

Summary
Plasma extracellular vesicles (EVs) represent a potential resource for biomarkers of multiple diseases. Here, we present a protocol for obtaining EVs from human plasma using asymmetrical flow field-flow fractionation technology. We describe steps for using tandem mass tags to perform comparative proteomic studies of a large clinical cohort. We then detail targeted quantitative analysis of differential proteins based on a parallel reaction monitoring technique.
For complete details on the use and execution of this protocol, please refer to Wu et al. (2020)1 and Li et al. (2023).2
Subject areas: Cell Biology, Cell Isolation, Cell Separation/Fractionation, Health Sciences, Clinical Protocol, Molecular/Chemical Probes, Proteomics, Protein Expression and Purification, Mass Spectrometry
Graphical abstract
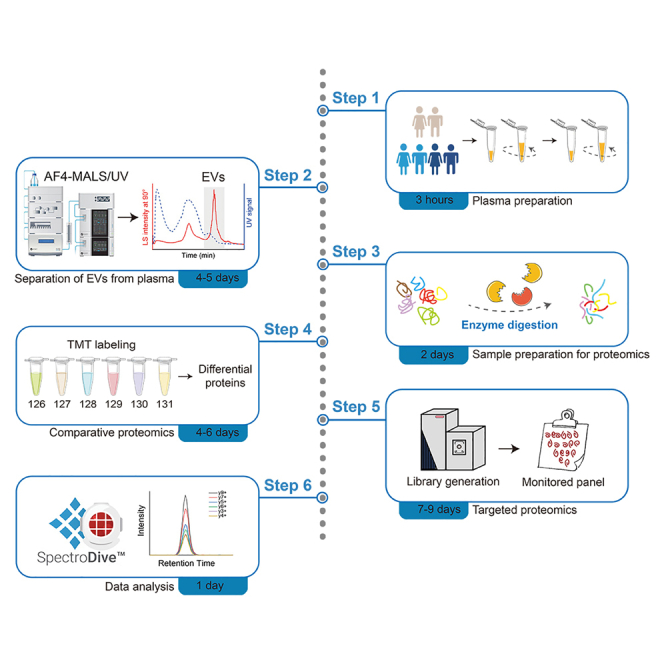
Highlights
-
•
Isolation of extracellular vesicles by asymmetrical flow field-flow fractionation
-
•
Detailed procedures of parallel reaction monitoring based on SpectroDive
-
•
An effective workflow for high-throughput screening of potential biomarkers
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Plasma extracellular vesicles (EVs) represent a potential resource for biomarkers of multiple diseases. Here, we present a protocol for obtaining EVs from human plasma using asymmetrical flow field-flow fractionation technology. We describe steps for using tandem mass tags to perform comparative proteomic studies of a large clinical cohort. We then detail targeted quantitative analysis of differential proteins based on a parallel reaction monitoring technique.
Before you begin
The AF4 technique for EVs isolation described in this scheme minimizes the used volume of plasma and experimental procedures and ensures the purity of plasma EVs. Therefore, it is suitable for clinical studies. The EVs isolated using this method can also be used for other subsequent studies. In addition, quantitative proteomics techniques based on TMT labeling and PRM are also applicable to studies of other sample types.
Institutional permissions
Written informed consent was obtained from the parents or legal guardians of patients under the age of consent. This study was approved by the Ethics Committee of The First Affiliated Hospital of Guangxi Medical University.
The AF4 system and instrument
The AF4 system used in this protocol is Eclipse 3 (Wyatt Technology, Germany). It consists of Agilent 1260 pump, on-line vacuum degasser, 280 nm UV detector, multi-angle light scattering (MALS) detector, and AF4 channel. In order to reduce impurities, a filter with 0.1 μm PVDF membrane is placed between the pump and the AF4 separation channel. The channel is equipped with a 10 kDa MWCO regenerated cellulose (RC) membrane (Milipore, USA) on the accumulation wall. The length of channel is 152 mm with an entrance width of 21.5 mm and an export of 3 mm. All data collection is addressed with Astra (version 5.3.4.20, Wyatt Technology).
Preparation of the AF4 instrument
Timing: 1 day
-
1.Preparation for the separation channel of AF4, see Figure 1.
-
a.Immerse a new RC membrane in deionized water for 12–18 h to activate it.
-
b.Replace the new membrane into the AF4 channel.
-
a.
CRITICAL: Hold the edge of the RC membrane with a tweezer, do not touch the inside of the membrane to avoid damaging it.
Note: The rough surface of the membrane should face the bottom wall of the channel.
-
2.Set the parameters of AF4.
-
a.Open the online software ChemStation instrument 1 and turn on the MALS and UV lights.
- b.
-
c.Click the “Instrument” on the menu → “More Wyatt Eclipse” → “Control” to open the flow control menu.
-
d.Wash the AF4 system for about 20 min by switching the “Focus Mode” and “Elution Mode”.
-
a.
Optional: When replacing a new RC membrane, one can load a sample of 5% BSA to coat it.
Figure 1.

Preparation for the AF4 channel
(A) Immerse a new 10 kD RC membrane in deionized water overnight.
(B) Replace the new membrane into the channel.
Table 1.
The flow rate of each control
| Time (min) | 0–1 | 1–2 | 2–7 | 7–9 | 9–14 | 14–29 | 29–49 | 49–54 |
|---|---|---|---|---|---|---|---|---|
| Detector flow | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Focus flow | 0 | 1.5 | 1.5 | 1.5 | 0 | 0 | 0 | 0 |
| Injection flow | 0 | 0 | 0.2 | 0 | 0 | 0 | 0 | 0 |
| Cross flow (Vx) | 0 | 0 | 0 | 0 | 3 | 3→1 | 1→0.1 | 0.1→0 |
Figure 2.

