

Abstract
Drug addiction, a phenomenon where cancer cells paradoxically depend on continuous drug treatment for survival, has uncovered cell signaling mechanisms and cancer co-dependencies. Here, we discover mutations that confer drug addiction to inhibitors of the transcriptional repressor Polycomb Repressive Complex 2 (PRC2) in diffuse large B-cell lymphoma (DLBCL). Drug addiction is mediated by hypermorphic mutations in the CXC domain of the catalytic subunit EZH2, which maintain H3K27me3 levels even in the presence of PRC2 inhibitors. Inhibitor treatment discontinuation leads to overspreading of H3K27me3, surpassing a repressive methylation ceiling compatible with lymphoma cell survival. Exploiting this vulnerability, we show that inhibition of SETD2 similarly induces H3K27me3 spreading and blocks lymphoma growth. Collectively, our findings demonstrate that constraints on chromatin landscapes can yield biphasic dependencies in epigenetic signaling in cancer cells. More broadly, we highlight how approaches to identify drug addiction mutations can be leveraged to discover cancer vulnerabilities.
Introduction
Drug resistance represents a major challenge for anti-cancer therapeutics. In some cases, drug resistance leads to drug addiction, where cancer cells depend on sustained drug treatment for survival. Drug addiction mechanisms have been most widely reported for kinase inhibitors and typically occur through activation of compensatory signaling pathways1–3. Due to the cumulative effects of bypass signaling and restored kinase signaling, kinase inhibitor withdrawal results in signaling overactivation and subsequent arrest in cell growth, revealing an upper limit for mitogenic signaling in cancer cells3,4. This mitogenic signaling ceiling likely reflects cellular safeguarding mechanisms such as oncogene-induced senescence, which can be triggered by overactivation of Ras, BRAF, and Myc signaling5–7. Drug addiction remains largely unexplored for other classes of drug targets.
Drug addiction may result from fine-tuned cell signaling thresholds inherent to the parental tissue of origin. As a key example, B cells are selected for an optimized level of signaling through the B-cell receptor (BCR), which promotes survival and positive selection during B-cell development. However, hyperactivation of BCR downstream kinases, for example from autoreactive B cells, results in excessive signaling and triggers negative selection8. Properly tuned signaling levels therefore equip B cells with an appropriately functional receptor that recognizes foreign antigens, but not self-antigens. This requirement for precise levels of B-cell receptor signaling is preserved in malignant B cells, with both the suppression and activation of B-cell kinase signaling leading to lymphoma cell death2,9. Whether similar thresholds exist in other pathways beyond kinase signaling in B-cell malignancies remain to be explored.
Notably, chromatin pathways are critical for B-cell maturation and are frequently dysregulated in lymphoma. Recurrent activating mutations in EZH2, the catalytic subunit of the transcriptional repressor PRC2, promote neoplastic growth in a wide range of lymphomas, including 20% of follicular lymphomas (FLs) and 10% of diffuse large B-cell lymphomas (DLBCLs)10,11. Within the germinal center, PRC2 represses cell cycle checkpoint genes, allowing for somatic hypermutation and expansion of B cells upon presentation of a foreign antigen12. In lymphoma, activating mutations in EZH2 repress these same cell cycle checkpoint genes to induce unrestrained, autonomous proliferation of lymphoma cells and expansion of germinal centers, predisposing cells to transformation13. The discovery of these activating mutations prompted the development of inhibitors targeting PRC2, leading to the approval of the EZH2 active-site inhibitor tazemetostat for FL and the clinical development of allosteric inhibitors targeting the PRC2 subunit, EED. Both active and allosteric site PRC2 inhibitors reduce H3K27me3 levels, derepress tumor suppressor genes, and promote lymphoma differentiation and ultimately cell death14,15.
Through the discovery of drug addiction mutations that hyperactivate PRC2, here we show that not only lower but also upper thresholds exist for the repressive PRC2 mark, H3K27me3, in EZH2-mutant DLBCL cells. We find that lymphoma cells survive in a ‘Goldilocks’ epigenetic state, which is bounded by a previously unknown repressive methylation ceiling. Pharmacological inhibition of SETD2, a PRC2 antagonist, can surpass this upper methylation limit and consequently block EZH2-mutant DLBCL growth, suggesting that PRC2 activation may represent a new therapeutic vulnerability in this context. Overall, our study reveals that upper limits on epigenetic modifications exist in cells and underscore the importance of methods to identify drug addiction mutations for discovery of new signaling thresholds.
Results
EZH2 CXC domain is a drug resistance hotspot
We surveyed the drug resistance landscape to PRC2 inhibitors in EZH2-mutant lymphoma using CRISPR-suppressor scanning, a method to identify drug resistance hot spots within a protein target16. Specifically, we performed CRISPR-suppressor scanning in the DLBCL cell line Karpas-422, which is heterozygous for the most common activating EZH2 mutation, EZH2 Y646N17. We used a pool of 650 single-guide RNAs (sgRNAs) spanning the coding sequence of the core members of PRC2 (EZH2, EED, and SUZ12) and selected for resistance to two PRC2 inhibitors: GSK343, an EZH2 active site inhibitor18; and EED226, an EED allosteric inhibitor14 that blocks PRC2 allosteric activation by H3K27me3 (Fig. 1a and Supplementary Dataset 1). Following 5 weeks of inhibitor treatment, we isolated genomic DNA from surviving cells and determined the enrichment of sgRNAs under each treatment condition. The top-enriched sgRNAs unique to each inhibitor treatment (EZH2 sgI109, EZH2 sgN699, and EED sgY365) targeted positions proximal to the inhibitor binding sites of the respective inhibitors (Fig. 1b–f, Extended Data Fig. 1a–e and Supplementary Dataset 2)14,19. Sequencing of the exons encompassing these positions revealed an enrichment of mutations that likely disrupt inhibitor binding, such as EZH2 I109_M110insM under GSK343 and EED Y365F/M366V under EED226 treatment (Fig. 1d–e and Extended Data Fig. 1d–e)19–21. The identification of inhibitor-binding site mutants supports that CRISPR-suppressor scanning of PRC2 identified sgRNAs leading to drug resistance.
Fig. 1 |. CRISPR-suppressor scanning identifies the EZH2 CXC domain as a drug resistance hotspot to PRC2 inhibitors.
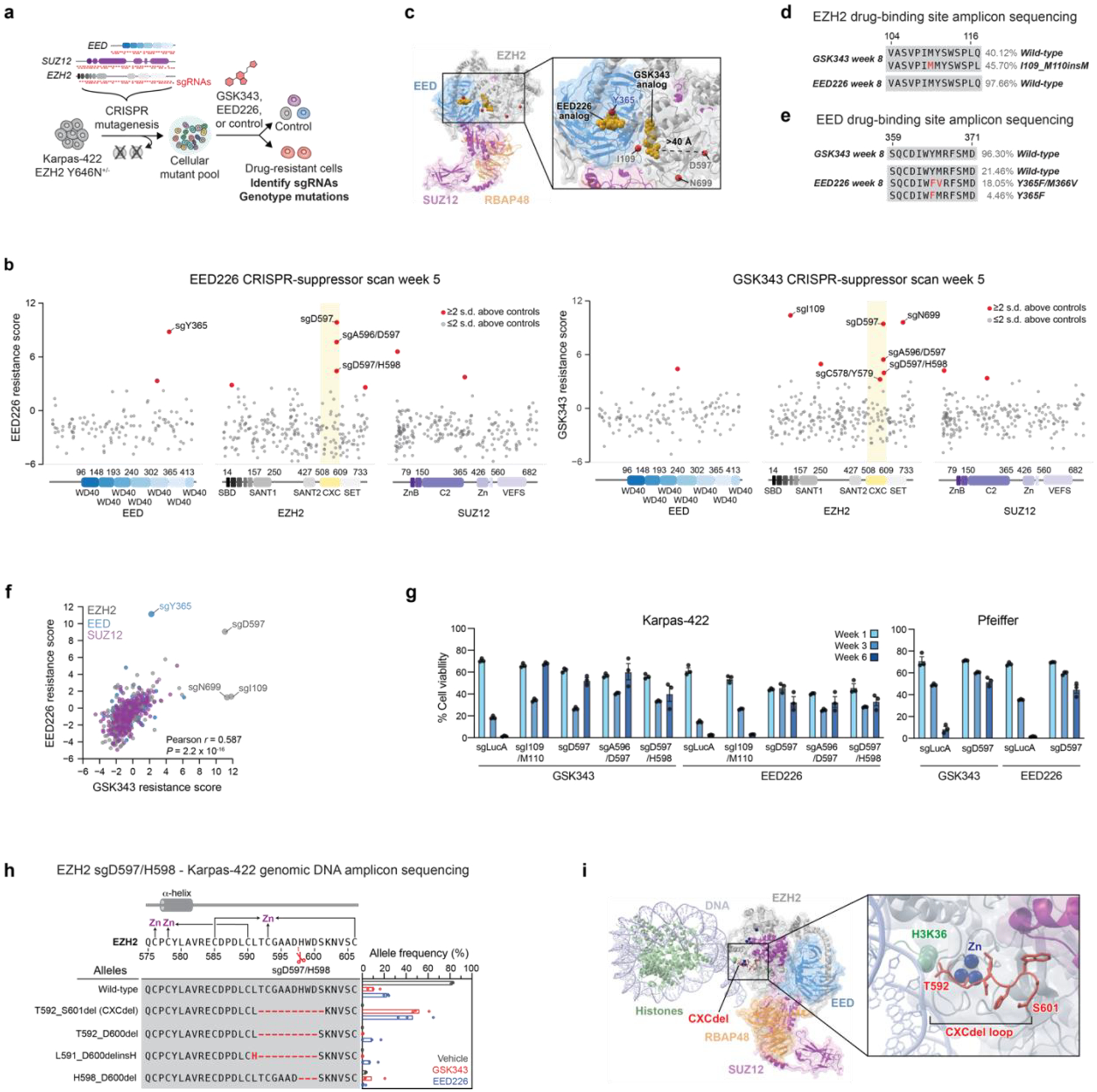
a. Overview of PRC2 CRISPR-suppressor scanning experiment.
b. Scatter plots showing resistance scores (y-axis) in Karpas-422 under 1 μM EED226 treatment (left) and 1 μM GSK343 treatment (right) at week 5. Resistance scores were calculated as the log2(fold-change) sgRNA enrichment under drug normalized to the mean of the negative control sgRNAs (n = 58). The PRC2-targeting sgRNAs (n = 650) are arrayed by their predicted cut sites in the coding sequences (x-axis). The location of the EZH2 CXC domain is shown by a yellow background. Data represent mean of n = 3 replicates.
c. Structural view showing enriched sgRNAs in EZH2 and EED drug binding sites identified in the GSK343 and EED226 CRISPR-suppressor scanning experiments. GSK343 and EED226 are shown in gold. Amino acids corresponding to enriched sgRNAs are shown as red spheres. PDB: 6WKR, 5K0M, 5LS6
d. Schematic showing EZH2 drug-binding mutation and wild-type allele frequencies from CRISPR-suppressor scanning under GSK343 or EED226 treatment.
e. Same as in d. but for EED drug-binding mutation and wild-type allele frequencies.
f. Scatter plot showing sgRNA resistance scores in the EED226 CRISPR-suppressor scan (y-axis) and GSK343 CRISPR-suppressor scan (x-axis). Data were analyzed by two-tailed Pearson correlation analysis.
g. Bar plots of cell viability following transduction of individual EZH2 sgRNAs and treatment with GSK343 (1 μM for Karpas-422 and incremental dosing up to 200 nM for Pfeiffer) or EED226 (1 μM for Karpas-422 and incremental dosing up to 500 nM for Pfeiffer) in Karpas-422 (left) and Pfeiffer cells (right).
h. Schematic showing genotypes and bar plots of allele frequencies (only alleles with >5% frequency shown) for mutations in the gDNA encoding EZH2 surrounding the cut site of CXC-targeting sgRNAs for GSK343 (red) or EED226 (blue) treatment at week 6 in Karpas-422 cells transduced with sgD597/H598. (top) Schematic depicts the secondary structure of C-terminal CXC domain and cysteine residues that coordinate the zinc ions.
i. Structural view of PRC2 engaged with a mononucleosome showing the location of CXCdel loop (red) and zinc ions (blue). EMDB-7306, PDB: 6WKR
Data in g-h are represented by mean ± s.e.m of n = 3. Results in d-e are representative of three replicates.
See also Extended Data Fig. 1.
Source data for Fig. 1 (statistical source data)
Unexpectedly, several sgRNAs targeting positions within the CXC domain of EZH2 were also highly enriched in both inhibitor treatment conditions (sgA596/D597, sgD597, sgD597/H598, Fig. 1b–c,f; Extended Data Fig. 1a and Supplementary Dataset 2). These sgRNAs target a loop in the CXC domain that lies 17 Å from the substrate nucleosome and over 40 Å from either inhibitor binding site (Fig. 1c)14,19,22,23. When individually transduced in Karpas-422 cells, these top-enriched EZH2 CXC-targeting sgRNAs conferred robust resistance to both inhibitor treatments, while cells transduced with EZH2 sgI109/M110 only survived in the presence of GSK343 (Fig. 1g, left panel), consistent with the selectivity observed in the CRISPR-suppressor scan. Additionally, transduction of EZH2 sgD597 also conferred robust resistance to PRC2 inhibitors in Pfeiffer, a DLBCL cell line heterozygous for a different PRC2 activating mutation, EZH2 A682G (Fig. 1g, right panel).
Genotyping the resultant bulk drug resistant cells from individual CXC-targeting sgRNA transductions revealed frequent in-frame deletion mutations within the EZH2 CXC domain. The most highly enriched mutation was a 10-amino acid (aa) deletion spanning EZH2 T592–S601, which was identified in all three CXC-targeting sgRNAs transductions across both inhibitor treatments (Fig. 1h and Extended Data Fig. 1f–i). The EZH2 T592–S601 deletion modifies a poorly conserved loop adjacent to a cluster of cysteine residues that coordinate zinc ions (Fig. 1i and Extended Data Fig. 1j–k)24. Hereafter, this mutation is referred to as CXCdel. Notably, EZH2 CXCdel is not enriched in the absence of PRC2 inhibitors (Fig. 1h and Extended Data Fig. 1f–i), suggesting that inhibitor treatment strongly selects for the deletion mutation. Taken together, these results demonstrate that mutations in the CXC domain of EZH2 confer drug resistance through an unknown mechanism in multiple DLBCL cell lines harboring distinct EZH2-activating mutations.
CRISPR-addiction scanning reveals drug addiction
We next sought to generate clonal Karpas-422 CXCdel cells for further study. However, despite the strong enrichment of EZH2 CXCdel in the presence of PRC2 inhibitors, clonal cell lines containing CXCdel could not be expanded from single cells when sorted into fresh media lacking inhibitor. We therefore considered whether the CXCdel-mutant cells have impaired growth in the absence of inhibitors. To test this possibility, the PRC2 CRISPR-suppressor scan was repeated and further extended by 3 weeks either in the continued presence of PRC2 inhibitors or after their withdrawal (Fig. 2a). Through this adapted method, which we term ‘CRISPR-addiction scanning’, we observed that the previously enriched CXC-targeting sgRNAs were highly depleted upon inhibitor removal, while the sgRNAs targeting the respective inhibitor binding sites remained enriched (Fig. 2b–d). Genotyping of bulk cells revealed that EZH2 CXCdel was highly depleted upon inhibitor withdrawal (Fig. 2e and Extended Data Fig. 2a), supporting the notion that cells containing this mutation are drug addicted (i.e., have reduced intrinsic fitness upon inhibitor removal)1,3.
Fig. 2 |. CRISPR-addiction scanning reveals that mutations in PRC2 can confer addiction to PRC2 inhibitors.
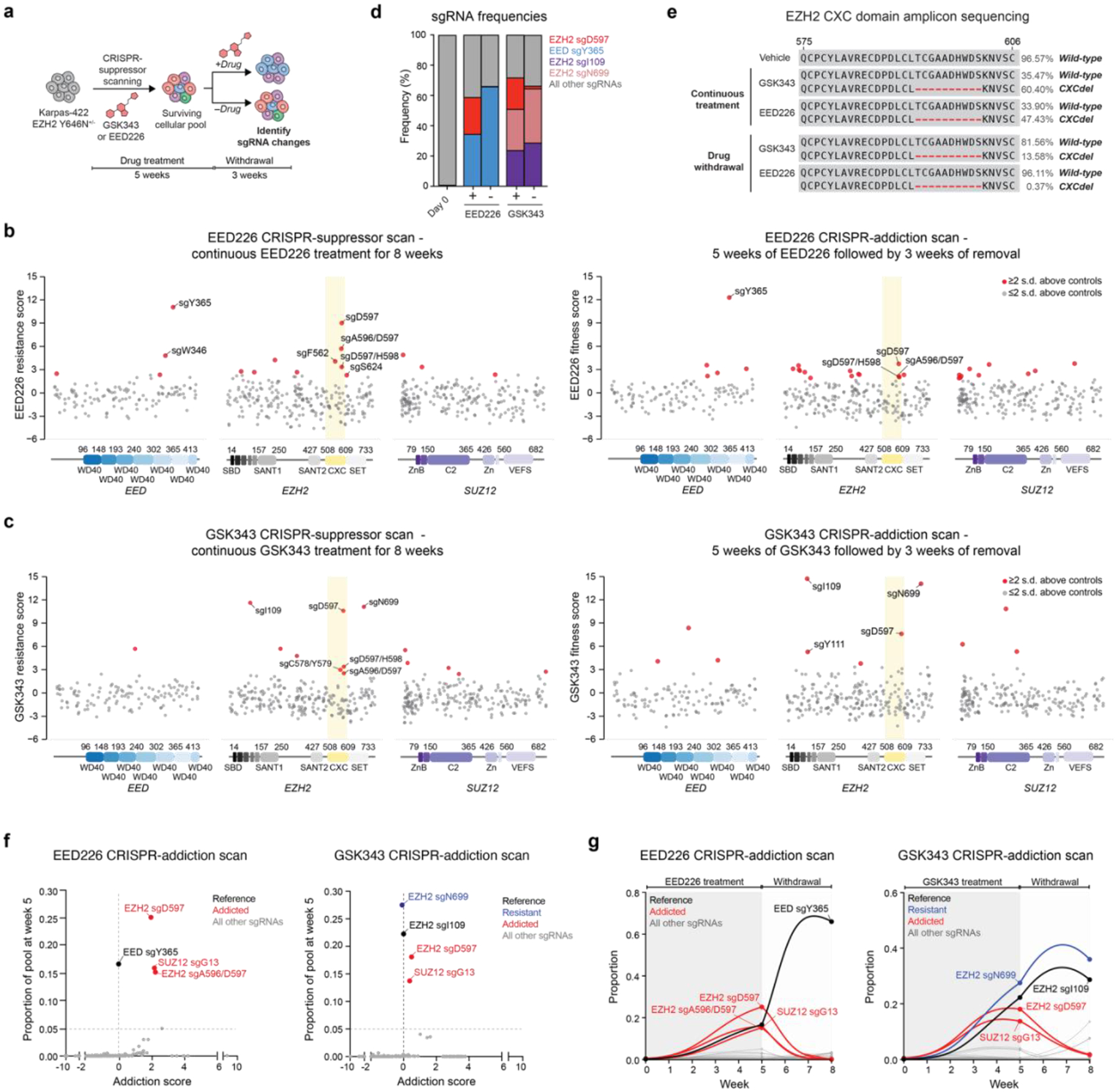
a. Overview of PRC2 CRISPR-addiction scanning experiment.
b. Scatter plots showing resistance or fitness scores (y-axis) in Karpas-422 following 8 weeks of continuous EED226 (1 μM) treatment (left) and 5 weeks of EED226 treatment followed by 3 weeks of inhibitor removal (right). Resistance or fitness scores were calculated as the log2(fold-change) sgRNA enrichment under drug normalized to the mean of the negative control sgRNAs (n = 58). The PRC2-targeting sgRNAs (n = 650) are arrayed by their predicted cut sites in the coding sequences (x-axis). The location of the EZH2 CXC domain is shown by a yellow background.
c. Same as in b. but for Karpas-422 following 8 weeks of continuous GSK343 treatment (1 μM) (left) and 5 weeks of GSK343 treatment followed by 3 weeks of inhibitor removal (right).
d. Stacked bar chart showing representation of top-enriched sgRNAs (>20% in frequency) in the surviving mutant pool under continuous EED226 or GSK343 (1 μM) treatments for 8 weeks versus 5 weeks of continuous inhibitor treatment followed by 3 weeks of inhibitor removal.
e. Schematic showing CXCdel mutation and wild-type allele frequencies from CRISPR-addiction scanning under vehicle, continuous GSK343, EED226 treatment or upon inhibitor withdrawal.
f. Scatter plot showing addiction score (x-axis) versus proportion of the sgRNA pool made up by each sgRNA at week 5 (y-axis) for the EED226 CRISPR-addiction scan (left) and GSK343 CRISPR-addiction scan (right).
g. Graph showing proportions of each sgRNA (y-axis) in the EED226 CRISPR-addiction scan (left) and GSK343 CRISPR-addiction scan (right) over time (x-axis). Curves were calculated using the estimated intrinsic growth rate r for each sgRNA-containing cell population under the assumption of competitive logistic growth. The reference sgRNA is shown in black, sgRNAs demonstrating resistance without addiction are in blue, and addicted sgRNAs are in red.
