

Abstract
Individuals with Prader-Willi syndrome (PWS) exhibit several metabolic and behavioral abnormalities associated with excessive food-seeking activity. PWS is thought to be driven in part by dysfunctional hypothalamic circuitry and blunted responses to peripheral signals of satiety. Previous work described a hypothalamic transcriptomic signature of individuals with PWS. Notably, PWS patients exhibited downregulation of genes involved in neuronal development and an upregulation of neuroinflammatory genes. Deficiencies of brain-derived neurotrophic factor (BDNF) and its receptor were identified as potential drivers of PWS phenotypes. Our group recently applied an adeno-associated viral (AAV)-BDNF gene therapy within a preclinical PWS model, Magel2-null mice, to improve metabolic and behavioral function. While this proof-of-concept project was promising, it remained unclear how AAV-BDNF was influencing the hypothalamic microenvironment and how its therapeutic effect was mediated. To investigate, we hypothalamically injected AAV-BDNF to wild type and Magel2-null mice and performed mRNA sequencing on hypothalamic tissue. Here, we report that (1) Magel2 deficiency is associated with neuroinflammation in the hypothalamus and (2) AAV-BDNF gene therapy reverses this neuroinflammation. These data newly reveal Magel2-null mice as a valid model of PWS-related neuroinflammation and furthermore suggest that AAV-BDNF may modulate obesity-related neuroinflammatory phenotypes through direct or indirect means.
Keywords: AAV, adeno-associated virus, gene therapy, BDNF, brain-derived neurotrophic factor, Prader-Willi syndrome, PWS, inflammation, neuroinflammation, hypothalamus
Graphical abstract
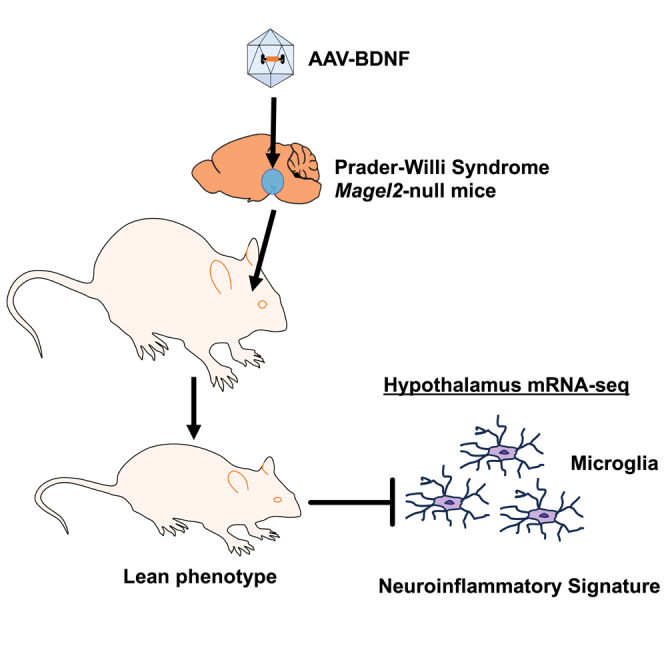
Cao and colleagues report that hypothalamic AAV-BDNF gene therapy ameliorates neuroinflammatory signature in a mouse model of PWS.
Introduction
Prader-Willi syndrome (PWS) is a contiguous gene syndrome that occurs in approximately 1 in 15,000 individuals.1 At its core, PWS is both a metabolic and behavioral disease. Individuals with PWS are continually occupied by food and the search for the next meal. As such, the syndrome manifests in excessive appetite, obesity, and metabolic syndrome in a characteristic progression.2 The patient population also exhibits developmental delays, cognitive impairment, and behavioral problems (e.g., verbal perseveration, temper tantrums, non-compliance, and psychosis) that intensify with worsened hyperphagia.2,3,4,5,6
PWS is driven by the loss of expression of several paternally expressed, imprinted genes located on chromosome 15q11-q13. Previous work used postmortem human hypothalamic samples to describe a transcriptomic signature of PWS.7 In short, Bochukova et al.7 observed that PWS was associated with a downregulation of genes involved in neuronal function and an upregulation of microglial genes and inflammatory markers.
The PWS transcriptomic signature was thought to be mediated in part by reduced expression of brain-derived neurotrophic factor (BDNF) and its receptor, tropomyosin receptor kinase B (TrkB).7 Within the brain, BDNF regulates synaptic plasticity, neuronal growth, survival, and differentiation.8 Genome-wide association studies have found that BDNF is 1 of 18 genetic loci associated with body mass index.9 Haploinsufficiency for BDNF or TrkB is associated with obesity and hyperphagia.10,11 Furthermore, BDNF polymorphisms and reductions in central/peripheral BDNF are linked to sedentary, anxiety-like, depression-like, and obsessive compulsive-like behaviors.12,13,14,15,16 Together, these data suggest modulation of BDNF or associated signaling may be of therapeutic benefit in the PWS patient population.
MAGE Family Member L2 (MAGEL2), is located within the PWS genetic region and its loss is thought to contribute to irregularities in energy homeostasis.17,18,19,20 Magel2-null mice mirror some aspects of PWS pathophysiology, including increased adiposity,20,21 impaired glycemic control,19,21 and alterations in neuroendocrine responses.17,18,19 Previous work by our lab applied an adeno-associated virus (AAV) based BDNF gene therapy to Magel2-null mice via hypothalamic injection to determine whether the vector could rescue metabolic function via the stimulation of various BDNF-related pathways.21 Hypothalamic BDNF gene transfer prevented weight gain, improved body composition, and increased relative energy expenditure in female Magel2-null mice. Moreover, BDNF gene therapy improved several measures of systemic metabolic function, including glucose metabolism, insulin sensitivity, and circulating adipokine levels. We observed no adverse behavioral effects, indicating high levels of efficacy and safety. Male Magel2-null mice also benefited from BDNF gene therapy, displaying improved body composition, insulin sensitivity, and glucose metabolism.21
While the previous work represented an important proof of concept, the molecular signature underlying the Magel2-null model and the ability of AAV-BDNF to treat metabolic dysfunction remained unclear. Here, we performed mRNA sequencing (mRNA-seq) to assess genotype- and gene-therapy driven alterations in hypothalamic gene expression. Importantly, this work represents the first transcriptomic analysis of the Magel2-null mouse model and describes a baseline level of inflammation mirroring that is observed in human individuals with PWS.7 Our data suggest that Magel2 loss of function is associated with heightened inflammation within the hypothalamus. Interestingly, AAV-BDNF induced a reversal of (1) the Magel2-null neuroinflammatory signature and (2) several genes involved in microglial activation/function. Altogether, our work suggests that AAV-BDNF may influence obesity-related neuroinflammatory phenotypes through direct or indirect means.
Results
Hypothalamic AAV-BDNF gene therapy improves body composition and circulating metabolic markers in Magel2-null mice
Previous work by our group showed that hypothalamic BDNF gene therapy improves metabolic and behavioral health in the Magel2-null mouse model.21 To pursue genetic signatures underlying these outcomes, we performed a similar short-term experiment and isolated hypothalamic tissue for downstream transcriptomic analyses. Female mice were injected with either AAV-YFP or AAV-BDNF vectors (Figure 1A) to the arcuate/ventromedial nuclei of the hypothalamus (ARC/VMH)—known sites of BDNF and pro-opiomelanocortin (POMC)/agouti-related peptide (AGRP) action.22,23 Body weight was monitored on a weekly basis. As soon as 2 weeks after AAV injection, Magel2-null mice injected with AAV-BDNF showed reduced body weight over AAV-YFP injected counterparts. At sacrifice, we observed AAV-BDNF reduced body weight and moderated weight gain in both wild type and Magel2-null mice as compared with AAV-YFP control counterparts (Figures 1B and 1C).
Figure 1.

AAV-BDNF gene therapy improves body composition in female Magel2-null mice
(A) Vector schematics. (B) Body weight. (C) Percent change in body weight. (D) Baseline relative fat mass. (E) Baseline relative lean mass. (F) Relative fat mass at 6 weeks post AAV injection. (G) Relative lean mass at 6 weeks post AAV injection. Data are means ± SEM. Sample size: n = 6 per group. ∗ or #p < 0.05, ∗∗ or ##p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. For time course measurements, # symbols denote WT YFP vs. Magel2-null YFP and ∗ symbols denote Magel2-null YFP vs. Magel2-null BDNF.