The control interface of the software shows the flow rate of each control during the separation of plasma EVs
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APOA1 (1:1000) | Abcam | Cat# ab52945 |
| APOB (1:1000) | Abcam | Cat# ab20737 |
| Albumin (1:1000) | Abcam | Cat# ab207327 |
| CD9 (1:1000) | Abcam | Cat# ab92726 |
| CD81 (1:1000) | Abcam | Cat# ab109201 |
| CD63 (1:1000) | Abcam | Cat# ab134045 |
| HSP90 (1:1000) | Abcam | Cat# ab34909 |
| TSG101 (1:1000) | Abcam | Cat# ab125011 |
| Goat anti-Rabbit IgG (H&L) [HRP] (1:10000) | Aksomics | Cat# KC-RB-035 |
| Goat anti-Mouse IgG (H&L) [HRP] (1:10000) | Aksomics | Cat# KC-MM-035 |
| Biological samples | ||
| Human plasma | The First Affiliated Hospital of Guangxi Medical University | This paper |
| Chemicals, peptides, and recombinant proteins | ||
| Iodoacetamide (IAM) | Sigma-Aldrich | Cat# I1149 |
| DL-Dithiothreitol (DTT) | Sigma-Aldrich | Cat# D9163 |
| Urea | Sigma-Aldrich | Cat# 51456 |
| Sequencing-grade modified trypsin, frozen | Promega | Cat# V5113 |
| Lysyl endopeptidase | WAKO | Cat# 125-05061 |
| Hydroxylamine solution | Sigma-Aldrich | Cat# 467804 |
| Methanol | J.T.Baker | Cat# 9093-68 |
| Acetonitrile | J.T.Baker | Cat# 9017-03 |
| Disodium hydrogen phosphate (Na2HPO4) | Sinopharm Chemical Reagent Co.,Ltd | Cat# XW755879402 |
| Sodium dihydrogen phosphate dihydrate (NaH2PO4·2H2O) | Xilong Scientific | Cat# 13472-35-0 |
| Sodium chloride (NaCl) | Sigma-Aldrich | Cat# S9888 |
| Tween-20 | Sinopharm Chemical Reagent Co.,Ltd | Cat# 30189328 |
| Nonfat dried milk powder | PanReac Application | Cat# A0830 |
| NH4HCO3 | Sigma-Aldrich | Cat# A6141 |
| 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) | Scientific Chemical | Cat# SH40067 |
| Tris | Sigma-Aldrich | Cat# 252859 |
| Formic acid (FA) | Fisher Scientific | Cat# A117-50 |
| 0.1% Formic acid (v/v) in acetonitrile, LC-MS Grade | Fisher Scientific | Cat# 85174 |
| 0.1% Formic acid (v/v) in water, LC-MS Grade | Fisher Scientific | Cat# 85170 |
| Ammonium hydroxide solution (NH4OH) | Sigma-Aldrich | Cat# 221228-500ML-PCA |
| Immobilon Western HRP Substrate | Millipore | Cat# WBKLS0500 |
| NuPAGE™ MOPS SDS running buffer (20×) | Invitrogen | Cat# NP0001 |
| NuPAGE™ MES SDS running buffer (20×) | Invitrogen | Cat# NP0002 |
| PageRuler Plus Prestained Protein Ladder | Thermo Scientific | Cat# 26619 |
| Critical commercial assays | ||
| TMT 6-plex reagents | Thermo Fisher Scientific | Cat# 90061 |
| The indexed retention time standards (iRT) peptides kit | Biognosys | Cat# Ki-300-2 |
| Pierce BCA kit | Thermo Scientific | Cat# 23227 |
| Pierce Quantitative Colorimetric Peptide Assay | Thermo Scientific | Cat# 23275 |
| Deposited data | ||
| Mass spectrometry data | http://www.proteomexchange.org | PXD034548 |
| Software and algorithms | ||
| Proteome Discoverer Version 2.2.0.388 | Thermo Fisher Scientific | N/A |
| Xcalibur | Thermo Fisher Scientific | N/A |
| SpectroDive 10.4 | Biognosys | N/A |
| ChemStation Instrument 1 online | Agilent Technology | N/A |
| ASTRA Version 5.3.4 | Wyatt Technology | N/A |
| Skanit RE for Varioskan Flash Version 2.4.5 | Thermo Scientific | N/A |
| Other | ||
| AF4 system Eclipse 3 | Wyatt Technology | N/A |
| Ultraviolet detector | Agilent Technology | N/A |
| Multi-angle light scattering (MALS) detectors | Wyatt Technology | https://www.wyatt.com/zh-hans/products/instruments/dawn-multi-angle-light-scattering-detector.html |
| Differential refraction detector | Wyatt Technology | N/A |
| High-speed refrigerated centrifuge | Thermo Scientific | N/A |
| Varioskan Flash spectral scanning multimode reader | Thermo Scientific | N/A |
| 10% NuPAGE Bis-Tris Gels | Thermo Scientific | Cat# NP0301BOX |
| 12% NuPAGE Bis-Tris Gels | Thermo Scientific | Cat# NP0341BOX |
| Immobilon-P membrane (PVDF membrane) | Millipore | Cat# IPVH00010 |
| Ultrafiltration centrifuge tube 10kD, 4ML | Millipore | Cat# UFC801096-1 |
| Regenerated cellulose membrane 10kD | SuperOn Technology | Cat# R7PA81151 |
| Visiprep™ SPE Vacuum Manifold standard | Supelco | Cat# 57030-U |
| Eppendorf ThermoMixer F1.5 | Eppendorf | https://www.eppendorf.com/gb-en/eShop-Products/Temperature-Control-and-Mixing/Instruments/Eppendorf-ThermoMixerF-p-5384000039 |
| Oasis HLB 1 cc Vac cartridge, 10 mg | Waters | Cat# 186000383 |
| ZipTip with 0.2 μL C18 resin | Waters | Cat# ZTC18M960 |
| Autosampler vial | Thermo Scientific | Cat# C5000-87 |
| XBridge peptide BEH C18 column (130 Å, 5 μm, 4.6 × 250 mm) | Waters | Cat# 186003565 |
| C18 trap column (Reprosil-Pur C18 AQ, 5 μm) | Dr. Maisch | https://dr-maisch.com/dr-maisch-phases/reprosil-pur/reprosil-pur-120-c18-aq |
