

Abstract
Successful infection strategies must balance pathogen amplification and persistence. In the obligate intracellular parasite Toxoplasma gondii, this is accomplished through differentiation into dedicated cyst-forming chronic stages that avoid clearance by the host immune system. The transcription factor BFD1 is both necessary and sufficient for stage conversion; however, its regulation is not understood. In this study, we examine five factors transcriptionally activated by BFD1. One of these is a cytosolic RNA-binding protein of the CCCH-type zinc finger family, which we name Bradyzoite Formation Deficient 2 (BFD2). Parasites lacking BFD2 fail to induce BFD1 and are consequently unable to fully differentiate in culture or in mice. BFD2 interacts with the BFD1 transcript under stress, and deletion of BFD2 reduces BFD1 protein levels, but not mRNA abundance. The reciprocal effects on BFD2 transcription and BFD1 translation outline a positive feedback loop that enforces the chronic-stage gene-expression program. Thus, our findings help explain how parasites both initiate and commit to chronic differentiation. This work provides new mechanistic insight into the regulation of T. gondii persistence, and can be exploited in the design of strategies to prevent and treat these key reservoirs of human infection.
Keywords: Toxoplasma gondii, bradyzoite, chronic infection, differentiation, feedback loop, RNA-binding protein
INTRODUCTION
Productive infection strategies require trade-offs between pathogen amplification and persistence. While replication increases the likelihood of transmission, the resultant tissue damage drives host immunity to promote pathogen clearance. At the same time, uncontrolled proliferation may be fatal to the host, altogether eliminating the reservoir of infection1. To balance these outcomes, many protozoan parasites have developed slow-growing chronic stages that can withstand unfavorable growth conditions and exhibit reduced immunogenicity2. An equilibrium between proliferation and latency can thereby promote the wellbeing of the host and ensure parasite transmission.
For apicomplexans of the Sarcocystidae family, persistence is achieved through the formation of large intracellular structures (tissue cysts) containing chronic stages known as bradyzoites. Whereas infected animals can survive long-term harboring these cysts, active replication of the proliferative forms (tachyzoites) results in severe, potentially deadly disease. The most widespread of these infections is caused by Toxoplasma gondii, which chronically infects up to one quarter of humans worldwide3. During acute infection, T. gondii tachyzoites disseminate throughout the body and cause pathology through host-cell lysis. A portion of tachyzoites give rise to bradyzoites, which encyst in brain and muscle tissue4. Most infections are controlled by the host immune response; however, reactivation of latent cysts can occur during periods of weakened immune function5–8. Additionally, there are currently no therapies that target chronic stages9, making it impossible to eradicate T. gondii from the host.
Although the physiological triggers of differentiation remain poorly understood, bradyzoites can be induced in cell culture by exogenous stressors like alkaline pH10,11. During this transition, the parasitophorous vacuole is remodeled into a glycan-rich cyst wall12–14. Bradyzoite metabolism shifts from aerobic respiration to anaerobic glycolysis and parasites accumulate cytoplasmic starch granules15–17. These changes coincide with a global restructuring of T. gondii gene expression18–23. Accordingly, proteins that bind chromatin24–27, RNA28–31, or DNA32–36 have been implicated in modulating stage conversion; however, it was not until the discovery of BFD1 that chronic differentiation could be shown to depend on the activity of a single factor.
Bradyzoite Formation Deficient 1 (BFD1), a Myb-like transcription factor, was discovered by screening ~200 T. gondii genes with putative nucleic acid-binding domains23. Despite being transcribed throughout the T. gondii asexual cycle, BFD1 protein is not detected in unstressed parasites, implying that its expression is translationally controlled. Parasites lacking BFD1 grow normally as tachyzoites, but enter a non-differentiated, G1-like state under alkaline stress. Exhibiting the behavior of a master regulator, conditional BFD1 expression is sufficient to induce differentiation in the absence of stress and closely recapitulates the transcriptional changes observed in naturally-derived bradyzoites. Yet it is unclear how BFD1 intersects with other chronic-stage regulators. We previously showed that a number of genes containing putative RNA- and DNA-binding domains are targets of BFD1, suggesting a cascade of downstream differentiation factors. In the present study, we set out to unravel the regulatory network extending from BFD1 by characterizing these effectors. In doing so, we uncover a positive feedback loop acting on BFD1 to reinforce commitment to chronic differentiation.
RESULTS
Analysis of BFD1-regulated gene-expression factors
Previous data sets revealed five putative nucleic acid-binding proteins that are both upregulated during differentiation and bound at their promoters by BFD1, making them likely secondary effectors of the bradyzoite program (Fig. 1a). Two of these—TGME49_306620 (AP2IX-9) and TGME49_208020 (AP2IB-1)—belong to the well-characterized ApiAP2 family of apicomplexan transcription factors. TGME49_311100, TGME49_224630, and TGME49_253790 encode CCCH-type zinc finger domains, involved in RNA-binding. Whereas AP2IX-9 was described previously as a regulator of early bradyzoite development33,35, the other four genes were functionally uncharacterized. We therefore generated conditional knockdown strains for each candidate to assess its role in differentiation. In a parental line expressing the heterologous TIR1 auxin receptor, each candidate was endogenously tagged with mNeonGreen (mNG) and the minimal auxin-inducible degron (mAID; Extended Data Fig. 1), allowing both detection and depletion of the gene product upon addition of indole-3-acetic acid (IAA; Fig. 1b)37,38. BFD1 was included as a control for its impact on differentiation.
Figure 1. Screening for downstream effectors of BFD1.
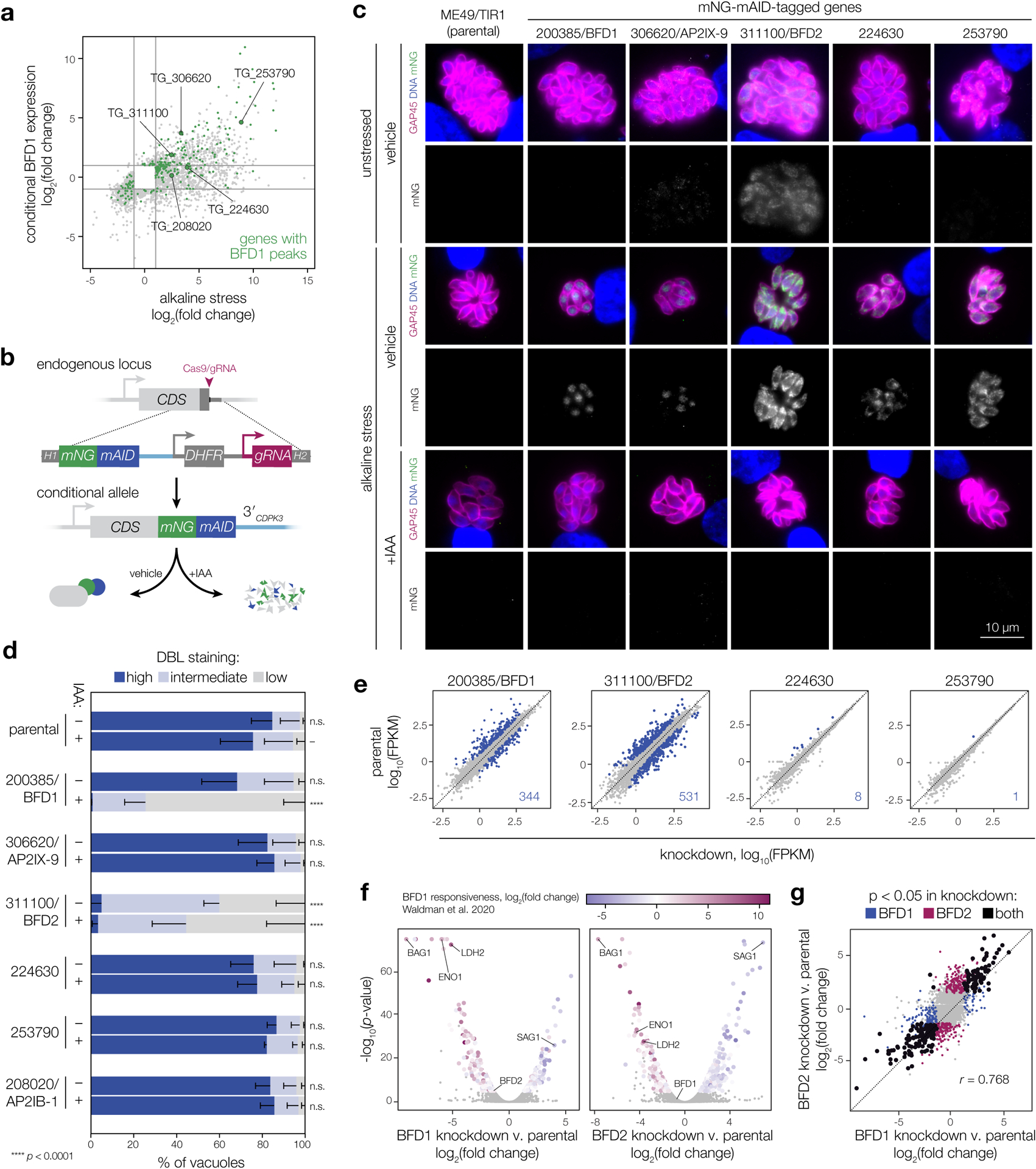
a, Comparison of genes significantly regulated after 48 h under alkaline stress or conditional BFD1 expression (≥2-fold; adjusted p < 0.001, Wald test in DESeq2). BFD1 targets identified by CUT&RUN chromatin profiling are in green23, with the five genes encoding predicted nucleic acid-binding domains indicated. b, Generation of conditional knockdown strains. Tagging with mNG-mAID targets proteins for degradation upon IAA treatment. Additional strain construction details are provided in Extended Data Fig. 8. c, Knockdown strains after 48 h under alkaline-stressed or unstressed conditions, treated with IAA or vehicle. Parasites are labeled by GAP45 (magenta), DNA with Hoechst (blue), and tagged genes by immunostaining for mNG (green). For clarity, mNG signal is shown below the corresponding merged image. Assay was performed twice with similar results. d, Differentiation of knockdown strains after 48 h of alkaline stress, assessed by cyst wall staining (DBL). Mean ± s.d. plotted for n = 3 biological replicates, with a minimum of 175 vacuoles counted per sample. ****p < 0.0001, One-way ANOVA with Dunnett’s correction performed on % DBL-high vacuoles. e–g, Effects of candidate knockdown on the chronic-stage transcriptome. Data reflect changes after 96 h of alkaline stress in the presence of IAA for n = 2 independent infections. Genes significantly affected by depletion of each factor (adjusted p-value < 0.05, Wald test in DESeq2) are highlighted and quantified in the respective plot (e). Complete lists of affected genes are provided in Supplementary Table 1. For BFD1- and BFD2- knockdown strains (f), significantly affected genes are colored by log2(fold change) during conditional BFD1 expression23. A comparison of the transcriptional effects of BFD1 versus BFD2 depletion is shown (g), highlighting genes meeting the significance threshold in both (black) or either sample independently (blue and magenta). Pearson correlation was performed on the union of statistically regulated points.
We first examined expression and localization of each factor. After 48 h of alkaline stress, we observed staining for BFD1 and four of the five candidates (Fig. 1c), with AP2IB-1 remaining below the limit of detection (not pictured). TGME49_311100 was uniquely expressed in acute stages, but upregulated in response to stress. The three predicted RNA-binding proteins localized to the cytosol while BFD1 and AP2IX-9 appeared nuclear, consistent with previous characterization23,35. All five detectable factors were depleted following treatment with IAA (Fig. 1c).
We examined the impact of each factor on stress-induced differentiation, by quantifying cyst wall staining with Dolichos biflorus lectin (DBL) using automated image analysis (Fig. 1d). Consistent with published results, loss of BFD1 blocked cyst formation under alkaline stress23, while AP2IX-9 had no impact33,35. TGME49_224630, TGME49_253790, and AP2IB-1 were similarly dispensable; however, parasites expressing the conditional allele of TGME49_311100 showed a marked decrease in DBL-positive vacuoles under both vehicle and IAA treatment. Given the insensitivity of this effect to IAA, we conclude TGME49_311100 is functionally inhibited by C-terminal tagging. Furthermore, our results implicate TGME49_311100 in the process of differentiation, leading us to name it Bradyzoite Formation Deficient 2 (BFD2).
BFD1 and BFD2 share a common transcriptional signature
Since DBL staining only describes one aspect of differentiation, we performed RNA sequencing on all knockdown strains following 96 h of stress. Only two strains exhibited obvious signatures of transcriptional reprogramming (Fig. 1e). AP2IX-9 and AP2IB-1 in particular had no effect when depleted from chronic stages (Extended Data Fig. 2), despite evidence for the former as an early antagonist of chronic-stage gene expression33,35. While we did identify a small regulon for TGME49_224630 (Supplementary Table 1), the affected transcripts are generally unresponsive to BFD123. Thus, this regulon likely represents a response to alkaline stress rather than a bona fide component of the core bradyzoite program. By contrast, BFD1 knockdown had the expected impact on the chronic-stage transcriptome, which inversely mirrored the effects of conditionally upregulating BFD1 under standard conditions (Fig. 1f). Strikingly, a near-identical response was observed in parasites depleted of BFD2 (Fig. 1f–g), suggesting that the two factors function closely within the differentiation pathway to reshape gene expression.
BFD2 is critical for differentiation
To rule out a dominant-negative effect from C-terminally tagging BFD2, we generated a knockout by replacing BFD2 with a TdTomato cassette (Δbfd2, Fig. 2a & Extended Data Fig. 3a). Knockouts were made in a tagged BFD1 background (Δbfd1::BFD1-TY) to facilitate detection of endogenously regulated BFD123. After 48 h of alkaline stress, DBL staining for Δbfd2 recapitulated our observations during knockdown; however, whereas deletion of BFD1 completely blocked cyst wall formation, a portion of Δbfd2 vacuoles stained faintly with DBL (Fig. 2b–c).
Figure 2. BFD2 is a CCCH-type zinc finger protein required for differentiation.
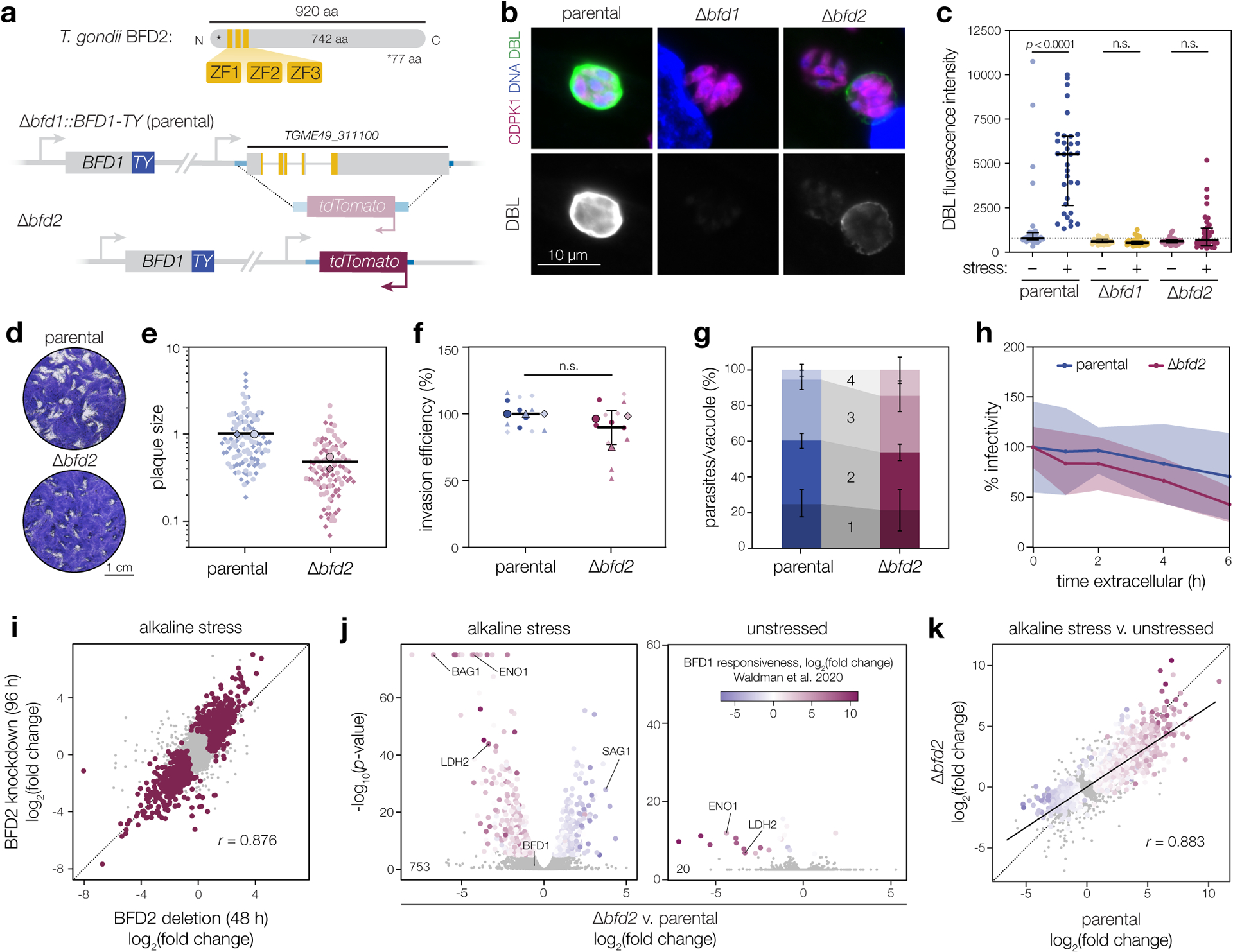
a, BFD2 with zinc finger domains indicated (top). A fluorescence cassette was integrated in place of BFD2 in the Δbfd1::BFD1-TY background (bottom). b, Representative vacuoles after 48 h of alkaline stress. Parasites are labeled by CDPK1 (magenta), DNA with Hoechst (blue), and differentiated vacuoles with DBL (green and below the corresponding merged image). Assay was performed three times with similar results. c, DBL staining intensity after 48 h under unstressed or alkaline-stressed conditions. Median ± IQR plotted for n = 34 vacuoles. n.s., p > 0.05 by One-way ANOVA with Tukey’s correction. d, Plaque assays after 16 days of growth. e, Plaque sizes were quantified for n = 2 independent infections, with replicate means (large points) calculated from at least 50 plaques (small points) and normalized to the parental mean. f, Invasion assay. Mean ± s.d. plotted for n = 3 infections in technical quadruplicate, normalized to the parental mean. n.s., p > 0.05 by Student’s two-tailed t-test. g, Parasites per vacuole after 24 hr of growth. Mean ± s.d. plotted for n = 3 biological replicates, with a minimum of 100 vacuoles counted per sample. p > 0.05 for all bins by Student’s two-tailed t-test. h, Parasite infectivity over extended extracellular stress. Mean ± s.d. for n = 5 fields counted from one infection series, normalized to infectivity at t = 0. i–k, Effects of BFD2 deletion. Data reflect n = 3 independent infections with significantly affected genes highlighted (adjusted p-value < 0.05, Wald test in DESeq2). Comparison of changes in Δbfd2 versus BFD2-mNG-mAID parasites after 48 and 96 h, respectively, under stress (i). Pearson correlation performed on the union of significant genes in both samples. Differential expression analysis (j) of parental versus Δbfd2 parasites after 48 h under alkaline-stressed and unstressed conditions. Significant points colored by log2(fold change) during conditional BFD1 expression23. Comparison of the effects of alkaline stress on parental versus Δbfd2 parasites (k). Trend line fit by linear regression and Pearson correlation performed on all significant points, colored as in j.