To confirm vector efficacy without the need for invasive profiling that might influence gene expression, body composition scans were performed. Consistent with previous reports,20,21 at baseline, we observed a genotype-driven increase in relative fat mass and decrease in relative lean mass within Magel2-null mice as compared with wild-type counterparts (Figures 1D and 1E). At 6 weeks post AAV injection, we observed AAV-BDNF treated Magel2-null mice exhibited a reduction in relative fat mass (Figure 1F) and an increase in relative lean mass (Figure 1G) as compared with genotype-matched AAV-YFP counterparts. Notably, these mice exhibited similar body composition to wild-type controls.
At 6 weeks after AAV injection, mice were humanely euthanized, and tissues were collected. Mice appeared physically distinct at time of euthanasia (Figure 2A). In three adipose depots—the brown adipose tissue, inguinal white adipose tissue (iWAT), and gonadal white adipose tissue—we observed a genotype-driven increase in absolute and relative tissue weight (Figures 2B and 2C), consistent with body composition observations. Magel2-null mice treated with AAV-BDNF showed a reduction in the adipose depot size as compared with genotype-matched AAV-YFP counterparts. We additionally observed AAV-BDNF treatment reduced absolute liver mass in Magel2-null mice as compared with AAV-YFP counterparts (Figure 2B). Interestingly, we observed an AAV-BDNF-driven decrease in absolute and relative pancreas mass in both wild-type and Magel2-null mice (Figures 2B and 2C), the functional significance of which is unknown.
Figure 2.

AAV-BDNF gene therapy influences adipose depot size and circulating metabolic markers in female Magel2-null mice
(A) Representative image of mice at 6 weeks post AAV injection. (B) Absolute tissue weight at 6 weeks post AAV injection. (C) Relative tissue weight at 6 weeks post AAV injection. (D) Serum leptin. (E) Serum insulin. (F) Serum glucose. (G) HOMA-IR index. Data are means ± SEM. Sample size: n = 5–6 per group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. BAT, brown adipose tissue; gWAT, gonadal white adipose tissue.
Serum was profiled to assess changes in systemic metabolism after AAV-BDNF gene therapy. Leptin serves as a central-peripheral messenger to maintain energy homeostasis and its production is positively correlated with adipose tissue mass.24 Similar to the previous experiment,21 Magel2-null mice exhibited hyperleptinemia as compared with wild-type controls. AAV-BDNF treatment completely reversed the hyperleptinemia in Magel2-null mice (Figure 2D). Similarly, Magel2-null mice displayed significantly higher fasting serum insulin level as compared with wild-type counterparts, which was completely rescued by AAV-BDNF treatment (Figure 2E). Contrary to the previous report, AAV-YFP treated Magel2-null mice displayed minor decreases in circulating glucose levels when compared with wild-type mice treated with AAV-YFP (Figure 2F). The Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) index was calculated from circulating insulin and glucose levels as a proxy for insulin sensitivity (Figure 2G). Interestingly, AAV-BDNF-treated Magel2-null mice exhibited a reduction in HOMA-IR score as compared with AAV-YFP genotype-matched counterparts, consistent with improved insulin sensitivity.
AAV-BDNF gene therapy alters the hypothalamic transcriptome in Magel2-null mice
Hypothalamic samples were subjected to mRNA-seq and differentially expressed genes (DEGs) were identified. We performed a principal component analysis (PCA) and identified four distinct clusters consistent with experimental group (Figure 3A). Two data subsets were compared to consider genotype (AAV-YFP Magel2-null vs. AAV-YFP wild type mice) and gene therapy (AAV-BDNF Magel2-null vs. AAV-YFP Magel2-null mice) driven influences on the transcriptome.
Figure 3.

AAV-BDNF gene therapy alters the hypothalamic transcriptome in female Magel2-null mice
(A) PCA plot. (B) Volcano plot depicting DEGs of the Magel2-null YFP vs. WT YFP comparison (genotype effect). (C) Volcano plot depicting DEGs of the Magel2-null BDNF vs. Magel2-null YFP comparison (gene therapy effect). (D) IPA; selected canonical pathways related to inflammatory processes. DEGs were identified using the following thresholds: log2[fold change] <−0.585 or >0.585 (corresponding with a fold change of 1.5), Q-value (or a false discovery rate p value) < 0.05. Sample size: WT YFP n = 6, WT BDNF n = 4, Magel2-null YFP n = 6, Magel2-null BDNF n = 6.
To understand how the transcriptome of Magel2-null mice differs from wild-type controls, we compared AAV-YFP treated Magel2-null and AAV-YFP-treated wild-type mice. We identified 472 upregulated and 95 downregulated DEGs (Figure 3B). As expected, Magel2 was among the listed DEGs (log2[fold change] = −1.54, Q < 0.001). Consistent with previous reports detailing altered circadian activity in Magel2-null mice,25 we observed a genotype-driven downregulation of Hcrt (encoding hypocretin or orexin). Crh (encoding corticotropin-releasing hormone) and Mc4r (encoding melanocortin 4 receptor) were downregulated, consistent with alterations in energy homeostasis circuitry.26 Socs3 (encoding suppressor of cytokine signaling 3), a negative regulator of insulin signaling,27 was upregulated in Magel2-null mice as compared with wild-type controls, consistent with the observed worsening of insulin sensitivity in the previous experiment.21 Interestingly, many inflammatory-related genes were among identified DEGs, including Cx3cr1 (encoding CX3C motif chemokine receptor 1), Cxcl10 (encoding C-X-C motif chemokine ligand 10), Nfkbia (encoding nuclear factor-kappa-B-inhibitor alpha), Pycard (encoding apoptosis-associated speck-like protein containing a CARD), Irf1 (encoding interferon regulatory factor 1), Tlr2 (encoding toll-like receptor 2), and Aif1 (encoding allograft inflammatory factor 1). Notably, Cx3cr1 is specifically expressed by microglia in the CNS,28,29 and Aif1 is expressed by microglia and peripheral macrophages.30 A full list of DEGs can be found in Table S1.
To understand how BDNF gene therapy influences the transcriptome in Magel2-null mice, we compared AAV-BDNF- and AAV-YFP-treated Magel2-null mice. We identified 114 upregulated and 266 downregulated DEGs (Figure 3C). Consistent with BDNF overexpression or administration of BDNF protein,31,32,33,34 we observed upregulation of Vgf (encoding VGF nerve growth factor inducible), the orexigenic peptides Npy (encoding neuropeptide Y) and Agrp (encoding AGRP), Vip (encoding vasoactive intestinal peptide), and Crh. BDNF gene therapy induced upregulation of Hctr1 (encoding hypocretin [orexin] receptor 1), which is interesting given the downregulation of Hcrt in naive Magel2-null mice.25 Notably, BDNF gene therapy induced a downregulation of several inflammatory-related markers in Magel2-null mice, including Ccl2 (chemokine [C-C motif] ligand 2), Il2ra (encoding IL-2 receptor alpha), Il12rb1 (encoding IL-12 receptor beta 1), Nlrp1b (encoding NLR family, pyrin domain containing 1B), Cxcr3 (encoding C-X-C motif chemokine receptor 3), Il1a (encoding IL-alpha), Il15 (encoding IL-15), Tlr2, Irf1, and Aif1. Consistent with observations of improved insulin sensitivity (Figure 2G), Socs3 was downregulated. Bdnf was flagged as a downregulated DEG, but this was unsurprising; endogenous mouse Bdnf was expected to be downregulated as a consequence of overexpression of the human form of BDNF via AAV vector. A full list of DEGs can be found in Table S2.
Ingenuity pathway analysis (IPA) was performed for the above two subsets (Magel2-null YFP vs. wild-type YFP mice and Magel2-null BDNF vs. Magel2-null YFP mice). Outputs were subjected to a comparison analysis to identify canonical pathways that are (1) influenced by genotype at baseline and (2) how the same pathways are altered after BDNF gene therapy in Magel2-null mice. Consistent with many of inflammatory-related DEGs, we observed activation of several inflammatory-related pathways (Figure 3D), including the phagosome formation, dendritic cell maturation, neuroinflammation, role of NFAT in regulation of the immune response, T cell receptor, macrophage classical activation, TREM1, pyroptosis, production of nitric oxide and reactive oxygen species in macrophages, and nuclear factor kappa B (NF-κB) signaling pathways. A full list of pathways can be found in Tables S3 and S4. Interestingly, we observed a marked reversal in the activation states of these signaling pathways in AAV-BDNF treated Magel2-null mice.