Data in b-d, f-g are represented by mean of n = 3. Results in e are representative of three replicates.
See also Extended Data Fig. 2.
Source data for Fig. 2 (statistical source data)
An alternative hypothesis for the depletion of CXC-targeting sgRNAs is that cells containing these sgRNAs were out-competed upon inhibitor removal by other cells containing previously suppressed sgRNAs. To test this, we fit a system of ordinary differential equations describing competitive logistic growth (the competitive Lotka-Volterra equations; see Supplementary Note for detailed description) to the sgRNA prevalence data at weeks 0, 5, and 825. By comparing the estimated intrinsic growth rate r for each sgRNA-containing population on-drug (ron) vs. off-drug (roff) relative to a drug resistant mutant that disrupts inhibitor binding, with intrinsic fitness that is assumed to be unaffected by drug (i.e., EZH2 sgI109 for GSK343 and EED sgY365 for EED226), we derived an addiction score, A = ron – roff. This score is positive when the sgRNA’s effect on intrinsic fitness is greater on-drug than off-drug (addiction), negative when the effect is greater off-drug than on-drug (expected wild-type behavior), and zero when the effect is equal on-drug and off-drug (resistance without addiction). We further restricted the inferred set of addicted sgRNAs (A > 0) to those that comprised at least 5% of the pool by week 5, indicating robust growth while on-drug (Fig. 2f–g and Extended Data Fig. 2b–h). By this metric, the fitness of the CXC-targeting sgRNAs as well as others declined upon inhibitor removal and were identified as addicted sgRNAs (Supplementary Dataset 3). Altogether, our CRISPR-addiction scan suggests that multiple resistance mutations enriched in the presence of PRC2 inhibitors may act as drug addiction mutations.
Drug addiction mutations hyperactivate PRC2
Consistent with results from CRISPR-addiction scanning, we successfully isolated clonal Karpas-422 CXCdel cells by single-cell sorting into media supplemented with 1 μM GSK343. Sanger sequencing of EZH2 complementary DNA (cDNA) using wild-type- and CXCdel-specific PCR primers showed that the isolated Karpas-422 clonal cell line contains the heterozygous EZH2 CXCdel mutation in trans with the Y646N mutation (Fig. 3a and Extended Data Fig. 3a,c). Karpas-422 CXCdel+/− clonal cells exhibited a bell-shaped dose-response curve, indicating a requirement for an optimal concentration of PRC2 inhibitors for proliferation (Fig. 3b). Indeed, Karpas-422 CXCdel+/− clonal cells do not proliferate in the absence of PRC2 inhibitors (Extended Data Fig. 3d). A similar bell-shaped dose-response curve was also observed for resistant Pfeiffer cells transduced with EZH2 sgD597 and maintained in the presence of GSK343 (200 nM) (Fig. 3c and Extended Data Fig. 3b–c). Cell cycle analysis of Karpas-422 CXCdel+/− revealed an accumulation of sub-G1 cells upon withdrawal of GSK343, indicating growth arrest (Fig. 3d). These data demonstrate that CXCdel-mutant lymphoma cells depend on PRC2 inhibitors and are drug addicted.
Fig. 3 |. EZH2 CXCdel mutant is addicted to PRC2 inhibitors.
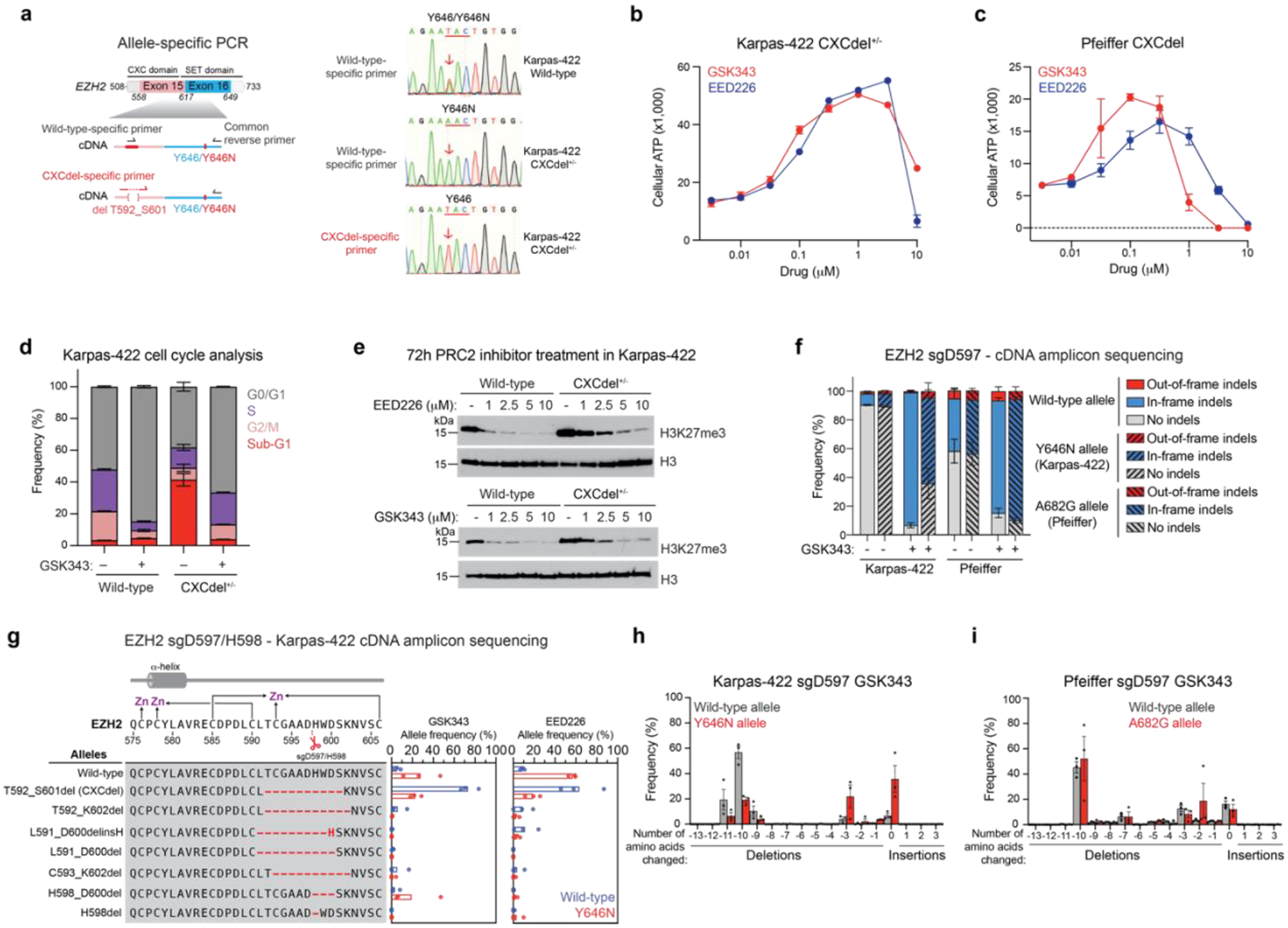
a. Allele-specific PCR followed by Sanger sequencing to determine if the CXCdel mutation resides in cis or in trans with respect to the Y646N mutation in the Karpas-422 CXCdel+/− clonal cell line. Schematic showing the experimental strategy to selectively amplify wild-type or CXCdel amplicon from cDNA template (left). Sanger sequencing traces of Karpas-422 wild-type or CXCdel+/− clonal cell lines amplified with the indicated primers (right).
b. Dose-response proliferation curves of Karpas-422 CXCdel+/− under GSK343 and EED226 treatment for 10 days.
c. Same as in b. but for Pfeiffer CXCdel.
d. Cell cycle analysis of Karpas-422 wild-type and CXCdel+/− cells under 1 μM GSK343 or vehicle treatment for 7 days.
e. Immunoblot showing H3K27me3 levels in wild-type and CXCdel+/− Karpas-422 cells after treatment with vehicle or indicated concentrations of GSK343 or EED226 for 72 hours.
f. Stacked bar plot showing the observed frequencies for out-of-frame, in-frame and no indels in the cDNA encoding EZH2 at week 6 in Karpas-422 and Pfeiffer cells transduced with sgD597 under vehicle or GSK343 (1 μM) treatment.
g. Schematic showing genotypes and bar plots of allele frequencies for mutations that are observed at frequencies of >5% in the cDNA encoding EZH2 surrounding the cut site of CXC-targeting sgRNA at week 6 in Karpas-422 cells transduced with sgD597/H598. Allele frequencies in the (blue) wild-type allele and (red) Y646N allele. (Top) Schematic depicts the secondary structure of C-terminal CXC domain and cysteine residues that coordinate the zinc ions.
h. Bar chart showing frequency of insertions or deletions of amino acids in the cDNA encoding EZH2 at week 6 in Karpas-422 transduced with sgD597 under GSK343 treatment. Deletions, no change and insertions are shown by red, yellow and blue backgrounds respectively.
i. Same as h. but for Pfeiffer.
Data in b-d, f-i are represented by mean ± s.e.m of n = 3. Results in a,e are representative of two independent experiments.
See also Extended Data Figs. 3–4.
See Supplementary Note for calculation of addiction score and Supplementary Fig. 1 for representative flow cytometry histograms for d.
Source data for Fig. 3 (statistical source data, uncropped immunoblots and flow cytometry gating strategy)
To investigate how EZH2 CXCdel impacts PRC2 activity, we assessed H3K27me3 levels in wild-type and CXCdel cells. A modest increase in global H3K27me3 levels was observed in CXCdel-mutant cells in the absence of PRC2 inhibitors. While wild-type cells showed reduced H3K27me3 levels under GSK343 or EED226 treatments, this reduction was attenuated in both Karpas-422 and Pfeiffer CXCdel cells at lower concentrations of PRC2 inhibitors (Fig. 3e and Extended Data Fig. 3e), suggesting that resistance is mediated by maintaining levels of H3K27me3 despite the presence of PRC2 inhibitors. At higher concentrations of PRC2 inhibitors, H3K27me3 was depleted in both wild-type and CXCdel cells, consistent with decreased cell viability observed in the growth assays (Fig. 3b and Extended Data Fig. 3f).
We considered if the CXCdel mutation requires or cooperates with the activating EZH2 SET domain mutations to maintain H3K27me3 levels and confer resistance. To assess EZH2 genotypes in a non-clonal setting, we performed targeted amplicon sequencing of the cDNA spanning the location of the CXCdel mutation and the SET domain DLBCL mutations (aa 560–711) in Karpas-422 and Pfeiffer cells transduced with CXC-targeting sgRNAs. While frameshift mutations are commonly observed in CRISPR/Cas9-induced mutagenesis experiments16,26, both EZH2 alleles remain largely in-frame within the CXC domain in both the bulk Karpas-422 and Pfeiffer populations, suggesting a strong selection pressure against EZH2 loss of heterozygosity (Fig. 3f and Extended Data Fig. 4a–i). CXCdel-mutation frequencies were higher in the wild-type EZH2 allele in Karpas-422 (Fig. 3g and Extended Data Fig. 4j–k). Notably, the most abundant deletion in Karpas-422 transduced with EZH2 sgD597 under GSK343 treatment was the 10-aa CXCdel mutation in the wild-type EZH2 allele whereas a smaller 3-aa deletion was more frequently observed in the EZH2 Y646N allele, suggesting a bias against larger deletions in the Y646N allele (Fig. 3g–h and Extended Data Fig. 4b–d, f–h, j–k). However, little allele preference for CXCdel was observed with respect to A682G in Pfeiffer, with a significant fraction of alleles containing CXCdel and A682G in cis (Fig. 3i and Extended Data Fig. 4e,i,l). Besides the CXCdel mutation, other large in-frame deletions were enriched in Pfeiffer cells treated with EED226, with little allele preference (Extended Data Fig. 4l). Smaller in-frame deletions were also enriched in Pfeiffer under vehicle treatment (Fig. 3f and Extended Data Fig. 4a,e,l). The contrasting genotypes between Karpas-422 and Pfeiffer suggest that mechanistic differences between their distinct SET domain activating mutations may influence allelic preference. Altogether, these findings support the notion that the EZH2 CXCdel mutation is predominantly heterozygous in DLBCL cell lines and acts synergistically with the SET domain activating mutations.
Given that CXCdel-mutant DLBCL cells can harbor the CXCdel mutation and activating SET domain mutation either in trans or in cis, we sought to characterize the impact of CXCdel on PRC2 activity in an otherwise wild-type EZH2 setting. First, we knocked-in CXCdel into the endogenous wild-type EZH2 locus in K562 cells (Extended Data Fig. 5a,d). Unlike Karpas-422 CXCdel cells, no significant changes in the fitness of K562 CXCdel cells were observed in the absence of PRC2 inhibitors (Extended Data Fig. 5b). H3K27me3 levels were increased in homozygous K562 CXCdel+/+/+ compared to K562 wild-type cells (Fig. 4a and Extended Data Fig. 5c), showing that CXCdel alone is sufficient to increase H3K27me3. We did not detect global changes in H3K36me327. Next, we biochemically assessed EZH2 CXCdel activity by purifying wild-type and CXCdel recombinant human PRC2 (EZH2 wild-type or CXCdel, EED, SUZ12, RBAP48, and AEBP2) (Extended Data Fig. 5g)28. PRC2 CXCdel exhibited a ten-fold increase in methyltransferase activity on unmodified mononucleosomes, demonstrating that the mutation’s hypermorphic effect is intrinsic to the PRC2 complex (Fig. 4b). The EZH2 CXC domain mediates interactions between PRC2 and the substrate nucleosome22,23, leading us to consider if enhanced PRC2 activity results from increased substrate affinity. In agreement, electrophoretic mobility shift assays (EMSA) demonstrated that PRC2 CXCdel has increased affinity towards mononucleosomes (Fig. 4c and Extended Data Fig. 5h). Taken together, these results support the notion that the CXCdel mutation activates PRC2 activity by increasing PRC2-nucleosome affinity.
Fig. 4 |. EZH2 CXC-mutants allosterically activate PRC2 methyltransferase activity by increasing substrate affinity.