Both functional inhibition (Extended Data Fig. 3b) and deletion of BFD2 (Fig. 2d–e) caused a reduction in plaque size, which could not be attributed to a change in host cell invasion (Fig. 2f), replication rate (Fig. 2g), or sensitivity to extracellular stress (Fig. 2h). Revisiting the results of our previous CRISPR-based screens, BFD2 appears dispensable under standard growth conditions in the RH strain39; however, in the ME49 screen that identified BFD1, four of the five guide RNAs targeting BFD2 decreased in abundance over serial passaging in standard media23 (Extended Data Fig. 4), and were further depleted in differentiating populations. The apparent cost to ME49 fitness led BFD2 to be excluded as a potential differentiation factor in our prior work.
To better understand the role of BFD2 throughout the T. gondii asexual cycle, we next performed RNA sequencing on parental and Δbfd2 parasites under alkaline-stressed or unstressed conditions. Reassuringly, Δbfd2 and the non-functional knockdown strain showed strong agreement under stress (Fig. 2i). Specifically, 753 genes were dysregulated in the absence of BFD2, reflecting a clear BFD1-responsive signature (Fig. 2j, left). The partial differentiation of Δbfd2 parasites was recapitulated in their transcriptional response to stress, which mirrored that of the parental strain but with a marked decrease in magnitude (Fig. 2k). By contrast, we observed only minor differences in unstressed samples (Fig. 2j, right), also limited to BFD1-regulated genes. This is consistent with an absence of spontaneously differentiating parasites in the Δbfd2 population, which normally form at low frequency in wild-type ME49 strains during routing culturing in fibroblasts (note DBL-positive vacuoles in unstressed samples, Fig. 2c). Proteome profiling further confirmed reduced levels for several canonical bradyzoite proteins in unstressed Δbfd2 (Extended Data Fig. 3c), none of which appeared fitness-conferring based on prior CRISPR-based screens39.
Although alkaline stress is a potent trigger of stage conversion, we sought to validate our findings in a more physiologically relevant model. T. gondii almost exclusively persists in neurons within infected animals14,40,41, and neurons support high rates of spontaneous differentiation in vitro42,43. Thus, we compared the transcriptomes of wild-type, Δbfd1, and Δbfd2 parasites—this time generated in the Pru genetic background—cultured for 48 h in mouse primary neurons. As predicted, both PruΔbfd1 and PruΔbfd2 showed a distorted transcriptional response consistent with a block in the BFD1 program (Extended Data Fig. 3d). Curiously, whereas stress-induced mutants had shown an inability to both fully induce and repress distinct gene sets (Fig. 1f & 2j), in neurons, loss of either factor primarily affected genes transcriptionally activated by BFD1. This could reflect a strain-specific effect or a difference in the parasite response to distinct differentiation stimuli. Nonetheless, as observed under stress (Fig. 1g), knockout of BFD1 and BFD2 had similar effects on the transcriptome, with loss of BFD1 causing changes of a greater magnitude (Extended Data Fig. 3e). These results further highlight the critical role of BFD2 in enforcing the BFD1 program.
Parasites lacking BFD2 are unable to generate cysts in mice
We examined how BFD2 impacts virulence and cyst formation in mice. Female CD-1 mice were injected intraperitoneally with 100 tachyzoites of either the parental or Δbfd2 strain (Fig. 3a). Despite the reduced fitness of Δbfd2 in vitro, weight loss and mortality were equivalent between the two cohorts (Fig. 3b–c), indicating that BFD2 is dispensable for the acute symptoms of T. gondii infection. Cyst burden was assessed for the brains of all mice at the time of euthanasia, based on coincidence of DBL with a general parasite marker (CDPK1; Fig. 3d). After 45 days, all surviving animals infected with the parental strain harbored hundreds of cysts; none were detected for Δbfd2 (Fig. 3e). A similar result was observed in moribund animals, wherein cysts were first detected 23 days post-injection with the parental strain and never with the mutant—eliminating the possibility that Δbfd2 cysts form and are cleared early in infection. Intriguingly, we did detect Δbfd2 parasites in low numbers in the brains of 45 day-infected mice, and both parental- and Δbfd2-infected animals exhibited pathology consistent with persistent immune activation (Fig. 3f). Nonetheless, our results suggest that BFD2 is dispensable for the acute infection in mice, yet necessary to produce chronic-stage cysts.
Figure 3. Parasites lacking BFD2 fail to generate brain cysts in mice.
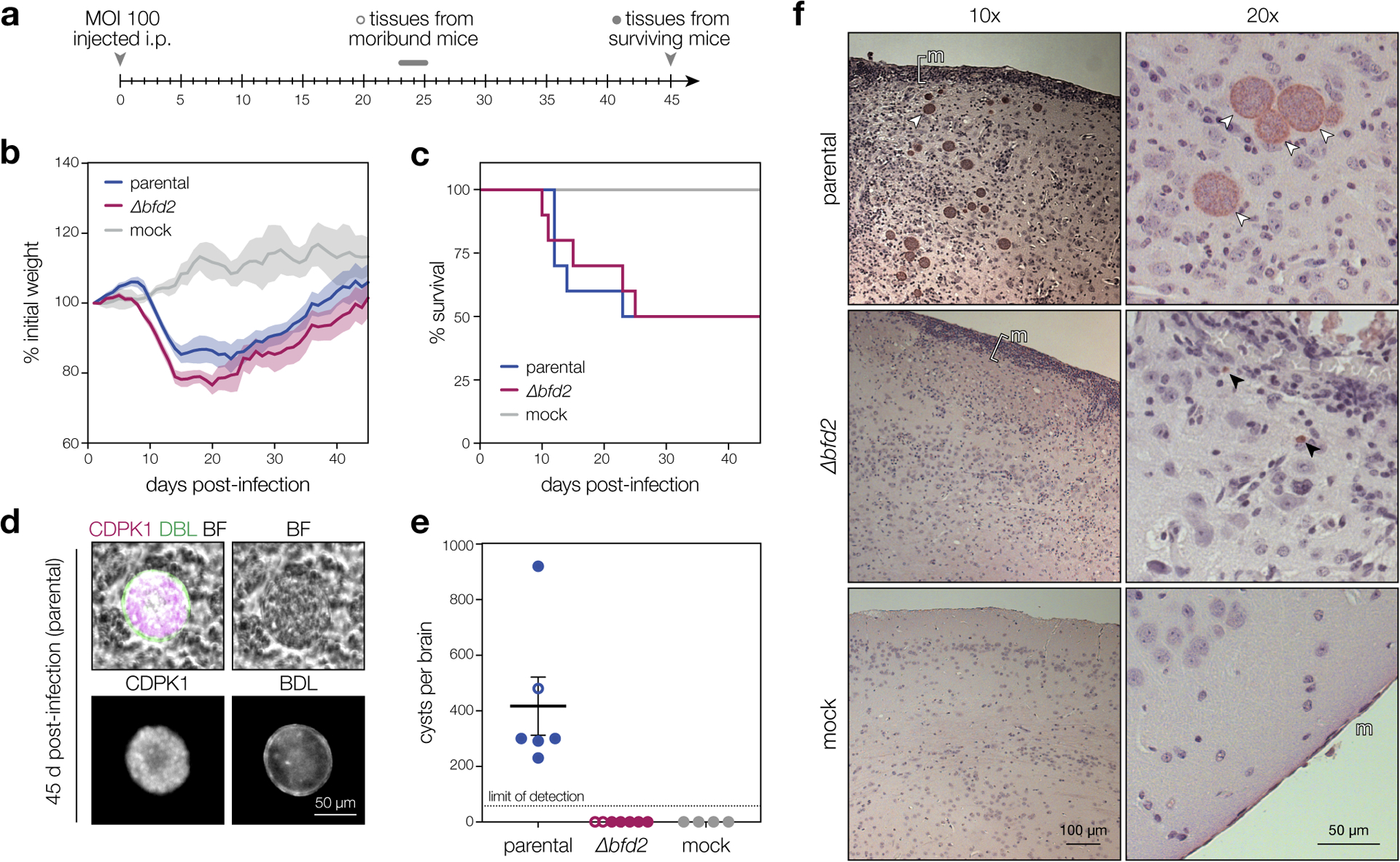
a, Timeline of mouse infections. CD-1 female mice were infected i.p. with 100 tachyzoites from each strain (n = 10) or buffer alone (n = 4). Cyst burdens were assessed in all moribund animals at the time of euthanasia and in surviving animals 45 days post-infection. b–c, Normalized weights (b) and survival (c) of animals in each infection group. Mean weight ± s.e.m. plotted for all surviving animals at a given time point. d, Representative cyst produced by the parental strain. The cyst wall is stained with DBL (green) and individual parasites are stained with CDPK1 (magenta). All structures scored as cysts in e were double-positive for DBL and CDPK1. e, Cyst burden per infected brain. Graphs reflect counts from surviving animals (closed circles) as well as all moribund animals sacrificed after 23 days of infection (open circles). Mean ± s.e.m. plotted for all cyst counts from parental-, Δbfd2-, and mock-infected animals (n = 6, 7, and 4, respectively) regardless of status at the time of brain harvest. f, Histology of brain sections from infected and mock-infected animals. Representative images at 10x and 20x magnification are shown for each strain. The meninges (m) are shown, with regions of inflammation highlighted with brackets. Cysts and individual parasites are indicated with white and black arrows, respectively. Samples were prepared from all surviving mice (closed circles in e) and selected images are representative of phenotypes observed throughout each infection cohort.
A feedback loop between BFD1 and BFD2 drives differentiation
BFD2 is induced by conditional BFD1 expression and its promoter shows evidence of BFD1 binding—characteristics that should place it downstream in the regulatory hierarchy. However, given the analogous effects of the two factors on differentiation, we sought to test this relationship directly. Stress-responsive changes in BFD2 transcript level were quantified in the presence (Δbfd1::BFD1-TY; wild-type) or absence of BFD1 (Δbfd1), as well as for parasites expressing a nonfunctional version lacking DNA-binding motifs (Δbfd1::BFD1ΔMYB-TY)23. Whereas BFD2 was upregulated in wild-type parasites (Fig. 4a), the effect was almost completely lost in Δbfd1 and the nonfunctional-complement, confirming that BFD2 is indeed induced by BFD1.
Figure 4. BFD1 and BFD2 comprise a positive-feedback loop.
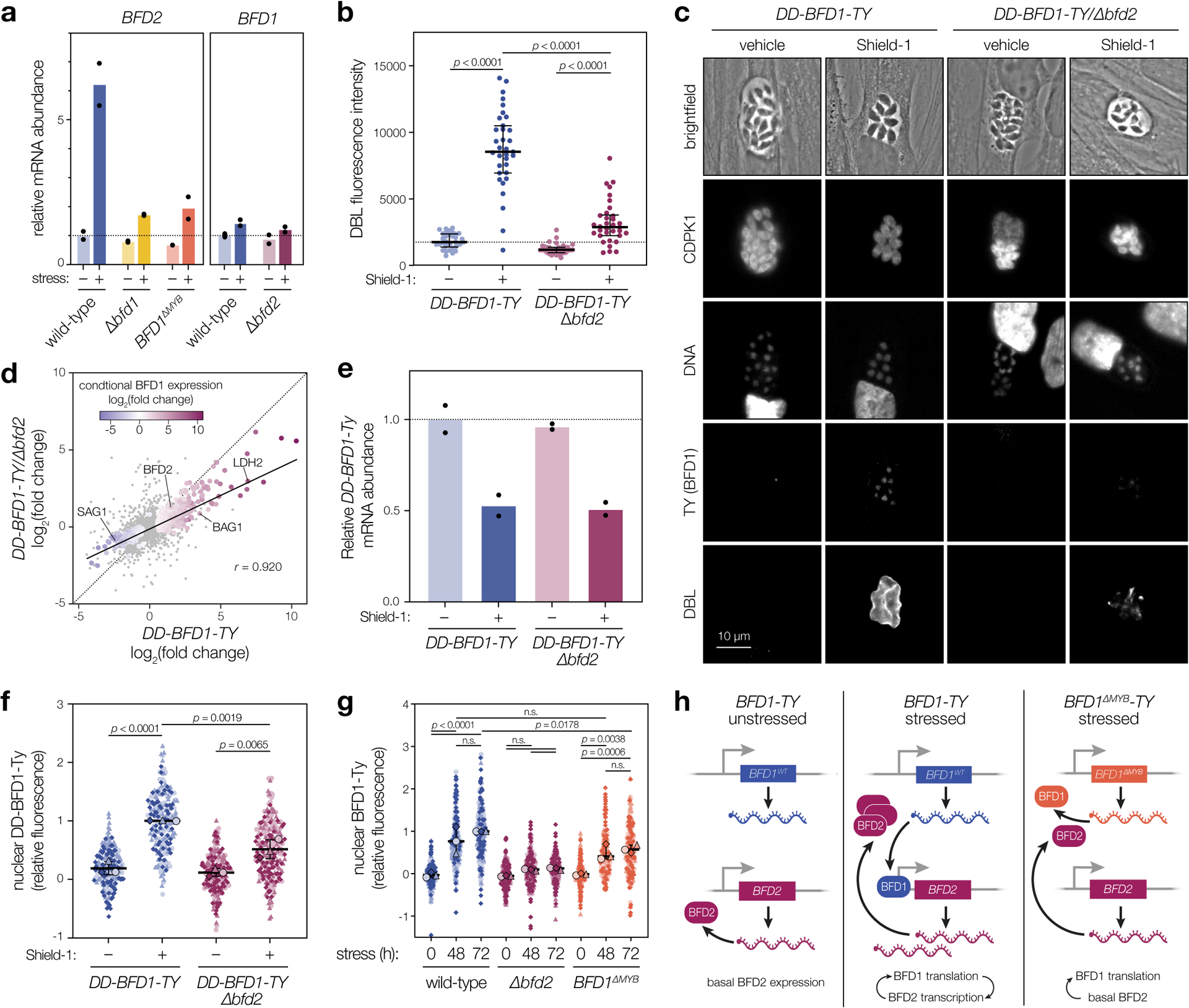
a, RT-qPCR of BFD1 and BFD2 mRNA after 48 h under alkaline-stressed or unstressed conditions. Wild-type and BFD1ΔMYB refer to the version of BFD1 expressed. Mean transcript abundance (n = 2 biological replicates) plotted relative to GCN5B, with normalization to the unstressed wild-type sample. See Supplementary Table 4 for CT values and analysis. b, DBL staining intensity after 48 h in Shield-1 or vehicle. Data quantified for a single replicate representative of assays performed in biological triplicate. Median ± IQR plotted for n = 34 vacuoles per condition. c, Representative vacuoles after 48 h in Shield-1 or vehicle. Parasites are labeled with CDPK1, DNA with Hoechst, BFD1 via the TY epitope, and differentiated vacuoles with DBL. d, Comparison of transcriptional changes in DD-BFD1-TY versus DD-BFD1-TYΔbfd2 parasites after 48 h of Shield-1 or vehicle treatment. Genes significantly affected in either sample (adjusted p < 0.05, Wald test in DESeq2) are colored by fold change during conditional BFD1 expression23. Pearson correlation performed on all significant points with trend line fit by linear regression. e, RT-qPCR of DD-BFD1-TY transcripts after 72 h in Shield-1 or vehicle. Relative quantification performed as in a, with normalization to vehicle-treated DD-BFD1-TY samples. Mean plotted for n = 2 independent infections. See Supplementary Table 5 for analysis. f, Quantification of BFD1 staining in c. Data are normalized to the mean of Shield-1-treated DD-BFD1-TY samples. Mean ± s.d. plotted for n = 3 infections, with replicate means (large points) generated from at least 59 parasite nuclei (small points) per strain. g, Quantification of BFD1 staining under alkaline-stressed or unstressed conditions. Mean ± s.d. plotted for n = 3 infections, with replicate data normalized to the 72 h-stressed wild-type mean. h, Model of BFD1-BFD2 feedback. In acute stages (left), BFD2 is basally expressed; BFD1 is transcribed but not translated. Under stress (center), BFD2 promotes BFD1 translation which, in turn, enhances BFD2 transcription. If BFD1 is nonfunctional (right), transcriptional activation of BFD2 does not occur, so BFD1 is not maximally induced. In b,f,g: n.s., p > 0.05; One-way ANOVA with Tukey’s correction.
