

Summary
Chromatin immunoprecipitation (ChIP) protocols have been used to reveal protein-DNA interactions of various cell types and tissues; however, optimization is required for each specific type of sample. Here, we present a ChIP protocol from murine inguinal white adipose tissue. We describe steps for tissue harvesting, crosslinking, chromatin extraction, shearing, immunoprecipitation, and purification. We then detail procedures for analysis including library preparation, sequencing, and qRT-PCR validation.
For complete details on the use and execution of this protocol, please refer to Antonia Katsouda et al. (2022).1
Subject areas: Sequence Analysis, Genomics, ChIP-seq, Metabolism, Molecular Biology
Graphical abstract
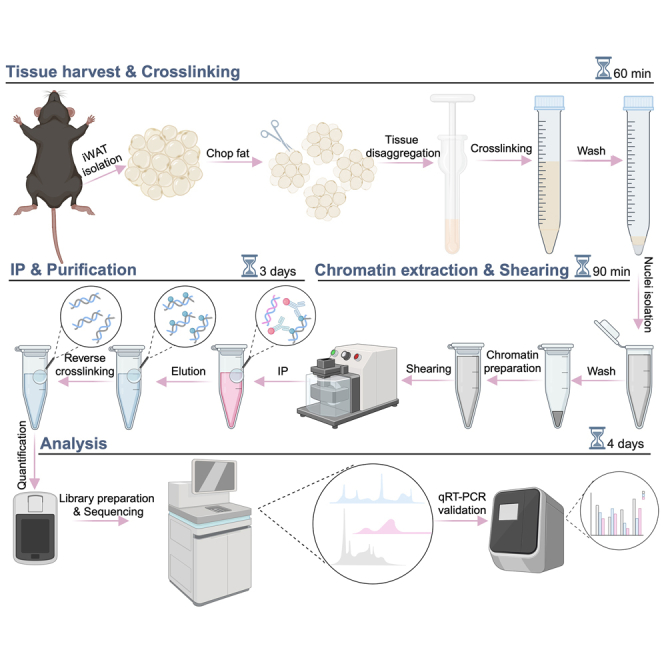
Highlights
-
•
Illustrates the steps for inguinal fat chromatin IP and analysis
-
•
Isolates chromatin from adipose tissue and uses for IP
-
•
Analyzes the purified protein-DNA complexes by deep sequencing and qRT-PCR
-
•
Provides optimized conditions to ensure high-quality data
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Chromatin immunoprecipitation (ChIP) protocols have been used to reveal protein-DNA interactions of various cell types and tissues; however, optimization is required for each specific type of sample. Here, we present a ChIP protocol from murine inguinal white adipose tissue. We describe steps for tissue harvesting, crosslinking, chromatin extraction, shearing, immunoprecipitation, and purification. We then detail procedures for analysis including library preparation, sequencing, and qRT-PCR validation.
Before you begin
The protocol was established using 10-week-old male C57BL6/J mice purchased from The Jackson Laboratory. All animals used for experimentation were bred/housed in individual ventilated cages, under specific pathogen–free, temperature-controlled (22°C) and 12-h light/ dark cycle conditions in full compliance with the guidelines of the Federation of Laboratory Animal Science Association recommendations in the Laboratory Animal Unit of Biomedical Research Foundation of the Academy of Athens (BRFAA) and allowed free access to diets and water.
Institutional permissions
All experimental procedures reported here were approved by the veterinary authority of the Prefecture of Athens and the Bioethics Committee, in accordance with the national Registration (Presidential Decree 56/ 2013) in harmony with the European Directive 63/2010.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-peroxisome proliferator-activated receptor (PPAR) gamma (PPARγ, 10 μg/IP) | Proteintech | 16643-1-AP |
| Anti-histone H3 (acetyl K27, H3K27ac, 10 μg/IP) | Abcam | ab4729 |
| Normal rabbit IgG (10 μg/IP) | Cell Signaling Technology | 2729s |
| Chemicals, peptides, and recombinant proteins | ||
| Sevoflurane | AbbVie | 4456 |
| Phosphate-buffered saline (PBS) | PAN-Biotech | P04-36500 |
| Sodium chloride (NaCl) | Calbiochem | 7760 |
| Nonidet P-40 (NP-40) | Sigma-Aldrich | 74385 |
| Sodium (Na)-deoxycholate | AppliChem | A1531,0025 |
| Sodium dodecyl sulfate (SDS) | PanReac AppliChem | A2572 |
| Protease inhibitors (PI) | Roche | 5892970001 |
| Phosphatase inhibitors (PhoI) | Roche | 4906837001 |
| 2,2′,2″,2‴-(Ethane-1,2-diyldinitrilo)tetraacetic acid (EDTA) | Merck | 4005 |
| Lithium chloride (LiCl) | AppliChem | A6286,0250 |
| 4-(2-Hydroxyethyl)piperazine-1-ethane-sulfonic acid (HEPES) | AppliChem | A3724,0250 |
| Octylphenol ethylene oxide condensate (Triton X-100) | Sigma-Aldrich | T9284 |
| Ethylene glycol tetraacetic acid (EGTA) | AppliChem | A0878,0025 |
| Ethanol | VWR | 20821.365 |
| Agarose | Nippon Genetics | AG02 |
| Tris-Base | Fisher BioReagents | BP152-5 |
| Glycerol | Melford | G1345 |
| Formaldehyde | AppliChem | A0877,0250 |
| Glycine | Sigma-Aldrich | 50046 |
| 1 Kb plus DNA ladder | NEB | N3200S |
| Ribonuclease (RNase) | Sigma-Aldrich | R6513 |
| Proteinase K | Roche | 031158360 |
| Critical commercial assays | ||
| KAPA SYBR Fast Master Mix | Kapa Biosystems | KK4618 |
| Dynabeads Protein G | Invitrogen | 10003D |
| NucleoMag NGS Beads | Macherey-Nagel | 15889167 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | NEB | E7645L |
| Qubit dsDNA HS Kit | Thermo Fisher Scientific | Q32854 |
