatory mechanisms of RV-provoked asthma exacerbations.
RV infections increase eosinophilic airway inflammation.
When RV infects the lower airways of asthma patients, there is increased airway inflammation, airway hyperresponsiveness and airflow obstruction to result in an exacerbation [17]. Central to an asthma exacerbation is the ability of infections to promote eosinophilic inflammation [70,71]. In 28 asthma subjects and 11 controls experimentally infected with RV16, lower airway symptoms were greater and associated with a significant fall in FEV1 in subjects with asthma. [9] Lower airway samples revealed significant increases in eosinophils that paralleled increases in nasal IL-4, IL-5, and IL-13, suggesting a link to lower airway T2 responses. There was also a significant generation of the epithelial alarmin IL-33, which correlated with increases in IL-5, lower airway symptoms, higher airway viral loads, and the presence of eosinophils. These patterns of RV-associated responses, including increased in IL-5 and IL-13 generation, point to an activation of ILC2 by IL-33 to generate IL-5 and direct eosinophil recruitment. An amplification of the T2 inflammatory signal also occurs as RV activates airway neutrophils to produce NETosis, the development of dsDNA nets, and activation of Th2 cells to produce a panel of T2 cytokines, including IL-5 (Figure 3) [72].
Figure 3.
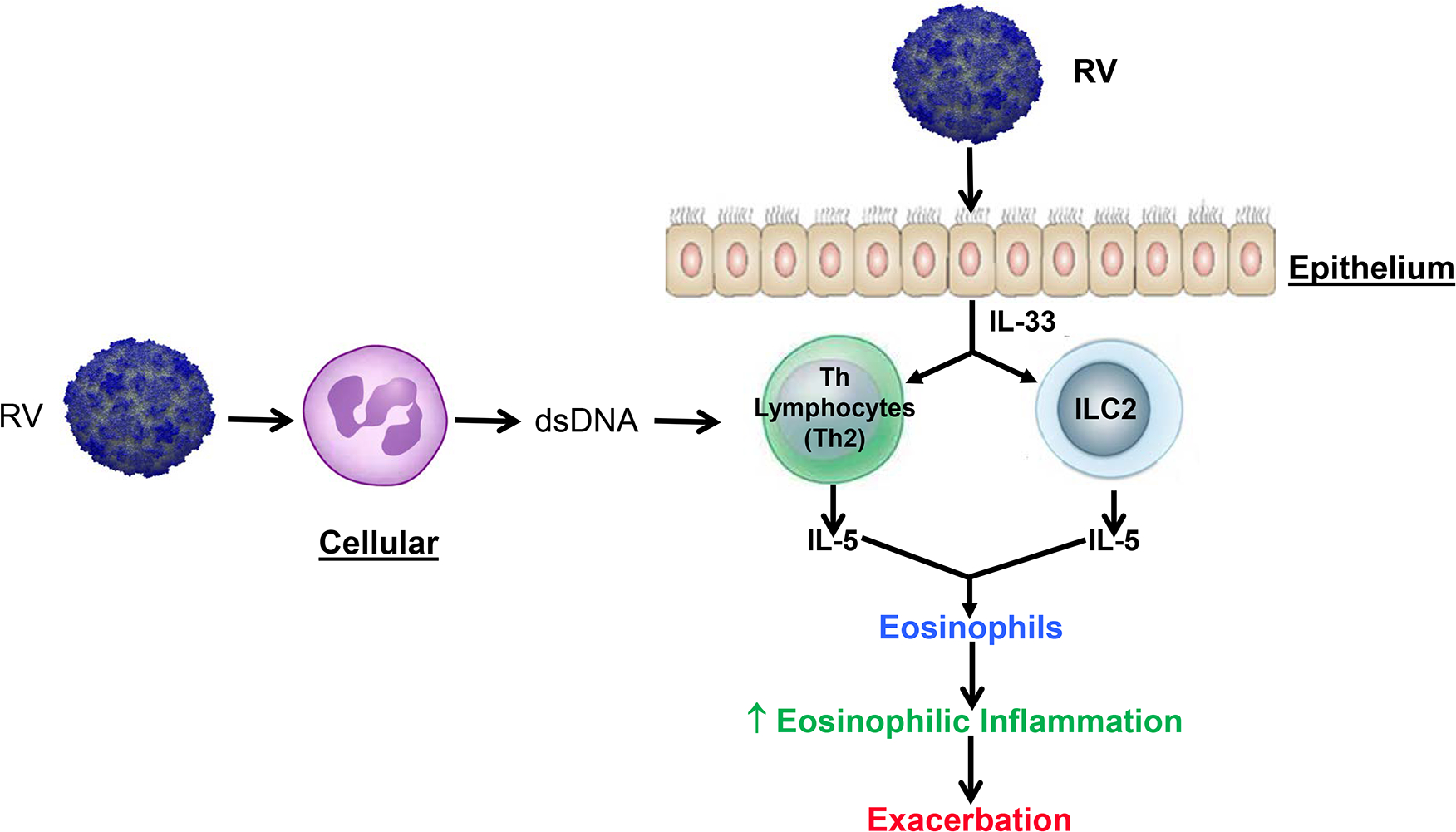
Rhinovirus promotion of eosinophilic inflammation involves epithelial cell activation to generate IL-33 and neutrophil NETosis to stimulate Th2 lymphocyte generation of IL-5 and other T2 cytokines [9, 72].
To further dissect the complexity and regulation of eosinophilic inflammation in RV-provoked asthma exacerbations, investigators performed kinetic analyses of the appearance of ILC1 and ILC2 cells in relationship to a RV16-induced asthma exacerbation [72]. In both asthma and control subjects, ILC2 increased with the RV infection, but to a greater degree in asthma. ILC1 cells contribute to host defense by generating antiviral interferons. In asthma, there was a delay in the airway increase of ILC1 cells to the RV16 inoculation suggesting a diminished or delayed antiviral response. These data suggest that changes in the reciprocal relationship, or balance, between ILC1 (protection):ILC2 (inflammation) in asthma with the consequences of diminished antiviral activity and enhanced eosinophil-associated inflammation; it is this imbalance that then leads to an exacerbation [74].
Transcriptomic analyses detect molecular pathways associated with exacerbation.
Transcriptomic analyses provide methods to explore the patterns of genetic pathway activation and to identify genes that ultimately translate to an exacerbation. Nasal cells were used as surrogates for the lung to identify molecular networks that participate in virus-induced asthma exacerbations [75]. Sixteen children with asthma [75] were assessed at baseline and during an asthma exacerbation. These inaugural studies found an “intricate, modular inflammatory program consisting of more than 1000 upregulated genes during an asthma exacerbation.” Of particular interest was the detection of interferon regulatory factor 7 as a potential genetic pathway associated with an asthma exacerbation.
To further identify the transcriptomic mechanisms of inflammation that determine whether a respiratory infection causes an exacerbation, 208 children, 6 to 17 years of age with moderate-to-severe asthma and a history of frequent exacerbations, were recruited for study [47]. Following determinations of baseline clinical characteristics and associated transcriptomic patterns of inflammation, the children were re-evaluated when they developed symptoms of a respiratory tract infection. From these analyses, it was possible to determine whether an exacerbation would occur and, if so, what were the inflammatory pathways activated. The kinetics and patterns of the inflammatory modules’ expression identified the molecular mechanisms and their kinetics that led to an exacerbation with a cold.