To assess the stress-dependency of this relationship, we asked whether loss of BFD2 could hamper differentiation induced by conditional BFD1 expression. Previously, we generated a regulatable version of BFD1 (DD-BFD1-TY, Extended Data Fig. 5a) that is constitutively degraded unless stabilized by the small molecule Shield-1. In the presence of Shield-1, BFD1 accumulates, causing uniform differentiation irrespective of stress23. In this genetic background, we knocked-out BFD2 and assessed differentiation by DBL staining, following 48 h of Shield-1 or vehicle treatment. Over 94% of Shield-1–treated DD-BFD1-TY vacuoles were DBL-positive (Fig. 4b–c), consistent with published results. Though we still observed a weak DBL signal for DD-BFD1-TYΔbfd2, addition of Shield-1 could not overcome the loss of BFD2—reminiscent of the incomplete differentiation observed for BFD2-knockouts under stress (Fig. 2c). This result was also mirrored at the transcriptome level (Fig. 4d). Thus, BFD2 seems to facilitate the BFD1 program regardless of whether it is triggered by stress or targeted induction of BFD1 alone.
Positive feedback is a genetic feature commonly associated with master regulators44. Despite being activated downstream of BFD1, we therefore considered that BFD2 may also be required for robust BFD1 induction. To test this, we initially compared conditional BFD1 expression in the presence or absence of BFD2. Loss of BFD2 had no effect on DD-BFD1-TY mRNA abundance (Fig. 4e)—the Shield-1–induced decrease in transcription can be ascribed to the TUB1 promoter driving the transgene23. Yet immunostaining revealed significantly lower DD-BFD1-TY protein levels in the nuclei of BFD2-knockouts (Fig. 4c & 4f). Encouraged by this result, we further examined endogenous BFD1 protein accumulation in alkaline-stressed wild-type, Δbfd2, and Δbfd1::BFD1ΔMYB-TY parasites, with the hypothesis that if BFD1 and BFD2 indeed promote one another’s expression, full induction of BFD1 should depend on its own activity. In parasites harboring functional copies of both factors (Δbfd1::BFD1-TY; wild-type), BFD1 was robustly detected after 48 h of stress and continued to accumulate over time (Fig. 4g). This effect was diminished in Δbfd2, with BFD1 levels reaching only ~7% of those measured for wild-type parasites after 72 h. Satisfyingly, Δbfd1::BFD1ΔMYB-TY—the nonfunctional BFD1 complement strain which cannot robustly induce BFD2 transcription—exhibited an intermediate level of BFD1 staining. Thus, we conclude that basal levels of BFD2 facilitate stress-responsive expression of BFD1. This in turn drives BFD2 upregulation, closing the feedback loop (Fig. 4h). In addition, since BFD1 mRNA levels were unaffected by the presence of BFD2 (Fig. 4a & 4e), regulation must occur either via BFD1 translation or protein turnover.
RNA-binding is necessary for the function of BFD2
Since C-terminal fusions had rendered the protein inactive (Fig. 1d), we sought to verify the localization of BFD2. We complemented Δbfd2 by introducing an N-terminally tagged cDNA copy back into the endogenous locus (HA-BFD2; Fig. 5a & Supplementary Figure 6a). HA-BFD2 parasites displayed normal plaque formation (Fig. 5b) and complete rescue of DBL staining under stress (Fig. 5c–d). By contrast, a version lacking the CCCH zinc fingers (HA-BFD2ΔZF) could not rescue either phenotype, confirming the importance of these domains for BFD2 function. Both HA-BFD2 and HA-BFD2ΔZF were detectable above background in unstressed parasites and localized to the cytosol, irrespective of culture condition (Fig. 5c). In addition, both were upregulated after 48 h of stress; however, the magnitude of this change was ~30% lower in the zinc finger mutant (Fig. 5e)—further evidence of positive feedback, since parasites cannot fully induce BFD1 nor maximally upregulate BFD2 without BFD2 activity.
Figure 5. BFD2 overexpression induces chronic differentiation.
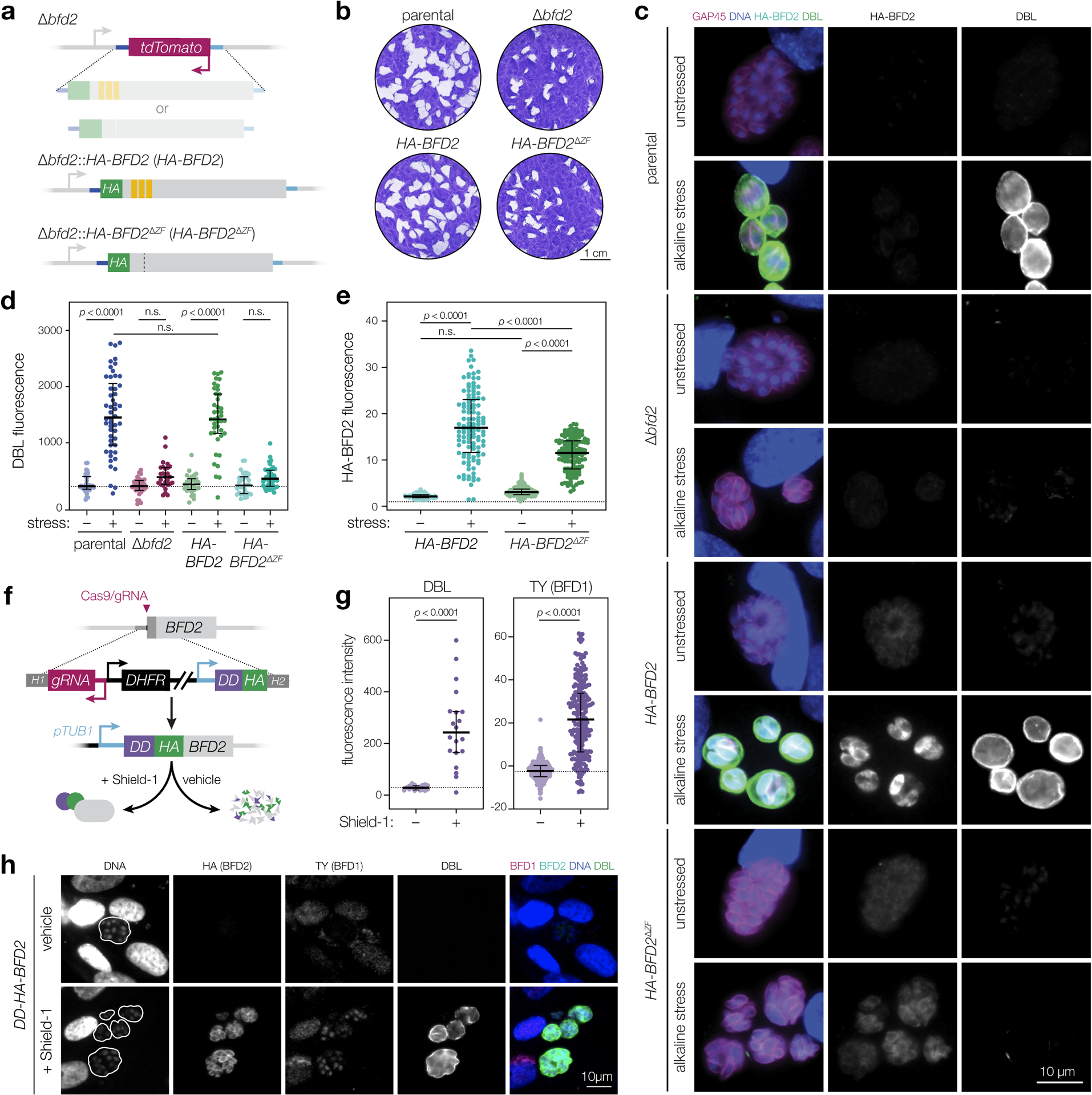
a, An HA-tagged cDNA copy of either wild-type (HA-BFD2) or non-RNA-binding (HA-BFD2ΔZF) BFD2 was integrated back into the endogenous locus for Δbfd2 complementation. b, Plaque assays after 15 days of growth. Parental refers to the Δbfd1::BFD1-TY strain used to construct Δbfd2. c–e, Immunofluorescence analysis after 48 h under alkaline-stressed or unstressed conditions. In representative images (c), parasites are labeled with GAP45 (magenta), DNA with Hoechst (blue), BFD2 via the HA epitope (cyan), and differentiated vacuoles with DBL (green). For clarity, HA-BFD2 and DBL channels are shown separately in grey scale. Quantification of DBL and HA-BFD2 staining are plotted in d and e, respectively. From left to right, data reflect the median ± IQR of measurements taken from n = 36, 50, 32, 27, 29, 41, 32, and 42 vacuoles (d) and n = 101, 114, 156, and 126 individual parasites (e) per sample. DBL signal is normalized to the unstressed parental strain. HA-BFD2 is normalized to background measured from an untagged control (dashed horizontal line). n.s., p > 0.05, One-way ANOVA with Tukey’s correction. f, Generation of a conditional BFD2 overexpression strain. The alpha tubulin promoter (pTUB1) drives transgene expression while an N-terminal destabilization domain (DD) enables regulatable induction of the gene product. Accumulated BFD2 is detected by the HA epitope. See Extended Data Fig. 8 for additional strain construction details. g, Analysis of DBL staining and BFD1 protein accumulation in DD-HA-BFD2 parasites grown for 48 h under Shield-1 or vehicle treatment. Median ± IQR plotted for n = 17 and 20 vacuoles (DBL), and n = 236 and 216 individual nuclei (BFD1), in vehicle- and Shield-1–treated samples, respectively. n.s., p > 0.05, Student’s two-tailed t-test. h, Representative vacuoles corresponding to the data plotted in g. DNA is stained with Hoechst (blue), BFD2 via the HA epitope (cyan), BFD1 by TY (magenta) and differentiated vacuoles with DBL (green). Vacuole boundaries are overlaid on the DNA channel.
Overexpression of BFD2 triggers differentiation
In keeping with our model of BFD2 as a positive regulator of BFD1, we asked whether its conditional overexpression is similarly sufficient for differentiation. In the Δbfd1::BFD1-TY background, we modified the native BFD2 locus to generate a regulatable allele, replacing the endogenous promoter with that of alpha tubulin while appending an N-terminal destabilization domain to the coding sequence (DD-HA-BFD2; Fig. 5f & Extended Data Fig. 6b). Treatment with the stabilizing Shield-1 ligand led to robust cytosolic accumulation of DD-HA-BFD2 and, by 48 h, 95% of vacuoles developed DBL positivity above the vehicle-treated control (Fig. 5g–h). Importantly, BFD1-TY also accumulated in the nuclei of parasites treated with Shield-1. Thus, overexpression of BFD2 is indeed sufficient for differentiation, likely through the induction of BFD1.
BFD2 binds to a cohort of transcripts that includes BFD1
We next asked whether BFD2 binds to BFD1 mRNA. HA-BFD2 immunoprecipitation (IP) was performed on alkaline-stressed and unstressed samples to measure BFD1 transcript abundance by RT-qPCR. HA-BFD2ΔZF samples were processed in parallel to control for non-specific interactions. Compared to total RNA, BFD1 was clearly enriched in IPs from stressed HA-BFD2 parasites (Fig. 6a, WT). No enrichment was detected for HA-BFD2ΔZF (Fig. 6a, ΔZF), confirming the specificity of the interaction. Although BFD1 was also lowly detected in the IP from unstressed cultures, this did not exceed background measured from HA-BFD2ΔZF under the same condition. This disparity could reflect a stress-dependent interaction between BFD2 and BFD1. Unexpectedly, however, two stage-independent transcripts (ASP5 and GCN5B) were also modestly enriched by IP under stress, possibly indicating a general capacity for BFD2 to bind RNA rather than preferential capture of certain transcripts.
Figure 6. BFD2 binds a cohort of transcripts during differentiation that includes BFD1.
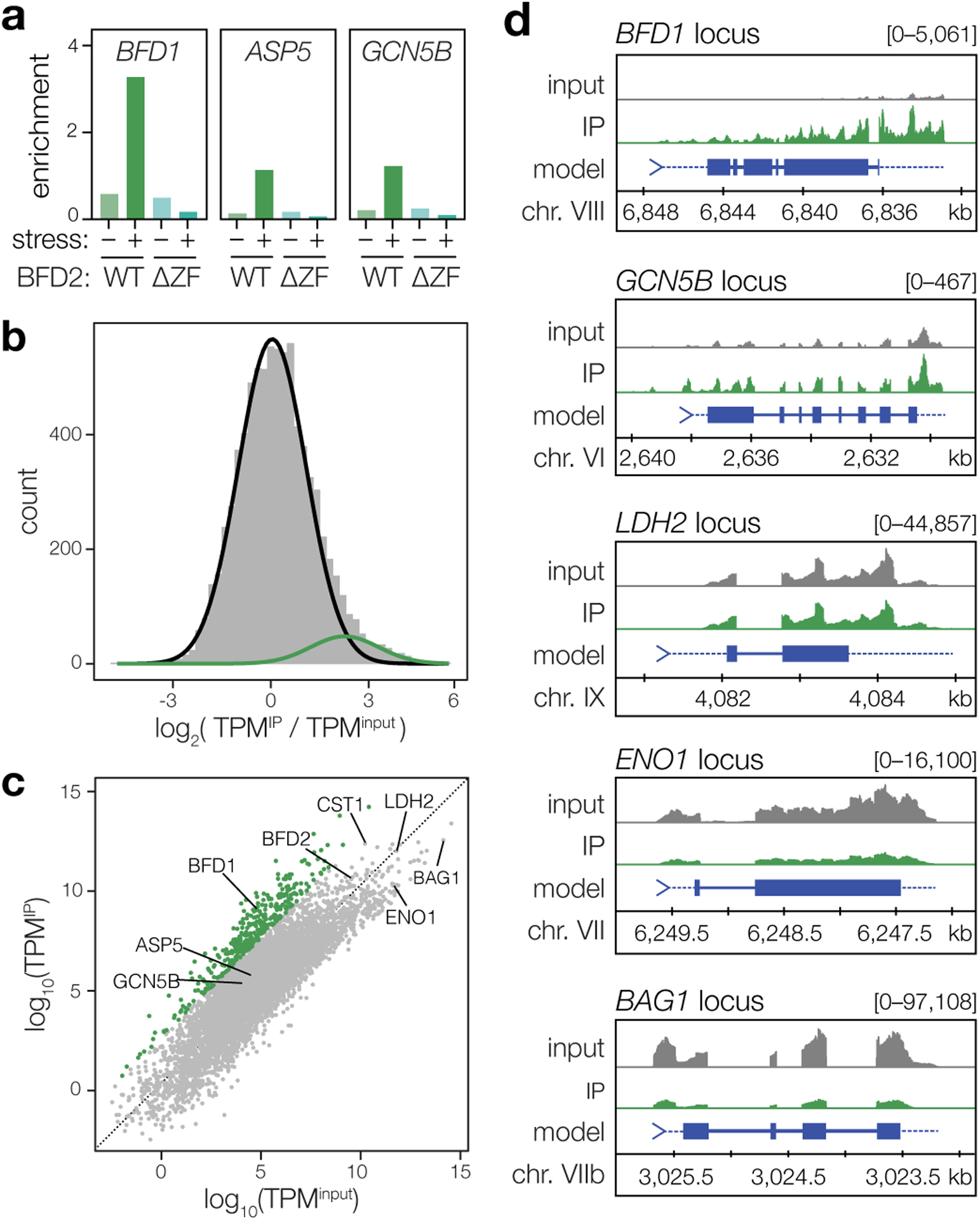
a, RT-qPCR analysis of BFD2-bound mRNAs after 72 h under alkaline-stressed or unstressed conditions. Pull-downs of HA-BFD2 and HA-BFD2ΔZF were performed using antibodies specific to the HA epitope, and enrichment of the indicated transcripts in IP versus input samples was assessed by the ercent Input method. For ΔCT values and analysis, refer to Supplementary Table 6. b, Log-transformed enrichment ratios for all transcripts detected in alkaline-stressed HA-BFD2 parasites. Using mixture modeling, the data were fitted with a combination of two Gaussian curves, one which corresponds to a broad range of non- or moderately interacting RNAs (black line), and one representing our conservative cutoff for enrichment (green). c, Relative abundance of mRNAs following immunoprecipitation of HA-BFD2, as compared to the input transcriptome. Enrichment was defined based on the log of the odds (LOD) ratio between the Gaussian functions plotted in b. Transcripts with a LOD > 0 (that is, having a greater likelihood of falling within the upper distribution) were considered highly enriched (green). Grey points represent all other RNAs not meeting this statistical cutoff. d, Representative transcripts reflecting varying degrees of enrichment following HA-BFD2 immunoprecipitation. Profiles display the total read count at each nucleotide position in either input (gray) or IP-enriched (green) samples, with paired tracks plotted on the same scale (denoted in brackets) for a given gene.
