

Summary
In the adult mammalian central nervous system (CNS), axons fail to regenerate spontaneously after injury because of a combination of extrinsic and intrinsic factors. Despite recent advances targeting the intrinsic regenerative properties of adult neurons, the molecular mechanisms underlying axon regeneration are not fully understood. Here, we uncover a regulatory mechanism that controls the expression of key proteins involved in regeneration at the translational level. Our results show that mRNA-specific translation is critical for promoting axon regeneration. Indeed, we demonstrate that specific ribosome-interacting proteins, such as the protein Huntingtin (HTT), selectively control the translation of a specific subset of mRNAs. Moreover, modulating the expression of these translationally regulated mRNAs is crucial for promoting axon regeneration. Altogether, our findings highlight that selective translation through the customization of the translational complex is a key mechanism of axon regeneration with major implications in the development of therapeutic strategies for CNS repair.
Keywords: axon regeneration, optic nerve, retinal ganglion cells, selective translation, ribosome, Huntingtin
Graphical abstract
Highlights
-
•
CNS axon regeneration depends on the selective translation of a subset of mRNAs
-
•
Translational complex activity changes when interacting with factors such as HTT
-
•
These factors regulate the translation of specific mRNAs in injured neurons
Schaeffer et al. demonstrate that the translational regulation of a specific subset of mRNAs is involved in the control of axon regeneration in the injured CNS. This selective translation process is controlled by the functional association of specific factors with ribosomes, such as the wild-type protein Huntingtin.
Introduction
In adult mammals, central nervous system (CNS) neurons are unable to regenerate after injury, leading to a permanent and irreversible loss of motor and/or cognitive functions. Although studies have focused on the growth-inhibitory role of the environment after injury,1,2 it is now clear that the regeneration failure of injured axons depends on the intrinsic properties of adult neurons.3 Large-scale analysis of neuronal populations allowed the identification of several factors essential for neuroprotection and regeneration and actively controlled during development, including mTOR,4 Krüppel-like factors,5 and JAK/STAT.6 Furthermore, the lesion itself triggers the regulation of molecular signaling pathways, which further impairs the intrinsic regeneration ability of injured neurons.7 In the visual system, proteomic and transcriptomic data analyses have established the impact of axon injury on retinal ganglion cells (RGCs), thereby highlighting how the injury blocks survival and growth programs and favors cell-death-associated programs.7,8,9
The modulation of several signaling pathways, alone or in combination, can promote long-distance CNS regeneration,4,5,7,10 yet only few neurons are able to respond to such modulation, and it remains challenging to obtain sufficient axon growth to rebuild a functional circuit. Therefore, there is a need to identify new candidates to promote axon regeneration. Previously, comparative proteomic analysis of RGCs in intact condition and after optic nerve injury highlighted the wild-type (WT) form of the protein Huntingtin (HTT) as a key regulator of neuronal injury response.7 Consistently, HTT has been identified as a central signaling hub of neural-progenitor-cell-grafting-induced regeneration in the lesioned spinal cord.11 However, the underlying mechanisms remain unknown, as are HTT’s physiological functions in the adult CNS. Indeed, HTT is best known for causing Huntington’s neurodegenerative disease when mutated.12 Interestingly, HTT controls axonal transport, specifically of brain-derived neurotrophic factor (BDNF),13 which is involved in neuroprotection14,15 and intrinsic regenerative properties of neurons.
To unlock CNS regenerative properties, many studies have focused on transcriptional regulation during development or after injury. Yet, transcriptional modulation is not sufficient to fully recapitulate axon growth. In contrast, the process of messenger RNA (mRNA) translation into proteins, the functional readout of gene expression in cells, remains poorly described in this context. Although mTOR activation triggers axon growth, its exact contribution to translational control in axon regeneration is not described. Protein synthesis is a highly ordered process that involves, along with mRNA and transfer RNA (tRNA), the translational complex, which is composed of the ribosome, translation factors, and non-canonical associated factors.16 Several studies point out that mRNA and protein levels correlate only partially in cells,17 depending on cell types and physio-pathological conditions.
