

Abstract
Efficient identification of epitopes is crucial for drug discovery and design as it enables the selection of optimal epitopes, expansion of lead antibody diversity, and verification of binding interface. Although high‐resolution low throughput methods like x‐ray crystallography can determine epitopes or protein–protein interactions accurately, they are time‐consuming and can only be applied to a limited number of complexes. To overcome these limitations, we have developed a rapid computational method that incorporates N‐linked glycans to mask epitopes or protein interaction surfaces, thereby providing a mapping of these regions. Using human coagulation factor IXa (fIXa) as a model system, we computationally screened 158 positions and expressed 98 variants to test experimentally for epitope mapping. We were able to delineate epitopes rapidly and reliably through the insertion of N‐linked glycans that efficiently disrupted binding in a site‐selective manner. To validate the efficacy of our method, we conducted ELISA experiments and high‐throughput yeast surface display assays. Furthermore, x‐ray crystallography was employed to verify the results, thereby recapitulating through the method of N‐linked glycans a coarse‐grained mapping of the epitope.
Keywords: computational design, epitope mapping, N‐linked glycans, protein–protein interactions
1. INTRODUCTION
The ability to rapidly identify epitopes and protein–protein interactions (PPIs) is of utmost importance not only in interrogating new biology (Nooren & Thornton, 2003), but also in drug design, where identification and selection of optimal epitopes represent a key element in the optimization of potency and safety (Wilson & Stanfield, 1994). It is important to have geometrically optimal epitopes to achieve the optimal potency of multispecific antibodies. This is particularly important in the case of bispecific antibodies like Mim8 (Ostergaard et al., 2021) and emicizumab (Kitazawa et al., 2012). Moreover, in de novo computational protein design, it is essential to rapidly determine whether the design is binding to the desired epitope. Here, the binding is often tested through methods like yeast surface display (YSD). Hence, there is a need for a fast method to structurally pinpoint the position of the epitope or determine if the binding interface is in accordance with computational design.
Traditional methods for mapping epitopes have certain limitations associated with them. One commonly used method is binning monoclonal antibodies (mAbs) into different epitope bins, thereby grouping the mAbs into overlapping or similar epitopes (Kohler & Milstein, 1975). Using full‐length mAbs can be misleading due to steric hindrance of the fragment crystallizable (Fc) region, which does not necessarily reflect the binding region of the epitope. Other common ways of determining epitopes include deep mutational scanning (Frank et al., 2022), peptide mapping (Zhao & Chalt, 1994), NMR spectroscopy (Becker et al., 2018; Monaco et al., 2017), x‐ray crystallography (Davies & Cohen, 1996; Kong et al., 2015), and cryo‐electron microscopy (Long et al., 2015). However, these are considered low throughput methods that require considerable effort or time and can test only a limited number of complexes. The first two methods have low resolution, while x‐ray and cryo‐EM of the co‐complex can provide atomistic details of the interaction. Another popular method that can be used to profile epitopes and PPIs is hydrogen‐deuterium exchange mass spectrometry (HDX‐MS), which has been used to accurately map interactions between antibodies and antigens (Jensen et al., 2015). The technique can accurately identify epitopes, but it is important to be cautious when analyzing the results as significant changes upon binding of the overall structure of the molecule can make it hard to interpret. In proteins with allosteric sites, it can be difficult to map the precise epitope using HDX‐MS due to conformational changes upon binding (Xu et al., 2019).
N‐linked glycosylation is a common post‐translational modification that occurs in many human proteins (Apweiler et al., 1999). It expands the functional properties of proteins by increasing the physico‐chemical properties, for example, stability (Sarkar & Wintrode, 2011). N‐linked glycosylation of proteins occurs through a consensus Asparagine(N)‐Xxx‐Serine(S)/Threonine(T) recognition motif. Here, the sidechain of the initial N represents the glycan acceptor site; the second position (Xxx) can be any amino acid except proline and is followed by either S or T at the third position. The efficiency of a glycosylated site depends on many factors, including the local three‐dimensional (3‐D) structure around the consensus motif (Suga et al., 2018) and surface accessibility. Introducing an N‐linked glycan within an epitope will disrupt ligand binding through simple steric hindrance. Others have investigated the idea of using an N‐linked glycan to mask or protect an epitope with great success. Lombana et al. (Lombana et al., 2019) developed the glycosylation‐engineered epitope mapping (GEM) method, which involves the insertion of N‐linked glycans at specific sites on the surface of the antigen to conceal localized regions. The method calculates the solvent‐accessible surface area (SASA) of the N and S/T residues and determines if they are exposed. There is no structural modeling of the N‐linked glycan, and the method has not incorporated the design of the S/T residue at the third position, which can have different preferences depending on local protein structure.
As discussed, identifying epitopes in a high throughput manner with high precision is desirable to understand biology and engineer optimal drugs targeting specific epitopes. Hence, we have improved on existing computational methods to insert and design the local sequence of the target protein such that it can accommodate an N‐linked glycan at scale. It was achieved through a combination of structural modeling and computational design techniques and can be fully automated. Further, it is possible to add N‐linked glycans at multiple positions of the human factor IXa (FIXa) without altering its overall structure (Brooks et al., 2013). Using this model system of FIXa, we were able to validate the method in accordance with a crystal structure of the complex using low‐ and high‐throughput experimental methods.