To better describe the specificity of BFD2 binding, we subjected pulldowns from stressed HA-BFD2 parasites to next generation sequencing (Fig. 6b–d). After excluding messages below a minimum threshold of detection, an enrichment ratio was calculated for each transcript based on representation in IP versus input samples (TPMIP/TPMinput, Fig. 6b). Reassuringly, relative mRNA abundance did not correlate with enrichment after pulldown (Extended Data Fig. 7a), indicative of specific capture of BFD2 targets rather than background from non-specific binding. Indeed, ENO1 and BAG1—two of the most abundant mRNAs in alkaline-stressed parasites—were not enriched in the immunoprecipitated material (Fig. 6c–d). Consistent with our results from RT-qPCR, both ASP5 and GCN5B appeared modestly enriched (~2-fold) by IP (Fig. 6c–d). A number of developmentally regulated genes, including LDH2, CST1, and BFD2, also showed varying degrees of enrichment; however, few exhibited the same magnitude of enrichment as BFD1, prompting us to seek a more rigorous approach to objectively stratify these interactions. Using Gaussian mixture modeling, we fit a combination of two Gaussian curves to the log-transformed enrichment ratios (Fig. 6b). A statistical cutoff was then set based on the log of the odds (LOD) of each gene appearing in the highly enriched (green) distribution. Based on this conservative metric, we defined a subset of 375 transcripts showing the strongest evidence of BFD2 binding (LOD > 0, Supplementary Table 7). BFD1 fell comfortably within this group, ranking in the top 0.5% of enriched messaged. Intriguingly, the majority of the cohort also displayed a stress-independent pattern of transcription, reminiscent of BFD1’s expression pattern (Extended Data Fig. 7b). Given the apparent role of BFD2 as a translational activator, this observation could point to BFD1 constituting part of a larger translational regulon. Moreover, it further emphasized the absence of confounding effects from differential transcription in the pulldown. Thus, our data provide strong support for the model of BFD2 binding to the BFD1 transcript under stress.
DISCUSSION
Encystation is a key feature of T. gondii persistence. We recently identified BFD1 as the master regulator of this process23. Now we expand this model with the discovery of BFD2, a second factor indispensable for the chronic stage. We show that basal levels of BFD2 are required for full induction of BFD1, placing it upstream in the regulatory hierarchy. Conversely, BFD1 transcriptionally activates BFD2, resulting in positive feedback. We show that BFD1 protein—but not mRNA—levels are affected by BFD2 knockout, implying translational regulation. In agreement with this, BFD2 interacts with BFD1 mRNA under alkaline stress. Together, our data support a model wherein BFD2 licenses BFD1 translation and acts through positive feedback to reinforce the BFD1 program.
Prior studies described RNA- and DNA-binding proteins that influence rates of stage conversion28–36; however, their relationship to BFD1 is unknown. Our own work identified several direct targets of BFD1 with nucleic acid-binding domains23, suggesting a hierarchy of differentiation-promoting factors. It was therefore unexpected that, of the five candidates examined, few exhibited obvious transcriptional signatures. AP2IX-9 in particular was previously shown to inhibit expression of bradyzoite-specific mRNAs33,35,36; yet, we observed no consequence from its depletion under alkaline stress. In this case, the function of AP2IX-9 may be overshadowed by other factors based on its transient expression, despite still being detectable in stressed parasites at 96 h35. Similarly, prior work implicated AP2IB-1 as transcriptionally upregulated in chronic forms; however, our inability to detect the tagged protein suggests that it may not be translated at the time points sampled. Subtle changes were detected for the other RNA-binding proteins, although our results do suggest that TGME49_224630 may be associated with alkaline stress responses outside of the BFD1 program. By far, the most obvious transcriptional signature was observed for BFD2.
BFD2 belongs to a widespread class of RNA-binding proteins defined by the presence of one or more CX7–8CX4–5CX3H (CCCH) zinc finger motifs45. These domains are found in proteins affecting virtually all stages of RNA metabolism46–48; however, few CCCH family members have been functionally characterized in apicomplexans. Two recent studies in Plasmodium spp. implicated ZFP4 and Pb103 in sexual reproduction due to gametocyte-specific effects49,50, yet the mechanism of regulation mediated by either factor was not definitive. Intriguingly, a CCCH protein in the non-apicomplexan parasite Trypanosoma brucei, was found to promote differentiation through association with polyribosomes51,52—consistent with positive translational control, as described for BFD2. At least 33 other genes in the T. gondii genome encode putative CCCH motifs (VEuPathDB.org). Given their importance across diverse parasite phyla, we suspect that further exploration of this protein family will expose new factors involved in developmental gene expression.
Our data implicate BFD2 as a core part of the T. gondii chronic differentiation program. While BFD2 orthologs are found in other closely related cyst-forming apicomplexans, such as Neospora and Hammondia, the factor appears restricted to the Sarcocystadae family (VEuPathDB.org). BFD1 displays a similar pattern of conservation, suggesting that the relationship between the two factors may be maintained in closely related species. Notably, although dogma suggests that an inability to produce cysts should render Δbfd2 incapable of long-term persistence, we could still detect parasites in brain sections from mice up to 45 days post-infection. This unexpected observation finds support in a parallel study, which showed that BFD1- and BFD2- (ROCY1-) knockouts are capable of reinitiating acute infection as late as five months post-inoculation, following immunosuppression53. Definitive characterization of these non-encysted forms will require further investigation and may expose novel mechanisms of T. gondii stress tolerance and persistence, unrelated to bradyzoite conversion.
Though not essential for tachyzoite growth, we observed a modest fitness cost to BFD2 deletion in cultured ME49 parasites. Puzzlingly, replication rates, invasion efficiency, sensitivity to extracellular stress, and acute-stage virulence were comparable between wildtype and Δbfd2 strains. Transcriptional differences in the knockout could be attributed to a lack of spontaneous differentiation and, at the protein level, we detected no deficiencies in known fitness-conferring proteins—although we acknowledge that the relevant players could be particularly low-abundance, and thereby missed by mass spectrometry. Given that BFD1 deletion did not elicit a similar reduction in fitness23, we hypothesize that minor defects in the Δbfd2 strain—subtle enough to be overlooked in cellular assays—ultimately result in reduced growth. Intriguingly, whereas loss of BFD2 in our prior ME49 screens also reflected compromised fitness23, deletion in Pru (type II) or RH (type I) Toxoplasma strains appears innocuous39,53. This suggests that strain-specific differences might account for the deficiencies of Δbfd2 tachyzoites.
BFD2 binding to BFD1 mRNA appears necessary for its translation. Given that BFD2 is expressed throughout the asexual cycle, it remains an open question how its stress-dependent effects are conferred. The observation that BFD1 is enriched by BFD2 pulldown under alkaline-stressed (but not unstressed) conditions seems to implicate differential binding as the event licensing translation of BFD2 targets. However, since BFD2 expression is substantially higher in differentiating parasites, differences in the amount of BFD2 captured by IP prevent us from unambiguously attributing increased association with BFD1 to changes in binding affinity. Alternatively, BFD2 may bind mRNA constitutively, such that its translational influence is regulated through other dynamic interactions. While this manuscript was under revision, BFD2 was found among several targets of a conserved serine/threonine phosphatase, protein phosphatase 2A (PP2A)53. Mutants of PP2A exhibited BFD2 hyperphosphorylation, which coincided with reduced DBL staining and incomplete induction of bradyzoite genes under stress—reminiscent of the partial differentiation of BFD2-knockouts. While the molecular consequences of these and other post-translational modifications on BFD2 have yet to be tested, this result could support a model wherein stress-dependent dephosphorylation of BFD2 enables the recruitment of factors necessary to translate its mRNA clients.
Conditional overexpression of BFD2 is sufficient to drive stage conversion irrespective of stress. This relationship does not preclude the function of BFD1 as master regulator of the chronic stage, since our collective data suggest that differentiation depends on the transcriptional changes induced by BFD1. However, the regulatory machinery involved appears to be responsive to the concentration of BFD2 and can be overwhelmed stoichiometrically. Based on this observation, we propose that once the amount of BFD2 exceeds a certain threshold (e.g., via transcriptional activation by BFD1), maintenance of the bradyzoite state becomes less dependent on the signals that initiated it. This phenomenon, known as hysteresis, is well-described in other developmental programs, such as cell-type determination in multicellular organisms and commitment to cell division54–58. In the context of the T. gondii chronic stage, such a circuit could help maintain parasites in a differentiated state in the presence of variable stimuli.
Feedback loops endow genetic circuits with several notable features. The discovery of a positive feedback loop acting on BFD1 therefore has fundamental implications for the properties of T. gondii latency. In addition to signal amplification, these circuits frequently exhibit bistability. That is, whereas hysteresis renders a genetic system less susceptible to noise, bistability confers switch-like binary control54. Previously, it was suggested that acute-to-chronic stage conversion in T. gondii involves the production of slow-growing intermediates, termed pre-bradyzoites33,35,36. This notion is not inconsistent with a binary model, since bistable systems can encode more than two cell states due to non-equilibrium59. Nevertheless, our observation that Δbfd2 parasites partially induce the chronic-stage program but cannot produce long-lived cysts in mice suggests that robust binary commitment is important. Perhaps more intriguingly, bistable systems exhibiting hysteresis are capable of ‘remembering’ a stimulus, long after it has been removed54. Network theory suggests that a double-positive loop such as we describe for BFD1 and BFD2 should lock into a self-perpetuating steady-state54. This raises the question of how the bradyzoite program is overturned during reactivation. It is possible that the absence of stress may be sensed directly by the circuit. Alternatively, there may be additional reactivation factors that antagonize BFD1- BFD2 feedback. Other groups have proposed that differentiation requires a balance of regulators, including proteins that repress the transcription of bradyzoite-specific genes33,35,36. Whether these or other factors directly inhibit BFD1 or BFD2 has yet to be examined.
In summary, our work shows that BFD2 is a major determinant of T. gondii persistence due its involvement in a positive feedback loop with the master regulator, BFD1. This discovery opens up exciting new questions surrounding the regulation of the BFD1 program, particularly as it relates to the maintenance and reactivation of chronic stages. Further exploration of the BFD1-BFD2 circuit will likely uncover heuristics that govern timing and commitment in differentiation. Together, these advances will aid in the design of therapeutic interventions to prevent or reverse chronic stages, bringing us closer to a radical cure against toxoplasmosis.
METHODS
Parasite and host cell culture
T. gondii parasites were maintained in human foreskin fibroblasts (HFFs) at 37°C under 5% CO2 in standard medium, consisting of DMEM (GIBCO) supplemented with 3% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine (GIBCO), and 10 μg/mL gentamicin (Thermo Fisher). Routine passaging was performed by scraping and transfer to fresh HFFs. For experimental infections, parasites were released from host cells by extrusion through a 27-gauge needle (i.e., syringe-release), and allowed to invade in standard medium containing 10% FBS. Alkaline-stress medium used for differentiation consists of RPMI 1640 supplemented with 1% FBS, 10 μg/mL gentamicin, 50 mM HEPES, and pH-adjusted to 8.1 with 10 N NaOH. IAA was used at 50 μM in standard medium and 500 μM under alkaline stress. Shield-1 was used at 3 μM in standard medium60.
T. gondii transfection
Syringe-released parasites were passed through 5 μm Millex filter units (Merck Millipore) to remove host cell debris, pelleted by centrifugation (1000 × g, 10 min), and electroporated with constructs in 400 μL of cytomix electroporation buffer (10mM KPO4, 120 mM KCl, 150 μM CaCl2, 5 mM MgCl2, 25 mM HEPES, 2mM EDTA) supplemented with 2 mM ATP and 5 mM glutathione. Electroporation was performed in 4mm cuvettes (400 μL final volume) using an ECM 830 Square Wave electroporator (BTX) with the following settings: 2 pulses, 1.7 kV, 176 μs pulse length, and 100 ms interval.
T. gondii strain generation
Unless otherwise stated (i.e., PruΔbfd2), strains were generated in the ME49 genetic background. Cloning was performed with Q5 polymerase (NEB) and NEBuilder HiFi DNA Assembly Master Mix. Oligos were ordered from IDT (full list in Supplementary Table 9). When needed, the following concentrations were used for selection: pyrimethamine (3 μM), chloramphenicol (20 μM), mycophenolic acid (25 μg/ml), and xanthine (50 μg/ml). Subcloning was performed in 96-well plates by sorting or plating at limiting dilution, with strains verified by at least two rounds of genotyping-PCR and sequencing.
TIR1-expressing ME49 (ME49/TIR1)
In ME49Δku80Δhxgprt parasites23, the heterologous TIR1 auxin receptor was introduced into a defined, neutral locus on chromosome VI61. Integration was aided by a previously validated gRNA/Cas9-expression plasmid targeting the neutral locus (Genbank #MN019116). A repair template encoding TIR1 driven by the alpha tubulin promoter (pTUB1-TIR1) and CAT expression cassette was amplified from Addgene plasmid #87258 using oligos P1/P2, which include regions of homology to integration site. Transfectants were selected for 2 weeks in chloramphenicol before subcloning, with single-plaque wells screened using P3/P4.
mNG-mAID-tagged (conditional knockdown) strains
In ME49/TIR1, genes were endogenously tagged with mNG-mAID using a previously described high-throughput (HiT) strategy38. Briefly, cutting units specific to each candidate were purchased as IDT gBlocks (P5–P10) and assembled with the empty mNG-mAID HiT vector (GenBank: OM640005, Extended Data Fig. 8A). Constructs were BsaI-linearized and co-transfected with pSS014 Cas9-expression plasmid (GenBank: OM640002), and the transfectants selected in pyrimethamine. Single-plaque wells of each strain were genotyped using a common strategy (P11–23, Extended Data Fig. 1), with PCRs to verify both integration of mNG-mAID and reciprocal loss of the wild-type untagged allele. Sequencing further verified maintenance of the reading frame across junctions where recombination occurred.
BFD2-knockout strains (Δbfd2, PruΔbfd2 and, DD-BFD1-TY/Δbfd2)
Oligonucleotides encoding gRNAs targeting the 5′ (P24/P25) and 3′ (P26/P27) ends of the BFD2 coding sequence were assembled with the pU6-Universal Cas9-expression plasmid (Genbank #OM640003) and sequenced with P28. A repair template consisting of pTUB1-TdTomato was amplified using P29/P30, which encode 40 bp of homology to regions immediately up- and downstream of the gRNA target sites. 25 μg each of the gRNAs and repair construct were transfected with into ME49ΔKu80Δbfd1::BFD1-TY and ME49ΔKu80Δbfd1/HXGPRT::pTUB1-DD-BFD1-TY parasites (described in Waldman et al., 2020) to generate Δbfd2 and DD-BFD1-TYΔbfd2, respectively. After 2–3 days, knockouts were sorted by FACS directly into 96-well plates and single-plaque wells were screened for integration of TdTomato (P31/P32) and reciprocal loss of BFD2 (P31/P33). For PruΔbfd2, the same gRNAs were used to target BFD2 in Pru-OVA-tdTomato62 parasites; however, BFD2 was replaced with an HXGPRT cassette to allow selection in mycophenolic acid and xanthine.
Deletion of BFD1 (PruΔbfd1)
PruΔbfd1 was generated with published reagents23, using the same FACS-based strategy as above. Briefly, gRNA/Cas9-expresson plasmids targeting either end of BFD1 were transfected into Pru-OVA-tdTomato parasites along with a repair template encoding pTUB1-mNG flanked by 40 bp of homology adjacent to the cut sites. Transfectants were sorted for mNeonGreen expression and subcloned, and a lineage lacking BFD1 was verified by PCR and sequencing. However, despite being mNeonGreen-positive, pTUB1-mNG was not found within the BFD1 locus, indicating that it had randomly integrated into the parasite genome.
BFD2-complemented strains (HA-BFD2WT and HA-BFD2ΔZF)
HA-tagged BFD2—either full-length or lacking the zinc finger domains—was reintroduced into ME49ΔKu80Δbfd1::BFD1-TYΔbfd2 at the endogenous locus, allowing FACS-enrichment of complemented parasites based on loss of TdTomato. gRNAs against the 5′ and 3′ ends of the fluorescence cassette (P34/P35 and P36/P37, respectively) were annealed and assembled with the pU6-Universal plasmid. For the full-length repair template, BFD2 was amplified from cDNA (P38/P39) and assembled with PCR fragments spanning ~500 nt immediately up- (P40/P41) and downstream (P42/P43) of the coding sequence. The HA epitope was introduced at the junction between the upstream amplicon and BFD2 via overhangs in the corresponding reverse and forward primers (i.e., P41 and P38). The ΔZF version was generated similarly, but with BFD2 amplified as two separate fragments (P38/P44 and P45/P39) to excluded the zinc finger domains. Annealed products were then used as templates for PCR to generate ~15 μg of linear repair construct (P46/P47), which was co-transfected with 20 μg of each gRNA plasmid into Δbfd2. After an initial round of FACS to enrich for low/non-fluorescent cells, complemented mutants were sorted directly into 96-well plates for subcloning. Single-plaque wells were screened by PCR for reintroduction of tagged BFD2 (P31/P32) and loss of TdTomato (P31/P23).
Conditional overexpression of BFD2 (DD-HA-BFD2)
DD-HA-BFD2 was generated using a derivation of the HiT vector strategy38, modified for manipulating N termini of targeted genes. A cutting unit specific to BFD2 was purchased as an IDT gBlock (P48) and Gibson-assembled with the empty pTUB1-DD HiT vector (GenBank accession number pending, Extended Data Fig. 8B). The resultant plasmid was sequence-verified with P49, then BsaI-linearized and transfected into ME49ΔKu80Δbfd1::BFD1-TY parasites with 50 μg of pSS014 Cas9-expression plasmid. Mutants were selected for 1 week in pyrimethamine before subcloning. Single-plaque wells were PCR-screened for successful integration of the construct (P51/P32) and reciprocal loss of the wild-type allele (P50/P32).
Criteria for identifying putative effectors of BFD1
Based on previous BFD1 CUT&RUN chromatin profiling23, we generated a list of genes which appeared direct targets of BFD1 (i.e., promoters encompass BFD1 peaks, called by MACS2). Of these, eight encoded putative RNA- or DNA-binding domains based on publicly available domain annotations and gene ontology. A final requirement of significant transcriptional induction (adjusted p-value < 0.05 calculated by DESeq2) in response to either alkaline stress or conditional BFD1 expression further narrowed the list to the five genes examined in this study.
