

Abstract
Background and Objectives
To elucidate current epidemiologic, clinical, and immunologic profiles and treatments of stiff-person syndrome (SPS) in Japan.
Methods
A nationwide mail survey was conducted using an established method. Data processing sheets were sent to randomly selected departments of internal medicine, neurology, pediatrics, psychiatry, and neurosurgery in hospitals and clinics throughout Japan to identify patients with SPS who were seen between January 2015 and December 2017.
Results
Thirty cases were identified as glutamic acid decarboxylase 65 (GAD65)–positive SPS cases on the basis of detailed clinical data of 55 cases. Four patients had α1 subunit of glycine receptor (GlyR) antibodies, and 1 patient had both GAD65 and GlyR antibodies. The total estimated number of patients with GAD65-positive SPS was 140, and the estimated prevalence was 0.11 per 100,000 population. The median age at onset was 51 years (range, 26–83 years), and 23 (76%) were female. Of these, 70% had classic SPS, and 30% had stiff-limb syndrome. The median time from symptom onset to diagnosis was significantly longer in the high-titer GAD65 antibody group than in the low-titer group (13 months vs 2.5 months, p = 0.01). The median modified Rankin Scale (mRS) at baseline was 4, and the median mRS at the last follow-up was 2. Among the 29 GAD65-positive patients with ≥1 year follow-up, 7 received only symptomatic treatment, 9 underwent immunotherapy without long-term immunotherapy, and 13 received long-term immunotherapy such as oral prednisolone. The coexistence of type 1 diabetes mellitus and the lack of long-term immunotherapy were independent risk factors for poor outcome (mRS ≥3) in the GAD65-positive patients (odds ratio, 15.0; 95% CI 2.6–131.6; p = 0.001; odds ratio, 19.8; 95% CI 3.2–191.5; p = 0.001, respectively).
Discussion
This study provides the current epidemiologic and clinical status of SPS in Japan. The symptom onset to the diagnosis of SPS was longer in patients with high-titer GAD65 antibodies than in those with low-titer GAD65 antibodies. The outcome of patients with SPS was generally favorable, but more aggressive immunotherapies are necessary for GAD65-positive patients with SPS.
Introduction
Stiff-person syndrome (SPS) is a rare autoimmune neurologic disorder characterized by progressive axial muscle stiffness, CNS hyperexcitability, and stimulus-sensitive painful muscle spasms.1 Women are predominantly affected (62–70% of cases), with most patients presenting in their 40s to 50s.2-4 SPS is classified into classic SPS and SPS variants, including stiff-limb syndrome (SLS) and progressive encephalomyelitis with rigidity and myoclonus (PERM), on the basis of clinical presentation.1,5 Most patients with SPS have antibodies against glutamic acid decarboxylase 65 (GAD65), the rate-limiting enzyme in the production of the inhibitory neurotransmitter γ-aminobutyric acid (GABA).1,6,7 Amphiphysin antibodies are also detected in some patients with paraneoplastic SPS.5 Patients with PERM may present with symptoms similar to classic SPS but with additional features, including brainstem symptoms, hyperekplexia, myoclonus, and dysautonomia.8,9 PERM is associated with glycine receptor (GlyR) α1 subunit antibodies and generally responsive to immunotherapy.4,10 Since the initial description of SPS in 1956, marked progress has been made in the clinical characterization of SPS.11 However, no large-scale epidemiologic studies have been conducted except for 1 clinic-based study that reported an estimated prevalence of one to two cases per million population.12 GAD65 antibodies are useful diagnostic markers, but their role in the pathogenesis of SPS is unclear.1,13 Moreover, their clinical relevance is questionable in patients with low GAD65 antibody titers.14 It has also been reported that the outcome is poor in patients with GAD65 antibodies.3,4 As some patients respond poorly to conventional immunotherapies, the exact nature of GAD65 antibody-associated SPS needs to be clarified. Against this backdrop, we conducted a nationwide epidemiologic survey of SPS in Japan and compared the clinical features among different immunologic groups (autoantibody-associated patients).
Methods
Epidemiologic Survey
A nationwide mail survey of SPS was conducted in 2018 in Japan.
The survey targeted 5 departments (internal medicine, neurology, pediatrics, psychiatry, and neurosurgery). First, the study centers were randomly selected from a complete list of hospitals and clinics in Japan in the Nationwide Epidemiologic Survey Manual issued by the Research Committee on Epidemiology of Intractable Disease.15 Selection rates were determined on the basis of 8 categories that were defined in accordance with the number of beds in a hospital: (I) university hospitals, 100%; (II) hospitals with ≥500 beds, 100%; (III) hospitals with 400–499 beds, 80%; (IV) hospitals with 300–399 beds, 40%; (V) hospitals with 200–299 beds, 20%; (VI) hospitals with 100–199 beds, 10%; (VII) private clinics or hospitals with <100 beds, 5%; and (VIII) specific neuromuscular centers dealing with intractable diseases, 100%.
We sent our first survey, which included a questionnaire and the diagnostic criteria for SPS adopted from the work of Dalakas1 (eTable 1, links.lww.com/NXI/A904), to each of the randomly selected study centers. The aim of the first survey was to obtain data on the number of patients with SPS who had visited the respective study centers from January 1, 2015, to December 31, 2017.
Second, the estimated number of patients was calculated for each category using the following formula: total estimated number of patients = reported number of patients/(selection rate × response rate) = reported number of patients/(number of responding centers/total number of study centers). Finally, the total estimated number of patients for all categories was determined. Ninety-five percent confidence intervals (95% CIs) were calculated, assuming a multinomial hypergeometric distribution. This method has been validated in previous nationwide surveys of intractable diseases in Japan.16-18
In the second survey sent to the study centers that reported the number of patients with SPS, a predefined format was used to collect the detailed clinical data of each patient (Figure 1).
Figure 1. Flowchart of the Nationwide Survey.
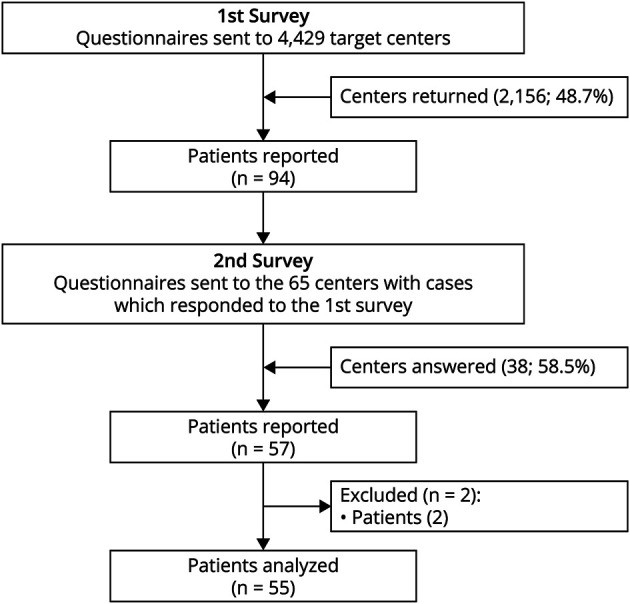
SPS = stiff-person syndrome.