| Qubit dsDNA BR Kit | Thermo Fisher Scientific | Q32853 |
| Agilent Bioanalyzer DNA1000 Kit | Agilent Technologies | 5067-1504 |
| NovaSeq 6000 SP Reagent Kit v1.5 (100 cycles) | Illumina | 20028401 |
| Experimental models: organisms/strains | ||
| C57BL/6J mice (10 weeks old, male) | Purchased from The Jackson Laboratory and bred in BRFAA | N/A |
| Oligonucleotides | ||
| Pdk4 F: GGCGGTGGTTAGGATCCG | Eurofins | N/A |
| Pdk4 R: GGGACCCTGGGACCACAA | Eurofins | N/A |
| Tmem 245 F: CGCTCACTGGAGGACCTTT | Eurofins | N/A |
| Tmem 245 R: AGCCTGTGGGTGAGTGAGAT | Eurofins | N/A |
| Dnajb1 F: TTTCGGAGAGGCCCGTCTC | Eurofins | N/A |
| Dnajb1 R: TCCGTAGCGGATCCAGCC | Eurofins | N/A |
| Cbx7 F: AAATTGGGCAGCTGGCAGC | Eurofins | N/A |
| Cbx7 R: GCCCAGAAGCCGAACTGAGAGAT | Eurofins | N/A |
| Software and algorithms | ||
| GraphPad Prism 7.0 | GraphPad Software | https://www.graphpad.com |
| FastQC | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Bowtie2 v2.5.1 | John Hopkins University | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| SAMtools | Massachusetts Institute of Technology | http://www.htslib.org/ |
| BEDtools | Quinlan Lab | https://bedtools.readthedocs.io/en/latest/index.html |
| deepTools | Max Planck Institute of Immunobiology and Epigenetics | https://deeptools.readthedocs.io/en/develop/ |
| MACS2 | N/A | https://chipster.csc.fi/manual/macs2.html |
| bedGraphtoBigWig | UCSC-ENCODE | https://www.encodeproject.org/software/bedgraphtobigwig/ |
| Integrated Genome Viewer | Broad Institute | https://software.broadinstitute.org/software/igv/ |
| BioRender | BioRender Software | https://app.biorender.com |
| Other | ||
| Tenbroeck tissue grinder-2 mL | Kimble | 057003 |
| Cell strainer 200 μm | pluriSelect | 43-50200-03 |
| S220 Focused-ultrasonicator | Covaris | 500217 |
| microTUBE AFA Fiber Pre-Slit Snap-Cap 6 × 16 mm | Covaris | 520045 |
| 16-Tube SureBeads Magnetic Rack | Bio-Rad | 1614916 |
| NovaSeq 6000 system | Illumina | N/A |
| Bioanalyzer 2100 instrument | Agilent Technologies | G2939BA |
| Qubit 4.0 fluorometer | Thermo Fisher Scientific | Q33238 |
| CFX96 real-time system | Bio-Rad | C1000 Touch |
| Hybridizer | UVP Laboratory Products | HB-1000 |
| NanoDrop spectrophotometer | Thermo Fisher Scientific | NanoDrop 2000 |
| Multifuge | Heraeus | 3S-R |
| Biofuge Fresco | Heraeus | fresco |
Materials and equipment
Buffers
Alternatives: Chemicals listed in the key resources table can all be replaced with the same chemicals from different suppliers, provided that they are of the equivalent grade. NP-40 can be replaced with IGEPAL CA-630 (Sigma-Aldrich).
Chemical solutions
-
•
1 M HEPES pH = 7.9 (11.92 HEPES, to 50 mL with ddH2O), store at 20°C–25°C up to 12 months
-
•
2.5 M Glycine (9.38 g Glycine, to 50 mL with ddH2O), store at 20°C–25°C up to 12 months
-
•0.5 M EDTA pH = 8 (7.31 g EDTA, to 50 mL with ddH2O), store at 20°C–25°C up to 12 months
-
○Prepare 40 mL of distilled water and place it on a magnetic stirring plate.
-
○Add 7.31 g of EDTA to the solution.
-
○Slowly adjust pH to 8.0 with sodium hydroxide (NaOH) 1 M. EDTA will be completely dissolved only when pH = 8
-
○Add distilled water until volume of 50 mL is reached.
-
○
-
•
0.5 M Tris-Hydrochloride (HCL) pH = 8.1 and pH = 8 (3.03 g Tris-Base, to 50 mL with ddH2O), store at 20°C–25°C up to 12 months
-
•
5 M NaCl (14.61 g NaCl, to 50 mL with ddH2O) store at 20°C–25°C up to 12 months
-
•
100 mM EGTA pH = 8 (7.31 g EGTA, to 50 mL with ddH2O), store at 20°C–25°C up to 12 months
-
•
10% SDS (5 g SDS, to 50 mL with ddH2O), store at 20°C–25°C up to 12 months
-
•
10% Triton-X (5 mL 100% Triton-X, to 50 mL with ddH2O), store at 20°C–25°C, up to 12 months
-
•
5 M LiCl (10.6 g LiCl to 50 mL with ddH2O), store at 20°C–25°C up to 12 months
-
•
10% Sodium deoxycholate (5 g sodium deoxycholate, to 50 mL with ddH2O), store at 20°C–25°C, up to 12 months
-
•
20 mg/μL RNase A (2 g RNase A, to 100 μL with ddH2O), store at -20°C up to 24 months
-
•
30 mg/μL proteinase K (3 g proteinase K, to 100 μL ddH2O), store at -20°C up to 24 months
-
•
80% ethanol (40 mL 100% ethanol, to 50 mL with ddH2O), store at 20°C–25°C up to 12 months
Note: All stock solutions and buffers are pH adjusted with either 1 M Hydrochloric acid or 1 M NaOH solutions.