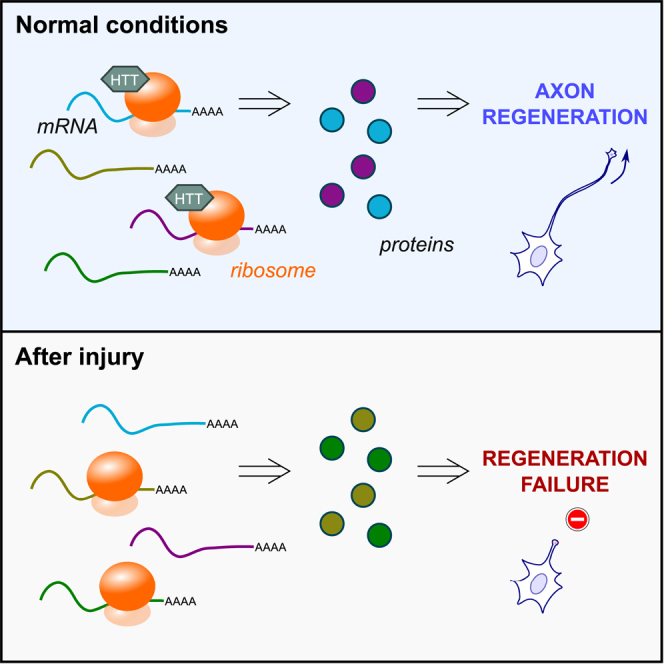
In this study, we show that HTT is dynamically regulated upon optic nerve injury and that it is indispensable for axon regeneration. Through the analysis of HTT phospho-mutant mouse lines, we show that HTT-mediated control of axon regeneration is not based on its role as a regulator of BDNF axonal transport. We further show that HTT interacts with the translational complex and that it does not control translation at the global level. Using comparative transcriptomic and translatomic analyses, we show that HTT specifically regulates the association of a subset of regeneration-associated mRNAs with ribosomes, thereby controlling their expression at the level of translation and not transcription. This is the case of the mRNA target Tox2 (thymocyte selection-associated HMG box), which is positively regulated by HTT at the translational level. Finally, the modulation of the HTT translationally regulated target Tox2 is critical to achieve the axon regeneration of lesioned RGCs in the adult CNS. Altogether, we provide evidence that the interaction of specific proteins such as HTT with ribosomes leads to the translation of specific mRNAs. Our results point out the key role of selective translation, through ribosome customization, in CNS axon regeneration.
Results
HTT is required for axon regeneration
Several studies have suggested that HTT is involved in CNS regeneration, although the underlying mechanisms remain unknown.7,11 HTT is ubiquitously expressed in the nervous system,18 but few data are available on the visual system. Looking at the mature retina, we found that HTT is robustly expressed in the RGC layer of adult mice, both at mRNA and protein levels (Figures 1A–1C). At 3 days post optic nerve crush injury (ONC) (3dpc), HTT expression is downregulated, both at transcript (Figures 1A and 1B) and protein levels (Figure 1C). The decrease in HTT expression correlates with the reduced regeneration capacity following CNS axon injury.
Figure 1.

HTT is required for axon regeneration
(A) In situ hybridization showing HTT mRNA expression in the adult retina in intact and injured (3dpc) conditions.
(B) Fluorescent in situ hybridization and quantification of HTT mRNA expression in intact and 3dpc RBPMS+ RGCs.
(C) Immunofluorescence and quantification of HTT protein expression in intact and 3dpc RBPMS+ RGCs.
Data are represented as mean +/− SEM. Mann-Whitney U tests, ∗∗∗p value < 0.001.
(D) Immunofluorescence and quantification showing HTT protein expression in Ptenfl/fl+AAV2-Cre or AAV2-Plap in intact and 3dpc RBPMS+ RGCs.