| Polymicro Flexible Fused Silica Capillary Tubing | Polymicro Technologies | https://www.molex.com/zh-cn/products/part-detail/1068150023 |
| C18 capillary column (Reprosil-Pur C18 AQ, 3 μm) | Dr. Maisch | https://dr-maisch.com/dr-maisch-phases/reprosil-pur/reprosil-pur-120-c18-aq |
| Polymicro Flexible Fused Silica Capillary Tubing | Polymicro Technologies | Cat# 1068150019 |
| P-2000 Laser-Based Micropipette Puller | Sutter instrument | https://www.sutter.com/MICROPIPETTE/p-2000.html |
| SevenCompact™ S210 | Mettler Toledo | Cat# 30130862 |
| FB-10T filter flask | AUTOSCIENCE | Cat# FB-10T |
| AP-01P vacuum pump | AUTOSCIENCE | Cat# AP-01P |
| Ultrasonic cleaner | Scientz | Cat# SB-5200D |
| Vacuum concentrator system | Labconco | Cat# 7310038 |
| Carbon-coated copper grids | Zhongjingkeyi Technology Co., Ltd | Cat# BZ110223a |
| 96-well plate | BRAND | Cat# BR781602 |
| Rigol L-3000 HPLC system | RIGOL | N/A |
| Nanodrop | Thermo Scientific | Cat# 2000 |
Materials and equipment
Phosphate buffer saline (PBS) buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Sodium chloride (NaCl) | 150 mM | 9.0 g |
| Disodium hydrogen phosphate (Na2HPO4) | 8 mM | 1.15 g |
| Sodium dihydrogen phosphate dihydrate (NaH2PO4·2H2O) | 2 mM | 0.3 g |
| ddH2O | N/A | 1 L |
| Total | N/A | 1 L |
Note: The PBS buffer is filtered with a 0.22 μm filter membrane and treated with sonication to eliminate air bubbles. The PBS buffer should be stored at 4°C before use.
Other important reagents
-
•
1 M dithiothreitol (DTT): add 0.154 g DTT in 1 mL ddH2O. After vortex, partition the solution into 20 μL/tube and store at −20°C for up to two years.
-
•
1 M iodoacetamide (IAM): add 0.185 g DTT in 1 mL ddH2O. After it is completely dissolved, the solution is divided into 40 μL per tube and store at −20°C for up to two years.
Note: This solvent is difficult to dissolve and requires vortex shock and sonication for 5 min.
-
•
100 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 8.5: add 0.238 g HEPES in 10 mL ddH2O, then adjust the pH to 8.5 with 10 M NaOH. Store at 4°C for up to 2 years.
-
•
0.1 μg/μL Lys C solution: take it from −80°C. After centrifugation, carefully open the bottle cap and add 200 μL ddH2O. Store the solution at −80°C.
-
•
10 × indexed retention time standards (iRT) solution: add 50 μL dissolution buffer to the iRT standard tube (iRT peptides kit, Biognosys, Switzerland). Vortex to mix, sonicate 5 min and store the 10 × iRT solution at 4°C and use within 12 weeks.
-
•
10 × Tris Buffered Saline (TBS): dissolve 121.14 g Tris-base, 87.66 g NaCl in 900 mL ddH2O, then adjust the pH to 7.5 with HCl and add ddH2O to 1 L. Store at 4°C.
-
•
1 × TBST: 100 mL 10 × TBS, 900 mL ddH2O, add 1 mL Tween-20. Store at 20°C–25°C.
-
•
5% w/v milk: dissolve 2.5 g non-fat dry milk into 1 × TBST for a final volume of 50 mL. Prepare this solution fresh.
-
•
2% acetonitrile (ACN)/0.1% ammonium hydroxide (NH4OH): 20 mL ACN, 980 mL ddH2O, add 1 mL NH4OH. Treat with sonication to eliminate air bubbles and store at 20°C–25°C for up to half a year.
-
•
98% ACN/0.1% NH4OH: 980 mL ACN, 20 mL ddH2O, add 1 mL NH4OH. Treat with sonication to eliminate air bubbles and store at 20°C–25°C for up to half a year.
Step-by-step method details
Plasma sample preparation for EVs isolation
This section describes the pretreatment steps before plasma EVs separation using the AF4 method. These steps could reduce the stickiness of plasma and remove cells or large debris in advance.
-
1.
Take the plasma from −80°C and thaw it at 4°C.
-
2.
Dilute 200 μL of the plasma with PBS at a 1:1 v/v ratio.
-
3.
Centrifuge the sample at 300 × g for 10 min at 4°C to remove intact cells.
-
4.
Carefully transfer the supernatant into a new 1.5 mL tube.
-
5.
Centrifuge the supernatant at 4200 × g for 20 min at 4°C to remove large cell debris, apoptosis body, and platelet.
-
6.
Carefully transfer the supernatant into a new 1.5 mL tube for the following enrichment of EVs.
Pause point: The supernatant can be stored at −80°C for several weeks.
Note: It is recommended to freeze and thaw the supernatant only once.
Isolation of plasma EVs using AF4 method
EVs are separated using AF4 according to the size differences. The real-time signals recorded in ASTRA are the instructions for effluent collection. The protein concentration of EVs fraction is determined by BCA assay and the enriched EVs are also characterized by Western blot and transmission electron microscopy (TEM).
-
7.
Open the monitoring software, ASTRA (version 5.3.4).
-
8.
Right-click on the “Experiments” and create a new template for the observation of signals.
-
9.
Set a duration time and check “LS 11”, “UV”, “dRI” on the right.
-
10.
Click the blue triangle on the control menu to start the sample injection.
