

Abstract
Protein aggregates are a common feature of nearly all neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS). Here we describe a method to quickly and accurately measure protein aggregation in cells expressing a fluorescently tagged aggregation-prone protein. This unbiased method obviates the need for manual scoring and facilitates the identification of factors governing protein self-assembly and its downstream consequences for cell heath.
Keywords: Protein aggregation, Neurodegeneration, Proteostasis, Single cell, Longitudinal microscopy
1. Introduction
Mutations in UBQLN2 cause X-linked ALS and ALS/frontotemporal dementia (FTD) [1–4]. UBQLN2-positive inclusions are observed in the brains and spinal cords of ALS/FTD patients with and without UBQLN2 mutations [1–5]. UBQLN2 also accumulates within the hippocampus and cerebellum of patients with ALS or frontotemporal dementia (FTD) due to C9ORF72 hexanucleotide expansion mutations, the most common cause of familial ALS and FTD [5]. Furthermore, UBQLN2 colocalizes with pathological aggregates characteristic of several neurodegenerative conditions, including huntingtin inclusion bodies seen in Huntington’s disease and Lewy bodies observed in synucleinopathies [6, 7]. The identification and quantification of UBQLN2 aggregates may therefore be of broad relevance to neurodegenerative disease research.
Similar to many other proteins implicated in ALS/FTD, UBQLN2 phase separates into liquid-like condensates, both in vitro and in cells [8–12]. How UBQLN2 phase separation impacts its function and pathogenicity remains unclear. Recent data suggest that UBQLN2 negatively regulates the assembly of stress granules (SGs), membraneless organelles composed of phase-separated proteins and RNA [13]. UBQLN2 phase separation may be a prerequisite for its interaction with SG proteins, as well as with components of other cytoplasmic and nuclear membraneless organelles.
We previously investigated the factors that govern UBQLN2 self-assembly by tagging UBQLN2 with an infrared fluorescent protein (iRFP) and expressing the fusion protein in primary rat cortical neurons [11]. We referred to neuronal iRFP-UBQLN2 assemblies simply as puncta, a word agnostic to quaternary structure, since these structures could realistically represent fluid-like complexes or more stable fibrillar structures. To quantify puncta in a high-throughput and unbiased way, we used the coefficient of variation (CV) of fluorescence intensity, hereafter referred to as CVintensity, of iRFP-UBQLN2 to predict puncta formation. This method builds off prior work showing that cells containing puncta exhibit distinct peaks in area-integrated fluorescence intensity that can reliably differentiate them from cells containing diffusely localized protein [14]. By extension, a neuron with such marked peaks in fluorescence will display a higher standard deviation of fluorescence intensity integrated over the cell area (SDintensity), compared to a neuron showing only a uniform, diffuse, and relatively dim signal. Therefore SDintensity should be similarly effective in discriminating between puncta-positive and puncta-negative cells. To account for differences in single-cell fluorescent intensity, CVintensity can be used instead, since it scales SDintensity by the mean intensity on a per-cell basis. By plotting a receiver operating characteristic (ROC) curve, we found that CVintensity was both a sensitive and a specific predictor of the presence of iRFP-UBQLN2 puncta in neurons [11]. This curve identified a CVintensity threshold value that could accurately segregate cells as those having puncta (above threshold) from those without (below threshold).
In this chapter, we describe how to calculate and use CVintensity in the quantification of UBQLN2 puncta. Although the initial experiments were conducted in iRFP-UBQLN2 expressing neurons, these methods could be readily applied in an analogous fashion to track the self-assembly of any aggregate-prone protein in neurons or other cell types. To facilitate this, we supply scripts to automate the generation of regions of interest (ROIs) that outline each cell, generate an ROC curve, calculate an optimal CVintensity threshold, and calculate/plot the percentage of puncta-positive cells in each experimental condition, thereby making each experiment as consistent and robust as possible.
2. Materials
2.1. Primary Neuron Transfection
Poly-D-Lysine plates and neurobasal complete medium are to be prepared prior to dissection. All other media are to be prepared, filter sterilized, and warmed to 37 °C prior to transfection on in vitro day 4 (DIV4). Each media volume listed below is sufficient for transfecting one 96-well plate.