AAV-BDNF gene therapy induces a reversal of inflammatory-related upstream regulators in Magel2-null mice
Next, we investigated upstream regulators that might explain broad alterations in the transcriptomic profiles. We did this by considering activation Z scores, which provide the directionality (e.g., activation or inhibition) and magnitude that a regulator may have on the observed downstream gene expression pattern. Activation Z scores are calculated algorithmically in the following manner: the network expression of multiple genes under the control of a putative regulator is considered and then compared to a random pattern of expression. The Z score determines likely regulators based upon the statistical significance of the pattern match.35 IPA identified 18 upstream regulators that (1) had positive activation Z scores as a consequence of genotype and (2) had negative activation Z scores when considering the effect of BDNF gene therapy in Magel2-null mice (Figure 4A). Several of these upstream regulators play roles in inflammation, including Infg (encoding interferon gamma), Tnf (encoding tumor necrosis factor), Myd88 (encoding myeloid differentiation primary response 88), Ccr2 (encoding C-C chemokine receptor type 2), Cxcr3, Il6 (encoding IL-6), and Trem2 (encoding triggering receptor expressed on myeloid cells 2). Notably, BDNF gene therapy in Magel2-null mice reversed the activation Z score of several of these inflammatory related markers, mirroring that which was observed in the canonical pathway analysis.
Figure 4.

AAV-BDNF gene therapy induces a reversal of inflammatory-related upstream regulators in female Magel2-null mice
(A) IPA upstream regulators with positive activation Z scores as a consequence of genotype and negative activation Z scores as a consequence of gene therapy. (B) IPA upstream regulators with negative activation Z scores as a consequence of genotype and positive activation Z scores as a consequence of gene therapy. Sample size: WT YFP n = 6, WT BDNF n = 4, Magel2-null YFP n = 6, Magel2-null BDNF n = 6.
In contrast, IPA identified 10 upstream regulators that (1) had negative activation Z scores as a consequence of genotype and (2) had positive activation Z scores when considering the effect of BDNF gene therapy in Magel2-null mice (Figure 4B). Several of these upstream regulators play roles in inflammatory processes, including Irgm1 (encoding immunity-related GTPase family M protein 1), Irf9 (encoding interferon regulatory factor 9), Ikbkb (encoding inhibitor of nuclear factor kappa-B kinase subunit beta), Il10 (encoding IL-10), and Il1r1 (encoding IL-1 receptor, type I). Taken together, these data indicate that AAV-BDNF gene therapy induces a reversal in inflammatory-related upstream regulators in Magel2-null mice.
AAV-BDNF gene therapy induces a reversal in microglial activation markers in Magel2-null mice
Given several indications of altered inflammatory processes, we began to consider cell-specific contributions. We used a set of previously identified putative genes for microglia, neurons, astrocytes, endothelial cells, and oligodendrocytes.36 We considered the overlap of the top 100 putative genes by cell type with our list of DEGs; 29 microglial markers were among both lists (Figure 5A). Furthermore, we observed a pattern of increased expression of these microglial markers when we compared Magel2-null mice treated with AAV-YFP with wild-type counterparts, indicating genotype-driven activation of microglial-related processes. Interestingly, AAV-BDNF gene therapy reversed the expression of microglial-related markers when applied in Magel2-null mice (Figure 5A). We performed this analysis for four additional cell types. For neuron (Figure S1A) and astrocyte (Figure S1B) putative markers, eight DEGs were flagged apiece; no clear expression shifts were observed between groups. Similar results were observed for endothelial cell (six DEGs; Figure S1C) and oligodendrocyte putative markers (one DEG; data not shown). Together, these data highlight microglia as a cellular population of interest in the hypothalamus for genotype- and gene therapy-related outcomes.
Figure 5.

AAV-BDNF gene therapy induces a reversal in microglial activation markers in female Magel2-null mice
(A) Gene expression of putative microglia markers; all listed genes were identified as DEGs. (B) IPA activation of microglia canonical pathway. DEGs were identified using the following thresholds: log2[fold change] < −0.585 or >0.585 (corresponding with a fold change of 1.5), Q-value (or a false discovery rate p value) < 0.05. Sample size: WT YFP n = 6, WT BDNF n = 4, Magel2-null YFP n = 6, Magel2-null BDNF n = 6.
To confirm microglial observations, we considered the IPA activation of microglia canonical pathway (Figure 5B). Several DEGs play roles in the pathway, including Cxcl10, Cybb, Ccl5, Tlr2, C3, Cd274, Ccr2, Ccl2, Cxc3r1, Trem2, and Nr4a1. We observed a genotype-related upregulation of these microglial activation markers that was reversed upon application of AAV-BDNF gene therapy.
AAV-BDNF gene therapy alters hypothalamic and adipose gene expression
Next, we probed hypothalamic gene expression using qRT-PCR as a secondary confirmation of DEGs identified through mRNA-seq analysis (Figure 6A). As expected, we observed an increase in Bdnf expression in AAV-BDNF treated wild-type and Magel2-null mice. As previously mentioned, Bdnf was identified as a downregulated DEG (AAV-BDNF vs. AAV-YFP wild-type mice) and counters qPCR observations. Notably, the primers used target the coding region of both the human and mouse form of the gene, suggesting AAV-BDNF increases the total amount of Bdnf transcript—whether endogenous or exogenous. Crh expression was downregulated as a consequence of genotype and was upregulated as a consequence of BDNF gene therapy, consistent with previous observations. Mc4r was downregulated and Socs3 was upregulated in AAV-YFP-treated Magel2-null mice as compared with wild-type counterparts. Various inflammatory markers, including Cx3cr1, Nfkbia, Aim2, Cxcl10, and Trem2, were upregulated in AAV-YFP treated Magel2-null mice when compared with wild-type counterparts.
Figure 6.

AAV-BDNF gene therapy alters hypothalamic and adipose gene expression in female Magel2-null mice
(A) Relative gene expression in the hypothalamus. (B) Relative gene expression in the iWAT. Data are means ± SEM. Sample size: WT YFP n = 6, WT BDNF n = 4, Magel2-null YFP n = 6, Magel2-null BDNF n = 6. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We additionally profiled the iWAT, a subcutaneous adipose depot, as a secondary confirmation of metabolic remodeling (Figure 6B). Consistent with adiposity and serum observations, AAV-YFP-treated Magel2-null mice exhibited increased Lep expression as compared with wild-type counterparts. AAV-BDNF reduced Lep expression in both Magel2-null and wild-type mice. We profiled various genes involved in adipose remodeling and maintenance. Ppargc1a (encoding peroxisome proliferator-activated receptor gamma coactivator 1-alpha), a transcriptional coactivator involved in mitochondrial biogenesis, was downregulated in AAV-YFP treated Magel2-null mice as compared with wild-type counterparts. Similar observations were made for Pparg (encoding peroxisome proliferator-activated receptor gamma) and Acacb (encoding acetyl-CoA carboxylase 2)—both of which are involved in fatty acid storage processes—and Adrb3 (encoding beta-3 adrenergic receptor), which regulates thermogenesis and lipolysis. No changes were observed in Adipoq (encoding adiponectin), Ucp1 (encoding uncoupling protein 1), Hsl (encoding hormone-sensitive lipase), or Glut4 (encoding glucose transporter type 4) gene expression. We observed increased Vegfa (encoding vascular endothelial growth factor A), which regulates adipose angiogenesis and browning,37 and Pten (encoding phosphatase and tensin homolog), which regulates adipocyte size and lipolysis,38 gene expression in AAV-BDNF treated Magel2-null mice when compared with AAV-YFP counterparts. No changes in Ccl2 expression were observed.
Long-term AAV-BDNF gene therapy modulates hypothalamic gene expression and signaling pathways in male Magel2-null mice
To further validate the hypothalamic gene expression signature revealed by the microarray in female Magel2-null mice, we newly examined the hypothalamic samples collected from the male cohorts in the previous long-term AAV-BDNF gene therapy study.21 Similar to observations in female mice, AAV-BDNF treatment rescued metabolic dysfunctions in male Magel2-null mice, as shown in a previous publication.21 qRT-PCR analysis of the male cohort revealed a hypothalamic gene expression signature (Figure 7A) similar to that identified in the female cohort of the current study (Figure 6A), characterized as a reversal of genotype-driven inflammatory/microglial markers by AAV-BDNF treatment.
Figure 7.

Long-term AAV-BDNF gene therapy alters hypothalamic gene expression and signaling pathways in male Magel2-null mice
(A) Relative gene expression in the hypothalamus. Data are means ± SEM. Sample size: n = 5 per group. (B) Western blotting and quantification. Data are means ± SEM. Sample size: WT YFP n = 4, Magel2-null YFP n = 5, Magel2-null BDNF n = 5. ∗p < 0.05, ∗∗p < 0.01.
To verify the transgene expression, we performed western blotting on the long-term male hypothalamic microdissections. The AAV-BDNF construct contains a hemagglutinin (HA) tag at the C-terminal of the human BDNF coding sequence. The BDNF transcript (pre-proBDNF) yields precursor (proBDNF) and mature (BDNF) form via intracellular and extracellular proteases. Western blotting using an HA antibody showed two bands corresponding with the precursor (28 kDa) and mature forms (12–14 KDa) of BDNF (Figure 7B).