Evaluation of Clinical Features, Treatment, and Outcome
We collected the detailed clinical data of patients with SPS through the second survey and classified the patients into 3 groups: (1) classic SPS: rigidity of the axial trunk, sometimes involving the proximal limbs, in association with muscle spasms resulting in abnormal axial posture19; (2) SLS: affecting one or more limbs with distal rigidity and abnormal posture of hands or feet20,21; and (3) PERM: progressive encephalomyelitis accompanied with rigidity and myoclonus3,22 (eTable 1, links.lww.com/NXI/A904). The detailed history, time from symptom onset to diagnosis, examination findings, and serologic and electrophysiologic data at the time of initial evaluation of all patients were documented. Treatments were classified as follows: (1) symptomatic treatment (e.g., GABAergic drugs), (2) first-line immunotherapy (IV methylprednisolone (IVMP), IV immunoglobulin (IVIg), or plasma exchange alone or in combination), (3) second-line immunotherapy (rituximab, cyclophosphamide), and (4) long-term oral immunotherapy (prednisolone (PSL), azathioprine, tacrolimus).4 The time from diagnosis to immunotherapy and/or IVIg and the improvement of symptoms were also documented. The disability level was assessed using the modified Rankin Scale (mRS) at baseline and at the last follow-up.23
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the Ethics Committee of Tokushima University Hospital. Written informed consent was obtained from the patients or their proxies.
Antibody Assays
GAD65 antibodies were measured by radioimmunoassay (RIA), enzyme immunoassay (EIA), or ELISA using commercially available kits (SRL, Inc. or BML, Inc., Tokyo, Japan). GAD65 antibodies were considered positive when the antibody titers were >1.4 U/mL with RIA, ≥5 U/mL with EIA, and ≥5 U/mL with ELISA.
Amphiphysin antibodies were measured using commercial immunoblotting assay (EUROIMMUN AG, Lübeck, Germany).
Dipeptidyl-peptidase–like protein 6 (DPPX) antibodies were measured with an IIFT Autoimmune Encephalitis Mosaic 6 test kit (EUROIMMUN AG, Lübeck, Germany) or an in-house cell-based assay (CBA) through the courtesy of Dr. Josep Dalmau (Barcelona, Spain).24
Both GlyR and GABAB receptor (GABABR) antibodies were measured with an established in-house CBA (eMaterial, links.lww.com/NXI/A910) through the courtesy of Dr. Keiko Tanaka (Niigata University, Japan).
Some GlyR antibody tests were performed at Oxford University, Oxford, and the Laboratory of Experimental Neuroimmunology, IDIBAPS-Hospital Clinic, Barcelona, using previously reported CBA techniques.8,25 Autoantibodies against other SPS-associated antigens, including GABABR, were also examined in IDIBAPS-Hospital Clinic.4,24,26
Statistical Analysis
Continuous and categorical variables were reported as median (range) and number (percentage), respectively. Comparisons across 2 or more groups were performed using the Kruskal-Wallis test (continuous variables) and the χ2 or Fisher exact test (categorical variables). Pairwise comparisons were performed using the Wilcoxon rank-sum test (continuous variables) and Fisher exact test (categorical variables). Factors that influenced the outcome of SPS were assessed using univariable binary logistic regression.
Odds ratios (profile likelihood 95% CIs) were used to measure the effect of predictors. SPSS version 19 and SAS version 9.4 were used for the analysis.
Data Availability
Any data not published in the article will be shared anonymously on request by any qualified investigator.
Results
First Survey
In the first survey, we randomly selected 4,429 (28.0%) study centers from a total of 15,848 hospitals and clinics with internal medicine, neurology, pediatrics, psychiatry, and neurosurgery departments across Japan, listed in the Nationwide Epidemiologic Survey Manual issued by the Research Committee on Epidemiology of Intractable Disease. Of these, 2,156 (48.7%) responded, reporting 94 patients with SPS in total (Figure 1).
Second Survey
In the second survey, we sent questionnaires to 65 study centers to collect detailed clinical data of patients with SPS. The clinical data of 57 patients were initially collected from 38 study centers (58%) that responded to the second survey. Of these, one patient for whom SPS could not be differentiated from other neurologic disorders and another patient with insufficient information were excluded. The study population thus consisted of 55 patients with the final diagnosis of SPS (Figure 1).
Antibody Findings
Thirty-three of the 55 patients tested positive for GAD65 antibodies in serum, and 16 of 24 patients tested positive for GAD65 antibodies in CSF. The antibody titers and the test methods for each case are presented in eFigure 1 (links.lww.com/NXI/A909) and eTable 2 (links.lww.com/NXI/A905).
Amphiphysin antibodies were examined in serum samples from 31 patients, whereas DPPX antibodies were examined in both serum and CSF samples from 7 patients or only serum samples from 18 patients. None of the patients was positive for amphiphysin or DPPX antibodies, and 1 patient had Ri antibodies (eTable 2, links.lww.com/NXI/A905).
GlyR and GABABR antibodies were examined in both serum and CSF samples from 8 patients or only serum samples from 18 patients. GlyR antibodies were identified in 5 patients (both serum and CSF [n = 4], serum only [n = 1]). One of the 5 patients positive for GlyR antibodies was also positive for GAD65 antibodies (eTable 2, links.lww.com/NXI/A905). We confirmed that the serum sample of 1 patient was positive for GABABR antibodies by IIFT Autoimmune Encephalitis Mosaic 6 and in-house CBA using GABABR stably expressing HEK293 cells (data not shown). This patient had no other SPS-related autoantibodies.
GAD65-Positive Patients
The 33 GAD65-positive patients were classified into 3 groups on the basis of clinical phenotype: classic SPS (23 patients, 70%), SLS (9 patients, 27%), and PERM (1 patient, 3%) (Figure 2). One patient with PERM who was positive for both GAD65 and GlyR antibodies was classified under another category. In this study, a high titer of GAD65 antibodies was defined as 1,800 U/mL with RIA, >10,000 U/mL with EIA, and >2,000 U/mL with ELISA on the basis of real data and a previous report.13 The 32 GAD65-positive patients were divided into 2 groups on the basis of antibody titer (8 patients with low-titer GAD65 antibodies and 24 patients with high-titer GAD65 antibodies) (eFigure 1, links.lww.com/NXI/A909, eMaterial, links.lww.com/NXI/A910). The range of low-titer GAD65 antibodies by measurement method is as follows: RIA: 2.6–1,023 U/mL; EIA: no applicable patient; and ELISA: 21.2–929 U/mL. Furthermore, low-titer GAD65-positive patients with SPS were defined on the basis of electrophysiology or CSF tests in addition to diagnostic criteria. On the basis of this low-titer criterion, 2 patients with a low titer of GAD65 antibodies were excluded. Therefore, of the 33 GAD65-positive patients, 30 were included in the GAD65-positive analysis. Twenty-three patients (76%) were female, the median age at onset of symptoms was 51 years (range, 26–83 years), and the median time of symptom onset to diagnosis was 12 months (range, 1–96 months). Finally, the 30 patients were classified into 2 groups on the basis of clinical phenotype: classic SPS (21 patients, 70%) and SLS (9 patients, 30%). Thirteen patients (43%) had type 1 diabetes mellitus (DM), and 11 (37%) had other autoimmune diseases, such as thyroid disease. Four (13%) had malignancies, including 2 with cancer (breast and lung) and 2 with thymoma. One patient was diagnosed before the onset of SPS symptoms; 1, during the course of evaluation of neurologic symptoms and 2, after the onset of SPS symptoms. These patients were scored for paraneoplastic neurological syndrome (PNS) according to the PNS care score, as follows: probable, 1 case, and non-PNS, 3 cases.27 EMG studies were performed in 15 patients (50%), and CSF was examined in 22 patients (73%). Thirteen (87%) of the 15 patients lacked antagonist inhibition on EMG. The median follow-up was 47 months (range, 1–216 months). The median mRS at baseline was 4 (1–5), and the median mRS at the last follow-up was 2 (0–6) (eTable 3, links.lww.com/NXI/A906).
