

Abstract
Background:
The CheckMate 274 trial demonstrated improved disease-free survival (DFS) with adjuvant nivolumab versus placebo in patients with muscle-invasive urothelial carcinoma at high risk of recurrence after radical surgery in both the intent-to-treat population and the subset with tumor programmed death ligand 1 (PD-L1) expression ≥1%.
Objective:
To analyze DFS using the combined positive score (CPS), which is based on PD-L1 expression in both tumor and immune cells.
Design, setting, and participants:
We randomized a total of 709 patients 1:1 to nivolumab 240 mg or placebo every 2 weeks intravenously for ≤1 year of adjuvant treatment.
Intervention:
Nivolumab 240 mg.
Outcome measurements and statistical analysis:
Primary endpoints were DFS in the intent-to-treat population and patients with tumor PD-L1 expression ≥1% using the tumor cell score (TC). CPS was determined retrospectively from previously stained slides. Tumor samples with both quantifiable CPS and TC were analyzed.
Results and limitations:
Of 629 patients evaluable for CPS and TC, 557 (89%) had CPS ≥1, 72 (11%) had CPS <1, 249 (40%) had TC ≥1%, and 380 (60%) had TC <1%. Among patients with TC <1%, 81% (n=309) had CPS ≥1. DFS was improved with nivolumab versus placebo in patients with TC ≥1% (hazard ratio [HR] 0.50; 95% confidence interval [CI] 0.35–0.71), CPS ≥1 (HR 0.62; 95% CI, 0.49–0.78), and with both TC <1% and CPS ≥1 (HR 0.73; 95% CI, 0.54–0.99).
Conclusion:
More patients had CPS ≥1 than TC ≥1%, and most patients who had TC <1% had CPS ≥1. In addition, patients with CPS ≥1 experienced improved DFS with nivolumab. These results may, in part, explain the mechanisms underlying a benefit with adjuvant nivolumab even in patients who had both TC <1% and CPS ≥1.
Patient summary:
We studied patients with bladder cancer after surgery to remove the bladder or components of the urinary tract in the CheckMate 274 trial to determine the impact of treatment with nivolumab versus placebo on the time patients live without cancer returning (disease-free survival; DFS) depending on the level of the biological marker programmed death ligand 1 (PD-L1) expressed either on their tumor cells (tumor cell score; TC) or on the tumor cells as well as the immune cells surrounding the tumor (combined positive score; CPS). We found that DFS was improved with nivolumab versus placebo in patients with TC ≥1%, CPS ≥1, and in patients with both TC <1% and CPS ≥1.
Keywords: CheckMate 274, Combined positive score, Nivolumab, PD-L1 expression
1. Introduction
Certain immune checkpoint inhibitors exploit a mechanism whereby tumor cells evade antitumor immune responses by expressing programmed death ligand 1 (PD-L1) on the cell surface. The programmed death 1 (PD-1) receptor mediates T-cell function via engagement of PD-L1, thereby enabling tumor survival. Increased PD-L1 expression on infiltrating immune cells may also be triggered by cytokines such as IFN-γ in the tumor microenvironment, contributing to adaptive immune resistance [1,2].
PD-L1 immunohistochemistry testing is commonplace in clinical trials evaluating immune checkpoint inhibitors, but the utility of PD-L1 expression as a predictive biomarker of response to these treatments has been limited [3,4]. The heterogeneity of PD-L1 expression assessment approaches used across clinical trials, including differing diagnostic assays, tissue preparation methods, sites of biopsy samples, and scoring techniques (tumor cells vs immune cells), plus variations in staining thresholds to define PD-L1 positivity, further confounds the lack of established clinical utility for PD-L1 testing [1,4,5]. Previous studies have measured PD-L1 expression using the combined positive score, which measures PD-L1 expression on both tumor and infiltrating immune cells (including lymphocytes and macrophages) [6], immunohistochemistry of tumor-infiltrating immune cells only [7], and the tumor cell score (TC) [8].
On the basis of CheckMate 274 primary disease-free survival (DFS) results, nivolumab (a fully human IgG4 PD-1 immune checkpoint inhibitor antibody) was approved by the US Food and Drug Administration (FDA) in August 2021 for treatment of patients with UC who are at high risk of recurrence after undergoing radical resection of UC, regardless of PD-L1 expression [9,10]. In CheckMate 274, adjuvant nivolumab improved DFS versus placebo in patients with high-risk muscle-invasive urothelial carcinoma (MIUC) after radical surgery. This benefit was observed both in the intent-to-treat (ITT) population (HR, 0.70; 98.22% CI, 0.55–0.90; P<0.001) and in patients with tumor PD-L1 expression ≥1% as assessed by the TC (HR 0.55; 98.72% CI, 0.35–0.85; p<0.001) in the primary analysis (minimum ITT population follow-up, 5.9 months). Beyond the benefit with nivolumab observed in the ITT population, an exploratory subgroup analysis indicated that patients with TC <1% show a numerical DFS benefit with nivolumab (HR 0.82; 95% CI, 0.63–1.06) [10], although this benefit did not meet conventional levels of statistical significance.
The relationship between cell types expressing PD-L1 (ie, tumor vs immune cells) and outcomes with adjuvant nivolumab could refine our understanding of the role of PD-L1 testing in informing clinical decisions and also offer further insights regarding the mechanism underlying antitumor activity with immune checkpoint blockade. Therefore, we conducted a post hoc exploratory DFS analysis based on PD-L1 expression levels assessed by TC (corresponding to PD-L1 expression in tumor cells) and by CPS (corresponding to PD-L1 expression in tumor cells and immune cells) in CheckMate 274 (median follow-up, 30 months in the ITT population).
2. Patients and methods
2.1. Study design and treatment
CheckMate 274 (ClinicalTrials.gov, NCT02632409) is a phase III, randomized, double-blind, multicenter trial of nivolumab versus placebo in patients with high-risk MIUC after radical surgery. The study design and methods have been reported previously [10]. Briefly, eligible patients had undergone radical surgery within 120 days before randomization, with or without neoadjuvant cisplatin-based chemotherapy. Patients had pathological evidence of UC (originating in the bladder, ureter, or renal pelvis), without clinical or radiologic recurrence and a high risk of recurrence. The latter was defined as pathological stage pT3-pT4a, or pN+ for patients who did not receive neoadjuvant therapy and were ineligible for or declined adjuvant cisplatin-based chemotherapy; and pathological stage ypT2-ypT4a or ypN+ for patients who received neoadjuvant cisplatin. Patients with upper tract disease were limited to 20% of total enrollment.