When an exacerbation occurred, there was an early increase in expression of SMAD3 modules, which represents genes that have a central role in airway inflammation and remodeling and are surrogates for TGF-β activity. The second, and later, phase associated with the development of an exacerbation was an increase in gene modules related to eosinophil activation and mucus hypersecretion (Figure 4a and 4b). These findings suggest a two-step process when an infection progresses to an exacerbation (Figure 5). Progression to an exacerbation and the identified gene modules was more likely when a respiratory virus was present (66.2% of the exacerbations were associated with RV-A and RV-C). Additional analyses evaluated whether there was a predictive relationship of T2 inflammation expression and the type 1 interferon modules expression. When there was an increase in genes associated with T2 inflammation vs. interferon, the probability of an exacerbation was significantly greater.
Figure 4a and 4b.
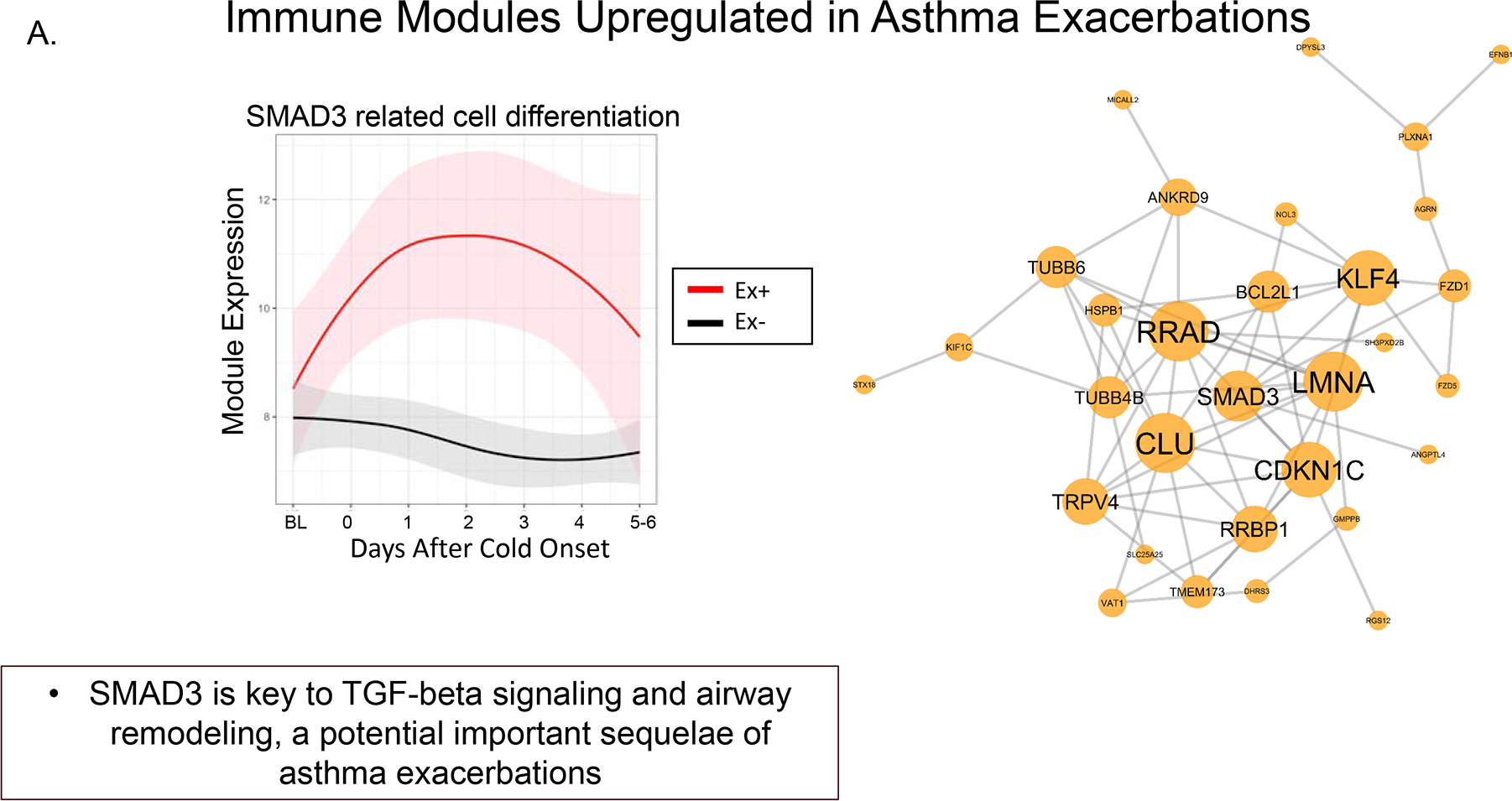
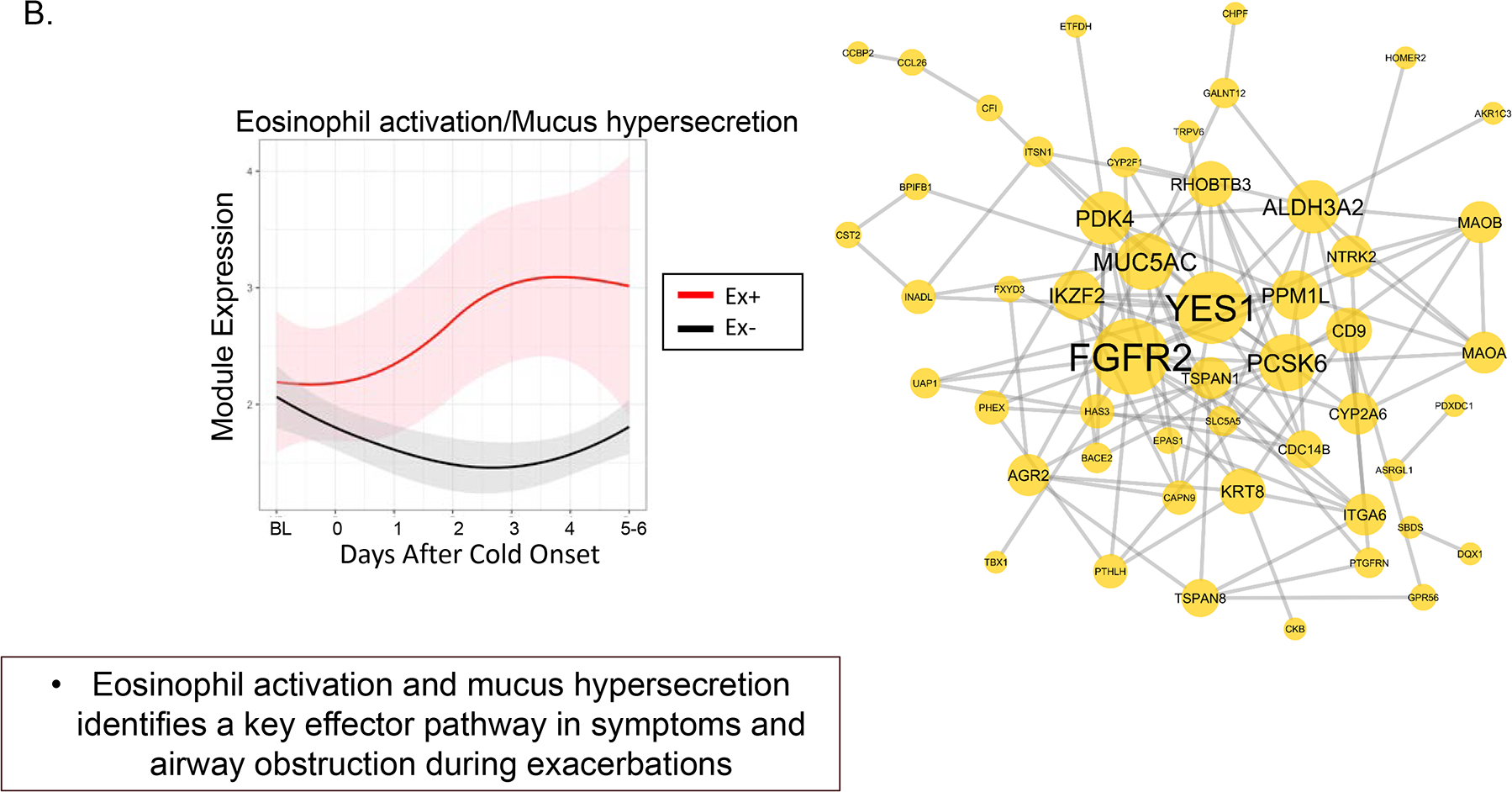
Longitudinal dynamics of inflammatory module expression and differential patterns of sequential module activation. 4a. Early inflammatory module activation includes SMAD3 to initiate inflammation and exacerbation. 4b. A second phase of inflammatory module activation includes genes associated with eosinophil activation and mucus hypersecretion, MUC5AC. The second phase leads to progression and persistent inflammation and airflow obstruction [47]. Reprinted with permission.