2. METHODS
2.1. Generation of anti‐FIXa antibodies
Antibodies specific for human FIXa were generated in Kymouse mice (Kymab Group Ltd) that comprise a human antibody repertoire. Kymouse mice were immunized with active‐site inhibited FIXa purchased from Haematologic Technologies Inc/Prolytics. After electrofusion, hybridomas were plated in microtiter plates and supernatants were screened by a fIXa ELISA. Hybridomas secreting antibodies specific for fIXa were propagated and their supernatants were subsequently used for protein A affinity purification. fIXa binding was confirmed on fortebio, and hybridoma hits were sequenced and recombinantly expressed in HEK293 cells using standard techniques.
2.2. N‐linked glycan library production and characterization
The DNA fragment encoding the amino acid sequence of HPC4 tagged human fIX without the gamma‐carboxyglutamic (GLA) domain was synthesized and cloned into the pJSV vector with CD33 signal peptide to replace the native signal peptide (Rezaie & Esmon, 1992). Ninety‐eight variants were created, each containing specific engineered mutations for N‐linked glycosylation. The mutations introduced were an asparagine (N) residue followed by any amino acid residue except proline (X) and a serine (S) or threonine (T) residue at different positions in the DNA sequence. Each of the 98 variants were individually cloned for further study or experimentation. HEK293 6E cells were used for transient production of these proteins following standard protocols for 293Fectin (Invitrogen). The cell cultures were harvested at 5 days post‐transfection by centrifugation at 6000 rpm for 15 min, and the supernatants were filtered using a 0.45 μm filter for purification with anti‐HPC4 sepharose 4FF affinity columns (anti‐HPC4 antibody coupled to the CNBr activated sepharose 4FF resin, GE), equilibrated in buffer (20 mM Tris–HCl, 100 mM NaCl, 1 mM CaCl2, pH 7.4). The bound proteins were eluted with buffer (20 mM Tris–HCl, 100 mM NaCl, pH 7.4, 1 mM EGTA) as final products. The variants were analyzed with SEC‐UPLC, LC–MS, and SDS‐PAGE, which were also digested with human factor XIa (Schmidt & Bajaj, 2003; Wolberg et al., 1997) and then analyzed with SDS‐PAGE to identify if the N‐linked glycosylation was inserted.
2.3. Fluorescence‐activated cell sorting analysis
The YSD plasmids were constructed to evaluate the binding of single‐chain variable fragments (scFv) to different factor fIXa variants. The AGA2 gene downstream of the GAL1 promoter was fused with a four‐part cassette encoding the VL, linker, VH, and FLAG tag fragments, forming an VL–Linker–VH‐FLAG‐Aga2 cassette. Two plasmids that contain (G4S)3 and (G4S)4 linker, respectively, were constructed.
The constructed plasmids were transformed into EBY100 cells (URA +, leu −, trp −) followed by cultivation and induction. The cells bearing different constructs were labeled with anti‐FLAG‐iFluor 488 and anti‐HPC4‐iFluor 647 antibodies (GenScript, China) followed by detection using similar protocols published previously (Yi et al., 2015) with the NovoCyte flow cytometer (Agilent, USA). The iFluor 647 fluorescent intensity was detected with the APC channel, 670/30 nm band pass, and iFluor 488 fluorescent intensity was detected with the FITC channel, 530/30 nm band pass. The binding efficiencies against different fIXa variants were evaluated as: Binding efficiency = [Mean Fluorescency Intensity of iFluor 647]/[Mean Fluorescency Intensity of iFluor 488], in which Mean Fluorescency Intensity (M.F.I.) of iFluor 647 and M.F.I. of iFluor 488 represents the binding of FIXa variants and surface display of scFv, respectively.
2.4. ELISA of N‐linked glycosylation library screening
An anti‐HPC4 monoclonal mouse antibody was coated onto 96‐well ELISA plates (9018, Corning) at 5 μg/mL, 100 μL/well. The plates were incubated at 4°C overnight. The plates were washed three times with PBS, blocked with 1% BSA for 3 h. The plates were washed three times before adding human FIX or human FIX with N‐linked glycosylation at 1 μg/mL, 100 μL/well. One hour later, the plates were washed three times before adding anti‐FIX antibodies to be assessed at 1 μg/mL, 100 μL/well. The plates were washed three times again before adding anti‐hIgG‐HRP (Invitrogen, Cat # A18823) antibody for detection. Thirty minutes later the plates were washed followed by measuring ELISA signaling at an absorbance of OD450.
2.5. Computational design of N‐linked glycans
The crystal structure of FIXa (PDB ID: 1RFN; Hopfner et al., 1999) was used as a starting point for calculating the Define Secondary Structure of Proteins (DSSP) (Kabsch & Sander, 1983) using RosettaScripts (Fleishman et al., 2011; Leaver‐Fay et al., 2011). A glycan model was generated by utilizing the crystal structure (PDB ID: 4Z7Q; Lam et al., 2013) with the glycan alpha‐d‐mannopyranose‐(1–3)‐[alpha‐d‐mannopyranose‐(1‐6)] beta‐d‐mannopyranose‐(1–4)‐2‐acetamido‐2‐deoxy‐beta‐d‐glucopyranose‐(1–4)‐2‐acetamido‐2‐deoxy‐beta‐d‐glucopyranose (chain K in crystal structure) creating the parameter file used in Rosetta. The N‐linked glycan was computationally modeled onto the protein structure using the RosettaMatch (Zanghellini et al., 2006), which identified multiple possible positions for the N‐linked glycan model. Subsequently, each of these matches was subjected to energy minimization using restraint‐based methods. To obtain the desired glycosylation motif, the N + 2 position was designed by repacking the position using a resfile which restricted substitutions to either S or T residues. All energies were scored using talaris2013 (Leaver‐Fay et al., 2013; O'Meara et al., 2015). Protein structures were analyzed and visualized using PyMOL (Schrodinger, 2015). For details and a list of commands and scripts used, please see Supporting Information Appendix.