Patients were randomized 1:1 (stratified permuted block randomization via interactive voice response system [IVRS]) to receive intravenous nivolumab 240 mg every 2 weeks or placebo. Patients were stratified according to PD-L1 expression level by TC (≥1% vs <1% or indeterminate), pathologic nodal status (N+ vs Nx or N0 with <10 nodes removed vs N0 with ≥10 nodes removed) and use of neoadjuvant cisplatin-based combination chemotherapy (yes vs no). The computer-generated randomization schedule was created by Clinical Supply Chain Technology based on the protocol requirements. System users were provisioned role-based access to the IVRS by the study manager. Site Investigators had access to screen and randomized patients at their sites. Unblinded pharmacists had access to treatment assignment modules of the IVRS system.
Patients were treated for up to 1 year of adjuvant therapy, or until disease recurrence or progression, unacceptable toxicity, or withdrawal of consent. Dose delays or discontinuations to manage adverse events were permitted. The study was conducted according to Good Clinical Practice guidelines and the Declaration of Helsinki. All patients provided written informed consent before enrollment.
2.2. Assessments
Tumor imaging assessments were performed continuously as described previously [10]. Tumor tissue from the most recently resected site of disease (preferred) or from the transurethral resection that yielded the initial diagnosis of muscle-invasive disease was required for biomarker analyses. To be randomized, patients had to have a PD-L1 expression level classification (≥1% vs <1% or indeterminate) as determined by the central laboratory.
PD-L1 immunohistochemistry was performed on formalin-fixed, paraffin-embedded tumor samples from the resected site of disease, obtained before randomization, using the Dako PD-L1 IHC 28–8 pharmDx assay and assessed by a pathologist. Specimens with ≥100 evaluable tumor cells were eligible for PD-L1 scoring. TC was determined from central laboratory testing before randomization, calculated as follows:
In this post hoc analysis, CPS was determined retrospectively at a central laboratory from the previously stained immunohistochemistry slides (using the Dako PD-L1 IHC 28–8 pharmDx assay). CPS was calculated as follows:
Patients enrolled in China were excluded owing to local regulatory restrictions. Our analysis includes only patients with both a quantifiable CPS and TC at baseline.
2.3. Outcomes
The primary endpoints of CheckMate 274 were DFS in the ITT population and in patients with tumor PD-L1 expression by TC ≥1%. DFS was defined as the time between the date of randomization and the date of first recurrence (local recurrence in the urothelial tract or in the non-urothelial tract [in pelvic soft tissue or involving pelvic nodes below the aortic bifurcation], or distant recurrence) or death, whichever occurred first. The primary definition of DFS accounts for subsequent anticancer therapy and new non-urothelial carcinoma primary cancer by censoring at the last evaluable disease assessment on or before the date of subsequent therapy/new non-urothelial carcinoma primary cancer.
This post hoc exploratory analysis evaluated DFS by treatment group per quantifiable CPS status (PD-L1 expression in tumor cells and immune cells) and TC status (PD-L1 expression in tumor cells) at baseline. Subpopulations of interest were patients with CPS <1, CPS ≥1, and those with both TC <1% and CPS ≥1. Our analysis differs from previous reports owing to the smaller analysis population of patients with TC who also had quantifiable CPS. Additionally, DFS was analyzed in prespecified subgroups among patients with CPS ≥1. An analysis was also conducted to evaluate the utility of both methods of PD-L1 scoring to predict DFS in patients with high-risk MIUC with prior radical surgery receiving nivolumab or placebo.
2.4. Statistical analyses
DFS was estimated using Kaplan-Meier methodology and compared between groups using a two-sided log-rank test. HRs and corresponding CIs were estimated using a Cox proportional-hazards model.
A sensitivity analysis of CPS and TC as continuous variables is described in the Appendix.
3. Results
3.1. Patients
Baseline characteristics of patients in the ITT population (all randomized patients) have been described previously [10]. Of the 709 patients in the ITT population, 629 (89%) had both quantifiable CPS and TC at baseline (nivolumab, n=315; placebo, n=314) (Fig. S1). Of these, 557 (89%) had CPS ≥1 (nivolumab, n=281; placebo, n=276), 72 (11%) had CPS <1 (nivolumab, n=34, placebo, n=38), 249 (40%) had TC ≥1% (nivolumab, n=124; placebo, n=125) and 380 (60%) had TC <1% (nivolumab, n=191; placebo, n=189). Among the 380 patients with TC <1%, 309 (81%) had CPS ≥1.
Baseline characteristics in patients with CPS ≥1 were generally balanced between treatment groups and comparable with the ITT population [10]; however, a greater proportion of patients with nivolumab versus placebo were aged <65 years and had N0 with ≥10 nodes resected. A generally similar distribution of baseline characteristics was observed in patients with CPS <1, although some imbalances were present in this group (Table 1). Exposure details are summarized in Table S1.