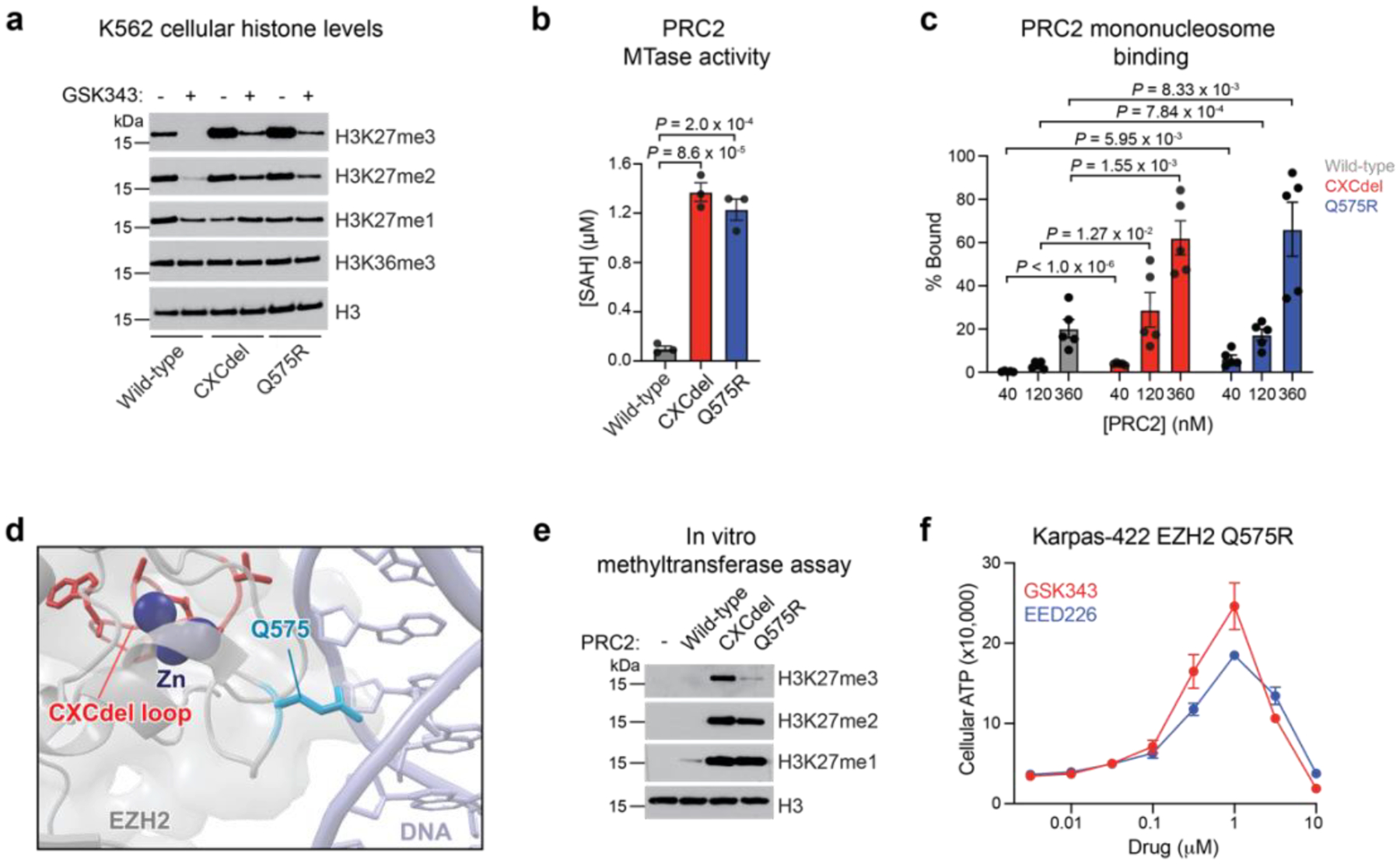
a. Immunoblot showing H3K27me1/2/3 levels in wild-type, CXCdel+/+/+ and Q575R K562 cells after treatment with vehicle or 5 μM GSK343 for 72 hours.
b. Barplot showing enzyme activity of wild-type, CXCdel, and Q575R PRC2 as measured by production of S-adenosyl homocysteine (SAH) by the MTaseGlo Methyltransferase Assay.
c. Quantification of the percentage of PRC2-nucleosome bound fraction across five EMSA replicates to measure binding of wild-type, CXCdel, and Q575R PRC2 to 185 bp mononucleosomes.
d. Structural view showing the location of EZH2 Q575 with respect to nucleosome DNA. The EZH2 CXCdel loop is shown in red, Q575 residue in light blue, and zinc ions are in dark blue. EMDB-7306, PDB: 6WKR
e. Immunoblot showing enzyme activity of wild-type, CXCdel, and Q575R PRC2 as measured by H3K27me1/2/3 levels.
f. Dose-response proliferation curves of Karpas-422 EZH2 Q575R under GSK343 and EED226 treatment for 10 days.
Data in b,f are represented by mean ± s.e.m of n = 3, data in c are represented by mean ± s.e.m of n = 5. Results in a,e are representative of two independent experiments. P values were calculated through two-tailed Student’s t-test for b-c.
See also Extended Data Fig. 5.
Source data for Fig. 4 (statistical source data, uncropped immunoblots)
While most cancer mutations in the EZH2 CXC domain abrogate PRC2 activity29, the hyperactivity of CXCdel suggests that some CXC domain mutations might also enhance PRC2 activity by increasing nucleosome binding. In support, EZH1 Q571R, a mutation present in 27% of thyroid adenomas, has been shown to activate PRC2 activity30. EZH1 is a homolog of EZH2 that is predominantly expressed in non-dividing cells31. The homologous residue in EZH2, Q575, inserts into the major groove of DNA wrapping the substrate nucleosome, suggesting that its mutation to a positively charged residue might increase nucleosome binding (Fig. 4d)22,23. Consistent with this notion, purified PRC2 containing EZH2 Q575R exhibited elevated methyltransferase activity and mononucleosome binding relative to wild-type PRC2 (Fig. 4b–c and Extended Data Fig. 5h). Notably, PRC2 CXCdel and Q575R exhibited increased activity across all three steps of methylation, from me0 to me3 (Fig. 4e). Moreover, K562 cells containing knock-in of EZH2 Q575R also exhibited elevated H3K27 methylation (Fig. 4a and Extended Data Fig. 5a,e). As such, we next considered if EZH2 Q575R confers drug addiction. We knocked in the EZH2 Q575R mutation into Karpas-422 cells in the presence of 1 μM EED226 (Extended Data Fig. 3c,5f). The EZH2 Q575R mutant cells displayed biphasic growth responses to increasing concentrations of GSK343 and EED226, demonstrating drug addiction (Fig. 4f). Thus, both deletions and point mutations in the EZH2 CXC domain can augment PRC2 nucleosome binding and methyltransferase activity to confer drug addiction in EZH2-mutant lymphoma cells.
Pervasive H3K27me3 spreading is a vulnerability of lymphoma
To investigate how CXCdel hyperactivity impacts the chromatin landscape, we profiled H3K27me3 using quantitative spike-in ChIP-seq (ChIP-Rx) in wild-type and CXCdel+/− Karpas-422 cells—continuously grown in GSK343 and after 72 hours of inhibitor withdrawal. Upon inhibitor withdrawal, H3K27me3 significantly increased and broadly spread across the genome in CXCdel+/− cells (Fig. 5a–d), suggesting that H3K27me3 might encroach into neighboring regions typically blocked by active histone modifications27,32,33. To determine which regions are most affected by H3K27me3 spreading upon inhibitor removal, we assessed the correlation between the levels of active histone modifications and the change in H3K27me3 signal at the borders of H3K27me3 domains upon inhibitor withdrawal. H3K36me3 was most associated with increases in H3K27me3 (Fig. 5e and Extended Data Fig. 6a–b). In agreement, we observed a greater increase in H3K27me3 within gene bodies compared to promoters of expressed genes for CXCdel+/− cells (Fig. 5f), consistent with the enrichment of H3K36me3 at gene bodies (Extended Data Fig. 6a)34. H3K27me3 ChIP-Rx also revealed spreading of H3K27me3 genome-wide and within gene bodies in CXCdel+/+/+ versus K562 wild-type cells (Fig. 5g–h and Extended Data Fig. 6c–e), showing that PRC2 CXCdel is sufficient for H3K27me3 domain spreading. Despite these global changes in H3K27me3 induced by CXCdel, genome-wide binding of EZH2 was largely unchanged between CXCdel and wild-type cells for both Karpas-422 and K562 (Extended Data Fig. 6f–g). Together, our data show that inhibitor withdrawal in CXCdel-mutant cells induced pervasive spreading of H3K27me3, which is pronounced at gene bodies marked with H3K36me3.
Fig. 5 |. EZH2 CXCdel hyperactivity induces pervasive spreading of H3K27me3.
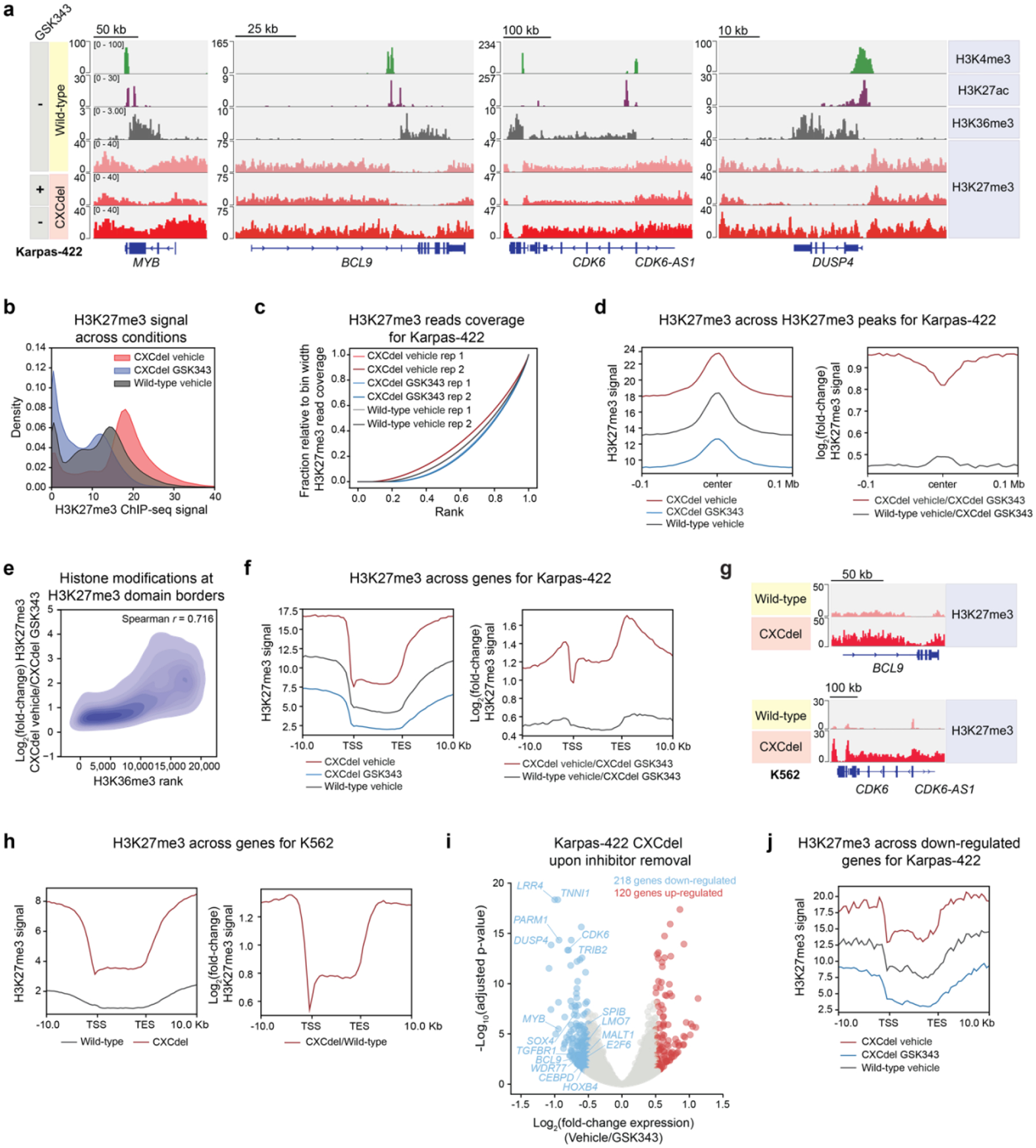
a. Genomic tracks showing histone modifications ChIP-seq in Karpas-422 wild-type and H3K27me3 ChIP-Rx performed in Karpas-422 wild-type and CXCdel+/− cells under continuous GSK343 (1 μM) treatment and 3 days post-inhibitor removal.
b. Density plot depicting the distribution of H3K27me3 ChIP-Rx signal (x-axis) of vehicle- and GSK343-treated (1 μM) Karpas-422 CXCdel+/− cells, and vehicle-treated Karpas-422 wild-type cells for 50-kb genomic bins.
c. Line plot depicting cumulative fraction H3K27me3 ChIP-seq coverage relative to that of the bin with the highest H3K27me3 read coverage (y-axis) as a function of the region’s relative genomic rank (x-axis) for Karpas-422 wild-type and CXCdel+/− cells.
d. Aggregate profile plots of Karpas-422 H3K27me3 ChIP-Rx signal (y-axis, left) and log2(fold-change) H3K27me3 ChIP-Rx signal relative to GSK343-treated (1 μM) CXCdel+/− cells (y-axis, right) centered around H3K27me3 domains in wild-type cells (x-axis).
e. Density plot depicting log2(fold-change) H3K27me3 ChIP-Rx signal between vehicle- and GSK343-treated (1 μM) Karpas-422 CXCdel+/− cells (y-axis) at Karpas-422 wild-type H3K27me3 domain borders relative to ChIP-seq signal rank for H3K36me3 (x-axis).
f. Aggregate profile plots of Karpas-422 H3K27me3 ChIP-Rx signal (y-axis, left) and log2(fold-change) H3K27me3 ChIP-Rx signal relative to GSK343 (1 μM)-treated CXCdel+/− cells (y-axis, right) centered around gene bodies (x-axis). (TSS: transcription start site; TES: transcription end site).
g. Genomic tracks showing H3K27me3 ChIP-Rx performed in K562 wild-type and CXCdel+/+/+ cells.
h. Aggregate profile plots of K562 H3K27me3 ChIP-Rx signal (y-axis, left) and log2(fold-change) H3K27me3 ChIP-Rx signal relative to wild-type and CXCdel+/+/+ cells (y-axis, right) centered around gene bodies (x-axis).
i. Volcano plot showing gene expression changes in Karpas-422 CXCdel+/− cells under continuous GSK343 (1 μM) treatment versus 3 days post-inhibitor removal. Genes with |log2(fold-change)| > 0.5 and adjusted P-value < 0.05 are defined as differentially expressed. P-values were computed with DESeq2 by Benjamini-Hochberg method, adjusted for multiple testing. Data were from three biological replicates.
j. Aggregate profile plot of Karpas-422 wild-type and CXCdel+/− H3K27me3 ChIP-Rx signal (y-axis) centered around gene bodies of genes down-regulated upon GSK343 withdrawal (x-axis).
Results in a,g are representative of two biological replicates. Data in b,d-f,h,j represent average of two biological replicates.
See also Extended Data Fig. 6–7.
RNA-seq before and after inhibitor withdrawal in Karpas-422 CXCdel+/− cells revealed 338 differentially expressed genes (|log2(fold-change)| > ± 0.5, padj < 0.05), of which 218 genes (64.5%) were down-regulated after 72 hours of inhibitor removal (Fig. 5i and Supplementary Dataset 4). For the down-regulated genes, an even greater increase in H3K27me3 within gene bodies was observed upon inhibitor withdrawal — with genes gaining the most H3K27me3 decreasing in expression to the greatest extent (Fig. 5j, Extended Data Fig. 7a–b). Of note, several key genes related to B-cell proliferation and survival, including MYB, BCL9, and CDK6, were down-regulated and gained H3K27me3 within their gene bodies (Fig. 5a, Extended Data Fig. 7c)35,36. In addition, we performed RNA-seq for both wild-type and CXCdel+/+/+ K562 cells and similarly observed the largest gains in H3K27me3 for down-regulated genes, along with a correlation between gene repression and increases in H3K27me3 signal (Extended Data Fig. 7d–f and Supplementary Dataset 5). Collectively, our observations support the notion that H3K27me3 overspreading upon inhibitor withdrawal contributes to silencing of genes critical for lymphoma growth.
Our findings demonstrate a biphasic dependency on H3K27me3 levels to maintain EZH2-mutant lymphoma proliferation, revealing that activating PRC2 is a potential avenue to block lymphoma growth. Active histone modifications, such as H3K36me3, can antagonize PRC2 activity32. Given the observation that H3K27me3 aberrantly spreads into H3K36me3-marked regions, we tested whether inhibition of SETD2, the only methyltransferase known to deposit H3K36me3, could facilitate H3K27me3 spreading and halt DLBCL proliferation. We treated a panel of EZH2-wild-type and -mutant lymphoma cell lines with SETD2-IN-1, a recently disclosed, selective SETD2 inhibitor37,38. Of the seven EZH2-mutant cell lines tested, six were sensitive to SETD2-IN-1 in comparison to only one of four wild-type EZH2 cell lines (Fig. 6a). SETD2-IN-1 treatment substantially reduced H3K36me3 levels across all cell lines, suggesting that the EZH2-mutant selectivity for the inhibitor is not correlated with a decrease in global H3K36me3 levels (Extended Data Fig. 8a). To independently validate the effects of SETD2 inhibition, we depleted SETD2 in wild-type EZH2 and mutant cell lines using short-hairpin RNAs (shRNAs) (Extended Data Fig. 8b–c). Pfeiffer and Karpas-422 (EZH2-mutant) showed the highest level of growth inhibition upon SETD2 knockdown, consistent with the observation that they are more sensitive to SETD2 inhibition than SUDHL4 and SUDHL5. As expected, SUDHL5, a EZH2 wild-type DLBCL cell line, did not respond to SETD2 knockdown (Extended Data Fig. 8c). Beside their antagonism with PRC2 activity, SETD2 and H3K36 trimethylation are also implicated in many other biological processes39–42 including DNA damage repair, and loss of SETD2 can led to genome instability40,42. However, treatment with SETD2-IN-1 had little effect on levels of phosphorylated histone variant H2AX (γH2AX), a marker for presence of DNA damage, suggesting that DNA damage is unlikely to be a major cause of sensitivity to SETD2-IN-1 in Karpas-422 cells (Extended Data Fig. 8d).
Fig. 6 |. SETD2 inhibition induces H3K27me3 spreading and halts lymphoma cell proliferation.