2.6. Production of NN‐8955Fab
The DNA fragments encoding the amino acid sequence of Fab (NN‐8955Fab) heavy chain and light chain were synthesized and cloned into the pJSV vector (generated in‐house using a CMV promoter) with CD33 signal peptide to replace the native signal peptide, respectively, which then were co‐transfected into HEK293 6E cells for expression together following standard protocols for 293Fectin (Invitrogen). The cell cultures were harvested at 5 days post‐transfection. Cells were removed from the culture by centrifugation at 6000 rpm for 15 min, and the supernatants were filtered using a 0.45 μm filter. The supernatants were loaded to HiTrap Protein G HP (5 mL, GE), equilibrated in PBS. The bound proteins were eluted with elution buffer (0.1 M glycine‐HCl, pH 2.7), the eluate was neutralized with buffer (1 M Tris, pH 9.0), which was further polished with HiLoad Superdex 200 16/60 (GE), equilibrated, and eluted with buffer (25 mM HEPES, 150 mM NaCl, pH 7.4).
2.7. Crystal structure determination of NN‐8955Fab in complex with des‐(GLA‐EGF1) fIXa
Crystals of NN‐8955Fab mixed in a 1:1 molar ratio with human EGR‐chloromethylketone active‐site inhibited des‐(GLA‐EGF1) fIXa from Cambridge ProteinWorks were grown using the sitting drop vapor diffusion technique at 18°C. A protein solution of 100 nL 6.9 mg/mL complex in 20 mM Tris–HCl, pH 7.4, 50 mM NaCl, 2.5 mM CaCl2 was mixed with 100 nL of 0.5 M ammonium sulfate, 0.1 M sodium citrate pH 5.6, 1.0 M lithium sulfate as precipitant and incubated over 60 μL precipitant. The crystal was cryo protected in a solution consisting of 0.375 M ammonium sulfate, 75 mM sodium citrate pH 5.6, 0.75 M lithium sulfate, 4% glycerol, 4% ethylene glycol, 4.5% sucrose, and 1% glucose prior to flash cooling in liquid nitrogen. Diffraction data were collected at 100 K at the Swiss Light Source beamline X06DA (1.00000 Å wavelength) using a Pilatus2M pixel detector from Dectris. Autoindexing, integration, and scaling of the data were performed with programs from the XDS package (Kabsch, 2010). The asymmetric unit contains one Fab:FIXa complexes as judged from Matthew's coefficient analysis. The structure was determined by molecular replacement with Phaser (McCoy et al., 2007) as implemented in the program suite Phenix (Liebschner et al., 2019) with the crystal structure of a homologous Fab and FIXa from PDB entry 3KCG (Johnson et al., 2010) as search models. The correct amino acid sequence for the Fab was model built using COOT (Emsley et al., 2010) and thereafter the structure was refined using steps of Phenix refinement (Afonine et al., 2012) and manual rebuilding in COOT. Diffraction data and refinement statistics are found in Table S1.
3. RESULTS
3.1. Computation‐guided epitope mapping of factor IXa
To profile the epitopes recognized by a panel of antibodies raised against fIXa with high throughput and coarse‐grained mapping, an N‐linked glycan variant library of fIXa was generated using a computational workflow (see Figure 1a). The computational protocol was generated to design N‐linked glycans for high‐throughput epitope profiling with coarse‐grained resolution (see Figure 1b). First, the crystal structure of fIXa, along with a model glycan was used as the input. The glycan was structurally inserted and modeled using RosettaMatch (Zanghellini et al., 2006) on positions that were considered loops according to the DSSP algorithm in RosettaScripts (Fleishman et al., 2011; Leaver‐Fay et al., 2011), resulting in 158 positions. From a statistical analysis, an epitope is comprised on average of 9–22 amino acid residues (Haste Andersen et al., 2006), and loop regions are often the most frequent regions used to insert N‐linked glycans in human proteins. From the computational screen, 158 positions were identified, all of which were designed using PackRotamer in Rosetta to obtain the N‐linked glycosylation motif N‐X‐S/T (where X represents all amino acids except proline). To increase the quality of the designed N‐linked glycans, the 3‐D structures were ranked using RosettaScripts to compute the cutoffs on the interface energies, <0 REU, to ensure energetically favorable interaction and geometrical restraints, <10, based on the structural models. This resulted in 98 positions that, according to the computational designs, could accommodate N‐linked glycosylation and were chosen for experimental verification (see Table S3 for a full list of sequences used in the study).
FIGURE 1.