(E) Whole optic nerve confocal images showing CTB+ regenerating axons in Ptenfl/fl+AAV2-Cre, Ptenfl/flHTTfl/fl+AAV2-Cre, and Ptenfl/flHTTfl/fl+AAV2-Plap conditions at 14dpc. The injury site is indicated by a red star.
(F) Quantification of integrated fluorescence intensity along the optic nerve. The top graph gives the multiple comparisons test between Ptenfl/fl+AAV2-Cre and Ptenfl/flHTTfl/fl+AAV2-Cre conditions.
(G) Whole-mount retina confocal images showing RBPMS+ and pS6+ RGCs in Ptenfl/fl+AAV2-Cre, Ptenfl/flHTTfl/fl+AAV2-Cre, and Ptenfl/flHTTfl/fl+AAV2-Plap intact conditions.
(H) Quantification of RBPMS+ RGCs per mm2 retina.
(I) Quantification of pS6+ RGCs per mm2 retina.
(J) Whole-mount retina confocal images showing RBPMS+ and pS6+ RGCs in Ptenfl/fl+AAV2-Cre, Ptenfl/flHTTfl/fl+AAV2-Cre, and Ptenfl/flHTTfl/fl+AAV2-Plap conditions at 14dpc.
(K) Quantification of RBPMS+ RGCs as a percentage of intact condition.
(L) Quantification of pS6+ RGCs per mm2 retina.
Data are represented as mean +/− SEM. One-way ANOVA with Bonferroni multiple comparisons test; ∗p value < 0.05, ∗∗p value < 0.01, and ∗∗∗p value < 0.001; ns, not significant.
To determine the role of HTT in axon regeneration, we deleted HTT specifically in RGCs by injecting Cre-expressing AAV2 (AAV2-Cre), or AAV2-placental alkaline phosphatase (AAV2-Plap) as a control, in the eyes of HTT-floxed (HTTfl/fl) mice19 (Figures S1A and S1B). Consistent with the decrease in HTT expression in lesioned, non-regenerative RGCs, we observed no regeneration in the HTT-deleted condition (Figures S1C and S1D). In addition, no significant difference in RGC numbers was detected between HTT-deleted and control RGCs, neither in intact condition (Figures S1E and S1F) nor at 14 days post injury (14dpc) (Figures S1H and S1I). As WT mice have a limited regenerative capacity, we used a regeneration-primed system to investigate HTT function in axon regrowth. The activation of mTOR pathway through the deletion of phosphatase and tensin homolog (Pten) is a well-described paradigm of axon regeneration.4 In contrast to WT RGCs, we found that, upon mTOR activation, HTT expression remained stable at 3dpc compared with the intact condition (Figure 1D). Therefore, to determine how HTT affects axon regeneration, we crossed HTTfl/fl mice with Ptenfl/fl mice and injected AAV2-Cre to co-delete Pten and HTT in RGCs (Figure S1K). Strikingly, axon regeneration at 14dpc was suppressed in Pten/HTT-deleted RGCs in contrast to Pten-deleted RGCs (Figures 1E and 1F). This effect was already observed at 3dpc (Figures S1L and S1M), indicating that HTT is required early in the regenerative process. These results demonstrate that HTT is essential for axon regeneration.
We then examined whether the loss of axon regeneration induced by HTT deletion was due to a decrease in RGC survival. We stained whole-mount retinas for RNA-binding protein with multiple splicing (RBPMS), a specific RGC marker.20 No significant difference was found between Pten/HTT-deleted and Pten-deleted RGCs in intact condition (Figures 1G and 1H) and at 14dpc (Figures 1J and 1K), which shows that HTT is necessary for axon regeneration but not for RGC survival. We then investigated whether the suppression of regeneration caused by HTT deletion is linked to an alteration in mTOR pathway. To do so, we analyzed the protein levels of phosphorylated ribosomal protein S6 (pS6), a functional readout of mTOR activation.21 The number of pS6-positive RGCs was similar in Pten/HTT-deleted and Pten-deleted RGCs, both in the intact condition (Figures 1G and 1I) and at 14dpc (Figures 1J and 1L). Similarly, no difference in pS6 level was observed between HTT-deleted and control RGCs in the intact condition (Figures S1E and S1G) and at 14dpc (Figures S1H and S1J). Therefore, HTT deletion does not impact mTOR activation induced by Pten deletion. Altogether, our results show that HTT is a critical intrinsic factor of CNS regeneration that acts independently of mTOR and neuronal survival.