Figure 5.
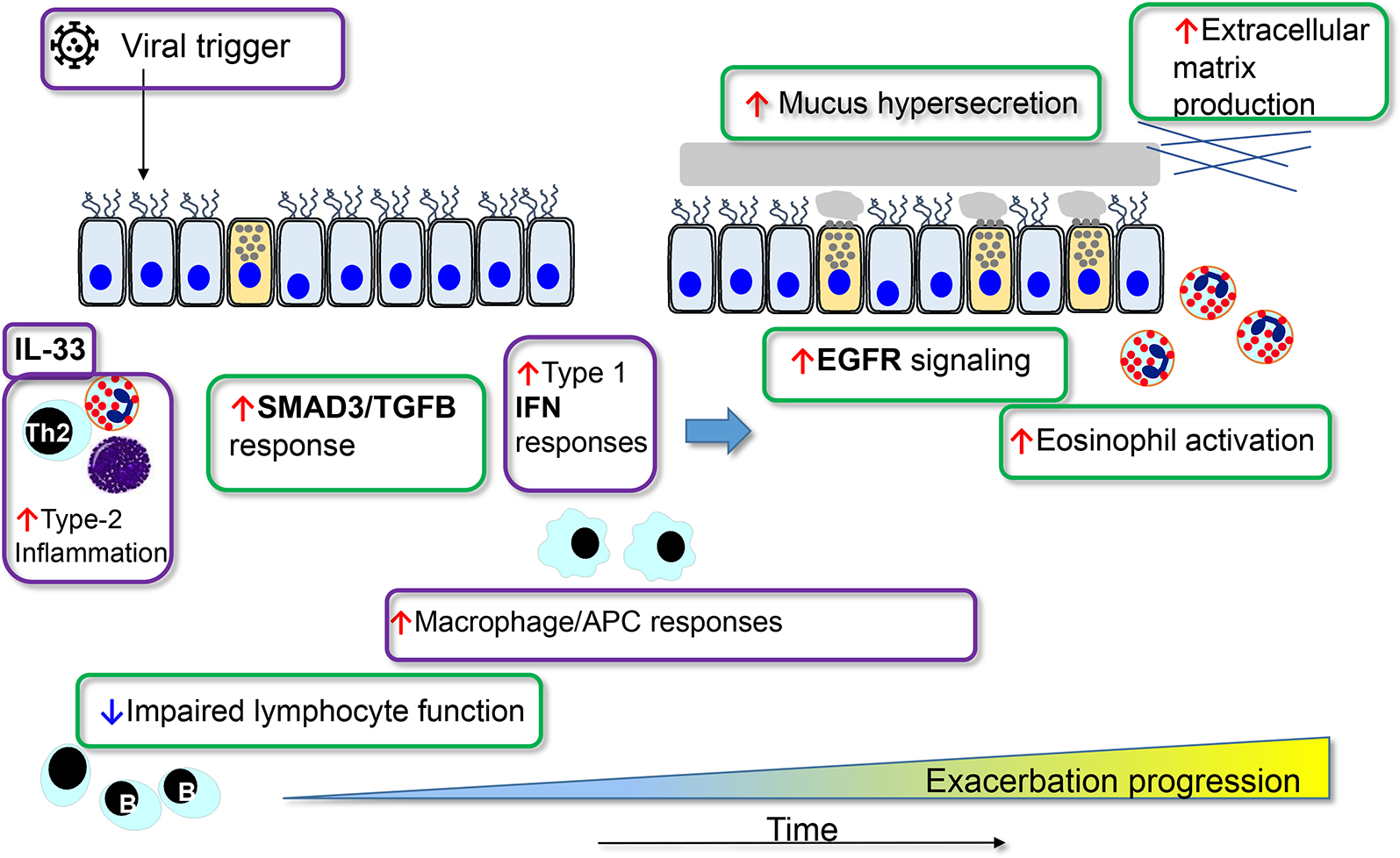
Longitudinal summary of respiratory virus activation of inflammatory immune modules leading to an exacerbation. An early response in activation of SMAD3 modules. This is followed in a few days by activation of T2 modules to generate eosinophilic inflammation and mucus hypersecretion (MUC5AC) [47].
These transcriptomic observations provide insight into key molecular pathways that determine whether a cold will provoke an exacerbation. There is an initial or early activation of epithelial cell inflammation (SMAD3) followed by an upregulation of eosinophil-associated and mucus hypersecretion pathways, the latter perhaps representing a more persistent and unresponsive inflammation. Although not cited, these observations also suggest that there is an imbalance in the expression of inflammation vs. antiviral activity in asthma. Therefore, risks for an asthma exacerbation include a loss of balance between inflammation and immune protection, thus allowing a respiratory virus a greater chance to provoke an exacerbation (Figures 2 and 5).
Asthma Therapy for Exacerbations
Limitations of current treatment approaches to prevent asthma exacerbations
Guidelines for asthma management recommend increased bronchodilator use and the initiation of systemic corticosteroids for acute exacerbations [76]. In the step-care management of asthma, increasing doses of combination ICS/LABA are recommended for both maintenance and rescue. This approach has proven effective in reducing the rates of exacerbation [77]. Corticosteroids are central to effective asthma management and relate to their regulation of airway inflammation. In severe asthma, which includes increased risks for exacerbations, step-care management has not proven sufficient to achieve disease control or prevent exacerbations. To overcome these treatment shortcomings, an expanding availability of biologics provides effective treatment for severe asthma and has become a standard of care [78].
