

ABSTRACT
Biofilm formation by the Gram-negative, Gammaproteobacteria Pseudomonas fluorescens relies on the repeats-in-toxin adhesins LapA and MapA in the cytoplasm, secretion of these adhesins through their respective type 1 secretion systems, and retention at the cell surface. Published work has shown that retention of the adhesins occurs via a post-translational mechanism involving the cyclic-di-GMP receptor LapD and the protease LapG. However, little is known about the underlying mechanisms that regulate the level of these adhesins. Here, we demonstrate that the master regulator FleQ modulates biofilm formation by both transcriptionally and post-transcriptionally regulating LapA and MapA. We find that a ΔfleQ mutant has a biofilm formation defect compared to the wild-type (WT) strain, which is attributed in part to a decrease in LapA and MapA abundance in the cell, despite the ΔfleQ mutant having increased levels of lapA and mapA transcripts compared to the WT strain. Through transposon mutagenesis and subsequent genetic analysis, we found that overstimulation of the Gac/Rsm pathway partially rescues biofilm formation in the ΔfleQ mutant background. Collectively, these findings provide evidence that FleQ regulates biofilm formation by both transcriptionally regulating the expression of the lapA and mapA genes and post-transcriptionally regulating the abundance of LapA and MapA, and that activation of the Gac/Rsm pathway can post-transcriptionally enhance biofilm formation by P. fluorescens.
IMPORTANCE
Biofilm formation is a highly coordinated process that bacteria undergo to colonize a variety of surfaces. For Pseudomonas fluorescens, biofilm formation requires the production and localization of repeats-in-toxin adhesins to the cell surface. To date, little is known about the underlying mechanisms that regulate biofilm formation by P. fluorescens. Here, we identify FleQ as a key regulator of biofilm formation that modulates both gene expression and abundance of LapA and MapA through both a transcriptional and post-transcriptional mechanism. We provide further evidence implicating activation of the Gac/Rsm system in FleQ-dependent regulation of biofilm formation. Together, our findings uncover evidence for a dual mechanism of transcriptional and post-transcriptional regulation of the LapA and MapA adhesins.
KEYWORDS: biofilm, regulation, Pseudomonas fluorescens, RTX adhesins
INTRODUCTION
Pseudomonas fluorescens is a Gram-negative, Gammaproteobacteria that is broadly distributed in the environment, and is commonly thought of as a plant commensal bacterium (1). P. fluorescens colonizes plant roots and survives on nutrients secreted by these plants while producing antimicrobial and antifungal metabolites that are secreted into the surrounding rhizosphere (2 - 4). While commonly associated with the environment, P. fluorescens has become a clinically relevant organism in that it has been associated with cases of hospital-acquired bacteremia via the contamination of hospital products (5 - 9) and has been associated with Crohn’s disease whereby P. fluorescens colonizes the intestines and increases the permeability of intestinal epithelial cells (10 - 14). To persist in these diverse environments, P. fluorescens relies on the highly coordinated transition from a motile to biofilm lifestyle (15, 16)
For P. fluorescens, biofilm formation is predominantly dependent on the repeats-in-toxin (RTX) adhesins LapA and MapA, which mediate surface attachment and contribute to biofilm initiation and maturation when localized to the cell surface (17, 18). Localization of the adhesins to the cell surface is mediated by each adhesin’s respective type 1 secretion systems, although recent evidence suggests that the Lap system secretes MapA in the absence of LapA (18). Cell surface retention of the adhesins is mediated by the cyclic-di-GMP sensing protein LapD, which sequesters the periplasmic protease LapG when levels of cyclic-di-GMP are high. When cyclic-di-GMP levels are depleted, LapG is free to cleave the N-terminus of LapA and MapA at their characteristic TAAG motif, which releases these adhesins from the cell surface where they no longer contribute to biofilm formation (19). Additionally, previous work has shown that levels of cyclic-di-GMP can be altered in response to specific metabolites, such as inorganic phosphate and citrate, via stimulation of the phosphodiesterase RapA and the diguanylate cyclase GcbC, respectively, which in turn affects LapD-dependent adhesin retention (20 - 23).
Apart from LapD-dependent regulation of adhesin retention, additional regulatory mechanisms affecting adhesin levels have been poorly characterized for P. fluorescens. The conserved transcriptional regulator FleQ has been shown to regulate a variety of targets related to biofilm formation, such as motility and exopolysaccharide synthesis, in multiple Pseudomonas spp. (24 - 29). FleQ has been associated with motility of P. fluorescens and there is some evidence that FleQ may regulate lapA gene expression (29, 30). Here, we demonstrate that FleQ both transcriptionally and post-transcriptionally regulates biofilm formation by simultaneously modulating lapA and mapA gene expression and overall levels of LapA and MapA, and we provide evidence that the post-transcriptional regulation occurs through the Gac/Rsm pathway.
RESULTS
FleQ regulates biofilm formation through the post-transcriptional regulation of the lapA and mapA genes
To assess the role of FleQ in biofilm formation, we made a chromosomal deletion of the fleQ gene in P. fluorescens Pf0-1 and probed the mutant for biofilm formation in a static biofilm assay using K-arginine(KA) medium, a minimal medium supplemented with arginine, which has previously been shown to support the formation of robust, LapA- and MapA-dependent biofilms (18). After 16 h of growth, the ΔfleQ mutant showed a significant decrease in biofilm formation compared to the wild-type (WT) strain (Fig. 1A). When the fleQ deletion mutation was complemented at the att site with the WT fleQ gene, biofilm formation was restored to levels similar to the WT strain (Fig. 1A). We additionally grew the WT and ΔfleQ mutant in a microfluidic device with continuous irrigation of KA medium and imaged the biofilm after 5 days of growth at room temperature. As with the static biofilm assay, the ΔfleQ mutant similarly has a biofilm formation defect under flow (Fig. S1).
Fig 1.

A FleQ-deficient strain has a biofilm formation defect due to a decrease in adhesin abundance. (A) Quantification of the biofilm formed by the WT strain, ΔfleQ mutant, and a ΔfleQ mutant complemented with the WT fleQ gene at the att site measured at an optical density of 550 nm (OD550) after 16 h of growth in KA minimal medium. Statistical significance was determined using an unpaired t-test. ****, P < 0.0001. (B) Swim zone (in millimeters) of the WT strain, ΔfleQ mutant, and a ΔfleQ mutant complemented with the WT fleQ gene at the att site after toothpick inoculation on KA medium supplemented with 0.3% agar after 24 h of growth at 30°C. Statistical significance was determined using an unpaired t-test. ****, P < 0.0001. (C) Gene expression of lapA, lapE, mapA, and mapE genes in the ΔfleQ mutant relative to the WT strain via quantitative reverse transcription PCR, after 16 h of growth on KA medium supplemented with 1.5% agar (KA agar), using the 2-ΔΔCt method, commonly referred to as the Livak method (31). For statistical significance, paired t-tests were conducted for each gene between the WT strain and ΔfleQ mutant. *, P < 0.05; **, P < 0.01. (D and F) Quantification of cell surface-associated LapA after 16 h of growth on KA agar (D) or MapA after 24 h of growth on KA agar (F) for the WT strain and ΔfleQ mutant using ImageJ by measuring the mean gray value of each spot using a pre-defined region of interest (ROI) and subtracting the background, which was determined by measuring the mean gray value of a section of the blot without any sample. Representative images are included above each graph. Statistical significance was determined using unpaired t-tests. *, P < 0.05; ***, P < 0.001. (E,G) Quantification of LapA after 16 h of growth on KA agar (E) or MapA after 24 h of growth on KA medium (G) from whole cell lysates that were prepared from 25 mL of cultures, concentrated to 100 µL in 3 mg/mL lysozyme with sonication, and quantified for total protein using the bicinchoninic acid (BCA) assay. Twenty-five micrograms (E) or 50 µg (G) of total protein was resolved on a 7.5% TGX gel and then blotted for LapA or MapA, respectively. Representative images are included above each graph. Statistical significance was determined using unpaired t-tests. *, P < 0.05; **, P < 0.01. All error bars represent standard deviation.
Previous work demonstrated that a P. fluorescens Pf0-1 transposon insertion mutant in FleQ is non-motile and that FleQ regulates motility through the transcriptional regulation of a subset of the flagellar assembly genes in Pseudomonas ogarae F113, a recently reclassified strain of P. fluorescens (30, 32 - 34). We thus assessed motility of the ΔfleQ mutant via swim assay using KA medium supplemented with 0.3% agar, which revealed that this mutant is unable to swim (Fig. 1B). When the fleQ deletion mutant was complemented at the att site with the WT fleQ gene, swimming motility was restored to levels similar to the WT strain (Fig. 1B).
Previous work showed that biofilm formation by P. fluorescens Pf0-1 is dependent on LapA and MapA and their localization to the cell surface (17, 18). To determine the effect(s) of FleQ on the abundance of these adhesins, we first assessed the expression levels of the genes encoding LapA and MapA, as well as the genes encoding their respective outer membrane porins LapE and MapE, in the WT and ΔfleQ mutant grown on KA agar. Interestingly, the ΔfleQ mutant shows modest (~1.8- to 5-fold) but significant increase in expression of all four genes compared to the WT strain (Fig. 1C).
Given the observed biofilm formation defect despite the increase in expression of the lap and map genes in the ΔfleQ mutant, we next assessed whether LapA or MapA protein level was negatively impacted by the absence of FleQ. To assess the impact on the LapA and MapA proteins, we probed for these adhesins on the cell surface using dot blot analysis and at the whole cell level using whole cell lysate Western blot, as described in the Materials and Methods. After 16 h of growth on KA agar, LapA is present at the cell surface and readily detected in whole cell lysates of the WT strain, while the ΔfleQ mutant has no detectable LapA at the cell surface or in whole cell lysates (Fig. 1D and E).