96-well plates
Poly-D-Lysine.
Neurobasal complete medium (NBC): Neurobasal medium (NBM), 1x Glutamax, 1x B27 supplement, 1x penicillin-streptomycin.
Neurobasal medium with kynurenic acid (NBKY): NBM, 1-mM kynurenic acid hydrate, 10-mM MgCl2, pH of 7.4.
Reduced-serum media-Opti-MEM.
Lipofectamine 2000.
pGW1-UBQLN2-iRFP plasmid,
pGW1-iRFP plasmid,
pGW1-mApple plasmid,
Eclipse Ti inverted microscope (Nikon) equipped with Perfect Focus System, a high aperture 20X objective lens, and Andor iXon3 897 EMCCD camera.
2.2. Software
Microsoft Excel or the free alternative—LibreOffice (https://www.libreoffice.org/download/download/).
FIJI—https://fiji.sc/.
Python3—https://www.python.org/downloads/. The following Python packages must also be installed: “numpy,” “pandas,” “matplotlib.pyplot” (see Note 1).
Sublime Text—https://www.sublimetext.com/ (recommended but not necessary) to edit and run python scripts.
2.3. Provided Files
The following files can be downloaded from https://github.com/BarmadaLab/puncta_id:
Figure 1 data.xlsx—Measurements corresponding to the images in Fig. 1.
experiment_template.xlsx—Provides a data organizing template that is compatible with downstream analyses.
ROI_generator.ijm—A FIJI macro demonstrating how to threshold images and generate ROIs.
ROC_data.csv—Template for organizing data and plotting an ROC curve. This is used as the input file in ROC curve.py.
ROC curve.py—Given a .csv file in the format of ROC_data. csv, will output a ROC curve plot and calculate an optimal CVintensity threshold.
representative_experiment.csv—The input file for puncta.py.
puncta.py—Python script that plots the number of puncta at each time point using the CVintensity threshold determined by the ROC curve. Outputs a modified spreadsheet with the suffix _punta.csv, listing whether each cell contains puncta at each time point as well as a table with the percent of neurons containing puncta at each time point with the suffix _percent.csv.
Fig. 1.
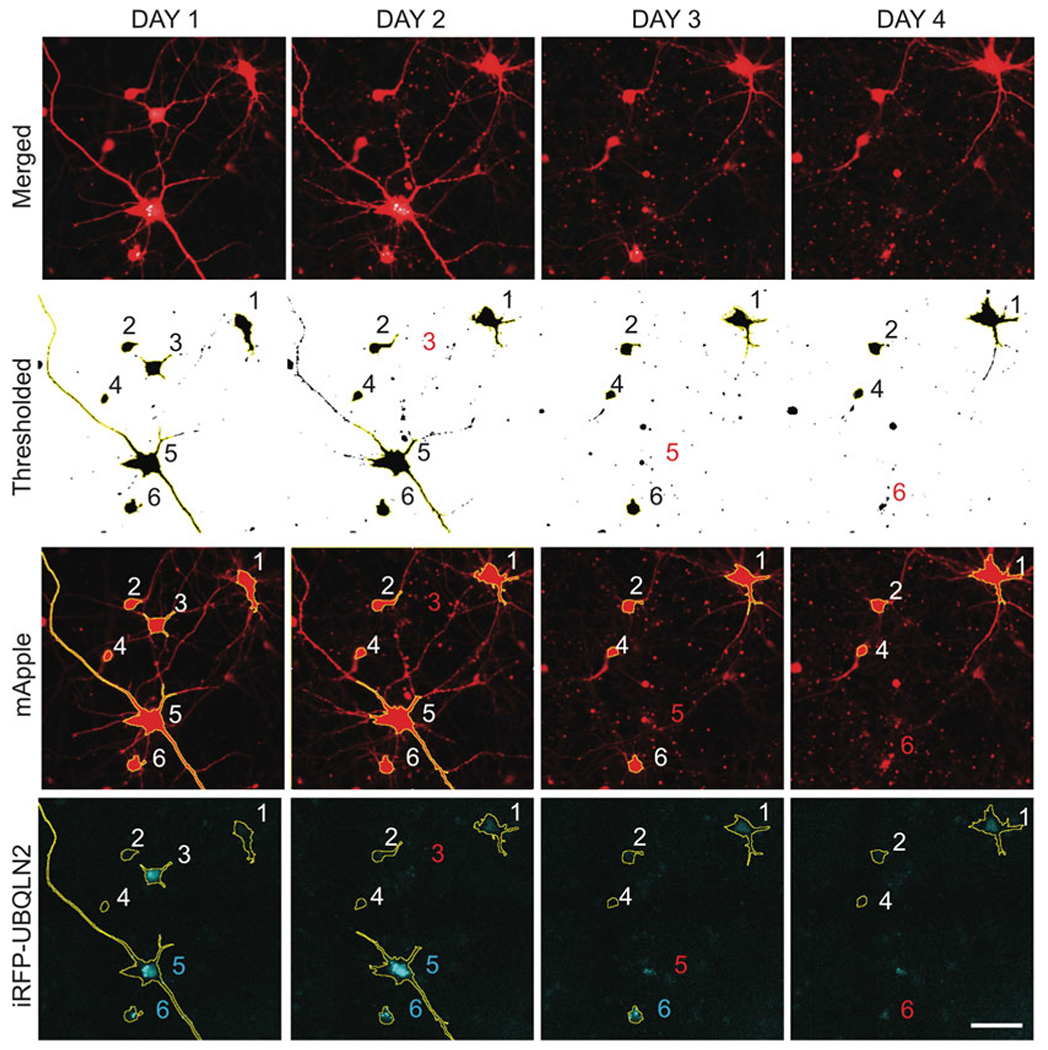
ROI identification for neurons coexpressing mApple (red) and iRFP-UBQLN2 (cyan). mApple channel images are thresholded using the “minimum dark” option. FIJI’s “Analyze Particles” tool is then used to superimpose ROIs onto objects larger than the “min_cell_area” specified by the user. These ROIs are saved to FIJI’s ROI manager and overlayed onto the iRFP channel to measure single-cell iRFP-UBQLN2 fluorescence intensity and standard deviation. Red numbers specify cell death. Cyan numbers indicate neurons with UBQLN2 puncta. Scale bar = 100 μm