We previously published that overexpressing the BDNF receptor TrkB full-length (TrkB.FL) in BTBR mice, a model of autism spectrum disorder and insulin resistance, improved metabolic outcomes. Profiling of signaling pathways downstream of BDNF/TrkB found that TrkB.FL overexpression increased phosphorylation of PLCγ in the hypothalamus.39 Here, we performed similar signaling pathway profiling in Magel2-null mice treated with AAV-BDNF (Figure 7B). Interestingly, Magel2-null mice treated with AAV-YFP displayed significantly lower Ras level compared with wild-type mice treated with AAV-YFP. The genotype-related reduction of Ras was normalized by AAV-BDNF treatment. In contrast, Magel2-null mice showed decreased phosphorylation of ERK, but AAV-BDNF treatment did not rescue this genotype-driven change in ERK signaling.
Discussion
Previous work by Bochukova et al. performed RNA sequencing on human postmortem hypothalamic samples to describe a transcriptomic signature of PWS. Interestingly, the group found that PWS upregulated genes overlapped with the transcriptome of orexigenic Agrp neurons and that downregulated genes overlapped with the expression of anorexigenic Pomc neurons. These observations remain consistent with the hyperphagic nature of PWS and the opposing, regulatory nature of Agrp/Pomc neurons toward the maintenance of feeding/fasting behavior. The group also described a downregulation of genes involved in neuronal function and an upregulation of microglial and inflammatory markers (including IRF1, BCL3, SGK1, and NFKBIA). Several inflammatory players, including TNF, TGFB1, IL1B, IFNG, NFKB, IL6, and IKBKB, were among the predicted activated upstream regulators. Furthermore, the group was the first to perform in situ hybridization of human PWS tissue, showing that individuals with PWS exhibited decreases in the number of cells expressing BDNF and NTRK2 (encoding TrkB) in the VMH. Ultimately, the authors concluded that BDNF plays a key role in PWS pathophysiology.7
Despite the value of this landmark study, the subsequent years failed to produce studies that investigated whether PWS preclinical models recapitulate the human transcriptomic profile and, in particular, the inflammatory signature observed in the hypothalamus of PWS patients. While our previous work highlighted the ability of AAV-BDNF gene therapy to ameliorate metabolic dysfunction in Magel2-null mice, data describing how BDNF overexpression altered the transcriptome remained lacking. Here, we observed that (1) Magel2 deficiency is associated with neuroinflammation in the hypothalamus and (2) AAV-BDNF gene therapy reverses this neuroinflammation. To this end, Magel2-null mice can be considered a valid model for probing PWS-related hypotheses relating to neuroinflammation processes. This finding provides a new avenue for basic scientific discoveries to further our knowledge of PWS pathophysiology and therapeutic development. It is important to note that MAGEL2 is but one gene within the PWS region; it remains possible that other genes contribute to neuroinflammation in a similar manner.
A great body of work suggests hypothalamic inflammation and metabolic syndrome are intertwined. Chronic overnutrition induces inflammation-like changes and low-degree activation of NF-κB and upstream regulator IKKβ (inhibitor of NF-κB kinase subunit beta).40 Both fatty acids and carbohydrates have been shown to increase NF-κB activation, which is a driving force for the expression of other inflammatory genes.40,41,42 Conversely, caloric restriction has been shown to reduce hypothalamic inflammation.43 Furthermore, inhibition of NF-κB/IKKβ via genetic and pharmacological means effectively reduces hypothalamic cytokine-driven microinflammation and improves metabolic function.41,44,45 Interestingly, NF-κB and IKKβ inhibit gonadotropin-releasing hormone,46 which is implicated in gonadal development and activity; PWS patients often exhibit hypogonadism.47 Further evidence suggests brain-specific activation of IKKβ leads to increased food intake, body weight gain, and interruptions of central insulin and leptin signaling.41 Likewise, NF-κB induces expression of SOCS3, which then inhibits neuronal insulin signaling.41 Interestingly, NF-κB is thought to be activated by leptin and mediates leptin-stimulated POMC transcription.48,49
Bochukova et al.7 report TNF-α and NF-κB among the top upstream regulators of the PWS transcriptomic signature. Interestingly, the NF-κB signaling pathway was activated within Magel2-null mice and we observed reduced activation of this pathway upon application of BDNF gene therapy. Furthermore, several upstream regulators known to influence NF-κB signaling, including Infg, Tnf, and Myd88, were reversed upon BDNF gene therapy application. Altogether, our observations of increased hypothalamic inflammation remain consistent with metabolic dysfunction observed in the Magel2-null model and individuals with PWS.
Both Bochukova et al. and we report an upregulation of microglial markers in PWS patients and the PWS murine model, Magel2-null mice. Importantly, microglia act as the resident immune cells of the CNS and play a critical role in regulation of neuronal activity and synaptic plasticity. Canonically, BDNF plays roles in synaptic plasticity, neuronal survival, differentiation, and maturation.50,51,52 Several reports53 raise the possibility that PWS-related genetic alterations result in neurodegeneration and neuronal loss, perhaps mediated in part via reduced expression of BDNF/NTRK2.7,54 It remains possible that microglia are activated in a protective manner to maintain and repair the remaining neural circuitry. Several microglial synaptic pruning markers,55 including C1qa, C1qb, C1qc (encoding complement component 1q subunits a/b/c), C3 (encoding complement component 3), and Trem2 were either upregulated at baseline in Magel2-null mice or were reduced following BDNF gene therapy (Tables S1 and S2). C1q and C3 are part of the complement cascade, which serves as a tagging mechanism for microglial pruning.56,57,58 Similarly, TREM2 is involved in microglial-dependent synapse elimination59 and was identified as an upstream regulator (Figure 4A). It remains feasible that overexpression of BDNF rescues normal synaptic processes, reducing the need for microglial pruning, thus reducing associated inflammation in Magel2-null mice. Alternative mechanisms remain plausible. The effects of neuronal BDNF on hypothalamic microglia could be complex and mediated by any/all of the following possibilities: (1) BDNF acts directly on microglia through microglial TrkB, (2) BDNF induces microenvironment changes in the hypothalamus, and (3) BDNF-driven systemic metabolic and immune changes drive feedback to the hypothalamus. Future work should specifically investigate the morphology and activation state of microglia and determine the role of hypothalamic microglia in mediating the metabolic dysfunctions in Magel2-deficiency. In addition to PWS, Schaaf-Yang syndrome is a genetic disorder caused by a disruption of MAGEL2.60,61 Another rare genetic syndrome, Chitayat-Hall syndrome, arises from pathogenic variants in MAGEL2.62,63 As such, mechanistic insights gained in Magel2-null model are highly relevant to these rare genetic diseases. Moreover, identifying the mechanisms of action underlying the hypothalamic BDNF gene therapy may pave ways to improve the efficacy and safety of gene therapy for PWS and other genetic forms of obesity.
Several questions remain following the current study. Importantly, PWS is described as a progressive disease. Individuals with PWS exhibit distinct nutritional phases, transitioning from a hypotonic and/or a failure to thrive period to subsequent periods where weight gain and hyperphagia occur progressively.47,64 Magel2-null mice mirror some aspects of this progression, having a period of impaired feeding and growth20,65 that later makes way to progressive failure of the hypothalamic leptin-POMC pathway,17,18 which underlies systemic metabolic dysfunction. Given the progressive nature of the disease, it would be interesting to determine whether neuroinflammation in the Magel2-null mice is present at a young age and whether it drives the development of metabolic syndrome. Future experiments must delineate (1) whether hypothalamic inflammation is causally connected with metabolic dysfunction in the Magel2-null model, (2) which cellular populations contribute to such processes, (3) whether inflammation is a primary defect or is secondary to neurodegenerative processes, and (4) whether microglia are a viable therapeutic target. Our laboratory will attempt to address some of these questions in the coming years.