Interestingly, MapA was not readily detected at the cell surface of the WT strain or the ΔfleQ mutant after 16 h of growth (Fig. S2). However, when growth on KA agar was increased to 24 h, MapA is detected at the cell surface in the WT strain, while MapA is decreased in the ΔfleQ mutant at the cell surface (Fig. 1F). Interestingly, despite scaling up the amount of cells harvested and increasing the protein concentration, levels of MapA in the WT strain were below the limit of detection in whole cell lysates when grown on KA agar at 24 h. However, when cultures were grown in KA liquid medium for 24 h in a volume of 25 mL and then concentrated to 100 µL as described in Materials and Methods, MapA is detected in whole cell lysate in the WT while this protein showed reduced levels in the ΔfleQ mutant (Fig. 1G).
Genetic studies reveal that LapA and MapA are produced at reduced levels in a FleQ-deficient strain
While we were unable to readily detect the LapA or MapA proteins in the ΔfleQ mutant, we sought to determine if a basal level of these adhesins is still produced in this mutant, and whether forcing these adhesins to the cell surface can restore biofilm formation in the ΔfleQ mutant. Since previous work has shown that loss of LapG in the WT strain leads to the retention of LapA and MapA on the cell surface (35 - 37), we first made a chromosomal deletion of the lapG gene in the ΔfleQ mutant and assessed the ΔlapGΔfleQ double mutant for its ability to form a biofilm. After 24 h of growth in KA medium, the ΔfleQ ΔlapG mutant shows a significant increase in biofilm formation compared to the ΔfleQ mutant (Fig. 2A), but the biofilm is still reduced compared to the WT.
Fig 2.

A FleQ-deficient strain produces adhesin sufficient to restore biofilm formation in a lapG mutant. (A) Quantification of the biofilm formed for the WT and the ΔfleQ, ΔlapG, and ΔfleQΔlapG mutant strains measured at OD550 after 24 h of growth in KA minimal medium. This time point was chosen because both LapA and MapA are detected at the cell surface at this time point. (B and C) Quantification of cell surface-associated LapA after 16 h of growth on KA agar (B) or MapA after 24 h of growth on KA agar (C) in the WT, ΔfleQ, ΔlapG, and ΔfleQΔlapG strains as described in Fig. 1. Representative images are included above each graph. (D) Swim zone (in millimeters) for the WT, ΔfleQ, ΔlapG, and ΔfleQΔlapG strains after toothpick inoculation on KA supplemented with 0.3% agar and 24 h of growth at 30°C. Statistical significance for this figure was determined using one-way analyses of variance (ANOVAs) with Tukey’s multiple comparisons tests. **, P < 0.01; ***, P < 0.001. All error bars represent standard deviation.
Given these findings, we then assessed the ΔfleQ ΔlapG mutant for surface-associated LapA and MapA using dot blot analysis at 16 h or 24 h, respectively, as described in Fig. 1. Here, we see that LapA and MapA are both detected at the cell surface in the ΔfleQ ΔlapG mutant but not in the ΔfleQ mutant (Fig. 2B and C). This result suggests that LapA and MapA are being produced, but below the limit of detection of the whole cell lysate Western blot in a FleQ-deficient strain. This finding indicates that these proteins can be detected if locked on the cell surface in the lapG mutant background (Fig. 2B and C). Consistent with this result, we find that over-expressing the LapBCE ABC transporter, required for LapA surface localization (17) and that we showed previously increases LapA surface localization (38), also partially rescues the biofilm defect of the fleQ mutant (Fig. S3).
While LapG-dependent regulation has not previously been associated with motility, we also assessed the ΔfleQ ΔlapG mutant for any impacts on motility using a swim assay. After 24 h of growth, the ΔfleQ ΔlapG mutant, like the ΔfleQ mutant, was unable to swim, while the WT strain and the ΔlapG mutant had similar swim zones (Fig. 2D).
To further demonstrate that levels of LapA are reduced in a FleQ-deficient strain, rather than being released from the cell surface by LapG, we quantified cyclic-di-GMP via liquid chromatography mass spectrometry (LC-MS/MS) from the WT strain and ΔfleQ mutant, that is, lower levels of cyclic-di-GMP would be expected to allow increased activity of the LapG protease. After 16 h of growth on KA agar, the WT strain and ΔfleQ mutant have similar levels of cyclic-di-GMP (Fig. S4A), indicating that the LapDG system was unlikely to be impacting adhesin localization.
Given the lack of a supernatant fraction when grown on agar, we assessed the WT strain and ΔfleQ mutant for secreted LapA by growing these strains in KA liquid medium and probing the supernatant fraction for LapA via Western blot, as described in the Materials and Methods. After 16 h of growth in KA liquid medium, LapA is readily detected in the supernatant fraction of the WT strain while the ΔfleQ mutant has little-to-no detectable LapA (Fig. S4B). To confirm that growth of the WT strain in KA liquid medium does not impact adhesin retention, we probed for LapA at the cell surface using dot blot analysis. After 16 h of growth in KA liquid medium, LapA is readily detected at the cell surface of the WT strain, whereas very little LapA is detected in the ΔfleQ mutant (Fig. S4C). Together, these data argue against a model whereby loss of biofilm formation is due to the inability to retain the LapA adhesin on the cell surface.
Mutational analysis of the lapA promoter reveals that FleQ transcriptionally regulates lapA
Previous work investigating FleQ-dependent gene regulation of Pseudomonas aeruginosa demonstrated that FleQ transcription occurs through the differential binding of the target promoter at two binding boxes. This work identified a binding box consensus sequence that was used to predict putative FleQ-regulated genes in other Pseudomonas spp. (28, 29). Interestingly, two FleQ binding boxes were identified within the predicted promoter region of the lapA gene of P. fluorescens Pf0-1 (29). To assess the function of these putative binding boxes, we mutagenized either Box 1 or Box 2 of the WT strain or the ΔfleQ mutant by altering the guanine in position 1 and cytosine in position 14 to adenine (Fig. 3A). We selected these nucleotides as they are the most conserved within the binding box consensus sequence (29).
Fig 3.

Mutagenizing the FleQ consensus sequences in the lapA promoter has a transcriptional effect that is masked in the ΔfleQ mutant. (A) Schematic showing the divergent lapA/lapE promoter. FleQ binding Box 1 and Box 2 are illustrated, and the respective sites of the point mutations are highlighted in bold. The lapA and lapE start codons are highlighted in bold and direction of translation is indicated by an arrow. The previously published FleQ binding consensus sequence and amino acid conservation of the two predicted boxes can be found in Baraquet and Harwood (29). (B) Quantification of the biofilm formed for the WT and the ΔfleQ mutant with a mutated FleQ binding Box 1 or Box 2, or the WT sequence at OD550 after 24 h of growth in KA minimal medium. (C) Swim zone (in millimeters) of WT and ΔfleQ strains with a mutated FleQ binding Box 1 or Box 2, or the WT sequence after toothpick inoculation on KA medium supplemented with 0.3% agar and 24 h of growth at 30°C. Statistical significance for this figure was determined using one-way ANOVAs with Tukey’s multiple comparisons tests. **, P < 0.01. All error bars represent standard deviation.
We then assessed these mutants for their ability to form a biofilm with the static biofilm assay. After 24 h of growth in KA medium, the Box 1 mutant in the WT and ΔfleQ mutant backgrounds showed no significant change in biofilm formation compared to their parent strains. However, the Box 2 mutant in the WT background showed a significant decrease in biofilm formation compared to the WT strain (Fig. 3B). The Box 2 mutant in the ΔfleQ mutant background showed no significant change in biofilm formation as compared to the ΔfleQ parent strain, but it is important to note that the ΔfleQ mutant already makes a much reduced biofilm compared to the WT. These data, along with the previously shown expression and protein data (Fig. 1D and E), suggest that FleQ may regulate lapA transcription via the Box 2 binding site.
We used the swim assay to verify that motility in these strains were unaffected, since altering the binding sites in the lapA promoter should not impact FleQ-dependent regulation on motility. As expected, the lapA Box 1 and Box 2 mutants had similar swim zones to their parent strains (Fig. 3C), demonstrating that the changes in biofilm formation were not a consequence of reduced motility. These data also support the conclusion that loss of FleQ impacts biofilm formation, in part, by reduced abundance of LapA.
Transposon mutagenesis reveals additional factors that contribute to FleQ-dependent regulation of biofilm formation
The observations above that mutating the fleQ gene results in a modest (but significant) increase in lapA and mapA gene expression, but a large reduction in whole cell (and thus surface) levels of the LapA and MapA proteins, suggest that FleQ plays a positive, post-transcriptional role in the regulation of LapA and MapA proteins. Given the hypothesized post-transcriptional effect observed for the ΔfleQ mutant, we sought to identify additional factors that contribute to FleQ-dependent biofilm formation by performing a genetic screen that restores biofilm formation to the fleQ mutant. Briefly, the ΔfleQ mutant was conjugated as recipient with an Escherichia coli strain donor containing a plasmid harboring the TnM mariner transposon, and ΔfleQ mutants with transposon insertion were selected by growth on lysogeny broth (LB) agar supplemented with gentamycin (selection) and chloramphenicol (counter-selection). Individual insertion mutants were screened for their ability to form a biofilm, and candidates with a significant increase in biofilm formation compared to the parent strain were chosen for further analysis.
The location of transposon insertion in these biofilm-forming candidate strains was determined via arbitrary primed PCR and sequencing of the flanking regions, as described in the Materials and Methods. From this screen, we identified four transposon insertions that mapped to the lapG gene coding sequence, the flhA gene coding sequence, intergenic region between the flhA and flhF genes, and the promoter region of the gacS gene (Fig. 4A). All four transposon insertions showed a significant increase in biofilm formation compared to the ΔfleQ parental strain after 24 h of growth on KA medium (Fig. 4B). We also assessed the motility of all four transposon insertions; none of the four mutants were able to swim on KA supplemented with 0.3% agar (Fig. 4C), suggesting that the increase in biofilm formation was not related to changes in motility.
Fig 4.