Figure 2. Flowchart of the Clinical Study.

GAD65 = glutamic acid decarboxylase 65; GlyR = glycine receptor α1 subunit. # Lack of antagonist inhibition on EMG is defined by continuous co-contraction of agonist and antagonist muscles (inhibitory to relax) as confirmed by EMG.
Epidemiologic Study of Patients With GAD65-Positive SPS
From the first survey, the total estimated number of patients with SPS was 257 (95% CI 161–354), with an estimated period prevalence of 0.2 (0.13–0.28) per 100,000 population using the background population from 2015 to 2017 reported by the Ministry of Internal Affairs and Communications of Japan. Thirty cases (55%) were identified as GAD65-positive SPS cases on the basis of detailed clinical data of 55 cases obtained in the second survey. Taken together, the total estimated number of patients with GAD65-positive SPS was 140, with a period prevalence of 0.11 per 100,000 population from 2015 to 2017.
Comparison Between Low-Titer and High-Titer GAD65 Antibody Groups
Six patients were assigned to the low-titer GAD65 antibody group, and 24 patients were assigned to the high-titer GAD65 antibody group. The high-titer GAD65 antibody group had significantly longer time from symptom onset to diagnosis than the low-titer GAD65 antibody group (13 vs 2.5 months, p = 0.01). We observed no significant differences in sex distribution, age at onset, clinical manifestations, comorbidity, EMG and CSF findings, and clinical improvement on the basis of mRS between the low-titer and high-titer GAD65 antibody groups (Table 1).
Table 1.
Clinical Characteristics of Patients With Low-Titer and High-Titer GAD65 Antibodies

| Clinical characteristic | Low-titer GAD65 Antibodies (n = 6) |
High-titer GAD65 Antibodies (n = 24) |
p Value |
| Female (%) | 3 (50) | 20 (83) | 0.12 |
| Age at onset, y, median (range) | 43.5 (26 to 83) | 52.5 (23 to 71) | 0.42 |
| Time from symptom onset to diagnosis, mo, median (range) | 2.5 (2 to 48) | 13 (1 to 96) | 0.01 |
| Clinical phenotype (%) | 1.00 | ||
| Classic SPS | 4 (67) | 17 (71) | |
| SLS | 2 (33) | 7 (29) | |
| PERM | 0 (0) | 0 (0) | |
| Type 1 DM (%) | 3 (50) | 10 (42) | 0.53 |
| Other autoimmune diseases (%) | 1 (17) | 10 (42) | 0.26 |
| Malignancy (%) | 2 (33) | 2 (8) | 0.17 |
| Lack of antagonist inhibition on EMGa | 5/5 (100) | 8/10 (80) | 0.43 |
| CSF pleocytosis | 0/6 (0) | 2/16 (13) | 0.52 |
| Elevated CSF protein | 2/5 (29) | 3/15 (20) | 0.37 |
| mRS at baseline, median (range) | 3.5 (2 to 4) | 4 (1 to 5) | 0.15 |
| Follow-up period, m, median (range) | 47 (2 to 120) | 44 (15 to 192) | 0.98 |
| mRS at the last follow-up, median (range) | 2.5 (0 to 6) | 2 (0 to 4) | 0.56 |
| Change in mRS, median (range) | 0.5 (−2 to 2) | 2 (0 to 5) | 0.09 |
Abbreviations: DM = diabetes mellitus; GAD65 = glutamic acid decarboxylase 65; mRS = modified Rankin Scale; PERM = progressive encephalomyelitis with rigidity and myoclonus; SLS = stiff-limb syndrome.
Lack of antagonist inhibition on EMG is defined by the continuous co-contraction of agonist and antagonist muscles (with inability to relax) as confirmed by EMG.
Clinical Characteristics, Treatment, and Longitudinal Outcomes of GAD65-Positive Patients
To evaluate clinical response to treatment, we excluded 1 patient without ≥1 year follow-up (Figure 2). Good outcome was defined by mRS of 0–2, and poor outcome was defined by mRS ≥3 after ≥1 year follow-up.3,4 We summarize the clinical characteristics, treatments, and longitudinal outcomes of 29 GAD65-positive patients with a median follow-up of 48 (15–180) months in eTable 4 (links.lww.com/NXI/A907).
Seven (39%) of 18 GAD65-positive patients having mRS ≥4 at baseline showed poor outcome. Among the 29 GAD65-positive patients, 7 were given only symptomatic treatment and 22 received immunotherapy and/or symptomatic treatment. On the basis of the immunotherapy regimen, we defined the effective long-term immunotherapy group as follows: first-line immunotherapy followed by maintenance therapy (oral prednisolone and/or tacrolimus; monthly IVIg; repeated IVIg within 4 months).
The types of therapy were finally classified as follows: (1) without immunotherapy, (2) immunotherapy without effective long-term immunotherapy, and (3) effective long-term immunotherapy. The time from diagnosis to immunotherapy and/or IVIg and the improvement of symptoms were also documented.
For the symptomatic treatment group, the median mRS was 4 (range, 1–5) at baseline and 1 (range, 1–3) at the last follow-up.
We further classified the 22 GAD65-positive patients who received immunotherapy into 2 groups as follows: 9 with immunotherapy but without effective long-term immunotherapy and 13 with effective long-term immunotherapy. The median mRS at baseline was the same at 4 (range, 2–5) for both immunotherapy groups, but the median mRS at the last follow-up was slightly lower for the effective long-term immunotherapy group (2; range, 0–4) than for the immunotherapy but without effective long-term immunotherapy group (3; range, 0–4) (eTable 4, links.lww.com/NXI/A907).
Clinical Outcomes of GAD65-Positive Patients
To identify the risk factors for the GAD65-positive patients, data of 19 (65.5%) patients with good outcome and 10 (34.5%) patients with poor outcome (mRS ≥3) were analyzed. In the univariable regression analysis, the risk factor that was significantly associated with poor outcome was the presence of type 1 DM (odds ratio 15.0; 95% CI 2.60–131.58; p = 0.002) and the lack of effective long-term immunotherapy (odds ratio 19.8; 95% CI 3.17–191.47; p = 0.001) (Table 2). One patient with type 1 DM had mild polyneuropathy, and another patient without type 1 DM had a history of cerebral infarction. However, neither had neurologic complications aside from SPS to explain the neurologic disability. There was no significant difference in the time from symptom onset to diagnosis and the time from diagnosis to immunotherapy including IVIg between the 2 groups (Table 2).