The current study has several limitations. Our hypothalamic dissection method includes various nuclei, including the VMH, PVH, ARC, and dorsomedial hypothalamus. Block dissection does not allow for analysis of discrete nuclei. Additional work could use laser capture to isolate individual hypothalamic nuclei for transcriptomic profiling. Alternatively, advanced technology can be applied to profile RNAs and proteins with high spatial resolution. With current bulk mRNA-seq data, we are unable to make discrete conclusions about the genetic signatures of certain cellular subpopulations. Future work using single-cell techniques will be necessary to increase biological resolution and drive investigations into these subpopulations. On a secondary note, we did not use male mice in the current mRNA-seq experiment. Previous work by our group noted similar metabolic responses in Magel2-null male and female mice to AAV-BDNF gene therapy; since our characterization in females was more comprehensive—and additionally included behavioral assessments—we chose them for transcriptomic analysis here. Gene expression profiling of selective inflammatory/microglial markers in the male cohort from the previous long-term AAV-BDNF gene therapy experiment (6 months) revealed a hypothalamic gene expression signature almost identical to what was found in the female cohort of current short-term AAV-BDNF gene therapy (6 weeks), suggesting the neuroinflammatory gene signature associated with Magel2 deficiency and its reversal by AAV-BDNF treatment likely to be independent of sex or duration of the gene therapy. It is important to highlight that male and female Magel2-null mice display some differences in neuroendocrine responses19 and future work will be needed to determine whether transcriptomic signatures differ at baseline and in response to BDNF gene therapy in males beyond the cluster of selected genes. As a final critique, all mice in the current experiment were subjected to surgical trauma and recovery. The current study cannot rule out the possibility that Magel2-null mice respond to injury differently—thus influencing acute inflammatory responses—than wild-type counterparts. As a counterpoints to these notions, (1) AAV-YFP treated Magel2-null mice regained their presurgical weight before the experimental endpoint and (2) the Bochukova et al. human PWS dataset remains complementary to our findings, despite this limitation.
Although BDNF is thought to be involved in PWS pathogenesis, little is known regarding the BDNF/TrkB signaling in the hypothalamus of Magel2-null mice. The profiling of various BDNF downstream signaling pathways, although preliminary, provides first evidence of altered signaling pathways in Magel2-null mice (Figure 7B). It is reported that TrkB receptor activation can activate three canonical pathways—MAPK (Ras/Raf/ERK), PI3K/AKT, and PLCγ.66 Magel2-null mice displayed reduced Ras and decreased phosphorylation of ERK in the hypothalamus, while the phosphorylation of AKT and PLCγ were not altered at baseline. Interestingly, AAV-BDNF treatment restored Ras levels, but not the phosphorylation of ERK. The implications of these genotype- and gene therapy-associated signaling changes remain unclear. GTPase is a downstream effector of BDNF-TrkB signaling in addition to PI3K/AKT, MAPK (Ras/Raf/ERK), and PLCγ. Ras binds GTPase and the complex becomes activated, thereby promoting neuronal growth, differentiation, and neuronal plasticity.67 The roles of Ras/GTPase complex in the phenotypes of Magel2-null mice as well as in the mechanisms of action of AAV-BDNF gene therapy warrant further investigation.
It is important to consider the challenges posed by a brain-directed gene therapy for PWS. Direct injections of a gene therapy to the hypothalamus—as in this manuscript—remain challenging because of the hypothalamus’ deep location within the brain. Alternative methods must be considered to facilitate translation to the clinic. AAV9 remains the gold standard for CNS targeting when using systemic delivery due in part to its characterized risk profile. Next-generation AAV serotypes (e.g., AAV.PHP.B, AAV-B1, and AAV-AS) have the capacity to cross the blood-brain barrier for broad transduction of the brain,68,69,70 but special considerations must be made if we wish to target discrete hypothalamic nuclei. Certain portions of the brain are accessible through endoscopic endonasal procedures, including the hypothalamus71; this route may be more attractive than invasive procedures using burr holes on the skull base. Optimization of such administration methods and serotype choice will be essential in the targeting of PWS-specific hypothalamic nuclei.
In summary, we present data that suggest Magel2-null mice recapitulate the neuroinflammatory genetic signature observed in the hypothalamus of individuals with PWS. Furthermore, we observed the upregulation of various microglial-specific markers in the hypothalamus of Magel2-null mice. Hypothalamic AAV-BDNF gene therapy ameliorated the hypothalamic inflammatory genetic signature associated with Magel2 deficiency, indicating that it may provide a beneficial reduction in inflammation concomitant with improvements in systemic metabolic function.
Materials and methods
Animals
Magel2-null mice harbor a maternally inherited imprinted/silenced wild type allele and a paternally inherited Magel2-lacZ knock-in allele that abolishes endogenous Magel2 gene function. Male mice containing the Magel2-lacZ allele (Jackson Laboratories #009062) were bred with female C57BL/6 mice to produce both wild-type mice and Magel2-null littermates. Mice were genotyped from ear notch biopsies. Identification of mutant offspring was performed by polymerase chain reaction genotyping with Magel2 and LacZ oligonucleotide primers (common forward, 5′-ATGGCTCCATCAGGAGAAC; Magel2 reverse, 5′-GATGGAAAGACCCTTGAGGT; and LacZ reverse, RW4237, 5′-GGGATAGGTCACGTTGGTGT).
Magel2-null mice develop metabolic deficiencies over time18,72; systemic manifestation occurs by 16 weeks of age.20 Therefore, all mice were older than 16 weeks of age at experiment start date (mean age, 30 ± 6 weeks). Female mice were used in this experiment, as previous experimentation by our group showed the ability of AAV-BDNF to improve metabolic and behavioral function in female Magel2-null mice.21
Mice were randomized by body weight and composition 1 week before hypothalamic injection. All mice had ad libitium access to food (normal chow diet, 11% fat, caloric density 3.4 kcal/g) (Teklad) and water. Mice were housed in standard laboratory cages (19.4 cm × 18.1 cm x 39.8 cm) within temperature (22°C–23°C) and humidity (30%–70%) controlled rooms under a 12:12 light:dark cycle. All animal experiments were in accordance with the regulations of The Ohio State University’s Institutional Animal Care and Use Committee.
Recombinant AAV design
The recombinant AAV (rAAV) vector (Figure 1A) was designed to be self-regulating in nature to allow for sustainable, safe weight loss. HA-tagged human BDNF (HA-BDNF, referred to as BDNF) or destabilized yellow fluorescent protein control (dsYFP, referred to as YFP) was inserted in a multiple cloning site following a cytomegalovirus enhancer and a chicken β-actin promoter. Woodchuck post-transcriptional regulatory element (WPRE) and a bovine growth hormone polyadenosine (BGH polyA) tail followed BDNF or YFP transgenes. In the BDNF vector, a second regulatory cassette included an AGRP promoter driving a microRNA targeting BDNF (miR-Bdnf) and WPRE/polyA elements. As body weight decreases and AGRP is physiologically induced, microRNA expression is activated to inhibit BDNF transgene expression; this leads to a sustainable plateau of body weight after a substantial weight loss is achieved, thus limiting the risk of excessive weight loss.32 All vectors were packaged into serotype 1 capsids and purified by iodixanol gradient centrifugation as previously described.32 Comprehensive metabolic modeling validating the vector was completed in male and female Magel2-null mice in a previous experiment.21
Hypothalamic injections of AAV
Mice received either AAV-BDNF-miR-Bdnf (denoted as AAV-BDNF throughout the text) or AAV-YFP to the hypothalamus, 1 × 1010 viral genomes per site, bilaterally. Before surgery, mice were treated with Ethiqua XR (3.25 mg/kg body weight), an extended-release buprenorphine solution, for pain management. Mice were anesthetized with a single intraperitoneal dose of ketamine/xylazine (80 mg/kg and 5 mg/kg) and secured via ear and incisor bars on a stereotaxic frame (Kopf). Anesthesia was further maintained with 2.5% isoflurane at 1 L/min during the injection procedure. A single midline incision was made through the scalp to expose the skull, and two small holes were made with a dental drill above the injection sites. rAAV vectors were administered bilaterally (0.5 μL per site) by a 10-μL Hamilton syringe and a Micro4 Micro Syringe Pump Controller (World Precision Instruments) at 200 nL/min to the ARC/VMH (anterior-posterior [AP], −1.1 mm; medial-laterale [ML], ±0.50 mm; dorsal-ventral [DV], −6.20 mm). When the infusion was finished, the syringe stayed for 5 min. Afterward, the syringe was slowly retracted from the brain and the scalp was sutured. Animals were returned to clean cages resting atop a heating pad. Mice were provided with supplemental care—HydroGel (ClearH2O) and mash—and were carefully monitored post-surgery until fully recovered.
Body composition assessment
An EchoMRI was used to measure fat and lean mass in live mice without anesthesia. At baseline and 6 weeks post AAV injection, body composition analysis was performed with a 3-in-1 Analyzer (EchoMRI LLC) according to manufacturer instructions. Mice were subjected to a 5-Gauss magnetic field and whole-body masses of fat, lean, free water, and total water were determined during separate cycles by manufacturer software comparison to a canola oil standard.