Table 1 –
Baseline demographic and clinical characteristics in patients with CPS ≥1 and CPS <1 (among all randomized patients with quantifiable CPS and TC at baseline)
| CPS ≥1 | CPS <1 | |||
|---|---|---|---|---|
| Nivolumab (N = 281) |
Placebo (N = 276) |
Nivolumab (N = 34) |
Placebo (N = 38) |
|
| Age | ||||
| Median (range), years | 66.0 (33–92) | 67.0 (42–88) | 66.5 (34–83) | 66.9 (47–81) |
| Male sex, n (%) | 214 (76) | 211 (76) | 23 (68) | 29 (76) |
| Race or ethnic group, n (%) a | ||||
| White | 236 (84) | 234 (85) | 25 (74) | 33 (87) |
| Asian | 39 (14) | 33 (12) | 6 (18) | 5 (13) |
| Black | 1 (0.36) | 3 (1.1) | 1 (2.9) | 0 |
| Other | 5 (1.8) | 5 (1.8) | 2 (5.9) | 0 |
| ECOG PS, n (%) a | ||||
| 0 | 180 (64) | 176 (64) | 23 (68) | 23 (61) |
| 1 | 95 (34) | 90 (33) | 11 (32) | 15 (39) |
| 2 | 6 (2.1) | 9 (3.3) | 0 | 0 |
| Tumor origin at initial diagnosis, n (%) | ||||
| Urinary bladder | 236 (84) | 222 (80) | 19 (56) | 26 (68) |
| Renal pelvis | 25 (8.9) | 32 (12) | 9 (26) | 11 (30) |
| Ureter | 20 (7.1) | 22 (8.0) | 6 (18) | 1 (2.6) |
| Minor histological variants present, n (%) | 122 (43) | 121 (44) | 14 (41) | 8 (21) |
| Less than 1 year from initial disease diagnosis to randomization, n (%) | 260 (93) | 251 (91) | 32 (94) | 35 (92) |
| Tumor cell PD-L1 expression of ≥1%, n (%) | 124 (44) | 124 (45) | 0 | 1 (2.6) |
| Previous cisplatin-based therapy, n (%) | 133 (47) | 126 (46) | 12 (35) | 20 (53) |
| Pathologic tumor stage at resection, n (%) b | ||||
| pTX | 5 (1.8) | 0 | 0 | 0 |
| pT0 | 4 (1.4) | 7 (2.5) | 0 | 0 |
| pTis | 3 (1.1) | 2 (0.72) | 0 | 1 (2.6) |
| pT1 | 7 (2.5) | 10 (3.6) | 5 (15) | 2 (5.3) |
| pT2 | 56 (20) | 53 (19) | 3 (8.8) | 9 (24) |
| pT3 | 158 (56) | 161 (58) | 23 (68) | 17 (45) |
| pT4 | 48 (17) | 42 (15) | 2 (5.9) | 9 (24) |
| Nodal status at resection, n (%) c | ||||
| N0 or NX with <10 nodes removed | 64 (23) | 72 (26) | 15 (44) | 12 (32) |
| N0 with ≥10 nodes resected | 80 (28) | 65 (24) | 3 (8.8) | 8 (21) |
| N1 | 57 (20) | 56 (20) | 4 (12) | 7 (18) |
| N2 | 68 (24) | 66 (24) | 12 (35) | 7 (18) |
| N3 | 11 (3.9) | 17 (6.2) | 0 | 3 (7.9) |
Not reported for one patient in the placebo arm with CPS ≥1.
Not reported for one patient in the placebo arm with CPS ≥1 and one patient in the nivolumab arm with CPS <1.
Not reported for one patient in the nivolumab arm with CPS ≥1 and one patient in the placebo arm with CPS <1.
CPS, combined positive score; ECOG PS, Eastern Cooperative Oncology Group performance status; N, node; p, pathologic; PD-L1, programmed death ligand 1; T, tumor; TC, tumor cell score; Tis, tumor in situ; X, cannot be assessed.
3.2. Disease-free survival
Median follow-up (defined as the time between randomization and the last known date alive for all randomized patients with quantifiable TC and CPS at baseline and without a DFS or OS event) was 30 months. In patients with CPS ≥1, median DFS was 25 (95% CI, 19-not estimable; n=281) months with nivolumab and 9.4 (95% CI, 8.2–15; n=276) months with placebo (Figure 1; Table S2). At 6 and 12 months, 77% and 67% of nivolumab-treated patients and 60% and 46% of placebo-treated patients were alive and disease-free. The HR for disease recurrence or death was 0.62 (95% CI, 0.49–0.78).
Fig. 1 – Kaplan–Meier plot of disease-free survival in patients with CPS ≥1 (among all randomized patients with quantifiable CPS and TC at baseline).
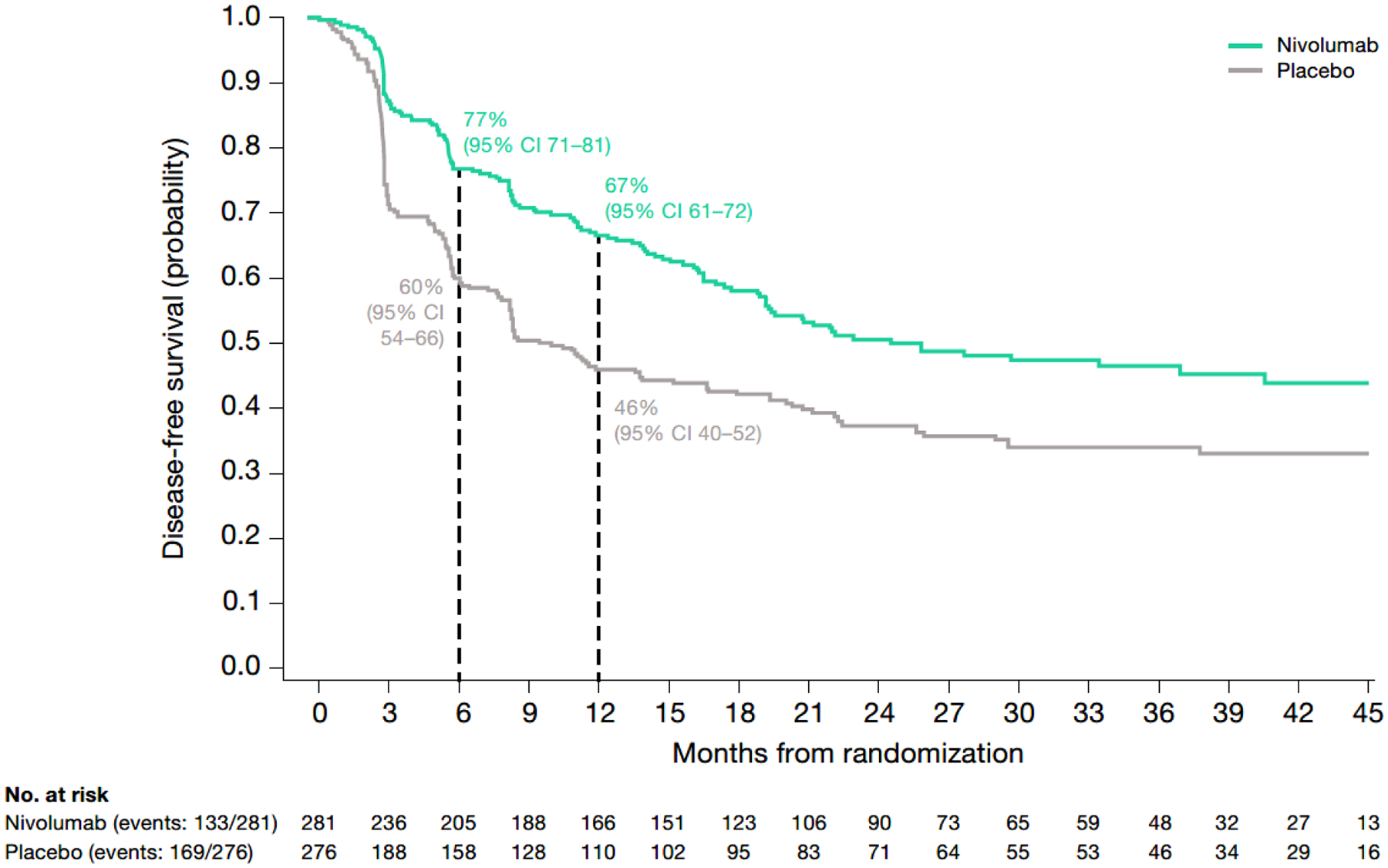
CPS, combined positive score; DFS, disease-free survival; HR, hazard ratio; NE, not estimable; TC, tumor cell score.