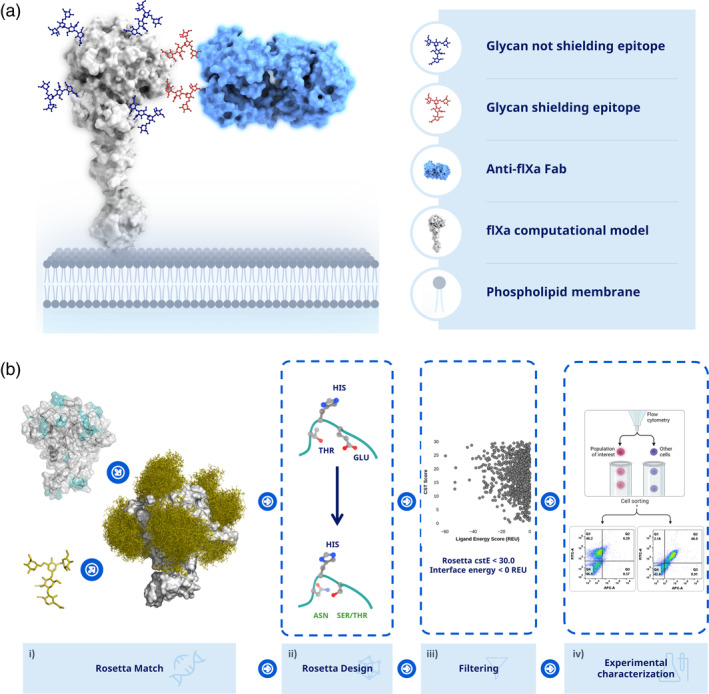
Epitope mapping through engineered N‐linked glycans. (a) The idea is to engineer N‐linked glycans into the protein of interest, which will mask the epitope/interface and thereby prevent binding/interaction. (b) Step (i) loop elements(cyan) are identified using DSSP on a crystal structure of fIXa, which returns potential positions to place the N‐linked glycan. Next, a model of glycan structure is generated, which is used by RosettaMatch to insert the N‐linked glycan onto the structure of fIXa. (ii) The sequence is designed by RosettaDesign to obtain the consensus motif N‐X‐S/T (X can be any amino acid except proline) and optimized according to the input geometry. (iii) Engineered variants are filtered based on deviations from the specified geometry, <10, and an interface energy <0 REU is required. (iv) The remaining designs are used to experimentally characterize binding epitopes using either yeast surface display or other binding methods, such as ELISA.
3.2. Expression and characterization of computational designed variants
It was decided to use fIXa without the gamma‐carboxyglutamic acid‐rich (GLA) domain, as the epitopes of interest were assumed to be distal to the membrane. Ninety‐nine variants, including the wild type (WT), were expressed in the mammalian cell line HEK293 to ensure that they were glycosylated. It was possible to purify 92 of the engineered variants, which was done in two steps. The first batch was produced in 30 mL cell culture while for variants not expressing here were upscaled to 300 mL cell culture. This resulted in concentrations ranging from 0.08 to 2.25 mg/mL for the 92 variants which were used for further characterization. To determine whether the engineered variants had successfully incorporated an N‐linked glycan, the level of N‐linked glycosylation of each variant was estimated by separating them based on their mass differences using SDS‐PAGE. This method was used to detect and compare the molecular weights of the different protein variants, confirming whether the N‐linked glycan had been successfully introduced. Ninety‐eight desGla‐hfIX variants were designed with a single de novo N‐linked glycan site, with 77 variants in the protease domain, 12 variants in the EGF2 domain, and 9 variants in the hinge region (see Figure S1). SDS‐PAGE analysis was suitable for characterizing the glycosylation level only for variants in the protease domain. SDS‐PAGE analysis showed that 53 out of the 77 variants were fully or partially glycosylated (for a comprehensive description, see Table S2). Since the mass difference of the EGF domains was too small to be detected using SDS‐PAGE, the EGF2 variants that were engineered with N‐linked glycosylation were tested using LC–MS. Seven of the variants in the EGF2 domain were shown to have incorporated N‐linked glycosylation (see Figure 2). Table 1 shows the range of the incorporation efficiencies from 0% to 100%, where five of the engineered variants were unquantifiable.
FIGURE 2.
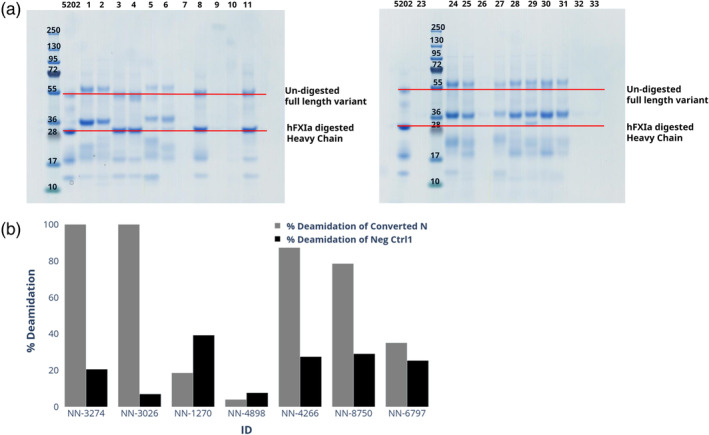
Characterization of N‐linked glycosylation in the EGF2 domain. (a) The engineered variants are characterized on a reducing SDS‐PAGE gel with NN‐7241 (fIXa without GLA domains) as reference. Shifts are indicated with respect to the heavy chain of fIXa (red line). (b) The glycosylation level of engineered fIXa variants could be characterized by LC–MS/MS by observing the convertion of asparagine to aspartic acid, which removes N‐linked glycosylation. To mitigate the false positive effect induced from chemical deamidation, deamidation ratio of N175 was calculated as a negative control. As an example, the LC–MS/MS analysis indicated that three of seven variants had 100% presence of the N‐linked glycosylation. One variant had partially (78.5% or lower) incorporated the N‐linked glycosylation, and three variants had not or limited (35.1%–4.0% or lower) incorporation of the N‐linked glycosylation.
TABLE 1.