-
11.
Suck up 300 μL of the prepared sample solution using a needle and insert the needle into the injector.
-
12.
Rotate the injector to "Load", then inject the sample solution and rotate the injector to "Inject".
-
13.
Collect the peak 4 effluent according to the signal showing on the collection panel including UV 280, LS 11, dRI, see Figure 3.
Note: The system needs to be washed for about 20 min before loading the next sample.
-
14.
Concentrate the collected effluent to about 150 μL using a 3 kDa MWCO membrane filter at 3000 × g for 3 h at 4°C.
Optional: Alternatively, concentrate with a 10 kDa MWCO membrane filter for about 30 min.
-
15.Quantify the protein concentration using a Pierce BCA Protein Assay Kit following the manufacturer’s instruction, please visit https://www.thermofisher.cn/document-connect/document-connect.html?url=https://assets.thermofisher.cn/TFS-Assets%2FLSG%2Fmanuals%2FMAN0011430_Pierce_BCA_Protein_Asy_UG.pdf.
-
a.Dilute the EVs sample twice with 10% SDS solution, then heat it at 95°C for 10 min.
-
b.Prepare appropriate amount of the BCA working reagent by mixing 50 parts of Reagent A with 1 part of Reagent B (A:B = 50:1, v/v). Calculate the volume required for Reagent A and Reagent B according to the following formula:Note: Three technical replicates are usually performed for each sample and standard. In addition, more working reagents are prepared in case of shortage. m is the number of extra samples, and 3–5 are recommended. The R2 value of the standard curve is required to be larger than 0.99.
-
c.Add 200 μL of the working reagent to each well of the 96-well plate.
-
d.Pipette 4 μL of water, 7 different concentrations of bovine serum albumin (BSA) standard solution, and EVs samples with triplicates into the 96-well plate successively.Note: BSA concentrations of 0 μg/μL, 0.125 μg/μL, 0.25 μg/μL, 0.5 μg/μL, 0.75 μg/μL, 1.0 μg/μL, 1.5 μg/μL, and 2.0 μg/μL are used to make the standard curve.CRITICAL: It is recommended to carefully pipet the sample up and down gently for several times to mix well.Optional: Instead of diluting the BSA standard (2 mg/mL) into seven concentrations according to the manufacturer protocol, one can use the protein standards available as a prediluted series of seven concentrations, such as Bio-Rad Cat#5000207.
-
e.Incubate the plate at 37°C for 30 min. After this, measure the absorbance values of samples at 562 nm.Note: After incubation, there may be air bubbles in sample wells. Use the tip of 10 μL pipette to pierce the air bubbles to prevent interference with quantification.
-
f.Calculate the protein concentration of each sample according to the standard curve.
-
a.
-
16.Western blot analysis of enriched EVs, see Figure 4A.
-
a.Separate 15 μg of proteins on SDS-PAGE gel at a constant voltage of 200 V.Note: Choose gel concentration according to the size of the target protein. Use 10% gel for proteins larger than 60 kDa and 12% gel for proteins less than 60 kDa.
-
b.Obtain the target protein stripe by cutting the gel. Try to keep the target protein in the center of the stripe.
-
c.Immerse the PVDF membrane in 100% methanol for 30 s to activate it.
-
d.Transfer the protein to the PVDF membrane at a constant current of 200 mA.Note: For apolipoprotein B-100 (APOB, 516 kDa), add 1% SDS to the transfer buffer and apply a constant current of 40 mA for 12–18 h transfer.
-
e.Wash the PVDF membrane using 1 × TBST for 5 min then block the membrane with 5% milk for at least 2 h.
-
f.Incubate the membrane with primary antibody (diluted with 5% milk in the ratio of 1:1000).
-
g.Wash the membrane using 1 × TBST for 10 min for 3 times.
-
h.Incubate the membrane with secondary antibody (diluted with 5% milk in the ratio of 1:10000)
-
i.Wash the membrane using 1 × TBST for 10 min for 3 times.
-
j.Mix the developer solution A and B in a ratio of 1:1, then drip the mixed solution to the membrane and develop the membrane.
-
a.
-
17.Prepare the EV sample for TEM analysis.
-
a.Put the carbon-coated copper grids into the glow instrument.
-
b.Vacuumize the instrument for 4 min.
-
c.Charge the carbon-coated copper grids with glow for 30 s.
-
d.5 μL of the EV sample is placed onto the coated side of the grid and allowed to adsorb for 1 min. Absorb away the drop using filter paper.
-
e.Wash the copper grid with the negative staining solution (2% uranyl acetate) and absorb away the drop using filter paper.
-
f.Add 5 μL of negative staining solution to the sample side of the grid and incubate for 1 min to stain the background around the EVs.
-
g.Air dry the grid and TEM imaging, see Figure 4B.
-
a.
Figure 3.

Signal recording during the sample separation
Blue: dRI. Red: MALS. Green: UV280.
Figure 4.

Characterization of separated EVs from human plasma using AF4 method
(A) Western blot analysis of EVs and whole plasma (each n=2) with a 10 μg protein loading volume.
(B) Transmission electron microscopy micrographs of EVs. Scaled bars: 100 nm, left; 200 nm, right. (A) and (B) are adapted with permission from Figure S1 (B) and Figure 1 (B) of Li et al.2 respectively.
Sample preparation for proteomic analysis
In this section, proteins are digested into tryptic peptides prior to the mass spectrometry (MS) analysis.
-
18.
Solubilize a total of 50 μg protein in a final concentration of 8 M urea/50 mM NH4HCO3, and reduce it with a final concentration of 20 mM DTT for 1 h at 37°C.
-
19.
For alkylation, add a final concentration of 40 mM IAM to the sample, and incubate the mixture in the dark for 45 min at 20°C–25°C.
-
20.
Add Lys C at an enzyme/protein ratio of 1:50 (w/w) for 3 h at 37°C.
-
21.