Tissue collection
At 6 weeks post AAV injection, mice were euthanized after a 4-h fast. Mice were anesthetized with 2.5% isoflurane (1.0 L/min) and then decapitated to collect trunk blood. Tissues to be used for mRNA analyses were flash frozen on dry ice and stored at −80°C until further processing. Hypothalamus was collected under a dissection microscope at sacrifice.
Serum harvest and analysis
Trunk blood was collected at euthanasia, clotted on ice, and centrifuged at 10,000 rpm for 10 min at 4°C. The serum component was collected and stored at −20°C until further analysis. ELISA kits were used to assay serum leptin (R&D Systems #DY498) and insulin (ALPCO #80-INSMSU-E01). A Caymen Chemical colorimetric assay was used to determine glucose levels (#10009582). HOMA-IR index was calculated as [fasting serum glucose (mmol/L) × fasting serum insulin (pmol/L)/22.5] as described elsewhere.73
Quantitative RT-PCR
Following tissue sonication, RNA was isolated using the RNeasy Mini kit (QIAGEN #74804) with RNase-free DNase treatment. cDNA was reverse transcribed using Taqman Reverse Transcription Reagents (Applied Biosystems #N8080234). qRT-PCR was completed on the StepOnePlus Real-Time PCR System using Power SYBR Green (Applied Biosystems #A25742) PCR Master Mix. Primer sequences are available in qRT-PCR data analysis. We calibrated data to endogenous controls—Hprt1 for hypothalamus and Actinb for iWAT—and quantified the relative gene expression using the 2−ΔΔCT method.74
mRNA sequencing
RNA was isolated from hypothalamic microdissection using the RNeasy Mini kit (QIAGEN #74804) with RNase-free DNase treatment. Aliquots of purified RNA samples of hypothalamic tissue were shipped to a research services laboratory (Novogene Corporation Inc.) for mRNA-seq. Two samples from the WT AAV-BDNF group were excluded before submission because RNA samples did not meet quality/purity thresholds set by Novogene. Upon arrival, samples were subjected to a quality control test followed by library preparation, sequencing by synthesis, and bioinformatic analysis. RNA-seq via Illumina NovaSeq platforms, which use a paired-end 150-bp sequencing strategy (short reads), was carried out by Novogene Corporation Inc. The single-stranded mRNAs were selectively captured or enriched and converted to cDNA for library preparation. Novogene provided quantification data analysis service (mapping, differential gene expression, etc.) for each sample. Further analysis was performed in house.
DEGs were identified using the following thresholds: log2[fold change] < −0.585 or >0.585 (corresponding with a fold change of 1.5), Q-value (or a false discovery rate p value) < 0.05. PCA was performed using the prcomp R package. Volcano and PCA plots were generated using the ggplot2 and ggrepel R packages. Cell-specific heatmaps were generated using the heatmap.2 R package. Functional and regulatory networks/pathways involving those DEGs were analyzed using the IPA software (QIAGEN, Ingenuity Systems; www.ingenuity.com). For downstream analyses, one sample from the Magel2-null AAV-YFP group was excluded as an outlier.
Cell type analysis
To investigate cell-type specific signatures, we used a previously published dataset of putative cell type-specific markers derived from purified neurons, astrocytes, microglia, oligodendrocytes, and endothelial cells from the mouse cortex.36 The top 100 putative genes were identified by ranking the fold enrichment in that cell types as compared with all other cell types (fragments per kilobase of transcript per million mapped reads [FPKM ]divided by the mean of FPKM values from all other cell types). This analysis mirrors that which was performed in Bochukova et al. Putative markers that were also considered DEGs were identified (Figures 4B and S1A–S1C).
Immunoblotting
One lobe of hypothalamus per mouse was homogenized in ice-cold Pierce RIPA buffer containing 1× Roche Phosstop and Calbiochem protease inhibitor cocktail III, then was subjected to a brief sonication on ice. Tissues lysates were spun at 13,000 rpm for 15 min at 4°C. The supernatant was collected, and the protein concentration was determined with a BCA protein assay kit (Pierce). Protein from each sample was loaded (15 μg) and separated by gradient gel (4%–20%, Mini-PROTEAN TGX, Bio-Rad), then transferred to a nitrocellulose membrane (Bio-Rad). Blots were incubated overnight at 4°C with the following primary antibodies: Beta-actin (Cell Signaling #4970, 1:1,000), phospho-AKT-Ser473 (p-AKT, Cell Signaling #9271, 1:1,000), total AKT (t-AKT, Cell Signaling #9272, 1:1,000), Phospho-p44/42 MAPK (T202/Y204) (p-ERK, Cell Signaling #9101, 1:1,000), total p44/42 MAPK (t-ERK, Cell Signaling #3965, 1:1,000), Phospho-PLCγ1 (Tyr783) (p-PLC γ1 (Tyr783), Cell Signaling # 14008, 1:500), Total PLCγ1 (t-PLCγ1, Cell Signaling # 2822, 1:1,000), Ras (Cell Signaling # 3965, 1:1,000). HA tag (Cell Signaling #3724T, 1:1,000). Chemiluminescence signal was detected and visualized by LI-COR Odyssey Fc imaging system (LI-COR Biotechnology). Quantification analysis was carried out with Image Studio software version 5.2 (LI-COR Biotechnology).
Statistical analysis
Data are expressed as means ± SEM. Microsoft Excel, IBM SPSS v.25, GraphPad Prism 9, and R v.4.1.3. software were used to analyze data. Two-way ANOVAs with Tukey’s post hoc test were used for comparisons between four groups. Normality was tested using the Shapiro-Wilk method. Outliers were determined and removed using the ROUT method. For Figure 7, a one-way ANOVA was used with a Tukey’s post hoc test for gene and protein expression analysis.
Acknowledgments
This work was supported by a grant from the Foundation for Prader-Willi Research (FPWR), NIH grant CA166590, and internal funding from The Ohio State University Comprehensive Cancer Center.
Author contributions
Conceptualization: N.J.Q. and L.C.; methodology, N.J.Q. W.H., and L.C.; validation, N.J.Q., X.M., and L.C.; formal analysis, N.J.Q., X.M., and L.C.; investigation, N.J.Q., X.Z., W.H., X.M., and L.C.; resources, N.J.Q. and L.C.; data curation, N.J.Q. and L.C.; writing – original draft, N.J.Q., and L.C.; writing – review and editing, N.J.Q., W.H., and L.C.; visualization, N.J.Q.; supervision, L.C.; project administration, L.C.; funding acquisition, L.C.
Declaration of interests
L.C. is an inventor of U.S. patent 9,265,843 B2 on the autoregulatory BDNF vector.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.09.004.
Supplemental information
Data and code availability
The authors confirm that data supporting the findings and conclusion of this study are presented within the article and Supplemental materials.