We then asked the extent of HTT-mediated control of regeneration and analyzed the outcome of HTT modulation in the peripheral nervous system (PNS). First, we assessed HTT expression in the PNS by focusing on the lumbar dorsal root ganglion (DRG) neurons, whose peripheral branch projects into the sciatic nerve. In contrast to the CNS, HTT expression remains stable in DRG neurons after sciatic nerve injury (Figure S1N). We then tested the effect of HTT deletion on these neurons via the intrathecal injection of a Cre-expressing AAV8 (AAV8-Cre) in HTTfl/fl mice to delete HTT in DRG neurons (Figures S1O and S1P). Interestingly, HTT-deleted DRG axons displayed shorter regeneration than control ones at 3dpc (Figures S1Q and S1R). This result demonstrates that HTT expression correlates with the capacity of DRG peripheral axons to regenerate after sciatic nerve injury and that HTT is crucial for regeneration in the PNS, suggesting a general contribution of HTT to axon regeneration.
HTT control of regeneration is not mediated by its role in BDNF axonal transport
We then sought to determine the molecular mechanism underlying the HTT-mediated control of CNS regeneration. We hypothesized that HTT controls regeneration through its role in BDNF axon trafficking.13,14,22,23,24 As previously demonstrated in cortical neurons,22 HTT phosphorylation state at serine 421 (S421) controls the directionality of BDNF axonal transport (Figure 2A). We verified that the phosphorylated state of HTT at S421 controls BDNF transport in adult RGC axons using an AAV2-expressing constitutively active Rheb1 (cRheb), which activates mTOR pathway and promotes mature RGC axon growth.25 We injected AAV2-cRheb in HTT S421A/A mice, where HTT unphosphorylable state at S421 promotes BDNF retrograde transport, and in HTT S421D/D mice, where HTT constitutively phosphorylated state at S421 promotes BDNF anterograde transport. 1 day after, we injected AAV2-BDNF-tdTomato to track BDNF-carrying vesicles. 2 weeks after virus injection, we set up adult retina explant cultures (Figure S2A).26 Live imaging allowed us to verify that the S421 point mutations of HTT control BDNF transport directionality in adult RGC axons, with an increase in BDNF retrograde transport in the HTT S421A/A condition and an increase in BDNF anterograde transport in the HTT S421D/D condition (Figure S2B), as described.22
Figure 2.

Axon regeneration and RGC survival are not modified by S421 phospho-mutations responsible for the HTT-mediated control of BDNF transport
(A) Schematic representation of transgenic mouse lines carrying S421 phospho-point mutations.22,24
(B) Whole optic nerve confocal images showing CTB+ regenerating axons in phospho-mutant mouse lines at 15dpc.
(C) Whole-mount retina confocal images showing RBPMS+ RGCs in phospho-mutant mouse lines in intact and 15dpc conditions.
(D) Quantification of integrated fluorescence intensity along the optic nerve.
(E) Quantification of RBPMS+ RGCs per mm2 retina in intact condition.
(F) Quantification of RBPMS+ RGCs as a percentage of intact RBPMS+ RGCs.
(G) Whole optic nerve confocal images showing CTB+ regenerating axons in phospho-mutant mouse lines injected with AAV2-cRheb or AAV2-Plap at 14dpc.
(H) Quantification of integrated fluorescence intensity along the optic nerve.
(I) Whole-mount retina confocal images showing RBPMS+ and pS6+ RGCs in phospho-mutant mouse lines injected with AAV2-cRheb or AAV2-Plap at 14dpc.