Biologics (see Glossary) prevent asthma exacerbations
Six monoclonal antibodies that target components of T2 inflammation, omalizumab, mepolizumab, reslizumab, benralizumab, dupilumab, and tezepelumab, are available for patient care (Table 1). These biologics are designed, at present, for treatment of severe asthma, which is uncontrolled asthma on high dose ICS/LABA, a history of frequent exacerbations, reduced lung function, and a T2 biomarker pattern of inflammation.
| Agent | ↓ Exacerbations | ↑ Lung Function | OCS ↓ | Special Feature |
|---|---|---|---|---|
| Omalizumab (IgE) | 25% | ± | — | >6 years of age, IgE |
| Mepolizumab (IL-5) | 50% | + | ++ | Greatest experience of anti-IL5s |
| Reslizumab (IL-5) | 50% | ++ | — | Weight — base dose (IV) |
| Benralizumab (IL-5R) | 50% | ++ | ++ | q 8 wks and IL-5R |
| Dupilumab (IL-4/IL-13) | 50% | ++ | ++ | Eosinophils, FeNO |
| Tezepelumab (anti-TSLA) | 50% | ++ | — | No biomarker necessary |
The biologic therapies for asthma are designed to regulate components of inflammation in severe disease, to improve disease control and reduce risks for exacerbations [78]. Not only have biologics transformed care of severe asthma and reduced the susceptibility for exacerbations, but the inflammatory sites targeted by these agents serve as “knockout models” and have provided insight into the pathophysiology of asthma exacerbations.
Anti-IgE (omalizumab).
Omalizumab is a monoclonal antibody directed against IgE to diminish its presence and function on immune cells. The Inner-City Ant-IgE Therapy for Asthma (ICATA) study (NCT00377572)I [79], a phase IV randomized clinical trial, evaluated omalizumab over 52 weeks in 419 inner-city subjects, ages 6 to 21 years, with persistent allergic asthma. Compared to placebo (standard-of-care), omalizumab significantly reduced symptoms (p <0.001) and exacerbations by 18.5% (p<0.001). Although not a prespecified outcome, the effects of omalizumab were evaluated on the seasonal patterns of exacerbations. The effects of omalizumab were seen almost exclusively on the prevention of spring and fall exacerbations (September-November), to parallel patterns of “September epidemics of asthma” [24,26].
These clinical observations indicated that IgE and allergic sensitization are significant contributors to exacerbations. In the ICATA study [79], RV was the most frequent virus detected in exacerbations (between 67%–74% of exacerbations). Omalizumab did not diminish the presence of a respiratory virus. These observations suggested that its protection against exacerbations was not related to eliminating respiratory viruses but rather modifying the generated inflammatory response.
In a subsequent phase IV randomized clinical trial, PROSE (NCT01430403)II [80], pre-seasonal treatment with omalizumab was designed to determine the effect of initiating omalizumab before children (6 to 17 years of age) returned to school in the fall, a prime time for exacerbations and risks for exacerbations, as well as the role of IgE [79]. Five hundred and thirteen children with moderate-to-severe asthma and one or more recent exacerbations in the preceding year were enrolled. Omalizumab reduced exacerbations by 52% (OR=0.48) for all subjects at all treatment levels but 67% (OR=0.73) in patients at step 5 care, i.e., high dose of ICS/LABA. Respiratory viruses were detected in 89% of exacerbations with RV dominant (81%).
To determine if reductions in IgE by omalizumab improved antiviral activity, blood samples were collected prior to randomization and at completion of the trial in 87 subjects. Peripheral blood mononuclear cells (PBMC) were isolated, cultured, cell-bond IgE was cross-linked, RV was added, and supernatants collected to measure interferon-α generation. PBMC from patients who received omalizumab had a significant increase of interferon-α generation to RV. Patients with the greatest interferon-α generation (> median values) had significantly fewer exacerbations [81]. Although these were in vitro assessments, they raise the possibility that reduction of cell-bound IgE by omalizumab restored antiviral generation of interferon-α and reflected a “normalization” of antiviral activity in asthma to prevent exacerbations as well as reducing mast cell generated inflammation (Figure 6). Omalizumab was the first biologic approved in asthma and has been shown to, primarily, prevent exacerbations. Experience with omalizumab over the past 20 years has demonstrated its efficacy and safety. It is the only biologic which is specifically designated for allergic asthma.
Figure 6.
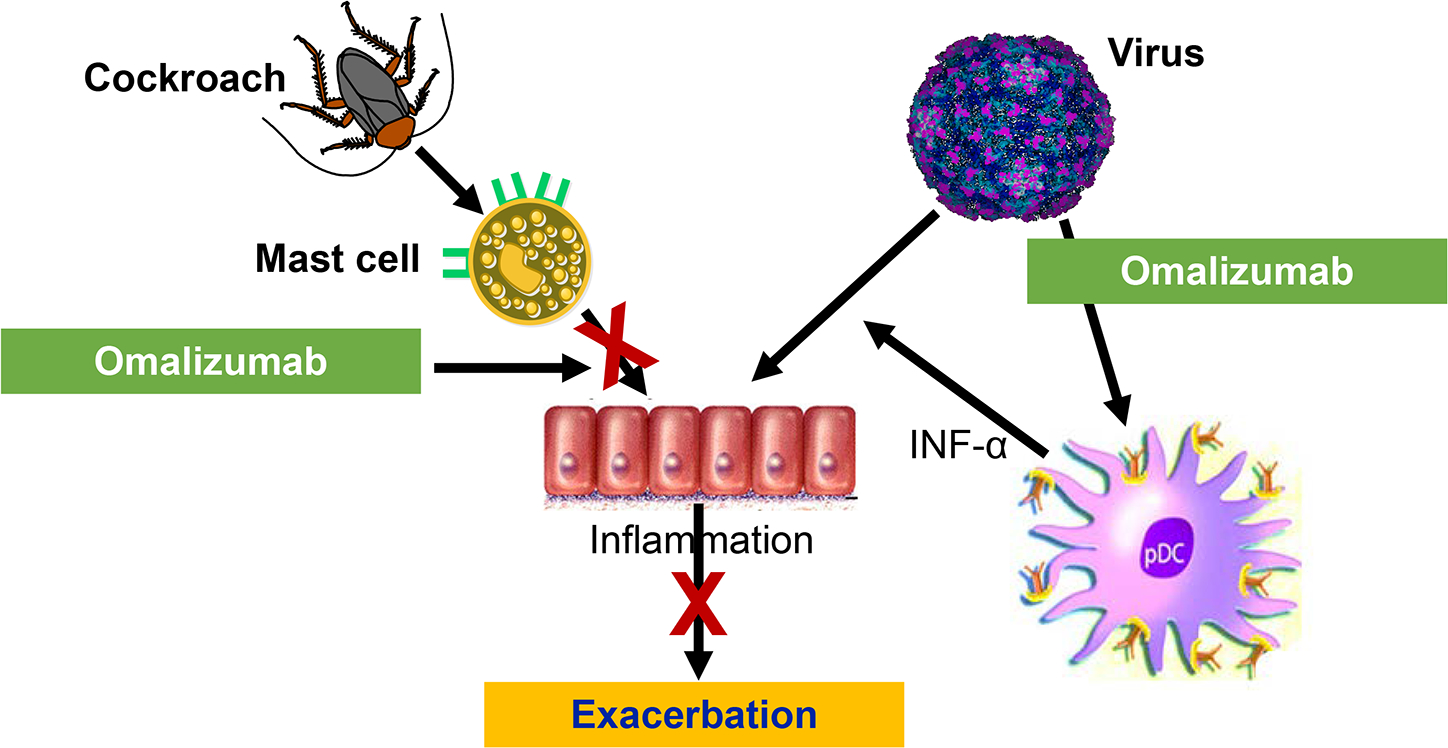
Prevention of asthma exacerbations by omalizumab, anti-IgE. Omalizumab reduces IgE including cell-bound IgE. With reduced cell-bound IgE, mast cell activation is reduced to diminish the development of inflammation. In addition, omalizumab removes IgE from the cell surface of dendritic cells, which allows dendritic cells to produce more antiviral interferon-α when exposed to respiratory viruses.