Lysis buffer
Prepare on the day of the experiment, keep on ice.
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES pH = 7.9 (1 M) | 50 mM | 1 mL |
| NaCl (5 Μ) | 140 mM | 560 μL |
| EDTA pH = 8 (0.5 M) | 1 mM | 40 μL |
| Glycerol (100%) | 10% | 2 mL |
| NP-40 (100%) | 0.5% | 100 μL |
| Triton-X (100%) | 0.25% | 50 μL |
| PI (100X) | 1X | 200 μL |
| PhoI (100X) | 1X | 200 μL |
| Nuclease-free water | N/A | 15.850 mL |
| Total | N/A | 20 mL |
Wash buffer
Prepare on the day of the experiment, keep on ice.
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl pH = 8.1 (0.5 M) | 10 mM | 400 μL |
| NaCl (5 M) | 200 mM | 800 μL |
| EDTA pH = 8 (0.5 M) | 1 mM | 40 μL |
| EGTA pH = 8 (100 mM) | 0.5 mM | 100 μL |
| Nuclease-free water | N/A | 18.660 mL |
| Total | N/A | 20 mL |
Sonication buffer
Prepare on the day of the experiment. Keep at 20–25°C to prevent precipitation of SDS.
| Reagent | Final concentration | Amount |
|---|---|---|
| SDS (10%) | 0.1% | 200 μL |
| EDTA pH = 8 (0.5 M) | 1 mM | 40 μL |
| Tris-HCl pH = 8.1 (0.5 M) | 10 mM | 400 μL |
| PI (100X) | 1X | 200 μL |
| PhoI (100X) | 1X | 200 μL |
| Nuclease-free water | N/A | 18.960 mL |
| Total | N/A | 20 mL |
Immunoprecipitation buffer
Prepare on the day of the experiment. Keep at 20–25°C to prevent precipitation of SDS.
| Reagent | Final concentration | Amount |
|---|---|---|
| SDS (10%) | 0.1% | 200 μL |
| EDTA pH = 8 (0.5 M) | 1 mM | 40 μL |
| Tris-HCl pH = 8.1 (0.5 M) | 10 mM | 400 μL |
| Triton X (100%) | 1% | 200 μL |
| NaCl (5 Μ) | 150 mM | 600 μL |
| Nuclease-free water | N/A | 18.560 mL |
| Total | N/A | 20 mL |
Wash buffer A
Store at 4°C up to 12 months. Keep on ice on the day of the protocol.
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl pH = 8 (0.5 M) | 10 mM | 400 μL |
| NaCl (5 Μ) | 150 mM | 600 μL |
| EDTA pH = 8 (0.5 M) | 2 mM | 80 μL |
| Triton-X (100%) | 1% | 200 μL |
| SDS (10%) | 0.1% | 200 μL |
| Nuclease-free water | N/A | 18.520 mL |
| Total | N/A | 20 mL |
Wash buffer B
Store at 4 °C up to 12 months. Keep on ice on the day of the protocol.
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl pH = 8 (0.5 M) | 10 mM | 400 μL |
| NaCl (5 Μ) | 150 mM | 600 μL |
| EDTA pH = 8 (0.5 M) | 1 mM | 40 μL |
| Triton-X (100%) | 1% | 200 μL |
| SDS (10%) | 0.1% | 200 μL |
| LiCl (5 M) | 250 mM | 1 mL |
| Sodium deoxycholate (10%) | 0.7% | 1.4 mL |
| Nuclease-free water | N/A | 16.160 mL |
| Total | N/A | 20 mL |
Tris-EDTA buffer
Prepare fresh. Add microliter amounts of high molarity HCl to lower the pH to 8.
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl pH = 8 (0.5 M) | 10 mM | 400 μL |
| EDTA pH = 8 (0.5 M) | 1 mM | 40 μL |
| Nuclease-free water | N/A | 19.560 mL |
| Total | N/A | 20 mL |
Elution buffer
Prepare fresh. Keep at 20–25°C to prevent precipitation of SDS.
| Reagent | Final concentration | Amount |
|---|---|---|
| SDS (10%) | 0.5% | 1 mL |
| HEPES pH = 7.9 (1 M) | 20 mM | 400 μL |
| EDTA pH = 8 (0.5 M) | 1 mM | 40 μL |
| Nuclease-free water | N/A | 18.560 mL |
| Total | N/A | 20 mL |
Equipment
-
•
2 mL-Tenbroeck tissue grinder, Kimble
Alternatives: Any other glass tissue grinder
-
•
S220 Focused-ultrasonicator, Covaris
Alternatives: Bioruptor Sonicator, Diagenode
-
•
microTUBE AFA Fiber Pre-Slit Snap-Cap 6 × 16 mm, Covaris
Alternatives: 0.2 mL microtubes for Bioruptor Pico, Diagenode
-
•
16-Tube SureBeads Magnetic Rack, Bio-Rad
Alternatives: Any Magnetic Rack suitable for separation of magnetic beads
-
•
NovaSeq 6000 system, Illumina
Alternatives: Any Illumina sequencing system capable of producing >10 million reads per sample, e.g. NextSeq 2000 system.