Quantification of N‐glycosylation in EGF2 variants by peptide mapping.
| NNCD | Serial number | Glycosylation (%) | Mutations | Position |
|---|---|---|---|---|
| NN‐5835 | 82 | 0 | T66N G68S | EGF2 |
| NN‐1265 | 18 | 27 | A72N N74S | EGF2 |
| NN‐7372 | 51 | 56.8 | S56N D58S | EGF2 |
| NN‐3972 | 74 | 57.3 | K60N V62S | EGF2 |
| NN‐7479 | 16 | 67.2 | Q75N | EGF2 |
| NN‐4800 | 19 | 100 | E73N Q75T | EGF2 |
| NN‐3045 | 41 | 100 | A57N N59S | EGF2 |
| NN‐1270 | 8 | ND | T41N N43S | EGF2 |
| NN‐3026 | 11 | ND | D58N K60T | EGF2 |
| NN‐3222 | 37 | ND | E67N Y69S | EGF2 |
| NN‐1358 | 50 | ND | E50N F52S | EGF2 |
| NN‐4898 | 71 | ND | S77N E79S | EGF2 |
Abbreviation: ND, not determined.
3.3. ELISA assay mapping of factor IXa epitope
To map the epitope of the anti‐fIXa NN‐8895 full‐length mAb, 56 purified variants were first tested in an ELISA assay along with the WT fIXa. Out of these, four N‐linked glycan variants (see Table 2) showed reduced signals in the binding assay comparable to the negative control buffer (see Figure 3a). This indicates that the N‐linked glycan in these variants interfered with binding and is part of the epitope. Loss of binding was defined as an ELISA signal equivalent to that of the buffer used. To verify if this was due to site‐selective masking of the epitope on fIXa, a crystal structure of the co‐complex was solved. From the co‐complex structure, it was observed that the inserted N‐linked glycans were indeed in the binding epitope (see Figure 3b), thus validating the reduced binding signal observed by the ELISA.
TABLE 2.
N‐linked glycosylated variants with reduced binding of NN‐8895.
| NNCD | Mutations | Epitope |
|---|---|---|
| NN‐5539 | F296N I298S | Yes |
| NN‐8759 | V285N R287T | Yes |
| NN‐6981 | T294N F296S | Yes |
| NN‐2887 | K295N | Yes |
FIGURE 3.
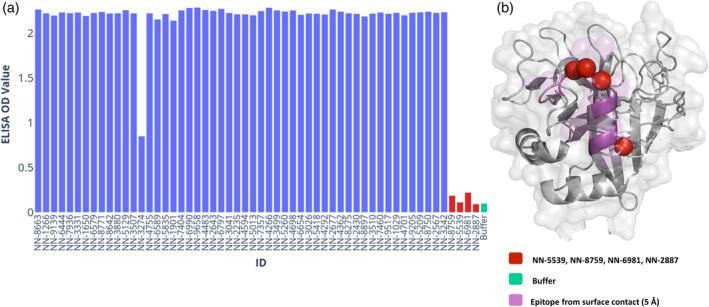
ELISA mapping of epitope. (a) Measuring ELISA signaling for each of the N‐linked glycosylated variants displaying the maximum value. Variants not showing reduced signal are in blue and the signal from the buffer (green) while variants with signal similar to the buffer are considered none binding (red). (b) The four variants with reduced signal and their positions (red spheres for C‐alpha positions) relative to the epitope as defined by the crystal structure (purple).
3.4. Epitope mapping using YSD
The first step in raising binders or verifying de novo‐designed proteins often involves relying on display technology, such as phage panning or yeast display. Compared to phage display, YSD can provide a more straightforward quantitative evaluation. In this study, we used two scFv versions of NN‐8895 with different linkers between Aga2 and scFv, G4Sx3 and G4Sx4 (NN‐8895‐L1 and NN‐8895‐L2, respectively), for epitope profiling using YSD (see Figure 4 and Figure S2). Four N‐linked glycosylated fIXa variants were chosen to evaluate the sensitivity of the method, with three N‐linked variants (NN‐8759, NN‐6981, and NN‐2887) in the epitope and one (NN‐5835) outside of the epitope. After the two scFvs (NN‐8895‐L1, NN‐8895‐L2) were displayed on the yeast surface, the cells were screened against the four variants under different concentrations (10 nM, 100 nM, and 1 μM) using FACS. Among the four variants, NN‐8759, NN‐6981, and NN‐2887 with N‐linked glycans in the epitope according to the crystal structure displayed a significantly reduced binding signal comparable to background levels under all three tested concentrations. However, in the case of NN‐5835, a binding signal was still observed in a concentration‐dependent manner. This is in accordance with the binding mode and epitope observed in the crystal structure between NN‐8895 and fIXa. In addition, surface displayed NN‐8895‐L1 showed a stronger binding against 1 μM NN‐5835 than that of N‐8895‐L2, indicating that the linker might affect the fIXa binding (Figure S2).
FIGURE 4.
Epitope mapping by yeast surface display. (a) Yeast surface displayed scFv NN‐8955‐L1 (FLAG coupled with anti‐FLAG‐iFluor 488, y‐axis) was screened against four fIXa variants with N‐linked glycans (coupled with anti‐HPC4‐iFluor 647, x‐axis) at three different concentrations: 10 nM, 100 nM, and 1 μM. (b) Crystal structure of the complex between NN‐8955 Fab (skyblue cartoon representation) and fIXa (white surface representation) with N‐linked glycans modeled computationally onto the structure where red colors represent epitopes being masked by N‐linked glycan while blue indicates no or little effect on binding.