3. Methods
3.1. Plating of Primary Rat Cortical Neurons (See Note 2)
Dissect mixed rat primary cortical neurons from embryonic day 20 rat pups, and plate at 0.5 × 10 [6] cells per mL on Poly-D-Lysine-coated 96-well plates. Fill each well with 200 uL of NBC medium (see Note 3).
3.2. Primary Neuron Transfection
A necessary step in this protocol is the specification of cellular regions of interest (ROIs) that outline the borders of each cell to be analyzed (Subheading 3.5). For this to be accomplished, it is essential to co-transfect a cellular morphology marker along with fluorescently labeled UBQLN2 or any protein of interest. A suitable morphology marker will exhibit diffuse localization throughout the entirety of a cell. Green fluorescent protein (GFP) and mApple are two commonly used fluorescent proteins that serve as good morphology markers.
Calculate the amount of DNA, Opti-MEM, and Lipofectamine 2000 needed for transfection master-mixes. Rather than making individual tubes for every well, it is advisable to make up two master-mixes, a DNA mix and a Lipofectamine 2000 mix. The Lipofectamine mix consists of 24.5 uL of Opti-MEM and .5 uL of Lipofectamine per well. For each well of a 96-well plate, the total amount of DNA should sum to 200 ng, and the amount of Opti-MEM should be [25-uL volume of DNA] (see Notes 4 and 5).
Make up DNA and Lipofectamine master-mixes, and incubate separately for 5 min.
Combine DNA and Lipofectamine mixes and incubate for 20 min.
During this incubation step, remove and collect conditioned medium in a sterile container. Wash neurons 2x with 100-uL Neurobasal medium, making sure to minimize the exposure of neurons to air. Place conditioned media in a 37 °C incubator with the lid ajar to allow for gas exchange.
Wash out the Neurobasal medium and replace with 100-uL NBKY medium.
Following the 20-min incubation of the DNA/Lipofectamine mixture (see step 3), add 50 uL of the mix dropwise to each well.
Place the plate in a 37 °C incubator for another 20 min.