-
•
Bioanalyzer 2100 instrument, Agilent Technologies
Alternatives: Any other DNA/RNA fragment analysis system (e.g. QIAxcel advanced system), or DNA gel electrophoresis equipment (a less sensitive/quantitative alternative)
-
•
NanoDrop 2000 Spectrophotometer, Thermo Fisher Scientific
Alternatives: Any other Spectrophotometer
-
•
Qubit 4.0 fluorometer, Thermo Fisher Scientific
Alternatives: Any other fluorescence-based quantification system with a dsDNA lower detection limit < 0.1 ng/mL
-
•
CFX96 Real-Time System, Bio-Rad
Alternatives: Any qPCR machine
-
•
HB-1000 hybridizer, UVP Laboratory Products
Alternatives: Any incubator or hybridization oven capable of maintaining 45οC–65οC temperature
-
•
Multifuge 3S-R, Heraeus
Alternatives: Any centrifuge with a falcon-rotor capable of maintaining 4οC temperature
-
•
Biofuge Fresco, Heraeus
Alternatives: Any centrifuge with an eppendorf-rotor capable of maintaining 4οC temperature
Others
-
•
A primary antibody against the protein of interest (ChIP-verified)
Note: A control ChIP performed in parallel is required. Prepare a ChIP reaction from an identical inguinal white adipose tissue (iWAT) sample using an equivalent amount (ng) of an isotype-matched non-specific antibody.
-
•
Sevoflurane
Alternatives: A similar inhaled anesthetic
Step-by-step method details
Tissue preparation and fixation
Timing: 60 min
The protocol below is for 1 ChIP assay. The starting material is 8 iWAT depots (coming from four 10-weeks old mice) with an average mass of 120 mg each.
CRITICAL: A parallel control ChIP assay using IgG is required. Double the starting material and the amounts of the buffers described below respectively.
Note: Ιn case that the starting material comes from an iWAT of different weight, the amounts of the following chemical solutions/ buffers must be re-calculated accordingly.
-
1.
Anaesthetize mice using sevoflurane, sacrifice them, remove iWAT depots surgically and place them in ice-cold PBS (Troubleshooting 1).
Note: Perform all of the following steps on ice unless otherwise stated.
-
2.
Transfer fat pads in a 1.5 mL eppendorf tube and dissect them into very small pieces (between 1-3 mm3) using a pair of scissors.
-
3.
Fat tissue pieces should be further processed in 1.5 mL PBS using a dounce glass grinder (perform 10 strokes with a tight pestle and for each stroke plunge the pestle to the bottom of the tube and rotate 180°).
-
4.
Pass sample through a 200 μm cell strainer and place it in a 15 mL conical tube.
-
5.
Add PBS up to 10 mL.
-
6.
Working in a fume hood, add 0.270 mL of 37% formaldehyde (final concentration 1%).
-
7.
Place on a rotating platform at 20°C–25°C for 15 min to perform chromatin crosslinking/ fixation.
-
8.
Quench crosslinking reaction by adding 0.513 mL 2.5 M glycine (final concentration 0.125 M).
-
9.
Place on a rotating platform at 20°C–25°C for 7 min.
-
10.
Centrifuge at 3.450 g for 5 min at 4°C, carefully discard the interphase and add 10 mL PBS.
-
11.
Repeat above wash step one more time for a total of 2 washes.
Chromatin extraction and shearing
Note: Perform all of the following steps on ice unless otherwise stated
-
12.
Add 400 μL of lysis buffer supplemented with protease inhibitors (PI) and phosphatase inhibitors (PhoI) and pipette to mix.
-
13.
Incubate for 15 min at 4°C.
-
14.
Meanwhile, mix the lysate by pipetting, using a 200 μL tip, every 5 min.
-
15.
Wash nuclei by centrifuging at 3.450 g for 10 min at 4°C, aspirate supernatant and re-suspend nuclei pellet in 1 mL wash buffer
-
16.
Repeat the above washing step two more times for a total of 3 washes.
Note: Nuclei can be stored at -80°C or continue with sonication steps below.
Pause point: Crosslinked nuclear pellets can be stored frozen by placing them directly at -80οC for several months.
-
17.
Turn on sonicator cooling system.
Note: Frozen nuclei pellets should be thawed on ice.
-
18.
Carefully add 0.5 mL sonication buffer and very gently turn tube sideways and twist to “wash” all the wash buffer off from the side of the tube while leaving the pellet completely intact.
-
19.
Centrifuge at 3.450 g for 5 min at 4°C and carefully discard the supernatant.
-
20.
Repeat steps 18 and 19 one more time.
-
21.
Resuspend nuclei pellet in 120 μL sonication buffer and incubate at 4°C for 10 min.
-
22.
Pipette to mix.
Note: A clumpy suspension is normal to be noticed.
-
23.
Centrifuge to collect the liquid at the bottom of the tube.
-
24.
Transfer sample to a microTUBE AFA Fiber Pre-Slit Snap-Cap 6 × 16 mm, being sure to fill tubes completely.
-
25.
Sonicate using the S220 Focused-ultrasonicator at 4°C for 12 min (duty factor, 75; peak power, 25; cycles per burst, 200) allowing the shearing of chromatin within a range of 250–500 bp DNA fragments.
-
26.
Transfer sample to a 1.5 mL eppendorf tube.
Note: In case that more than one ChIP reaction is performed, repeat sonication using the rest of the sample and combine them.
-
27.
Centrifuge sample at 16.060 g for 30 min at 4°C and collect supernatant in a new 1.5 mL eppendorf tube. This step is required to remove non-sonicated and non-fragmented chromatin.
Note: Chromatin samples can be stored at -80οC or continue with the immunoprecipitation steps below.
-
28.
Isolate 10 μL of chromatin preparation for reverse crosslink reaction (see below), quantitation and electrophoresis analysis with a 2% agarose gel.
Quality validation and quantification of sheared chromatin
Note: It is highly recommended to examine the fragmentation of chromatin prior to continuing with the IP.
-
29.
Incubate chromatin samples (step: 28, section: chromatin extraction and shearing) with 20 mg RNase A (add 1 μL of 20 mg/μL RNase A) at 37°C for 30 min.
-
30.
Add 30 mg proteinase K (1 μL of 30 mg/μL proteinase K) and incubate the sample at 50°C for 30 min.
-
31.
Add 8 μL of 5 M NaCl (final concentration 0.8 M NaCl) and incubate for 16 h at 65°C.