References
- 1.Resnick J.L., Nicholls R.D., Wevrick R., Prader-Willi Syndrome Animal Models Working Group Recommendations for the investigation of animal models of Prader-Willi syndrome. Mamm. Genome. 2013;24:165–178. doi: 10.1007/s00335-013-9454-2. [DOI] [PubMed] [Google Scholar]
- 2.Elena G., Bruna C., Benedetta M., Stefania D.C., Giuseppe C. Prader-willi syndrome: clinical aspects. J. Obes. 2012;2012 doi: 10.1155/2012/473941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verhoeven W.M., Curfs L.M., Tuinier S. Prader-Willi syndrome and cycloid psychoses. J. Intellect. Disabil. Res. 1998;42:455–462. doi: 10.1046/j.1365-2788.1998.4260455.x. [DOI] [PubMed] [Google Scholar]
- 4.Verhoeven W.M.A., Tuinier S. In: Catatonia in Autism Spectrum Disorders. Dhossche D.M., Wing L., Ohta M., Neumärker K.J., editors. Academic Press; 2006. Prader–willi Syndrome: Atypical Psychoses and Motor Dysfunctions; pp. 119–130. [DOI] [Google Scholar]
- 5.Feighan S.M., Hughes M., Maunder K., Roche E., Gallagher L. A profile of mental health and behaviour in Prader-Willi syndrome. J. Intellect. Disabil. Res. 2020;64:158–169. doi: 10.1111/jir.12707. [DOI] [PubMed] [Google Scholar]
- 6.Steinhausen H.C., Eiholzer U., Hauffa B.P., Malin Z. Behavioural and emotional disturbances in people with Prader-Willi Syndrome. J. Intellect. Disabil. Res. 2004;48:47–52. doi: 10.1111/j.1365-2788.2004.00582.x. [DOI] [PubMed] [Google Scholar]
- 7.Bochukova E.G., Lawler K., Croizier S., Keogh J.M., Patel N., Strohbehn G., Lo K.K., Humphrey J., Hokken-Koelega A., Damen L., et al. A Transcriptomic Signature of the Hypothalamic Response to Fasting and BDNF Deficiency in Prader-Willi Syndrome. Cell Rep. 2018;22:3401–3408. doi: 10.1016/j.celrep.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reichardt L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speliotes E.K., Willer C.J., Berndt S.I., Monda K.L., Thorleifsson G., Jackson A.U., Lango Allen H., Lindgren C.M., Luan J., Mägi R., et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kernie S.G., Liebl D.J., Parada L.F. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyons W.E., Mamounas L.A., Ricaurte G.A., Coppola V., Reid S.W., Bora S.H., Wihler C., Koliatsos V.E., Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc. Natl. Acad. Sci. USA. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim Y.-K., Lee H.-P., Won S.-D., Park E.-Y., Lee H.-Y., Lee B.-H., Lee S.-W., Yoon D., Han C., Kim D.-J., Choi S.H. Low plasma BDNF is associated with suicidal behavior in major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2007;31:78–85. doi: 10.1016/j.pnpbp.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 13.Chen Z.Y., Jing D., Bath K.G., Ieraci A., Khan T., Siao C.J., Herrera D.G., Toth M., Yang C., McEwen B.S., et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall D., Dhilla A., Charalambous A., Gogos J.A., Karayiorgou M. Sequence variants of the brain-derived neurotrophic factor (BDNF) gene are strongly associated with obsessive-compulsive disorder. Am. J. Hum. Genet. 2003;73:370–376. doi: 10.1086/377003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Júdice P.B., Magalhães J.P., Hetherington-Rauth M., Correia I.R., Sardinha L.B. Sedentary patterns are associated with BDNF in patients with type 2 diabetes mellitus. Eur. J. Appl. Physiol. 2021;121:871–879. doi: 10.1007/s00421-020-04568-2. [DOI] [PubMed] [Google Scholar]
- 16.Wendland J.R., Kruse M.R., Cromer K.R., Murphy D.L. A large case-control study of common functional SLC6A4 and BDNF variants in obsessive-compulsive disorder. Neuropsychopharmacology. 2007;32:2543–2551. doi: 10.1038/sj.npp.1301394. [DOI] [PubMed] [Google Scholar]
- 17.Mercer R.E., Michaelson S.D., Chee M.J.S., Atallah T.A., Wevrick R., Colmers W.F. Magel2 is required for leptin-mediated depolarization of POMC neurons in the hypothalamic arcuate nucleus in mice. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pravdivyi I., Ballanyi K., Colmers W.F., Wevrick R. Progressive postnatal decline in leptin sensitivity of arcuate hypothalamic neurons in the Magel2-null mouse model of Prader-Willi syndrome. Hum. Mol. Genet. 2015;24:4276–4283. doi: 10.1093/hmg/ddv159. [DOI] [PubMed] [Google Scholar]
- 19.Tennese A.A., Wevrick R. Impaired hypothalamic regulation of endocrine function and delayed counterregulatory response to hypoglycemia in Magel2-null mice. Endocrinology. 2011;152:967–978. doi: 10.1210/en.2010-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bischof J.M., Stewart C.L., Wevrick R. Inactivation of the mouse Magel2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum. Mol. Genet. 2007;16:2713–2719. doi: 10.1093/hmg/ddm225. [DOI] [PubMed] [Google Scholar]
- 21.Queen N.J., Zou X., Anderson J.M., Huang W., Appana B., Komatineni S., Wevrick R., Cao L. Hypothalamic AAV-BDNF gene therapy improves metabolic function and behavior in the Magel2-null mouse model of Prader-Willi syndrome. Mol. Ther. Methods Clin. Dev. 2022;27:131–148. doi: 10.1016/j.omtm.2022.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu B., Goulding E.H., Zang K., Cepoi D., Cone R.D., Jones K.R., Tecott L.H., Reichardt L.F. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cone R.D., Cowley M.A., Butler A.A., Fan W., Marks D.L., Low M.J. The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int. J. Obes. Relat. Metab. Disord. 2001;25:S63–S67. doi: 10.1038/sj.ijo.0801913. [DOI] [PubMed] [Google Scholar]
- 24.Abella V., Scotece M., Conde J., Pino J., Gonzalez-Gay M.A., Gómez-Reino J.J., Mera A., Lago F., Gómez R., Gualillo O. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 2017;13:100–109. doi: 10.1038/nrrheum.2016.209. [DOI] [PubMed] [Google Scholar]
- 25.Kozlov S.V., Bogenpohl J.W., Howell M.P., Wevrick R., Panda S., Hogenesch J.B., Muglia L.J., Van Gelder R.N., Herzog E.D., Stewart C.L. The imprinted gene Magel2 regulates normal circadian output. Nat. Genet. 2007;39:1266–1272. doi: 10.1038/ng2114. [DOI] [PubMed] [Google Scholar]
- 26.Correa-da-Silva F., Fliers E., Swaab D.F., Yi C.X. Hypothalamic neuropeptides and neurocircuitries in Prader Willi syndrome. J. Neuroendocrinol. 2021;33 doi: 10.1111/jne.12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carow B., Rottenberg M.E. SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 2014;5:58. doi: 10.3389/fimmu.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolf Y., Yona S., Kim K.-W., Jung S. Microglia, seen from the CX3CR1 angle. Front. Cell. Neurosci. 2013;7:26. doi: 10.3389/fncel.2013.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishiyori A., Minami M., Ohtani Y., Takami S., Yamamoto J., Kawaguchi N., Kume T., Akaike A., Satoh M. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett. 1998;429:167–172. doi: 10.1016/s0014-5793(98)00583-3. [DOI] [PubMed] [Google Scholar]
- 30.Jurga A.M., Paleczna M., Kuter K.Z. Vol. 14. 2020. (Overview of General and Discriminating Markers of Differential Microglia Phenotypes). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foglesong G.D., Huang W., Liu X., Slater A.M., Siu J., Yildiz V., Salton S.R.J., Cao L. Role of hypothalamic VGF in energy balance and metabolic adaption to environmental enrichment in mice. Endocrinology. 2016;157:983–996. doi: 10.1210/en.2015-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao L., Lin E.J.D., Cahill M.C., Wang C., Liu X., During M.J. Molecular therapy of obesity and diabetes by a physiological autoregulatory approach. Nat. Med. 2009;15:447–454. doi: 10.1038/nm.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeanneteau F.D., Lambert W.M., Ismaili N., Bath K.G., Lee F.S., Garabedian M.J., Chao M.V. BDNF and glucocorticoids regulate corticotrophin-releasing hormone (CRH) homeostasis in the hypothalamus. Proc. Natl. Acad. Sci. USA. 2012;109:1305–1310. doi: 10.1073/pnas.1114122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pellegri G., Magistretti P.J., Martin J.L. VIP and PACAP potentiate the action of glutamate on BDNF expression in mouse cortical neurones. Eur. J. Neurosci. 1998;10:272–280. doi: 10.1046/j.1460-9568.1998.00052.x. [DOI] [PubMed] [Google Scholar]