(J) Quantification of RBPMS+ RGCs per mm2 retina at 14dpc.
(K) Quantification of pS6+ RGCs per mm2 retina at 14dpc.
The injury site is indicated by a red star. Data are represented as mean ± SEM. One-way ANOVA with Bonferroni multiple comparisons test; ∗p value < 0.05 and ∗∗∗p value < 0.001; ns, not significant.
Next, to explore the potential role of HTT-mediated transport of BDNF in axon regeneration, we performed ONC and analyzed axon regeneration in HTT S421A/A and S421D/D mutant mice (Figure 2A).13,22 This analysis revealed no difference between heterozygous (S/A, S/D), homozygous (A/A, D/D), and WT (S/S) mice (Figures 2B and 2D). We concluded from these data that enhancing HTT-mediated retrograde (S421A) or anterograde (S421D) BDNF transport did not improve or alter axon regeneration. We also analyzed RGC survival in these mutant mice and found no difference in RGC numbers in the intact and injured conditions (Figures 2C, 2E, and 2F). Moreover, we performed the same set of experiments in other HTT phospho-mutant mouse lines, where serines 1181 and 1201 (S1181/S1201) are mutated to control the dynamics of BDNF axonal transport (Figure S2C).13,27 We found no change in axon regeneration or in RGC survival in these mutant mice (Figures S2D–S2H).
As WT mice have a limited regenerative capacity, we assessed the effect of the S421 point mutations in a regenerative context. To do so, we activated mTOR pathway via cRheb overexpression25 in the RGCs of S421 phospho-mutant mice (Figure 2G). We found that AAV2-cRheb significantly enhanced axon regeneration, with no difference among heterozygous (S/A, S/D), homozygous (A/A, D/D), and WT (S/S) mice. Hence, no difference was observed in the regenerative effect of mTOR activation when the HTT-mediated retrograde (S421A) or anterograde (S421D) transport of BDNF was modulated (Figures 2G and 2H). We analyzed RGC survival and pS6 protein levels and found no difference in RGC survival between S421 phospho-mutant and WT mice (Figures 2I–2K). Altogether, these results suggest that HTT is involved in axon regeneration independently of BDNF axonal transport.
HTT interacts with ribosomes
To identify the molecular mechanism by which HTT controls axon regeneration, we analyzed WT HTT-binding partners identified by Shirasaki and colleagues in the mouse brain.28 We submitted a list of 747 putative HTT-interacting proteins to network clustering analysis.29 A dense cluster of ribosomal components was highlighted, as well as translation-associated factors, RNA-binding proteins, and aminoacyl tRNA synthetases (Figure S3A; Table S1). This analysis suggested that HTT interacts with the translational complex. We performed HTT immunoprecipitation using mouse embryonic fibroblasts (MEFs). We found that ribosomal proteins of the small subunit RPS6 and of the large subunits RPL3 and RPL22 immunoprecipitate with HTT. This is also the case of the translation initiation factor eIF4G and the RNA-binding protein Pabp1, supporting HTT interaction with the translational complex (Figures S3B and S3C).
Subsequently, we performed purification of ribosomes from MEFs and whole retina lysates30 (Figure 3A). The ribosomal proteins RPL22 and RPS6 are enriched in the ribosomal fraction, whereas we observed a minimal contamination of nuclear and mitochondrial components (Figure 3B). We detected the protein HTT in the ribosomal fraction, both in MEFs and in the retina, further confirming that HTT interacts with the translational complex (Figure 3B). In particular, HTT co-fractionates with ribosomes even in increasingly stringent conditions (Figure 3C), meaning that HTT strongly interacts with ribosomes. Additionally, RNase I treatment of cell lysate did not remove HTT from the ribosomal fraction, unlike the RNA-binding protein Pabp1 (Figure 3D). Thus, our results suggest that RNA degradation and ribosome dissociation do not affect HTT’s presence in the ribosomal fraction, showing that HTT is a ribosome-interacting protein independent of RNA.