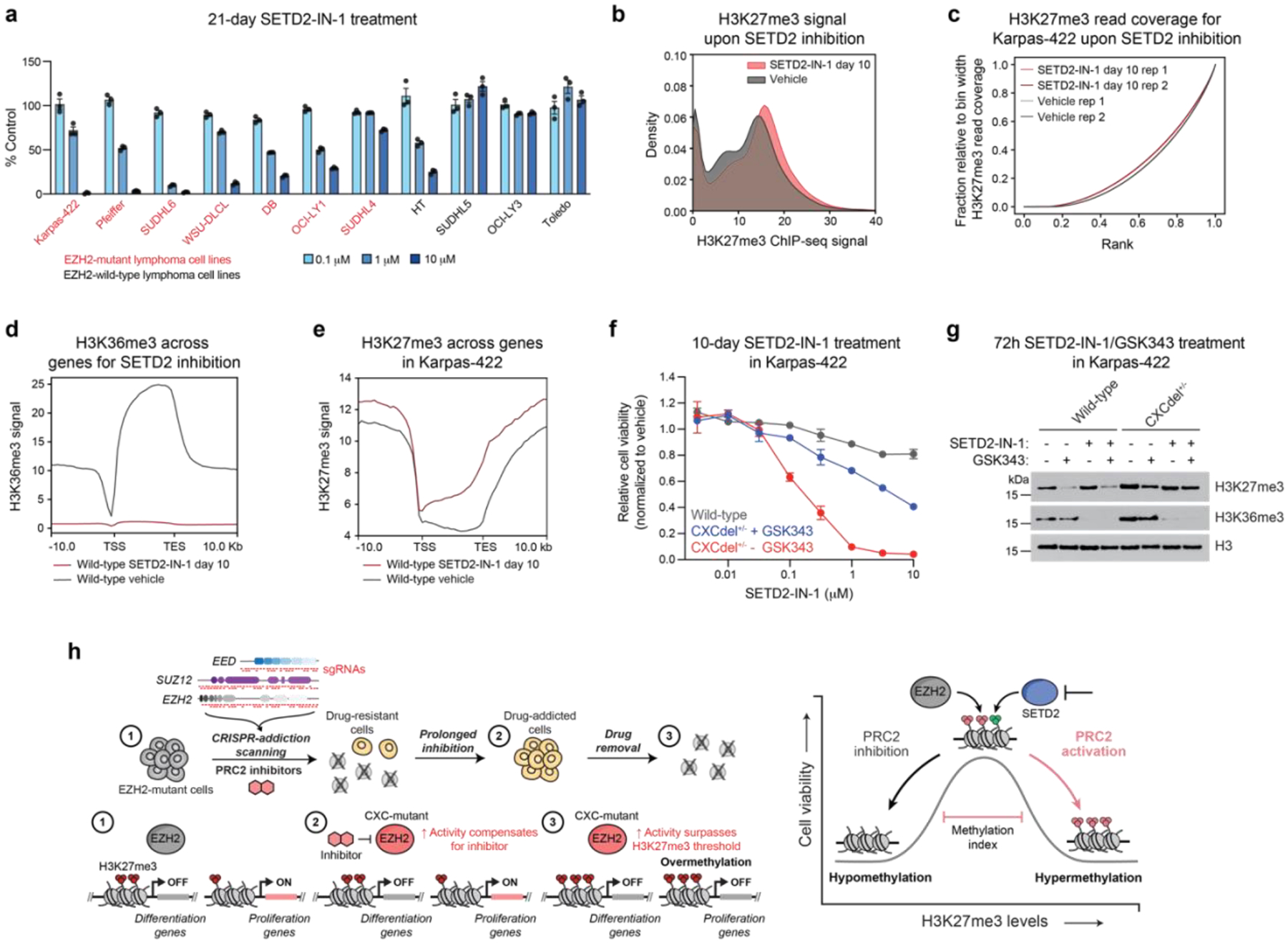
a. Bar plots showing cell proliferation in the presence of SETD2-IN-1 at 0.1, 1, or 10 μM after 21-day treatment.
b. Density plot depicting the distribution of H3K27me3 ChIP-Rx signal (x-axis) of SETD2-IN-1-treated (5 μM) and vehicle-treated Karpas-422 wild-type cells for 50-kb genomic bins.
c. Line plot depicting cumulative fraction H3K27me3 ChIP-seq coverage relative to that of the bin with the highest coverage (y-axis) as a function of the region’s relative genomic rank (x-axis) for vehicle-treated and SETD2-IN-1-treated (5 μM) Karpas-422 wild-type cells.
d. Aggregate profile plot of wild-type Karpas-422 H3K36me3 ChIP-Rx signal (y-axis) for SETD2-IN-1 (5 μM) treatment centered around gene bodies (x-axis).
e. Same as in d. but for H3K27me3 ChIP-Rx signal.
f. Dose-response proliferation curves of Karpas-422 wild-type and CXCdel+/− ± GSK343 (0.5 μM) cells under SETD2-IN-1 treatment for 10 days.
g. Immunoblot showing H3K27me3 and H3K36me3 levels in Karpas-422 wild-type and CXCdel+/− cells after treatment with vehicle, SETD2-IN-1 (5 μM) and/or GSK343 (0.5 μM) for 72 hours.
h. Model illustrating the repressive methylation index in EZH2-mutant lymphoma. (Left) Schematic showing the CRISPR-addiction scanning workflow applied to identify drug addiction mutations in PRC2 (EED, SUZ12 and EZH2) to PRC2 inhibitors. (1) Lymphoma cells with activating EZH2 mutations have elevated H3K27me3 levels which keep the expression of differentiation-related genes repressed while proliferation-related genes turned on. (2) PRC2 inhibitors can reactivate differentiation genes, while CXC mutations further elevate PRC2 activity to compensate for the loss of activity from PRC2 inhibition to maintain repression of differentiation genes. (3) The removal of inhibitor results in PRC2 over-activation that results in widespread H3K27 trimethylation that turns off proliferation genes, revealing a H3K27me3 methylation ceiling exists in the lymphoma cells. (Right) An optimal level of H3K27me3 is required for lymphoma cell growth.
Data in a,f are represented by mean ± s.e.m of n = 3. Results in b,d-e are representative of two biological replicates. Results in g are representative of two independent experiments.
See also Extended Data Fig. 8–9.
Source data for Fig. 6 (statistical source data and uncropped immunoblots)
Profiling H3K36me3 and H3K27me3 in Karpas-422 using ChIP-Rx revealed that SETD2 inhibition substantially decreased levels of H3K36me3 and led to concomitant H3K27me3 spreading (Fig. 6b–e and Extended Data Fig. 9a–b). This spreading was pronounced in gene bodies of expressed genes, particularly those down-regulated in Karpas-422 CXCdel+/− cells upon GSK343 withdrawal (Extended Data Fig. 7f). RNA-seq upon SETD2 inhibition revealed 225 differentially expressed genes, of which 137 genes (60.8%) were down-regulated (Extended Data Fig. 9c and Supplementary Dataset 6). For genes that were down-regulated, a greater increase in H3K27me3 was observed for both Karpas-422 CXCdel+/− cells upon GSK343 withdrawal and SETD2-inhibited wild-type Karpas-422 cells (Extended Data Fig. 9d–e). Consistent with the observation for K562 and Karpas-422 CXCdel cells, gene repression and increases in H3K27me3 were modestly correlated (Extended Data Fig. 9e). More broadly, within gene bodies, changes in H3K27me3 observed upon SETD2 inhibition were correlated with changes in H3K27me3 observed for Karpas-422 CXCdel+/− cells upon inhibitor withdrawal, and this trend was even more evident for SETD2-IN-1 down-regulated genes (Extended Data Fig. 9f). Loss of H3K36me3 was correlated with increases in H3K27me3 (Extended Data Fig. 9g). Gene ontology analysis of down-regulated genes upon SETD2 inhibition revealed enrichment in pathways associated with cell division and apoptosis (Extended Data Fig. 9h). Altogether, these data suggest that SETD2 inhibition can induce epigenomic changes that partially phenocopy those induced by CXCdel.
Lastly, we reasoned that if CXCdel and SETD2 inhibition both lead to H3K27me3 spreading and growth inhibition, then CXCdel+/− cells should be more sensitive to SETD2-IN-1. Indeed, CXCdel+/− cells were most sensitive to SETD2-IN-1 in the absence of GSK343 while wild-type cells were the least sensitive (Fig. 6f). We also observed higher levels of H3K27me3 in CXCdel+/− cells, particularly in the absence of GSK343 (Fig. 6g). Collectively, these data suggest that SETD2 inhibition selectively blocks the growth of EZH2-mutant DLBCL cell lines, in part by inducing H3K27me3 spreading, nominating PRC2 activation as a strategy to block EZH2-mutant lymphoma cell growth.
Discussion
PRC2 is not only an important drug target for anti-cancer therapeutics but has also served as an instructive paradigm for the study of chromatin regulation15,43. By adapting CRISPR-scanning approaches, here we discover mutations in PRC2 that unexpectedly confer drug addiction in EZH2-mutant lymphoma and explore their broader implications on chromatin regulation. To the best of our knowledge, our study presents the first example of drug addiction to epigenetic inhibitors, revealing chromatin thresholds in specific cancer-mutant contexts.
Through genomic and biochemical approaches, we show that drug addiction mutations in the EZH2 CXC domain cooperate with activating cancer mutations in the EZH2 SET domain to maintain H3K27me3 levels in the presence of mechanistically distinct PRC2 inhibitors. The CXC domain mutations increase PRC2 binding to mononucleosomes, which allosterically activates PRC2 by enhancing substrate affinity. Consistent with this idea, the incorporation of cofactors like AEBP2 and PHF1 into the PRC2 complex has also been found to stimulate its activity by increasing substrate binding affinity44–46. While it remains to be determined whether drug resistance or addiction mutations within the EZH2 CXC domain may arise in a clinical setting, our work nominates this region as a potential hotspot for PRC2-activating mutations in cancer. Indeed, we show that the homologous Q571R mutation found in EZH1 enhances EZH2 activity and confers drug addiction in EZH2-mutant lymphoma30. The selective occurrence of CXC- and SET-domain activating mutations in EZH1 and EZH2, respectively, in different cancer contexts suggests that their distinct mechanisms of PRC2 activation are functionally distinct for tumorigenesis and their possible differences remain to be explored.
While the mechanism underlying the activating CXC point mutation Q575R is intuitive, how deletion of the CXCdel loop enhances EZH2 nucleosome binding remains unclear. Rather than CXCdel being neomorphic, we speculate that it instead disables a yet to be identified allosteric mechanism that inhibits PRC2 activity. Notably, while the CXC domain is highly evolutionarily conserved, the CXCdel loop is considerably less so (Extended Data Fig. 1j–k), suggesting that it is not necessary for protein folding but is instead involved in regulation. Furthermore, the loop directly contacts SUZ12. We speculate that deletion of the CXCdel loop reduces SUZ12 contact to confer greater conformational flexibility to the CXC domain23,47, which may subsequently enhance nucleosome binding. Although the precise mechanism of EZH2 activation remains to be determined, our studies highlight how drug resistance mutations can be employed to uncover distal protein sites involved in potential allostery48.
Beyond clinical implications, drug resistance mutations can reveal new target biology48. Notably, drug addiction mutations in PRC2 enabled the discovery of an H3K27me3 ceiling in EZH2-mutant lymphoma, informing our understanding of constraints on chromatin landscapes. This H3K27me3 ceiling forms the upper limit of a “repressive methylation index” – a window of H3K27me3 levels permissible for lymphoma cell proliferation (Fig. 6h). Activating EZH2 SET domain mutations in lymphoma elevate H3K27me3 levels to block B-cell differentiation but in doing so create a ‘Goldilocks’ epigenetic state near this repressive methylation ceiling. Whether a H3K27me3 ceiling exists in non-EZH2-mutant tumors or regulates normal B-cell development remain to be further explored. These findings raise the possibility that other epigenetic signaling pathways may exhibit biphasic dependencies in cancer contexts.
Our study demonstrates how PRC2 activation represents an approach to target EZH2-mutant lymphoma by driving the cells beyond this H3K27me3 repressive ceiling. While direct pharmacological activation PRC2 has not been achieved, our studies show that indirect PRC2 activation via SETD2 inhibition could be used to surpass the H3K27me3 ceiling. Indeed, our results show that EZH2-mutant lymphoma cell lines are more sensitive to SETD2 inhibition. Overspreading of H3K27me3 upon inhibitor withdrawal and SETD2 inhibition is pronounced at the borders of H3K27me3 domains and within gene bodies, suggesting that subsequent down-regulation of cancer growth genes may mediate the anti-proliferative effects. However, H3K27me3 overspreading may also alter physicochemical properties of chromatin and lead to broader biophysical consequences on chromatin organization (e.g., hyper-compaction)49. Consequently, this anti-proliferative mechanism may starkly differ from cell growth inhibition caused by kinase inhibitor drug addiction, which activates pre-existing negative feedback circuits. Regardless of the underlying mechanisms, our findings suggest that strategies to activate PRC2, either directly or indirectly, may be promising therapeutic opportunities for EZH2-mutant lymphoma.
The paradoxical phenomenon of drug addiction has uncovered new cell signaling thresholds and therapeutic vulnerabilities, yet remains largely unexplored with most drug target classes beyond kinases1,2. To explore these possibilities, we developed CRISPR-addiction scanning, which leverages a drug withdrawal arm with accompanying analytical frameworks to identify drug addiction mutations across protein targets. While simple to implement, CRISPR-addiction scanning offers a conceptual advance to study underappreciated dimensions of drug target relationships. More broadly, our work provides an instructive case study into how identification of drug addiction mutations may be exploited to reveal new cancer vulnerabilities. Consequently, intentionally searching for drug addiction mutations using methods like CRISPR-addiction scanning may be warranted to interrogate these possibilities across broader drug target classes.
Methods
Cell lines and chemical compounds
The following cell lines were used: K562 (ATCC, CCL-243), 293FT (Thermo Fisher Scientific, R70007), Karpas-422 and Pfeiffer (gifts from B.E. Bernstein, Dana-Farber Cancer Institute, SU-DHL6, WSU-DLCL, DB, OCI-LY1, SU-DHL4, HT, SU-DHL5, OCI-LY3 and Toledo (gifts from R. Ryan, University of Michigan and SF9 (Expression Systems, 94-001F). All cell lines were authenticated by Short Tandem Repeat profiling (Genetica) and routinely tested for mycoplasma (Sigma-Aldrich). All media for human cell lines were supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco) and FBS (Peak Serum). All human cell lines were cultured in RPMI-1640 (Gibco) supplemented with 10% FBS, except for Karpas-422 with 20% FBS and 293FT cells in DMEM (Gibco) supplemented with 10% FBS, 0.1 mM MEM Non-Essential Amino Acids, 6 mM L-glutamine and 1 mM MEM Sodium Pyruvate. All human cell lines were cultured in humidified 5% CO2 incubator at 37 °C. SF9 was cultured in a non-humidified, non-CO2 incubator at 27 °C shaking at 120 rpm in ESF921 media (Expression Systems) supplemented with 50 U/mL penicillin and 50 μg/mL streptomycin.
GSK343, EED226 and SETD2-IN-1 were purchased from MedChemExpress.
Lentiviral infection
For lentivirus production, plasmids were co-transfected with GAG/POL and VSVG plasmids into 293FT using FuGENE HD (Promega). Media was exchanged after 7 h and the viral supernatant was collected 48–72 h after transfection and filtered (0.45 μm). Cells were transduced by spinfection at 1,070 × g for 90 min at 37 °C with 8 μg/ml polybrene (Santa Cruz Biotechnology). After 48 h post-transduction, media was changed and puromycin (Thermo Fisher Scientific) selection was carried out for 7 days at 1–2 μg/mL.
sgRNA pooled cloning and CRISPR-suppressor scanning experiments
The PRC2 tiling library included all sgRNA with an NGG PAM cleavage site and off-target score (MIT Specificity Score) greater than 20 within the coding sequence of the three members of the core PRC2 complex: EZH2 (NP_004447.2), EED (NP_001294936.1) and SUZ12 (NP_056170.2)50. These sgRNA sequences are listed in Supplementary Table 2. Oligos containing the sgRNA sequences were cloned into pLentiCRISPR.v2, a gift from F. Zhang (Addgene no. 52961), in a pooled fashion50,51. Lentiviral particles carrying the resultant PRC2 tiling library were generated as described above and titered51,52. Karpas-422 cells (4.8 × 107) were transduced at a multiplicity of infection < 0.3. After puromycin selection, cells were split into pools and treated with 1 μM GSK343, 1 μM EED226 or vehicle. Genomic DNA was isolated at specified time points. To measure the composition of the population, sgRNA sequences for all replicates were amplified with PCR primers and sequenced16.
Validation of enriched sgRNAs from CRISPR-suppressor scan
sgRNAs were cloned into pLentiCRISPR.v2; Karpas-422 and Pfeiffer cells were transduced with the resultant plasmids and selected with puromycin. Transduced Karpas-422 cells were treated with 1 μM of GSK343 or 1 μM of EED226 for 6 weeks. Transduced Pfeiffer cells were treated with incremental concentrations of GSK343 or EED226 (for GSK343 - 50 nM for 2.5 weeks, 100 nM for 1.5 weeks and 200 nM for 2 weeks; for EED226 - 200 nM EED226 for 2.5 weeks and 500 nM EED226 for 3.5 weeks). Viability was monitored by flow cytometry with Helix NP™ NIR viability dye (BioLegend).
Generation of clonal drug-resistant EZH2 mutant and knock-in cell lines
To generate drug-resistant mutant cells, Karpas-422 cells were transduced with pLentiCRISPR.v2 EZH2 sgD597 and treated with 1 μM GSK343 for 6 weeks to enrich for drug-resistant mutant cells. Surviving cells were single cell-sorted into media containing 1 μM GSK343.
To generate CXCdel, Q575R Karpas-422 or K562 knock-in cell lines, electroporation was performed using NEON system with ribonucleoprotein (RNP) complex. gRNA complex was formed by reconstituting gene-specific crRNA and tracrRNA (IDT) at 1:1 ratio, hybridized by incubation at 95°C for 5 min, followed by cooling to room temperature. RNP complex was formed by mixing gRNA complex with Alt-R Cas9 enzyme (IDT) and incubation at room temperature for 15 min. 2 × 105 cells were washed twice with PBS and resuspended in buffer R. RNP complex, single-stranded oligodeoxynucleotides (ssODNs) and Alt-R Cas9 electroporation enhancer (IDT) were added to the cell suspension and electroporated at 1000V (50 ms pulse width for 1 pulses) for Karpas-422, 1450V (10 ms pulse width for 3 pulses) for Karpas-422. After electroporation, cells were immediately transferred to prewarmed media supplemented with HDR Enhancer (IDT). To generate single-cell clones, cells were sorted on a MoFlo Astrios EQ cell sorter.
Generation of inducible SETD2 knockdown cell lines
shRNAs targeting SETD2 were cloned into Tet-regulated pRRL.RTREVIR miR-E shRNA expression vector. DLBCL cell lines Karpas-422, Pfeiffer, SUDHL4 and SUDHL5 were transduced with shRen.713 or shSETD253. Transduced cell populations were sorted based on Venus fluorescence by flow cytometry. Expression of shRNA was induced with 1 μg/mL doxycyline. Knockdown efficiency was determined by quantification of SETD2 transcript levels. Total RNA was isolated from cells using RNeasy Mini Kit (Qiagen). cDNA synthesis was carried out using SuperScript III First-Strand Synthesis Supermix (Thermo Fisher). Quantitative PCR analysis was performed with SYBR Select Master Mix (Thermo Fisher Scientific) on a Bio-Rad CFX96 real-time PCR system. All signals were quantified using the delta Cq method and normalized to levels of GAPDH. The shRNA sequences and primers for amplification of SETD2 transcripts are listed in Supplementary Table 2.
Genotype determination
Genomic DNA was isolated using the QIAamp® Blood Mini Kit (Qiagen). For library preparation, genomic PCR primers with Illumina adapter sequences were used to amplify specified regions of EZH2 as previously described16. Samples were sequenced on a MiSeq instrument (Illumina). The sequencing reads were analyzed using CRISPResso2 (v.2.0.40)54.
Cell growth assays
Cells were seeded in 96-well plates with 45,000 cells per well in triplicate with drug or vehicle treatments. The number of viable cells was quantified at days 3, 7, 10 and 14 with CellTiter-Glo (Promega) on a SpectraMax i3x plate reader (Molecular Devices). ATP standard curve was prepared using known concentrations of ATP and used to calculate the ATP content of cells. On the days of measurement, cells were replated with fresh media containing drug or vehicle.
Cell cycle analysis
Wild-type and CXCdel+/− Karpas-422 cells were treated with 1 μM GSK343 or vehicle for 7 days. Cells were washed with PBS and then fixed with cold 70% ethanol and incubated for a minimum of 2 h at −20°C. After fixing, cells were washed twice with PBS and then resuspended in Propidium Iodide (PI) staining solution (1x PBS supplemented with 50 μg/mL PI, Invitrogen; 100 μg/mL RNase A, Qiagen; and 2 mM MgCl2) and incubated for 30 min at room temperature in the dark. Data acquisition was performed on ACEA Novocyte flow cytometer (Agilent) using NovoExpress software (version 1.5.6). Data was analyzed with FlowJo™ (version 10.7.1).