Immunofluorescence assays
IFAs were performed on coverslips pre-seeded with HFFs. Samples were fixed with 4% formaldehyde for 20 min, permeabilized with 1% Triton X-100 for 8 min, and blocked for 25 min with 5% IFS and 5% goat serum in PBS (blocking buffer). All antibody incubations were performed for 1 h in a humidified chamber, with coverslips inverted on 30 μL of antibody diluted in blocking buffer spotted onto Parafilm. All steps were performed at room temperature, with three PBS washes between. After the final wash, coverslips were rinsed in deionized water, mounted onto slides with 5 μL of Prolong Diamond (Thermo Fisher), and cured for at least 45 min at 37°C before imaging. Antibody and stain dilutions were prepared as follows: rabbit anti-GAP45 (gift from Dominique Soldati, University of Geneva), 1:10,000; guinea pig anti-CDPK1, custom antibody (Corvance), 1:10,000; mouse anti-mNeonGreen, clone 32F6 (ChromoTek), 1:500; mouse anti-SAG1 (gift from David Sibley)63, 1:500; mouse anti-TY64, 1:1000; rabbit anti-HA, clone SG77 (Invitrogen), 1:1000; mouse anti-HA, clone 16B12 (Biolegend), 1:1000; Hoechst 33258 (Santa Cruz), 1:20,000; DBL-488 (Vector Labs), 1:1000. Secondary antibodies labeled with Alexa Fluor 488, 594, or 647 (Thermo Fisher) were all used at 1:1000. Images were acquired by Eclipse Ti (Nikon) microscope with NIS Elements software.
Plate-based differentiation screen
Parasites were inoculated onto HFFs in black-wall 96-well microplates for 4 h in standard media, then washed twice with PBS and switched to alkaline medium containing 500 μL IAA or an equivalent volume of PBS (vehicle). After 48 h, plates were fixed and permeabilized as described above. Wells were treated sequentially with 50 μL of primary (guinea pig anti-CDPK1) and secondary (anti-guinea pig-594, DBL-488, and Hoechst 33258) staining solutions for 1 h each, with three PBS washes between. Fluorescence images were collected at 20x magnification with a Cytation3 Cell Imaging Multi-Mode Plate Reader (Aligent Techonologies).
For blinded scoring of DBL staining, a mask was created for each image in Fiji (v. 2.1.0/1.53c) based on CDPK1 to delineate individual vacuoles, regardless of differentiation status. This was applied to the corresponding DBL scan from each set, creating a second mask to enable visualization of the isolated DBL-staining profile for each vacuole. Using the Interactive Learning and Segmentation Toolkit (ilastik v. 1.3.3)65, vacuoles were categorized as negative- intermediate- or high, based on the intensity and variance of the DBL signal.
RNA-seq analysis of conditional knockdown strains
Syringe-released parasites were allowed to invade HFFs in 15 cm dishes under standard conditions for 4 h, then monolayers were rinsed with PBS and switched to alkaline-stress media containing either 500 μM IAA or PBS (vehicle). After 96 h, parasites were harvested by sequential extrusion through 27- and 30-gauge needles followed by passage through a 5 μm filter, pelleted by centrifugation (1,200 × g for 10 min) and flash-frozen in TRIzol (Thermo Fisher). Total RNA was isolated by Zymo Direct-zol RNA Miniprep Kit and RNA quality was assessed by BioAnalyzer or Fragment analyzer. All samples were split in two at the time of RNA isolation and treated as technical replicates in downstream processing. Libraries were generated using the Swift Rapid RNA Library Kit for stranded RNA-seq (Swift Biosciences) and subjected to 150×150 paired-end sequencing on the NovaSeq 6000 platform (Illumina). Reads were trimmed using Cutadapt v.3.6 and aligned to the ME49 genome (ToxoDB v.56 assembly) with STAR v.2.7.0a. Differential expression analysis was done using the DESeq2 package (v. 1.12.3) in R. Adjusted p-value of 0.05 was used as a statistical cutoff for differential expression.
RNA-seq analysis of BFD2 deletion
In human foreskin fibroblasts
HFF monolayers in 15 cm dishes were infected with syringe-released parasites for 4 h, then rinsed with PBS and either returned to standard medium or treated with one of two differentiation triggers. For Δbfd2 and its parental strain (Δbfd1::BFD1-TY; Fig. 2i–k), stage-conversion was induced by alkaline stress. For DD-BFD1-TY and DD-BFD1-TYΔbfd2 (Fig. 4d), parasites received standard media supplemented with 3 μM Shield-1 or the equivalent volume of 100% ethanol (vehicle). Parasites were cultured for an additional 48 h before being released from host cells by scraping, syringe-release, and filtering as described above, then collected by centrifugation (1,000 x g for 10 min) and flash-frozen in liquid nitrogen. Total RNA was isolated by Qiagen RNeasy Plus Micro Kit following the protocol for animal and human cells, with cell pellets split in two and treated as technical replicates for downstream processing. Library preparation, sequencing, and analysis were performed with the same pipeline as was applied to conditional knockdown strains.
In mouse primary neurons
Primary mouse neurons were generated as described previously66, with all procedures approved by the University of Arizona’s Institutional Animal Care and Use Committee (12–391). Briefly, fetal pups were isolated from pregnant Cre reporter mice67 (Jacksons Laboratories, #007906) and dissected to recover the cerebral hemispheres, regardless of sex. Cortical and hippocampal neurons were recovered by established protocols68, and seeded onto poly-L-lysine-coated 6-well plates in DMEM supplemented with 0.6% D-glucose (Sigma), L-Glutamine (Thermo Fisher), and 5% FBS. After 4 h, media was changed to complete Neurobasal™ media (Thermo Fisher) supplemented with B27 (Thermo Fisher), L-Glutamine and Penicillin/Streptomycin antibiotic cocktail. On day 4, neurons received 5 μM cytosine arabinoside to stop glial proliferation. On day 10, neurons were infected (MOI of ~2) with parental, PruΔbfd1, or PruΔbfd2 parasites and left for an additional 48 h in culture before being dissociated with accutase (5 mins at 37°C), pelleted by centrifugation, and flash-frozen in liquid nitrogen. Subsequent steps (RNA isolation through analysis) were performed identically to the above workflow.
Plaque assays
HFFs in 6-well plates were inoculated with 1×103 parasites and allowed to sit undisturbed in standard media containing 10% IFS. After 14–16 days (indicated in the respective figures), plates were rinsed with PBS, fixed with 100% ethanol for 10 min, and dried. The following day, wells were stained for 5 min with crystal violet solution (12.5 g crystal violet, 125 mL 100% ethanol, 500 mL 1% ammonium oxalate), rinsed with water, and imaged. Plaques were manually delineated and their areas quantified using Fiji69.
Host cell invasion assays
Assays were performed in glass-bottom 24-well plates seeded with HFFs. Immediately prior to infection, wells were rinsed with PBS and filled with 500 μL of pre-warmed invasion medium (DMEM without sodium bicarbonate, supplemented with 20 mM HEPES, 1% IFS, and pH-adjusted to 7.4). 7×105 syringe-released parasites were added to each well and settled onto the monolayer by centrifugation (290 × g for 5 min), then allowed to invade at 37°C for 20 min before fixing in 4% formaldehyde for 10 min. Wells were incubated for 10 min in blocking solution (see Immunofluorescence assays) and stained for SAG1 for 30 min. After permeabilizing with 0.25% TritonX-100 for 8 min, wells were blocked a second time and stained for DNA and CDPK1, allowing discrimination between intracellular (CDPK1+, SAG1−) and extracellular (CDPK1+, SAG1+) parasites. Biological replicates were performed on different days, with technical replicates being individually infected wells. At least five fields were imaged per well by Nikon Eclipse Ti microscope and counts were averaged to create an overall measure of invasion, expressed as the number of intracellular parasites per host cell nucleus.
Intracellular replication assays
Δbfd1::BFD1-TY and Δbfd2 parasites were infected onto coverslips seeded with HHFs in standard media and allowed to grow for 24 h. Samples were fixed, permeabilized, and stained for DNA and CDPK1 as described under Immunofluorescence assays. Numbers of parasites per vacuole was calculated from a minimum of 100 vacuoles per strain.
Extracellular stress assays
Single-cell suspensions of extracellular Δbfd1::BFD1-TY and Δbfd2 parasites were generated by scraping, extruding cultures through 27-gauge needles, and passing through 5 μm filters. 50 μL of suspension was inoculated onto HFFs on coverslips in standard media, and the remaining volume was maintained at 37°C. At intermittent timepoints thereafter, additional coverslips were inoculated with the suspensions at an equivalent MOI. After 24 h, all coverslips were fixed and stained for DNA and CDPK1 as described in Immunofluorescence assays. Infectivity at each timepoint was quantified the number of vacuoles containing 2 or more parasites, normalized to the number of host nuclei per field. Within strains, data were normalized to infectivity at t = 0.
Quantitative analysis of immunofluorescence assays
All analyses were performed using Fiji69. For DBL quantification, individual vacuoles were manually defined as regions of interest (ROIs) based on a CDPK1 counterstain. ROIs were saved with the ROI Manager function, applied to the corresponding DBL channel, and their pixel intensity (Integrated Density) quantified. As a measure of background, pixel intensity was quantified over non-infected regions of the monolayer. Corrected Total Fluorescence (CTF) for individual ROIs was calculated using the following function: CTF = [Integrated Density – (ROI area × mean background)]/ROI area. Cytosolic HA-BFD2 accumulation was determined similarly, with individual parasites delineated based on the HA signal itself. For BFD1-TY, parasite nuclei were defined as ROIs based on Hoechst and the background measured from non-nuclear regions of intravacuolar parasites.
Acute-stage quantitative proteomics
Sample harvest and processing
Δbfd1::BFD1-TY and Δbfd2 parasites were grown in HFFs in 15 cm dishes under standard conditions for 48 h, then harvested by scraping, extrusion through a 27-gauge needle, and passage through 5 μm filters. Samples were pelleted by centrifugation at 1,200 × g for 10 min, and resuspended in lysis buffer (10% SDS, 100 mM TEAB pH 7.5, 2 mM MgCl2, and 2X HALT protease inhibitor) for storage at −80°C until further steps. Proteins were prepared for quantitative mass spectrometry exactly as described previously70, using a modified version of the S-trap protocol (Protifi) following by lyophilization, labeling with TMT reagents (Thermo Fisher), and fractionation.
MS acquisition
Samples were analyzed using an Orbitrap Eclipse Tribrid mass spectrometer equipped with a FAIMS Pro source71 connected to an Vanquish HPLC chromatography system, using 0.1% formic acid as Buffer A and 80% acetonitrile/0.1% formic acid as Buffer B. Peptides were loaded onto a heated analytical column (ES902, Thermo, PepMap Neo Trap Cartridge C18 2 μm, 100 Å, 75 μm × 25 cm, 40°C) via trap column (174500, Thermo, Acclaim PepMap C18 5 μm, 100 Å, 300 μm × 5 cm nanoViper) and separated at 300 nl/min on a gradient of 3–25% B for 90 min, 25–40% B for 30 min, 40–95% B for 10 min, and 95% B for 6 min. The orbitrap and FAIMS were operated in positive ion mode with a positive ion voltage of 2100V, ion transfer tube temperature of 300°C, standard FAIMS resolution, and compensation voltage of −45, −55 and −65V with 4.2 mL/min carrier gas. Full scan spectra were acquired in profile mode at a resolution of 120,000, with scan range of 400–1600 m/z, 300% AGC target, automatically determined maximum injection time, intensity threshold of 2 × 104, 2–6 charge state, dynamic exclusion of 60 sec, and mass tolerance of 10 ppm. MS2 spectra were generated in centroid mode using quadrupole isolation, an isolation window of 0.7 m/z, CID collision energy of 35 h, activation time of 10 ms, ion trap detection with a turbo scan rate, standard AGC target, and maximum injection time set to automatic. Real-Time Search was performed using the ToxoDB GT1 protein database (release 49), including static methylthio (+45.9877 Da; C), static TMT6plex (+229.1629; K and any N termini), and variable oxidation (+15.9949; M). TMT SPS MS3 mode was used, enabling close-out, a maximum peptides per protein of 5, and maximum search time of 40 ms. Scoring thresholds were set for Xcorr (2), dCn (0.1), precursor ppm (10), and charge state (2). MS3 spectra were generated in centroid mode, with a synchronous precursor selection of 10, MS isolation window of 2 m/z, MS2 isolation window of 2 m/z, HCD collision energy of 55 at a resolution of 50,000, a mass range of 100–500 m/z, 200% AGC target, for 10 data dependent scans.
Proteomic analysis
Raw files were analyzed in Proteome Discoverer (v. 2.4) to generate peak lists and protein and peptide IDs using Sequest HT (Thermo Fisher) and the ToxoDB ME49 protein database (release 43). The following modifications were included in the search: dynamic oxidation (+15.995 Da; M), dynamic acetylation (+42.011 Da; N-terminus), static TMT6plex (+229.163 Da; any N-terminus), static TMT6plex (+229.163 Da; K), and static methylthio (+45.988 Da; C). Exported peptide and protein abundance files were loaded into R for further analysis.
Assessment of acute pathogenicity and brain cyst formation
Mice
Six-week-old female CD-1 mice (Charles River) were maintained in the Whitehead Institute for three weeks prior to their use in experiments. Mouse facilities were kept on a 12 h dark-light cycle under ambient temperature and humidity. All animal protocols were approved by the Institutional Animal Care and Use Committee at the Massachusetts Institute of Technology (0220-004-23).
Mouse infections
Mice were infected intraperitoneally by injecting 100 acute-stage parasites of either strain in 100 μL of PBS (n = 10 each) or PBS alone (mock; n = 4). Weights were recorded daily. Animals displaying excessive distress or morbidity were euthanized with CO2 followed by cervical dislocation. Surviving animals were sacrificed 45 days post-infection.
Brains were harvested from all mice at euthanasia. Brains from moribund animals were used in their entirety for cyst quantification. For animals surviving to day 45, brains were bisected at the time of harvest and one hemisphere was set aside for immunohistochemistry (below).
Ex vivo quantification of cysts
Brains were dissected into 2 mL PBS and homogenized by repeated extrusion through an 18-gauge needle until uniformity was achieved. Homogenates were brought to 10 mL with PBS, pelleted by centrifugation (1,000 × g for 5 min), and the supernatant discarded. Homogenates were resuspended in 700 μL PBS and the final volume recorded, then 100 μL was combined with 900 μL of ice-cold 100% methanol to fix at room temperature. After 5 min, samples were centrifuged at 5,200 × g for 5 min (conditions used for all subsequent centrifugation steps), washed with 1 mL PBS, centrifuged again, and the supernatant was removed by aspiration. To label cysts, pelleted samples were resuspended in 500 μL of primary staining solution (1:150 DBL-488 and 1:1000 guinea pig anti-CDPK1 in PBS) and incubated overnight at 4°C with rotation. The following day, samples were centrifuged, washed with 1 mL PBS, and incubated in secondary staining solution (anti-guinea pig-594, diluted 1:1000 in PBS) rotating at room temperature. After 1 h, homogenates were washed again and resuspended in 1 mL PBS. For each sample, four 50 μL aliquots were plated into a clear-bottomed 96-well microplate and examined at 20x magnification by fluorescence microscopy. Cysts were identified by double-positivity for both DBL and CDPK1. With the exception of those prepared from moribund mice, all samples were quantified blindly. Cyst burdens represent the mean of the four counts collected for each sample, multiplied by the appropriate dilution factor.
Immunohistochemistry
Brain tissue was fixed in 10% formalin, embedded in paraffin and sectioned. Deparaffinized sections were hydrated and antigen was retrieved in 0.01 M sodium citrate buffer at pH 6.0 followed by blocking of endogenous peroxidase with 0.3% H2O2. Tissues were blocked with 2% goat serum (Invitrogen). T. gondii parasites were detected with rabbit anti-Toxoplasma antibody72 (gift from Fausto Araujo, Palo Alto Medical Foundation, 1:100) followed by biotinylated goat anti-rabbit IgG (Vector Laboratories). ABC reagent (Vector Laboratories) was then applied and DAB substrate (Vector Laboratories) was added to visualize T. gondii staining. Slides were counterstained with hematoxylin to visualize nuclei. Images were acquired at 10x magnification with a Leica DM6000 Widefield Fluorescence Microscope.
Quantitative reverse transcription PCR (RT-qPCR)
Parasites were inoculated onto HFFs in T12.5 flasks for 4 h, then monolayers were rinsed with PBS and returned to standard medium or treated to induce differentiation, either by stress (Δbfd2 and Δbfd1::BFD1-TY; Fig. 4a) or 3 μM Shield-1 (DD-BFD1-TY and DD-BFD1-TYΔbfd2; Fig. 4e). After 48 h, cultures were scraped and collected by centrifugation (1,000 × g for 10 min), and flash-frozen in liquid nitrogen. Total RNA was isolated with the Qiagen RNeasy Plus Micro Kit, DNaseI-treated with the TURBO DNA-free kit (Thermo Fisher), then quantified by NanoDrop spectrophotometer for use in first-strand cDNA synthesis with SuperScriptIII (Thermo Fisher) and random hexamer priming. Dye-based qPCR was performed using PowerUp SYBR Green Master Mix (Thermo Fisher) with ‘No-RT’ controls included for each sample. Prior to use, all qPCR primers (Supplementary Table 3) were verified for amplification efficiency in the 95–105% range and specificity by melt curve analysis. Reactions were run on a QuantStudio6 Real-Time PCR system (Applied Biosystems) using “Standard” ramp speed with the following parameters: 2 min at 50°C; 2 min at 95°C; 40 cycles of 15 s at 95°C, followed by 1 min at 60°C. Transcripts of interest were quantified by comparative CT (ΔΔCT), using GCN5B as an internal control and normalizing to uninduced samples.