-
32.
To purify the DNA sample, add 100 μL of NucleoMag beads and incubate at 20°C–25°C.
-
33.
Place tube in the magnetic holder until all the beads are separated to the edge of the tube.
-
34.
While keeping the tubes in the holder, carefully remove the supernatant with a pipette.
-
35.
Wash by adding 300 μL of 80% ethanol.
-
36.
Remove tube from the magnetic holder and invert tube for 30 s.
-
37.
Place tube into magnetic holder and repeat the above wash step one more time for a total of 2 washes.
-
38.
Discard the final wash and add 25 μL ddH2O.
-
39.
Mix by pipetting and incubate for 5 min at 20°C–25°C.
-
40.
Place tube into magnetic holder and incubate for 2 min.
-
41.
Collect the supernatant in to a new 1.5 mL eppendorf tube.
-
42.
Quantify DNA concentration using a NanoDrop spectrophotometer (Troubleshooting 2).
-
43.
Load 500 ng of DNA on a 2% agarose gel with 1 Kb plus ladder (Troubleshooting 3).
Note: To proceed with IP, DNA should be sheared in size ranging from 250–500 bp (Figure 1)
Figure 1.

Shearing DNA sample run on a 2% agarose gel with 1 Kb ladder
Fragments between 250 and 500 bp are required for ChIP.
Immunoprecipitation and purification
The following steps detail the immunoprecipitation of protein/DNA complexes from sonicated nuclear extracts. Using the starting material of this protocol (8 inguinal iWAT depots with an average mass of 120 mg each), 50 μg of sheared chromatin could be isolated. This protocol is standardized for 1 ChIP reaction.
Note: Frozen sheared chromatin samples should be thawed on ice.
-
44.
Add 11 μL 10% Triton-X (final concentration 1% Triton-X) and 3.3 μL 5 M NaCl (final concentration 150 mM NaCl) in the sheared chromatin solution (Use sheared chromatin from step: 27, section: chromatin extraction and shearing)
Note: The starting material of this section is 50 μg of sheared chromatin in a final volume of 110 μL.
-
45.
Aliquot 50 μL protein G-Dynabeads into 1.5 mL eppendorf tube.
-
46.
Place tube on the magnetic rack and allow beads to separate for 2 min.
-
47.
Carefully discard supernatant using a pipette.
-
48.
Add 50 μL of immunoprecipitation buffer and mix by inverting for 30 s.
Note: 50 μL of protein G-Dynabeads corresponds to 10 μg of antibody according to the manufacturer’s guidelines (https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.com/TFS-Assets%2FLSG%2Fmanuals%2FMAN0015809_Dynabeads_Protein_G.pdf).
-
49.
Place tube on magnetic rack and repeat the above 2 steps for one more time.
-
50.
Remove tube from magnetic rack and resuspend beads in 50 μL immunoprecipitation buffer.
-
51.
Add 50 μg of sheared chromatin (step 44, section: immunoprecipitation and purification) and place tube on an orbital mixer at 4°C for 90 min.
Note: Pre-clearing step is used to minimize non-specific binding of protein-G beads.
-
52.
Place sample on the magnetic rack for 2 min
-
53.
Collect supernatant into a new 1.5 mL eppendorf tube.
-
54.
Remove 10 μL of the sample and store at -80°C.
-
55.
Add 10 μg of the antibody and incubate at an orbital mixer at 4°C for 16 h.
Note: Using ChIP-grade antibodies is highly recommended. If a ChIP-grade antibody is not available, the ability of the antibody to pull down the target protein of interest under crosslinking conditions should be previously tested. In this protocol, we used antibodies against PPARγ and H3K27ac.
Note: The amount of primary antibody is required for a high signal-to-noise ratio varies and should be tested in case that a different antibody from the ones described in this protocol is used.
-
56.
Repeat steps: 45–50, section: immunoprecipitation and purification.
-
57.
Incubate beads with the chromatin-antibody solution (step: 12, section: immunoprecipitation and purification) in an orbital mixer at 4°C for 4 h.
-
58.
Place tube on the magnetic rack and allow beads to separate for 2 min.
-
59.
Discard supernatant and wash the recovered resin with 500 μL wash buffer A by pipetting.
-
60.
Move the resin with wash buffer from the previous step to a new eppendorf tube, incubate for 2 min on ice, place the tube on the magnetic rack and discard supernatant.
-
61.
Repeat the above washing step for 3 more times for a total of 4 washes.
-
62.
Discard supernatant and add 500 μL of wash buffer B.
-
63.
Mix by pipetting and place the resin on a new eppendorf tube.
-
64.
Place tube on the magnetic rack and discard supernatant.
-
65.
Repeat the above washing step for one more time, for a total of 2 washes.
-
66.
Discard supernatant and add 500 μL of TE buffer.
-
67.
Place the resin with TE buffer in a new eppendorf tube.
-
68.
Place tube on the magnetic rack and discard supernatant.
-
69.
Add 25 μL of elution buffer.
-
70.
Elute the captured chromatin fragments by incubating sample at 65°C for 30 min with occasional rigorous 10 s vortexing every 5 min.
-
71.
Proceed to the reverse cross-linking reaction and purification of the eluted DNA, following the 29–41 steps of “quality validation and quantification of sheared chromatin” section.
-
72.
Quantify DNA concentration using a Qubit fluorometer (or an equivalent technology), according to manufacturer’s protocol (https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.com/TFS-Assets%2FLSG%2Fmanuals%2FMAN0017209_Qubit_4_Fluorometer_UG.pdf, Troubleshooting 4).
-
73.
Store at -20οC or continue with the library preparation and sequencing.
Downstream assays for analysis
Analysis of ChIP results can be achieved using qRT-PCR and/or NGS sequencing. In case that several DNA targets of the protein of interest have been previously reported, the relative enrichment of these areas can be checked by qRT-PCR to confirm a successful ChIP reaction, before proceeding to sequencing.
