

Abstract
Genetic testing was completed on 1294 deafness persons referred to the Molecular Otolaryngology Research Laboratories to establish a diagnosis of DFNB1. Exon 2 of GJB2 was screened for coding sequence allele variants by denaturing high-performance liquid chromatography (DHPLC) complemented by bi-directional sequencing. If two deafness-causing mutations of GJB2 were identified further screening was not performed. If only a single deafness-causing mutation was identified, we screened for the g.1777179_2085947del (hereafter called del(GJB6-D13S1830)) and mutations in the non-coding region of GJB2.
Phenotype-genotype correlations were evaluated by categorizing mutations as either protein truncating or non-truncating. 205 persons carried two GJB2 exon 2 mutations and were diagnosed as having DFNB1; 100 persons carried only a single deafness-causing allele variant of exon 2. Thirty-seven of these persons were c.35delG carriers, and 51 carried other allele variants of GJB2. Persons diagnosed with DFNB1 segregating two truncating/nonsense mutations had a more severe phenotype than persons carrying two missense mutations, with mean hearing impairments being 88% and 37%, respectively (P<0.05). The number of deaf c.35delG carriers was greater than expected when compared to the c.35delG carrier frequency in normal-hearing controls (P<0.05), suggesting the existence of at least one other mutation outside the GJB2 coding region that does not complement GJB2 deafness-causing allele variants.
Keywords: GJB2, c.35delG, del(GJB6-D13S1830), DFNB1 phenotype
Introduction
Congenital deafness affects 0.05 to 0.1% of children in developed countries (Mehl and Thomson, 1998; 2002). Usually there is neither a history of familial hearing loss nor other physical abnormalities, making attempts to identify causality difficult. However over the past few years genetic testing has dramatically changed the clinical evaluation of the deaf child. Following identification of the first recessive deafness locus, DFNB1 (Guilford, et al., 1994; MIM#220290), and cloning of the associated gene, GJB2 (Kelsell, et al., 1997; MIM#121011), researchers showed that mutations in the encoded protein, Connexin 26 (Cx26), account for half of recessive deafness in many different populations (Estivill, et al., 1998; Green, et al., 1999; Kelsell, et al., 1997; Scott, et al., 1998; Zelante, et al., 1997). Cx26 deafness is now recognized as one of the world’s most common genetic conditions, with mutation carrier rates exceeding 4% in some ethnic groups (Morrell, et al., 1998).
Establishing a molecular diagnosis of Cx26 deafness is important clinically since these children can avoid further diagnostic tests and are not at increased risk for medical co-morbidity. Bony abnormalities of the cochlea are not part of the deafness phenotype (Cohn, et al., 1999; Denoyelle, et al., 1999; Green, et al., 2003) and developmental motor milestones and vestibular function are normal. The rare exceptions include a child with bony overgrowth of the cochlea noted at surgery and another with asymmetry of the right modiolus (Kenna, et al., 2001), a child with vertigo, migraine and unilateral weakness (Cohn, et al., 1999), and a child with marked prematurity and maturational vestibular weakness (Denoyelle, et al., 1999).
Cx26 deafness occasionally co-segregates with skin abnormalities like keratitis-ichthyosis-deafness syndrome (KID; MIM#148210) and palmoplantar keratoderma (PPK; MIM#148350). The former is inherited as an autosomal dominant trait, although it is rare to find large pedigrees with several affected persons; in most cases, the affected person presents with a de novo mutation. Of the reported KID-causing GJB2 mutations, D50N is most commonly identified (Richard, et al. 2002). PPK is characterized by hyperkeratosis of the palms and soles, often with peeling (Hohl, 2000). It is rare and has been reported with only four allele variants of GJB2 (p.Arg75Gln, p.Glu42del, p.Gly59Ala, p.Arg75Trp; Heathcote, et al., 2000; Richard, et al., 1998; Rouan, et al., 2001; Uyguner, et al., 2002). Vohwinkel syndrome (VS; MIM#124500), a specific form of PPK and deafness caused by the p.Asp66His mutation (Maestrini, et al., 1999), has the additional component of auto-amputation secondary to band-like circumferential constrictions of the digits. As a general rule, however, persons with Cx26 deafness are unlikely to present with co-morbid conditions, and tests of vision, intelligence, cardiac conductivity and thyroid function are normal (Cohn, et al., 1999; Green, et al., 1999; Fukushima, et al., 2002; Green, et al., 2002).
Genetic screening of GJB2 to establish a diagnosis of DFNB1 has important limitations. First among these limitations is the genetic testing itself (Smith, 2001). Although GBJ2 is a small gene with only a single coding exon, the 90+ different allele variants associated with autosomal recessive non-syndromic deafness (ARNSD) are scattered throughout the gene making mutation screening of the entire coding sequence essential. In spite of this approach, the identification of a single deafness-causing allele variant is not uncommon, implying the presence of a ‘missed’ mutation in a non-coding region or coincidental carrier status in a person with deafness of another etiology.
A second limitation is in predicting the degree of deafness. Among persons with Cx26 deafness, the degree of deafness can vary from mild to profound (mild (<40 dB), 1.7%; moderate (40–55 dB), 10.3%; moderately severe (56–70 dB), 7.8%; severe (71–90 dB), 30.2%; profound (>90 dB), 50.0%) even among persons with the same mutations (Cohn, et al., 1999, Denoyelle, et al., 1999, Green, et al., 1999, Mueller, et al., 1999, Sobe, et al., 2000). Typically, the audiogram has a down-sloping or flat pattern (Green, et al., 2003). Selective mid-frequency loss is rare (Mueller, et al., 1999) and selective low frequency loss has not been described. Symmetry between ears is normal, although one-fourth of individuals have intra-aural differences of up to 20dB (Cohn, et al., 1999; Denoyelle, et al., 1999). The loss tends to be stable, with neither improvement nor fluctuation in hearing level in the majority of cases over the long term (Green, et al., 2003).