Remove the media and replace with 100 uL of NBKY. Perform a second wash with 100-uL NBKY per well.
Replace the NBKY with 100 uL of fresh NBC media and 100 uL of conditioned media per well.
Place the plate in a 37 °C incubator. Fluorescence is typically visible as early as 14–24 h after transfection.
3.3. Imaging
Establish a fiduciary mark to serve as a reference point. This can be any constant mark on the bottom of the plate. By measuring the x and y distances of each acquired image relative to this mark, it is possible to align images over multiple time points.
Find a field of neurons. Record the x and y coordinates relative to the fiduciary.
For each fluorescent channel, focus and determine the proper imaging exposure time. It is important to ensure that the intensity of imaged fluorophores remains within the dynamic range of the detector. Make sure that no images display saturated signals.
Acquire images. For each image, record the x and y coordinates relative to the fiduciary. At subsequent time points, align each field to the coordinates recorded at time point 1.
3.4. Image Processing (See Note 6)
If multiple adjacent images were acquired in a well, stitching can be used to combine individual images into a single contiguous image. Stitching can be performed in FIJI by clicking on the Plugins dropdown menu and then selecting Stitching and then Grid/Collection Stitching. The order that images were acquired will determine which selections are made in the Type and Order dropdown menus. Hit the OK button. The number of images acquired in the well will dictate the Grid size x and Grid size y values. The degree of overlap [%], representing the amount of overlap between two adjacent images, will also need to be adjusted accordingly. The Directory specifies the folder that contains the images that are to be stitched. File names for tiles need to match the names of the images (see Note 7). Make sure that the Display fusion button is checked and Compute overlap is unchecked. Hit OK. Depending on the appearance of the stitched image, the Type, Order, and Degree of overlap [%] may need to be adjusted.
Background subtraction can be used to boost the signal-to-noise ratio of images. To perform background subtraction, maneuver the mouse to the Process dropdown menu. Then select Subtract Background. … Set the Rolling ball radius to at least the diameter of the largest foreground object present in the image.
3.5. Region of Interest (ROI) Identification
The FIJI macro ROI_generator.ijm will help specify cellular ROIs in a semi-automated fashion. Figure 1 illustrates the process this script uses in identifying ROIs. However, it is also possible to manually identify each cellular ROI by thresholding and using the magic wand function in FIJI. The instructions below outline how to use the macro.
Open ROI_generator.ijm by dragging the script into the FIJI toolbar.
In line 2 write the location of the folder that contains images corresponding to the cell marker channel (i.e., GFP or mApple; see Note 8).
In line 4 specify the location of the folder to save ROIs. This folder must be created before running the script to avoid crashing.
In line 8 input the minimum cell area. This should be the smallest area in pixels that a healthy neuron could span (see Note 9).
Hit Run. A series of .zip files will populate the ROI folder specified as the “out_dir.”
3.6. Collecting Single-Cell Measurements
The provided file Figure 1 data.xlsx serves as a template for how to organize measurements generated in FIJI. These measurements correspond to the ROIs in Fig. 1. It is important to follow the formatting of this template for puncta.py to function properly.
From the Analyze dropdown menu, choose Select Measurements, and then place checkmarks beside Standard deviation and Mean gray value.
Open the morphology marker channel time point 1 image in FIJI.
Open the ROI set corresponding to this time point by dragging the .zip file into the FIJI toolbar. A series of ROIs will appear in the ROI manager. To see all ROIs superimposed, check the Show All and Labels buttons on the bottom right corner of the ROI manager.
Ensure that all of the ROIs correspond to neurons.
Open the images for the subsequent time points and accompanying ROI sets. Determine when cells die, and mark the last time point they were seen alive in the column last_tp (see Note 10).
With transient transfection, the expression of exogenous proteins tends to increase over time. As a result, dim cells that were not identified by ROI_generator.ijm at time point 1 may be picked up at later time points. These cells should be deleted from the ROI set to ensure that all of the cells analyzed were present from the beginning of the time series. In addition, it is common for FIJI to assign different labels to the same cell at different time points. All measurements should be recorded in the experiment_template.xlsx file using the ROI labels from the first time point.
Open the corresponding time point 1 image in the channel corresponding to the protein of interest (here Cy5 for iRFP-UBQLN2).