4. DISCUSSION
Rapid identification and characterization of epitopes are of utmost importance to optimizing drugs so that they can interact optimally with their targets. Rapidly profiling epitopes with high resolution can, for example, aid in drug discovery to also ensure that lead compounds have diverse interactions with their target, and lastly, validate de novo–designed proteins. The ability to map epitopes rapidly and accurately on protein structures is essential for accelerating the engineering and design of proteins, giving confidence in the epitope selection. Here, we have created a fast, reliable, and fine‐grained method to insert N‐linked glycans using in silico methods that map them directly onto a crystal structure or a structural model of the target protein with the necessary sequence modifications to incorporate the N‐linked glycan. We used a model system with the human coagulation fIXa to verify the protocol. It was shown that the method was robust and could be applied in both low‐ and high‐throughput screening assays, such as ELISA and YSD. In the process of inserting the N‐linked glycans, a conservative approach was chosen to mainly focus on loops as defined computationally by DSSP. It has been shown that loops have a higher probability of N‐linked glycosylation in human proteins (Lam et al., 2013) but N‐linked glycans are also observed in other secondary elements such as sheets and helices. This method can be easily extended to other secondary elements like helices or sheets by modifying the computational method to include these elements. In this study, to achieve full coverage of the fIXa surface, N‐linked glycans were introduced with a maximum spacing of 19 residues between each glycan. As this distance has been observed in other studies of epitopes to ensure coverage of the area, it ensures full coverage of the target protein and increases the likelihood of masking all potential epitopes, even on large proteins like fIXa. Linear epitopes can be in the range of 4–12 amino acids (Buus et al., 2012) and one might need to expand the frequency of N‐linked glycans to other secondary elements in the protein of interest. We did observe that not all the engineered N‐linked glycans were incorporated into fIXa. Not all designed positions can sustain the incorporation of an N‐linked glycan, and this needs to be considered when applying the method. In a few cases, the site was also not 100% glycosylated, which could potentially make interpretation difficult if the glycosylation level was not properly analyzed. Through the utilization of computational methods, we successfully reduced the initially tested positions from 158 to 98 for further experimental validation. This reduction of positions is particularly important as purification and validation of many variants is resource intensive. Our primary objective was to accurately map the epitope of fIXa while also assessing the robustness of the chosen methodology hence we expressed all engineered N‐linked glycans. Prediction algorithms like NetNGlyc (Gupta & Brunak, 2002) could be employed to estimate the level of N‐linked glycosylation and quality of the site. Overall, the combination of computational methods and experimental testing allowed us to streamline the number of positions to be analyzed, assess the reliability of our approach, and effectively but incorporation of prediction algorithms could improve the workflow. This would still need benchmarking as there might be difference between engineered sites and native ones.
Multi‐targeting mAbs toward nonoverlapping epitopes gain more popularity especially in fields like cancer and infectious diseases like covid19 (Oostindie et al., 2022). Our method could in principle be expanded to screen for multi‐epitope targeting but was not tested. The structural models should be able to ensure compatibility between the different engineered N‐linked glycans increasing the success of incorporating multiple insertions.
The N‐linked glycosylation‐dependent method is constrained by the requirement that the target proteins must be expressed in host systems that can perform N‐linked glycosylation. Therefore, proteins expressed only in Escherichia coli are unsuitable for this method. However, this computational approach is adaptable to any chemical modifications, such as PEGylation, which may offer greater flexibility in some cases. The method's scope is generalizable to these modifications, making it a versatile technique.
Modification of protein sequences is a complex task that can lead to destabilization or changes in protein topology. While adding N‐linked glycans can improve biophysical properties such as solvation and low aggregation propensity to mention a few (Hanson et al., 2009; Hebert et al., 2014), it is also possible that these modifications may destabilize the protein. The process of engineering N‐linked glycans relies on the assumption that the target protein maintains its 3D topology after the modification where native N‐linked glycans often do not perturb the structure (Lee et al., 2015). In our method, we model a full N‐linked glycan and use interface energy from Rosetta to evaluate the designs, considering the potential destabilization caused by the modification. Moreover, the method uses Rosetta to select optimal residues at the N + 2 position, which attempts to minimize the total energy of the protein. However, it is possible that some proteins may not be amenable to N‐linked glycan engineering, making our method unusable in such cases. To handle the N‐linked glycan engineering process more conservatively, we used loops to ensure the maximum incorporation of N‐linked glycans while minimizing destabilization since asparagine and serine residues can affect secondary elements such as helices through capping (Doig & Baldwin, 1995).
In the ELISA experiment, variability of the signal was observed, indicating heterogeneity for some of the variants. The heterogeneity observed in the binding signal can also be attributed to the occupancy of N‐linked glycans, which may result in a reduced binding signal, although this does not necessarily mean the complete removal of the signal. This means that even if an N‐linked glycan is present on the protein surface and partially masking the epitope, the scFv may still be able to bind to the protein with reduced affinity. However, we were able to get 59/92 N‐linked glycosylated variants with N‐linked glycans inserted into fIXa. Using ELISA from the binding results, we were able to determine the right epitope according to the crystal structure with a clear signal of where the epitope was located. Four of the N‐linked glycan variants showed a reduced signal, and according to the crystal structure, they were the only variants that should disrupt binding. The computational de‐novo design of protein binders is often evaluated using yeast display, but confirmation of epitope binding can be a time limitation. It was not clear whether applying the epitope mapping using YSD would be as sensitive as the ELISA. However, the results showed that we were able to map the epitope using YSD by using four variants with engineered N‐linked glycans close to the epitope. Three of the N‐linked glycans showed reduced signals in ELISA, and a similar result was observed using titrations in YSD. The results were encouraging, as there were multiple nonspecific interactions between the engineering protein and its N‐linked glycans that could be anticipated.