BFD2-RNA immunoprecipitation (RIP)
Parasites were inoculated onto HFFs in 15 cm dishes for 4 h then switched to alkaline stress or kept under standard conditions. After 48 h, unstressed cultures were harvested by scraping and extrusion through a 27-gauge needle and 5 μm filter, pelleted by centrifugation (1,200 × g for 10 min) and frozen at −80°C in complete Magna RIP lysis buffer (proprietary, below). Stressed samples were processed similarly after 72 h, but with an additional extrusion through 30-gauge needles before filtering. Approximately 2×107 parasites was harvested for each strain and condition. Unenriched ‘input’ samples were generated by reserving back 10% of starting lysate; the remaining volume was subjected to immunoprecipitation (IP) using the Magna RIP kit (Millipore) with 5 μg of mouse anti-HA IgG (Biolegend, clone 16B12), coated onto protein A/G magnetic beads as described in the Magna RIP technical manual. RNA purified from both IP and input samples was concentrated by ethanol precipitation and resuspended in equivalent volumes of RNAse-free water for use in downstream analyses.
Detection of BFD2-associated mRNAs
Analysis of specific targets by RT-qPCR
Samples were treated with TURBO DNaseI (Thermo Fisher) and cDNA was generated from equivalent volumes of IP and input RNA using SuperScriptIII with random hexamer priming. Dye-based qPCR was performed as described above using primers from Supplementary Table 3. Enrichment of transcripts in IP samples was calculated by the Percent of Input method.
Unbiased discovery of BFD2 targets by next generation sequencing
Input and IP samples from stressed HA-BFD2 parasites were poly(A)-selected using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB) and libraries generated with the SMARTer Pico Input Stranded Total RNA-seq Kit (Takara Bio), excluding the step for mammalian rRNA depletion. Samples were run with 100×100 paired-end sequencing on the NovaSeq 6000 platform (Illumina). Reads were trimmed and mapped to the ME49 genome as described above.
To call BFD2-enriched messages, we adapted a previously described RIP-seq enrichment analysis pipeline73. For each transcript, raw read counts were used to calculate a read coverage (d), defined as the square root of the sum of squares for reads in both the IP and input control: d = sqrt(IP × IP + input × input). Only transcripts with greater than 5 mapped reads in both IP and input samples and d >10 were included in downstream analysis. To normalize for sequence depth, we converted raw reads to Transcripts Per Million (TPM) for both IP and input samples. This was then used to generate an enrichment ratio for each gene, defined as (TPMIP/TPMinput).
We performed Gaussian mixture modeling on log2-transformed enrichment ratios using the normalmixEM function in the R package mixtools74. Informed by prior studies73, model fitting was restrained to two distributions, assuming equal variance. Convergence was reached within 250 iterations, producing the Gaussian curves plotted in Fig. 6b. Posterior probabilities were then generated from the mixture model and used to calculate an odds ratio for each gene. A log of the odds (LOD) ratio greater than 0 indicates a higher probability of falling within the upper—rather than lower—distribution; thus, LOD > 0 was used as a threshold for enrichment. Note that several genes with evidence of BFD2 binding (i.e. GCN5B and ASP5) do not meet this statistical threshold. Thus, we consider this a conservative cutoff for highly enriched mRNAs.
Statistical analysis
Information about biological replicates, the number of observations, and exact statistical tests performed is in figure legends. For RNA-seq analysis, adjusted p-values were calculated by DESeq2, which uses the Wald test as a default. All other statistical tests were performed in Prism (GraphPad).
Extended Data
Extended Data Fig. 1. Genotyping conditional depletion strains.
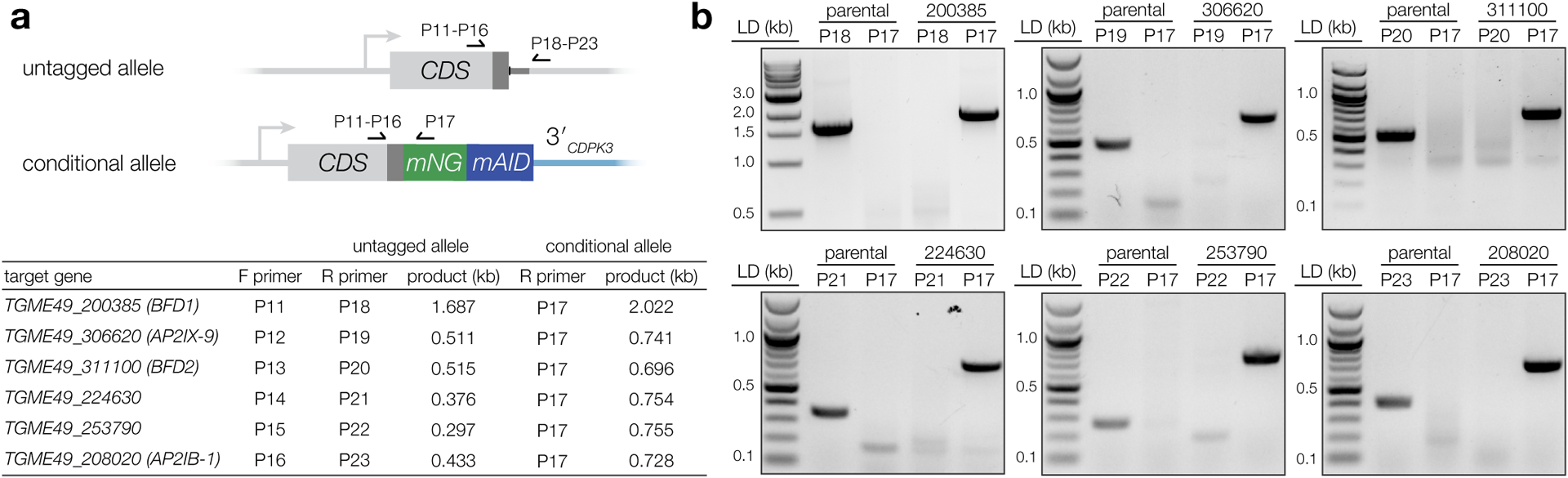
a, Clones were screened to verify integration of mNG-mAID downstream of the targeted coding sequence (CDS) and the reciprocal loss of the untagged allele. Diagram shows binding sites for primers listed in the table below, with regions used to direct construct integration (i.e., H1 & H2, Fig. 1b) in dark gray. In each case, a common gene-specific forward primer (P11–16) was paired with reverse primers against either mNG (P17) or the respective endogenous 3′ UTR (P18–23), downstream of the integration site. Expected PCR product size given for each template and primer combination. Refer to Supplementary Table 9 for a complete list of primer sequences. b, PCRs were performed on ME49/TIR1 gDNA (parental) as a control, in addition to the respective tagged strain. Lanes are labeled with the reverse primer and gDNA template used in each reaction. For positive clones, bands were extracted and subjected to Sanger sequencing to verify in-frame integration of the tag.
Extended Data Fig. 2. Effects of AP2IX-9 and AP2IB-1 knockdown on the chronic-stage transcriptome.
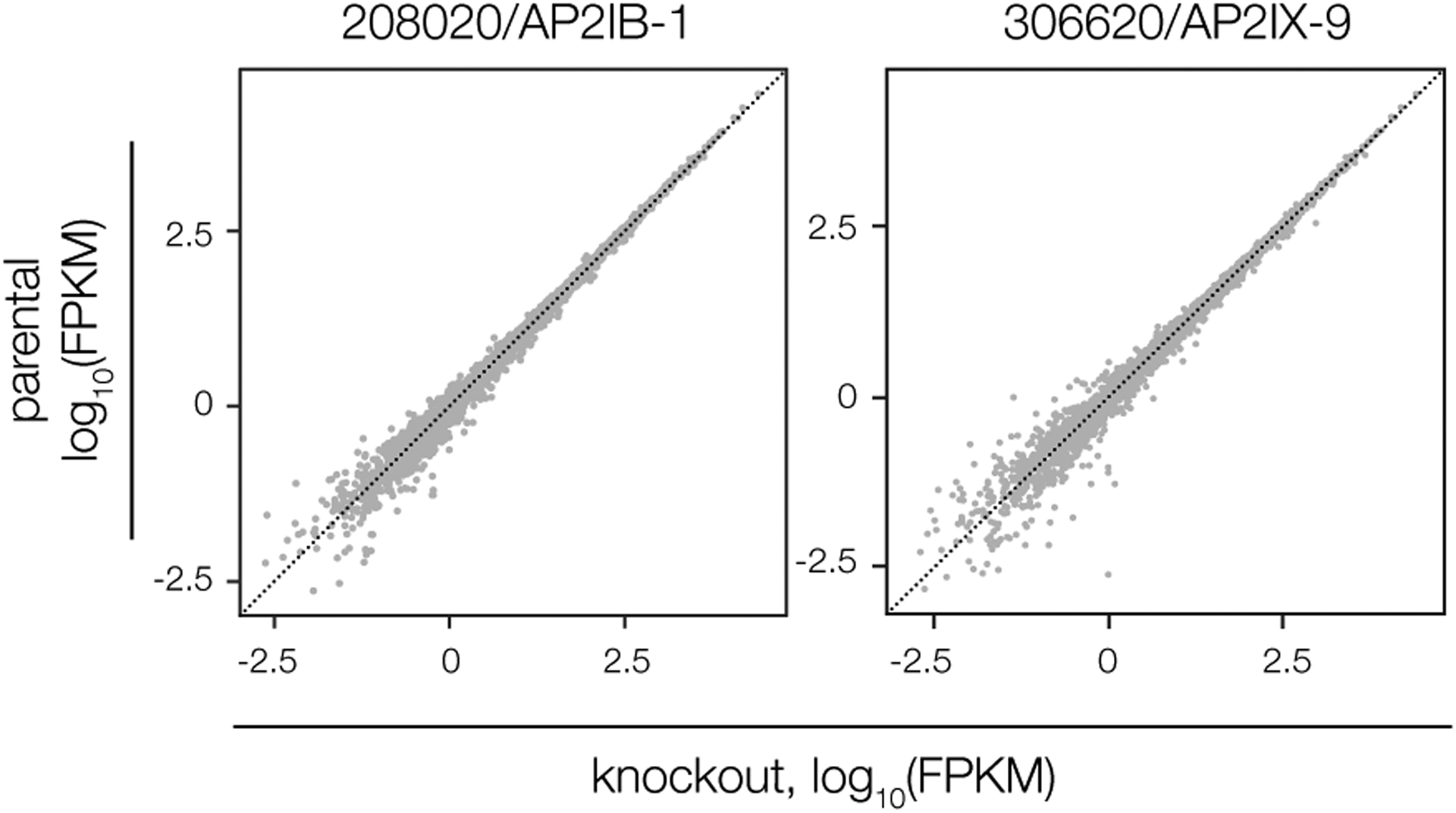
Data reflect changes in knockdown strains (relative to the parental) after 96 h in alkaline-stress medium containing IAA. Differential expression analysis was performed as in Fig. 1e, based on n = 2 biological replicates. No genes are significantly affected (adjusted p < 0.05, calculated by DESeq2) by depletion of either factor.
Extended Data Fig. 3. Genotyping and characterization of BFD2-deficient parasites.
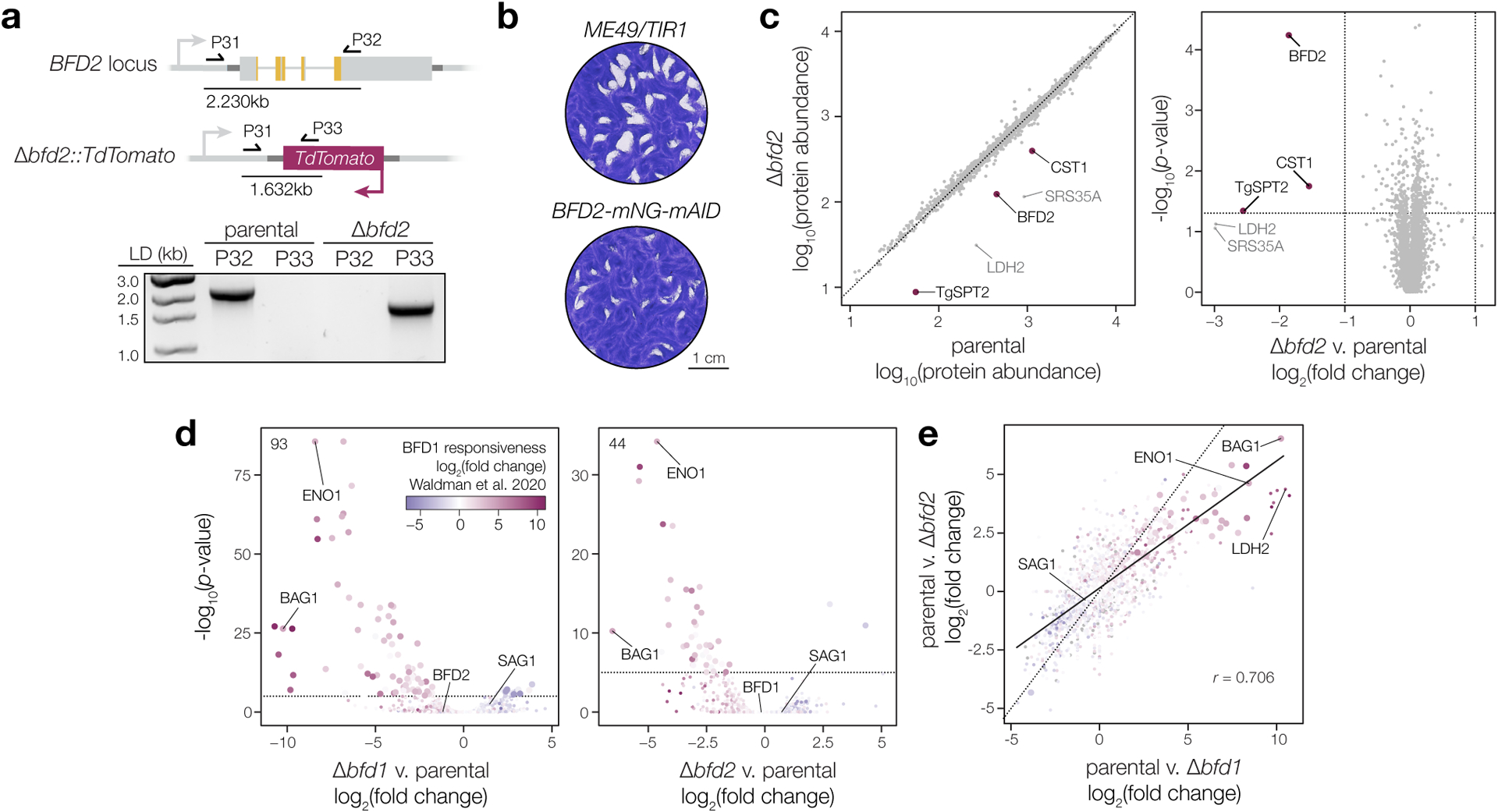
a, Δbfd2 clones were screened by PCR for replacement of the endogenous coding sequence with TdTomato using a common forward primer in the BFD2 5′ UTR (P31) and reverse primers against either BFD2 (P32) or the fluorescent reporter (P33). Diagram shows priming in the parental strain (top) and at the modified allele in Δbfd2 (below), with expected product sizes indicated. Reverse primers and gDNA template used in each reaction are listed above the respective lane. b, Plaque assays after 16 days of undisturbed growth. c, Comparison of protein abundance in unstressed parental (Δbfd1::BFD1-TY) and Δbfd2 parasites. Quantitative proteomics identified a total of 29,806 unique peptides corresponding to 4,303 individual proteins. Significantly affected proteins (magenta) were defined as those meeting three criteria: (i) a minimum of two unique peptides, (ii) absolute fold-change > 2, and (iii) p-value < 0.05. Differences are limited to canonical bradyzoite markers (CST1, LDH2, and SRS35A) or other developmentally regulated genes (TgSPT2)78. d–e, Effects of BFD1 or BFD2 deletion on the parasite transcriptome during infection of mouse primary neurons. Data are based on n = 3 independent infections with color assigned based on log2(fold change) during conditional BFD1 expression23. Significantly affected genes (adjusted p-value < 0.05 by Wald test with DESeq2) are indicated by larger point size. Differential expression analysis was performed for parasites lacking either factor, as compared to the parental strain. Number of genes meeting the cutoff for statistical significance is indicated (d). Comparison of the effects of BFD1- versus BFD2-deletion (e) reveals a comparatively larger impact for the former. Pearson correlation performed on all significant points with trend line fit by linear regression.
Extended Data Fig. 4. Re-analysis of BFD2-deficient parasites in previous ME49 screens.