Identifying other factors that contribute to FleQ-dependent regulation of adhesins. (A) Schematic showing the location of FleQ-deficient transposon mutants that restore biofilm formation. Triangles indicate insertion into the genome and the arrow above each triangle indicates directionality of the Ptac promoter. Genomic coordinates are included for reference. (B) Quantification of the biofilm formed by the WT strain, the ΔfleQ mutant and the indicated ΔfleQ derivatives at OD550 after 24 h of growth in KA minimal medium. (C) Swim zone (in millimeters) measurements of the WT strain, the ΔfleQ mutant, and ΔfleQ derivatives after toothpick inoculation on KA medium supplemented with 0.3% agar and 24 h of growth at 30°C. Statistical significance for this figure was determined using one-way ANOVAs with Tukey’s multiple comparisons tests. ****, P < 0.0001. All error bars represent standard deviation.
Stimulation of the Gac/Rsm pathway rescues biofilm formation in a FleQ-deficient strain
Given our findings from the genetic screen, we hypothesized that the changes in biofilm formation for the four mutants identified were likely due to changes in adhesin abundance or their localization to the cell surface. For the lapG insertion mutant, we have already demonstrated that the loss of LapG in a ΔfleQ mutant leads to an increase in biofilm formation, which is due to increased localization of LapA and MapA to the cell surface (Fig. 2A through C). Thus, identifying a mutation in the lapG gene served to help validate the screening approach.
Our preliminary genetic analysis suggests that the flhAF operon may be involved in biofilm formation in a FleQ-independent manner (data not shown). Analysis of the flhAF operon and its role in biofilm formation will be addressed in a separate study.
We further analyzed the impact of the insertion mutation in the promoter of the gacS gene. First, given that the transposon carries a constitutive Ptac promoter oriented in a manner that could drive expression of the gacS gene (Fig. 4A), we assessed the impact of the insertion on gacS gene expression through quantitative reverse transcription PCR. The insertion mutation in the promoter of the gacS gene results in expression of this gene to a level that is ~20 times higher than the gacS gene expressed in the ΔfleQ mutant or the WT strain (Fig. S5).
Previous work in P. aeruginosa and P. fluorescens (now P. protegens) demonstrated that the Gac/Rsm pathway is regulated by the GacS-GacA two component system. The sensor kinase GacS phosphorylates its response regulator GacA, which then binds to the upstream activating sequence of its targets and upregulates transcription of small regulatory RNAs (39 - 44). For P. fluorescens, these small regulatory RNAs are known to bind the RsmA, RsmE, and RsmI proteins (collectively, the “Rsm proteins”) and thereby prevent the Rsm proteins from binding to the mRNA of their target genes (42, 45 - 47).
Given the increased biofilm formation of the ΔfleQ mutant carrying an insertion mutation in the promoter of the gacS gene, and the increased level of gacS expression in this genetic background, we hypothesized that increased activation of the Gac/Rsm pathway might result in increased biofilm formation. To simulate constitutive overexpression of the Gac/Rsm pathway, we made chromosomal deletions of the rsmA, rsmE, and rsmI genes, which code for the three Rsm proteins, in both the WT strain and ΔfleQ mutant. We assessed the ΔrsmA ΔrsmE ΔrsmI triple mutant as well as individual deletion mutants for biofilm formation. After 24 h of growth in KA medium, the ΔrsmA ΔrsmE ΔrsmI triple mutant shows a significant increase in biofilm formation in the ΔfleQ background (Fig. 5A). The ΔrsmE single mutant also shows a significant increase in the ΔfleQ background, whereas the ΔrsmA and ΔrsmI single mutants show no significant difference in biofilm formation in either strain background (Fig. S6A), suggesting that this phenotype may be largely RsmE-dependent. We then overexpressed RsmE from the arabinose-inducible high-copy plasmid pMQ72 in the ΔrsmE single mutant and ΔrsmA ΔrsmE ΔrsmI triple mutant backgrounds and assessed these strains for biofilm formation. After 24 h of growth in KA medium in the absence of inducer, biofilm formation was significantly reduced in the ΔrsmE and ΔfleQ ΔrsmE strains with the RsmE overexpression construct (Fig. S7A). Similarly, overexpression of RsmE in the ΔrsmA ΔrsmE ΔrsmI and ΔfleQ ΔrsmA ΔrsmE ΔrsmI mutants leads to significant reduction in biofilm formation after 24 h of growth in KA medium without inducer (Fig. S7B).
Fig 5.
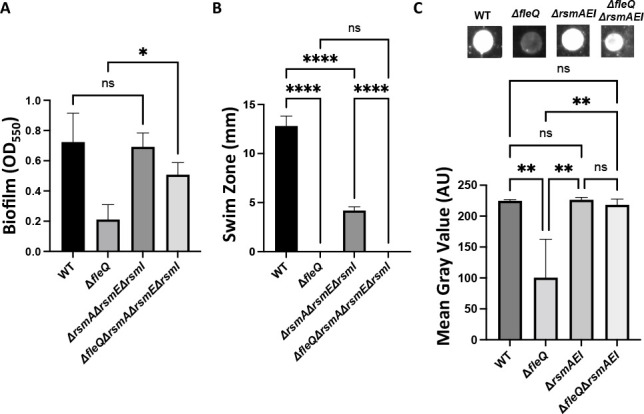
Modulation of the Gac/Rsm system increases biofilm formation in a WT and FleQ-deficient strain. (A) Quantification of the biofilm formed by the WT, ΔfleQ, ΔrsmAΔrsmEΔrsmI, and ΔfleQ ΔrsmAΔrsmEΔrsmI strains measured at OD550 after 24 h of growth in KA minimal medium. (B) Swim zone (in millimeters) of the WT, ΔfleQ, ΔrsmAΔrsmEΔrsmI, and ΔfleQ ΔrsmAΔrsmEΔrsmI strains after toothpick inoculation on KA medium supplemented with 0.3% agar and 24 h of growth at 30°C. (C) Quantification of cell surface-associated LapA after 16 h of growth on KA agar as described in Fig. 1. Representative images are included above each graph. Statistical significance for this figure was determined using one-way ANOVAs with Tukey’s multiple comparisons tests. *, P < 0.05; **, P < 0.01; ****, P < 0.0001. All error bars represent standard deviation.
Given that the Gac/Rsm pathway has been shown to regulate motility in various pseudomonads, we assessed these mutants for motility on KA medium supplemented with 0.3% agar. After 24 h of growth, the ΔrsmA, ΔrsmE, and ΔrsmI single mutants show no significant difference in motility compared to the WT or ΔfleQ parental strains (Fig. S6B). Interestingly, the ΔrsmA ΔrsmE ΔrsmI triple mutant in the WT background showed a significant decrease in motility compared to the parental strain, whereas the triple mutant in the ΔfleQ background was non-motile like the parental strain (Fig. 5B). Growth curves in KA minimal medium revealed that the ΔrsmA ΔrsmE ΔrsmI triple mutant in the WT background has a growth defect when grown planktonically (Fig. S8), which likely explains the strain’s decrease in motility compared to the WT strain.
Given these biofilm and motility results, we then assessed LapA localization on the cell surface of the ΔrsmE single mutant and ΔrsmA ΔrsmE ΔrsmI triple mutant via dot blot analysis. After 16 h of growth on KA agar, the ΔrsmA ΔrsmE ΔrsmI and ΔrsmE mutants showed a significant increase in LapA surface levels in the ΔfleQ background, but not the WT background (Fig. 5C; Fig. S6C).
Since the biofilm results suggest specificity among the Rsm proteins, we assessed the specificity of the small regulatory RNAs by overexpressing the RsmX, RsmY, or RsmZ sRNAs in a WT strain or ΔfleQ mutant from the isopropyl β- d-1-thiogalactopyranoside (IPTG)-inducible plasmid pMQ123 and assessed the strains for biofilm formation. After 24 h of growth in KA medium supplemented with 1 mM IPTG for induction, the ΔfleQ mutant expressing RsmZ showed a modest but statistically significant increase in the biofilm formed compared to growth in KA alone (Fig. 6A). However, no impact on biofilm formation was observed when any of the small regulatory RNAs were overexpressed in the WT strain (Fig. S9A).
Fig 6.

Overexpressing the small regulatory RNA RsmZ in a FleQ-deficient increases biofilm formation. (A) Quantification of the biofilm formed by the ΔfleQ strain with and without pMQ123, pMQ123-RsmX, pMQ123-RsmY, or pMQ123-RsmZ measured at OD550 after 24 h of growth in KA minimal medium with or without 1 mM IPTG for induction. (B) Quantification of the biofilm formed by the ΔfleQΔrsmAΔrsmI strain with and without the pMQ123-RsmZ construct measured at OD550 after 24 h of growth in KA minimal medium with or without 1 mM IPTG for induction. (C) Quantification of cell surface-associated LapA after 16 h of growth on KA agar with or without 1 mM IPTG for induction as described in Fig. 1. Statistical significance for this figure was determined using two-way ANOVAs with Tukey’s multiple comparisons tests. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All error bars represent standard deviation.
Given this finding, we then overexpressed RsmZ in a ΔrsmA ΔrsmI or ΔfleQ ΔrsmA ΔrsmI mutant, which still has the ability to produce RsmE, and assessed the strain for biofilm formation. After 24 h of growth in KA medium supplemented with 1 mM IPTG for induction, the ΔfleQ ΔrsmA ΔrsmI mutant overexpressing RsmZ showed a significant increase in the biofilm formed compared to growth in KA alone (Fig. 6B), whereas overexpressing RsmZ had little effect on the ΔrsmA ΔrsmI mutant strain (Fig. S9B). We then assessed the FleQ-deficient strains for cell surface levels of LapA via dot blot analysis. After 16 h of growth on KA agar supplemented with 1 mM IPTG, the ΔfleQ ΔrsmA ΔrsmI mutant overexpressing RsmZ showed a significant increase in LapA surface levels compared to growth on KA agar alone (Fig. 6C).
A mutation in the gacA gene does not impact early biofilm formation in KA medium
Previous work in P. aeruginosa demonstrated that the Gac/Rsm pathway regulates biofilm formation in both the PA01 and PA14 strains and that deletion of either gacS or gacA, which disrupts the Gac/Rsm pathway, leads to a large reduction in biofilm formation (39, 44). To assess the impact of disrupting the Gac/Rsm pathway in P. fluorescens Pf0-1, we made a chromosomal deletion of gacA in the WT and ΔfleQ strains and assessed these mutants for biofilm formation. Interestingly, after 24 h of growth in KA medium, the ΔgacA mutant showed no difference in biofilm formation compared to the WT strain and the ΔgacAΔfleQ mutant showed no difference in biofilm formation compared to the ΔfleQ mutant (Fig. 7A).