Anti-IL-5/IL-5R biologics reduce eosinophils.
IL-5 is the pivotal cytokine for bone marrow production of eosinophils. Mepolizumab and reslizumab are humanized antibodies that target IL-5 to diminish eosinophil production. Benralizumab is a monoclonal antibody that targets the IL-5R on eosinophils to cause eosinophil apoptosis. Both strategies significantly diminish peripheral blood eosinophils and, as a group, reduce exacerbations by approximately 50%. [78]
MENSA (NCT01691521) III, Mepolizumab as adjunctive therapy in patients with severe asthma, is a phase III randomized clinical trial that enrolled patients 12 to 82 years of age with moderate-to-severe asthma, uncontrolled disease, and two or more exacerbations in the previous year. Two doses of mepolizumab, either 75 mg intravenously or 100 mg subcutaneously, were given for 32 weeks vs. placebo. A total of 576 subjects underwent randomization. Both mepolizumab dosing strategies reduced peripheral blood eosinophils and exacerbations by 47% (intravenous) and 53% (subcutaneous). Lung functions did not significantly change, but symptoms, as measured by St. George’s Respiratory Questionnaire, improved [82]. Reduction of blood eosinophils is associated with a significant, but not complete, elimination of exacerbations. These data point to the participation of eosinophils as an important component of exacerbations but also suggesting that pathways in addition to eosinophils contribute to exacerbations. However, the fall in eosinophils does not indicate that exacerbations will be prevented. Exacerbation prevention occurs at only 50%, but eosinophil reduction is nearly 100%. This suggests pathways other than IL-5 are involved in exacerbations.
IL-4/IL-13 biologics.
The IL-4/IL-13 pathways contribute broadly to inflammation including IgE production, promoting of eosinophil migration via expression of vascular adhesion molecules, disruption of the airway epithelium, airway remodeling and enhancement of airway smooth muscle contractility [5]. Dupilumab is a monoclonal antibody that targets the IL-4Rα and antagonizes the IL-4/IL-13 pathway. In a phase III randomized clinical trial, QUEST (NCT02414854)IV [83], dupilumab reduced the rate of asthma exacerbations by over 50% and also significantly improved FEV1 values by over 200 ml in patients with moderate-to-severe asthma and underlying T2 inflammation. The improved FEV1 occurred within two weeks of initiating treatment and baseline levels of FeNO fell rapidly. In VOYAGE (NCT02948959)V [84], a phase III randomized clinical trial, dupilumab showed similar reductions in asthma exacerbations and improved lung function in children 6 to 11 years of age. Children of this age are especially at risk for exacerbations and long-term side effects from systemic corticosteroid use [85]. In both children and adults, dupilumab prevents exacerbations in the presence of T2 inflammation, characterized by increases in either FeNO (≥ 20 ppb) or eosinophils (≥300 cells/μL) or both. The mechanisms by which dupilumab reduces exacerbations are not established. The rapid fall in FeNO signals diminished airway inflammation and a possible restoration of epithelial cell function [86], raising the possibility that one of dupilumab’s benefits involves improved epithelial cell function.
In TRAVERSE (NCT02134028)VI, a phase III open-label extension study, [87] it was determined that dupilumab provides a sustained benefit with an improved FEV1 and a progressive reduction of asthma exacerbations. At week 96 of dupilumab treatment, subjects from the Phase 2b randomized clinical trial (NCT01854047) VII and QUEST (NCT02414854) IV showed a year-by-year progressive reduction in exacerbations to eventually being nearly 90% exacerbation-free. These findings suggest that long-term prevention of exacerbations reduces risks for exacerbations, possibly by diminishing airway inflammation as reflected by a “normalization” of FeNO values to make the airways less susceptible to respiratory virus infections. As only 50% of exacerbations were prevented, pathways other than IL-4/IL-13 are involved.
Anti-TSLP.
Anti-TSLP binds thymic stromal lymphopoietin (TSLP), which is an epithelial cell-derived, innate cytokine that is generated in response to environmental triggers, such as respiratory viruses, pollutants, and aeroallergens. The release of TSLP activates ILC2s to potentiate T2 inflammation via production of IL-5 and IL-13. In NAVIGATOR (NCT03347279)VII [88], a phase III randomized control trial, tezepelumab decreased asthma exacerbations by over 50% and improved FEV1. Although outcome benefits from tezepelumab were greater in the presence of T2 inflammation, efficacy was also noted in non-T2 inflammation (eosinophils <150 cells/μL). Tezepelumab also lowered T2-high biomarkers, including IgE, peripheral blood eosinophils, and FeNO. Tezepelumab is the first approved biologic therapy that targets the non-T2 inflammatory pathways (eosinophil counts <150 cells/μL). As with the other inflammatory targeted cytokines, tezepelumab significantly reduced, but did not eliminate, exacerbations.
Positioning of biologics in asthma treatment
Six biologics (Table 1) are approved for treatment of severe asthma. It is estimated that this phenotype affects 10 to 15% of all asthma patients. Current indications for biologics include patients at GINA step-care 4 or 5 (high dose therapy), either not controlled or requiring high dose treatment to maintain control. A common component of severe disease is frequent exacerbations. Other than tezepelumab and omalizumab, patients under consideration for a biologic need to demonstrate a T2-high phenotype, e.g., increased blood eosinophils and/or FeNO (GINA). A major objective in treatment with current biologic is to prevent exacerbations, thus achieving disease control and reducing or eliminating the need for systemic corticosteroids.
As a group, the available biologics for severe asthma reduce exacerbations by approximately 50%. Predicting which severe asthma patient is most likely to benefit from a specific biologic has not been fully established. However, a trial of 3 to 4 months with the selected biologic has proven beneficial to determine efficacy, which is usually reflected by a reduced frequency of exacerbations. Finally, as a group, safety with approved biologics has been good. As a therapeutic group, biologics have transformed asthma treatment and provided reductions in asthma burdens faced by patients. For many, it has established a new lifestyle free of symptoms and risks for exacerbation.