The purpose of this paper is to describe our experience with mutation screening of GJB2 in families segregating ARNSD. We report the relative frequency of GBJ2 allele variants in this population, the frequency of non-coding GJB2 mutations associated with deafness at the DFNB1 locus, and the phenotypic-genotypic correlations we have observed. Based on difference between observed and expected numbers of deaf persons carrying a single deafness-causing allele variant of Cx26, we posit the existence of at least one additional mutation associated with the DFNB1 phenotype outside the coding region of GJB2.
Materials and Methods
Patient Recruitment
Persons with congenital deafness were sequentially accrued from hearing loss referrals to the Molecular Otolaryngology Research Laboratories, specifically excluding individuals with syndromic, unilateral, acquired or dominant types of hearing loss. Evaluation included a complete history and physical examination, audiometry, and in 90% of cases, computed temporal bone tomography. The University of Iowa Human Subjects Committee approved all procedures.
GJB2 Mutation Screening
Determination of GJB2-related deafness is dependent on the identification of mutations in the DNA of affected individuals. For this analysis, DNA was extracted from peripheral whole blood (approximately 10 cc) using established procedures (Grimberg, et al. 1989). We completed mutation screening of GJB2 using denaturing high-performance liquid chromatography (DHPLC) complemented by direct sequencing after first verifying the sensitivity and specificity of DHPLC.
To determine the sensitivity and specificity of DHPLC, we screened a GJB2 Mutation Panel and compared single-strand conformational polymorphism (SSCP) analysis and DHPLC with direct sequencing. The GJB2 Mutation Panel was derived from 55 individuals segregating 52 distinct sequence variants and combinations of variants as determined by direct sequencing. Sequencing was completed on an Applied Biosystems (ABI) model 3700 automated sequencer. Sequence data were compared to published sequence for GJB2 using the Sequencer 4.1.2 software program package (Gene Codes).
SSCP analysis was performed as described previously (Green, et al., 1999). For DHPLC, we completed polymerase chain reaction (PCR) amplification of exon 2 of GJB2 using 20ng of genomic DNA in a 50 μl reaction containing 5 μl buffer (160 mM (NH4)2SO4, 670 mM Tris-HCl pH8.8, 0.1% Tween-20), 1.65 μl of 50 mM MgCl2, 0.5 μl of 2.5 mM each dATP, dCTP, dTTP and dGTP, 10 pmol each forward and reverse primer, 5% w/v Betaine (Sigma Cat # B0300) and 0.5 U Taq polymerase, with primers selected to optimize PCR amplification and mutation detection based on analysis of melting domains on Wavemaker 4.1.34 software (Transgenomic, Inc.) (Table 1). Amplification conditions were 95° C for 1 min, followed by three sets of 3 cycles each of 95° C for 10 sec, 62° C (60° C, set 2; 58° C, set 3) for 20 sec, and 72° C for 40 sec, ending with an extension cycle of 72° C for 10 min. Heteroduplex analysis was performed by pooling mutant samples with a sequence-verified wild type sample at a ratio of 2:1, respectively, and denaturing in a thermal cycler for 5 min at 95° C followed by slow cooling to 25° C at a rate of 1° C/minute. DHPLC analysis of each amplicon was performed at three different temperatures using the Wavemaker 4.1.34 software package (Transgenomic, Inc.) to estimate the optimal temperature and bracketing this temperature by +/− 2° C to maximize sensitivity. Sensitivity and specificity of SSCP and DHPLC were calculated by comparing samples showing an abnormal migration (SSCP) or elution profile (DHPLC) with sequencing data.
Table 1.
GJB2 primer pairs tested by DHPLC against the GJB2 Mutation Panel
| Name | Sequence |
|---|---|
| Cx26-F | CGTCTTTTCCAGAGCA AACCGC |
| Cx26-R | CGCCGCCGCGCATCCCTCTCATGCTGTCT |
| Cx26-PGC-F | CCCGCCGCCCCCGCCGCGTCTTTTCCAGAGCAAACCGCCCA |
| Cx26-PGC-R | CCGCGCCCGCCCTGCCGCCCCCGCCCATCCCTCTCATGCTG TCT |
Persons with congenital deafness of unknown etiology were diagnosed as having DFNB1 if mutation screening by DHPLC showed an abnormal elution profile and bi-directional sequencing confirmed the presence of two deafness-causing allele variants of GJB2. In persons with a single deafness-causing allele variant of GJB2, we screened exon 1 for the splice donor mutation and the GJB6-D13S1830 interval for del(GJB6-D13S1830). Exon 1 was screened as described previously (Green, et al., 1999); to screen for del(GJB6-D13S1830), we used primers GJB6-1R and BKR-1 to amplify the break-point of the deletion (del Castillo, et al., 2002) and primers GJB6-1R-del [5’-AGG GGT CGC TGT GGT GGA C-3’] and BKR-1-del[5’-GGT GGT TGT CAG GGG CTG TG-3’] paired with GJB6-1R and BKR-1, respectively, to amplify both ends of the deletion of the wild-type allele. If either of these mutations was found, the affected person was diagnosed with DFNB1. Persons without a second identifiable DFNB1-causing mutation were considered to be deaf and coincidental carriers of a GJB2 deafness-causing allele variant.