The computational method is fast, but it can be memory‐intensive depending on the setup. The protein expression, purification, and characterization steps can take weeks and can be resource intensive depending on setup. For large proteins like coagulation factor IXa, many variants may be needed to thoroughly mask the epitope. The experimental method does not require any specialized equipment, such as x‐ray crystallography or cryoEM. Additionally, it does not require any additional setup beyond binding studies, such as ELISA or YSD. The throughput of the method would be like peptide microarrays and there could be different pros and cons depending on the problem at hand. This method is especially useful for newer methods, such as computational de novo design, or cases where a specific epitope is desired. In these cases, only a few N‐linked variants may be needed, and multiple binders can easily be screened.
To conclude, we have developed an in silico method to insert N‐linked glycans using structural modeling. The method was validated using fIXa as a model system, and successfully performed epitope mapping using experimental methods, such as ELISA and YSD.
AUTHOR CONTRIBUTIONS
Per Jr. Greisen: Conceptualization; methodology; software; data curation; formal analysis; writing—review and editing; writing—original draft. Li Yi: Methodology; writing—review and editing; formal analysis. Rong Zhou: Conceptualization; methodology; validation; writing—review and editing; formal analysis. Jian Zhou: Supervision; writing—review and editing. Eva Johansson: Methodology; validation; writing—review and editing. Tiantang Dong: Resources; data curation. Haimo Liu: Resources; data curation. Laust B. Johnsen: Resources; data curation; methodology. Søren Lund: Resources; data curation; methodology. L. Anders Svensson: Resources; data curation; methodology. Haisun Zhu: Resources; data curation. Nidhin Thomas: Writing—review and editing; visualization; validation; software. Zhiru Yang: Resources; supervision. Henrik Østergaard: Conceptualization; writing—review and editing; methodology; project administration; supervision; formal analysis.
Supporting information
Data S1: Supporting Information.
ACKNOWLEDGMENTS
The authors thank the entire Novo Nordisk Mim8 project team for contributions, in particular, Xiaoli Yan for expression of these variants in the library, Xinping Qu, Lin Li, and Chenxi Shen for purification of these variants, Xiaoai Wu for collecting data. We would like to express our gratitude to Bjarne Gram Hansen and Ida Hilden for enabling and guiding us in exploring scientific avenues.
Greisen PJ, Yi L, Zhou R, Zhou J, Johansson E, Dong T, et al. Computational design of N‐linked glycans for high throughput epitope profiling. Protein Science. 2023;32(10):e4726. 10.1002/pro.4726
Review Editor: Nir Ben‐Tal
Contributor Information
Per Jr. Greisen, Email: pgreisen@gmail.com.
Rong Zhou, Email: rzhou01@foxmail.com.
Henrik Østergaard, Email: henrik3601@gmail.com.
REFERENCES
- Afonine PV, Grosse‐Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 4):352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS‐PROT database. Biochim Biophys Acta. 1999;1473(1):4–8. [DOI] [PubMed] [Google Scholar]
- Becker W, Bhattiprolu KC, Gubensak N, Zangger K. Investigating protein‐ligand interactions by solution nuclear magnetic resonance spectroscopy. ChemPhysChem. 2018;19(8):895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AR, Sim D, Gritzan U, Patel C, Blasko E, Feldman RI, et al. Glycoengineered factor IX variants with improved pharmacokinetics and subcutaneous efficacy. J Thromb Haemost. 2013;11(9):1699–1706. [DOI] [PubMed] [Google Scholar]
- Buus S, Rockberg J, Forsstrom B, Nilsson P, Uhlen M, Schafer‐Nielsen C. High‐resolution mapping of linear antibody epitopes using ultrahigh‐density peptide microarrays. Mol Cell Proteomics. 2012;11(12):1790–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DR, Cohen GH. Interactions of protein antigens with antibodies. Proc Natl Acad Sci U S A. 1996;93(1):7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig AJ, Baldwin RL. N‐ and C‐capping preferences for all 20 amino acids in alpha‐helical peptides. Protein Sci. 1995;4(7):1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleishman SJ, Leaver‐Fay A, Corn JE, Strauch EM, Khare SD, Koga N, et al. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PloS One. 2011;6(6):e20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank F, Keen MM, Rao A, Bassit L, Liu X, Bowers HB, et al. Deep mutational scanning identifies SARS‐CoV‐2 nucleocapsid escape mutations of currently available rapid antigen tests. Cell. 2022;185(19):3603–16 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Brunak S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput. 2002:310–322. [PubMed] [Google Scholar]
- Hanson SR, Culyba EK, Hsu TL, Wong CH, Kelly JW, Powers ET. The core trisaccharide of an N‐linked glycoprotein intrinsically accelerates folding and enhances stability. Proc Natl Acad Sci U S A. 2009;106(9):3131–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haste Andersen P, Nielsen M, Lund O. Prediction of residues in discontinuous B‐cell epitopes using protein 3D structures. Protein Sci. 2006;15(11):2558–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Lamriben L, Powers ET, Kelly JW. The intrinsic and extrinsic effects of N‐linked glycans on glycoproteostasis. Nat Chem Biol. 2014;10(11):902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner KP, Lang A, Karcher A, Sichler K, Kopetzki E, Brandstetter H, et al. Coagulation factor IXa: the relaxed conformation of Tyr99 blocks substrate binding. Structure. 1999;7(8):989–996. [DOI] [PubMed] [Google Scholar]