Immunoblotting
For whole cell extracts, cells were lysed on ice using radioimmunoprecipitation assay (RIPA) buffer (Boston BioProducts) supplemented with fresh HALT™ protease inhibitor cocktail (Thermo Fisher Scientific). The lysate was clarified by centrifugation at 18,000 x g for 15 min. For isolation of core histones, lysates were prepared using the Histone Extraction Kit (Active Motif). Protein concentration of the lysates was measured using Bradford Assay (Bio-rad). Immunoblotting was performed according to standard procedures. The primary antibodies used are as follows: EZH2 (Cell Signaling Technology, #5246; 1:5,000 dilution); H3K27me1 (Active Motif, cat no. 61015; 1:5,000 dilution); H3K27me2 (Cell Signaling Technology, #9728; 1:5,000 dilution); H3K27me3 (Cell Signaling Technology, #9733; 1:5,000 dilution); H3K36me3 (Cell Signaling Technology, #9763); Histone 3 (Active Motif, cat no. 39763; 1:10,000 dilution); GAPDH (Santa Cruz Biotechnology, sc-477724; 1:10,000 dilution).
Purification of recombinant PRC2
Human recombinant PRC2 used contained 5 members: EZH2, SUZ12, EED, RBAP48 and AEBP2 (UniProtDB entry isoform sequences Q15910-2, Q921E6-3, O75530-1, Q09028-1, Q6ZN18-1 respectively). Recombinant CXCdel PRC2 contained the same 5 complex members, with a 10-amino acid deletion in EZH2 from T592-S601. Recombinant Q575R PRC2 contained the same 5 complex members. All members were tagged with an N-terminal MBP tag cleavable by PreScission protease. pFastBac plasmids for the five wild-type PRC2 members were gifts from T. Cech. The proteins were co-expressed in insect cells as according to the Bac-to-Bac baculovirus expression system. Detection of gp64 was used to determine baculovirus titer (Expression Systems). For expression, SF9 cells were grown to a density of 2 × 106 cells/mL and infected with PRC2 baculovirus at a MOI of 1.2 for AEBP2 and 0.7 for all other subunits. The cells were incubated for 72 h (27 °C, 120 rpm), harvested, and then frozen with liquid nitrogen for future purification.
PRC2 complexes were purified according to a published procedure28,55. All protein purification steps were performed at 4 °C. Insect cells were first lysed by incubating for 1 h with lysis buffer (10 mM Tris-HCl, pH 7.9, 250 mM NaCl, 0.5% Nonidet P-40 Substitute, 1 mM TCEP) at a ratio of 8 mL of buffer per gram of cell pellet. The lysate was clarified by centrifugation at 29,000 x g for 40 min. The clarified lysate was added to equilibrated Amylose resin (NEB Cat no. E8021S, 0.3 mL of resin per gram of cell pellet) and bound in batch for 2 h. The resin was then washed with >10 column volumes (CV) of lysis buffer, followed by >16 CV of Wash buffer 1 (10 mM Tris-HCl, pH 7.9, 500 mM NaCl, 1 mM TCEP) and lastly >16 CV Wash buffer 2 (10 mM Tris-HCl, pH 7.9, 150 mM NaCl, 1 mM TCEP). PRC2 was eluted by addition of 3 CV of Wash buffer 2 + 10 mM Maltose. Protein was concentrated to 15 mg/mL using a Amicon Ultra-15 Centrifugal Filter Unit, 30 kDa molecular weight cut off (MWCO). In-house made PreScission protease was added at a ratio of 1:100 (protease:PRC2) and the NaCl concentration was adjust to 250 mM by addition of the appropriate amount of 5 M NaCl. After overnight incubation, cleavage of the MBP tags was confirmed by SDS-PAGE. Next, the cleaved protein was subjected to a 5mL HiTrap Heparin column at a flow rate of 1.5 mL/min with a gradient elution from Buffer A (10 mM Tris-HCl, pH 7.9, 150 mM NaCl, 1 mM TCEP) to Buffer B (10 mM Tris-HCl, pH 7.9, 2 M NaCl, 1 mM TCEP) over 35 CV. The salt concentration was diluted to 350 mM and the protein was concentrated and subjected to size-exclusion over a Superose 6 Increase 10/300 GL column with running buffer (20 mM HEPES, pH 7.9, 150 mM NaCl, 1 mM TCEP) at a flow rate of 0.5 mL/min. PRC2-peak fractions were verified with SDS-PAGE. The correct fractions were pooled and concentrated as indicated above. The final concentration was determined by UV absorbance at 280 nm measured by Nanodrop. The ratio of absorbance at 260 nm/280 nm ratio was less than 0.7 indicating minimal nucleic acid contamination.
Purification of mononucleosomes
Xenopus histones were expressed in E. coli and purified as previously described with minor modifications56,57. Briefly for the histone purification, inclusion bodies were prepared and extracted as described previously56,57. These pellets were subsequently dissolved into unfolding buffer (7 M guanidinium HCl, 20 mM Tris-HCl, pH 7.5, 10 mM DTT), dialyzed into SAU-200 buffer (7 M Urea, 20 mM sodium acetate pH 5.2, 200 mM NaCl, 5 mM 2-mercaptoethanol, 1 mM EDTA) and loaded onto a 5mL HiTrap SP HP column, eluted by gradient elution over 30 CV from SAU-200 buffer to SAU-600 buffer (7 M Urea, 20 mM sodium acetate pH 5.2, 600 mM NaCl, 5 mM 2-mercaptoethanol, 1 mM EDTA). Histone-containing fractions were pooled, dialyzed into distilled water containing 2 mM 2-mercaptoethanol (at least 3 changes) and lyophilized for storage at −20°C58. For histone octamer assembly, the histones were first dissolved in in unfolding buffer (20 mM Tris-HCl pH 7.5, 6 M guanidinium HCl, 5 mM DTT), combined in equimolar amounts and dialyzed into refolding buffer (10 mM Tris-HCl pH 7.5, 2 M NaCl, 1 mM EDTA, 5 mM b-ME) and purified over a Superdex 200 column (GE Healthcare). Octamer-containing fractions were pooled according to purity and histone stoichiometry, and then flash-frozen for storage at −80 °C after addition of glycerol to a final concentration of 10% (v/v).
The 185-base pair (bp) nucleosome DNA used with Xenopus nucleosomes was atcgctgttcaatacatgcacaggatgtatatatctgacacgtgcctggagactagggagtaatccccttggcggttaaaacgcgggg gacagcgcgtacgtgcgtttaagcggtgctagagctgtctacgaccaattgagcggcctcggcaccgggattctccagggcggccgc gtatagggat. The 601-nucleosome positioning sequence is underlined. The plasmid containing the 185-bp nucleosomal DNA sequence was a gift from Philip Cole (Harvard Medical School). The 185-bp nucleosomal DNA sequence was prepared via large scale plasmid preparation followed by cleavage with the EcoRV-HF restriction enzyme. The cleavage product was purified with Q Sepharose HP resin with a stepwise elution from Buffer A (20 mM Tris pH 8.0, 1 mM EDTA) to 40% Buffer B (20 mM Tris pH 8.0, 1 mM EDTA, and 1 M NaCl) and elution with 70% Buffer B. Eluted DNA was subsequently purified by size-exclusion on a Superdex 200 column in Buffer B. The resulting product was buffer-exchanged into Buffer A and stored at −20 °C. Nucleosomes were reconstituted by mixing the DNA and the octamer in a 1:1.1 ratio and performing gradient dialysis as described previously57. Reconstituted nucleosomes were purified by anion exchange with a TSKgel DEAE-5PW (7.5 mm × 7.5 cm) column; their composition was confirmed by native gel electrophoresis and were subsequently concentrated and stored as described previously56. All dialysis steps employed 6–8 kDa MWCO dialysis tubing (Spectra-Por) and 10 kDa MWCO Amicon centrifugal filter units were used for concentration and desalting.
Electrophoretic mobility shift assay (EMSA) with mononucleosomes
45 nM of mononucleosomes purified as above were incubated with increasing amounts of protein in 20 μL of assay buffer (50 mM Tris-HCl, pH 7.5, at 25 °C, 25 mM KCl, 2.5 mM MgCl2, 0.1 mM ZnCl2, 2 mM 2-mercaptoethanol, 0.1mg/mL BSA, 10% v/v glycerol). After incubation at 30 °C for 30 min, 15 μL of the reaction was loaded onto a 3.5% TBE polyacrylamide gel. Gel electrophoresis was performed at 140 V for 1 h in cold 1 × TBE at 4 °C. The gel was stained with SYBR Green I (Thermo Fisher Scientific) and fluorescence images were acquired by an Azure c200 imager. Densitometry of the bound PRC2-mononucleosome band was performed by ImageJ.
MTase-Glo enzyme assay
Endpoint methyltransferase measurements were performed in a 20 μL reaction volume utilizing 80 nM of PRC2 enzyme with 1 μM of mononucleosomes and 40 μM SAM in reaction buffer (50 mM Tris-HCl, pH 8 at 30°C, 10 mM KCl, 2.5 mM MgCl2, 0.1 mM ZnCl2, 2 mM 2-mercaptoethanol, 0.1 mg/mL BSA, 5% v/v glycerol). Reactions were incubated at 30 °C for 2 h and quenched with 0.1 % TFA. Enzyme activity was assessed by SAH production as determined by luminescence through the MTase-Glo assay (Promega). Briefly, SAH production for each reaction was calculated by correlating the raw luminescence to a SAH standard curve correcting for basal luminescence of mononucleosome and buffer to determine SAH production.
ChIP-seq
Karpas-422 and K562 cells were crosslinked with 1% formaldehyde (Sigma-Aldrich) for 10 min and quenched with 125 mM glycine. Cells were lysed, sonicated (Branson sonifier, 0.7s on / 1.3s off, 5 min total on, 50% amplitude) and clarified by centrifugation. Lysates were incubated overnight at 4°C with the following antibodies: anti-EZH2 (Cell signaling Technology, #5246); anti-H3K27me3 (Cell signaling Technology, #9733S); anti-H3K36me3 (Cell signaling Technology, #9763). For quantitative ChIP, drosophila chromatin (Active Motif cat no. 08221011) and spike-in antibody (Active Motif cat no. AB27377370) were added to sonicated chromatin59. Dynabeads Protein G (Thermo Fisher) were added on the next day. Following incubation, the beads were isolated and washed. Immunoprecipitated chromatin was eluted, treated with RNase A and Proteinase K and purified by SPRI beads. To prepare ChIP-seq libraries, the chromatin was subjected to end-repair (End-It DNA End-Repair Kit, Lucigen), A-tailing (Klenow fragment, 3’−5’ exo-, NEB), ligation to barcoded adapters (KAPA) and PCR library amplification (KAPA), followed by SPRI-size selection and purification. Libraries were sequenced using a NovaSeq SP kit (Illumina) on a Novaseq instrument (Illumina).
RNA-seq
Total RNA was isolated from cells in triplicate separate cultures using RNeasy Mini Kit (Qiagen). Library preparation was performed using the QuantSeq 3’ mRNA-Seq Library Prep Kit FWD (Lexogen). Libraries were sequenced using a NovaSeq SP kit (Illumina) on a Novaseq instrument (Illumina).
ChIP-seq data analysis
For quantitative ChIP-seq analysis, reads were aligned to a composite reference genome for dm6 and hg38 utilizing bowtie2 (version 2.3.2) according to the Spiker analysis framework (version 1.03). Aligned reads were sorted using samtools (version 0.1.19) and the Spiker split_bam.py script was run to calculate scaling factors and to split reads into an alignment file containing reads specifically aligning to the hg38 genome (Spiker, version 1.0.3)60. This alignment file was then converted into spike-in normalized bigwig file for further analysis and visualization utilizing the deepTools bamCoverage command with the following parameters: -binSize 50 --ignoreDuplicates --normalizeUsing RPKM --ignoreForNormalization chrM --extendReads –scaleFactor. Peaks for H3K27me3 and H3K36me3 were called using Sicer with the parameters: -e 8000 -s hg38 -g 12000 -w 4000 (version 2.0). Individual replicate peaks were subsequently combined utilizing Bedtools2 merge -d 12000 (version 2.26.0). Border regions of peaks were called by using the Bedtools2 “flank” command with the parameter -b 50000 (version 2.26.0). For bin level analysis, the H3K27me3 signal value was averaged across 50 kb bins utilizing the multiBigwigSummary function (deepTools, version 3.4.3). To generate metaprofile plots comparing between treatment conditions the log2 option in the bigwigCompare function was used prior to plotting with the plotHeatmap function (deepTools, version 3.4.3). To generate fingerprint plots investigating H3K27me3 spreading, the plotFingerprint function was used on the hg38 split alignment files for individual replicates with the parameters: --ignoreDuplicates --extendReads --skipZeros (deepTools, version 3.4.3). Subsequent data processing was performed in Python (version 3.9.6) using standard plotting functions.
For EZH2 ChIP-seq analysis, reads were aligned to the hg38 genome utilizing bowtie2 (version 2.3.2). Aligned reads were sorted using samtools (version 0.1.19). This alignment file was then converted into bigwig format for further analysis and visualization utilizing the deepTools bamCoverage command with the following parameters: -binSize 50 --ignoreDuplicates --normalizeUsing RPKM --ignoreForNormalization chrM –extendReads --blackListFileName. Specifically, the file “ENCFF356LFX.bed” downloaded from Encode was used to define blacklisted regions for hg38. For bin level analysis, the EZH2 signal value was averaged across 50 kb bins utilizing the multiBigwigSummary function (deepTools, version 3.4.3). Subsequent data processing was performed in Python (version 3.9.6) using standard plotting functions.
H3K4me3, H3K27ac and H3K36me3 data from ENCODE ENCFF932BQZ, ENCFF435PXJ and ENCFF603BHK was used in Fig. 5 and Extended Data Fig. 6.
RNA-seq data processing and differential gene expression analysis
RNA-seq data was processed according to the QuantSeq 3′ mRNA-Seq Library Prep recommended analysis pipeline through alignment to the Ensembl transcriptome (GRCh38). DESeq2 with R (v.3.5.1) was employed for the differential expression analysis. All genes with adjusted P < 0.05, calculated using the Benjamini–Hochberg correction, and |log2(fold-change)| > 0.5 were considered to be differentially expressed. The location of the transcription start site (TSS) and transcription end sites (TES) of genes was determined using the biomaRt package (version 2.41.1) and only genes with a defined hgnc symbol name were included in the annotation list. When multiple isoforms were present, the TSS and TES were defined to be at the minimum and maximum positions, respectively. A table of significantly differentially expressed genes is provided in Supplementary Datasets 4–6.
Quantification and statistical analysis
Microsoft excel (version 16.69.1), PRISM (version 9.3.1) (Graphpad) and R (version 3.6.2) software packages are used to perform statistical analyses. Statistical parameters including the exact value and definition of n, the definition of center, dispersion, precision measures (mean ± s.d. or s.e.m), statistical significance, and statistical tests used are reported in figures and figure legends. Protein structures were visualized using PyMOL (version 2.5.2) (Schrödinger).
Extended Data
Extended Fig. 1 |. sgRNAs targeting EZH2 CXC domain and the drug-binding pockets are the most enriched in CRISPR-suppressor scanning.
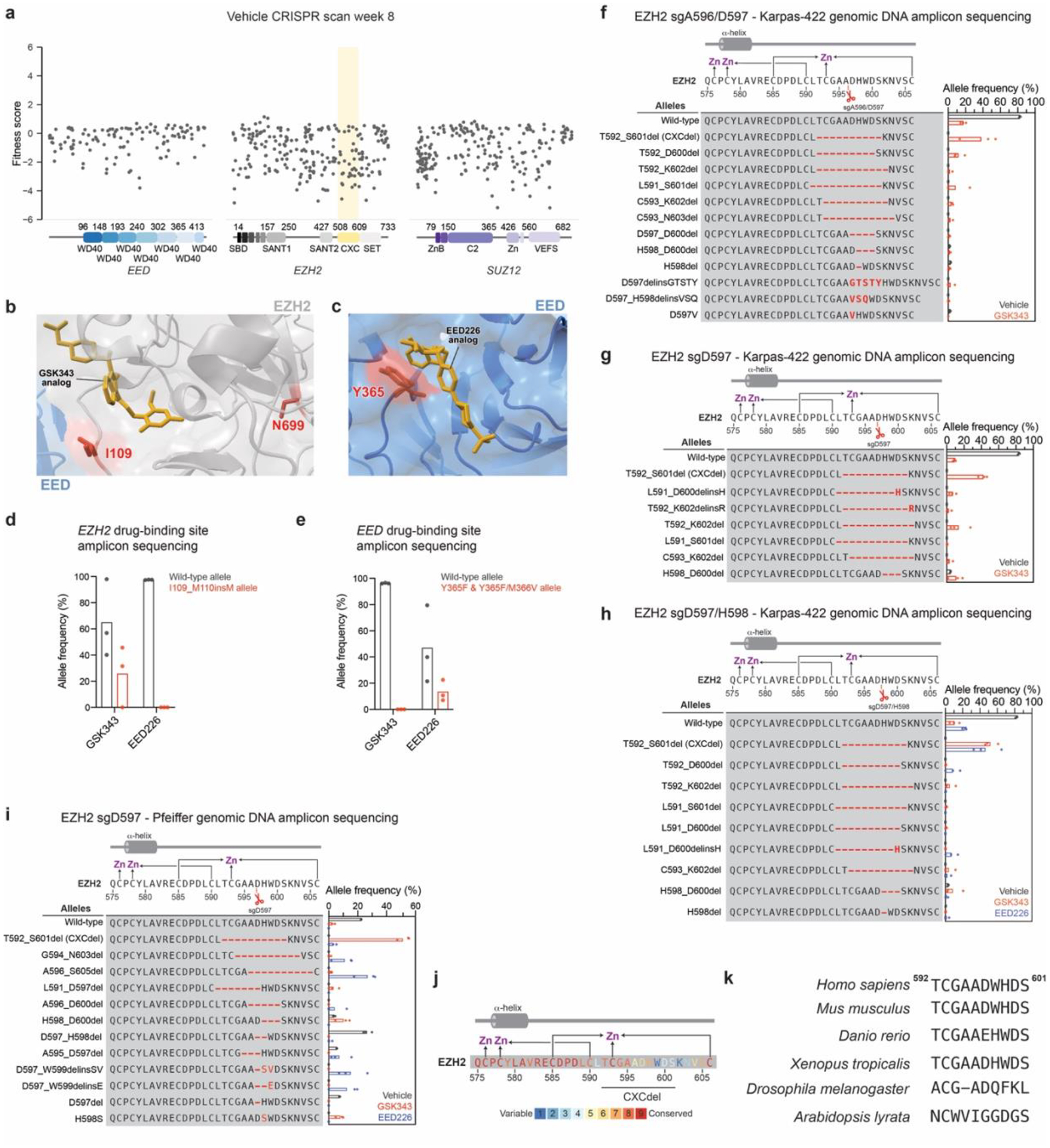
a. Scatter plots showing fitness scores (y-axis) in Karpas-422 under vehicle treatment at week 8. Fitness scores were calculated as the log2(fold-change) sgRNA enrichment under vehicle normalized to the mean of the negative control sgRNAs (n = 58). The PRC2-targeting sgRNAs (n = 650) are arrayed by their predicted cut sites in the coding sequences (x-axis). Data represent mean of n = 3 replicates.
b. Structural view of the GSK343 binding site with EZH2 residues I109 and N699 highlighted in red. GSK343 analog is shown in gold. PDB: 5LS6
c. Structural view of the EED226 binding site with EED residue Y365 highlighted in red. EED226 analog is shown in gold. PDB: 5K0M
d. Bar plots of wild-type (grey) and EZH2 I109_M110insM mutant (red) allele frequencies from CRISPR-suppressor scanning under 5-week GSK343 (1 μM) or EED226 (1 μM) treatment.
e. Same as in d. but for wild-type (grey) and EED Y365F and Y365F/M366V mutant (red) allele frequencies.
f. Schematic showing genotypes and bar plots of allele frequencies for mutations that are observed at frequencies of >2% in the gDNA encoding EZH2 surrounding the CXCdel loop for 6-week GSK343 (1 μM) treatment after transduction with sgA596/D597 in Karpas-422. Allele frequencies under vehicle (grey) or GSK343 (red). (top) Schematic depicts the secondary structure of C-terminal CXC domain and cysteine residues that coordinate the zinc ions.
g. Same as in f. but for sgD597 transduction in Karpas-422.
h. Same as in f. but for sgD597/H598 transduction in Karpas-422.
i. Same as in f. but for sgD597 transduction in Pfeiffer.
j. The conservation scores of each amino acid within and surrounding the EZH2 CXCdel loop are shown in colors ranging from variable (blue) to conserved (red) as calculated by ConSurf-DB24. (Top) Schematic depicts the secondary structure of C-terminal CXC domain and cysteine residues that coordinate the zinc ions.
k. Comparison of amino acids homologous to the EZH2 CXCdel loop in various model organisms.