Calculation of Residual Hearing and Determining Phenotype-Genotype Correlations
To compare audiograms, audiometric data were converted into a numerical score (percent deafness, %D) using the American Medical Association Deafness Disability Index. To obtain this score for each ear, the pure tone average (PTA) at 0.5, 1, 2, 3 (or 4) kHz was determined, 25dB was subtracted from this number, and the result was multiplied by 1.5%. Overall %D was calculated by weighting the better hearing ear five times the poorer hearing ear, yielding a final score based on hearing in speech frequencies in both ears that is a highly meaningful quantification of functional hearing. This measure de-emphasizes the usually clinically and physiologically unimportant differences between a 100 dB vs. 120 dB hearing loss, but emphasizes the differences between a 65dB and an 80dB hearing loss. For example, a PTA of 65dB is equivalent to %D of 60, a PTA of 78dB is equivalent to a %D of 80, and a PTA of 91dB is equivalent to a %D of 100. For moderate losses, mean threshold levels and the disability index can be readily converted back and forth.
If precise audiometric data were unavailable, the patient’s available audiometric data were interpolated to derive the degree of residual hearing. To evaluate phenotype-genotype correlations, mutations were categorized as either protein truncating (nonsense mutations and deletions and insertions leading to frame shifts) or non-truncating (missense mutations).
Additional Non-Coding GJB2 Mutations
To assess the possibility of additional DFNB1 deafness-causing mutations outside the GJB2 coding region, we compared the observed c.35delG carrier frequency in our study population to the expected number of c.35delG carriers based on the c.35delG carrier frequency in normal-hearing controls. As the c.35delG carriers in our study population tested negative for a second DFNB1-causing mutation (we excluded all known coding and non-coding DFNB1-causing mutations), their carrier state was considered independent of their hearing impairment. They were considered deaf and coincidentally c.35delG carriers.
Results
GJB2 Mutation Panel
The panel of GJB2 deafness-causing allele variants (including 4 novel mutations shown in bold in Table 3) was amplified with several different primer pairs to identify the best primers for DHPLC analysis: Cx26-PGC-F and -R primers have 20bp-GC clamps on their 5’ ends to enhance the melt profile for DHPLC detection; Cx26-A160/B944 primers have been described by Lalwani, et al; and Cx26-F-R primers have a 10-bp GC clamp on the 5’ end of the reverse primer to increase the GC content of the amplicon at the 3’ end of exon 2 (Table 1). The GC clamp helps to retain the 3’ end of the amplified product in the DNA-SEP column for a longer time, thus eluting the first and second halves of exon 2 at similar times.
Table 3.
GJB2 coding sequence mutations detected by SSCP, DHPLC (primer pair Cx26-F-R) and direct sequencing.
| Allele 1 | Allele 2 | Screening techniques | |||||
|---|---|---|---|---|---|---|---|
| Nucleotide change | Amino Acid change | Nucleotide change | Amino Acid change | SSCP | DHPLC | SEQ | |
| 1 | c.551G>C | p.Arg184Pro | - | - | +* | +# | + |
| 2 | c.35delG | Frameshift | c.35delG | Frameshift | + | + | + |
| 3 | c.35delG | Frameshift | c.339T>G | p.Ser113Arg | +/− | + | + |
| 4 | c.101T>C | p.Met34Thr | c.101T>C | p.Met34Thr | + | + | + |
| 5 | c.35delG | Frameshift | c.167delT | Frameshift | +/+ | + | + |
| 6 | c.35delG | Frameshift | c.298C>T | p.His100Tyr | +/+ | + | + |
| 7 | c.457G>A | p.Val153Ile | - | - | + | + | + |
| 8 | c.35delG | Frameshift | c.269insT | Frameshift | +/+ | + | + |
| 9 | c.35delG | Frameshift | c.598G>T | p.Gly200X | +/− | + | + |
| 10 | c.101T>C | p.Met34Thr | - | - | + | + | + |
| 11 | c.167delT | Frameshift | - | - | + | +/− | + |
| 12 | c.79G>A | p.Val27Ile | - | - | − | + | + |
| 13 | c.109G>A | p.Val37Ile | c.109G>A | p.Val37Ile | + | + | + |
| 14 | c.35delG | Frameshift | [c.35delG;c.153delT] | Frameshift | + | + | + |
| 15 | c.35delG | Frameshift | c.94C>T | p.Arg32Cys | +/− | + | + |
| 16 | c.35delG | Frameshift | c.645_648delTAGA | Frameshift | +/+ | + | + |
| 17 | c.269T>C | p.Leu90Pro | c.283G>A | p.Val95Met | +/+ | + | + |
| 18 | c.35delG | Frameshift | c.269insT | Frameshift | +/+ | + | + |
| 19 | c.101T>C | p.Met34Thr | c.298C>T | p.His100Tyr | +/+ | + | + |