Highlight all ROIs in the ROI manager window and click Measure. This will output the fluorescence intensity and standard deviation of the fluorescently labeled protein in each neuron.
Copy and paste the mean intensity and standard deviation values into the experiment_template.xlsx file under the appropriate columns. Populating these two columns will also fill the CV column corresponding to the time point by using the following formula to calculate CVintensity: SDintensity / meanintensity.
Repeat steps 7–9 for each time point and field of view.
3.7. Making a Receiver Operating Characteristic (ROC) Curve to Determine the Optimal CVintensity Threshold
To use single-cell CVintensity as a metric of protein self-assembly, it is essential to determine if CVintensity can accurately predict the presence of puncta in a particular set of images (see Note 11). To do this the user must score a representative dataset by eye and record the cells that contain puncta (true positives) and those that do not (true negatives). After measuring the CVintensity for each cell, the distribution of CVintensity in these two groups can be compared, and a receiver operating characteristic (ROC) curve can be generated to evaluate the suitability of CVintensity as a proxy for manual scoring. This curve is crucial for identifying the optimal CVintensity threshold value that can be used to automate the analysis of subsequent datasets.
Select a set of images from an experimental condition that is expected to have a high frequency of puncta. In the example provided in ROC_data.csv, neurons expressing iRFP-UBQLN2-P506T-ΔUBL were chosen due to the preponderance of cells with puncta observed in this condition (11) (see Note 12).
After collecting single-cell measurements as outlined in Subheading 3.7, manually score each cellular ROI for the presence or absence of puncta. The ability to confidently classify puncta by eye will vary depending on the particular protein of interest. For this reason, it is advisable to not just specify whether a cell has puncta or not but also the level of confidence in that determination. Therefore, cells without puncta should be scored as “0,” any cells that likely contain puncta should be scored as “1,” and those cells with definite puncta should be scored as “2.” Record these scores in the puncta column in ROC_data.csv (see Note 13). The precision of the ROC curve in pinpointing a suitable threshold to use increases with the number of scored cells and the confidence of each assignment. In the example provided in ROC_data.csv, 552 neurons were scored. With a large difference in CVintensity between puncta(−) and puncta(+) cells, fewer cells need to be scored to achieve sufficient precision.
Open ROC curve.py in a Python integrated development environment (IDE) (see Note 14).
Specify the address of the directory that has the file ROC_data.csv in line 9.
In line 14, specify whether to generate an ROC curve using all puncta(+) cells (scored as “1” or “2”) by inputting “no” or just high confidence cells (scored as “2” only) by inputting “yes.”
In line 16, specify the upper bound of CVintensity values to evaluate. This value should be near the max CVintensity value in your dataset, excluding outliers.
In line 18, specify the number of CVintensity thresholds to evaluate (see Note 15).
In line 20, assign the channel corresponding to the protein of interest. This should be whatever text is in the column headers (if the header is labeled “Cy5-CV-1,” write “Cy5”).
In line 22, write the time point when puncta were manually scored.
Build (run) ROC curve.py. This will produce an ROC curve with the optimal CVintensity threshold displayed at the top, representing the CVintensity threshold value with the highest score determined by the following equation: true positive % + (100-false positive %). The area under the curve (AUC), a measure of how capable CVintensity is in distinguishing cells with and without aggregates, is also reported. The plot can be saved by clicking the disk icon.
Select a CVintensity threshold. Based on the ROC curves in Fig. 2, a CVintensity of 0.51 is expected to accurately identify 95.7% of cells that the user would have high confidence in being puncta(+) (4.3% false-negative rate), with a false-positive rate of only 4.8%. Depending on the experiment, it may be preferable to sacrifice a degree of sensitivity to increase selectivity and reduce the false-positive rate. In this regard, the ROC_table.csv file that is outputted may be particularly useful for selecting an appropriate CVintensity threshold.
Fig. 2.