Fig 7.

The Gac/Rsm system contributes to LapA abundance. (A) Quantification of the biofilm formed by the WT, ΔfleQ, ΔgacA, and ΔfleQΔgacA strains measured at OD550 after 24 h of growth in KA minimal medium. (B) Quantification of cell surface-associated LapA after 16 h of growth on KA agar as described in Fig. 1. Statistical significance for this figure was determined using one-way ANOVAs with Tukey’s multiple comparisons tests. *, P < 0.05; **, P < 0.01; ***, P < 0.001. All error bars represent standard deviation. (C) Swim zone (in millimeters) of the WT, ΔfleQ, ΔgacA, and ΔfleQΔgacA strains after toothpick inoculation on KA supplemented with 0.3% agar and 24 h of growth at 30°C.
The ΔgacA and ΔgacAΔfleQ mutants were assessed for cell surface levels of LapA via dot blot analysis. After 16 h of growth on KA agar, the ΔgacA mutant showed no significant difference in surface LapA compared to the WT strain, and the ΔfleQ ΔgacA mutant shows no significant difference in surface LapA compared to ΔfleQ strain (Fig. 7B).
Given that the Gac/Rsm pathway has been implicated in regulating motility, we next assessed the motility of these mutants on KA medium supplemented with 0.3% agar. The ΔgacA mutant showed a modest but significant increase in swimming motility compared to the WT strain, which is consistent with previous work in other pseudomonads. The ΔfleQ ΔgacA mutant also showed no significant difference in swimming motility from the parental ΔfleQ strain; both strains were non-motile (Fig. 7C). Together, these results suggest that the Gac/Rsm pathway is not strictly required for biofilm formation in P. fluorescens Pf0-1, but that activation of the pathway can enhance biofilm formation.
Replacing the native lapA promoter with the constitutive Plac promoter partially overcomes FleQ-dependent regulation of LapA
Given that FleQ post-transcriptionally regulates LapA abundance, we posited that increasing lapA expression by replacing the native lapA promoter with the non-native, constitutively active Plac promoter would partially rescue biofilm formation in a FleQ-deficient strain.
We inserted the constitutive Plac promoter in front of the lapA open reading frame in the WT and ΔfleQ strains, replacing the regulatory and the 5´ untranslated regions of the lapA gene, and assessed these strains for biofilm formation. After 24 h of growth in KA medium, the strain carrying the Plac promoter in the ΔfleQ background showed significantly more biofilm formed compared to the parental strain (Fig. 8A). In contrast, the strain carrying the Plac promoter in WT background showed no difference in the biofilm formed compared to the parent strain (Fig. 8A).
Fig 8.

Replacing the native lapA promoter with a non-native FleQ-independent promoter partially restores biofilm formation in a FleQ-deficient strain. (A) Quantification of the biofilm formed by the WT and ΔfleQ strains with the native lapA or Plac promoter measured at OD550 after 24 h of growth in KA minimal medium. (B) Quantification of cell surface-associated LapA after 16 h of growth on KA agar as described in Fig. 1. (C) Swim zone (in millimeters) of the WT and ΔfleQ strains with the native lapA or Plac promoter strains after toothpick inoculation on KA supplemented with 0.3% agar and 24 h of growth at 30°C. Statistical significance for this figure was determined using one-way ANOVAs with Tukey’s multiple comparisons tests. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All error bars represent standard deviation.
We next assessed the Plac promoter variants for LapA levels on the cell surface via dot blot analysis. After 16 h of growth on KA agar, the WT Plac promoter variant showed no significant difference in cell surface-associated LapA as compared to the WT parental strain (Fig. 8B). In contrast, the ΔfleQ Plac::lapA strain showed significantly more cell surface-associated LapA as compared to the ΔfleQ parental strain. This increased LapA likely accounts for the increased biofilm formation observed for the ΔfleQ Plac::lapA strain. Collectively, these data indicate that sequences within the lapA promoter region are required to mediate FleQ-dependent regulation of this adhesin, a finding consistent with the phenotypes of the Box 2 mutants described above.
To confirm that driving lapA expression from the constitutive Plac promoter does not impact motility, we assessed these strains on motility agar. As expected, the WT carrying the Plac promoter swam like its parental strain, whereas the Plac promoter variant of the ΔfleQ background was non-motile, like the ΔfleQ mutant (Fig. 8C).
DISCUSSION
Our data show that FleQ both transcriptionally and post-transcriptionally regulates biofilm formation by P. fluorescens. Our genetic analyses show that the loss of FleQ leads to a decrease in biofilm formation, which is attributed, at least in part, to the reduction of LapA and MapA abundance, and thus the lack of these adhesins on the cell surface. We also note that the FleQ-deficient strain is unable to swim, which also likely contributes to the biofilm defect. Whereas FleQ-dependent regulation is typically thought of as being completely transcriptional (26, 28, 29), our qRT-PCR analysis reveal that transcription of lapA and mapA genes are increased in a FleQ-deficient strain compared to a WT strain despite a reduction in LapA/MapA protein level, which suggests that FleQ-dependent regulation of these adhesins is, at least in part, post-transcriptional. Our subsequent mutagenic study of the predicted FleQ binding boxes in the lapA promoter (29), along with the qRT-PCR data, suggests that FleQ does have a transcriptional effect on the lapA gene, but this alone is not sufficient to explain the decrease in biofilm formation in the FleQ-deficient strain. Despite the loss of biofilm formation, additional genetic and biochemical analyses utilizing our knowledge of LapD-dependent biofilm formation (35) show that a basal amount of LapA and MapA is still produced in a FleQ-deficient strain, which is sufficient to promote biofilm formation in LapG-deficient background. Interestingly, we did not observe restoration of biofilm formation to WT levels in the ΔlapG mutant, which we attribute to the loss of motility in a FleQ-deficient strain, as motility plays a key positive role in early biofilm formation by this organism (48).
Our transposon mutagenesis identified four candidates that were able to restore biofilm formation in a FleQ-deficient strain, but did not restore swimming motility. These candidates included the histidine-kinase GacS that has been previously implicated in post-transcriptional regulation. This mutant showed high levels of gacS gene expression, which suggests overstimulation of the Gac/Rsm pathway. We subsequently found that mimicking an overstimulation of this pathway by deleting all of the target downstream regulators RsmA, RsmE, and RsmI restored biofilm formation in a FleQ-deficient strain, much like the gacS::TnM mutant from the transposon screen. Interestingly, when deleting these regulators individually, we only observed a restoration of biofilm formation when the rsmE gene is deleted. This observation is likely due to the nature of the Rsm proteins having distinct regulomes with a subset of overlapping targets, which has been previously demonstrated in Pseudomonas putida (49). We similarly stimulated the Gac/Rsm pathway by overexpressing the small regulatory RNAs and found that biofilm formation was only restored in a FleQ-deficient strain when RsmZ was expressed from a multi-copy plasmid. This specificity is likely due to the variability of binding affinities between the small regulatory RNAs and Rsm proteins, which has previously been demonstrated for P. aeruginosa (46).
Interestingly, previous findings demonstrated that the P. fluorescens Pf0-1 gacA gene contains the point mutation N109P and that this strain is deficient in certain phenotypes commonly regulated by the Gac/Rsm pathway, such as biosurfactant or siderophore production. A merodiploid strain was then generated by inserting the gacA gene from P. protegens Pf-5 at a neutral att site of P. fluorescens Pf0-1, thus producing a P. fluorescens Pf0-1 strain containing both the native gacA and non-native gacAPf-5. The addition of gacAPf-5 notably restored these Gac/Rsm-associated phenotypes in the P. fluorescens Pf0-1 merodiploid strain, which led to the initial conclusion that the P. fluorescens Pf0-1 Gac/Rsm system was completely deficient (50). A subsequent study revealed that inserting the gacAPf-5 allele, and generating the previously mentioned merodiploid strain, in a P. fluorescens Pf0-1 ΔfleQ (previously ΔadnA) mutant restored biofilm formation relative to the ΔfleQ strain, which initially linked the Gac/Rsm pathway to biofilm formation (51). These published findings along with our results suggest that the native P. fluorescens Pf0-1 Gac/Rsm system is attenuated rather than completely deficient compared to other P. fluorescens strains, likely due to the previously described N109P point mutation, and that overstimulation of the pathway via overexpressing the native gacS gene or modulating the native downstream regulators is sufficient to induce the Gac/Rsm pathway and restore biofilm formation in a ΔfleQ mutant. These findings provide further evidence that the Gac/Rsm pathway directly regulates biofilm formation via modulation of the adhesins in a FleQ-dependent manner. The conclusion of an attenuated P. fluorescens Pf0-1 Gac/Rsm pathway would additionally explain why loss of GacA function is not sufficient to reduce biofilm formation in an otherwise WT strain. A recently published RNA-seq data set revealed that the gacA gene is significantly downregulated and the downstream regulators are marginally but not significantly different in a P. ogarae F113 (formerly P. fluorescens F113) FleQ-deficient strain compared to a WT strain (32), suggesting that the Gac/Rsm pathway activity is attenuated in a P. ogarae F113 FleQ-deficient strain. This published finding further implicates the Gac/Rsm pathway in the FleQ-dependent post-transcriptional regulation of biofilm formation and suggests that the loss of FleQ in P. fluorescens Pf0-1 likely further attenuates the Gac/Rsm pathway.
Given the complex transcriptional, post-transcriptional, and post-translational regulation among the individual components of the Gac/Rsm pathway and overlapping but distinct nature of the individual RsmA, RsmE, and RsmI regulomes, the FleQ-dependent regulation of specific components of the Gac/Rsm system in the context of biofilm formation and dynamics of the Rsm proteins and small regulatory RNAs in the context of biofilm formation will be explored further in subsequent studies. Collectively, our findings demonstrate that FleQ regulates biofilm formation via controlling the levels of the RTX adhesins, whereby FleQ both directly regulates gene expression of the adhesins and post-transcriptionally regulates protein abundance via the Gac/Rsm system.