| 20 | c.511G>A | p.Ala171Thr | - | - | + | + | + |
| 21 | c.551G>C | p.Arg184Pro | - | - | + | + | + |
| 22 | c.35delG | Frameshift | - | - | + | + | + |
| 23 | c.139G>A | p.Glu47Lys | c.380G>A | p.Arg127His | +/+ | + | + |
| 24 | c.249C>G | p.Phe83Leu | - | - | + | + | + |
| 25 | c.35delG | Frameshift | c.596C>T | p.Ser199Phe | +/− | + | + |
| 27+ | c.35delG | Frameshift | c.71G>A | p.Trp24X | +/− | + | + |
| 28 | c.109G>A | p.Val37Ile | c.109G>A | p.Val37Ile | + | + | + |
| 29 | c.35delG | Frameshift | c.311_324del | Frameshift | +/+ | + | + |
| 30 | c.269T>C | p.Leu90Pro | - | - | + | +/− | + |
| 31 | c.380G>A | p.Arg127His | - | - | + | + | + |
| 32 | c.35delG | Frameshift | c.184G>C | p.Arg184Pro | +/+ | + | + |
| 33 | c.235delC | Frameshift | c.257G>C | p.Ser86Thr | +/+ | + | + |
| 35+ | c.35delG | Frameshift | c.269T>C | p.Leu90Pro | +/+ | + | + |
| 36 | c.235delC | Frameshift | c.558_603dup | Frameshift | +/+ | + | + |
| 37 | c.95G>A | p.Arg32His | c.313_326del | Frameshift | +/− | + | + |
| 38 | c.167delT | Frameshift | c.167delT | Frameshift | + | +/− | + |
| 39 | c.427C>T | p.Arg143Trp | c.313_326del | Frameshift | +/− | + | + |
| 40 | c.109G>A | p.Val37Ile | c.250G>C | p.Val84Leu | +/− | + | + |
| 41 | c.35delG | Frameshift | c.169C>T | p. Gln57X | +/+ | + | + |
| 42 | c.35delG | Frameshift | c.313_326del | Frameshift | +/+ | + | + |
| 43 | c.380G>A | p.Arg127His | - | - | + | + | + |
| 44 | c.109G>A | p.Val37Ile | c.427C>T | p.Arg143Trp | −/− | + | + |
| 45 | c.608T>C | p.Ile203Thr | - | - | + | + | + |
| 46 | c.506G>A | p.Cys169Tyr | - | - | + | + | + |
| 47 | c.479G>A | p.Gly160Ser | - | - | + | +/− | + |
| 49+ | [c.79G>A;c.341A>G] | [p.Val27Ile;p.Glu114Gly] | [c.79G>A;c.341A>G] | [p.Val27Ile;p.Glu114Gly] | −/− | + | + |
| 50 | c.139G>T | p.Glu47X | - | - | + | + | + |
| 51 | c.35delG | Frameshift | c.380G>A | p.Arg127His | +/+ | + | + |
| 52 | c.564_565delGA | Frameshift | - | - | + | + | + |
| 53 | c.109G>A | p.Val37Ile | c.645_648delTAGA | Frameshift | +/− | + | + |
| 54 | c.35G>T | p.Gly12Val | - | - | + | + | + |
| 55 | c.313_326del | Frameshift | - | - | + | + | + |
| Mutation detection accuracy | 82.3% | 98.1 % | 100% | ||||
Novel mutations in bold; SSCP, single-strand conformational polymorphism analysis; DHPLC, denaturing high performance liquid chromatography; SEQ, sequencing
Runs 26, 34 and 48 were wild-type controls
SSCP: +, mutation detected; −, mutation not detected; +/+, both mutations detected; +/−, first mutation detected, second mutation not detected
DHPLC: +, mutation detected in all four runs (each run completed at 3 temperatures); +/−, mutation detected in at least 1 DHPLC run but not in all runs
DNA numbering based on cDNA sequence GenBank NM_004004.3 with +1 as A of ATG start codon.
GJB2-reference: Locus link ID 2706, cDNA AF479776.1; contig NT_024524.13 (1741611..1743919)
All variants were analyzed at 62°C, 60°C and 58°C to increase DHPLC detection efficiency since some variants were not detected at 62°C (p.Leu90Pro and p.Ile230Thr). Wave profiles were analyzed for differences in shape and retention times between wild type and mutant profiles. To test detection efficiency and reproducibility, four separate amplifications and DHPLC runs were completed with each primer pair (Table 2).
Table 2.
GJB2 Mutation Panel detection rates based on primer pair selection
| Experiment | Cx26-PGC-F-R | Cx26-A160-B944 | Cx26-F-R |
|---|---|---|---|
| Run 1 | 50/52 | 51/52 | 52/52 |
| Run 2 | 45/52 | 46/52 | 51/52 |
| Run 3 | 47/52 | 51/52 | 52/52 |
| Run 4 | 46/52 | 51/52 | 49/52 |
| Total | 188/208 | 199/208 | 204/208 |
| % detected | 90.4% | 95.7% | 98.1% |
| Detection at predicted optimal temperature | 45/52 | 46/52 | 49/52 |
| Detection only by completing a 3-temperature run | 5/52 | 5/52 | 3/52 |
| Never detected | 2/52 | 1/52 | 0/52 |
The ability of DHPLC to detect these variants was significantly greater than SSCP analysis (98.1% vs 82.6%, respectively; p value=0.0001), with the detection rate of DHPLC approaching that of direct sequencing. We found that the amplicon generated with primer pair Cx26-F-R offers the best possible detection rate, producing an abnormal DHPLC elution profile for at least one temperature during at least one of four runs for all sequence variants tested (Table 3). On this basis, we estimate the sensitivity of DHPLC at 98.1% for mutation screening of exon 2 of GJB2 with primer pair Cx26F-R.