Dilute the sample with 25 mM NH4HCO3 to a final concentration of <1.5 M urea.
-
22.
Add trypsin at an enzyme/protein ratio of 1:50 (w/w). Incubate the sample in a ThermoMixer F1.5 for heating and mixing at 37°C for 12–18 h.
-
23.
Use a final concentration of 0.1% formic acid (FA) to stop the protein digestion.
-
24.Desalt the sample with an Oasis HLB 1 cc Vac cartridge using a vacuum manifold. The detailed procedure is as follows:
-
a.Wash, 1 mL methanol and 1 mL 75% acetonitrile (ACN)/0.1% FA.
-
b.Equilibrium, 1 mL 0.1% FA for twice.
-
c.Loading the sample solution for 3–4 times.
-
d.Wash, 1 mL 0.1% FA for twice.
-
e.Elution, 400 μL of 20% ACN/0.1% FA, 40% ACN/0.1% FA, and 60% ACN/0.1% FA successively.
-
a.
-
25.
Dry the sample in a centrifugal vacuum concentrator (Labconco, USA).
Tandem mass tags (TMT)-based quantitative proteomics
This section includes TMT labeling, detection of labeling efficiency, adjustment of sample mixing ratio, and high pH reversed-phase HPLC fractionation.
-
26.
Redissolve the desalted peptides in 50 μL of 100 mM HEPES (pH 8.5).
Optional: Measure the peptide concentration using Nanodrop or Pierce quantitative colorimetric peptide assay.
-
27.
Equilibrate the TMT6-plex label reagents to 20°C–25°C for about 30 min.
Optional: Alternatively, other plex isobaric mass tags are also available.3 For example, 16-plex TMT reagents4 are available commercially, and even 29-plex can be achieved by combinatorial use of labelling channels.5
-
28.
Label the 50 μL peptides with a TMT6-plex kit according to the manufacturer’s protocol, please visit https://www.thermofisher.cn/document-connect/document-connect.html?url=https://assets.thermofisher.cn/TFS-Assets%2FLSG%2Fmanuals%2FMAN0011639_TMT_Mass_Tagging_Reag_UG.pdf.
Note: TMT labeling at a final concentration of 25%–30% ACN is recommended.
-
29.Calculate the labeling efficiency.
-
a.Mix 2 μL of each labeled sample. After drying, redissolve the sample in 10 μL of 0.1% FA.
-
b.Desalt the sample using μC18 ZipTip, then dry again and redissolve the sample with 15 μL of 0.1% FA.
-
c.Centrifuge the sample at 20,000 × g for about 30 min, and transfer the supernatant into a new autosampler vial.
-
d.Load sample into an EASY-nLC 1000 HPLC system coupled to Nanospray Flex Ion Source and Q Exactive Mass Spectrometer.
-
e.Elute peptides in an in-house packed C18 trap column (100 μm × 2 cm, Reprosil-Pur C18 AQ, 5 μm, Dr. Maisch GmbH, Germany), and then separate peptides on an in-house packed C18 capillary column (75 μm × 20 cm, Reprosil-Pur C18 AQ, 3 μm, Dr. Maisch GmbH, Germany) using mobile phase A (0.1% FA) and B (ACN/0.1% FA) at a flow rate of 310 nL/min with the following 78-min gradient:
-
i.4%–10% solvent B for 5 min.
-
ii.10%–22% solvent B for 50 min.
-
iii.22%–32% solvent B for 15 min.
-
iv.32%–90% solvent B for 1 min.
-
v.90% solvent B for 7 min.
-
i.
-
f.Operate Q Exactive in data-dependent MS/MS mode using 20 most intense precursors detected in survey scan from 300 to 1600 m/z performed at 70,000 resolution. Also include as settings:
-
i.Select precursor ions with an isolation width of 2 m/z and the dynamic exclusion time of 50 s for fragmentation with normalized collision energy of 27%.
-
ii.Acquire MS/MS spectra at a resolution of 17,500 at m/z 200 and set the target value as 5e4 with a maximum injection time of 80 ms.
-
i.
-
g.Process the obtained raw data in Proteome Discoverer 2.2 using a regular workflow.Search the raw data against the Uniprot human protein database together with a common contaminant list (download from http://www.coxdocs.org/doku.php?id=maxquant:start_downloads.htm) using the Sequest HT search engine for protein identification. Set other parameters as follows.
-
i.Select trypsin as the enzyme and allow two miss cleavage.
-
ii.Set the mass tolerance of precursor and product ions to 10 ppm and 0.02 Da, respectively.
-
iii.Specify carbamidomethylation on cysteine as fixed modifications; specify TMT-labeling (lysine and peptide N-terminus), methionine oxidation, and acetylation of the N-terminus as variable modifications.
-
iv.Set the false discovery rate (FDR) to 1% for both protein and peptide identifications.
-
v.Remove contaminant proteins before downstream analysis.Note: In this step, the TMT labeling of lysine and peptide N-terminus should be designated as dynamic modifications, not fixed modifications.
-
i.
-
h.Calculate the labeling efficiency of TMT on peptides.CRITICAL: The labeling efficiency should be higher than 95%.
-
a.
-
30.
Calculate the total signal intensity at peptide or peptide spectral matches (PSMs) levels. Then, adjust the mixing ratio of each sample making the total signal intensity of each sample equally.
-
31.
Combine the labeled samples according to the adjusted mixing ratio of each sample.
-
32.
Dry the mixture in a centrifugal vacuum concentrator.
-
33.
Redissolve the mixture with 200 μL of 0.1% FA.
-
34.
Desalt the mixture using an Oasis HLB 1 cc Vac cartridge.
-
35.
Dry the elution in a centrifugal vacuum concentrator.
-
36.
Redissolve the dried elution with 100 μL of buffer A (2% ACN/0.1% NH4OH).
-
37.
Centrifuge the sample at 20,000 × g for 20 min, then transfer the supernatant into a new tube.