MATERIALS AND METHODS
Strains and media used in this study
The strains and plasmids used in this study are listed in Table S1. P. fluorescens Pf0-1 and E. coli S17-1 λ-pir, SM10 λ-pir, and JM109 were used throughout this study. E. coli was routinely grown in LB and P. fluorescens was routinely grown on LB or on KA minimal medium, as previously defined by Collins et al. (18), containing 50 mM Tris-HCl (pH 7.4), 0.61 mM MgSO4, 1 mM K2HPO4, and 0.4% (wt/vol) L-arginine HCl, or KA supplemented with 1.5% agar. Medium was supplemented with 50 µg/mL carbenicillin for E. coli strains harboring the transposon-containing shuttle vector pBT20, miniTn7/pMQ56, or the Tn7 transposase-containing helper plasmid pTns3, or with 10 µg/mL gentamycin when harboring the allelic exchange plasmid pMQ30 or the expression plasmids pMQ72 and pMQ123. For P. fluorescens, the medium was supplemented with 150 µg/mL kanamycin when harboring pSMC21 or 30 µg/mL gentamycin when harboring pMQ72, pMQ123, or miniTn7 integrated into the att site.
Construction of in-frame chromosomal deletion mutants
For all chromosomal deletions, the allelic exchange vector pMQ30 was first digested with SmaI (New England BioLabs) and ~1.5 kb flanking regions of the targeted gene were amplified using Phusion polymerase (New England BioLabs) with primers containing 20 bp of homology to the pMQ30 SmaI cut-site at their 5´ end. All primers used in the study are listed in Table S2. These amplicons were inserted into pMQ30 using the NEBuilder HiFi DNA Assembly kit (New England BioLabs) according to the manufacturer specifications, and the constructs were electroporated in E. coli S17-1 λ-pir. After recovery in LB for 1 h at 30°C, cells were plated on LB agar with 10 µg/mL gentamycin; candidates were sequenced to confirm fragment insertion into the plasmid. Constructs were introduced into P. fluorescens through conjugation, whereby 1 mL of P. fluorescens and 1 mL of E. coli were mixed in a 2 mL microcentrifuge tube, pelleted, washed in LB, and then plated on LB agar supplemented with 30 µg/mL gentamycin and 30 µg/mL chloramphenicol to select for P. fluorescens merodiploids. Cells were then plated on LB agar without sodium chloride supplemented with 10% sucrose to facilitate looping out of the drug resistance cassette. Deletions were confirmed with PCR amplification and Sanger sequencing.
Construction of fleQ complementation mutant
For fleQ complementation, the vector miniTn7/pMQ56 was first digested with SmaI (New England BioLabs) and the fleQ gene and its native promoter were amplified using Phusion polymerase (New England BioLabs) with primers containing 20 bp of homology to the mTn7/pMQ56 SmaI cut-site at their 5´ end. All primers used here are listed in Table S2. The amplicon was inserted into miniTn7/pMQ56 using the NEBuilder HiFi DNA Assembly kit (New England BioLabs) according to the manufacturer specifications, and the constructs were electroporated in E. coli S17-1 λ-pir. After recovery in LB for 1 h at 30°C, cells were plated on LB agar supplemented with 10 µg/mL gentamycin; candidates were sequenced to confirm fragment insertion into the plasmid. Plasmid DNA was isolated using the QIAprep Spin Miniprep Kit (Qiagen) and introduced into P. fluorescens through conjugation with the construct and the helper plasmid pTns3 containing the miniTn7 transposase machinery, whereby 500 µL of the three strains was mixed in a 2 mL microcentrifuge tube. Cells were pelleted, washed in LB, and then plated on LB agar supplemented with 30 µg/mL gentamycin and 30 µg/mL chloramphenicol to select for miniTn7 insertion into P. fluorescens. Incorporation onto the genome was confirmed via PCR amplification and Sanger sequencing.
Construction of rsmE overexpression construct
For rsmE overexpression, the arabinose-inducible shuttle vector pMQ72 was first digested with SmaI (New England BioLabs) and the rsmE gene was amplified using Phusion polymerase (New England BioLabs) with primers containing 20 bp of homology to the pMQ72 SmaI cut-site at their 5´ end. The forward primer additionally contained the high-affinity T7 phage gene 10 ribosomal binding site (52) and 8 bp spacer between the pMQ72 homology and start codon. All primers used are listed in Table S2. The amplicons were inserted into pMQ72 downstream of the PBAD promoter using the NEBuilder HiFi DNA Assembly kit (New England BioLabs) according to the manufacturer specifications, and the constructs were electroporated in E. coli S17-1 λ-pir. After recovery in LB for 1 h at 30°C, cells were plated on LB agar supplemented with 10 µg/mL gentamycin; candidates were sequenced to confirm fragment insertion into the plasmid. Plasmid DNA was isolated using the QIAprep Spin Miniprep Kit (Qiagen) and introduced into P. fluorescens through electroporation. Cells were allowed to recover in LB for 1 h at 30°C and then were plated on LB agar supplemented with 30 µg/mL gentamycin to select for retention of the construct.
Construction of small regulatory RNA overexpression constructs
For all overexpression constructs, the lacI-containing shuttle vector pMQ123 was first digested with BamHI (New England BioLabs) and the targeted small regulatory RNAs were amplified using Phusion polymerase (New England BioLabs) with primers containing 20 bp of homology to the pMQ123 BamHI cut-site at their 5´ end. All primers used in the study are listed in Table S2. The amplicons were inserted into pMQ123 downstream of the Ptac promoter using the NEBuilder HiFi DNA Assembly kit (New England BioLabs) according to the manufacturer specifications, and the constructs were electroporated in E. coli S17-1 λ-pir. After recovery in LB for 1 h at 30°C, cells were plated on LB agar supplemented with 10 µg/mL gentamycin; candidates were sequenced to confirm fragment insertion into the plasmid. Plasmid DNA was isolated using the QIAprep Spin Miniprep Kit (Qiagen) and introduced into P. fluorescens through electroporation. Cells were allowed to recover in LB for 1 h at 30°C and then were plated on LB agar supplemented with 30 µg/mL gentamycin to select for retention of the construct.
Biofilm formation in static 96-well plate
P. fluorescens Pf0-1 strains were grown in 5 mL of LB at 30°C for ~16 h with agitation. 1.5 µL of inoculum was added to 100 µL of KA minimal medium in each well of a 96-well round bottom polypropylene plate, and the plate was incubated for the indicated time at 30°C in a humidified chamber. After incubation, supernatants were discarded and wells were washed once in water. One hundred twenty-five microliters of 0.1% (wt/vol) crystal violet was pipetted into each well and the plate was incubated for 45 min at room temperature. Wells were then washed twice in water to remove excess crystal violet, and then the plate was incubated at 37°C for 1 h to dry the wells. One hundred fifty microliters of a 45% methanol, 45% water, and 10% glacial acetic acid solution (destain solution) was added to each well and the plate was incubated for 5 min at 25°C. One hundred microliters was then transferred from each well to a 96-well flat bottom polystyrene plate and measured on a spectrophotometer at an optical density of 550 nm (OD550).
Biofilm formation in microfluidic devices
Bacterial strains were grown in 5 mL LB supplemented with 150 µg/µL kanamycin to select for the pSMC21 plasmid overnight at 30°C with agitation. One milliliter of overnight culture was transferred to a 1.5 mL microcentrifuge tube and the cells pelleted. Cell pellets were washed twice in KA medium and pellets resuspended in 100 µL of KA medium, and cultures were normalized to an OD600 of 1.5 in a 96-well plate. Microfluidic devices were assembled and operated as previously described in Collins et al. (18), except that in these studies, the medium used was KA. After 120 h of growth at room temperature, biofilms were imaged using a Nikon Eclipse Ti inverted microscope equipped with a Plan Fluor 40 DIC M N2 objective and Hamamatsu ORCA-Flash 4.0 camera. GFP was excited using a 488 nm laser and images were quantified using Fiji (ImageJ) (53), as reported (54). We imaged the base of the biofilm near the glass substratum.
Quantitative reverse transcription PCR
Bacterial strains were grown in 5 mL LB overnight at 30°C with agitation and then 400 µL of inoculum was pipetted onto KA medium supplemented with 1.5% agar. Following incubation, cells were scraped into 1 mL fresh KA medium using disposable cell scrapers with 20 mm blade width (VWR). Cells were pelleted with the respective supernatants discarded and then flash frozen in a dry ice-ethanol bath to preserve RNA integrity and stored at −80°C. In preparation for RNA extraction, pellets were thawed and resuspended in 100 µL of 3 mg/mL lysozyme and incubated at room temperature for 3 min. RNA was then extracted and isolated from cells using the Zymo Direct-zol RNA miniprep kit with TRI Reagent according to the manufacturer specifications. The isolated RNA was then treated twice with the TURBO DNA-free kit (Invitrogen) according to manufacturer specifications, to remove contaminating genomic DNA from the samples. The isolated RNA was used to generate complementary DNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientifc) according to manufacturer specifications. Quantitative PCR was conducted using the complementary DNA and SsoFast EvaGreen Supermix (Bio-Rad) using the manufacturer specifications for reaction preparation, primer design, and thermocycler profile, and reactions were run in a CFX 96 Touch Real-Time PCR Detection System (Bio-Rad). All primers used for quantitative PCR are listed in Table S2. Relative gene expression was analyzed using the 2-ΔΔCt method, commonly referred to as the Livak method, which utilizes a qRT-PCR-based method (31).