Patient Recruitment and GJB2 Mutation Screening
GJB2 mutation screening was completed on 1294 persons with congenital deafness of unknown etiology. Two hundred five patients carried two non-complementary GJB2 deafness-causing allele variants of exon 2 and were diagnosed as having DFNB1 (Table 4). One patient was diagnosed with DFNA3. In the remaining 1088 persons, two non-complementary GJB2 mutations were not identified. One hundred persons carried a single allele variant of exon 2 and 988 persons carried two wild-type alleles. Within the group of 100 persons carrying a single allele variant of exon 2, we found compound heterozygosity for c.1–3172G>A and del(GJB6-D13S1830) in 5 and 7 persons, respectively [c.1–3172G>A]+[c.35delG], n=3; [c.1–3172G>A]+[c.167delT], n=1; [c.1–3172G>A]+[c.551G>C], n=1; [del(GJB6-D13S1830)]+[c.35delG], n=5; [del(GJB6-D13S1830)]+[c.139G>T], n=1; [del(GJB6-D13S1830)]+[c.167delT], n=1), leaving 88 of 1076 deaf persons (8.2 %) as carriers of a single GJB2 allele variant (Tables 4, 5; 4 novel mutations were identified and are shown in bold in Table 5). Thirty-seven of 88 were c.35delG heterozygotes; the remainder carried other allele variants of GJB2.
Table 4.
DFNB1 genotypes occurring more than once
| Allele Pairs | N (217) | Percent |
|---|---|---|
| [c.35delG]+[c.35delG] | 103 | 47.47 |
| [c.35delG]+[c.167delT] | 13 | 5.99 |
| [c.35delG]+[c.101T>C] | 8 | 3.69 |
| [c.35delG]+[c.312_325del] | 8 | 3.69 |
| [c.109G>A]+[c.109G>A] | 6 | 2.76 |
| [c.35delG]+[del(GJB6-D13S1830)] | 5 | 2.30 |
| [c.35delG]+[c.269insT] | 5 | 2.30 |
| [c.79G>A;c.341A>G]+[c.79G>A;c.341A>G] | 3 | 1.38 |
| [c.101T>C]+[c.101T>C] | 3 | 1.38 |
| [c.35delG]+[c.1–3172G>A] | 3 | 1.38 |
| [c.35delG]+[c.71G>A] | 3 | 1.38 |
| [c.35delG]+[c.551G>C] | 3 | 1.38 |
| [c.167delT]+[c.167delT] | 3 | 1.38 |
| [c.101T>C]+[c.298C>T] | 2 | 0.92 |
| [c.35delG]+[c.94C>T] | 2 | 0.92 |
| [c.35delG]+[c.169C>T] | 2 | 0.92 |
| [c.35delG]+[c.269T>C] | 2 | 0.92 |
| [c.35delG]+[c.427C>T] | 2 | 0.92 |
| [c.35delG]+[c.596C>T] | 2 | 0.92 |
GJB2-reference: Locus link ID 2706, cDNA AF479776.1; contig NT_024524.13 (1741611..1743919)
Table 5.
Persons carrying a single GJB2 allele variant
| Alleles | N (88) | Percent |
|---|---|---|
| c.35delG | 37 | 42.05 |
| c.101T>C | 17 | 19.32 |
| c.380G>A | 7 | 7.95 |
| c.109G>A | 6 | 6.82 |
| c.167delT | 3 | 3.41 |
| c.269T>C | 2 | 2.27 |
| c.31_44del | 1 | 1.14 |
| c.34G>T | 1 | 1.14 |
| c.35G>T | 1 | 1.14 |
| c.71G>A | 1 | 1.14 |
| c.110T>C | 1 | 1.14 |
| c.246C>G | 1 | 1.14 |
| c.298C>T | 1 | 1.14 |
| c.331A>G | 1 | 1.14 |
| c.355G>A | 1 | 1.14 |
| c.365A>T | 1 | 1.14 |
| c.416G>A | 1 | 1.14 |
| c.511G>A | 1 | 1.14 |
| c.516G>A | 1 | 1.14 |
| c.551G>C | 1 | 1.14 |
| c.564_565del | 1 | 1.14 |
| c.596C>T | 1 | 1.14 |
GJB2-reference: Locus link ID 2706, cDNA AF479776.1; contig NT_024524.13 (1741611..1743919); novel mutations in bold.
Residual Hearing and Phenotype Genotype Correlations
Percent deafness in persons with a genetic diagnosis of DFNB1 varied from 1% to 100%. The most common genotype, [c.35delG]+[c.35delG], was associated with severe-to-profound deafness (hearing impairment of >80%) in nearly 90% of affected persons. In compound heterozygotes carrying one c.35delG allele and any other GJB2 deafness-causing allele variant, severe-to-profound deafness was observed in only 60% of affected persons, and in persons carrying two GJB2 deafness-causing missense mutations severe-to-profound deafness was never observed (P<0.05; Fisher exact test). Persons with a genotype of two truncating or nonsense mutations were more likely to have severe-to-profound deafness than persons with a genotype that included one or two missense mutations (P<0.05; Fisher exact test). Similarly, persons with one truncating or nonsense mutation and a missense mutation on the other allele were more likely to have severe-to-profound deafness than persons with two missense mutations (P<0.05; Fisher exact test) (Fig 1).
Figure 1.