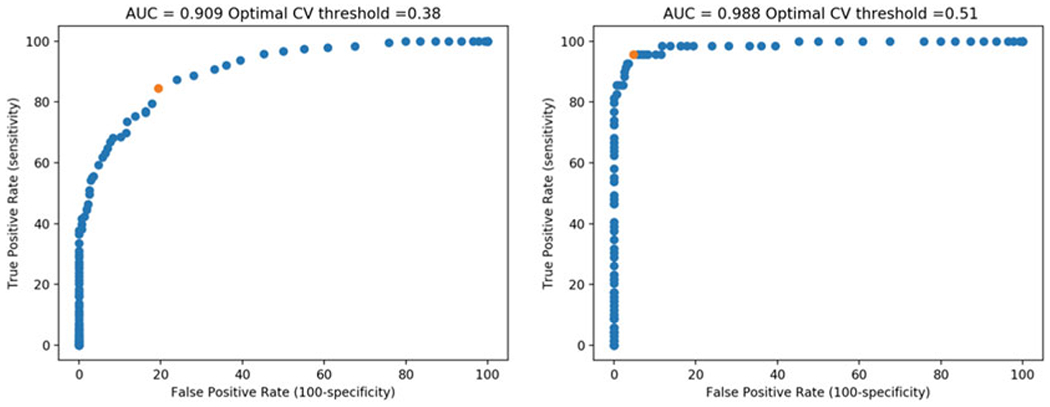
ROC curves generated from analyzing ROC_data.csv using ROC curve.py. The plot on the left incorporates all cells identified as having puncta, regardless of the confidence level; the plot on the right shows only cells with definite puncta. Red dots highlight the calculated optimal CVintensity thresholds
3.8. Calculating and Plotting Puncta Over Time
A python script puncta.py is provided that imports single-cell CVintensity data in the format of representative_experiment.csv and outputs a graph plotting the percentage of cells containing puncta at each time in each condition. In addition, a table is generated with the suffix _percent.csv as well as another file that identifies the time points showing the predicted puncta for each cell. This file has the suffix _puncta.csv.
Open puncta.py in a Python IDE.
In line 7 write the folder location containing the .csv file containing single-cell CVintensity values. The name of this file can be changed from representative_experiment.csv so long as the new name is written in line 8. However, the formatting must be the same as representative_experiment.csv.
Write the optimal CVintensity threshold value (determined in Subheading 3.7) in line 14.
Write the fluorescent channel corresponding to the protein of interest (here, Cy5) in line 16.
Build puncta.py. A plot of puncta over time is displayed (see Fig. 3). This window allows the user to save the plot, but if desired, the data can be plotted in other software by using the data exported in the _percent.csv file.
Fig. 3.
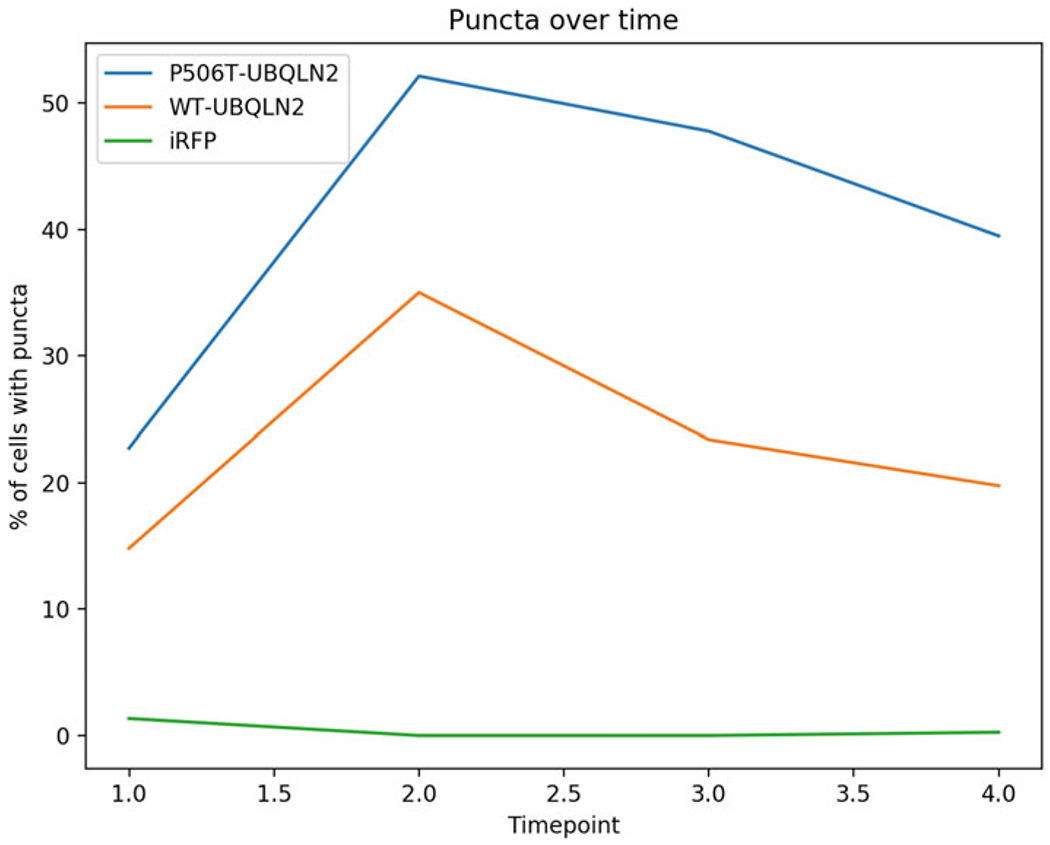
Percentage of neurons containing UBQLN2 puncta over time. Using a CVintensity threshold of 0.51 as determined in Subheading 3.7, the percentage of puncta(+) neurons is plotted for neurons expressing iRFP-WT-UBQLN2, iRFP-P506T-UBQLN2 (mutant), and iRFP alone. The absence of puncta in the iRFP alone control shows that the chosen CVintensity threshold produces few false positives
4 Notes
A tutorial on how to install Python packages can be found here: https://packaging.python.org/tutorials/installing-packages/
Additional descriptions of transfection, imaging, and image processing techniques can be found in a separate publication [15].
Video protocols illustrating primary rat cortical neuron dissections [16, 17] are an excellent means of improving the yield and health of cultured neurons.
The total amount of DNA per well is constant, regardless of whether one or multiple plasmids are used. An example would be co-transfecting 100 ngpGW1-UBQLN2-iRFP with 100 ng pGW1-mApple.