Data in a, d-e, f-i are represented by mean of n = 3.
Extended Fig. 2 |. CRISPR-addiction scanning reveals drug addiction mutations.
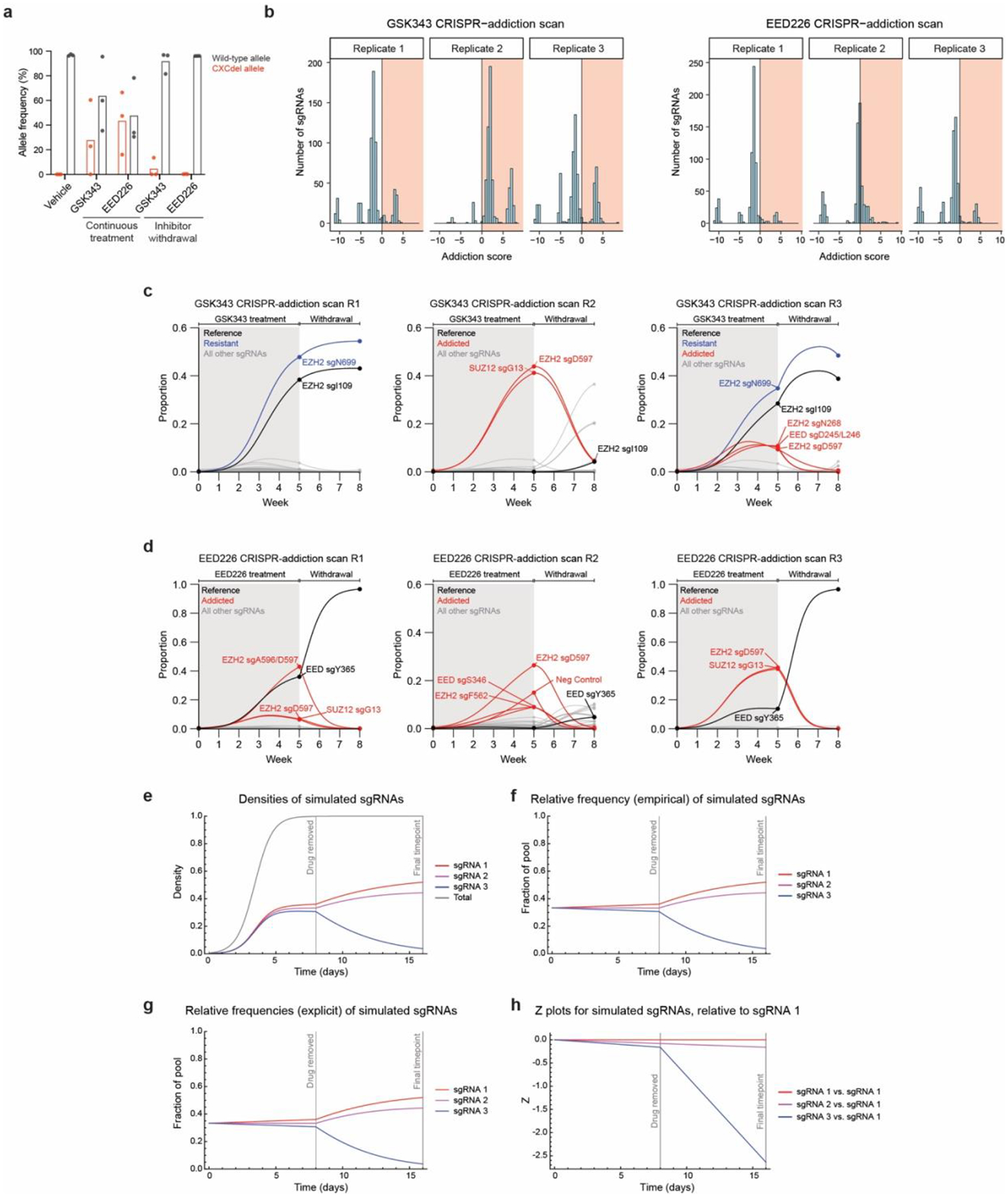
a. Bar plot of wild-type (grey) and CXCdel (red) allele frequencies (y-axis) from CRISPR-addiction scanning. Data are represented by mean of n = 3.
b. Histogram showing distribution of addiction scores (x-axis) among the three replicates of the GSK343 (left) and EED226 (right) CRISPR-addiction scanning experiments.
c. Graph showing proportions of each sgRNA (y-axis) in each replicate of the GSK343 CRISPR-addiction scan over time (x-axis). Curves were calculated using the estimated intrinsic growth rate r for each sgRNA-containing cell population under the assumption of competitive logistic growth. The reference sgRNA is shown in black, sgRNAs demonstrating pure resistance are in blue and addicted sgRNAs are in red.
d. Same as in c. but for each replicate of the EED226 CRISPR-addiction scan.
e. Plots showing simulated densities for three hypothetical sgRNA populations. sgRNA densities begin at 1/500. The intrinsic fitness (uninhibited growth rates) for sgRNA populations 1–3 are 1.51, 1.50, and 1.49, respectively. When drug is removed, the fitness of sgRNA populations 1 and 2 remain unchanged, while the fitness of sgRNA 3 decreases to 1.20. The sgRNA densities over time are described by Equation 7. The total density is equal to the sum of the densities of the three sgRNAs and is described by Equation 2.
f. Plots showing simulated sgRNA proportions according to Equation 7. Trajectories are derived by dividing the sgRNA densities depicted in (E) by the total population size.
g. Plots showing simulated sgRNA proportions according to Equation 9. sgRNA proportions start at 0.33. The intrinsic fitness (uninhibited growth rates) for sgRNA populations 1–3 are 1.51, 1.50, and 1.49, respectively. When drug is removed, the fitness of sgRNAs 1 and 2 remain unchanged, while the fitness of sgRNA 3 decreases to 1.20.
h. Plots showing Z-values for simulated sgRNAs relative to sgRNA population 1. The definition of Z is given in Equation 10. The slopes of these lines give may be used to compute the change in intrinsic fitness for each sgRNA when on drug vs. off drug.
Extended Fig. 3 |. CXCdel lymphoma cells are addicted to PRC2 inhibitors.
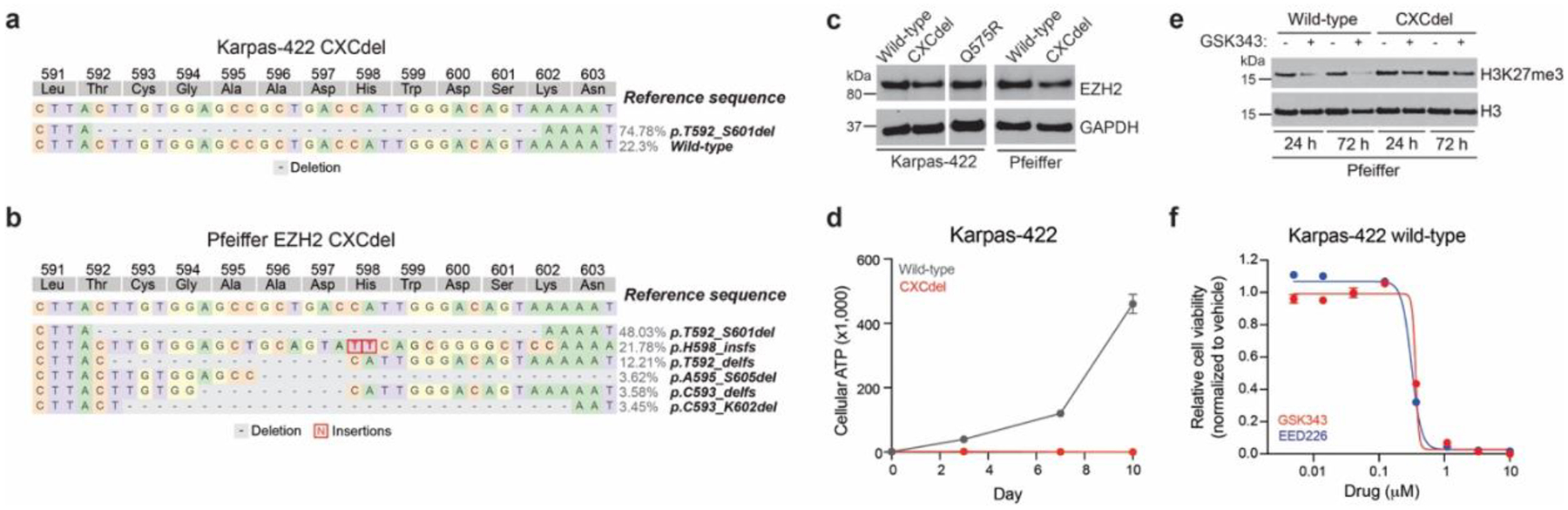
a. Schematic showing genotypes and allele frequencies of the Karpas-422 CXCdel+/− clonal line. We believe that the over-representation of the CXCdel allele in the clonal cell line can be attributed to PCR bias. The heterozygous nature of Karpas-422 CXCdel is confirmed in Fig. 3a.
b. Schematic showing genotypes and allele frequencies for mutations that are observed at frequencies of > 2% in the gDNA encoding the EZH2 CXC domain in Pfeiffer CXCdel.
c. Immunoblots showing EZH2 levels in Karpas-422 wild-type, CXCdel+/− and Q575R (left); and Pfeiffer wild-type, CXCdel (right).
d. Line plot depicting cumulative growth of Karpas-422 wild-type and CXCdel+/− over 10 days.
e. Immunoblot showing H3K27me3 levels in wild-type and CXCdel Pfeiffer cells after treatment with vehicle or 200 nM GSK343 for 24 or 72 hours.
f. Dose-response proliferation curves of Karpas-422 wild-type under GSK343 or EED226 treatment for 10 days.
Results in a-c,e are representative of two independent experiments. Data in d,f are represented by mean ± s.e.m of n = 3.
Extended Fig. 4 |. CXCdel lymphoma cells are depleted upon drug withdrawal.
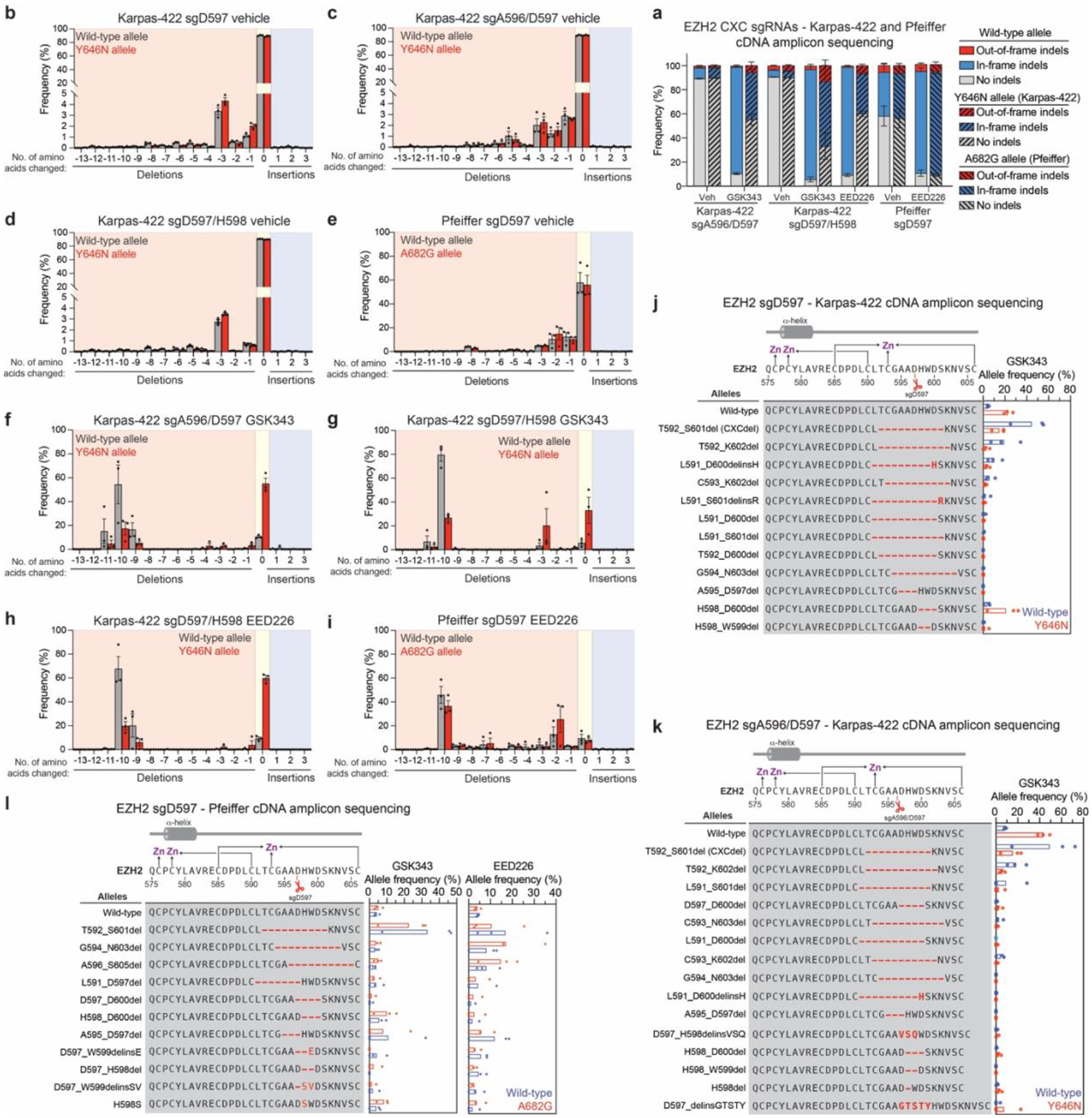
a. Stacked bar plot showing the observed frequencies for out-of-frame, in-frame and no indels in the cDNA encoding EZH2 at week 6 in Karpas-422 and Pfeiffer cells transduced with sgA596/D597, sgD597/H598 or sgD597 under vehicle, GSK343 (1 μM) or EED226 (1 μM) treatment.
b. Bar chart showing frequency of insertions or deletions of amino acids in the cDNA encoding EZH2 at week 6 in Karpas-422 transduced with sgD597 under vehicle treatment.
c. Same as b. but for sgA596/D597 transduction in Karpas-422 under vehicle treatment.
d. Same as b. but for sgD597/H598 transduction in Karpas-422 under vehicle treatment.
e. Same as b. but for sgD597 transduction in Pfeiffer under vehicle treatment.
f. Same as b. but for sgA596/D597 transduction in Karpas-422 under GSK343 (1 μM) treatment.
g. Same as b. but for sgD597/H598 transduction in Karpas-422 under GSK343 (1 μM) treatment.
h. Same as b. but for sgD597/H598 transduction in Karpas-422 under EED226 (1 μM) treatment.
i. Same as b. but for sgD597 transduction in Pfeiffer under EED226 (incremental doses up to 500 nM) treatment.
j. Schematic showing genotypes and bar plots of allele frequencies for mutations that are observed at frequencies of >2% in the cDNA encoding EZH2 encompassing the CXC and SET domains for GSK343 treatment at week 6 for sgD597transduction in Karpas-422. Allele frequencies in the (blue) wild-type allele and (red) Y646N allele. (Top) Schematic depicts the secondary structure of C-terminal CXC domain and cysteine residues that coordinate the zinc ions.
k. Same as in j. but for sgA596/D597 transduction in Karpas-422.
l. Same as in j. but for sgD597 transduction in Pfeiffer. The wild-type allele is shown in blue and the A682G allele is shown in red.
Data in a-k are represented by mean ± s.e.m of n = 3.
Extended Fig. 5 |. EZH2 CXC-mutants elevate PRC2 methyltransferase activity in vitro and in cells.