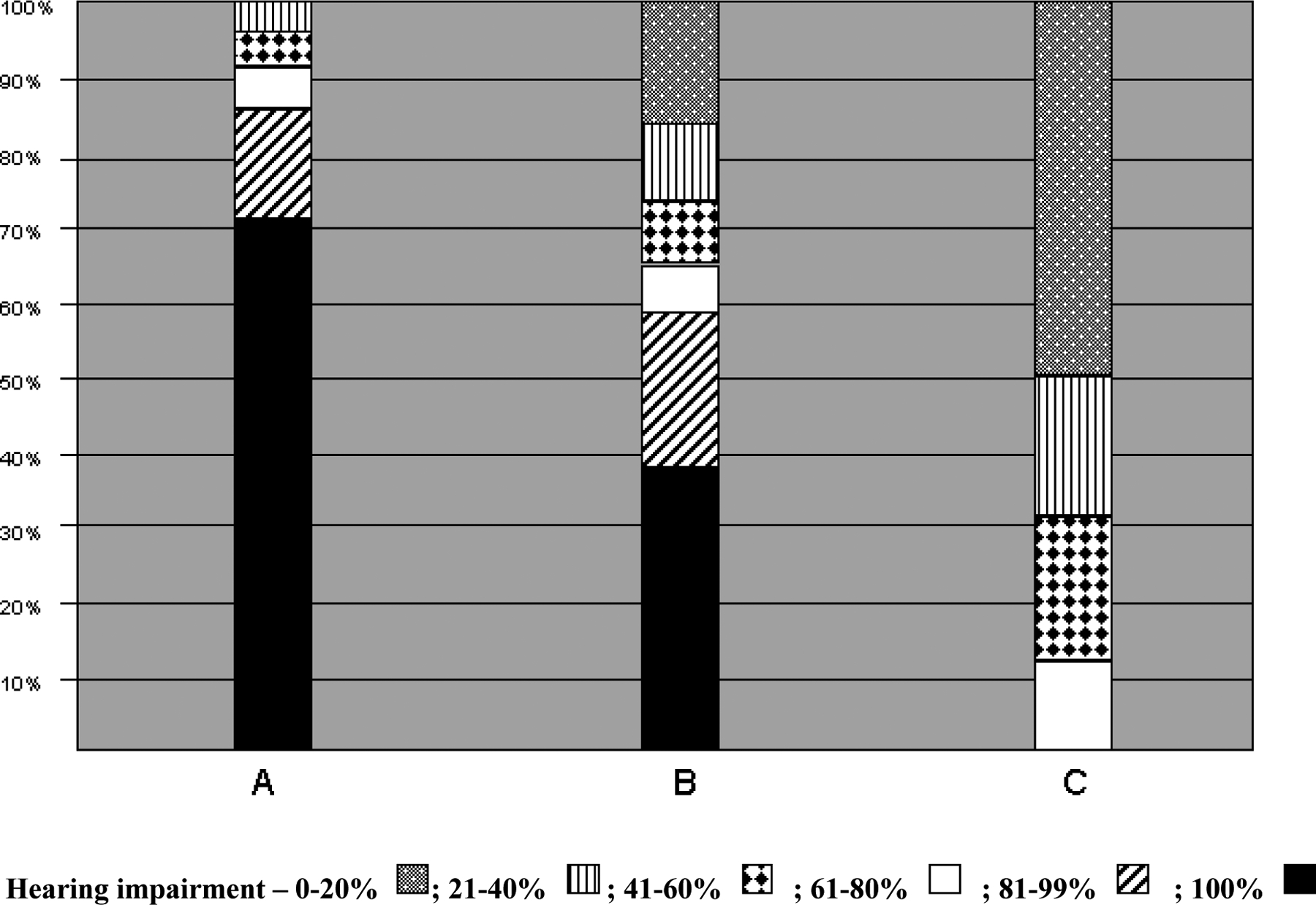
Genotype-phenotype correlations for biallelic GJB2 mutation carriers show that percent hearing impairment is highest in persons who are [c.35delG]+[c.35delG] homozygotes and lowest in persons who carry two missense mutations of GJB2 (P<0.05; Fisher exact test). In persons who are compound heterozygotes and carry a single c.35delG allele, severe-to-profound deafness is more common than in persons who carry two missense mutations of GJB2 (P<0.05; Fisher exact test) (A, [c.35delG]+[c.35delG] homozygotes; B, [c.35delG]+[non-c.35delG] compound heterozygotes; C, biallelic missense mutations of GJB2).
Additional Non-coding GJB2 Mutations
The identification of 37 deaf c.35delG carriers in the cohort of 1076 non-DFNB1 deaf persons was greater than expected based on a combined estimated c.35delG carrier rate of 4 per 369 (Kelley, et al., 1998; Morell, et al., 1998; Scott, et al., 1998) (P<0.05; Fisher exact test). This discrepancy suggests the presence of additional DFNB1-causing mutations outside the coding region of GJB2.
Discussion
The contribution GJB2 makes to the ARNSD genetic load has had a major impact on clinical medicine. Prior to 1997, ARNSD in a deaf child of hearing parents was an exclusionary diagnosis made only after other etiologies of deafness had been eliminated. In an attempt to facilitate the early identification of infants ‘at-risk’ for deafness, a list of high-risk factors was used to trigger auditory testing in select infants. Antenatal factors such as family history, neonatal complications like hyperbilirubinemia and apnea, congenital infections such as toxoplasmosis, syphilis, rubella, cytomegalovirus and herpes, and parental concern were indications sufficient to warrant auditory testing. Unfortunately, the majority of infants with ARNSD had none of these risk factors and their hearing loss typically remained unrecognized until the second or third years of life.
Universal screening has obviated the need for this ‘risk-driven’ approach and with newborn physiologic tests of hearing, early identification of hearing loss is becoming routine. By coupling this screening strategy with genetic testing limited to mutational analysis of GBJ2, an unequivocal diagnosis of inherited deafness can be made in approximately 50% of babies with ARNSD. Currently, 19 laboratories in the USA offer mutation screening of GJB2.