- Jensen PF, Larraillet V, Schlothauer T, Kettenberger H, Hilger M, Rand KD. Investigating the interaction between the neonatal Fc receptor and monoclonal antibody variants by hydrogen/deuterium exchange mass spectrometry. Mol Cell Proteomics. 2015;14(1):148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DJ, Langdown J, Huntington JA. Molecular basis of factor IXa recognition by heparin‐activated antithrombin revealed by a 1.7‐A structure of the ternary complex. Proc Natl Acad Sci U S A. 2010;107(2):645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Igawa T, Sampei Z, Muto A, Kojima T, Soeda T, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18(10):1570–1574. [DOI] [PubMed] [Google Scholar]
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–497. [DOI] [PubMed] [Google Scholar]
- Kong L, Torrents de la Pena A, Deller MC, Garces F, Sliepen K, Hua Y, et al. Complete epitopes for vaccine design derived from a crystal structure of the broadly neutralizing antibodies PGT128 and 8ANC195 in complex with an HIV‐1 Env trimer. Acta Crystallogr D Biol Crystallogr. 2015;71(Pt 10):2099–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam PV, Goldman R, Karagiannis K, Narsule T, Simonyan V, Soika V, et al. Structure‐based comparative analysis and prediction of N‐linked glycosylation sites in evolutionarily distant eukaryotes. Genomics Proteomics Bioinformatics. 2013;11(2):96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver‐Fay A, O'Meara MJ, Tyka M, Jacak R, Song Y, Kellogg EH, et al. Scientific benchmarks for guiding macromolecular energy function improvement. Methods Enzymol. 2013;523:109–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver‐Fay A, Tyka M, Lewis SM, Lange OF , Thompson J, Jacak R, et al. ROSETTA3: an object‐oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Qi Y, Im W. Effects of N‐glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci Rep. 2015;5:8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, et al. Macromolecular structure determination using x‐rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol. 2019;75(Pt 10):861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombana TN, Matsumoto ML, Berkley AM, Toy E, Cook R, Gan Y, et al. High‐resolution glycosylation site‐engineering method identifies MICA epitope critical for shedding inhibition activity of anti‐MICA antibodies. MAbs. 2019;11(1):75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F, Fong RH, Austin SK, Chen Z, Klose T, Fokine A, et al. Cryo‐EM structures elucidate neutralizing mechanisms of anti‐chikungunya human monoclonal antibodies with therapeutic activity. Proc Natl Acad Sci U S A. 2015;112(45):13898–13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco S, Tailford LE, Juge N, Angulo J. Differential epitope mapping by STD NMR spectroscopy to reveal the nature of protein‐ligand contacts. Angew Chem Int ed Engl. 2017;56(48):15289–15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nooren IM, Thornton JM. Diversity of protein‐protein interactions. EMBO J. 2003;22(14):3486–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meara MJ, Leaver‐Fay A, Tyka MD, Stein A, Houlihan K, DiMaio F, et al. Combined covalent‐electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J Chem Theory Comput. 2015;11(2):609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostindie SC, Lazar GA, Schuurman J, Parren P. Avidity in antibody effector functions and biotherapeutic drug design. Nat Rev Drug Discov. 2022;21(10):715–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergaard H, Lund J, Greisen PJ, Kjellev S, Henriksen A, Lorenzen N, et al. A factor VIIIa‐mimetic bispecific antibody, Mim8, ameliorates bleeding upon severe vascular challenge in hemophilia A mice. Blood. 2021;138(14):1258–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaie AR, Esmon CT. The function of calcium in protein C activation by thrombin and the thrombin‐thrombomodulin complex can be distinguished by mutational analysis of protein C derivatives. J Biol Chem. 1992;267(36):26104–26109. [PubMed] [Google Scholar]
- Sarkar A, Wintrode PL. Effects of glycosylation on the stability and flexibility of a metastable protein: the human serpin alpha(1)‐antitrypsin. Int J Mass Spectrom. 2011;302(1–3):69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AE, Bajaj SP. Structure‐function relationships in factor IX and factor IXa. Trends Cardiovasc Med. 2003;13(1):39–45. [DOI] [PubMed] [Google Scholar]
- Schrodinger, LLC . The PyMOL molecular graphics system, Version 1.8. 2015.
- Suga A, Nagae M, Yamaguchi Y. Analysis of protein landscapes around N‐glycosylation sites from the PDB repository for understanding the structural basis of N‐glycoprotein processing and maturation. Glycobiology. 2018;28(10):774–785. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Stanfield RL. Antibody‐antigen interactions: new structures and new conformational changes. Curr Opin Struct Biol. 1994;4(6):857–867. [DOI] [PubMed] [Google Scholar]
- Wolberg AS, Morris DP, Stafford DW. Factor IX activation by factor XIa proceeds without release of a free intermediate. Biochemistry. 1997;36(14):4074–4079. [DOI] [PubMed] [Google Scholar]
- Xu J, Cui K, Shen L, Shi J, Li L, You L, et al. Crl activates transcription by stabilizing active conformation of the master stress transcription initiation factor. Elife. 2019;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L, Taft JM, Li Q, Gebhard MC, Georgiou G, Iverson BL. Yeast endoplasmic reticulum sequestration screening for the engineering of proteases from libraries expressed in yeast. Methods Mol Biol. 2015;1319:81–93. [DOI] [PubMed] [Google Scholar]
- Zanghellini A, Jiang L, Wollacott AM, Cheng G, Meiler J, Althoff EA, et al. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006;15(12):2785–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Chalt BT. Protein epitope mapping by mass spectrometry. Anal Chem. 1994;66(21):3723–3726. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting Information.