Table 2.
Univariable Regression Analysis Assessing Predictors of Poor Outcome (mRS ≥3) at the Last Clinical Follow-up in 29 Patients With GAD65 Antibodies

| Variable | Good outcome (n = 19) | Poor outcome (n = 10) | Univariable analysis | |
| OR (95% CI) | p Value | |||
| Female (%) | 15/20 (79) | 8/10 (25) | 0.94 (0.11–5.98) | 0.94 |
| Age at onset, y, median (range) | 50 (23–71) | 52 (25–67) | 0.98 (0.92–1.04) | 0.53 |
| Time from symptom onset to diagnosis, mo, median (range) | 12 (1–84) | 12 (2–96) | 0.99 (0.96–1.02)) | 0.48 |
| Clinical phenotype (%) | ||||
| Classic SPS | 13/19 (68) | 7/10 (70) | — | |
| SLS | 6/19 (32) | 3/10 (30) | — | |
| PERM | 0 (0) | 1 (0) | — | |
| Type 1 DM (%) | 4/19 (21) | 8/10 (80) | 15.00 (2.60–131.58) | 0.002 |
| Other autoimmune diseases (%) | 9/19 (47) | 2/10 (20) | 0.28 (0.04–1.48) | 0.14 |
| Malignancy (%) | 1/19 (5) | 2/10 (13) | 4.50 (0.38–105.43) | 0.23 |
| mRS ≥4 at baseline | 11/19 (58) | 7/10 (63) | 0.59 (0.10–2.88) | 0.52 |
| Time from diagnosis to immunotherapy, mo, median (range) | 0 (0–7) | 1 (0–9) | 0.85 (0.51–1.24) | 0.38 |
| Time from diagnosis to IVIg, mo, median (range) | 1 (0–7) | 1 (0–3) | 1.01 (0.54–2.02) | 1.00 |
| Type of therapy | ||||
| Without immunotherapy | 6/19 (32) | 1/10 (10) | 0.24 (0.01–1.76) | 0.17 |
| Without effective long-term immunotherapy | 2/19 (11) | 7/10 (70) | 19.83 (3.17–191.47) | 0.001 |
| With effective long-term immunotherapy | 11/19 (58) | 2/10 (20) | 0.18 (0.02–0.96) | 0.05 |
Abbreviations: DM = diabetes mellitus; GAD65 = glutamic acid decarboxylase 65; mRS = modified Rankin Scale; PERM = progressive encephalomyelitis with rigidity and myoclonus; SLS = stiff-limb syndrome.
All p values were calculated by the likelihood ration test.
GlyR-Positive, Seronegative, and Other Categories
The 22 GAD65-negative patients were grouped according to clinical phenotype as follows: classic SPS (10 patients, 45%), SLS (5 patients, 23%), and PERM (7 patients, 32%). One patient with classic SPS who was Ri-positive and 8 who did not undergo antibody screening were excluded. Of the 8 GAD65-negative patients, 3 who did not take an EMG test or did not show lack of antagonist inhibition on EMG were excluded. Of the 10 patients screened for antibodies and/or lack of antagonist inhibition on EMG, 4 had autoantibodies against GlyR and 1 had autoantibodies against GABABR. One GABABR-positive patient had SLS but not limbic encephalitis or status epilepticus. We excluded this GABABR-positive patient from the SPS category. The Ri-positive patient was classified into other categories (Figure 2, eTable 5, links.lww.com/NXI/A908).
The 9 GAD65-negative patients were finally classified into 3 groups on the basis of clinical phenotype: classic SPS (2 patients, 22%), SLS (2 patients, 22%), and PERM (5 patients, 56%).
The patients with classic SPS were seronegative. One patient with SLS was seronegative, whereas the other patient with SLS had GlyR antibodies. Of the 5 patients with PERM, 3 were positive for GyR antibodies and 2 were seronegative. Among the 9 GAD65-negative patients, 5 (56%) had malignancies (lung cancer, tongue cancer, lymphoma). Patients in the GlyR-positive group and the seronegative group showed severe symptoms at baseline, but those in the GlyR-positive group showed great improvement after treatment compared with those in the seronegative group. Half of the patients in the GlyR-positive group received first-line and second-line immunotherapies (eTable 3, links.lww.com/NXI/A906).
The clinical characteristics of the GlyR-positive group, the seronegative group, and the other categories are summarized in eTable 5 (links.lww.com/NXI/A908).
Discussion
We elucidated the current epidemiologic, clinical, and immunologic profiles of GAD65-positive SPS in Japan and the treatments given. The estimated prevalence of GAD65-positive SPS was 0.11 per 100,000 population. The prevalence, sex distribution, age at onset, and predominance of classic SPS were similar to those reported previously.2-4,12,28 The outcome of GAD65-positive SPS was generally favorable compared with those of previous studies.3,4,28
Patients with GAD65 antibodies, regardless of titer, had a high frequency of type 1 DM, other autoimmune diseases, and CSF oligoclonal bands (OCBs), although no significant differences were observed between the high-titer and low-titer GAD65 antibody groups. The time from symptom onset to diagnosis was significantly longer in the high-titer GAD65 antibody group than in the low-titer GAD65 antibody group. The fact that 2 patients in the low-titer GAD65 antibody group showed high-titer GAD65 antibodies while their progress was being monitored (data not shown) underscored the need to re-examine GAD65 titer after a period of time. Although the median mRS at baseline was slightly higher in the high-titer GAD65 antibody group than in the low-titer GAD65 antibody group, there was no significant difference in the median mRS at the last follow-up between the 2 groups. These findings are in line with previous studies indicating that anti-GAD65 antibody titers are not correlated with response to therapies, such as IVIg and rituximab.29,30
Interestingly, a high titer of intrathecal GAD65 antibodies was detected in 1 patient with a low titer of serum GAD65 antibodies (eFigure 1, links.lww.com/NXI/A909, eMaterial, links.lww.com/NXI/A910). This finding of intrathecal production of GAD65 antibodies is considered the strongest evidence linking a neurologic syndrome to autoimmunity.31,32 This means that even when serum GAD65 antibody levels are low, it is important to test CSF for GAD65 antibodies, as recommended in previous studies.13,31
We also found an independent association between type 1 DM and the lack of effective long-term immunotherapy, and these risk factors were related to poor outcome in patients with GAD65 antibodies. Baseline disease severity and the presence of GAD65 antibodies are poor prognostic factors in SPS.4 However, the frequency of poor outcome (mRS ≥3 at the last follow-up in GAD65-positive patients with high disease severity (mRS ≥4 at baseline)) of 39% was lower than that (80%) of a previous study.3
In our cohort, type 1 DM was detected only in the GAD65-positive group. Type 1 DM and SPS share a common immunogenetic background.2 In this study, 11 (61%) patients with high disease severity underwent long-term immunotherapy. However, poor-outcome patients who had type 1 DM tended to avoid long-term immunotherapy, such as oral prednisolone, although they had severe symptoms at baseline (eTable 4, links.lww.com/NXI/A907).