Transposon mutagenesis
P. fluorescens Pf0-1 ΔfleQ mutant strain and E. coli JM109 strain carrying the pBT20 plasmid were grown in LB (no antibiotics) for P. fluorescens and 50 µg/mL carbenicillin for E. coli overnight at 30°C, with agitation. One milliliter of each culture was mixed in a 2.0 mL microcentrifuge tube and centrifuged for 3 min at 13,000 rpm. Cell pellets were washed in fresh LB and then resuspended in 100 µL of LB. The mixture was plated on LB agar to facilitate conjugation of the pBT20 plasmid into P. fluorescens. Plates were incubated for 90 min at 30°C, and the cell mixture was scraped up in 1 mL LB with a disposable cell scraper with 20 mm blade width. The cell pellet was washed once in fresh LB, and then dilutions were plated on LB agar supplemented with 30 µg/mL gentamycin and 30 µg/mL chloramphenicol to select for P. fluorescens mutants with chromosomal transposon insertions and against the E. coli donor, respectively. Individual candidates were picked with sterile pipette tips and inoculated in LB in sterile 96-well flat bottom polystyrene plates, and the plates were incubated at 30°C for 16 h. One hundred microliters of 20% sterile glycerol was added to each well and plates were frozen at −80°C. For every independent replicate of a conjugation, several random wells were chosen for arbitrary primed PCR analysis (see below) to verify the presence of P. fluorescens Pf0-1 containing a transposon. Mutants were then screened for their ability to form biofilm by using a 96-pin replicator (Dankar) to transfer inoculum from the frozen plates to 96-well round bottom polypropylene plates (Corning) containing 100 µL of fresh KA medium. Plates were incubated and wells were quantified according to the static biofilm formation crystal violet (CV) assay described above. Candidates with increased biofilm formation compared to the ΔfleQ mutant parental strain were struck out for single colonies from the freezer plate stock onto fresh LB agar with 30 µg/mL gentamycin and incubated overnight at 30°C. A single colony was picked with a sterile pipette tip and used to inoculate LB supplemented with 30 µg/mL gentamycin, which was incubated overnight at 30°C with agitation. Seven hundred fifty microliters of inoculum and 750 µL of sterile 20% glycerol were mixed in 2 mL cryovials (Nunc) and stored at −80°C. From these frozen stocks, candidates were rescreened for biofilm formation in KA medium for 24 h according to the biofilm formation method described above. For candidates with increased biofilm formation compared to the ΔfleQ mutant parental strain, transposon chromosomal insertion sites were determined using arbitrary primed PCR as described by O’Toole et al. (48). Based on these results, primers were designed to amplify and sequence the chromosomal flanks of the predicted insertion site to confirm the exact insertion location.
Swim assay
P. fluorescens Pf0-1 strains were initially grown overnight in 5 mL of LB at 30°C with agitation, and then 1 mL aliquots were transferred to 1.5 mL microcentrifuge tubes. Sterile toothpicks were used to stab inoculate KA supplemented with 0.3% agar (KA Swim Agar), and plates were incubated for 24 h at 30°C. The diameter of resulting swim zones was measured using a ruler.
Lap and MapA cell surface levels via dot blot analysis
P. fluorescens Pf0-1 strains were initially grown in 5 mL of LB at 30°C for ~16 h with agitation, and then 400 µL of inoculum was pipetted onto KA medium supplemented with 1.5% agar. For liquid-grown experiments, 100 µL of inoculum was pipetted into 5 mL of KA liquid medium. The plates or tubes were incubated at 30°C for either 16 h or 24 h for LapA-HA- and MapA-HA-tagged strains, respectively. Following incubation, cells were scraped into 1 mL fresh KA medium using disposable cell scrapers with 20 mm blade width (VWR). Cell pellets were washed once and then resuspended in 110 µL of fresh KA medium. In a 96-well plate, cultures were normalized and diluted to a final OD600 value of 10. Ten microliters of normalized cell culture was spotted on 0.2 µm pore-size nitrocellulose membrane (Bio-Rad) and allowed to dry. The membrane was then incubated in Tris-buffered saline (TBS; Bio-Rad) supplemented with 3% bovine serum albumin (BSA) (Sigma) and 0.1% Tween 20 (TBST; Sigma) for 1 h at 25°C and then transferred to TBST supplemented with purified anti-human influenza hemagglutinin (HA) antibody clone 16B12 (Biolegend) at a 1:2,000 dilution for 1 h at 25°C. The nitrocellulose membrane was washed three times in TBST for 5 min at 25°C, and then transferred to TBST supplemented with rabbit anti-mouse IgG antibody conjugated to horseradish peroxidase (Bio-Rad) at a 1:15,000 dilution for 1 h at 25 ˚C. The membrane was then washed three times in TBST for 5 min at 25°C and then once in TBS for 5 min at 25°C to wash away excess Tween 20. The nitrocellulose was overlaid with a mixture of 1 mL enhanced luminol and 1 mL oxidizing reagent for 30 s at 25°C per specification of the Western Lightning ECL Pro Kit (Perkin-Elmer), and then the blots were exposed on a BioRad ChemiDoc MP Imaging System. Images were imported to ImageJ (NIH) and the dots were quantified as previously described in Collins et al. (18).
Whole cell lysate Western blot analysis
P. fluorescens Pf0-1 strains were initially grown in 5 mL of LB at 30°C for ~16 h with agitation. For LapA-HA tagged strains, 400 µL of inoculum was pipetted onto KA medium supplemented with 1.5% agar and incubated at 30°C for 16 h. Following incubation, cells were scraped into 1 mL fresh KA medium using disposable cell scrapers with 20 mm blade width (VWR). For MapA-HA tagged strains, 1 mL of inoculum was pipetted into 25 mL of KA medium and incubated at 30°C for 24 h with agitation at 200 rpm. After incubation, cultures were transferred to 50 mL conical centrifuge tubes. Cells were centrifuged and then resuspended in 100 µL of 3 mg/mL lysozyme and incubated at room temperature for 5 min. Reactions were incubated on ice and sonicated four times for 5 s at 30% amplitude, with incubation on ice between replicates. Cell slurries were spun down at 4°C and cell lysates were transferred to fresh 1.5 mL centrifuge tubes. Serial dilutions of cell lysates were made, and protein was quantified using the Pierce BCA Protein Assay Kit (Thermo Fisher) according to the manufacturer specifications. Twenty-five micrograms or 50 µg of normalized protein for LapA-HA or MapA-HA tagged strains, respectively, was mixed with 4× Laemmli Sample Buffer (Bio-Rad) and 2-mercaptoethanol to a 1× working dilution and samples were boiled for 5 min. Samples were loaded in 7.5% Mini-Protean TGX precast gels (Bio-Rad) with either 3 µL HiMark Pre-Stained Protein Standard (Thermo Fisher) for LapA or 3 µL Spectra Multicolor High Range Protein Ladder (Thermo Fisher) for MapA and resolved for 4 h at 120V for LapA and 3 h at 115V for MapA in Mini-PROTEAN Tetra Vertical Electrophoresis Cells (Bio-Rad) with 1× Tris/glycine/SDS buffer (Bio-Rad). The gel was transferred to 0.2 µm pore-size nitrocellulose membrane (Bio-Rad) using the Transblot Turbo Transfer System (Bio-Rad) according to manufacturer specifications, using the high molecular weight setting. The membrane was then incubated in TBS (Bio-Rad) supplemented with 3% BSA (Sigma) and 0.1% Tween 20 (TBST; Sigma) for 1 h at 25°C and then transferred to TBST supplemented with purified anti-HA antibody clone 16B12 (Biolegend) at a 1:2,000 dilution for 1 h at 25°C. The nitrocellulose membrane was washed three times in TBST for 5 min at 25°C, and then transferred to TBST supplemented with rabbit anti-mouse IgG antibody conjugated to horseradish peroxidase (Bio-Rad) at a 1:15,000 dilution for 1 h at 25°C. The membrane was then washed three times in TBST for 5 min at 25°C and then once in TBS for 5 min at 25°C to wash away excess Tween 20. The nitrocellulose was overlaid with a mixture of 1 mL enhanced luminol and 1 mL oxidizing reagent for 30 s at 25°C per specification of the Western Lightning ECL Pro Kit (Perkin-Elmer), and then the blots were exposed on a BioRad ChemiDoc MP Imaging System. Images were imported to ImageJ (NIH) and blots were quantified by defining a region of interest (ROI) and using that object to measure each band for mean gray value. The background signal was determined by measuring an area of the blot without a band and subtracting this value from the measured bands.
Supernatant Western blot analysis
P. fluorescens Pf0-1 strains were initially grown in 5 mL of LB at 30°C for ~16 h with agitation. One hundred microliters of inoculum was pipetted into 5 mL of KA liquid medium and incubated at 30°C for 16 h. Following incubation, cultures were measured for bacterial density (OD600) and then transferred to 15 mL conical centrifuge tubes and centrifuged at 5,000 rpm for 20 min at 4°C. Supernatants were transferred to Ambicon Ultra 30 kDa MWCO Centrifugal Filter concentrators (Millipore Sigma) and concentrated through centrifugation according to the manufacturer specifications. The concentrated high molecular weight fractions (>30 kDa) were transferred to fresh microcentrifuge tubes and normalized based on bacterial density. Ten microliters of these normalized fractions were mixed with 4× Laemmli Sample Buffer (Bio-Rad) and 2-mercaptoethanol to a 1× working dilution and samples were boiled for 5 min. Samples were loaded in 7.5% Mini-Protean TGX precast gels (Bio-Rad) with 3 µL HiMark Pre-Stained Protein Standard (Thermo Fisher) and resolved for 4 h at 120V in Mini-PROTEAN Tetra Vertical Electrophoresis Cells (Bio-Rad) with 1× Tris/glycine/SDS buffer (Bio-Rad). The gel was transferred to 0.2 µm pore-size nitrocellulose membrane (Bio-Rad) using the Transblot Turbo Transfer System (Bio-Rad) according to manufacturer specifications, using the high molecular weight setting. The membrane was then incubated in TBS (Bio-Rad) supplemented with 3% BSA (Sigma) and 0.1% Tween 20 (TBST; Sigma) for 1 h at 25°C and then transferred to TBST supplemented with purified anti-HA antibody clone 16B12 (Biolegend) at a 1:2,000 dilution for 1 h at 25°C. The nitrocellulose membrane was washed three times in TBST for 5 min at 25°C, and then transferred to TBST supplemented with rabbit anti-mouse IgG antibody conjugated to horseradish peroxidase (Bio-Rad) at a 1:15,000 dilution for 1 h at 25°C. The membrane was then washed three times in TBST for 5 min at 25°C and then once in TBS for 5 min at 25°C to wash away excess Tween 20. The nitrocellulose was overlaid with a mixture of 1 mL enhanced luminol and 1 mL oxidizing reagent for 30 s at 25°C per specification of the Western Lightning ECL Pro Kit (Perkin-Elmer), and then the blots were exposed on a BioRad ChemiDoc MP Imaging System. Images were imported to ImageJ (NIH) and blots were quantified by defining a ROI and using that object to measure each band for mean gray value. The background signal was determined by measuring an area of the blot without a band and subtracting this value from the measured bands.