-
74.Sequencing.
-
a.Optimally begin NGS library preparation with 10 ng for each ChIP / input reaction, with the NEBNext Ultra II DNA Library Prep Kit, following the manufacturer’s protocol (https://international.neb.com/-/media/nebus/files/manuals/manuale7103-e7645.pdf?rev=de09eaf8fcdf45e0ac8a66bf6fee75fb&hash=4617FF2DC70A34462B8C24A16123353B).
-
b.Quantify libraries using the Qubit dsDNA HS fluorometric assay.CRITICAL: Minimum material required for library prep is 500 pg and maximum is 1 μg ChIP/input DNA.Note: Each ChIP-seq library that will be sequenced in the same sequencing run must be ligated with a different indexed adapter, in order to distinguish the raw fastq data of each sample.
-
c.Check the quality and size distribution of the libraries with Agilent Bioanalyzer DNA1000 (https://www.agilent.com/cs/library/usermanuals/public/G2938-90014_DNA1000Assay_KG.pdf) or HS kit (https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.com/TFS-Assets%2FLSG%2Fmanuals%2FQubit_dsDNA_HS_Assay_UG.pdf), following the manufacturer’s instructions.CRITICAL: Electropherogram of the libraries should optimally depict a distribution from 200–700 bp, with maximum peak at 250–300 bp and no visible peaks of primer dimers (∼120 bp) and adapter dimers (∼140 bp) (Figure 2). Maximum peak distribution can vary from 250–700 bp, but not exceed that range, in order for Illumina sequencing to be performed successfully.
-
d.Pool indexed libraries together in equimolar amounts.
-
e.Sequence the pool of indexed libraries at the Illumina NovaSeq 6000 sequencer.Note: Sequencing mode can vary depending on the sequencer and the sequencing cartridge used, but in general Single End, 75–100 bp read length is more than enough for ChIP-seq analysis.CRITICAL: Ideally, at least 20 Million 75–100 bp Single End Reads per sample should be obtained.
-
a.
-
75.Sequencing analysis. (Troubleshooting 5, 6)
-
a.Perform Quality Control using FastQC using the following steps:Command.>fastqc ∗.gzNote: FastQC is a quality control tool for high-throughput sequence data, which allows the performance of quality control checks on raw sequence data. Herein, we used the fastq.gz files.Assessment of results is performed with Per Base Sequence quality module of FASTQC. BoxWhisker type plot is drawn for each position (x-axis), while the y axis represents the quality scores (Q score). Ideally, mean quality should be over 28, which represents the green area of the graph. Positions with Quality score below 20 must be removed by trimming, which can be performed either from the alignment tool or from specific trimming tools like fastx_trimmer (FASTX-Toolkit, Cold Spring Harbor).
-
b.Build the genome index following the steps below.Command.>bowtie2-build mm10.fa mm10Note: Bowtie2 is a memory efficient tool for aligning the short reads to reference genomes.2 Input file should be in fasta format. Herein, the mouse genome downloaded from UCSC (mm10 version) is used to build the genome index.
-
c.Perform alignment using Bowtie2Command.>bowtie2 -p 6 -5 3 -3 3 -x mm10 -U sample.fastq.gz -Ssample.sam;Note: In this example, 6 threads are used, and every read is trimmed in 5′ and 3′ prime removing 3 bases, to increase sensitivity.
-
d.Preprocess the aligned files.Note: This step is performed with SAMtools3 and BEDtools4 commands. Both are tool packages to handle SAM, BAM and BED files. Briefly, low-quality reads, duplicates, mitochondrial DNA and blacklist region annotations are removed. The SAM file is converted to BAM file and eventually to BED file. Before every step, the BAM file is indexed and sorted. The final BED file is also sorted.
-
i.Convert SAM file to BAM file.Command.>samtools view -bS -F 4 -q 20 -@ 8 sample.sam >sample.bam
-
ii.Sort BAM file.Command.>samtools sort -@ 5 -m 4G sample.bam -o samplesort.bam
-
iii.Remove duplicate reads.Command.>samtools rmdup -s samplesort.bam sampledup.bam
-
iv.Index new file.Command.>samtools index sampledup.bam
-
v.Generate stats for file and remove Mitochondrial DNA annotations.Command.>samtools idxstats sampledup.bam | cut -f 1 | grep -vchrM | xargs samtools view -@ 5 -b sampledup.bam > xh.bam
-
vi.Index new file.Command.>samtools index xh.bam
-
vii.Remove blacklist region annotations.Command.>bedtools intersect -abam xh.bam -b mm10.blacklist.bed.gz-v > final.bam
-
viii.Index new file.Command.> samtools index final.bam
-
ix.Sort final BAM file.Command.>samtools sort -n -@ 5 -m 4G final.bam -o finalsort.bam
-
x.Convert BAM file to BED file.Command.>bamToBed -i finalsort.bam > final.bed
-
xi.Sort final BED file.Command.>sort -k1,1 -k2,2n h5.bed > finalsort.bed
-
i.
-
e.After the completion of the preprocessing for both Treatment and Input files, perform peak calling using MACS2.5
-
i.For transcription factors, the following command is used, generating short peaks.>macs2 callpeak -f BED -g mm --keep-dup all --call-summits -t IP.bed -c Input.bed -p 0.01 --bdg -npeaks
-
ii.For histone modification marks, the following command is used, generating broad peaks.>macs2 callpeak -f BED -g mm --keep-dup all -tIP.bed -c Input.bed --broad --broad-cutoff 0.1 --bdg-n peaks
-
i.
-
f.Generate bigwig files.