Screening can be accomplished using a variety of techniques, although all are not equally accurate, timely or cost effective (Kristensen, et al., 2001). One of the more popular methods is SSCP. Mutations are detected by migration differences in single-stranded DNA, with confirmatory sequencing as required. Although fast and cost effective, we found SSCP to be only 83% effective in detecting a panel of different GJB2 allele variants.
At the other extreme is direct sequencing. While this approach represents the gold standard against which other mutation screening strategies should be compared, it is more consuming and expensive, especially for large genes. DHPLC offers an attractive alternative between SSCP and direct sequencing. It is accurate, reliable, timely, and cost effective. We found that the amplicon generated with primer pair Cx26-F-R produced an abnormal DHPLC elution profile for at least one temperature during at least one of four runs for all sequence variants we tested, giving DHPLC a sensitivity of at least 98.1% for mutation screening of exon 2 of GJB2 (Table 3).
Although Cx26 deafness is reportedly most often severe to profound, many studies suffer from ascertainment bias caused by selecting families segregating this degree of hearing impairment at the exclusion of milder losses. This study avoided this type of bias by including all persons with congenital or presumed congenital losses irrespective of degree, permitting us to identify several important genotype-phenotype relationships. First, it is clear that genotype-phenotype associations do exist for Cx26 deafness. Percent deafness is significantly greater in persons segregating two truncating/nonsense mutations as compared to persons segregating two missense mutations (P<0.05; Fisher exact test). The four most common missense mutations in our series were [p.Val37Ile] + [p.Val37Ile], n=6; [p.Met34Thr] + [p.Met34Thr], n=3; [p.Val27Ile;p.Glu114Gly] + [p.Val27Ile;p.Glu114Gly], n=3; [p.Met34Thr] + [p.His100Tyr], n=2). The consistently greatest percent deafness was found in persons carrying del(GJB6-D13S1830) [del(GJB6-D13S1830)] + [c.35delG], n=5; [del(GJB6-D13S1830)] + [p.Glu47X], n=1; [del(GJB6-D13S1830)] + [c.167delT], n=1). Our data suggest that with sufficient numbers of persons, it will be possible to make accurate phenotype predictions for several specific genotypes.
The number of deaf c.35delG carriers we identified was greater than the expected carrier frequency (P<0.05; Fisher exact test), supporting the existence of other DFNB1-causing mutations outside the GJB2 coding region. Identifying these mutations will do much to clarify our understanding of Cx26 deafness.
Acknowledgments
This study is supported by grant G.0277.01 from the FWO (Flemish Fonds voor Wetenschappelijk Onderzoek) (GVC) and grant RO1-DC02842 from the NIDCD (RJHS).
References
- Cohn ES, Kelley PM, Fowler TW, Gorga MP, Lefkowitz DM, Kuehn HJ, Schaefer GB, Gobar LS, Hahn FJ, Harris DJ, Kimberling WJ. 1999. Clinical studies of families with hearing loss attributable to mutations in the connexin 26 gene (GJB2/DFNB1). Pediatrics 103:546–50. [DOI] [PubMed] [Google Scholar]
- Dahl E, Manthey D, Chen Y, Schwarz HJ, Chang YS, Lalley PA, Nicholson BJ, Willecke K. 1996. Molecular cloning and functional expression of mouse connexin-30, a gap junction gene highly expressed in adult brain and skin. J Biol Chem 271:17903–17910. [DOI] [PubMed] [Google Scholar]
- del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, Menendez I, Moreno F. 2002. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 346(4):243–249. [DOI] [PubMed] [Google Scholar]
- Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, Garabedian EN, Petit C. 1999. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: implications for genetic counselling. Lancet, 353(9161):1298–1303. [DOI] [PubMed] [Google Scholar]
- Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D’Agruma L, Mansfield E, Rappaport E, Govea N, Mila M, Zelante L, Gasparini P. 1998. Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet 351(9100):394–398. [DOI] [PubMed] [Google Scholar]
- Fukushima K, Sugata K, Kasai N, Fukuda S, Nagayasu R, Toida N, Kimura N, Takishita T, Gunduz M, Nishizaki K. 2002. Better speech performance in cochlear implant patients with GJB2-related deafness. Int J Pediatr Otorhinolaryngol 62:151–7. [DOI] [PubMed] [Google Scholar]
- Green GE, Mueller RF, Cohn ES, Avraham KB, Smith RJH. 2003. Audiologic Manifestations and Features of Connexin 26 Deafness. J Audiol Med (In press). [Google Scholar]
- Green GE, Scott DA, McDonald JM, Teagle HFB, Tomblin BJ, Spencer LJ, Woodworth GG, Knutson JF, Gantz BJ, Sheffield VC, Smith RJH. 2002. Performance of Cochlear Implant Recipients with GJB2-related Deafness. Am J Med Genet, 109:167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJ. 1999. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. Jama, 281(23):2211–2216. [DOI] [PubMed] [Google Scholar]
- Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A. 1989. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucl Acid Res. 17(20):8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilford P, Ben Arab S, Blanchard S, Levilliers J, Weissenbach J, Belkahia A, Petit C. 1994. A non-syndrome form of neurosensory, recessive deafness maps to the pericentromeric region of chromosome 13q. Nat Genet 6(1):24–28. [DOI] [PubMed] [Google Scholar]