In patients with CPS <1, median DFS was 6.4 (95% CI, 5.1–13; n=34) months with nivolumab and 8.4 (95% CI, 5.4–14; n=38) months with placebo. The HR for disease recurrence or death was 1.22 (95% CI, 0.67–2.20) (Fig. 2; Table S2). At 6 and 12 months, 52% and 36% of nivolumab-treated patients and 60% and 37% of placebo-treated patients were alive and disease-free.
Fig. 2 – Kaplan–Meier plot of disease-free survival in patients with CPS <1 (among all randomized patients with quantifiable CPS and TC at baseline).
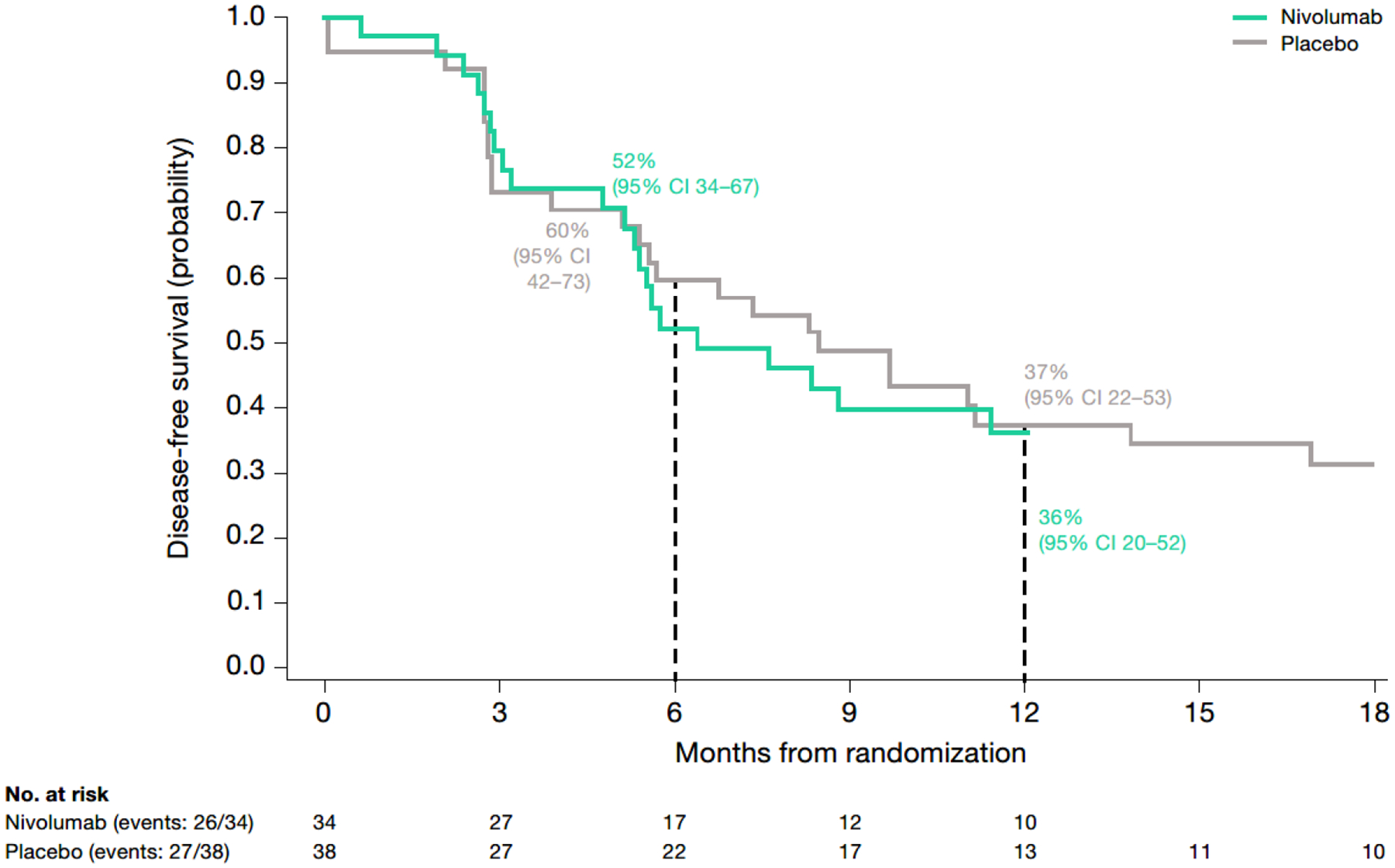
CPS, combined positive score; DFS, disease-free survival; HR, hazard ratio; TC, tumor cell score.
In patients with TC ≥1%, median DFS was not reached (95% CI, 25-not estimable; n=124) with nivolumab and 8.4 (95% CI, 5.6–18; n=125) months with placebo (Table S2). The HR for disease recurrence or death was 0.50 (95% CI, 0.35–0.71). At 6 and 12 months, 75% and 69% of nivolumab-treated patients and 56% and 45% of placebo-treated patients were alive and disease-free.