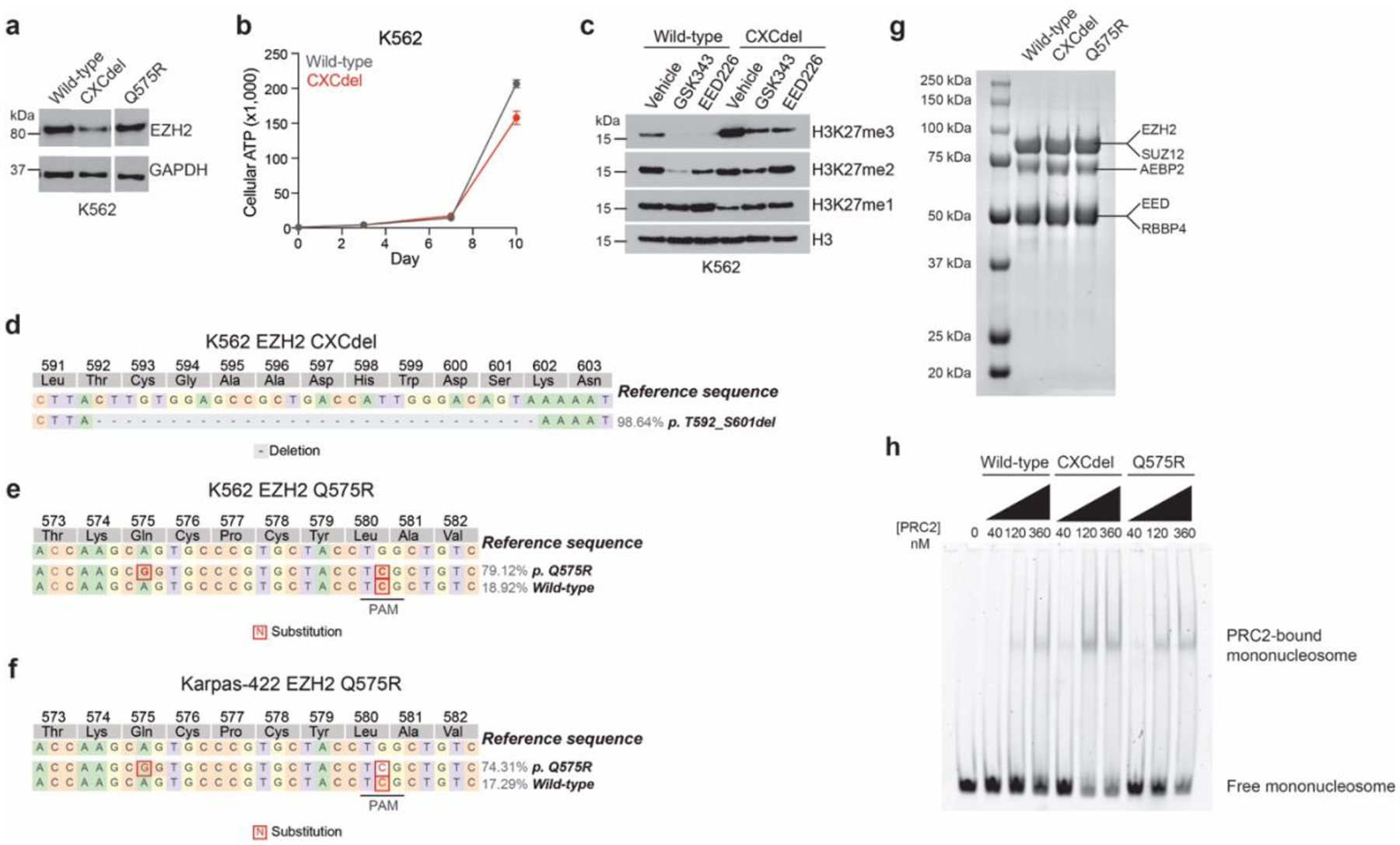
a. Immunoblot showing EZH2 levels in K562 wild-type, CXCdel+/+/+ and Q575R.
b. Line plot depicting cumulative growth of K562 wild-type and CXCdel+/+/+ over 10 days.
c. Immunoblot showing H3K27me1/2/3 levels in K562 wild-type and CXCdel+/+/+ treated with 5 μM GSK343 or EED226 for 72 h.
d. Schematic showing genotypes and allele frequencies for mutations that are observed at frequencies of > 2% in the gDNA encoding the EZH2 CXC domain in K562 CXCdel+/+/+.
e. Same as d. but for K562 Q575R.
f. Same as d. but for Karpas-422 Q575R.
g. Purification of recombinant 5-member PRC2 complex. SDS-PAGE stained with Coomassie blue showing purified wild-type, CXCdel, and Q575R PRC2 complexes.
h. Representative EMSA showing binding of wild-type, CXCdel and Q575R PRC2 to 185-bp mononucleosomes. The upper band corresponds to the PRC2-bound mononucleosome and bottom band corresponds to the free, unbound mononucleosome.
Results in a,c-g are representative of two independent experiments, h is representative of five independent experiments.
Extended Fig. 6 |. PRC2 hyperactivity leads to H3K27me3 spreading on chromatin.
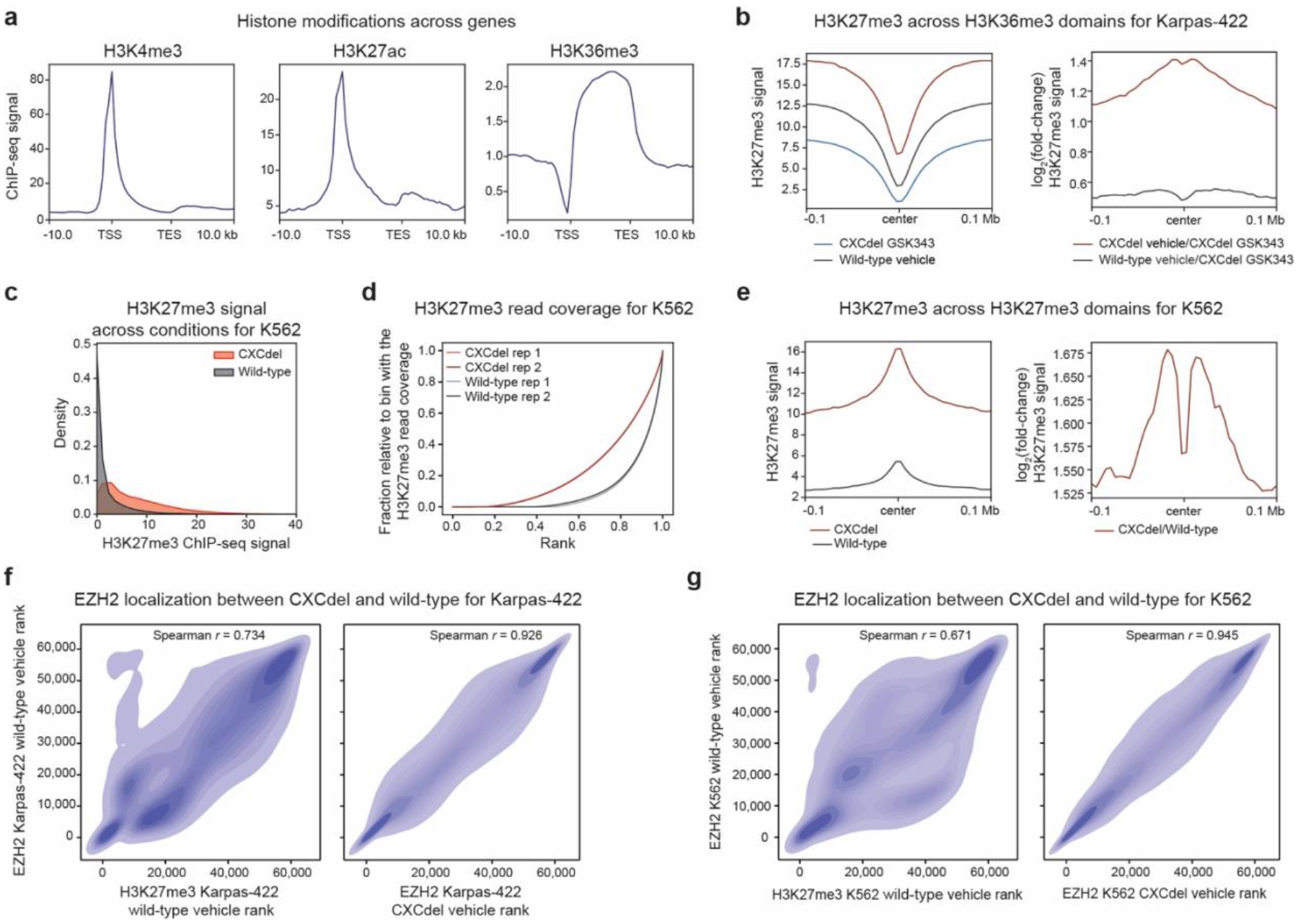
a. Aggregate profile plots of Karpas-422 ENCODE ChIP-seq signal (y-axis) for the specified histone modification centered around gene bodies (x-axis).
b. Aggregate profile plots of Karpas-422 H3K27me3 ChIP-Rx signal (y-axis, left) and log2(fold-change) H3K27me3 ChIP-Rx signal relative to GSK343-treated CXCdel+/− cells (y-axis, right) centered around H3K36me3 domains in wild-type cells (x-axis).
c. Density plot depicting the distribution of H3K27me3 ChIP-Rx signal (x-axis) of K562 wild-type and CXCdel+/+/+ cells for 50-kb genomic bins.
d. Line plot depicting cumulative fraction H3K27me3 ChIP-seq coverage relative to that of the bin with the highest coverage (y-axis) as a function of the region’s relative genomic rank (x-axis) for K562 wild-type and CXCdel+/+/+ cells.
e. Same as b. but for K562.
f. Density plot depicting EZH2 ChIP-seq signal rank for vehicle-treated Karpas-422 wild-type cells (y-axis) relative to H3K27me3 ChIP-Rx signal rank for vehicle-treated wild-type cells (x-axis, left) and EZH2 ChIP-seq signal rank for vehicle-treated CXCdel+/− (x-axis, right).
g. Same as f. but for K562.
Results in b-c,e-g represent average of two biological replicates.
Extended Fig. 7 |. H3K27me3 overspreading silences genes related to lymphoma growth.
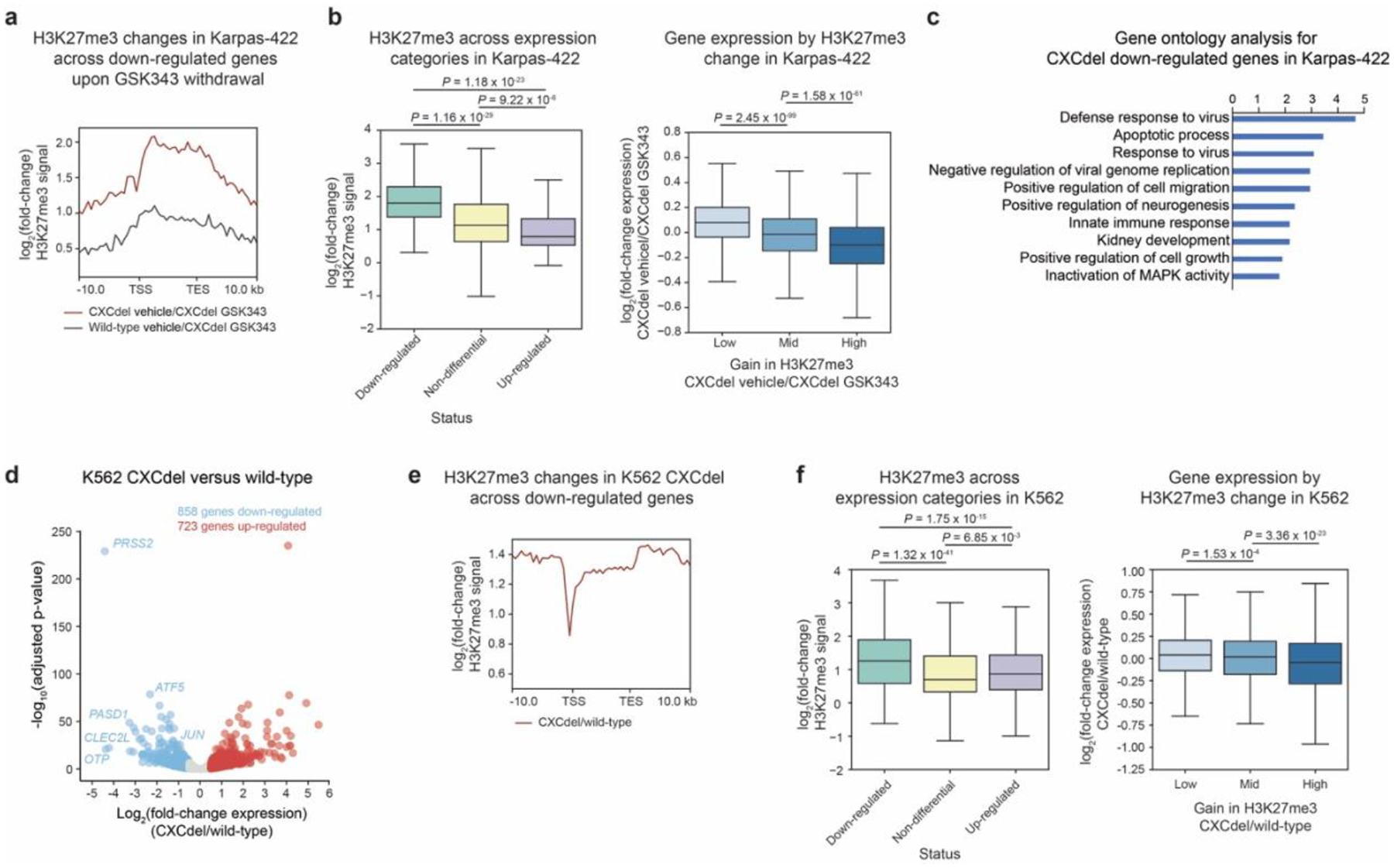
a. Aggregate profile plots of log2(fold-change) H3K27me3 ChIP-Rx signal relative to GSK343-treated Karpas-422 CXCdel+/− (y-axis) centered around gene bodies (x-axis) of downregulated genes in CXCdel+/− upon drug withdrawal.
b. Boxplot depicting log2(fold-change) H3K27me3 ChIP-Rx signal across gene bodies (y-axis) with down-regulated, non-differential and up-regulated genes in Karpas-422 CXCdel+/− upon drug withdrawal annotated (x-axis) (left). Boxplot depicting log2(fold-change) gene expression (y-axis) by change in H3K27me3 (x-axis) for CXCdel+/− vehicle-treated relative to GSK343-treated (right).
c. Top 10 enriched pathways within the downregulated genes upon GSK343 withdrawal in Karpas-422 CXCdel+/− cells. Functional enrichment analysis of differentially expressed genes was performed using DAVID and P values were computed with modified Fisher’s Exact test.
d. Volcano plot showing gene expression changes in K562 CXCdel+/+/+ versus wild-type cells. Genes with |log2(fold-change)| > 0.5 and adjusted P-value < 0.05 are defined as differentially expressed. P-values were computed with DESeq2 by Benjamini-Hochberg method, adjusted for multiple testing. Data were from three biological replicates.
e. Aggregate profile plots of K562 CXCdel log2(fold-change) H3K27me3 ChIP-Rx signal relative to wild-type cells (y-axis) centered around gene bodies (x-axis) of downregulated genes in CXCdel+/+/+ cells.
f. Same as i. but for K562 CXCdel+/+/+ relative to wild-type K562.
Results in a-c,e-f, represent average of two biological replicates.
For b,f, the interquartile range (IQR) is depicted by the box with the median represented by the center line. Whiskers maximally extend to 1.5 × IQR (with outliers excluded). P values were calculated by a Mann-Whitney-Wilcoxon two-sided test.
Extended Fig. 8 |. SETD2 depletion halts lymphoma growth.
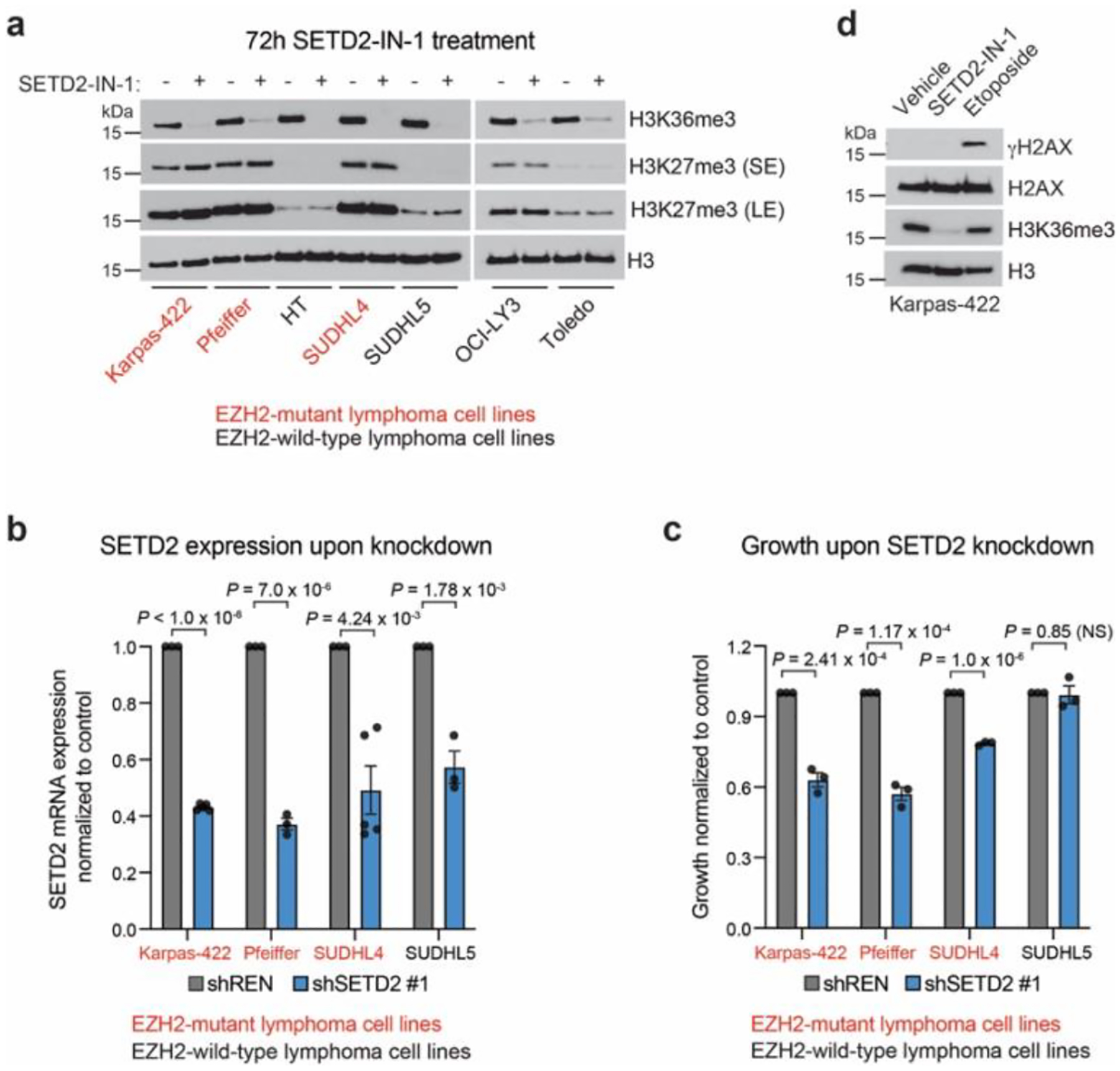
a. Immunoblot showing H3K27me3 and H3K36me3 levels in lymphoma cell lines treated with 5 μM SETD2-IN-1 for 72 h.
b. Bar plots showing SETD2 expression at the transcript level after SETD2 knockdown for 72 h.
c. Bar plots showing cell proliferation after SETD2 knockdown for 10 days.
d. Immunoblot showing γH2AX and H3K36me3 levels in Karpas-422 treated with 5 μM SETD2-IN-1 for 10 days or 10 μM etoposide for 24 h.