-
i.Subtract input signal from IP signal, using the macs2 bdgcompare tool.Command.>macs2 bdgcompare -t IP.bdg -c Input.bdg -msubstract -o IPsubstract.bdg
-
ii.Sort the generated bdg files and then convert them to bigwig with bedGraphToBigWig tool.Note: For the last step, the mm10.chrom.sizes file is needed from the UCSC. The generated files can be loaded at a Genome Viewer (herein, IGV).Command.>sort -k1,1 -k2,2n IPsubstract.bdg > IPsubsorted.bdg>bedGraphToBigWig IPsubsorted.bdg mm10.chrom.sizesIpfinal.bw
-
i.
-
g.Visualize the IP signal at selected regions (Figure 3).Note: Herein, the PPARγ and H3K27ac signal is depicted at their respective peaks with descending order in terms of signal power.
-
i.Generate matrices using the deepTools6 command computeMatrix. Reference-point mode is used, with the window of base pairs defined ± from the peak center.Command.>computeMatrix reference-point -b 5000 -a 5000--SortRegions descend -S Ipfinal.bw -R peakfile.bedNote: This command generates a matrix.mat.gz file which is used at the next command
-
ii.Generate summary and tornado plot with plotHeatmap.Command.>plotHeatmap -m matrix.mat.gz --heatmapWidth 8 --heatmapHeight 24 --dpi 300 --ColorMap ‘Blues’ -outplot.pngNote: At this step, you can add in the command the option --outFileSortedRegions which generates a list of the coordinates used for the visualization.
-
i.
-
a.
-
76.qRT-PCR.Note: This step is recommended to validate the ChIP results.
-
a.Design and test qRT-PCR primers.Note: Use primerBlast to design pairs of primers targeting the area of interest. Ideally, Tm of primers should be over 60°C and not exceeding 72°C, with minimal difference (0.5°C–1°C) between the forward and the reverse primer. Self-complementarity scores should be retained below 2. The amplicon should not exceed 150 bp to achieve maximum efficiency.
-
b.Set up qRT-PCR reactions using primers targeting the region of interest.Note: The samples to be analyzed include the eluted DNA from IP, the eluted DNA from IgG antibody, and the Input DNA.
-
i.Use 1 μL of each sample for every qRT-PCR reaction (step 72, section: immunoprecipitation and purification) as templates and perform qRT-PCR reactions using KAPA SYBR Fast Master Mix and a Bio-Rad CGC96 Real-Time System, following the manufacturer’s protocol (https://www.sigmaaldrich.com/deepweb/assets/sigmaaldrich/product/documents/418/169/sflckb.pdf).Note: Input sample can be accordingly diluted to adjust into the detection Ct value range of the primers.PCR reactions should be set in at least duplicates, and Ct values should have minimal variance across samples.
-
ii.Perform a reaction consisting of 40 cycles followed by melting curve generation, using the following PCR reaction conditions:3 min initial denaturation at 95°C.Start of Cycle.15 s at 95°C.30 s at 60°C.Fluorescence identification.End of Cycle.Melting curve step.0.5°C increments with Fluorescence identification, from 60°C to 95°C.
-
i.
-
c.Analyze qRT-PCR resultsNote: In this paper, we used the Input percentage method for the quantitation of ChIP-PCR. The Input percentage analysis shows the amount of DNA pulled down by the antibody of interest used in the IP reaction, compared to the amount of starting sample (Input sample)The formula for the calculation is:
ΔCt=CtIP - CtInput - Log2 (Dilution Factor). Dilution Factor is defined by the percentage of starting material which is used as an Input, multiplied by the times the input sample is diluted.For example, we used 10 μL out of 120 μL (thus, the Input fraction is 8.33% times of the starting material) and we performed a ten-fold dilution for the qPCR. Therefore, the dilution factor is 12 × 10 = 120 and the log2(120) = 6.9 must be subtracted from the Ct value of input.Finally, the formula is:Input percentage = 2-ΔCt ∗ 100.
-
a.
Figure 2.

Analysis of the quality and size distribution of the ChIP-seq library
(A and B) Electropherogram and (B) gel image coupled with the Agilent DNA1000 ladder for size reference. Those results were obtained using the Agilent Bioanalyzer DNA1000 kit.
Figure 3.

ChIP-seq analysis depicting the genome-wide and locus-specific binding of PPARγ and H3K27ac
ChIP-seq was performed using the iWAT of WT mice (A) Tornado plot depicting PPARγ DNA binding and H3K27ac distribution throughout their respective peaks (±3 kb from peak center) and (B) chromatin binding of PPARγ and H3K27ac modification at the promoters of Pdk4, Tmem245, Dnajp1 and Cbx7. The relative length of the represented genomic loci is also indicated.
Expected outcomes
ChIP-ed and purified genomic fragments are expected to yield accurate and high-quality ChIP-seq data. We have used this protocol to identify the genome-wide occupancy of PPARγ, which is considered to be an abundantly expressed transcription factor in iWAT depots and the distribution of H3K27ac, a histone modification indicating transcriptionally active chromatin elements. The conditions described herein result in proper chromatin fragment length (Figure 1) and in sufficient quantities to be used for both sequencing and ChIP-PCR assays, thus surpassing both the problems of tissue handling and low cell number. This approach results in high quality libraries (Figure 2) and eventually in creating datasets to perform the wide range of ChIP-seq analysis pipeline like Tornado plots (Figure 3A) and bigwig files (Figure 3B) to inspect the binding of the target of interest in selected loci. These results are in agreement with ChIP-PCR assays (Figure 4), thus providing the opportunity, at least for some cases, to skip the sequencing step and proceed immediately to the study of specific targets throughout the genome, which are either previously known from other studies or established among research groups.
Figure 4.