- 35.Krämer A., Green J., Pollard J., Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y., Chen K., Sloan S.A., Bennett M.L., Scholze A.R., O'Keeffe S., Phatnani H.P., Guarnieri P., Caneda C., Ruderisch N., et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.During M.J., Liu X., Huang W., Magee D., Slater A., McMurphy T., Wang C., Cao L. Adipose VEGF Links the White-to-Brown Fat Switch With Environmental, Genetic, and Pharmacological Stimuli in Male Mice. Endocrinology. 2015;156:2059–2073. doi: 10.1210/en.2014-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang W., Queen N.J., McMurphy T.B., Ali S., Cao L. Adipose PTEN regulates adult adipose tissue homeostasis and redistribution via a PTEN-leptin-sympathetic loop. Mol. Metab. 2019;30:48–60. doi: 10.1016/j.molmet.2019.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson J.M., Boardman A.A., Bates R., Zou X., Huang W., Cao L. Hypothalamic TrkB.FL overexpression improves metabolic outcomes in the BTBR mouse model of autism. PLoS One. 2023;18 doi: 10.1371/journal.pone.0282566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cai D., Khor S. "Hypothalamic Microinflammation" Paradigm in Aging and Metabolic Diseases. Cell Metab. 2019;30:19–35. doi: 10.1016/j.cmet.2019.05.021. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X., Zhang G., Zhang H., Karin M., Bai H., Cai D. Hypothalamic IKKβ/NF-κB and ER Stress Link Overnutrition to Energy Imbalance and Obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Souza C.T., Araujo E.P., Bordin S., Ashimine R., Zollner R.L., Boschero A.C., Saad M.J.A., Velloso L.A. Consumption of a Fat-Rich Diet Activates a Proinflammatory Response and Induces Insulin Resistance in the Hypothalamus. Endocrinology. 2005;146:4192–4199. doi: 10.1210/en.2004-1520.J.Endocrinology. [DOI] [PubMed] [Google Scholar]
- 43.Sadagurski M., Cady G., Miller R.A. Anti-aging drugs reduce hypothalamic inflammation in a sex-specific manner. Aging Cell. 2017;16:652–660. doi: 10.1111/acel.12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Purkayastha S., Zhang G., Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-β and NF-κB. Nat. Med. 2011;17:883–887. doi: 10.1038/nm.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benzler J., Ganjam G.K., Pretz D., Oelkrug R., Koch C.E., Legler K., Stöhr S., Culmsee C., Williams L.M., Tups A. Central Inhibition of IKKβ/NF-κB Signaling Attenuates High-Fat Diet–Induced Obesity and Glucose Intolerance. Diabetes. 2015;64:2015–2027. doi: 10.2337/db14-0093.J.Diabetes. [DOI] [PubMed] [Google Scholar]
- 46.Zhang G., Li J., Purkayastha S., Tang Y., Zhang H., Yin Y., Li B., Liu G., Cai D. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature. 2013;497:211–216. doi: 10.1038/nature12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emerick J.E., Vogt K.S. Endocrine manifestations and management of Prader-Willi syndrome. Int. J. Pediatr. Endocrinol. 2013;2013:14. doi: 10.1186/1687-9856-2013-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jang P.-G., Namkoong C., Kang G.M., Hur M.-W., Kim S.-W., Kim G.H., Kang Y., Jeon M.-J., Kim E.H., Lee M.-S., et al. NF-κB activation in hypothalamic pro-opiomelanocortin neurons is essential in illness-and leptin-induced anorexia. J. Biol. Chem. 2010;285:9706–9715. doi: 10.1074/jbc.M109.070706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi X., Wang X., Li Q., Su M., Chew E., Wong E.T., Lacza Z., Radda G.K., Tergaonkar V., Han W. Nuclear factor κB (NF-κB) suppresses food intake and energy expenditure in mice by directly activating the Pomc promoter. Diabetologia. 2013;56:925–936. doi: 10.1007/s00125-013-2831-2. [DOI] [PubMed] [Google Scholar]
- 50.Marosi K., Mattson M.P. BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol. Metab. 2014;25:89–98. doi: 10.1016/j.tem.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vanevski F., Xu B. Molecular and neural bases underlying roles of BDNF in the control of body weight. Front. Neurosci. 2013;7:37. doi: 10.3389/fnins.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu B., Xie X. Neurotrophic factor control of satiety and body weight. Nat. Rev. Neurosci. 2016;17:282–292. doi: 10.1038/nrn.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manning K.E., Holland A.J. Puzzle Pieces: Neural Structure and Function in Prader-Willi Syndrome. Diseases. 2015;3:382–415. doi: 10.3390/diseases3040382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bochukova E.G. Transcriptomics of the Prader–Willi syndrome hypothalamus. Handb. Clin. Neurol. 2021;181:369–379. doi: 10.1016/B978-0-12-820683-6.00027-0. [DOI] [PubMed] [Google Scholar]
- 55.Cornell J., Salinas S., Huang H.Y., Zhou M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 2022;17:705–716. doi: 10.4103/1673-5374.322423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Györffy B.A., Kun J., Török G., Bulyáki É., Borhegyi Z., Gulyássy P., Kis V., Szocsics P., Micsonai A., Matkó J., et al. Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proc. Natl. Acad. Sci. USA. 2018;115:6303–6308. doi: 10.1073/pnas.1722613115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fonseca M.I., Chu S.H., Hernandez M.X., Fang M.J., Modarresi L., Selvan P., MacGregor G.R., Tenner A.J. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflammation. 2017;14:48. doi: 10.1186/s12974-017-0814-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stevens B., Allen N.J., Vazquez L.E., Howell G.R., Christopherson K.S., Nouri N., Micheva K.D., Mehalow A.K., Huberman A.D., Stafford B., et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 59.Filipello F., Morini R., Corradini I., Zerbi V., Canzi A., Michalski B., Erreni M., Markicevic M., Starvaggi-Cucuzza C., Otero K., et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity. 2018;48:979–991.e8. doi: 10.1016/j.immuni.2018.04.016. [DOI] [PubMed] [Google Scholar]
- 60.McCarthy J., Lupo P.J., Kovar E., Rech M., Bostwick B., Scott D., Kraft K., Roscioli T., Charrow J., Schrier Vergano S.A., et al. Schaaf-Yang syndrome overview: Report of 78 individuals. Am. J. Med. Genet. 2018;176:2564–2574. doi: 10.1002/ajmg.a.40650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schaaf C.P., Marbach F. In: Schaaf-Yang Syndrome. GeneReviews((R)), Adam M.P., Everman D.B., Mirzaa G.M., Pagon R.A., Wallace S.E., Bean L.J.H., Gripp K.W., Amemiya A., editors. 1993. [Google Scholar]
- 62.Jobling R., Stavropoulos D.J., Marshall C.R., Cytrynbaum C., Axford M.M., Londero V., Moalem S., Orr J., Rossignol F., Lopes F.D., et al. Chitayat-Hall and Schaaf-Yang syndromes:a common aetiology: expanding the phenotype of MAGEL2-related disorders. J. Med. Genet. 2018;55:316–321. doi: 10.1136/jmedgenet-2017-105222. [DOI] [PubMed] [Google Scholar]
- 63.Patak J., Gilfert J., Byler M., Neerukonda V., Thiffault I., Cross L., Amudhavalli S., Pacio-Miguez M., Palomares-Bralo M., Garcia-Minaur S., et al. MAGEL2-related disorders: A study and case series. Clin. Genet. 2019;96:493–505. doi: 10.1111/cge.13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller J.L., Lynn C.H., Driscoll D.C., Goldstone A.P., Gold J.A., Kimonis V., Dykens E., Butler M.G., Shuster J.J., Driscoll D.J. Nutritional phases in Prader-Willi syndrome. Am. J. Med. Genet. 2011;155a:1040–1049. doi: 10.1002/ajmg.a.33951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schaller F., Watrin F., Sturny R., Massacrier A., Szepetowski P., Muscatelli F. A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum. Mol. Genet. 2010;19:4895–4905. doi: 10.1093/hmg/ddq424. [DOI] [PubMed] [Google Scholar]
- 66.Pradhan J., Noakes P.G., Bellingham M.C. The Role of Altered BDNF/TrkB Signaling in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2019;13:368. doi: 10.3389/fncel.2019.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang E.J., Reichardt L.F. Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 68.Choudhury S.R., Harris A.F., Cabral D.J., Keeler A.M., Sapp E., Ferreira J.S., Gray-Edwards H.L., Johnson J.A., Johnson A.K., Su Q., et al. Widespread Central Nervous System Gene Transfer and Silencing After Systemic Delivery of Novel AAV-AS Vector. Mol. Ther. 2016;24:726–735. doi: 10.1038/mt.2015.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choudhury S.R., Fitzpatrick Z., Harris A.F., Maitland S.A., Ferreira J.S., Zhang Y., Ma S., Sharma R.B., Gray-Edwards H.L., Johnson J.A., et al. In Vivo Selection Yields AAV-B1 Capsid for Central Nervous System and Muscle Gene Therapy. Mol. Ther. 2016;24:1247–1257. doi: 10.1038/mt.2016.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.L., Yang B., Huber N., Pasca S.P., Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wolf D.A., Hanson L.R., Aronovich E.L., Nan Z., Low W.C., Frey W.H., 2nd, McIvor R.S. Lysosomal enzyme can bypass the blood-brain barrier and reach the CNS following intranasal administration. Mol. Genet. Metab. 2012;106:131–134. doi: 10.1016/j.ymgme.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 72.Maillard J., Park S., Croizier S., Vanacker C., Cook J.H., Prevot V., Tauber M., Bouret S.G. Loss of Magel2 impairs the development of hypothalamic Anorexigenic circuits. Hum. Mol. Genet. 2016;25:3208–3215. doi: 10.1093/hmg/ddw169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fraulob J.C., Ogg-Diamantino R., Fernandes-Santos C., Aguila M.B., Mandarim-de-Lacerda C.A. A Mouse Model of Metabolic Syndrome: Insulin Resistance, Fatty Liver and Non-Alcoholic Fatty Pancreas Disease (NAFPD) in C57BL/6 Mice Fed a High Fat Diet. J. Clin. Biochem. Nutr. 2010;46:212–223. doi: 10.3164/jcbn.09-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif.) 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that data supporting the findings and conclusion of this study are presented within the article and Supplemental materials.