Results in a,d are representative of two independent experiments. Data in b-c are are represented by mean ± s.e.m of n = 3, except for b Karpas-422 and SUDHL4 shSETD2 #1 n = 5.
Extended Fig. 9 |. SETD2 inhibition leads to H3K27me3 spreading on chromatin.
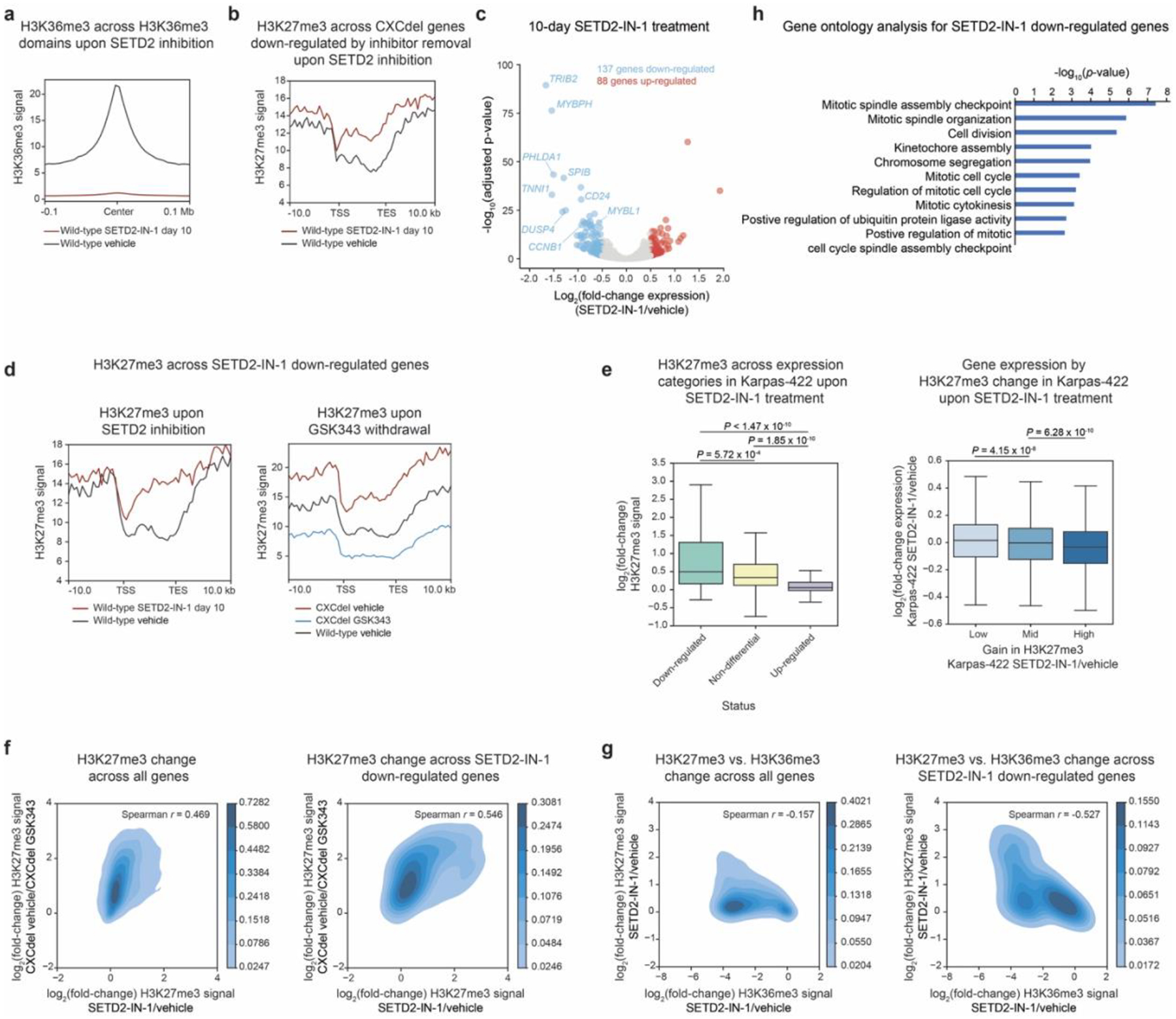
a. Aggregate profile plot of wild-type Karpas-422 H3K36me3 ChIP-Rx signal (y-axis) for SETD2-IN-1 (5 μM) treatment centered around H3K36me3 domains in wild-type cells (x-axis).
b. Aggregate profile plot of wild-type Karpas-422 H3K27me3 ChIP-Rx signal (y-axis) for SETD2-IN-1 (5 μM) treatment centered around gene bodies of genes downregulated upon GSK343 withdrawal in CXCdel+/− cells (x-axis).
c. Volcano plot showing gene expression changes in gene expression in Karpas-422 wild-type cells under SETD2-IN-1 (5 μM) treatment for 10 days versus vehicle. Genes with |log2(fold-change)| > 0.5 and adjusted P-value < 0.05 are defined as differentially expressed. P-values were computed with DESeq2 by Benjamini-Hochberg method, adjusted for multiple testing. Data were from three biological replicates.
d. Aggregate profile plot of wild-type Karpas-422 H3K27me3 ChIP-Rx signal (y-axis) for SETD2-IN-1 (5 μM) treatment (left) and for vehicle- or GSK343-treated wild-type and CXCdel+/− cells (right) centered around gene bodies of genes downregulated upon SETD2-IN-1 (5 μM) treatment (x-axis).
e. Boxplot depicting log2(fold-change) H3K27me3 ChIP-Rx signal across gene bodies (y-axis) with downregulated, non-differential and upregulated genes in SETD2-IN-1 (5 μM) treatment annotated (x-axis) (left). Boxplot depicting log2(fold-change) gene expression (y-axis) by change in H3K27me3 (x-axis) for Karpas-422 wild-type cells treated with SETD2-IN-1 relative to vehicle (right).
f. Density heatmap comparing log2(fold-change) H3K27me3 ChIP-Rx signal of Karpas-422 CXCdel vehicle relative to Karpas-422 CXCdel GSK343 treatment (y-axis) versus log2(fold-change) H3K27me3 ChIP-Rx signal of Karpas-422 SETD2-IN-1 (5 μM) relative to vehicle treatment (x-axis) across all genes (left) and across downregulated upon SETD2-IN-1 (5 μM) treatment (right).
g. Same as j. but for H3K27me3 ChIP-Rx signal of Karpas-422 SETD2-IN-1 treatment relative to vehicle (y-axis) versus log2(fold-change) H3K36me3 ChIP-Rx signal of Karpas-422 SETD2-IN-1 (5 μM) relative vehicle treatment (x-axis).
h. Top 10 enriched pathways within the downregulated genes upon SETD2 inhibition in Karpas-422 wild-type cells. Functional enrichment analysis of differentially expressed genes was performed using DAVID and P values were computed with modified Fisher’s Exact test.
Results in a-b, d-h represent average of two biological replicates. For e, the interquartile range (IQR) is depicted by the box with the median represented by the center line. Whiskers maximally extend to 1.5 × IQR (with outliers excluded). P values were calculated by a Mann-Whitney-Wilcoxon two-sided test.
Supplementary Material
Acknowledgments
We thank P. Gosavi, members of T. Cech laboratory especially A. Gooding and Y. Long, and members of B. Kingston laboratory especially S. Marr and T. Oei for guidance on protein biochemistry experiments; P. Cole for providing the nucleosomal DNA sequence for binding studies; K. Ngan for guidance on computational analysis of CRISPR-suppressor scans; S. Araten for aiding in protein conservation analysis; M. Quezada for aiding in genomics analysis; D. Youmans, D. Narducci and A. Hansen for guidance on live-cell imaging experiments; R. Ryan for providing cell lines for SETD2 inhibitor studies; the Bauer Core Facility at Harvard University, particularly Z. Nizioleck and J. Nelson for their assistance with cell sorting. We thank members of the Liau laboratory for helpful discussions and comments on the manuscript. This work was supported by award no. 1DP2GM137494 (B.B.L.) from the National Institute of General Medical Sciences, American Cancer Society Research Scholar Grant, RSG-22-083-01-DMC (B.B.L.). startup funds from Harvard University (B.B.L), award no. T32GM007753 (A.M.F.) from the National Institute of General Medical Sciences, Charles A. King Trust Postdoctoral Research Fellowship (H.S.K.) from Sara Elizabeth O’Brien Trust/Simeon J. Fortin Charitable Foundation, Bank of America Private Bank, Co-Trustees.
Footnotes
Competing Interests
B.B.L. holds a sponsored research project with AstraZeneca, is a scientific consultant for Imago BioSciences, and is a shareholder and member of the scientific advisory board of Light Horse Therapeutics. The remaining authors declare no competing interests.
Code availability
Computer code employed in Fig. 2 for calculation of the addiction score is publicly available at https://github.com/skissler/EZH2 under the GNU General Public License, Version 3.0 (details included in code repository). Custom code used to analyze CRISPR-suppressor scanning data is available at https://github.com/liaulab/CRISPR-suppressor_scanning. All other computer code employed are available upon request.
Data availability
ChIP–seq and RNA-seq data have been deposited to NCBI GEO (GSE199889). Transformed CRISPR-suppressor scanning reads (log2 + 1) used for Fig. 1 and Extended Data Fig. 1 are supplied in Supplementary Dataset 2. Addiction scores and growth rates used for Fig. 2 and Extended Data Fig. 2 are supplied in Supplementary Dataset 3. Oligonucleotide sequences are provided as Supplementary Information. Unprocessed gel and immunoblot images, as well as additional data generated in this study, are provided as Source Data with this paper. The following publicly available datasets were used PDB accession codes 6WKR, 5K0M, 5LS6, EMDB-7306; and ENCODE datasets ENCFF932BQZ, ENCFF435PXJ and ENCFF603BHK.
References
- 1.Thakur MD et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494, 251–255 10.1038/nature11814 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Z et al. Signaling thresholds and negative B cell selection in acute lymphoblastic leukemia. Nature 521, 357–361 10.1038/nature14231 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kong X et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature 550, 270–274 10.1038/nature24037 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajan SS et al. The mechanism of cancer drug addiction in ALK-positive T-Cell lymphoma. Oncogene 39, 2103–2117 10.1038/s41388-019-1136-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serrano M, Lin AW, McCurrach ME, Beach D & Lowe SW Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 88, 593–602 10.1016/s0092-8674(00)81902-9 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Michaloglou C et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720–724 10.1038/nature03890 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Reimann M et al. Tumor stroma-derived TGF-beta limits myc-driven lymphomagenesis via Suv39h1-dependent senescence. Cancer Cell 17, 262–72 10.1016/j.ccr.2009.12.043 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Nemazee D Mechanisms of central tolerance for B cells. Nat Rev Immunol 17, 281–294 10.1038/nri.2017.19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ecker V et al. Targeted PI3K/AKT-hyperactivation induces cell death in chronic lymphocytic leukemia. Nat Commun 12, 3526 10.1038/s41467-021-23752-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reddy A et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 171, 481–494.e15 10.1016/j.cell.2017.09.027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmitz R et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. New Engl J Medicine 378, 1396–1407 10.1056/nejmoa1801445 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Béguelin W et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat Commun 8, 877 10.1038/s41467-017-01029-x (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Béguelin W et al. EZH2 Is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell 23, 677–692 10.1016/j.ccr.2013.04.011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qi W et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol 13, 381–388 10.1038/nchembio.2304 (2017). [DOI] [PubMed] [Google Scholar]
- 15.McCabe MT et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112 10.1038/nature11606 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Vinyard ME et al. CRISPR-suppressor scanning reveals a nonenzymatic role of LSD1 in AML. Nat Chem Biol 15, 529–539 10.1038/s41589-019-0263-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bödör C et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 122, 3165–3168 10.1182/blood-2013-04-496893 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verma SK et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. Acs Med Chem Lett 3, 1091–1096 10.1021/ml3003346 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooun A et al. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat Commun 7, 11384 10.1038/ncomms11384 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibaja V et al. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene 35, 558–566 10.1038/onc.2015.114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker T et al. Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors. Oncotarget 6, 32646–32655 10.18632/oncotarget.5066 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poepsel S, Kasinath V & Nogales E Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat Struct Mol Biol 25, 154–162 10.1038/s41594-018-0023-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kasinath V et al. JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science 371, eabc3393 10.1126/science.abc3393 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chorin AB et al. ConSurf-DB: An accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein Sci Publ Protein Soc 29, 258–267 10.1002/pro.3779 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murray JD Mathematical Biology. Springer, 10.1007/b98868. (2002). [DOI] [Google Scholar]
- 26.Gosavi PM et al. Profiling the Landscape of Drug Resistance Mutations in Neosubstrates to Molecular Glue Degraders. Acs Central Sci 8, 417–429 10.1021/acscentsci.1c01603 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan W et al. H3K36 Methylation Antagonizes PRC2-mediated H3K27 Methylation*. J Biol Chem 286, 7983–7989 10.1074/jbc.m110.194027 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Youmans DT, Gooding AR, Dowell RD & Cech TR Competition between PRC2.1 and 2.2 subcomplexes regulates PRC2 chromatin occupancy in human stem cells. Mol Cell 81, 488–501.e9 10.1016/j.molcel.2020.11.044 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ernst T et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 42, 722–726 10.1038/ng.621 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Calebiro D et al. Recurrent EZH1 mutations are a second hit in autonomous thyroid adenomas. J Clin Invest 126, 3383–3388 10.1172/jci84894 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Margueron R et al. Ezh1 and Ezh2 Maintain Repressive Chromatin through Different Mechanisms. Mol Cell 32, 503–518 10.1016/j.molcel.2008.11.004 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmitges FW et al. Histone Methylation by PRC2 Is Inhibited by Active Chromatin Marks. Mol Cell 42, 330–341 10.1016/j.molcel.2011.03.025 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Laugesen A, Højfeldt JW & Helin K Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol Cell 74, 8–18 10.1016/j.molcel.2019.03.011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ernst J et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 10.1038/nature09906 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Velichutina I et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 116, 5247–5255 10.1182/blood-2010-04-280149 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caganova M et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest 123, 5009–5022 10.1172/jci70626 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lampe JW et al. Discovery of a First-in-Class Inhibitor of the Histone Methyltransferase SETD2 Suitable for Preclinical Studies. Acs Med Chem Lett 10.1021/acsmedchemlett.1c00272 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lampe J et al. Substituted Indoles and methods of use thereof. (2020). [Google Scholar]
- 39.Carvalho S et al. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res 41, 2881–2893 10.1093/nar/gks1472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leung W et al. SETD2 Haploinsufficiency Enhances Germinal Center-Associated AICDA Somatic Hypermutation to Drive B-cell Lymphomagenesis. Cancer Discov 12, 1782–1803 10.1158/2159-8290.cd-21-1514 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carvalho S et al. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife 3, e02482 10.7554/elife.02482 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfister SX et al. SETD2-Dependent Histone H3K36 Trimethylation Is Required for Homologous Recombination Repair and Genome Stability. Cell Reports 7, 2006–2018 10.1016/j.celrep.2014.05.026 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim KH & Roberts CWM Targeting EZH2 in cancer. Nat Med 22, 128–134 10.1038/nm.4036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi J et al. DNA binding by PHF1 prolongs PRC2 residence time on chromatin and thereby promotes H3K27 methylation. Nat Struct Mol Biol 24, 1039–1047 10.1038/nsmb.3488 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Lee C-H et al. Distinct Stimulatory Mechanisms Regulate the Catalytic Activity of Polycomb Repressive Complex 2. Mol Cell 70, 435–448.e5 10.1016/j.molcel.2018.03.019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Son J, Shen SS, Margueron R & Reinberg D Nucleosome-binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Gene Dev 27, 2663–2677 10.1101/gad.225888.113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kasinath V et al. Structures of human PRC2 with its cofactors AEBP2 and JARID2. Science 359, eaar5700 10.1126/science.aar5700 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Freedy AM & Liau BB Discovering new biology with drug-resistance alleles. Nat Chem Biol 17, 1219–1229 10.1038/s41589-021-00865-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boettiger AN et al. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529, 418–422 10.1038/nature16496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 50.Hsu PD et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31, 827–832 10.1038/nbt.2647 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ngan KC et al. CRISPR-Suppressor Scanning for Systematic Discovery of Drug-Resistance Mutations. Curr Protoc 2, e614 10.1002/cpz1.614 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joung J et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 12, 828–863 10.1038/nprot.2017.016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fellmann C et al. An Optimized microRNA Backbone for Effective Single-Copy RNAi. Cell Reports 5, 1704–1713 10.1016/j.celrep.2013.11.020 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol 37, 224–226 10.1038/s41587-019-0032-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davidovich C, Goodrich KJ, Gooding AR & Cech TR A dimeric state for PRC2. Nucleic Acids Res 42, 9236–9248 10.1093/nar/gku540 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luger K, Rechsteiner TJ & Richmond TJ Preparation of nucleosome core particle from recombinant histones. Methods Enzymol 304, 3–19 10.1016/s0076-6879(99)04003-3 (1999). [DOI] [PubMed] [Google Scholar]
- 57.Dyer PN et al. Reconstitution of Nucleosome Core Particles from Recombinant Histones and DNA. Methods Enzymol 375, 23–44 10.1016/s0076-6879(03)75002-2 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Clapier CR, Längst G, Corona DFV, Becker PB & Nightingale KP Critical Role for the Histone H4 N Terminus in Nucleosome Remodeling by ISWI. Mol Cell Biol 21, 875–883 10.1128/mcb.21.3.875-883.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Orlando DA et al. Quantitative ChIP-Seq Normalization Reveals Global Modulation of the Epigenome. Cell Reports 9, 1163–1170 10.1016/j.celrep.2014.10.018 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Wu D, Wang L & Huang H Protocol to apply spike-in ChIP-seq to capture massive histone acetylation in human cells. Star Protoc 2, 100681 10.1016/j.xpro.2021.100681 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP–seq and RNA-seq data have been deposited to NCBI GEO (GSE199889). Transformed CRISPR-suppressor scanning reads (log2 + 1) used for Fig. 1 and Extended Data Fig. 1 are supplied in Supplementary Dataset 2. Addiction scores and growth rates used for Fig. 2 and Extended Data Fig. 2 are supplied in Supplementary Dataset 3. Oligonucleotide sequences are provided as Supplementary Information. Unprocessed gel and immunoblot images, as well as additional data generated in this study, are provided as Source Data with this paper. The following publicly available datasets were used PDB accession codes 6WKR, 5K0M, 5LS6, EMDB-7306; and ENCODE datasets ENCFF932BQZ, ENCFF435PXJ and ENCFF603BHK.