pGW1 plasmids express well in neurons.
For simplicity, all image processing is done manually on a per-image basis in this protocol. An automated FIJI macro can be found here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6594158/bin/NIHMS1036131-supplement-Sup_File_2.ijm
For example, for a 2 × 2 montage made up of individual frames labeled GFP_01.tif, GFP_02.tif, GFP_03.tif, and GFP_04.tif, grid size (for x and y) should be 2, and the File names for tiles = GFP_{ii}.tif.
This folder should include only image files; otherwise the macro will crash.
This is an important step to filter out cellular debris within each image that can confound the discrimination of neuronal soma.
It is advisable to record the time of death as the last time point a cell was observed alive (left censoring), rather than the first time at which a cell was noted to be dead (right censoring). This is because the cell could have died at any point between the last time it was observed to be alive and when it is visibly dead.
Given that CVintensity is directly proportional to the area-integrated standard deviation of fluorescence intensity, this variable best predicts the presence of puncta when these are relatively bright and occupy a small fraction of the total cellular ROI area, both of which maximize measurements of standard deviation. Dim puncta that barely stand out from the background are not identified as efficiently. For this reason, it is important to ensure that (a) the images do not contain any saturated pixels (see step 3 in Subheading 3.3) and (b) background subtraction (see step 2 in Subheading 3.4) is applied to boost signal to noise.
These neurons express iRFP-UBQLN2 carrying an ALS-linked mutation (P506T) and a deletion of UBQLN2’s ubiquitin-like (UBL) domain. Each of these mutations increases UBQLN2’s propensity to self-assemble [11].
Either replace the values in the ROC_data.csv file provided or rename the file. Alternatively, score cells in a.csv file with any name, and specify this file as the input in lines 9 and 10 of ROC_curve.py.
This can be Python IDLE (https://docs.python.org/3/library/idle.html) or a text editor such as Sublime Text (https://www.sublimetext.com/). If using Sublime Text, go to the Tools dropdown menu, and select Build system and then choose Python.
If an upper bound of 3 is selected and 300 breaks are specified, an interval of 0.01 (i.e., 0, 0.01, 0.02, 0.03, etc.) will be plotted.