-
38.Fractionate the sample into 10 fractions using high pH reversed-phase HPLC on a RIGOL L-3000 series HPLC system (RIGOL, China). Inject the sample onto a C18 column (130 Å, 5 μm, 4.6 × 250 mm, Waters, USA) and elute it using buffers A and B (98% ACN/0.1% NH4OH) at a constant flow rate of 0.7 mL/min with a 76 min gradient as follows:
-
a.5%–8% solvent B for 5 min.
-
b.8%–18% solvent B for 35 min.
-
c.18%–32% solvent B for 22 min.
-
d.32%–95% solvent B for 2 min.
-
e.95% solvent B for 4 min.
-
a.
Note: All fractions are collected at 90 s intervals and concatenated into 10 post-fractions.
Optional: Alternatively, other fractionation methods, such as strong cation exchange10 and off-gel electrophoresis fractionation,11 and Dionex UltiMate 3000 HPLC system (Thermo Fisher Scientific, USA) are also available.
-
39.
Dry all fractions in a centrifugal vacuum concentrator.
-
40.
Redissolve each sample with 50 μL of 0.1% FA.
-
41.
Centrifuge samples at 20,000 × g for 30 min, and transfer supernatants into new autosampler vials.
-
42.
Analyze the fractionated peptide samples using LC-MS/MS, see detailed parameters in step 29d-f.
Optional: Several approaches are available to alleviate the ratio compression problem of the isobaric labeling methods, such as synchronous precursor selection (SPS)12 and real-time search (RTS)13 acquisition methods.
-
43.
Search the raw data using Proteome Discoverer 2.2. Designate the TMT labeling of lysine and peptide N-terminus as fixed modifications. For details about other parameters, see step 29g.
Note: The scaled abundance of proteins is used for quantitative analysis.
Targeted proteomics, parallel reaction monitoring (PRM)
After identified from comparative proteomics, the differential proteins are verified using PRM. This section contains the generation of spectral library and assay panel. Spectra are acquired with an Orbitrap Eclipse Tribrid mass spectrometer coupled with an EASY-nLC 1200 HPLC system (Thermo Fisher Scientific). The criteria for selecting quantotypic target peptides and typical values of instrument parameters used in PRM experiments have been reported in detail by Rauniyar14 and Gallien et al.15
-
44.
Select proteins from differential proteins for the following monitoring.
-
45.For peptide quantitation, Pierce quantitative colorimetric peptide assay is used according to the manufacturer’s protocol, please visit https://www.thermofisher.cn/document-connect/document-connect.html?url=https://assets.thermofisher.cn/TFS-Assets%2FLSG%2Fmanuals%2F23275_quantpeptide_color_UG.pdf.
-
a.Tryptic peptides digested from 50 μg protein are redissolved with 100 μL 0.1% FA, of which 50 μL is transferred to a new tube and 50 μL 0.1% FA is added to dilute the peptide concentration.
-
b.Prepare a dilution series of the peptide standard to generate a standard curve.
-
c.Prepare appropriate amount of working reagent by mixing 50 parts of Reagent A with 48 part of Reagent B and 2 part of Reagent C (A:B:C = 50:48:2, volume ratio). After vortex and mixture, add 180 μL working reagent to each well of the 96-well plate.
-
d.Pipette 20 μL of water, 7 different concentrations of peptide standard, and peptide samples with triplicates into the 96-well plate successively.Note: Peptide concentrations of 0 μg/μL, 0.0156 μg/μL, 0.0313 μg/μL, 0.0625 μg/μL, 0.125 μg/μL, 0.25 μg/μL, 0.5 μg/μL, and 1.0 μg/μL are used to make the standard curve.
-
e.Incubate the plate at 22°C for 30 min or 37°C for 15 min.
-
f.Measure the absorbance value at 480 nm using a Varioskan Flash spectral scanning multimode reader (Thermo Fisher Scientific, USA) and calculate the peptide concentration of each sample according to the standard curve.
-
a.
-
46.Acquire the MS data using data-dependent acquisition mode.
-
a.Take 1 μg peptide of each sample and mix together, then 10 × iRT peptide is spiked into the mixed peptide sample.
-
b.1 μg of mixture is used for data-dependent MS/MS analysis. Detailed parameters are exampled as follows:
-
i.Acquire full MS scans at a resolution of 60,000 at m/z 200 across the mass range of 350–1500 m/z with an AGC of 4e5 and maximum injection time of 50 ms.
-
ii.Acquire MS/MS spectra at a resolution of 15,000 at m/z 200, an isolation window of 1.6, a collision energy of 30% and an AGC target value of 5e4 with a maximum injection time of 22 ms.
-
i.
-
a.
-
47.Set up library generation from Pulsar search engine in the SpectroDive™ 10.4.
-
a.First, click “Generate Library from Pulsar / Search Archives…”.
-
b.Click “Add Runs from File…”, and upload the obtained DDA raw data.
-
c.Select a FASTA file by clicking “Fasta File…”, such as human Swiss-Prot FASTA database.
-
d.Click ‘Next’ in the lower right corner. The emerged Pulsar search parameters are set as follows by clicking on the option “Search Settings…”:
-
i.Select the enzyme as trypsin/P.
-
ii.Set carbamidomethyl (C) to a fixed modification, and set acetyl (Protein N-term) and oxidation (M) to variable modifications.
-
iii.Ion types include b, y.
-
iv.Other settings are default and follow the wizard.
-
i.
-
e.After the search finished, one spectral library will be generated.
-
a.
Optional: Spectral library can also be generated from results of external search engines, such as MaxQuant, Proteome Discoverer, ProteinPilot, Mascot. The search result need be imported into the SpectroDive from “Import Spectral Library…”.
-
48.Generate an assay panel based on the spectral library.
-
a.Choose ‘Prepare’ and click ‘Generate New Panel…’.
-
b.Select the just established spectral library and click “Next”.
-
c.Paste the differential protein annotations into the ‘Filter’ and click ‘Check All’.