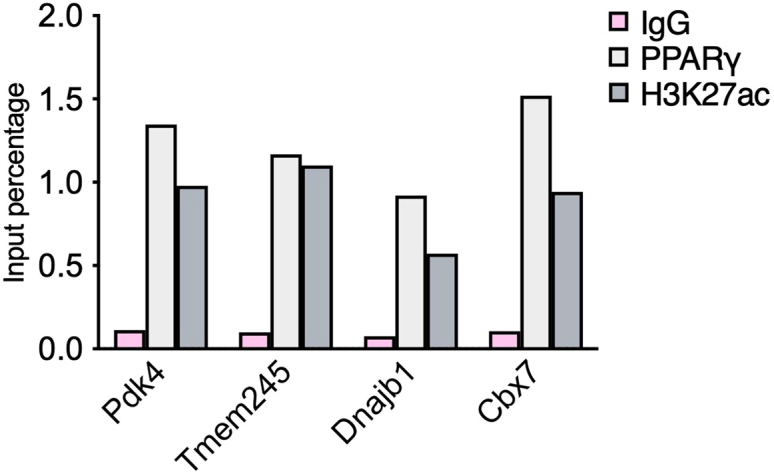
An example of qPCR analysis of chromatin immunoprecipitated samples using antibodies against PPARγ and H3K27ac in iWAT of WT mice
Results were obtained using sets of primers that target the promoters of Pdk4, Tmem245, Dnajp1 and Cbx7 genes. Signal is expressed as input percentage. Data are presented as means, N = 2.
Limitations
This protocol has been used to carry out successful ChIP-seq experiments for several transcription factors and histone modifications. However, the specificity and the quality of the antibodies used in the immunoprecipitation reactions are of paramount importance for the success of this technique. Higher amount of starting material may be required for proteins expressed at low levels.
We have obtained consistent results from chromatin shearing of iWAT of C57BL6 under several conditions (e.g., genetic manipulation, high fat diet feeding),1 while we have no experience using other inguinal fat tissue from other species or other depots of white adipose tissue of mice. We anticipate that this protocol could be applied to mouse gonadal WAT and white adipose depots isolated from other mammals. However, further optimization of the chromatin shearing conditions may be required.
Troubleshooting
Problem 1
Fresh tissue samples are not immediately available (Related to section: “tissue preparation and fixation”, step: 1).
Potential solution
Snap-frozen tissues could also be used in this protocol. However, it is crucial to thaw them on ice avoiding high temperature in tissue samples and thus, preventing sample degradation by proteases.
Problem 2
Concentration of the fragmented chromatin is too low (Related to section: “quality validation and quantification of sheared chromatin”, step: 42).
Potential solution
Immunoprecipitation could be successfully achieved using lower amounts of sheared chromatin.1 However, in case that less than 15 μg of DNA is quantified following the sonication, ensure that the disassociation and lysis steps were performed as described in this protocol and increase the starting material. Alternatively, the fixation time can be increased to form more stable DNA-protein interactions or a greater amount of antibody can be used.
Problem 3
Chromatin is under-fragmented and fragments are too large (Related to section: “quality validation and quantification of sheared chromatin”, step: 43).
Potential solution
Sonication conditions to generate fragments in the range of 250–500 bp could vary in case that a different sonication set up is used. In this case, it is highly recommended to perform shearing efficiency test experiments before you proceed with the immunoprecipitation. To optimize chromatin fragmentation, during sonication remove 10 μL aliquots of chromatin at various time points. Add 90 μL of immunoprecipitation buffer, spin-down for 10 min using a microfuge, transfer supernatant to new eppendorf tube and proceed with reverse cross-linking, RNAse/proteinase K treatment and purification of DNA (section: quality validation and quantification of sheared chromatin). Alternatively, fixation time can be reduced to 10 min and sonication parameters in Covaris software can be altered to produce more powerful sonication conditions, followed by titration and agarose gel analysis.
Problem 4
No DNA detected following the immunoprecipitation. (Related to section: immunoprecipitation and purification, step: 72).
Potential solution
Increase the amount of chromatin used for every reaction. Check the suitability and the quality of DNA purification Dynabeads. Decrease the number of washes during IP step. Use another antibody with greater specificity and sensitivity.
Problem 5
No enrichment is obtained following the sequencing. (Related to section: “downstream assays for analysis”, step 75).
Potential solution
Increase the amount of antibody used for IP. Decrease the number of washes during IP. Decrease the concentration of NaCl, Sodium-deoxycholate and LiCl at the wash buffer during IP. Increase the amount of chromatin per reaction.
Problem 6
Noisy signal after ChIP sequencing (Related to section: “downstream assays for analysis”, step 75).
Potential solution
Increase the number of washes during the IP procedure. Increase the concentration of NaCl, Sodium-deoxycholate and LiCl at the wash buffer. Increase the amount of incubation time of every washing buffer during washing steps of IP. Decrease the amount of shearing time at the sonication step.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Andreas Papapetropoulos (apapapet@pharm.uoa.gr).
Materials availability
This study did not generate any unique reagents.
Data and code availability
This study did not generate new code. Raw ChIP-seq data can be obtained from the lead contact.
Acknowledgments
The authors would like to thank the Greek Genome Center of the Biomedical Research Foundation of the Academy of Athens for carrying out the sequencing experiments. This work was co-financed by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – CREATE – INNOVATE (project code: T2EΔK-00843, to A.P.).
Author contributions
A.K. and D.V. optimized the protocol and wrote the manuscript. G.V. performed the sequencing experiments. A.P. and D.T. supervised the work and provided resources and funding. All authors reviewed and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
References
- 1.Katsouda A., Valakos D., Dionellis V.S., Bibli S.I., Akoumianakis I., Karaliota S., Zuhra K., Fleming I., Nagahara N., Havaki S., et al. MPST sulfurtransferase maintains mitochondrial protein import and cellular bioenergetics to attenuate obesity. J. Exp. Med. 2022;219 doi: 10.1084/jem.20211894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinlan A.R., Hall I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate new code. Raw ChIP-seq data can be obtained from the lead contact.