References
- 1.Deng H-X, Chen W, Hong S-T et al. (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477(7363):211–215. 10.1038/nature10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams KL, Warraich ST, Yang S et al. (2012) UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol Aging 33(10):2527.e3–2527.e10. 10.1016/j.neurobiolaging.2012.05.008 [DOI] [PubMed] [Google Scholar]
- 3.Teyssou E, Chartier L, Amador MDM et al. (2017) Novel UBQLN2 mutations linked to amyotrophic lateral sclerosis and atypical hereditary spastic paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiol Aging 58:239.e11–239.e20. 10.1016/j.neurobiolaging.2017.06.018 [DOI] [PubMed] [Google Scholar]
- 4.Gkazi SA, Troakes C, Topp S et al. (2019) Striking phenotypic variation in a family with the P506S uBqLN2 mutation including amyotrophic lateral sclerosis, spastic paraplegia, and frontotemporal dementia. Neurobiol Aging 73:229.e5–229.e9. 10.1016/j.neurobiolaging.2018.08.015 [DOI] [PubMed] [Google Scholar]
- 5.Brettschneider J, Van Deerlin VM, Robinson JL et al. (2012) Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol 123(6):825–839. 10.1007/s00401-012-0970-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mori F, Tanji K, Odagiri S et al. (2012) Ubiquilin immunoreactivity in cytoplasmic and nuclear inclusions in synucleinopathies, polyglutamine diseases and intranuclear inclusion body disease. Acta Neuropathol 124(1): 149–151. 10.1007/s00401-012-0999-z [DOI] [PubMed] [Google Scholar]
- 7.Safren N, Chang L, Dziki KM et al. (2015) Signature changes in ubiquilin expression in the R6/2 mouse model of Huntington’s disease. Brain Res 1597:37–46. 10.1016/j.brainres.2014.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dao TP, Kolaitis RM, Kim HJ et al. (2018) Ubiquitin modulates liquid-liquid phase separation of UBQLN2 via disruption of multivalent interactions. Mol Cell 69(6):965–978.e6. 10.1016/j.molcel.2018.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dao TP, Martyniak B, Canning AJ et al. (2019) ALS-linked mutations affect UBQLN2 Oligomerization and phase separation in a position- and amino acid-dependent manner. Structure 27(6):937–951. 10.1016/J.STR.2019.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Jones HB, Dao T et al. (2019) Single amino acid substitutions in stickers, but not spacers, substantially alter UBQLN2 phase transitions and dense phase material properties. J Phys Chem B 123(17):3618–3629. 10.1021/acs.jpcb.9b01024 [DOI] [PubMed] [Google Scholar]
- 11.Sharkey LM, Safren N, Pithadia AS et al. (2018) Mutant UBQLN2 promotes toxicity by modulating intrinsic self-assembly. Proc Natl Acad Sci U S A 115(44):E10495–E10504. 10.1073/pnas.1810522115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higgins N, Lin B, Monteiro MJ (2019) Lou Gehrig’s disease (ALS): UBQLN2 mutations strike out of phase. Structure 27(6):879–881. 10.1016/j.str.2019.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Nirujogi RS, Myong S et al. (2018) Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proc Natl Acad Sci U S A 115(49):E11485–E11494. 10.1073/pnas.1811997115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramdzan YM, Polling S, Chia CPZ et al. (2012) Tracking protein aggregation and mislocalization in cells with flow cytometry. Nat Methods 9(5):467–470. 10.1038/nmeth.1930 [DOI] [PubMed] [Google Scholar]
- 15.Weskamp K, Safren N, Miguez R et al. (2019) Monitoring neuronal survival via longitudinal fluorescence microscopy. J Vis Exp 143. 10.3791/59036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viesselmann C, Ballweg J, Lumbard D et al. (2010) Nucleofection and primary culture of embryonic mouse hippocampal and cortical neurons. J Vis Exp 47. 10.3791/2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pacifici M, Peruzzi F (2012) Isolation and culture of rat embryonic neural cells: a quick protocol. J Vis Exp 63. 10.3791/3965 [DOI] [PMC free article] [PubMed] [Google Scholar]