At present, there is no evidence that biologics lead to disease modification or can be discontinued after 3 to 5 years and yet sustain benefit. Nonetheless, for high-risk patients with severe asthma and frequent use of systemic corticosteroids, biologics have provided benefit and potential for clinical remission of asthma with continued use of a biologic. [88]
Concluding remarks
Asthma exacerbations remain a major health care burden for many patients, especially those with severe disease. Significant advances have led to a greater recognition of at-risk patients and an availability for more specific interventions and improved control with current biologics (see Clinician’s corner). Other key advances to improved care will require identification of how the predominant causative microbe, rhinovirus, increases in T2 inflammation and burgeoning characterizations of the molecular pathways that lead to exacerbations. A next pivotal step will be to identify patient endotypes, e.g., molecular pathways that put them at risk for exacerbations and to then use precision-directed interventions to block these processes. Achieving this advance holds the promise that exacerbations will be more effectively controlled and no longer contribute to the disease burden that they do today (see Outstanding questions). When these new interventions are developed, it will be akin to Achilles’ mother being able to more fully emerge her infant son into the River Styx to provide more complete protection from any risks, which, in our case, will be asthma exacerbations.
Clinician’s corner.
Exacerbations are major contributors to the health burden of asthma patients. Although any patient with asthma is at risk for an exacerbation, the risk and burden falls primarily on those with severe disease. Asthma guidelines recommend a step-care approach to achieve disease control in asthma; the step-care approach uses increasing doses of inhaled corticosteroids, often in combination with a second controller, e.g., long-acting beta agonists. In severe asthma, this approach does not consistently lead to disease control or prevent exacerbations or the need for systemic corticosteroids.
Viral respiratory infections are the principal trigger of asthma exacerbations. Although any respiratory virus can provoke an exacerbation, the common cold virus, rhinovirus, is the major provoker of asthma attacks. Rhinoviruses infect the lower airway and accentuate existing inflammation with an increase in airway eosinophilia that leads to increased airflow obstruction and symptoms of asthma. Current treatment with inhaled corticosteroids, in combination with LABA, do not consistently prevent exacerbations. New treatment approaches are necessary and now available.
To fill this unmet need, biologics for the treatment of severe asthma have proven efficacious and prevent exacerbations. Biomarkers are helpful to identify patients with T2 inflammation and include increased peripheral blood eosinophils and FeNO. Treatment of severe asthma patients with biologics blocks key inflammatory pathways to prevent viral respiratory infections to further increase inflammation and provoke exacerbations. Regulating the molecular components of inflammation in asthma and during exacerbations has proved to be effective to mitigate risks for exacerbations and reduce the burdens of asthma.
Outstanding questions.
What are the specific molecular pathways that respiratory viruses activate to increase inflammation and cause an exacerbation?
What are the genetic factors, both of the patient and the virus, that lead to an increased susceptibility for asthma exacerbations? Are these factors different in children vs. adults?
What are the molecular pathways that are activated in an exacerbation to cause a rapid loss of lung function? If these pathways are identified early in the course of asthma, can long-term consequences and complications of exacerbations be prevented?
Are there biomarkers with greater specificity, predictability and mechanistic insight than blood eosinophils and FeNO to predict risks for exacerbations and identify the pathogenic pathways of an exacerbation?
Can biologics be developed to effectively prevent all exacerbations or used at the time of an exacerbation?
Resources
-
https://clinicaltrials.gov/ct2/show/NCT00377572?term=00377572&draw=2&rank=1
-
https://clinicaltrials.gov/ct2/show/NCT01430403?term=01430403&draw=2&rank=1
-
https://clinicaltrials.gov/ct2/show/NCT01691521?term=01691521&draw=2&rank=1
-
https://clinicaltrials.gov/ct2/show/NCT02414854?term=02414854&draw=2&rank=1
-
https://clinicaltrials.gov/ct2/show/NCT02948959?term=02948959&draw=2&rank=1
-
https://clinicaltrials.gov/ct2/show/NCT02134028?term=02134028&draw=2&rank=1
-
https://clinicaltrials.gov/ct2/show/NCT01854047?term=01854047&draw=2&rank=1
-
https://clinicaltrials.gov/ct2/show/NCT03347279?term=03347279&draw=2&rank=1
Glossary
- Allergic sensitization
refers to the production and expression of IgE antibodies to environmental triggers such as pollens, house dust mite or animal dander. IgE attaches to mast cells, which are activated with exposure to sensitizing allergens
- Asthma exacerbation
episodes of increased severity of asthma with difficulty breathing and decline in pulmonary function that can result in respiratory failure and death when severe
- Biologics
monoclonal antibodies that target a pathway implicated in disease pathogenesis
- Non-T2 inflammation
less well-characterized immune response that is associated with Th17 cells, IL-17 production, and the presence of neutrophilic inflammation
- Severe asthma
asthma characterized by persistent and uncontrolled symptoms, frequent exacerbations, and decreased pulmonary functions despite optimal therapy
- T2 high inflammation
associated with overexpression of IL-4, IL-5, IL-13, and eosinophils; seen in the majority of asthma patients. Th2 and ILC2 cell activation are the primary source of these cytokines
- Transcriptome
mRNA transcripts expressed following immune activation and identify the genes and gene modules that are activated and lead to either upregulation of inflammation or modulation of the inflammatory response that regulates these processes and responses
References
- 1.Vos T et al. (2020) Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396, 1204–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pate CA et al. (2021) Asthma Surveillance — United States, 2006–2018. MMWR Surveill. Summ. 70, 1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denlinger LC et al. (2017) Inflammatory and Comorbid Features of Patients with Severe Asthma and Frequent Exacerbations. Am. J. Respir. Crit. Care Med. 195, 302–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore WC et al. (2010) Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am. J. Respir. Crit. Care Med. 181, 315–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pelaia C et al. (2020) Molecular Targets for Biological Therapies of Severe Asthma. Front. Immunol. 11, 603312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar RK et al. (2014) Respiratory viral infection, epithelial cytokines, and innate lymphoid cells in asthma exacerbations. J. Leukoc. Biol. 96, 391–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karta MR et al. (2016) Insights into Group 2 Innate Lymphoid Cells in Human Airway Disease. Curr. Allergy Asthma Rep. 16, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duerr CU and Fritz JH (2016) Regulation of group 2 innate lymphoid cells. Cytokine 87, 1–8 [DOI] [PubMed] [Google Scholar]