The outcome of GAD65-positive patients with SPS is expected to worsen when the diagnosis is made at a very late stage or effective immunotherapy is not initiated early on.28 We suppose that the outcome of GAD65-positive patients with SPS is favorable because the time from onset to diagnosis and the time from diagnosis to initiation of treatment are shorter than those previously reported.4,28
Improvement brought about by IVIg infusion may last for 1–4 months, and repeated infusions are required if there are benefits after the first infusion.33 A study of GAD65-positive patients has demonstrated the long-term efficacy of IVIg maintenance therapy; however, monthly IVIg maintenance therapy is infrequently prescribed in Japan.34 In this study, 1 good-outcome patient received monthly IVIg, and 3 good-outcome patients received an IVIg induction dose at approximately 4- to 7-month intervals, whereas 5 poor-outcome patients received an IVIg induction dose at approximately 4- to 12-month intervals (eTable 4, links.lww.com/NXI/A907). Taken together, the long-term benefits of IVIg may not be sufficiently gained in GAD65-positive patients with poor outcome.
Autopsies performed on patients with GAD65-positive SPS have demonstrated the infiltration of cytotoxic T cells or focal perivascular lymphocyte cuffing in the spinal cord and neuronal loss.35,36 Because GAD65 is an intracellular protein and GAD65 antibodies may not be the direct causative factor, patients with GAD65 antibodies are believed to be in an immunologically activated condition. Our observations suggest the need to consider early aggressive immunotherapy including IVIg maintenance therapy for GAD65-positive patients with high disease severity to prevent immune reactions and neuronal reactions.
Patients with GlyR antibodies were more frequently found in the PERM group than in the classic SPS group, consistent with a previous report.4 The age at onset in the GlyR-positive group was also similar to that reported previously, namely most patients with PERM presented with symptoms in their fifth or sixth decade.5,9 We found anti-GlyR antibodies in 8% (one patient) of GAD65-positive patients who were screened for GlyR antibodies, confirming the previous finding that GlyR antibodies were also found in 10–15% of GAD65-positive patients.37,38 Our data are in agreement with the results of the previous study describing that seronegative and GlyR-positive patients showed higher disease severity than GAD65-positive patients, although the outcomes of the GlyR-positive patients were better than those of the GAD65-positive patients.4 This finding could be explained as follows: more rapid and severe symptom onset in PERM than in SPS, GlyR-positive patients received more aggressive treatment than GAD65-positive patients, and the nature of GlyR, that is, it is a cell surface antigen.4 Indeed, in this study, half of patients in the GlyR-positive group underwent a combination of first-line and second-line immunotherapies.
Patients in the seronegative group showed poorer outcome than those in the GAD65-positive or GlyR-positive group. The poor outcome might be explained by the absence of immunotherapy or insufficient immunotherapy as previously reported.4 Moreover, half of the seronegative patients were associated with malignancies. There may be another etiology in seronegative SPS because the GAD65-seronegative patients had a higher frequency of malignancies than the GAD65-seropositive patients.3 In addition, there was a coincidental finding that could not be related to PNS.
Autoantibodies against 6 antigens, including GAD65, GlyR, amphiphysin, gephyrin, DPPX, and GABAAR, but not GABABR, have been reported in patients with SPS.6-8,26,39,40 Most patients with GABABR antibodies develop early seizures or status epilepticus as a component of limbic encephalitis and rapidly progressive cognitive decline.41-44 Our patient with GABABR antibodies did not show well-known clinical features, such as seizures and cognitive impairment; instead, this patient showed painful spasms of the legs that might lead to misdiagnosis. The detection of GABABR antibodies in SPS requires careful interpretation.
This study has the following limitations:
We used a random sampling method; the reason being that enormous effort was required to conduct a survey of not only neurologists but also internists in Japan. The estimated response rate of the first survey was 48.7%, and this estimate was based on a method in which nonresponse study centers were taken into consideration.15
From the 94 patients reported in the first survey, the final number of patients from whom detailed clinical data could be obtained decreased to 55. We could not derive conclusive information from this small data size. In addition, because we could not statistically confirm whether the presence of GAD65 antibodies had an effect on poor outcome, we focused our prognostic analysis on only GAD65-positive patients.
Because the anti-GlyR antibody test was recently established in Japan, this study might have been skewed toward GAD65-positive patients.
We did not perform CBA to detect GAD65 antibodies nor did we find any correlation between GAD65 pattern on immunohistochemistry and GAD65 antibody levels, as previously reported.45
Many clinical variables potentially associated with poor outcome were examined in patients with GAD65 antibodies (number of patients = 10). Multiple testing may have led to the increase in alpha error. However, because of the hypothesis-generating nature of this study, no correlation of alpha error was conducted. Therefore, the p values between 0.05 and 0.004 (Table 2) should be considered carefully.
Because outcome measures other than median mRS were lacking, we decided to focus on detailed clinical information including treatment content.
The maintenance therapy varied. Corticosteroids were most commonly used, although one anecdotal report showed limited benefits. Case reports showed that tacrolimus was effective when used after steroid therapy.46 Because there were cases in which prognosis was good with oral steroids and/or tacrolimus after IVMP or an IVIg induction dose, steroid and tacrolimus were included in the effective treatment category. Immunotherapeutic options other than IVIg require further evidenced-based consideration.
In conclusion, our study demonstrated that the estimated prevalence of GAD65-positive SPS is 0.1 per 100,000 population, and the outcome of GAD65-positive patients is generally favorable. To establish a clinical correlation with GAD65 antibodies, further investigation including newly identified autoantibodies in a larger sample size is necessary.
Acknowledgment
The authors thank Dr. Josep Dalmau (Service of Neurology, IDIBAPS Hospital Clinic, University of Barcelona, Barcelona, Spain) and Dr. Angela Vincent (Nuffield Department of Clinical Neurosciences, John Radcliffe Hospital, Oxford University, Oxford, UK) for examining the results of antibody tests against GlyR and other inhibitory synaptic proteins in some patients.