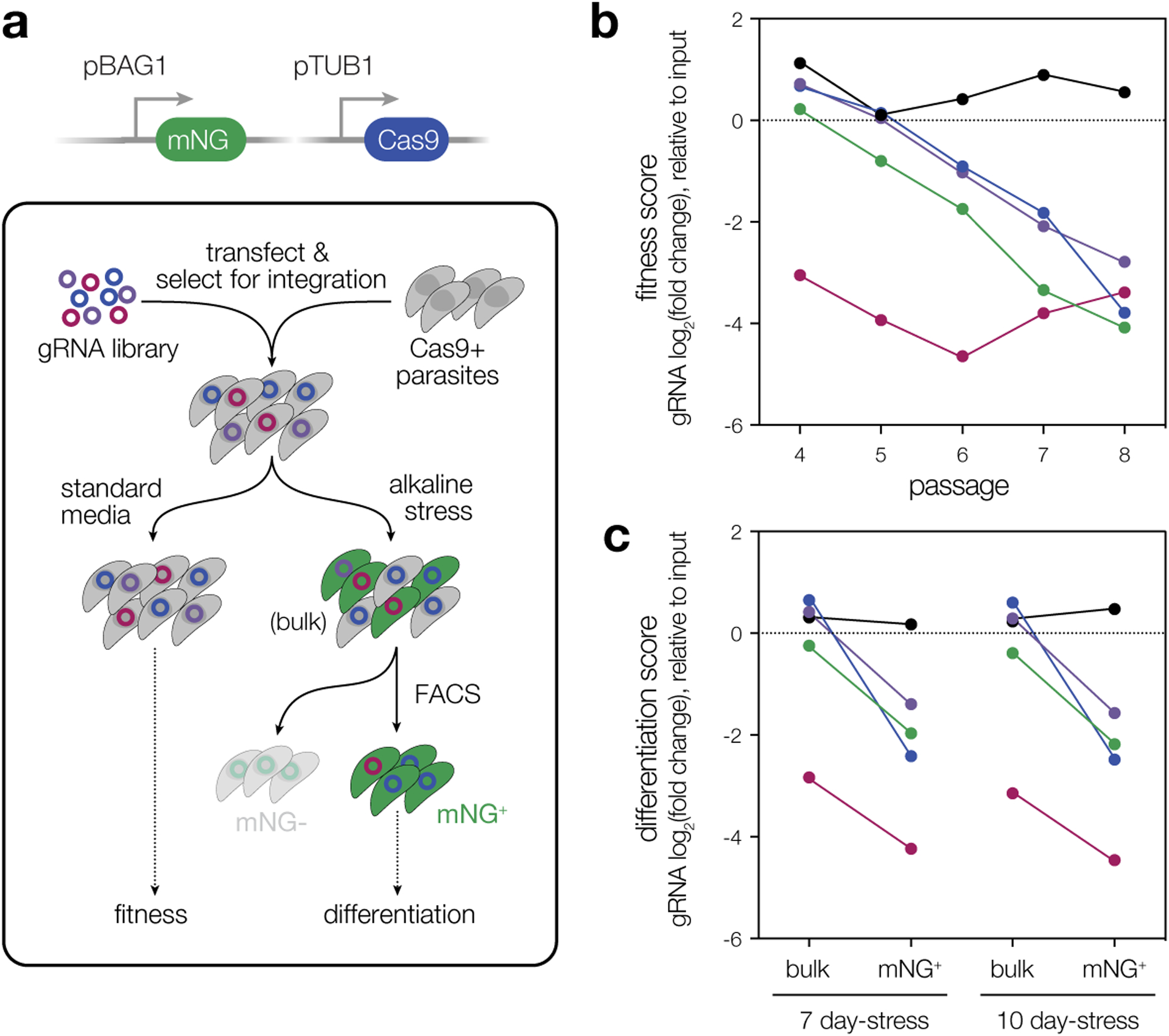
a, Overview of the CRISPR-based screen that identified BFD123. A CRISPR-compatible ME49 strain was modified to express mNeonGreen (mNG) under the bradyzoite-specific BAG1 promoter (pBAG1), enabling isolation of chronic stages by fluorescence-activated cell sorting (FACS). The reporter strain was transfected with guide RNA (gRNA) libraries targeting ~200 predicted nucleic acid-binding proteins with five gRNAs per gene. After initial passages allowing for guide integration and gene disruption, transfectants were split between alkaline-stressed and unstressed (standard media) conditions. Samples were collected from each population over a 10-day period, with bradyzoites (mNG+-stressed parasites) isolated by FACS. Integrated gRNAs from all samples were enumerated by next-generation sequencing and the abundance of each guide was assessed relative to the input library. The log2(fold change) for guides targeting each gene is referred to as its fitness or differentiation score, based on representation in unstressed or bradyzoite samples, respectively. b,c. Analysis of BFD2-targeting gRNAs from the screen described in a. Four of the five guides targeting BFD2 were lost from the transfectant pool under standard conditions over the course of serial passaging (b). Subsequent sequence-level analysis revealed that the single guide that remains abundant (black) is likely non-cutting due to a mismatch in the protospacer and its intended genomic target. Among alkaline-stressed cultures (c), at both time points examined—with the exception of the non-cutting guide (black)—gRNAs targeting BFD2 are underrepresented in bradyzoite samples (mNG+) relative to the unsorted alkaline-stressed population (bulk).
Extended Data Fig. 5. BFD2 deletion in a conditional BFD1 strain.
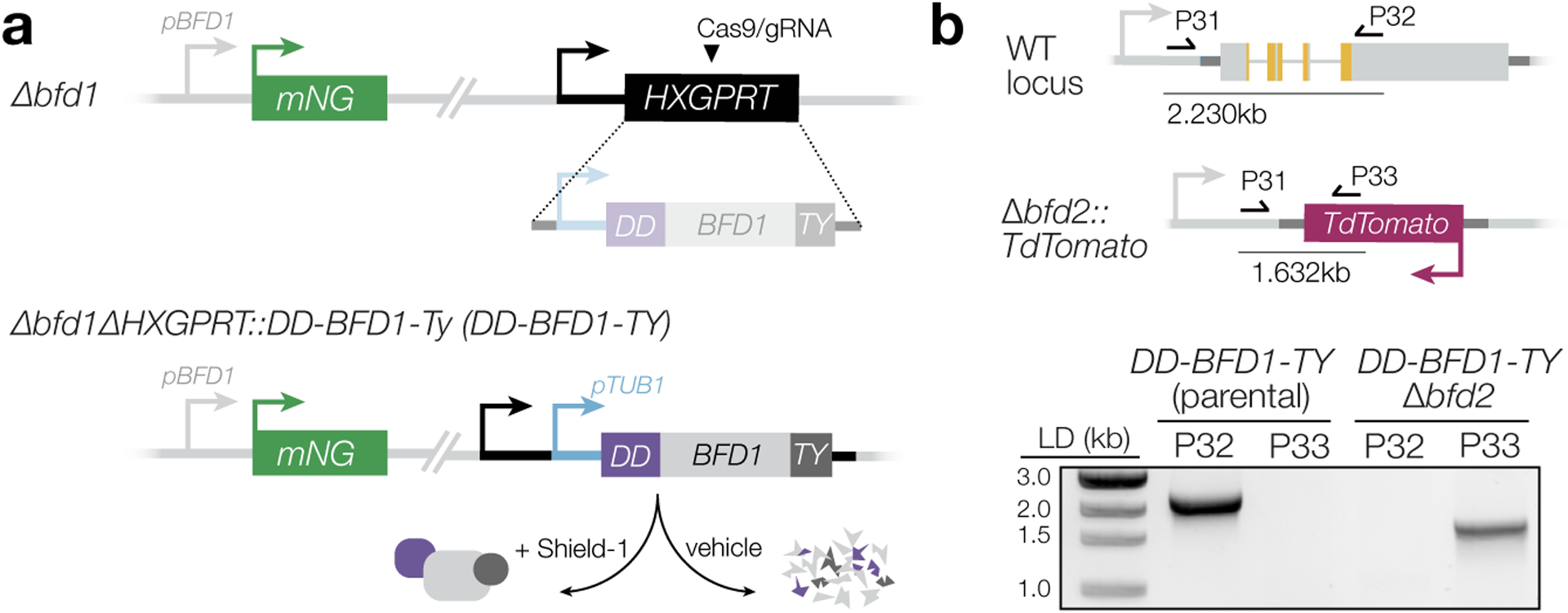
a, Schematic of ligand-inducible BFD1. The DD-BFD1-TY strain was constructed previously23, by integration at the HXGPRT locus in the Δbfd1 genetic background. A heterologous promoter (pTUB1) drives expression of the transgene, but DD-BFD1-TY protein is only stabilized upon treatment with Shield-1. b, Validation of BFD2-knockout in DD-BFD1-TY. Selected clones with screened using the same strategy described in Extended Data Fig. 3, verifying both loss of endogenous BFD2 and replacement with the fluorescence cassette. Gel shows PCRs performed on gDNA from both the parental (DD-BFD1-TY) and DD-BFD1-TYΔbfd2 strains. Reverse primers used are listed over the respective well, with expected product sizes indicated in the diagram above.
Extended Data Fig. 6. Genotyping BFD2-complemented and conditional overexpression strains.
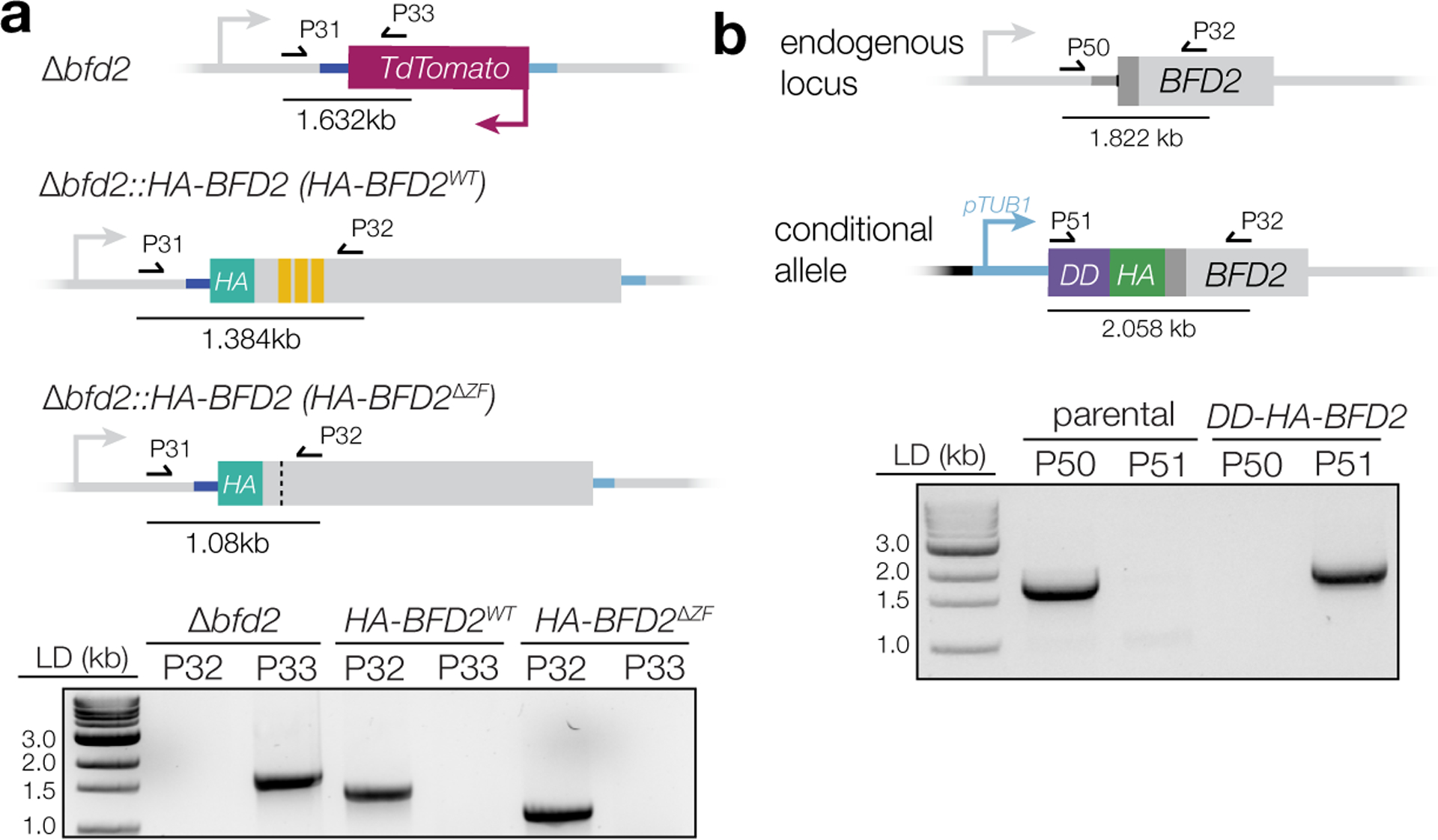
a, Complementation with wild-type (HA-BFD2) or non-RNA-binding (HA-BFD2ΔZF) BFD2 was verified by PCR using the same primers as in Extended Data Fig. 3, screening for reintroduction of the coding sequence and reciprocal loss of TdTomato at the endogenous BFD2 locus. Diagram shows PCR priming in Δbfd2 (top) versus the complemented loci (below), with expected product sizes indicated. Reverse primers and gDNA template used in each reaction are listed above the respective lane. b, For conditional BFD2 expression, pTUB1-DD-HA was integrated at the endogenous BFD2 locus (P51/P32), resulting in coincident loss of the untagged allele (P50/P32). As in a, diagram shows the relative positions of primer binding with expected product sizes, and lanes are labeled above with the gDNA template and forward primer used.
Extended Data Fig. 7. Neither mRNA abundance nor differential expression are predictive of interaction with BFD2.
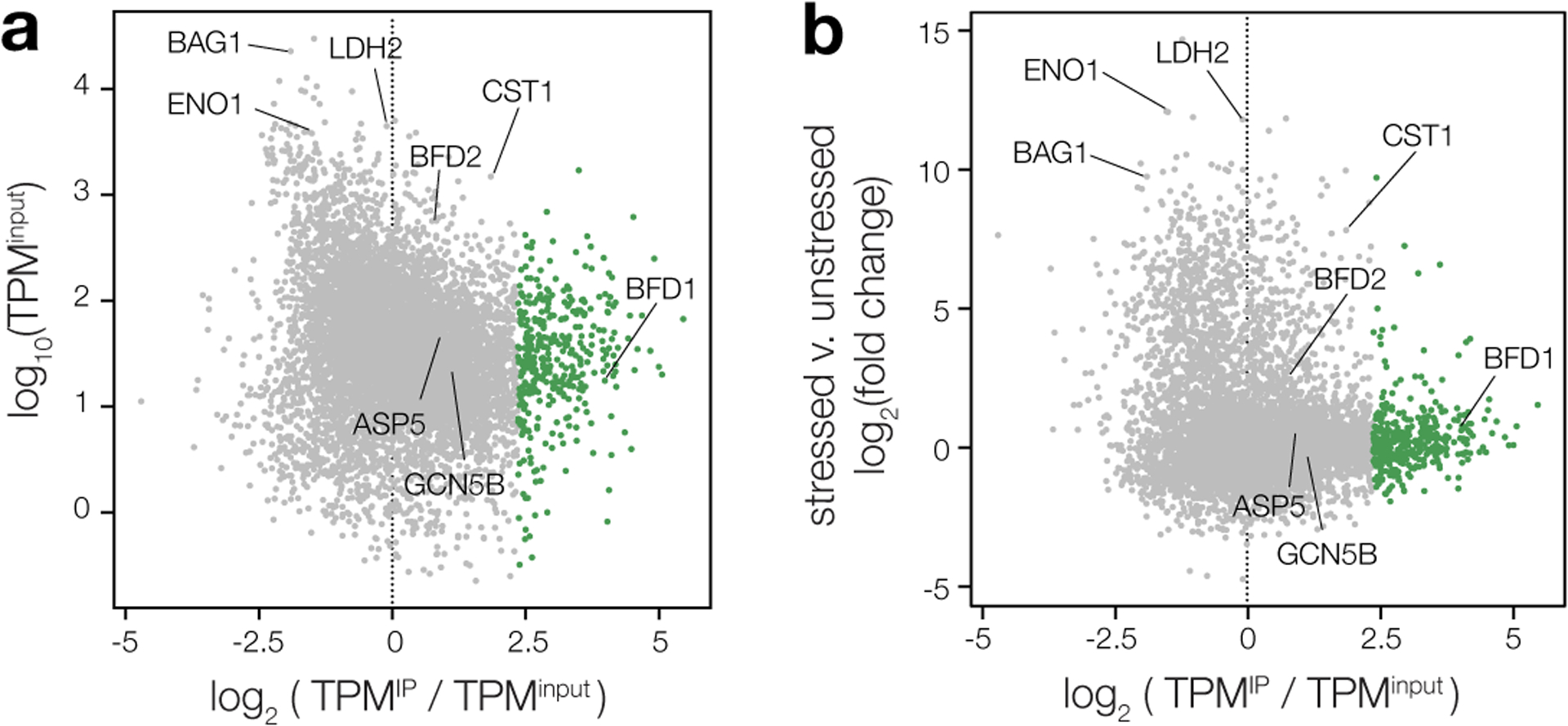
Comparison of log-transformed enrichment ratios (TPMIP/TPMinput) for all transcripts detected in stressed HA-BFD2 parasites and either abundance in the unenriched input (a) or change in expression after 48 h of stress, based on a previously published dataset (b)23. The 375 most highly enriched genes identified by Gaussian mixture modeling are highlighted in green.
Extended Data Fig. 8. Strain construction using the HiT vector strategy.