-
d.Next, precursors are filtered as follows: precursor m/z, 350–1500; precursor charge, 2–3; missed cleavages, 0; peptide length, 8–25; Only proteotypic; allowed modification, carbamidomethyl (C).
-
e.Click “Check All” and “Next”.
-
f.Filter the fragments of the selected precursors as follows: fragment m/z, 300–1800; max fragment charge, 2; Ion types, b and y; allowed loss types: noloss; min fragment length, 3; Top N fragments, 6.
-
g.Click “Apply Settings” and follow the wizard of “Next”, then the panel is created.
-
a.
-
49.Modify the established panel. Precursors from the differential proteins will be further filtered in this step.
-
a.Click “Analysis” and “Set up Targeted Analysis from File…”.
-
b.Upload the DDA data and assign the established panel from the left corner. Follow the wizard and finish this process.
-
c.A tree view shows the details of the panel. Right-click on the precursor to be deleted and choose “Reject”.Note: The tree view can be organized by Protein or Panel. Right-click on the name of the experiment and move the mouse over “Group By…”, then one can choose how the data is presented.CRITICAL: The MS2 XIC and more detailed information of transitions should be checked one by one. Precursors with Q value < 0.01 but poor signal intensity and peak shape should be discarded in the panel. At least two precursors per protein are recommended for quantitative analysis.
-
d.Choose “Report” module and click “BGS Factory Report” under “Run Pivot Report” and make sure “EG.PrecursorId” is checked.
-
e.Click “Export report” and save the result, then copy the column of “EG.PrecursorId”.
-
f.Go back to the “Prepare”, and right-click on the established panel and click “Generate Modified Panel”.
-
g.To filter precursors, paste the precursor id into the “Filter” of “Peptide Sequences”.
-
h.Continue to filter the fragments of the precursors as described above. Then follow the wizard to complete this process.
-
a.
-
50.
Create the calibration run. Click “Import from Library…”, and choose the spectral library and the calibration run. Then click “Finish”.
-
51.
Build the PRM data acquisition method. Concurrent select the “PRM iRT-Kit” below “LC Calibration”, the modified panel below “Panels”, and the generated calibration run below “Choose RT Calibration Run”, then modify the specific parameters in “3) Export Method”. As such, the new method can be exported by clicking “Export Method”.
-
52.Import the target list in Xcalibur and analyze each sample using PRM.
-
a.Take 1 μg of each sample for monitoring according to the exported precursor list.
-
b.The data acquisition parameters are exampled as follows: full MS scans are acquired at a resolution of 60,000 at m/z 200 with an AGC of 4e5 and maximum injection time of 20 ms; MS/MS spectra are acquired at a resolution of 30,000 at m/z 200, an isolation window of 1, a collision energy of 30% and an AGC target value of 2e5 with a maximum injection time of 80 ms.
-
a.
PRM data analysis using SpectroDive
This section mainly contains the selection of transitions for protein quantitation. The confidence and quantitative area of the spectrum need to be confirmed manually.
-
53.
Click “Analysis” and “Set up Targeted Analysis from File…”. Upload the PRM raw data and assign the established panel.
-
54.
Select the search and extraction settings schema for the PRM analysis. The default BGS factory settings are used in this protocol.
-
55.
Follow the wizard and specify the condition, replicates, and other information of the raw data. After clicking “Finish”, all the data in this protocol will be loaded within about 1 h.
Optional: Right-click on the file name and select "Group by protein" to facilitate data verification based on the protein name. In order to analyze a specific protein conveniently, one can filter the data by entering the protein/gene name in the Node Text Filter. In addition, the more convenient way to screen multiple data is to display MS2 XIC and MS2 XIC Alignment in the upper and lower panels.
-
56.Manually screen the precursors and their fragments in the MS2 XIC panel.
-
a.For precursors and fragments to exclude, right-click on the fragment and select “Exclude” or “Remove” in the node or all runs.CRITICAL: Precursors and fragments with bad elution peak shape should be excluded. The fragment ions in the following cases need to be excluded: different peak patterns from other b+, y+ ions; interfered by other ions. Product ions with high intensity and an m/z above the precursor m/z are preferred, see Figure 5.
-
b.For precursors that require adjustment of the quantitative area range, manually adjust the green dotted lines representing the start and end points of the peak area calculations.Note: In some cases, the intensity alignment of the fragment ions from a precursor given by the software is very consistent across all runs, but there is an obvious error in the selection of the elution peak range, which may only select part of the region with high consistency of fragment ion intensity alignment or the wrong peak.
-
a.
-
57.
Then, right-click on the name of the experiment to recalculate the Q values by clicking “Recalculate Qvalues” for qualitative analysis, and then refresh the post analysis by clicking “Refresh Post Analysis”.
Note: After screening, some excluded precursors also pass the threshold of Q value, which may be false positives because their signal may be very low, close to noise.
-
58.
After completing all data filtering and adjustment, save the result as a .spe file.
-
59.
Go to the “Report” module and export the “Run Pivot Report” and the “Normal Report”. The default export parameters are sufficient for subsequent quantitative analysis.
Figure 5.

Fragments with bad elution peak shapes need be excluded (red arrow)
Expected outcomes
Figure 3 shows the AF4 separation fractogram obtained from one sample. The red solid lines and green solid lines represent the LS intensity at 90°C and the UV signal, respectively. EVs fraction is collected at Peak 4, the duration time of which is about 42–52 min. Typical EV proteins yield from 150 μL of plasma is about 150 μg. Figure 4 shows the characterization results of EVs separated from human plasma by Western blot and TEM analysis, proving that high-purity EVs are obtained by the AF4 method. This protocol is designed to identify potential protein biomarkers of disease. Differential proteins are first identified through comparative proteomics, then PRM is used to further filter out potential biomarkers among a large number of differential proteins. This protocol can be used to narrow down the range of potential biomarkers and facilitate the subsequent large-scale population validation.