In patients with TC <1%, median DFS was 17 (95% CI, 13–21; n=191) with nivolumab and 9.6 (95% CI, 8.2–14; n=189) months with placebo. At 6 and 12 months, 74% and 60% of nivolumab-treated patients and 63% and 45% of placebo-treated patients were alive and disease-free (HR 0.80; 95% CI, 0.61–1.04; Table S2).
In patients with TC <1% and CPS ≥1 (representing 81% of all patients with TC <1%), median DFS was 19 (95% CI, 16–33; n=157) months with nivolumab and 10 (95% CI, 8.2–19; n=152) months with placebo (Fig. 3; Table S2). The HR for disease recurrence or death was 0.73 (95% CI, 0.54–0.99). At 6 and 12 months, 78% and 65% of nivolumab-treated patients and 64% and 46% of placebo-treated patients were alive and disease-free.
Fig. 3 – Kaplan–Meier plot of disease-free survival in patients with TC <1% and CPS ≥1 (among all randomized patients with quantifiable CPS and TC at baseline).
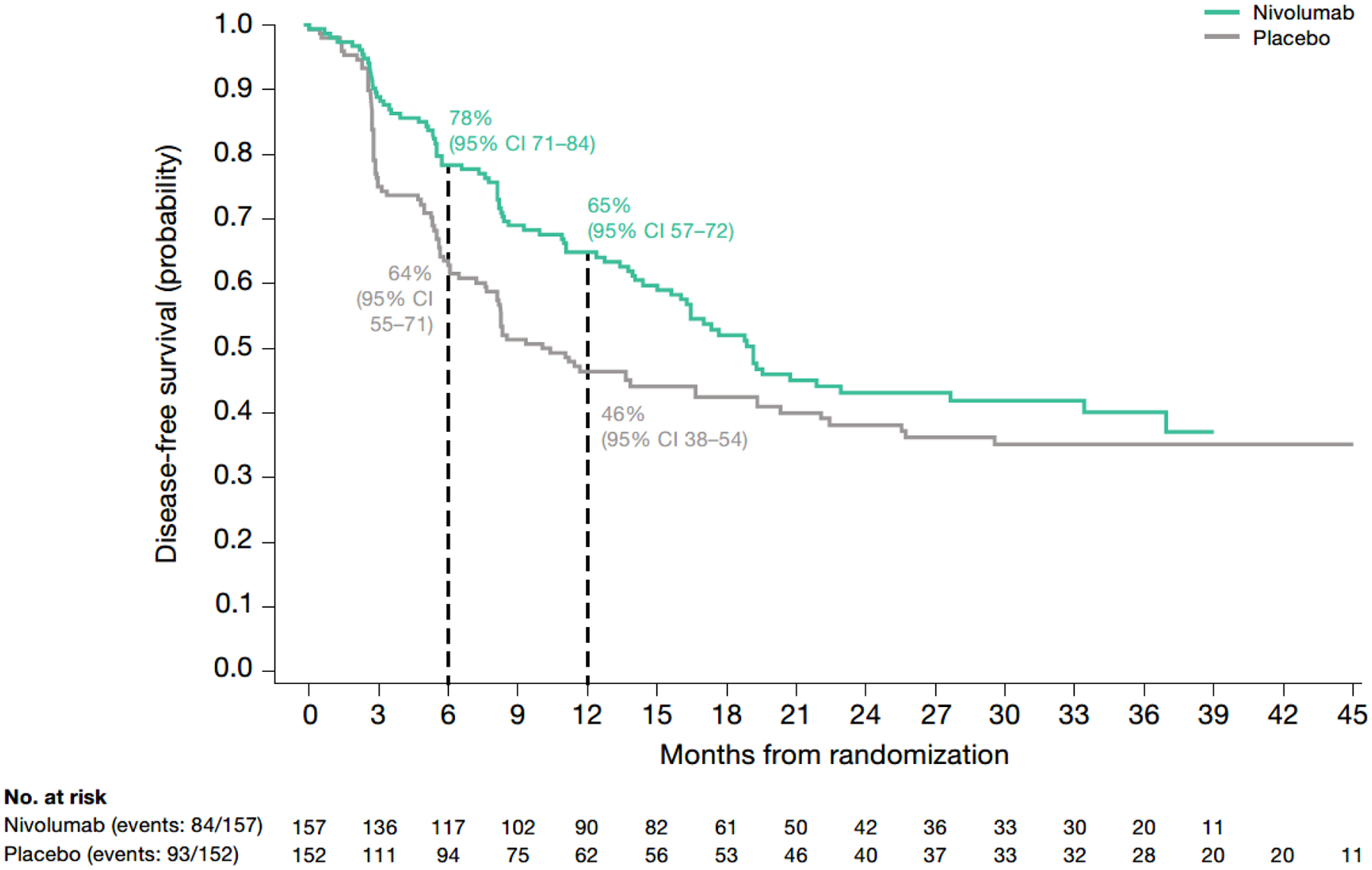
CPS, combined positive score; DFS, disease-free survival; HR, hazard ratio; TC, tumor cell score.
The DFS analysis by subgroup in patients with CPS ≥1 is shown in Figure 4. DFS hazard ratios favored nivolumab over placebo in most subgroups analyzed.