Cyclic-di-GMP quantification
P. fluorescens Pf0-1 strains were initially grown in 5 mL of LB at 30°C for ~16 h with agitation, and then 400 µL of inoculum was pipetted onto KA medium supplemented with 1.5% agar and incubated at 30°C for 16 h. Nucleotides were extracted as previously described in Webster et al. (55). Extracts were quantified for cyclic-di-GMP by LC-MS/MS at the Michigan State University and normalized by dry cell pellet weight.
Growth curves
P. fluorescens Pf0-1 strains were initially grown in 5 mL of LB at 30°C for ~16 h with agitation. Cultures were used to inoculate 100 µL of KA medium in sterile 96-well polystyrene clear flat bottom plates (Corning) at a starting OD600 ~0.01. Plates were continually incubated at 30°C in a BioTek Synergy Neo2-multimode microplate reader (Agilent) and OD600 measurements were taken every 15 min for 24 h, with 10 s of linear shaking before each measurement.
ACKNOWLEDGMENTS
We would like to thank Dr. Shanice Webster and Dr. Sherry Kuchma for numerous helpful discussions. We would especially like to thank Dr. Fabrice Jean-Pierre for many insightful conversations regarding the Gac/Rsm system in multiple Pseudomonas spp. We thank Dr. Carey Nadell and his laboratory for their generous gift of microfluidic devices.
Liquid chromatography with tandem mass spectrometry services were provided by Lijun Chen and the Mass Spectrometry and Metabolomics Core Facility at Michigan State University. Sequencing services were provided by the Dartmouth Molecular Biology Core. Growth curves were generated using equipment from the BioMT Molecular Interactions and Imaging Core (NIH/NIGMS P20-GM113132).
This work was supported by NIH grants R01GM123609 and R01AI168017 to G.A.O. The Molecular Biology Core was supported by NIH grant P30CA023108. The bioMT scientific cores were supported by NIH grant P20GM113132.
Contributor Information
George A. O’Toole, Email: georgeo@dartmouth.edu.
Michael Y. Galperin, NCBI, NLM, National Institutes of Health, Bethesda, Maryland, USA
DATA AVAILABILITY
This manuscript has no large data sets.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jb.00152-23.
Fig. S1 to S9.
Tables S1 and S2.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Paulsen IT, Press CM, Ravel J, Kobayashi DY, Myers GSA, Mavrodi DV, DeBoy RT, Seshadri R, Ren Q, Madupu R, Dodson RJ, Durkin AS, Brinkac LM, Daugherty SC, Sullivan SA, Rosovitz MJ, Gwinn ML, Zhou L, Schneider DJ, Cartinhour SW, Nelson WC, Weidman J, Watkins K, Tran K, Khouri H, Pierson EA, Pierson LS, Thomashow LS, Loper JE. 2005. Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol 23:873–878. doi: 10.1038/nbt1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Howell CR. 1979. Control of Rhizoctonia solani on cotton seedlings with Pseudomonas fluorescens and with an antibiotic produced by the bacterium. Phytopathology 69:480. doi: 10.1094/Phyto-69-480 [DOI] [Google Scholar]
- 3. Keel C. 1992. Suppression of root diseases by Pseudomonas fluorescens CHA0: importance of the bacterial secondary metabolite 2,4-diacetylphloroglucinol. MPMI 5:4. doi: 10.1094/MPMI-5-004 [DOI] [Google Scholar]
- 4. Haas D, Défago G. 2005. Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat Rev Microbiol 3:307–319. doi: 10.1038/nrmicro1129 [DOI] [PubMed] [Google Scholar]
- 5. Khabbaz RF, Arnow PM, Highsmith AK, Herwaldt LA, Chou T, Jarvis WR, Lerche NW, Allen JR. 1984. Pseudomonas fluorescens bacteremia from blood transfusion. Am J Med 76:62–68. doi: 10.1016/0002-9343(84)90751-4 [DOI] [PubMed] [Google Scholar]
- 6. Hsueh P-R, Teng L-J, Pan H-J, Chen Y-C, Sun C-C, Ho S-W, Luh K-T. 1998. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol 36:2914–2917. doi: 10.1128/JCM.36.10.2914-2917.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gershman MD, Kennedy DJ, Noble-Wang J, Kim C, Gullion J, Kacica M, Jensen B, Pascoe N, Saiman L, McHale J, Wilkins M, Schoonmaker-Bopp D, Clayton J, Arduino M, Srinivasan A, Pseudomonas fluorescens Investigation Team . 2008. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis 47:1372–1379. doi: 10.1086/592968 [DOI] [PubMed] [Google Scholar]
- 8. Wong V, Levi K, Baddal B, Turton J, Boswell TC. 2011. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol 49:2093–2096. doi: 10.1128/JCM.02559-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benito N, Mirelis B, Luz Gálvez M, Vila M, López-Contreras J, Cotura A, Pomar V, March F, Navarro F, Coll P, Gurguí M. 2012. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect 82:286–289. doi: 10.1016/j.jhin.2012.09.008 [DOI] [PubMed] [Google Scholar]
- 10. Landers CJ, Cohavy O, Misra R, Yang H, Lin Y-C, Braun J, Targan SR. 2002. Selected loss of tolerance evidenced by Crohn’s disease–associated immune responses to auto- and microbial antigens. Gastroenterology 123:689–699. doi: 10.1053/gast.2002.35379 [DOI] [PubMed] [Google Scholar]
- 11. Wei B, Huang T, Dalwadi H, Sutton CL, Bruckner D, Braun J. 2002. Pseudomonas fluorescens encodes the Crohn’s disease-associated I2 sequence and T-cell superantigen. Infect Immun 70:6567–6575. doi: 10.1128/IAI.70.12.6567-6575.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Madi A, Svinareff P, Orange N, Feuilloley MG, Connil N. 2010. Pseudomonas fluorescens alters epithelial permeability and translocates across Caco-2/TC7 intestinal cells. Gut Pathog 2:16. doi: 10.1186/1757-4749-2-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Madi Amar, Lakhdari O, Blottière HM, Guyard-Nicodème M, Le Roux K, Groboillot A, Svinareff P, Doré J, Orange N, Feuilloley MGJ, Connil N. 2010. The clinical Pseudomonas fluorescens MFN1032 strain exerts a cytotoxic effect on epithelial intestinal cells and induces interleukin-8 via the AP-1 signaling pathway. BMC Microbiol 10:215. doi: 10.1186/1471-2180-10-215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alnabhani Z, Montcuquet N, Biaggini K, Dussaillant M, Roy M, Ogier-Denis E, Madi A, Jallane A, Feuilloley M, Hugot J-P, Connil N, Barreau F. 2015. Pseudomonas fluorescens alters the intestinal barrier function by modulating IL-1β expression through hematopoietic NOD2 signaling. Inflamm Bowel Dis 21:543–555. doi: 10.1097/MIB.0000000000000291 [DOI] [PubMed] [Google Scholar]
- 15. Sauer K, Camper AK, Ehrlich GD, Costerton JW, Davies DG. 2002. Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J Bacteriol 184:1140–1154. doi: 10.1128/jb.184.4.1140-1154.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sauer K, Stoodley P, Goeres DM, Hall-Stoodley L, Burmølle M, Stewart PS, Bjarnsholt T. 2022. The biofilm life cycle: expanding the conceptual model of biofilm formation. Nat Rev Microbiol 20:608–620. doi: 10.1038/s41579-022-00767-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hinsa SM, Espinosa-Urgel M, Ramos JL, O’Toole GA. 2003. Transition from reversible to irreversible attachment during biofilm formation by Pseudomonas fluorescens WCS365 requires an ABC transporter and a large secreted protein. Mol Microbiol 49:905–918. doi: 10.1046/j.1365-2958.2003.03615.x [DOI] [PubMed] [Google Scholar]
- 18. Collins AJ, Pastora AB, Smith TJ, O’Toole GA. 2020. MapA, a second large RTX adhesin conserved across the pseudomonads, contributes to biofilm formation by Pseudomonas fluorescens. J Bacteriol 202:e00277-20. doi: 10.1128/JB.00277-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Collins AJ, Smith TJ, Sondermann H, O’Toole GA. 2020. From input to output: the Lap/c-di-GMP biofilm regulatory circuit. Annu Rev Microbiol 74:607–631. doi: 10.1146/annurev-micro-011520-094214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Monds RD, Newell PD, Schwartzman JA, O’Toole GA. 2006. Conservation of the Pho regulon in Pseudomonas fluorescens Pf0-1. Appl Environ Microbiol 72:1910–1924. doi: 10.1128/AEM.72.3.1910-1924.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Monds RD, Newell PD, Gross RH, O’Toole GA. 2007. Phosphate-dependent modulation of c-di-GMP levels regulates Pseudomonas fluorescens Pf0-1 biofilm formation by controlling secretion of the adhesin LapA. Mol Microbiol 63:656–679. doi: 10.1111/j.1365-2958.2006.05539.x [DOI] [PubMed] [Google Scholar]
- 22. Giacalone D, Smith TJ, Collins AJ, Sondermann H, Koziol LJ, O’Toole GA. 2018. Ligand-mediated biofilm formation via enhanced physical interaction between a diguanylate cyclase and its receptor. mBio 9:e01254-18. doi: 10.1128/mBio.01254-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dahlstrom KM, Collins AJ, Doing G, Taroni JN, Gauvin TJ, Greene CS, Hogan DA, O’Toole GA. 2018. A multimodal strategy used by a large c-di-GMP network. J Bacteriol 200:e00703-17. doi: 10.1128/JB.00703-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pérez-Mendoza D, Felipe A, Ferreiro MD, Sanjuán J, Gallegos MT. 2019. AmrZ and FleQ co-regulate cellulose production in Pseudomonas syringae pv. tomato DC3000. Front Microbiol 10:746. doi: 10.3389/fmicb.2019.00746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Molina-Henares MA, Ramos-González MI, Daddaoua A, Fernández-Escamilla AM, Espinosa-Urgel M. 2017. FleQ of Pseudomonas putida KT2440 is a multimeric cyclic diguanylate binding protein that differentially regulates expression of biofilm matrix components. Res Microbiol 168:36–45. doi: 10.1016/j.resmic.2016.07.005 [DOI] [PubMed] [Google Scholar]