Limitations
The PRM method developed in this protocol is based on the commercial software of SpectroDive, the steps of which are not applicable to the free software of Skyline. In addition, the comparative proteomics used in this protocol utilizes a prefractionation technique, achieving an extensive identification of the EV proteome. However, this makes the identification of low-abundance differential proteins difficult in PRM.
Troubleshooting
Problem 1
Signals of AF4 analysis is messy (step 13).
Potential solution
-
•
Check if there are air bubbles in the AF4 channel.
-
•
Place the RC membrane in the right direction.
-
•
Fully wash the system and use fresh buffers.
Problem 2
The analysis does not start immediately after switching the injector to INJECT (step 12).
Potential solution
Swiftly rotate the knob to LOAD and re-rotate to INJECT.
Problem 3
The protein or peptide concentrations of samples varied widely in the replicates (step 15 and 45).
Potential solution
Check if there are air bubbles in the wells or the working reagents and the sample are well mixed.
Problem 4
The identified number of proteins was too small (step 43).
Potential solution
-
•
Check if the protein cleavage efficiency is low, which may be caused by excessive volume of the protein solution system.
-
•
Search the raw data using pFind to check if the carbamylation of proteins/peptides is increased. If so, process the enzymatic digestion step as soon as possible to reduce the time the protein/peptide sample stays in the urea solution.
Problem 5
Some precursors cannot be monitored (step 52).
Potential solution
Check if the status of the LC-MS/MS instrument has changed or the retention time range of precursors is set properly.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Fuquan Yang (fqyang@ibp.ac.cn).
Materials availability
No newly generated materials are associated with this protocol.
Data and code availability
Original data have been deposited to ProteomeXchange: PXD034548.
Acknowledgments
This study was supported by the National Key Research and Development Program of China under grant 2018YFA0507801. We thank Xiaoqing Wang and Xufei Wang at the Shanghai Omicsolution Co., Ltd for their suggestions on SpectroDive procedure.
Author contributions
N.L. and Y.Y. wrote the manuscript. B.W. and J.W. contributed with comments to the manuscript. F.Y. supervised the project. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
References
- 1.Wu B., Chen X., Wang J., Qing X., Wang Z., Ding X., Xie Z., Niu L., Guo X., Cai T., et al. Separation and characterization of extracellular vesicles from human plasma by asymmetrical flow field-flow fractionation. Anal. Chim. Acta. 2020;1127:234–245. doi: 10.1016/j.aca.2020.06.071. [DOI] [PubMed] [Google Scholar]
- 2.Li N., Wu B., Wang J., Yan Y., An P., Li Y., Liu Y., Hou Y., Qing X., Niu L., et al. Differential proteomic patterns of plasma extracellular vesicles show potential to discriminate beta-thalassemia subtypes. iScience. 2023;26 doi: 10.1016/j.isci.2023.106048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X., Sun Y., Zhang T., Shu L., Roepstorff P., Yang F. Quantitative Proteomics Using Isobaric Labeling: A Practical Guide. Dev. Reprod. Biol. 2021;19:689–706. doi: 10.1016/j.gpb.2021.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J., Van Vranken J.G., Pontano Vaites L., Schweppe D.K., Huttlin E.L., Etienne C., Nandhikonda P., Viner R., Robitaille A.M., Thompson A.H., et al. TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods. 2020;17:399–404. doi: 10.1038/s41592-020-0781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun H., Poudel S., Vanderwall D., Lee D.G., Li Y., Peng J. 29-Plex tandem mass tag mass spectrometry enabling accurate quantification by interference correction. Proteomics. 2022;22 doi: 10.1002/pmic.202100243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Codrea M.C., Nahnsen S. Platforms and Pipelines for Proteomics Data Analysis and Management. Adv. Exp. Med. Biol. 2016;919:203–215. doi: 10.1007/978-3-319-41448-5_9. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J., Xin L., Shan B., Chen W., Xie M., Yuen D., Zhang W., Zhang Z., Lajoie G.A., Ma B. PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Djomehri S.I., Gonzalez M.E., da Veiga Leprevost F., Tekula S.R., Chang H.Y., White M.J., Cimino-Mathews A., Burman B., Basrur V., Argani P., et al. Quantitative proteomic landscape of metaplastic breast carcinoma pathological subtypes and their relationship to triple-negative tumors. Nat. Commun. 2020;11:1723. doi: 10.1038/s41467-020-15283-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tyanova S., Temu T., Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 10.Chan K.C., Issaq H.J. Fractionation of peptides by strong cation-exchange liquid chromatography. Methods Mol. Biol. 2013;1002:311–315. doi: 10.1007/978-1-62703-360-2_23. [DOI] [PubMed] [Google Scholar]
- 11.Macron C., Lane L., Núñez Galindo A., Dayon L. Identification of Missing Proteins in Normal Human Cerebrospinal Fluid. J. Proteome Res. 2018;17:4315–4319. doi: 10.1021/acs.jproteome.8b00194. [DOI] [PubMed] [Google Scholar]
- 12.McAlister G.C., Nusinow D.P., Jedrychowski M.P., Wühr M., Huttlin E.L., Erickson B.K., Rad R., Haas W., Gygi S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erickson B.K., Mintseris J., Schweppe D.K., Navarrete-Perea J., Erickson A.R., Nusinow D.P., Paulo J.A., Gygi S.P. Active Instrument Engagement Combined with a Real-Time Database Search for Improved Performance of Sample Multiplexing Workflows. J. Proteome Res. 2019;18:1299–1306. doi: 10.1021/acs.jproteome.8b00899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rauniyar N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 2015;16:28566–28581. doi: 10.3390/ijms161226120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallien S., Domon B. Detection and quantification of proteins in clinical samples using high resolution mass spectrometry. Methods. 2015;81:15–23. doi: 10.1016/j.ymeth.2015.03.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Original data have been deposited to ProteomeXchange: PXD034548.