Fig. 4 –
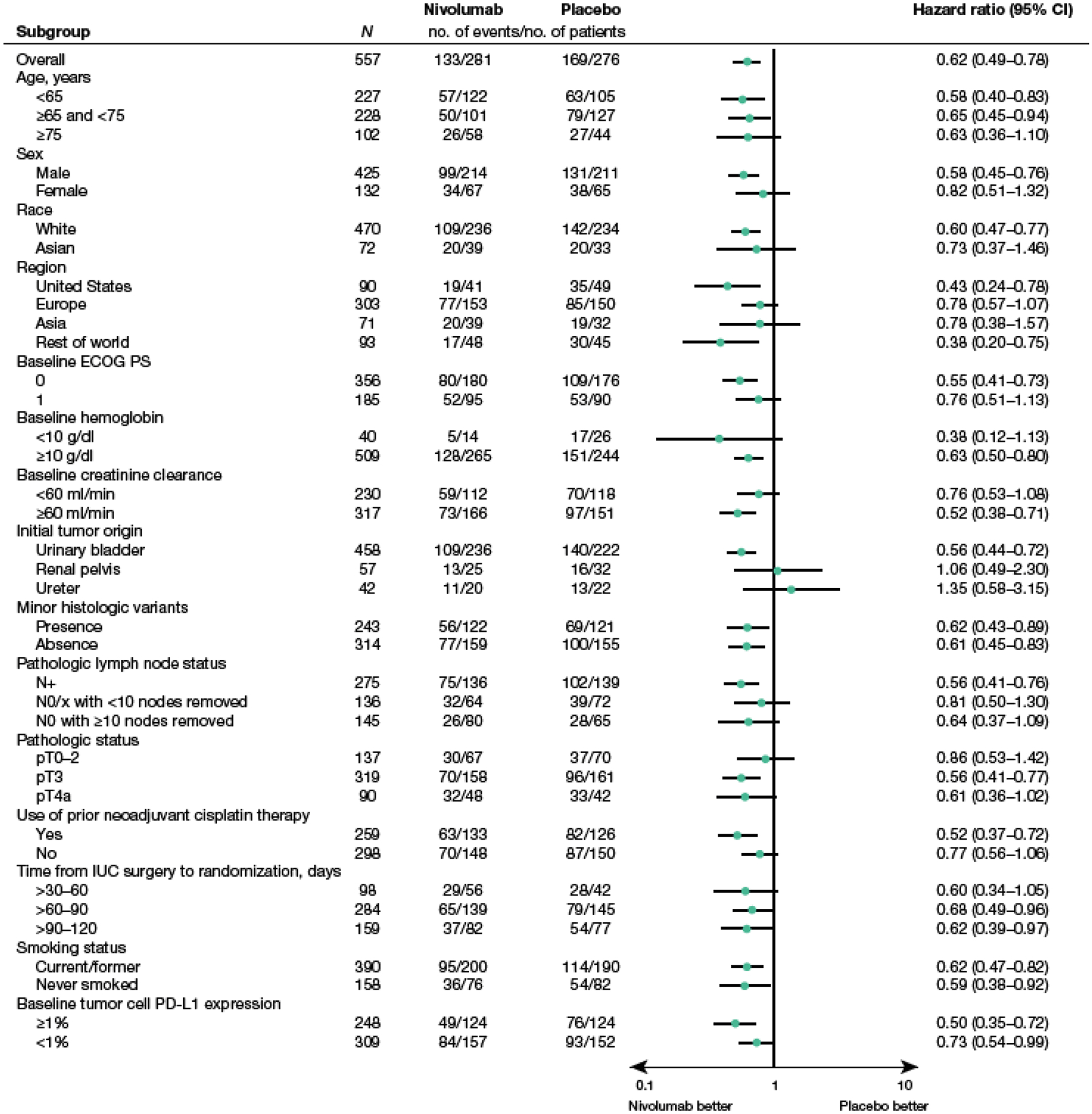
Forest plot of DFS by clinical and demographic subgroup in patients with CPS ≥1 (among all randomized patients with quantifiable CPS and TC at baseline).
Median DFS is based on Kaplan–Meier estimates. Hazard ratio calculated with stratified Cox proportional hazard model.
Hazard ratio is not computed for subset (except age, region, and sex) category with less than 10 patients per treatment group.
CPS, combined positive score; DFS, disease-free survival; ECOG PS, Eastern Cooperative Oncology Group performance status; IUC, invasive urothelial carcinoma; N, node; P, pathologic; PD-L1, programmed death ligand 1; T, tumor; TC, tumor cell score; X, cannot be assessed
CPS and TC were moderately correlated (Kendall’s tau = 0.62). For both CPS and TC as continuous variables, association with DFS appeared to be consistent with the dichotomous analysis. These associations were stronger in the nivolumab than in the placebo arm. For TC, estimated HR (95% CI) was 0.92 (0.87–0.98) and 1.03 (0.98–1.08), respectively. The ratio of these two HRs was 0.90 (95% CI, 0.83–0.97). For CPS, estimated HR (95% CI) was 0.67 (0.54–0.83) and 0.92 (0.78–1.08), respectively. The ratio of these two HRs was 0.73 (95% CI, 0.56–0.95).
4. Discussion
This analysis identifies limitations of using diagnostic PD-L1 expression assays to select patients with the greatest clinical benefit from PD-1/PD-L1–specific antibody therapy. Differences in patient assignment to “PD-L1-positive” status and in clinical benefit were observed when using the TC and CPS calculation methodologies in existing immunohistochemistry-stained tumor specimens from patients in CheckMate 274. One major difference comparing the TC and CPS calculations was that a higher proportion of patients in CheckMate 274 had CPS ≥1 than had TC ≥1%. In fact, most patients with TC <1% had CPS ≥1. However, this was not the case for all patients with TC <1%. The DFS benefit with nivolumab versus placebo was observed in the CPS ≥1, TC ≥1%, and TC <1% subpopulations. Within the CPS ≥1 subpopulation, this DFS benefit with nivolumab was also observed across most clinically relevant subgroups analyzed, consistent with primary results in the ITT population [10]. Although a DFS benefit with nivolumab versus placebo was not observed in patients with CPS <1, this subpopulation comprised only approximately 11% of the total analysis population; as such, interpretation of results in this population is limited by the small number of patients. The small proportion of CPS-negative patients therefore precludes definitive conclusions. Potential imbalances in baseline characteristics between treatment arms in the CPS <1 group might also be attributable to the small number of patients.
In our analysis, median DFS with nivolumab in patients with TC <1% and CPS ≥1 was nearly double that with placebo. As 81% of patients in the TC <1% subpopulation had CPS ≥1, these results suggest that most patients with TC <1% may also benefit from adjuvant treatment with nivolumab, provided they have CPS ≥1. This observation must be considered within the known limitations of PD-L1 use as a biomarker [3]. Nevertheless, this finding is consistent with a previous subgroup analysis that showed a trend toward a DFS benefit in patients with TC <1% (HR 0.82, 95% CI, 0.63–1.06) [10]. Although PD-L1 expression in immune cells only was not assessed, the CPS data suggest that patients with positive PD-L1 expression on immune cells alone would also benefit from nivolumab treatment.