Glossary
- CBA
cell-based assay
- EIA
enzyme immunoassay
- GAD65
glutamic acid decarboxylase 65
- GlyR
glycine receptor
- IVIg
IV immunoglobulin
- IVMP
IV methylprednisolone
- mRS
modified Rankin Scale
- PERM
progressive encephalomyelitis with rigidity and myoclonus
- PNS
paraneoplastic neurological syndrome
- RIA
radioimmunoassay
- SLS
stiff-limb syndrome
- SPS
stiff-person syndrome
Appendix 1. Authors

| Name | Location | Contribution |
| Naoko Matsui, MD, PhD | Department of Neurology, Tokushima University Graduate School of Biomedical Sciences, Tokushima, Japan | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Keiko Tanaka, MD, PhD | Department of Animal Model Development, Brain Research Institute, Niigata University; Department of Multiple Sclerosis Therapeutics, Fukushima Medical University, School of Medicine, Fukushima, Japan | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data |
| Mitsuyo Ishida, PhD | Department of Neurology, Tokushima University Graduate School of Biomedical Sciences, Tokushima, Japan | Analysis or interpretation of data |
| Yohei Yamamoto, MD, PhD | Department of Neurology, Tokushima University Hospital, Tokushima, Japan | Major role in the acquisition of data |
| Yuri Matsubara, MD, MPH | Department of Public Health, Jichi Medical University, Shimotsuke, Japan | Analysis or interpretation of data |
| Reiko Saika, MD, PhD | Department of Neurology, National Institute of Neuroscience, National Center of Neurology and Psychiatry, Tokyo, Japan | Major role in the acquisition of data |
| Takahiro Iizuka, MD, PhD | Department of Neurology, Kitasato University School of Medicine, Sagamihara, Japan | Major role in the acquisition of data; analysis or interpretation of data |
| Koshi Nakamura, MD, PhD | Department of Public Health and Hygiene, Graduate School of Medicine, University of the Ryukyus, Okinawa, Japan | Analysis or interpretation of data |
| Nagato Kuriyama, MD, PhD | Department of Epidemiology for Community Health and Medicine, Kyoto Prefectural University of Medicine; Department of Social Health Medicine, Shizuoka Graduate University of Public Health, Shizuoka, Japan | Analysis or interpretation of data |
| Makoto Matsui, MD, PhD | Department of Neurology, Kanazawa Medical University, Ishikawa, Japan | Analysis or interpretation of data |
| Kokichi Arisawa, MD, PhD | Department of Preventive Medicine, Tokushima University Graduate School of Biomedical Sciences, Tokushima, Japan | Analysis or interpretation of data |
| Yosikazu Nakamura, MD, MPH | Department of Public Health, Jichi Medical University, Shimotsuke, Japan | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Ryuji Kaji, MD, PhD | National Hospital Organization Utano Hospital, Kyoto, Japan | Major role in the acquisition of data |
| Satoshi Kuwabara, MD, PhD | Department of Neurology, Graduate School of Medicine, Chiba University, Chiba, Japan | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design |
| Yuishin Izumi, MD, PhD | Department of Neurology, Tokushima University Graduate School of Biomedical Sciences, Tokushima, Japan | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design |
Appendix 2. Coinvestigators
| Name | Location | Role | Contribution |
| Nagaaki Katoh, MD, PhD | Shinshu University School of Medicine, Matsumoto, Japan | Site Investigator | Major role in the acquisition of data |
| Shinji Kondo, MD, PhD | Sanin-Rosai Hospital, Yonago, Japan | Site Investigator | Major role in the acquisition of data |
| Hiroshi Otani, MD, PhD | Kuwamizu Hospital, Kumamoto, Japan | Site Investigator | Major role in the acquisition of data |
| Misa Nakano, MD, PhD | Suita Municipal Hospital, Suita, Japan | Site Investigator | Major role in the acquisition of data |
| Akiko Ishii, MD, PhD | University of Tsukuba, Tsukuba, Japan | Site Investigator | Major role in the acquisition of data |
| Masashi Watanabe, MD | Ehime Prefectural Central Hospital, Matsuyama, Japan | Site Investigator | Major role in the acquisition of data |
| Kengo Maeda, MD, PhD | Vories Memorial Hospital, Omihachiman, Japan | Site Investigator | Major role in the acquisition of data |
| Tomoyuki Miyamoto, MD, PhD | Dokkyo Medical University Saitama Medical Center, Koshigaya, Japan | Site Investigator | Major role in the acquisition of data |
| Michiaki Koga, MD, PhD | Yamaguchi University Graduate School of Medicine, Ube, Japan | Site Investigator | Major role in the acquisition of data |
| Yuko K. Takahashi, MD, PhD | Yokohama City Minato Red Cross Hospital, Yokohama, Japan | Site Investigator | Major role in the acquisition of data |
| Yuko Takeuchi, MD, PhD | Masuko Memorial Hospital, Nagoya, Japan | Site Investigator | Major role in the acquisition of data |
| Yoshitaka Yamanaka, MD, PhD | Chiba University, Chiba, Japan | Site Investigator | Major role in the acquisition of data |
| Kazumoto Shibuya, MD, PhD | Chiba University, Chiba, Japan | Site Investigator | Major role in the acquisition of data |
| Toshio Fukutake, MD, PhD | Kameda Medical Center, Kamogawa, Japan | Site Investigator | Major role in the acquisition of data |
| Keisuke Tokui, MD, PhD | Aichi Medical University, Nagakute, Japan | Site Investigator | Major role in the acquisition of data |
| Chikako Kaneko, MD, PhD | Southern TOHOKU Research Institute for Neuroscience, Southern TOHOKU General Hospital, Koriyama, Japan | Site Investigator | Major role in the acquisition of data |
| Nobuhiro Ogawa, MD, PhD | Shiga University of Medical Science, Otsu, Japan | Site Investigator | Major role in the acquisition of data |
| Masataka Kitaguchi, MD, PhD | Baba Memorial Hospital, Sakai, Japan | Site Investigator | Major role in the acquisition of data |
| Kishin Koh, MD, PhD | University of Yamanashi, Chuo, Japan | Site Investigator | Major role in the acquisition of data |
| Hidenori Ogata, MD, PhD | Kyushu University, Fukuoka, Japan | Site Investigator | Major role in the acquisition of data |
| Norihito Yoshida, MD, PhD | Saitama Medical Center, Kawagoe, Japan | Site Investigator | Major role in the acquisition of data |
| Hitoki Nanaura, MD, PhD | Nara Medical University, Kashihara, Japan | Site Investigator | Major role in the acquisition of data |
| Takao Kiriyama, MD, PhD | Nara Medical University, Kashihara, Japan | Site Investigator | Major role in the acquisition of data |
| Yoshio Tsuboi, MD, PhD | Fukuoka University, Fukuoka, Japan | Site Investigator | Major role in the acquisition of data |
| Shinsuke Fujioka, MD, PhD | Fukuoka University, Fukuoka, Japan | Site Investigator | Major role in the acquisition of data |
| Kentaro Deguchi, MD, PhD | Okayama City Hospital, Okayama, Japan | Site Investigator | Major role in the acquisition of data |
| Sachiko Hasebe, MD | Osaka Red Cross Hospital, Osaka, Japan | Site Investigator | Major role in the acquisition of data |