- 9.Jackson DJ et al. (2014) IL-33–Dependent Type 2 Inflammation during Rhinovirus-induced Asthma Exacerbations In Vivo. Am. J. Respir. Crit. Care Med. 190, 1373–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castillo JR et al. (2017) Asthma Exacerbations: Pathogenesis, Prevention, and Treatment. J. Allergy Clin. Immunol. Pract. 5, 918–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altman MC et al. (2020) Evolving concepts in how viruses impact asthma: A Work Group Report of the Microbes in Allergy Committee of the American Academy of Allergy, Asthma & Immunology. J. Allergy Clin. Immunol. 145, 1332–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makris S and Johnston S (2018) Recent advances in understanding rhinovirus immunity. F1000Res. 7, F1000 Faculty Rev-1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortega H et al. (2021) Rhinovirus and asthma: Challenges and opportunities. Rev. Med. Virol. 31, e2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim CK et al. (2009) Clinical and Epidemiological Comparison of Human Metapneumovirus and Respiratory Syncytial Virus in Seoul, Korea, 2003–2008. J. Korean Med. Sci. 25, 342–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuse H et al. (2005) Naturally Occurring Parainfluenza Virus 3 Infection in Adults Induces Mild Exacerbation of Asthma Associated with Increased Sputum Concentrations of Cysteinyl Leukotrienes. Int. Arch. Allergy Immunol. 138, 267–272 [DOI] [PubMed] [Google Scholar]
- 16.Kim C-K et al. (2018) Viral Infections and Associated Factors That Promote Acute Exacerbations of Asthma. Allergy Asthma Immunol. Res. 10, 12–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Message SD et al. (2008) Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proceedings of the National Academy of Sciences 105, 13562–13567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papi A and Johnston SL (1999) Rhinovirus Infection Induces Expression of Its Own Receptor Intercellular Adhesion Molecule 1 (ICAM-1) via Increased NF-κB-mediated Transcription*. J. Biol. Chem. 274, 9707–9720 [DOI] [PubMed] [Google Scholar]
- 19.Nicholson KG et al. (1993) Respiratory viruses and exacerbations of asthma in adults. Br. Med. J. 307, 982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bønnelykke K et al. (2014) A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat. Genet. 46, 51–55 [DOI] [PubMed] [Google Scholar]
- 21.Bashir H et al. (2018) Association of rhinovirus species with common cold and asthma symptoms and bacterial pathogens. J. Allergy Clin. Immunol. 141, 822–824.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwane MK et al. (2011) Human Rhinovirus Species Associated With Hospitalizations for Acute Respiratory Illness in Young US Children. J. Infect. Dis. 204, 1702–1710 [DOI] [PubMed] [Google Scholar]
- 23.Minor TE et al. (1974) Viruses as Precipitants of Asthmatic Attacks in Children. JAMA 227, 292–298 [PubMed] [Google Scholar]
- 24.Johnston SL et al. (1995) Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ 310, 1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson DJ et al. (2015) The influence of asthma control on the severity of virus-induced asthma exacerbations. J. Allergy Clin. Immunol. 136, 497–500.e3 [DOI] [PubMed] [Google Scholar]
- 26.Johnston NW et al. (2006) The September epidemic of asthma hospitalization: School children as disease vectors. J. Allergy Clin. Immunol. 117, 557–562 [DOI] [PubMed] [Google Scholar]
- 27.Jackson DJ et al. (2020) Association of respiratory allergy, asthma, and expression of the SARS-CoV-2 receptor ACE2. J. Allergy Clin. Immunol. 146, 203–206.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruano FJ et al. (2020) Impact of the COVID-19 pandemic in children with allergic asthma. J. Allergy Clin. Immunol. Pract. 8, 3172–3174.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beken B et al. (2021) Asthma and allergic diseases are not risk factors for hospitalization in children with coronavirus disease 2019. Ann. Allergy Asthma Immunol. 126, 569–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terry PD et al. (2021) Asthma in Adult Patients with COVID-19. Prevalence and Risk of Severe Disease. Am. J. Respir. Crit. Care Med. 203, 893–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao L et al. (2021) Asthma in patients with suspected and diagnosed coronavirus disease 2019. Ann. Allergy Asthma Immunol. 126, 535–541.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi L et al. (2021) Asthma in patients with coronavirus disease 2019. Ann. Allergy Asthma Immunol. 126, 524–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grandbastien M et al. (2020) SARS-CoV-2 Pneumonia in Hospitalized Asthmatic Patients Did Not Induce Severe Exacerbation. J. Allergy Clin. Immunol. Pract. 8, 2600–2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho KS et al. (2021) The relationship between asthma, eosinophilia, and outcomes in coronavirus disease 2019 infection. Ann. Allergy Asthma Immunol. 127, 42–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sunjaya AP et al. (2021) Asthma and risk of infection, hospitalization, ICU admission and mortality from COVID-19: Systematic review and meta-analysis. J. Asthma 59, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang BZ et al. (2021) Asthma Disease Status, COPD, and COVID-19 Severity in a Large Multiethnic Population. J. Allergy Clin. Immunol. Pract. 9, 3621–3628.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bisgaard H et al. (2007) Childhood Asthma after Bacterial Colonization of the Airway in Neonates. N. Engl. J. Med. 357, 1487–1495 [DOI] [PubMed] [Google Scholar]
- 38.Durack J et al. (2020) Distinct associations of sputum and oral microbiota with atopic, immunologic, and clinical features in mild asthma. J. Allergy Clin. Immunol. 146, 1016–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Durack J et al. (2017) Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J. Allergy Clin. Immunol. 140, 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kloepfer KM et al. (2014) Detection of pathogenic bacteria during rhinovirus infection is associated with increased respiratory symptoms and asthma exacerbations. J. Allergy Clin. Immunol. 133, 1301–7, 1307.e1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jackson DJ et al. (2012) Evidence for a Causal Relationship between Allergic Sensitization and Rhinovirus Wheezing in Early Life. Am. J. Respir. Crit. Care Med. 185, 281–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soto-Quiros M et al. (2012) High titers of IgE antibody to dust mite allergen and risk for wheezing among asthmatic children infected with rhinovirus. J. Allergy Clin. Immunol. 129, 1499–1505.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wark PAB et al. (2005) Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 201, 937–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gill MA et al. (2010) Counterregulation between the FcεRI Pathway and Antiviral Responses in Human Plasmacytoid Dendritic Cells. The Journal of Immunology 184, 5999–6006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rich HE et al. (2020) Insights Into Type I and III Interferons in Asthma and Exacerbations. Front. Immunol. 11, 574027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altman MC et al. (2019) Transcriptome networks identify mechanisms of viral and nonviral asthma exacerbations in children. Nat. Immunol. 20, 637–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tiotiu AI et al. (2020) Impact of Air Pollution on Asthma Outcomes. Int. J. Environ. Res. Public Health 17, 6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wenzel SE et al. (2007) IL4Rα Mutations Are Associated with Asthma Exacerbations and Mast Cell/IgE Expression. Am. J. Respir. Crit. Care Med. 175, 570–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hunninghake GM et al. (2007) Polymorphisms in IL13, total IgE, eosinophilia, and asthma exacerbations in childhood. J. Allergy Clin. Immunol. 120, 84–90 [DOI] [PubMed] [Google Scholar]