PD-L1 scoring varies across clinical trials in oncology, with both the TC and CPS calculations widely utilized and yielding conflicting results [4]. Clinicians should also be aware that differences in the predictive value of PD-L1 staining may be a function of both the specific malignancy and the specific diagnostic assay. For example, the FDA-approved companion diagnostic assay to determine patients eligible for pembrolizumab as first-line treatment of metastatic head and neck squamous cell cancer used the CPS methodology, while a tumor cell expression assay is approved to determine eligibility for pembrolizumab as first-line treatment for non-small cell lung cancer [11].
A more nuanced understanding of the impact of PD-L1 expression in different cellular subsets and their association with the efficacy of immune checkpoint inhibitors is increasingly important in clinical practice because of differing methodologies and results. In contrast to our analysis in this UC study, a previous study in patients with non-small cell lung cancer showed a similar frequency of PD-L1 positivity when using either the CPS or tumor cell expression scoring [12]. Conversely, another study evaluating CPS and tumor cell expression scoring in patients with gastric cancer also reported that PD-L1 positivity was more frequent with CPS (58%) versus with TC (13%) [13]. Differences in CPS and TC scoring across tumor types may result from differing levels of PD-L1 expression on immune cells. These differing outcomes highlight the importance of our findings for clinical decision-making specific to patients with UC. Although PD-L1 positivity appears to be more frequent based on CPS ≥1 than TC ≥1%, our results support the efficacy benefit of nivolumab in the ITT population and suggest that evaluating PD-L1 expression by CPS confirms and expands the benefit of nivolumab beyond the TC ≥1% population to the TC <1% population.
Three markedly different PD-L1 calculation methodologies have been used in clinical trials of immune checkpoint inhibitors in patients with previously treated metastatic UC: TC (nivolumab; CheckMate 275), CPS (pembrolizumab; KEYNOTE-045), and expression based solely on immune cells (atezolizumab; IMvigor 210) [6,8,14]. Additionally, in the IMvigor010 trial of adjuvant atezolizumab in patients with MIUC, PD-L1 status was also based on expression on immune cells only [7]. Consequently, it is not unexpected that observed associations between PD-L1 expression and clinical outcomes also differed across these trials. In IMvigor 210, PD-L1 expression on immune cells was associated with response to atezolizumab while in KEYNOTE-045 and CheckMate 275, clinical activity of pembrolizumab and nivolumab, respectively, was observed regardless of tumor PD-L1 expression [6,8,14]. These findings underscore the variability in available data describing the relationship between PD-L1 expression and efficacy of immune checkpoint inhibitors in UC.
This study has several limitations. First, this analysis was not prespecified before commencement of the trial and excluded 80 patients who lacked either quantifiable CPS and/or TC at baseline. However, patient specimens were ascertained in a prospective manner. Second, the small number of patients in the CPS <1 subpopulation especially limits the comparison versus the CPS ≥1 subpopulation and conclusions regarding the DFS results. We also did not examine PD-L1 expression only on immune cells, thus precluding direct comparisons between tumor expression and immune cell PD-L1 expression. Finally, evaluation of other biomarkers (such as circulating tumor DNA, tumor mutational burden, and gene and immune cell infiltration signatures) in this setting will be required to identify patients who will derive the most benefit from treatment with adjuvant nivolumab.
In summary, this post hoc analysis indicated that most patients in CheckMate 274 had CPS ≥1, and that most patients with TC <1% had CPS ≥1. DFS was improved with nivolumab versus placebo in patients with TC ≥1%, CPS ≥1, and with both TC <1% and CPS ≥1. The results of this post hoc analysis suggest that most patients with TC <1% may also benefit from adjuvant treatment with nivolumab for high-risk MIUC after radical resection, provided they have CPS ≥1. These results provide additional insight into the differences between CPS and TC as measures of PD-L1 expression. Further prospective studies will be needed to evaluate the impact of these PD-L1 expression measures on clinical decision making.
Supplementary Material
Take home message.
We analyzed disease-free survival by tumor cell score (TC) and combined positive score PD-L1 expression in CheckMate 274. The results provide insights on the role of adjuvant nivolumab in patients with muscle-invasive urothelial carcinoma who have low PD-L1 by TC.
Acknowledgments:
The authors would like to acknowledge the patients and families who made this study possible, the clinical study teams who participated in the study, Dako, an Agilent Technologies, Inc. company, for collaborative development of the PD-L1 IHC 28-8 pharmDx assay (Santa Clara, CA, USA), Bristol Myers Squibb (Princeton, NJ, USA), and Ono Pharmaceutical Company Ltd. (Osaka, Japan). The authors acknowledge Scott D. Chasalow for advice on statistical methods and contributions to the interpretation of results. The study was supported by Bristol Myers Squibb. All authors contributed to and approved the manuscript; writing and editorial assistance were provided by Nicolette Belletier, PhD, of Parexel, funded by Bristol Myers Squibb.
Funding/Support and role of the sponsor:
Supported by Bristol Myers Squibb in collaboration with Ono Pharmaceutical. Bristol Myers Squibb sponsored the study, contributed to its design, and participated in the collection, analysis, and interpretation of the data and in the writing, reviewing, and approval of the manuscript. All authors had access to all relevant data and participated in writing, review, and approval of this manuscript, with editorial assistance funded by the study funder. No honoraria or payments were made for authorship.