- Heathcote K, Syrris P, Carter ND, Patton MA. 2000. A connexin 26 mutation causes a syndrome of sensorineural hearing loss and palmoplantar hyperkeratosis (MIM 148350). J Med Genet 37(1):50–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl D 2000. Towards a better classification of erythrokeratodermias. Br J Dermatol 143(6):1133–1137. [DOI] [PubMed] [Google Scholar]
- Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, Kimberling WJ. 1998. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet 62(4):792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM. 1997. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387(6628):80–83. [DOI] [PubMed] [Google Scholar]
- Kenna MA, Wu BL, Cotanche DA, Korf BR, Rehm HL. 2001. Connexin 26 studies in patients with sensorineural hearing loss. Arch Otolaryngol Had Neck Surg 127(9):1037–42. [DOI] [PubMed] [Google Scholar]
- Kristensen VN, Kelefiotis D, Kristensen T, Borresen-Dale A-L. 2001. High-throughput methods for detection of genetic variation. Biotechniques 30:318–332. [DOI] [PubMed] [Google Scholar]
- Lautermann J, Frank HG, Jahnke K, Traub O, Winterhager E. 1999. Developmental expression patterns of connexin-26 and −30 in the rat cochlea. Dev Genet 25:306–311. [DOI] [PubMed] [Google Scholar]
- Lautermann J, ten Cate WJ, Altenhoff P, Grümmer R, Traub O, Frank HG, Jahnke K, Winterhager E. 1998. Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res 294:415–420. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu XS, Wei L, Altman RB, Batzoglou S. 2004. Eukaryotic regulatory element conservation analysis and identification using comparative genomics. Genome Res. 14(3):451–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestrini E, Korge BP, Ocana-Sierra J, Calzolari E, Cambiaghi S, Scudder PM, Hovnanian A, Monaco AP, Munro CS. 1999. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families. Hum Mol Genet 8(7):1237–1243. [DOI] [PubMed] [Google Scholar]
- Mehl AL, Thomson V. 1998. Newborn hearing screening: the great omission. Pediatrics 101:E4–E4. [DOI] [PubMed] [Google Scholar]
- Mehl AL, Thomson V. 2002. The Colorado newborn hearing screening project, 1992–1999: on the threshold of effective population-based universal newborn hearing screening. Pediatrics 109:E7–E7. [DOI] [PubMed] [Google Scholar]
- Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, Fisher R, Van Camp G, Berlin CI, Oddoux C, Ostrer H, Keats B, Friedman TB. 1998. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med 339(21):1500–1505. [DOI] [PubMed] [Google Scholar]
- Mueller RF, Nehammer A, Middleton A, Houseman M, Taylor GR, Bitner-Glindzciz M, Van Camp G, Parker M, Young ID, Davis A, Newton VE, Lench NJ. 1999. Congenital non-syndromal sensorineural hearing impairment due to connexin 26 gene mutations--molecular and audiological findings. Int J Pediatr Otorhinolaryngol 50:3–13. [DOI] [PubMed] [Google Scholar]
- Morton NE. 1991. Genetic epidemiology of hearing impairment. Ann N Y Acad Sci 630: 16–31. [DOI] [PubMed] [Google Scholar]
- Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynänen M, Jabs EW, Bale SJ, DiGiovanna, Uitto J, Russell L. 2002. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafenss syndrome. Am J Hum Genet 70(5):1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, Bale SJ. 1998. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet 103(4):393–399. [DOI] [PubMed] [Google Scholar]
- Rouan F, White TW, Brown N, Taylor AM, Lucke TW, Paul DL, Munro CS, Uitto J, Hodgins MB, Richard G. 2001. trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci 114(Pt 11):2105–2113. [DOI] [PubMed] [Google Scholar]
- Scott DA, Kraft ML, Carmi R, Carmi R, Ramesh A, Elbedour K, Yairi Y, Srikumari Srisailapathy CR, Rosengren SS, Markham AF, Mueller RF, Lench NJ, Van Camp G, Smith RJH, Sheffield VC. 1998. Identification of mutations in the Connexin 26 gene that cause autosomal recessive non-syndromic hearing loss. Hum Mutation 11(5):387–394. [DOI] [PubMed] [Google Scholar]
- Scott DA, Kraft ML, Stone EM, Sheffield VC, Smith RJ. 1998. Connexin mutations and hearing loss. Nature 391(6662):32. [DOI] [PubMed] [Google Scholar]
- Smith RJ. 2001. Mutation screening for deafness: more than simply another diagnostic test. Arch Otolaryngol Head Neck Surg 127(8):941–942. [DOI] [PubMed] [Google Scholar]
- Sobe T, Vreugde S, Shahin H, Davis N, Berlin M, Kanaan M, Yaron Y, Orr-Urtreger A, Frydman M, Shohat M, Avraham KB. 2000. The prevalence and expression of inherited connexin 26 mutations associated with nonsyndromic hearing loss in the Israeli population. Hum Genet 106:50–57. [DOI] [PubMed] [Google Scholar]
- Uyguner O, Tukel T, Baykal C, Eris H, Emiroglu M, Hafiz G, Ghanbari A, Baserer N, Yuksel-Apak M, Wollnik B. 2002. The novel R75Q mutation in the GJB2 gene causes autosomal dominant hearing loss and palmoplantar keratoderma in a Turkish family. Clin Genet 62(4):306–309. [DOI] [PubMed] [Google Scholar]
- Zelante L, Gasparini P, Estivill X, Melchionda S, D’Agruma L, Govea N, Mila M, Monica MD, Lutfi J, Shohat M, Mansfield E, Delgrosso K, Rappaport E, Surrey S, Fortina P. 1997. Connexin26 mutations associated with the most common form of non- syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum Mol Genet 6(9):1605–1609. [DOI] [PubMed] [Google Scholar]