- 50.Bochkov YA et al. (2015) Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proceedings of the National Academy of Sciences 112, 5485–5490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharma S et al. (2009) Variants in TGFB1, Dust Mite Exposure, and Disease Severity in Children with Asthma. Am. J. Respir. Crit. Care Med. 179, 356–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sordillo JE et al. (2015) Genome-wide expression profiles identify potential targets for gene-environment interactions in asthma severity. J. Allergy Clin. Immunol. 136, 885–892.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu AC et al. (2010) Fungal Exposure Modulates the Effect of Polymorphisms of Chitinases on Emergency Department Visits and Hospitalizations. Am. J. Respir. Crit. Care Med. 182, 884–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bukvic BK et al. (2013) Asthma severity, polymorphisms in 20p13 and their interaction with tobacco smoke exposure. Pediatr. Allergy Immunol. 24, 10–18 [DOI] [PubMed] [Google Scholar]
- 55.Kljaic-Bukvic B et al. (2014) Genetic variants in endotoxin signalling pathway, domestic endotoxin exposure and asthma exacerbations. Pediatr. Allergy Immunol. 25, 552–557 [DOI] [PubMed] [Google Scholar]
- 56.Du R et al. (2012) Genome-wide association study reveals class I MHC–restricted T cell–associated molecule gene (CRTAM) variants interact with vitamin D levels to affect asthma exacerbations. J. Allergy Clin. Immunol. 129, 368–373.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forno E and Celedón JC (2019) Epigenomics and Transcriptomics in the Prediction and Diagnosis of Childhood Asthma: Are We There Yet? Frontiers in Pediatrics 7, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Demenais F et al. (2018) Multiancestry association study identifies new asthma risk loci that colocalize with immune cell enhancer marks. Nat. Genet. 50, 42–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sordillo JE et al. (2021) A polygenic risk score for asthma in a large racially diverse population. Clinical & Experimental Allergy 51, 1410–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pandey G et al. (2018) A Nasal Brush-based Classifier of Asthma Identified by Machine Learning Analysis of Nasal RNA Sequence Data. Sci. Rep. 8, 8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hammar KS et al. (2018) Reduced CDHR3 expression in children wheezing with rhinovirus. Pediatr. Allergy Immunol. 29, 200–206 [DOI] [PubMed] [Google Scholar]
- 62.Pech M et al. (2018) Rhinovirus infections change DNA methylation and mRNA expression in children with asthma. PLoS One 13, e0205275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choy DF et al. (2016) Peripheral blood gene expression predicts clinical benefit from anti–IL-13 in asthma. J. Allergy Clin. Immunol. 138, 1230–1233.e8 [DOI] [PubMed] [Google Scholar]
- 64.DiMango E et al. (2018) Risk Factors for Asthma Exacerbation and Treatment Failure in Adults and Adolescents with Well-Controlled Asthma during Continuation and Step Down Therapy. Ann. Am. Thorac. Soc. 15, 955–961 [DOI] [PubMed] [Google Scholar]
- 65.Nakagome K and Nagata M (2018) Involvement and Possible Role of Eosinophils in Asthma Exacerbation. Front. Immunol. 9, 2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pijnenburg MW (2019) The Role of FeNO in Predicting Asthma. Frontiers in Pediatrics 7, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Busse WW et al. (2021) Baseline FeNO as a prognostic biomarker for subsequent severe asthma exacerbations in patients with uncontrolled, moderate-to-severe asthma receiving placebo in the LIBERTY ASTHMA QUEST study: a post-hoc analysis. The Lancet Respiratory Medicine 9, 1165–1173 [DOI] [PubMed] [Google Scholar]
- 68.Yuan YL et al. (2021) Total IgE Variability Is Associated with Future Asthma Exacerbations: A 1-Year Prospective Cohort Study. J. Allergy Clin. Immunol. Pract. 9, 2812–2824 [DOI] [PubMed] [Google Scholar]
- 69.Lemanske RF et al. (1989) Rhinovirus upper respiratory infection increases airway hyperreactivity and late asthmatic reactions. J. Clin. Invest. 83, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Calhoun WJ et al. (1994) A common cold virus, rhinovirus 16, potentiates airway inflammation after segmental antigen bronchoprovocation in allergic subjects. J. Clin. Invest. 94, 2200–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Toussaint M et al. (2017) Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat. Med. 23, 681–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dhariwal J et al. (2021) Pulmonary Innate Lymphoid Cell Responses during Rhinovirus-induced Asthma Exacerbations In Vivo : A Clinical Trial. Am. J. Respir. Crit. Care Med. 204, 1259–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Busse WW and Gern JE (2022) Weaving innate lymphoid cells (ILC) into the fabric of asthma exacerbations. J. Allergy Clin. Immunol. 149, 1579–1581 [DOI] [PubMed] [Google Scholar]
- 74.Bosco A et al. (2012) Interferon regulatory factor 7 is a major hub connecting interferon-mediated responses in virus-induced asthma exacerbations in vivo. J. Allergy Clin. Immunol. 129, 88–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.2022 GINA Report, Global Strategy for Asthma Management and Prevention. . Global Initiative for Asthma. [Google Scholar]
- 76.O’Byrne PM et al. (2018) Inhaled Combined Budesonide-Formoterol as Needed in Mild Asthma. N. Engl. J. Med. 378, 1865–1876 [DOI] [PubMed] [Google Scholar]
- 77.Brusselle GG and Koppelman GH (2022) Biologic therapies for severe asthma. N. Engl. J. Med. 386, 157–171 [DOI] [PubMed] [Google Scholar]
- 78.Busse WW et al. (2011) Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N. Engl. J. Med. 364, 1005–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Esquivel A et al. (2017) Effects of Omalizumab on Rhinovirus Infections, Illnesses, and Exacerbations of Asthma. Am. J. Respir. Crit. Care Med. 196, 985–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gill MA et al. (2018) Enhanced plasmacytoid dendritic cell antiviral responses after omalizumab. J. Allergy Clin. Immunol. 141, 1735–1743.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ortega HG et al. (2014) Mepolizumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. 371, 1198–1207 [DOI] [PubMed] [Google Scholar]
- 82.Castro M et al. (2018) Dupilumab Efficacy and Safety in Moderate-to-Severe Uncontrolled Asthma. N. Engl. J. Med. 378, 2486–2496 [DOI] [PubMed] [Google Scholar]
- 83.Bacharier LB et al. (2021) Dupilumab in Children with Uncontrolled Moderate-to-Severe Asthma. N. Engl. J. Med. 385, 2230–2240 [DOI] [PubMed] [Google Scholar]
- 84.Bleecker ER et al. (2020) Systematic Literature Review of Systemic Corticosteroid Use for Asthma Management. Am. J. Respir. Crit. Care Med. 201, 276–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Couillard S et al. (2021) Fractional Exhaled Nitric Oxide Nonsuppression Identifies Corticosteroid-Resistant Type 2 Signaling in Severe Asthma. Am. J. Respir. Crit. Care Med. 204, 731–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wechsler ME et al. (2022) Long-term safety and efficacy of dupilumab in patients with moderate-to-severe asthma (TRAVERSE): an open-label extension study. Lancet Respir Med 10, 11–25 [DOI] [PubMed] [Google Scholar]
- 87.Menzies-Gow A et al. (2021) Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma. N. Engl. J. Med. 384, 1800–1809 [DOI] [PubMed] [Google Scholar]
- 88.Menzies-Gow A et al. (2020) An expert consensus framework for asthma remission as a treatment goal. J. Allergy Clin. Immunol. 145, 757–765 [DOI] [PubMed] [Google Scholar]
- 89.Israel E and Reddel HK (2017) Severe and Difficult-to-Treat Asthma in Adults. N. Engl. J. Med. 377, 965–976 [DOI] [PubMed] [Google Scholar]