Financial disclosures:
MDG reports consulting or advisory fees from BioMotiv, Janssen, Dendreon, Merck, GlaxoSmithKline, Lilly, Astellas Pharma, Genentech, Bristol Myers Squibb (BMS), Novartis, Pfizer, EMD Serono, AstraZeneca, Seattle Genetics, Incyte, Aileron Therapeutics, Dracen, Inovio Pharmaceuticals, NuMab, Dragonfly Therapeutics, Basilea, UroGen Pharma, Infinity Pharmaceuticals, and Gilead Sciences; patent: methods and compositions for treating cancer and related methods, Mount Sinai School of Medicine July 2012, application number 20120322792; stock ownership in Rappta Therapeutics; and research funding (institutional) from Janssen Oncology, Dendreon, Novartis, BMS, Merck, AstraZeneca, and Genentech/Roche. DFB reports consulting or advisory fees from Merck, Dragonfly Therapeutics, Fidia Farmaceutici S.p.A., and BMS; travel accommodations, expenses from Merck; and research funding (institutional) from Novartis, Merck, BMS, AstraZeneca, and Seattle Genetics/Astellas. JAW reports consulting or advisory fees from Nucleix, BMS, MSD, Ipsen, Sanofi-Aventis, Janssen Oncology, and OncoDiag; and honoraria from Astellas Pharma, BeiGene, Ferring, AstraZeneca, Janssen, MSD, and BMS/Pfizer. JEG reports consulting or advisory fees and honoraria from Janssen-Cilag, Bayer Schering Pharma, and BMS. YT reports honoraria from BMS, Pfizer, Ono Pharmaceutical, and Astellas; and research funding (institutional) from Ono Pharmaceutical, Takeda, and Astellas. BPV reports consulting or advisory fees from Astellas Pharma, Bayer, Sanofi, BMS, Roche, Ipsen, MSD Oncology, Novartis, EUSA Pharma, and Pfizer; travel accommodations, expenses from BMS, Pfizer, Roche, Ipsen, Astellas Pharma, and MSD Oncology; and honoraria from Astellas Pharma, BMS, Ipsen, Roche, Bayer, EUSA Pharma, Novartis, and Pfizer. M-OG reports consulting fees from AstraZeneca, BMS, Ipsen, MSD, Pfizer, Astellas Pharma, EUSA Pharma, Merck Serono, Roche Pharma AG, Takeda, Eisai, and Bayer Vital; honoraria from Astellas Pharma, AstraZeneca, BMS, MSD, Pfizer, Ipsen, Merck Serono, and EUSA Pharma; travel accommodations, expenses from BMS and Merck Serono; and research funding (institutional) from BMS and Intuitive Surgical. LA reports consulting or advisory fees from Aadi Bioscience; other fees from Pfizer; and research funding (institutional) from Pfizer, Exelixis, BMS, Astellas Pharma, Acerta Pharma, Novartis, Bayer, Agensys, Merck, Genentech/Roche, Tokai Pharmaceuticals, AVEO, Peloton Therapeutics, Calithera Biosciences, Seattle Genetics, Inovio Pharmaceuticals, Eisai, Lilly, Amgen, Surface Oncology, and BioNTech AG. GG reports speakers bureau fees (institutional) from Janssen Oncology, Ipsen, BMS, Amgen, Sanofi-Aventis, MSD Oncology, and Astellas Pharma; travel accommodations, expenses from Janssen Oncology, BMS, Astellas Pharma, Pfizer, Ipsen, Sanofi, and AstraZeneca; and advisory board fees (institutional) from BMS, Janssen, Ipsen, Sanofi-Aventis, MSD Oncology, Pfizer, Bayer, and AstraZeneca. AN reports consulting or advisory fees from MSD, Roche, Bayer, AstraZeneca, Clovis Oncology, Janssen, Incyte, Seattle Genetics/Astellas, BMS, Rainier Therapeutics, GlaxoSmithKline, and Ferring; travel accommodations, expenses from Roche, MSD, AstraZeneca, Janssen, and Rainier Therapeutics; honoraria from Roche, Merck, AstraZeneca, Janssen, Foundation Medicine, and BMS; and research funding (institutional) from MSD, AstraZeneca, Ipsen, and Seagen. FS reports consulting or advisory fees from BMS, Roche, MSD, and Sanofi; research funding from BMS; travel accommodations, expenses from BMS, Celgene, and Sanofi; and royalties from Cureab GmbH. MW-R is employed by and has stockownership in BMS and Agios; and reports PCT patent publication WO2020/198676 (institutional). FN, JL, SC, JZ, and KÜ-K are employed by and have stock ownership in BMS. DY declared no conflicts of interest.
Footnotes
Data were presented in part at the ASCO Genitourinary Cancers Symposium; February 17–19, 2022; San Francisco, CA, USA. Abstract number 491.
Data sharing:
Bristol Myers Squibb’s policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html.
References
- [1].Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 2015;14:847–56. [DOI] [PubMed] [Google Scholar]
- [2].Chen S, Crabill GA, Pritchard TS, et al. Mechanisms regulating PD-L1 expression on tumor and immune cells. J Immunother Cancer 2019;7:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Doroshow DB, Bhalla S, Beasley MB, et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol 2021;18:345–62. [DOI] [PubMed] [Google Scholar]
- [4].Zhu J, Armstrong AJ, Friedlander TW, et al. Biomarkers of immunotherapy in urothelial and renal cell carcinoma: PD-L1, tumor mutational burden, and beyond. J Immunother Cancer 2018;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hirsch FR, McElhinny A, Stanforth D, et al. PD-L1 immunohistochemistry assays for lung cancer: results from phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J Thorac Oncol 2017;12:208–22. [DOI] [PubMed] [Google Scholar]
- [6].Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med 2017;376:1015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bellmunt J, Hussain M, Gschwend JE, et al. Adjuvant atezolizumab versus observation in muscle-invasive urothelial carcinoma (IMvigor010): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2021;22:525–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sharma P, Retz M, Siefker-Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 2017;18:312–22. [DOI] [PubMed] [Google Scholar]
- [9].OPDIVO (nivolumab) [package insert]. Bristol Myers Squibb, Princeton, NJ; 2022. [Google Scholar]
- [10].Bajorin DF, Witjes JA, Gschwend JE, et al. Adjuvant nivolumab versus placebo in muscle-invasive urothelial carcinoma. N Engl J Med 2021;384:2102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].KEYTRUDA (pembrolizumab) [package insert]. Merck, Whitehouse Station, NJ; 2021. [Google Scholar]
- [12].De Marchi P, Leal LF, Duval da Silva V, et al. PD-L1 expression by tumor proportion score (TPS) and combined positive score (CPS) are similar in non-small cell lung cancer (NSCLC). J Clin Pathol 2021;74:735–40. [DOI] [PubMed] [Google Scholar]
- [13].Kulangara K, Zhang N, Corigliano E, et al. Clinical utility of the combined positive score for programmed death ligand-1 expression and the approval of pembrolizumab for treatment of gastric cancer. Arch Pathol Lab Med 2019;143:330–7. [DOI] [PubMed] [Google Scholar]
- [14].Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016;387:1909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bristol Myers Squibb’s policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html.