| Jun Ochiai, MD, PhD | Nagoya-Ekisaikai Hospital, Nagoya, Japan | Site Investigator | Major role in the acquisition of data |
| Shunichi Satoh, MD, PhD | Satou Clinic, Nagano, Japan | Site Investigator | Major role in the acquisition of data |
| Masafumi Sanefuji, MD, PhD | Kyushu University, Fukuoka, Japan | Site Investigator | Major role in the acquisition of data |
| Yoshito Ishizaki, MD, PhD | Kyushu University, Fukuoka, Japan | Site Investigator | Major role in the acquisition of data |
| Fumihiro Yanagimura, MD, PhD | National Hospital Organization Niigata Hospital, Niigata, Japan | Site Investigator | Major role in the acquisition of data |
| Toshiyuki Nakanishi, MD, PhD | Nerima Hikarigaoka Hospital, Tokyo, Japan | Site Investigator | Major role in the acquisition of data |
| Hiroyuki Saito, MD, MBA | Yamaguchi University Hospital, Ube, Japan | Site Investigator | Major role in the acquisition of data |
| Kazuhiro Horiuchi, MD | Hakodate Municipal Hospital, Hakodate, Japan | Site Investigator | Major role in the acquisition of data |
| Satoshi Orimo, MD, PhD | Kamiyoga Setagaya-Dori Avenue Clinic, Tokyo, Japan | Site Investigator | Major role in the acquisition of data |
| Noritoshi Arai, MD, PhD | National Center for Global Health and Medicine, Tokyo, Japan | Site Investigator | Major role in the acquisition of data |
| Juntaro Kaneko, MD, PhD | Kitasato University School of Medicine, Sagamihara, Japan | Site Investigator | Major role in the acquisition of data |
| Shigeaki Suzuki, MD, PhD | Keio University School of Medicine, Tokyo, Japan | Site Investigator | Major role in the acquisition of data |
| Yuki Tajiri, MD, PhD | Tottori University, Yonago, Japan | Site Investigator | Major role in the acquisition of data |
| Hiroshi Takigawa, MD, PhD | Tottori University, Yonago, Japan | Site Investigator | Major role in the acquisition of data |
| Ritsuko Hanajima, MD, PhD | Tottori University, Yonago, Japan | Site Investigator | Major role in the acquisition of data |
| Etsuro Nakanishi, MD, PhD | Kyoto University Graduate School of Medicine, Kyoto, Japan | Site Investigator | Major role in the acquisition of data |
| Kimitoshi Kimura, MD, PhD | Kyoto University Graduate School of Medicine, Kyoto, Japan | Site Investigator | Major role in the acquisition of data |
| Hiroki Yamazaki, MD | Tokushima University, Tokushima, Japan | Site Investigator | Major role in the acquisition of data |
| Yoshihiko Nishida, MD, PhD | Itsuki Hospital, Tokushima, Japan | Site Investigator | Major role in the acquisition of data |
Study Funding
The Japanese Society for the Promotion of Science Grant-in-Aid for Scientific Research C (Grant No. 20K0778400) and the Health and Labor Sciences Research Grant on Intractable Diseases (Neuroimmunologic Diseases) from the Ministry of Health, Labour and Welfare of Japan (20FC1030).
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NN for full disclosures.
References
- 1.Dalakas MC. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol. 2009;11(2):102-110. doi: 10.1007/s11940-009-0013-9 [DOI] [PubMed] [Google Scholar]
- 2.Dalakas MC, Fujii M, Li M, McElroy B. The clinical spectrum of anti-GAD antibody-positive patients with stiff-person syndrome. Neurology. 2000;55(10):1531-1535. doi: 10.1212/wnl.55.10.1531 [DOI] [PubMed] [Google Scholar]
- 3.McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. 2012;69(2):230-238. doi: 10.1001/archneurol.2011.991 [DOI] [PubMed] [Google Scholar]
- 4.Martinez-Hernandez E, Ariño H, McKeon A, et al. Clinical and immunologic investigations in patients with stiff-person spectrum disorder. JAMA Neurol. 2016;73(6):714-720. doi: 10.1001/jamaneurol.2016.0133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baizabel-Carvallo JF, Jankovic J. Stiff-person syndrome: insights into a complex autoimmune disorder. J Neurol Neurosurg Psychiatry. 2015;86(8):840-848. doi: 10.1136/jnnp-2014-309201 [DOI] [PubMed] [Google Scholar]
- 6.Solimena M, Folli F, Aparisi R, Pozza G, De Camilli P. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in stiff-man syndrome. N Engl J Med. 1990;322(22):1555-1560. doi: 10.1056/nejm199005313222202 [DOI] [PubMed] [Google Scholar]
- 7.Solimena M, De Camilli P. Autoimmunity to glutamic decarboxylase (GAD) in stiff-man syndrome and insulin-dependent diabetes mellitus. Trends Neurosci. 1991;14(10):452-457. doi: 10.1016/0166-2236(91)90044-u [DOI] [PubMed] [Google Scholar]
- 8.Hutchinson M, Waters P, McHugh J, et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology. 2008;71(16):1291-1292. doi: 10.1212/01.wnl.0000327606.50322.f0 [DOI] [PubMed] [Google Scholar]
- 9.Carvajal-González A, Leite MI, Waters P, et al. Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain. 2014;137(8):2178-2192. doi: 10.1093/brain/awu142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinson SR, Lopez-Chiriboga AS, Bower JH, et al. Glycine receptor modulating antibody predicting treatable stiff-person spectrum disorders. Neurol Neuroimmunol Neuroinflamm. 2018;5(2):e438. doi: 10.1212/nxi.0000000000000438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moersch FP, Woltman HW. Progressive fluctuating muscular rigidity and spasm (“stiff-man” syndrome); report of a case and some observations in 13 other cases. Proc Staff Meet Mayo Clin. 1956;31(15):421-427. [PubMed] [Google Scholar]
- 12.Meinck HM, Thompson PD. Stiff man syndrome and related conditions. Mov Disord. 2002;17(5):853-866. doi: 10.1002/mds.10279 [DOI] [PubMed] [Google Scholar]
- 13.Graus F, Saiz A, Dalmau J. GAD antibodies in neurological disorders—insights and challenges. Nat Rev Neurol. 2020;16(7):353-365. doi: 10.1038/s41582-020-0359-x [DOI] [PubMed] [Google Scholar]
- 14.Muñoz-Lopetegi A, de Bruijn MAAM, Boukhrissi S, et al. Neurologic syndromes related to anti-GAD65: clinical and serologic response to treatment. Neurol Neuroimmunol Neuroinflamm. 2020;7(3):e696. doi: 10.1212/nxi.0000000000000696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura Y, Matsumoto T, Tamakoshi A, et al. Prevalence of idiopathic hypoparathyroidism and pseudohypoparathyroidism in Japan. J Epidemiol. 2000;10(1):29-33. doi: 10.2188/jea.10.29 [DOI] [PubMed] [Google Scholar]
- 16.Miyamoto K, Fujihara K, Kira JI, et al. Nationwide epidemiological study of neuromyelitis optica in Japan. J Neurol Neurosurg Psychiatry. 2018;89(6):667-668. doi: 10.1136/jnnp-2017-317321 [DOI] [PubMed] [Google Scholar]
- 17.Suichi T, Misawa S, Beppu M, et al. Prevalence, clinical profiles, and prognosis of POEMS syndrome in Japanese nationwide survey. Neurology. 2019;93(10):e975-e983. doi: 10.1212/wnl.0000000000008062 [DOI] [PubMed] [Google Scholar]
- 18.Matsubara Y, Nakamura Y, Tamura N, et al. A nationwide questionnaire survey on the prevalence of ankylosing spondylitis and non-radiographic axial spondyloarthritis in Japan. Mod Rheumatol. 2022;32(5):960-967. doi: 10.1093/mr/roab096 [DOI] [PubMed] [Google Scholar]