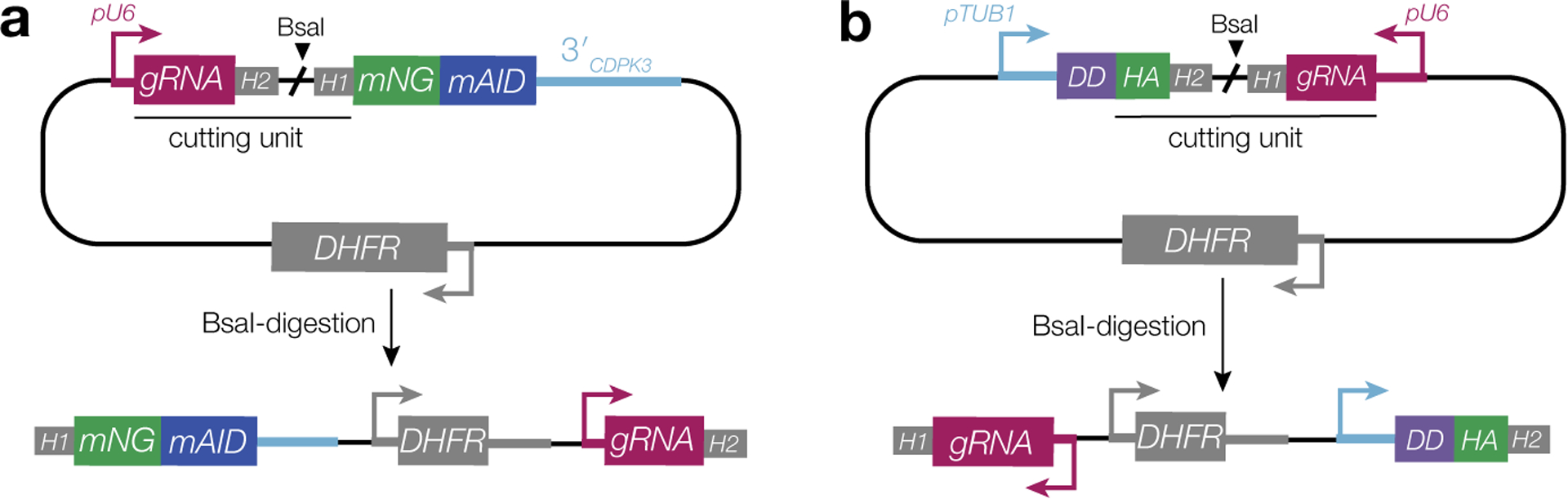
a, Schematic of C-terminal tagging HiT vectors (top), as described previously79. Targeted integration of BsaI-linearized constructs (bottom) is facilitated by a guide RNA (gRNA) specific to the 3′ end of the coding sequence and 40 bp homology regions (H1, H2), both encoded in the gene-specific cutting unit. Transcription of the gRNA is driven by a type III promoter (pU6). A heterologous 3′ untranslated region (3′CDPK3) allows expression of the gene product. DHFR denotes a pyrimethamine resistance cassette to enable mutant selection. b, N-terminal HiT vector configuration for generation of conditional overexpression strains. Construct integration endogenously tags the targeted gene with the Shield-1-stabilized degradation domain (DD) and replaces the native promoter with that of alpha tubulin (pTUB1). Cutting units are designed similarly to those in a, with a gRNA that targets the coding sequence 5′ end encoded in reverse orientation. For the inducible BFD2 strain in particular, HA was also designed into the cutting unit to enable detection of the protein by the same epitope used for examination of endogenously regulated BFD2.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dominique Soldati for the GAP45 antibody; David Sibley for the SAG1 antibody; Fausto Araujo for the anti-Toxoplasma antibody; the Whitehead Institute Genome Technology Core, particularly Tom Volkert, Jennifer Love, and Sumeet Gupta for their expertise with library preparation and next-generation sequencing; and the Whitehead Bioinformatics and Research Computing Core for consultation and software support. This work relied on VEupathDB, and we thank all contributors to this resource. This project was supported by grants from the NIH (R01AI158501) and the Smith Family Foundation (Odyssey Award) to S.L..
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing interests.
DATA AVAILABILITY
For BFD1, BFD2, TGME49_253790, and TGME49_224630, the subsets of genes identified as significantly affected by depletion are listed in Supplementary Table 1. The results of quantitative proteome profiling in Δbfd2 parasites are provided in Supplementary Table 2. Primers used for RT-qPCR analysis of BFD1, BFD2, GCN5B, and ASP5 are available in Supplementary Table 3. CT values and analyses of RT-qPCR experiments are provided in Supplementary Tables 4–6. Raw reads and analysis of RNA immunoprecipitation and sequencing (RIP-seq) for BFD2 are provided in Supplementary Table 7. Minimally processed results from all bulk RNA sequencing performed in this study are provided in Supplementary Table 8. Oligonucleotides used in this study for molecular cloning are in Supplementary Table 9. Unprocessed data from the transcriptional and proteome profiling described herein are available through Gene Expression Omnibus (GEO) and the Proteomics Identification Database (PRIDE), respectively, with the following accession numbers: GSE223819 (Fig. 1e–g & Extended Data Fig. 2); GSE223869 (Fig. 2i–k); PXD039648 (Extended Data Fig. 3c); GSE223621 (Extended Data Fig. 3d-e); GSE223877 (Fig. 4d); GSE223620 (Fig. 6b–d & Extended Data Fig. 7). Additional unprocessed data is provided as Source Data.
REFERENCES
- 1.Cressler CE, McLEOD DV, Rozins C, VAN DEN Hoogen J & Day T The adaptive evolution of virulence: a review of theoretical predictions and empirical tests. Parasitology 143, 915–930 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett MP, Kyle DE, Sibley LD, Radke JB & Tarleton RL Protozoan persister-like cells and drug treatment failure. Nat. Rev. Microbiol 17, 607–620 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montoya JG & Liesenfeld O Toxoplasmosis. Lancet 363, 1965–1976 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Dubey JP, Lindsay DS & Speer CA Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin. Microbiol. Rev 11, 267–299 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grant IH, Gold JW, Rosenblum M, Niedzwiecki D & Armstrong D Toxoplasma gondii serology in HIV-infected patients: the development of central nervous system toxoplasmosis in AIDS. AIDS 4, 519–521 (1990). [PubMed] [Google Scholar]
- 6.Luft BJ & Remington JS Toxoplasmic encephalitis in AIDS. Clin. Infect. Dis 15, 211–222 (1992). [DOI] [PubMed] [Google Scholar]
- 7.Jones JL et al. Ocular toxoplasmosis in the United States: recent and remote infections. Clin. Infect. Dis 60, 271–273 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Derouin F, Pelloux H & ESCMID Study Group on Clinical Parasitology. Prevention of toxoplasmosis in transplant patients. Clin. Microbiol. Infect 14, 1089–1101 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Alday PH & Doggett JS Drugs in development for toxoplasmosis: advances, challenges, and current status. Drug Des. Devel. Ther 11, 273–293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soête M, Camus D & Dubremetz JF Experimental induction of bradyzoite-specific antigen expression and cyst formation by the RH strain of Toxoplasma gondii in vitro. Exp. Parasitol 78, 361–370 (1994). [DOI] [PubMed] [Google Scholar]
- 11.Skariah S, McIntyre MK & Mordue DG Toxoplasma gondii: determinants of tachyzoite to bradyzoite conversion. Parasitol. Res 107, 253–260 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tu V et al. The Toxoplasma gondii Cyst Wall Interactome. MBio 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tu V et al. Enrichment and proteomic characterization of the cyst wall from in vitro Toxoplasma gondii cysts. MBio 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferguson DJ & Hutchison WM An ultrastructural study of the early development and tissue cyst formation of Toxoplasma gondii in the brains of mice. Parasitol. Res 73, 483–491 (1987). [DOI] [PubMed] [Google Scholar]
- 15.Denton H, Roberts CW, Alexander J, Thong KW & Coombs GH Enzymes of energy metabolism in the bradyzoites and tachyzoites of Toxoplasma gondii. FEMS Microbiol. Lett 137, 103–108 (1996). [DOI] [PubMed] [Google Scholar]
- 16.Fox BA, Gigley JP & Bzik DJ Toxoplasma gondii lacks the enzymes required for de novo arginine biosynthesis and arginine starvation triggers cyst formation. Int. J. Parasitol 34, 323–331 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Shukla A et al. Glycolysis is important for optimal asexual growth and formation of mature tissue cysts by Toxoplasma gondii. Int. J. Parasitol 48, 955–968 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Behnke MS, Radke JB, Smith AT, Sullivan WJ Jr & White MW The transcription of bradyzoite genes in Toxoplasma gondii is controlled by autonomous promoter elements. Mol. Microbiol 68, 1502–1518 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radke JR et al. The transcriptome of Toxoplasma gondii. BMC Biol. 3, 26 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pittman KJ, Aliota MT & Knoll LJ Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genomics 15, 806 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buchholz KR et al. Identification of tissue cyst wall components by transcriptome analysis of in vivo and in vitro Toxoplasma gondii bradyzoites. Eukaryot. Cell 10, 1637–1647 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cleary MD, Singh U, Blader IJ, Brewer JL & Boothroyd JC Toxoplasma gondii asexual development: identification of developmentally regulated genes and distinct patterns of gene expression. Eukaryot. Cell 1, 329–340 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waldman BS et al. Identification of a Master Regulator of Differentiation in Toxoplasma. Cell 180, 359–372.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saksouk N et al. Histone-modifying complexes regulate gene expression pertinent to the differentiation of the protozoan parasite Toxoplasma gondii. Mol. Cell. Biol 25, 10301–10314 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bougdour A et al. Drug inhibition of HDAC3 and epigenetic control of differentiation in Apicomplexa parasites. J. Exp. Med 206, 953–966 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maubon D et al. Activity of the Histone Deacetylase Inhibitor FR235222 on Toxoplasma gondii: Inhibition of Stage Conversion of the Parasite Cyst Form and Study of New Derivative Compounds. Antimicrob. Agents Chemother 54, 4843–4850 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farhat DC et al. A MORC-driven transcriptional switch controls Toxoplasma developmental trajectories and sexual commitment. Nat Microbiol 5, 570–583 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gissot M et al. Toxoplasma gondii Alba proteins are involved in translational control of gene expression. J. Mol. Biol 425, 1287–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Liu M et al. Characterization of TgPuf1, a member of the Puf family RNA-binding proteins from Toxoplasma gondii. Parasit. Vectors 7, 141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gissot M et al. An evolutionary conserved zinc finger protein is involved in Toxoplasma gondii mRNA nuclear export. Cell. Microbiol 19, (2017). [DOI] [PubMed] [Google Scholar]
- 31.Vanchinathan P, Brewer JL, Harb OS, Boothroyd JC & Singh U Disruption of a locus encoding a nucleolar zinc finger protein decreases tachyzoite-to-bradyzoite differentiation in Toxoplasma gondii. Infect. Immun 73, 6680–6688 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker R et al. The Toxoplasma nuclear factor TgAP2XI-4 controls bradyzoite gene expression and cyst formation. Mol. Microbiol 87, 641–655 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong D-P, Radke JB & White MW Opposing Transcriptional Mechanisms Regulate Toxoplasma Development. mSphere 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang S et al. Toxoplasma gondii AP2IX-4 Regulates Gene Expression during Bradyzoite Development. mSphere 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radke JB et al. ApiAP2 transcription factor restricts development of the Toxoplasma tissue cyst. Proc. Natl. Acad. Sci. U. S. A 110, 6871–6876 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Radke JB et al. Transcriptional repression by ApiAP2 factors is central to chronic toxoplasmosis. PLoS Pathog. 14, e1007035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown KM, Long S & Sibley LD Conditional Knockdown of Proteins Using Auxin-inducible Degron (AID) Fusions in Toxoplasma gondii. Bio Protoc 8, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith TA, Lopez-Perez GS, Shortt E & Lourido S High-throughput functionalization of the Toxoplasma kinome uncovers a novel regulator of invasion and egress. bioRxiv 2021.09.23.461611 (2021) doi: 10.1101/2021.09.23.461611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sidik SM et al. A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell 166, 1423–1435.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferguson DJ & Hutchison WM The host-parasite relationship of Toxoplasma gondii in the brains of chronically infected mice. Virchows Arch. A Pathol. Anat. Histopathol 411, 39–43 (1987). [DOI] [PubMed] [Google Scholar]
- 41.Melzer TC, Cranston HJ, Weiss LM & Halonen SK Host Cell Preference of Toxoplasma gondii Cysts in Murine Brain: A Confocal Study. J. Neuroparasitology 1, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mouveaux T et al. Primary brain cell infection by Toxoplasma gondii reveals the extent and dynamics of parasite differentiation and its impact on neuron biology. Open Biol. 11, 210053 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lüder CG, Giraldo-Velásquez M, Sendtner M & Gross U Toxoplasma gondii in primary rat CNS cells: differential contribution of neurons, astrocytes, and microglial cells for the intracerebral development and stage differentiation. Exp. Parasitol 93, 23–32 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Davis TL & Rebay I Master regulators in development: Views from the Drosophila retinal determination and mammalian pluripotency gene networks. Dev. Biol 421, 93–107 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hall TMT Multiple modes of RNA recognition by zinc finger proteins. Curr. Opin. Struct. Biol 15, 367–373 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Hajikhezri Z, Darweesh M, Akusjärvi G & Punga T Role of CCCH-Type Zinc Finger Proteins in Human Adenovirus Infections. Viruses 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maeda K & Akira S Regulation of mRNA stability by CCCH-type zinc-finger proteins in immune cells. Int. Immunol 29, 149–155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu M & Blackshear PJ RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat. Rev. Immunol 17, 130–143 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanhsen B, Farrukh A, Pradel G & Ngwa CJ The Plasmodium falciparum CCCH zinc finger protein ZNF4 plays an important role in gametocyte exflagellation through the regulation of male gametocyte enriched transcripts. bioRxiv 2022.03.08.483571 (2022) doi: 10.1101/2022.03.08.483571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hirai M, Maeta A, Mori T & Mita T Pb103 Regulates Zygote/Ookinete Development in Plasmodium berghei via Double Zinc Finger Domains. Pathogens 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paterou A, Walrad P, Craddy P, Fenn K & Matthews K Identification and stage-specific association with the translational apparatus of TbZFP3, a CCCH protein that promotes trypanosome life-cycle development. J. Biol. Chem 281, 39002–39013 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walrad P, Paterou A, Acosta-Serrano A & Matthews KR Differential trypanosome surface coat regulation by a CCCH protein that co-associates with procyclin mRNA cis-elements. PLoS Pathog. 5, e1000317 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J-L et al. The protein phosphatase 2A holoenzyme is a key regulator of starch metabolism and bradyzoite differentiation in Toxoplasma gondii. Nat. Commun 13, 1–16 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferrell JE Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr. Opin. Cell Biol 14, 140–148 (2002). [DOI] [PubMed] [Google Scholar]
- 55.Lek M et al. A homeodomain feedback circuit underlies step-function interpretation of a Shh morphogen gradient during ventral neural patterning. Development 137, 4051–4060 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Sha W et al. Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts. Proc. Natl. Acad. Sci. U. S. A 100, 975–980 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moses K & Rubin GM Glass encodes a site-specific DNA-binding protein that is regulated in response to positional signals in the developing Drosophila eye. Genes Dev. 5, 583–593 (1991). [DOI] [PubMed] [Google Scholar]
- 58.Solomon MJ Hysteresis meets the cell cycle. Proc. Natl. Acad. Sci. U. S. A 100, 771–772 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fang X et al. Cell fate potentials and switching kinetics uncovered in a classic bistable genetic switch. Nat. Commun 9, 1–9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Banaszynski LA, Chen L-C, Maynard-Smith LA, Ooi AGL & Wandless TJ A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Markus BM, Bell GW, Lorenzi HA & Lourido S Optimizing Systems for Cas9 Expression in Toxoplasma gondii. mSphere 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shallberg LA et al. Impact of secondary TCR engagement on the heterogeneity of pathogen-specific CD8+ T cell response during acute and chronic toxoplasmosis. PLoS Pathog. 18, e1010296 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burg JL, Perelman D, Kasper LH, Ware PL & Boothroyd JC Molecular analysis of the gene encoding the major surface antigen of Toxoplasma gondii. J. Immunol 141, 3584–3591 (1988). [PubMed] [Google Scholar]
- 64.Bastin P, Bagherzadeh Z, Matthews KR & Gull K A novel epitope tag system to study protein targeting and organelle biogenesis in Trypanosoma brucei. Mol. Biochem. Parasitol 77, 235–239 (1996). [DOI] [PubMed] [Google Scholar]
- 65.Berg S et al. ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Chandrasekaran S et al. IFN-γ stimulated murine and human neurons mount anti-parasitic defenses against the intracellular parasite Toxoplasma gondii. Nat. Commun 13, 1–13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Madisen L et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaech S & Banker G Culturing hippocampal neurons. Nat. Protoc 1, 2406–2415 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chan AW et al. Analysis of CDPK1 targets identifies a trafficking adaptor complex that regulates microneme exocytosis in Toxoplasma. bioRxiv 2023.01.11.523553 (2023) doi: 10.1101/2023.01.11.523553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bekker-Jensen DB et al. A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol. Cell. Proteomics 19, 716–729 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Conley FK & Jenkins KA Immunohistological study of the anatomic relationship of toxoplasma antigens to the inflammatory response in the brains of mice chronically infected with Toxoplasma gondii. Infect. Immun 31, 1184–1192 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu Z, Guan X, Schmidt CA & Matera AG RIP-seq analysis of eukaryotic Sm proteins identifies three major categories of Sm-containing ribonucleoproteins. Genome Biol. 15, R7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Benaglia T, Chauveau D, Hunter DR & Young DS mixtools: An R Package for Analyzing Mixture Models. J. Stat. Softw 32, 1–29 (2010). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For BFD1, BFD2, TGME49_253790, and TGME49_224630, the subsets of genes identified as significantly affected by depletion are listed in Supplementary Table 1. The results of quantitative proteome profiling in Δbfd2 parasites are provided in Supplementary Table 2. Primers used for RT-qPCR analysis of BFD1, BFD2, GCN5B, and ASP5 are available in Supplementary Table 3. CT values and analyses of RT-qPCR experiments are provided in Supplementary Tables 4–6. Raw reads and analysis of RNA immunoprecipitation and sequencing (RIP-seq) for BFD2 are provided in Supplementary Table 7. Minimally processed results from all bulk RNA sequencing performed in this study are provided in Supplementary Table 8. Oligonucleotides used in this study for molecular cloning are in Supplementary Table 9. Unprocessed data from the transcriptional and proteome profiling described herein are available through Gene Expression Omnibus (GEO) and the Proteomics Identification Database (PRIDE), respectively, with the following accession numbers: GSE223819 (Fig. 1e–g & Extended Data Fig. 2); GSE223869 (Fig. 2i–k); PXD039648 (Extended Data Fig. 3c); GSE223621 (Extended Data Fig. 3d-e); GSE223877 (Fig. 4d); GSE223620 (Fig. 6b–d & Extended Data Fig. 7). Additional unprocessed data is provided as Source Data.