- 26. Banerjee P, Kumawat C, Jain D. 2019. Sensor I regulated ATPase activity of FleQ is essential for motility to biofilm transition in Pseudomonas aeruginosa. ACS Chem Biol 14:1515–1527. doi: 10.1021/acschembio.9b00255 [DOI] [PubMed] [Google Scholar]
- 27. Hickman JW, Harwood CS. 2008. Identification of FleQ from Pseudomonas aeruginosa as a c-di-GMP-responsive transcription factor. Mol Microbiol 69:376–389. doi: 10.1111/j.1365-2958.2008.06281.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baraquet C, Murakami K, Parsek MR, Harwood CS. 2012. The FleQ protein from Pseudomonas aeruginosa functions as both a repressor and an activator to control gene expression from the pel operon promoter in response to c-di-GMP. Nucleic Acids Res 40:7207–7218. doi: 10.1093/nar/gks384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baraquet C, Harwood CS. 2016. FleQ DNA binding consensus sequence revealed by studies of FleQ-dependent regulation of biofilm gene expression in Pseudomonas aeruginosa. J Bacteriol 198:178–186. doi: 10.1128/JB.00539-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blanco-Romero E, Redondo-Nieto M, Martínez-Granero F, Garrido-Sanz D, Ramos-González MI, Martín M, Rivilla R. 2018. Genome-wide analysis of the FleQ direct regulon in Pseudomonas fluorescens F113 and Pseudomonas putida Kt2440. Sci Rep 8:13145. doi: 10.1038/s41598-018-31371-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 32. Blanco-Romero E, Durán D, Garrido-Sanz D, Rivilla R, Martín M, Redondo-Nieto M. 2022. Transcriptomic analysis of Pseudomonas ogarae F113 reveals the antagonistic roles of AmrZ and FleQ during rhizosphere adaption. Microb Genom 8:000750. doi: 10.1099/mgen.0.000750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garrido-Sanz D, Redondo-Nieto M, Martin M, Rivilla R. 2021. Comparative genomics of the Pseudomonas corrugata subgroup reveals high species diversity and allows the description of Pseudomonas ogarae sp. nov. Microb Genom 7:000593. doi: 10.1099/mgen.0.000593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robleto EA, López-Hernández I, Silby MW, Levy SB. 2003. Genetic analysis of the AdnA regulon in Pseudomonas fluorescens: nonessential role of flagella in adhesion to sand and biofilm formation. J Bacteriol 185:453–460. doi: 10.1128/JB.185.2.453-460.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boyd CD, Chatterjee D, Sondermann H, O’Toole GA. 2012. LapG, required for modulating biofilm formation by Pseudomonas fluorescens Pf0-1, is a calcium-dependent protease. J Bacteriol 194:4406–4414. doi: 10.1128/JB.00642-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boyd CD, Smith TJ, El-Kirat-Chatel S, Newell PD, Dufrêne YF, O’Toole GA. 2014. Structural features of the Pseudomonas fluorescens biofilm adhesin LapA required for LapG-dependent cleavage, Biofilm formation, and cell surface localization. J Bacteriol 196:2775–2788. doi: 10.1128/JB.01629-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith TJ, Font ME, Kelly CM, Sondermann H, O’Toole GA. 2018. An N-terminal retention module anchors the giant adhesin LapA of Pseudomonas fluorescens at the cell surface: a novel subfamily of type I secretion systems. J Bacteriol 200:e00734-17. doi: 10.1128/JB.00734-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Monds RD, Newell PD, Wagner JC, Schwartzman JA, Lu W, Rabinowitz JD, O’Toole GA. 2010. Di-adenosine tetraphosphate (Ap4A) metabolism impacts biofilm formation by Pseudomonas fluorescens via modulation of c-di-GMP-dependent pathways. J Bacteriol 192:3011–3023. doi: 10.1128/JB.01571-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brencic A, McFarland KA, McManus HR, Castang S, Mogno I, Dove SL, Lory S. 2009. The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol Microbiol 73:434–445. doi: 10.1111/j.1365-2958.2009.06782.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heeb S, Blumer C, Haas D. 2002. Regulatory RNA as mediator in GacA/RsmA-dependent global control of exoproduct formation in Pseudomonas fluorescens CHA0. J Bacteriol 184:1046–1056. doi: 10.1128/jb.184.4.1046-1056.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Humair B, Wackwitz B, Haas D. 2010. GacA-controlled activation of promoters for small RNA genes in Pseudomonas fluorescens. Appl Environ Microbiol 76:1497–1506. doi: 10.1128/AEM.02014-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Valverde C, Heeb S, Keel C, Haas D. 2003. RsmY, a small regulatory RNA, is required in concert with RsmZ for GacA-dependent expression of biocontrol traits in Pseudomonas fluorescens CHA0. Mol Microbiol 50:1361–1379. doi: 10.1046/j.1365-2958.2003.03774.x [DOI] [PubMed] [Google Scholar]
- 43. Kay E, Dubuis C, Haas D. 2005. Three small RNAs jointly ensure secondary metabolism and biocontrol in Pseudomonas fluorescens CHA0. Proc Natl Acad Sci U S A 102:17136–17141. doi: 10.1073/pnas.0505673102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Davies JA, Harrison JJ, Marques LLR, Foglia GR, Stremick CA, Storey DG, Turner RJ, Olson ME, Ceri H. 2007. The GacS sensor kinase controls phenotypic reversion of small colony variants isolated from biofilms of Pseudomonas aeruginosa PA14. FEMS Microbiol Ecol 59:32–46. doi: 10.1111/j.1574-6941.2006.00196.x [DOI] [PubMed] [Google Scholar]
- 45. Reimmann C, Valverde C, Kay E, Haas D. 2005. Posttranscriptional repression of GacS/GacA-controlled genes by the RNA-binding protein RsmE acting together with RsmA in the biocontrol strain Pseudomonas fluorescens CHA0 . J Bacteriol 187:276–285. doi: 10.1128/JB.187.1.276-285.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Janssen KH, Diaz MR, Golden M, Graham JW, Sanders W, Wolfgang MC, Yahr TL. 2018. Functional analyses of the RsmY and RsmZ small noncoding regulatory RNAs in Pseudomonas aeruginosa. J Bacteriol 200:e00736-17. doi: 10.1128/JB.00736-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sorger-Domenigg T, Sonnleitner E, Kaberdin VR, Bläsi U. 2007. Distinct and overlapping binding sites of Pseudomonas aeruginosa Hfq and RsmA proteins on the non-coding RNA RsmY. Biochem Biophys Res Commun 352:769–773. doi: 10.1016/j.bbrc.2006.11.084 [DOI] [PubMed] [Google Scholar]
- 48. O’Toole GA, Kolter R. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol Microbiol 28:449–461. doi: 10.1046/j.1365-2958.1998.00797.x [DOI] [PubMed] [Google Scholar]
- 49. Huertas-Rosales Ó, Romero M, Chan K-G, Hong K-W, Cámara M, Heeb S, Barrientos-Moreno L, Molina-Henares MA, Travieso ML, Ramos-González MI, Espinosa-Urgel M. 2021. Genome-wide analysis of targets for post-transcriptional regulation by Rsm proteins in Pseudomonas putida. Front Mol Biosci 8:624061. doi: 10.3389/fmolb.2021.624061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Loper JE, Hassan KA, Mavrodi DV, Davis EW, Lim CK, Shaffer BT, Elbourne LDH, Stockwell VO, Hartney SL, Breakwell K, Henkels MD, Tetu SG, Rangel LI, Kidarsa TA, Wilson NL, van de Mortel JE, Song C, Blumhagen R, Radune D, Hostetler JB, Brinkac LM, Durkin AS, Kluepfel DA, Wechter WP, Anderson AJ, Kim YC, Pierson LS, Pierson EA, Lindow SE, Kobayashi DY, Raaijmakers JM, Weller DM, Thomashow LS, Allen AE, Paulsen IT. 2012. Comparative genomics of plant-associated Pseudomonas spp: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet 8:e1002784. doi: 10.1371/journal.pgen.1002784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seaton SC, Silby MW, Levy SB. 2013. Pleiotropic effects of GacA on Pseudomonas fluorescens Pf0-1 in vitro and in soil. Appl Environ Microbiol 79:5405–5410. doi: 10.1128/AEM.00819-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Olins PO, Devine CS, Rangwala SH, Kavka KS. 1988. The T7 phage gene 10 leader RNA, a ribosome-binding site that dramatically enhances the expression of foreign genes in Escherichia coli. Gene 73:227–235. doi: 10.1016/0378-1119(88)90329-0 [DOI] [PubMed] [Google Scholar]
- 53. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Katharios-Lanwermeyer S, Koval SA, Barrack KE, O’Toole GA. 2022. The diguanylate cyclase YfiN of Pseudomonas aeruginosa regulates biofilm maintenance in response to peroxide. J Bacteriol 204:e0039621. doi: 10.1128/JB.00396-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Webster SS, Lee CK, Schmidt WC, Wong GCL, O’Toole GA. 2021. Interaction between the type 4 pili machinery and a diguanylate cyclase fine-tune c-di-GMP levels during early biofilm formation. Proc Natl Acad Sci U S A 118:e2105566118. doi: 10.1073/pnas.2105566118 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S9.
Tables S1 and S2.
Data Availability Statement
This manuscript has no large data sets.