- 19.Lorish TR, Thorsteinsson G, Howard FM Jr. Stiff-man syndrome updated. Mayo Clin Proc. 1989;64(6):629-636. doi: 10.1016/s0025-6196(12)65339-7 [DOI] [PubMed] [Google Scholar]
- 20.Brown P, Rothwell JC, Marsden CD. The stiff leg syndrome. J Neurol Neurosurg Psychiatry. 1997;62(1):31-37. doi: 10.1136/jnnp.62.1.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saiz A, Graus F, Valldeoriola F, Valls-Solé J, Tolosa E. Stiff-leg syndrome: a focal form of stiff-man syndrome. Ann Neurol. 1998;43(3):400-403. doi: 10.1002/ana.410430322 [DOI] [PubMed] [Google Scholar]
- 22.Barker RA, Revesz T, Thom M, Marsden CD, Brown P. Review of 23 patients affected by the stiff man syndrome: clinical subdivision into stiff trunk (man) syndrome, stiff limb syndrome, and progressive encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry. 1998;65(5):633-640. doi: 10.1136/jnnp.65.5.633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banks JL, Marotta CA. Outcomes validity and reliability of the modified Rankin scale: implications for stroke clinical trials: a literature review and synthesis. Stroke. 2007;38(3):1091-1096. doi: 10.1161/01.str.0000258355.23810.c6 [DOI] [PubMed] [Google Scholar]
- 24.Boronat A, Gelfand JM, Gresa-Arribas N, et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol. 2013;73(1):120-128. doi: 10.1002/ana.23756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Hernandez E, Sepulveda M, Rostásy K, et al. Antibodies to aquaporin 4, myelin-oligodendrocyte glycoprotein, and the glycine receptor alpha1 subunit in patients with isolated optic neuritis. JAMA Neurol. 2015;72(2):187-193. doi: 10.1001/jamaneurol.2014.3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petit-Pedrol M, Armangue T, Peng X, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 2014;13(3):276-286. doi: 10.1016/s1474-4422(13)70299-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graus F, Vogrig A, Muñiz-Castrillo S, et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neuroimmunol Neuroinflamm. 2021;8(4):e1014. doi: 10.1212/nxi.0000000000001014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rakocevic G, Alexopoulos H, Dalakas MC. Quantitative clinical and autoimmune assessments in stiff person syndrome: evidence for a progressive disorder. BMC Neurol. 2019;19:1. doi: 10.1186/s12883-018-1232-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dalakas MC, Fujii M, Li M, Lutfi B, Kyhos J, McElroy B. High-dose intravenous immune globulin for stiff-person syndrome. N Engl J Med. 2001;345(26):1870-1876. doi: 10.1056/nejmoa01167 [DOI] [PubMed] [Google Scholar]
- 30.Dalakas MC, Rankocevic G, Dambrosia JM, Alexopoulos H, McElroy B. A double-blind, placebo-controlled study of rituximab in patients with stiff-person syndrome. Ann Neurol. 2017;82(2):271-277. doi: 10.1002/ana.25002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsiortou P, Alexopoulos H, Dalakas MC. GAD antibody-spectrum disorders: progress in clinical phenotypes, immunopathogenesis and therapeutic interventions. Ther Adv Neurol Disord. 2021;14:17562864211003486. doi: 10.1177/17562864211003486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chéramy M, Hampe CS, Ludvigsson J, Casas R. Characteristics of in-vitro phenotypes of glutamic acid decarboxylase 65 autoantibodies in high-titre individuals. Clin Exp Immunol. 2013;171(3):247-254. doi: 10.1111/cei.12026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dalakas MC. Advances in the pathogenesis and treatment of patients with stiff person syndrome. Curr Neurol Neurosci Rep. 2008;8(1):48-55. doi: 10.1007/s11910-008-0009-y [DOI] [PubMed] [Google Scholar]
- 34.Yi J, Dalakas MC. Long-term effectiveness of IVIg maintenance therapy in 36 patients with GAD antibody-positive stiff-person syndrome. Neuroimmunol Neuroinflamm. 2022;9(5):e200011. doi: 10.1212/nxi.0000000000200011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holmøy T, Skorstad G, Røste LS, Scheie D, Alvik K. Stiff person syndrome associated with lower motor neuron disease and infiltration of cytotoxic T cells in the spinal cord. Clin Neurol Neurosurg. 2009;111(8):708-712. doi: 10.1016/j.clineuro.2009.06.005 [DOI] [PubMed] [Google Scholar]
- 36.Ishizawa K, Komori T, Okayama K, et al. Large motor neuron involvement in Stiff-man syndrome: a qualitative and quantitative study. Acta Neuropathol. 1999;97(1):63-70. doi: 10.1007/s004010050956 [DOI] [PubMed] [Google Scholar]
- 37.McKeon A, Martinez-Hernandez E, Lancaster E, et al. Glycine receptor autoimmune spectrum with Stiff-man syndrome phenotype. JAMA Neurol. 2013;70(1):44-50. doi: 10.1001/jamaneurol.2013.574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alexopoulos H, Akrivou S, Dalakas MC. Glycine receptor antibodies in stiff-person syndrome and other GAD-positive CNS disorders. Neurology. 2013;81(22):1962-1964. doi: 10.1212/01.wnl.0000436617.40779.65 [DOI] [PubMed] [Google Scholar]
- 39.De Camilli P, Thomas A, Cofiell R, et al. The synaptic vesicle-associated protein amphiphysin is the 128-kD autoantigens of Stiff-man syndrome with breast cancer. J Exp Med. 1993;178(6):2219-2223. doi: 10.1084/jem.178.6.2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butler MH, Hayashi A, Ohkoshi N, et al. Autoimmunity to gephyrin in stiff-man syndrome. Neuron. 2000;26(2):307-312. doi: 10.1016/s0896-6273(00)81165-4 [DOI] [PubMed] [Google Scholar]
- 41.Lancaster E, Lai M, Peng X, et al. Antibodies to the GABAB receptor in limbic encephalitis with seizures: case series and characterization of the antigen. Lancet Neurol. 2010;9(1):67-76. doi: 10.1016/s1474-4422(09)70324-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Höftberger R, Titulaer MJ, Sabater L, et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology. 2013;81(17):1500-1506. doi: 10.1212/wnl.0b013e3182a9585f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Coevorden-Hameete MH, de Bruijn MAAM, de Graaff E, et al. The expanded clinical spectrum of anti-GABABR encephalitis and added value of KCTD16 autoantibodies. Brain. 2019;142(6):1631-1643. doi: 10.1093/brain/awz094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bastiaansen AEM, van Steenhoven RW, de Bruijn MAAM, et al. Autoimmune encephalitis resembling dementia syndromes. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1039. doi: 10.1212/nxi.0000000000001039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saiz A, Blanco Y, Sabater L, et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain. 2008;131(10):2553-2563. doi: 10.1093/brain/awn183 [DOI] [PubMed] [Google Scholar]
- 46.Nakane S, Fujita K, Shibuta Y, et al. Successful treatment of stiff person syndrome with sequential use of tacrolimus. J Neurol Neurosurg Psychiatry. 2013;84(10):1177-1180. doi: 10.1136/jnnp-2013-305425 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Any data not published in the article will be shared anonymously on request by any qualified investigator.