Figure 3.

HTT interacts with ribosomes but does not control global translation
(A) Schematic representation of ribosome purification procedure.
(B) Immunoblot of the subcellular fractions from WT MEF lysate and adult WT mouse retina lysate.
(C) Immunoblot of the total and purified ribosomal fractions of MEF lysate with increasing KCl concentrations.
(D) Immunoblot of the total and purified ribosomal fractions of MEF lysate, following RNase I treatment of the post-mitochondrial fraction.
(E) Representative polysome profile of WT MEFs on a 15%–50% sucrose gradient. Immunoblot showing the presence of HTT in the 40S, 60S, monosome, and light polysome fractions.
(F) Schematic representation of PLA experiment. PLA staining confocal images on retina sections, showing interaction between HTT and RPS6 or rRNA in RGCs.
(G) PLA staining confocal images showing interaction between HTT and RPS6 or rRNA in WT retinas, and controls with individual antibodies alone.
To determine the nature of HTT interaction with ribosomes, we performed a polysome profiling on MEF lysate. This method allows one to distinguish among the small subunit (40S), large subunit (60S), monosome (80S), and polysomes (fractions where mRNAs carry multiple ribosomes), which are heavier with increasing translational activity. Interestingly, we found that HTT is present in both 40S and 60S fractions; in the monosome fraction; and, to a lesser extent, in the light polysome fractions (Figure 3E). HTT interaction with ribosomal subunits was not altered by the chemical disruption of the ribosome (EDTA treatment), the blockage of translation initiation (harringtonin treatment), the blockage of translation elongation (puromycin treatment), and RNA degradation (RNase I treatment) (Figures S3D and S3E). mRNA is necessary for the formation of active 80S ribosomes,31 and the degradation of ribosomal RNA (rRNA) can dissociate ribosomes.32 Thus, our results are consistent with the fact that HTT interacts with ribosomes in an RNA-independent manner. Altogether, these results demonstrate that HTT can bind to both subunits, independently of ongoing translation.
We further validated this interaction specifically in RGCs using in situ proximity ligation assay (PLA) staining of HTT and components of the ribosomal complex (Figure 3F). Using PLA on WT mouse retina sections, we showed that HTT interacts with rRNA and RPS6 (Figures 3F and 3G). These results demonstrate that HTT associates with the translational complex and suggest a role for HTT in translation regulation during CNS regeneration.
HTT controls mRNA-specific translation
Based on the interaction between HTT and the translational complex, we further investigated the role of HTT in controlling protein synthesis. We first examined whether HTT controls global protein translation. We used a surface sensing of translation (SUnSET) technique to compare global protein synthesis in WT and HTT-deleted samples. Using puromycin incorporation in MEFs, we found that HTT deletion had no effect on the level of global protein synthesis (Figure 4A). This result was confirmed in vivo in RGCs, using fluorescent detection of protein synthesis with the SUnSET approach by O-propargyl-puromycin (OPP) incorporation. As a control, the protein synthesis inhibitor anisomycin inhibited OPP incorporation in vivo (Figure 4B). Consistent with previous results,4 we observed that new protein synthesis significantly decreases at 3dpc in WT RGCs (Figure 4C). Moreover, HTT deletion in intact RGCs did not affect the global level of protein synthesis (Figure 4D), suggesting that HTT does not control global translation in vivo.
Figure 4.

HTT deletion does not modify global translation
(A) SUnSET assay showing puromycin incorporation revealed by immunoblotting in HTT-deleted MEFs (MEF HTTfl/fl+Cre) versus control MEFs. Unpaired t test, ∗∗∗p value < 0.001.
(B) Representative images and quantification of OPP incorporation in WT RGCs with DMSO or anisomycin injection.
(C) Representative images and quantification of OPP incorporation in intact and 3dpc RGCs.
(D) Representative images and quantification of OPP incorporation in HTTfl/fl+AAV2-Plap and HTTfl/fl+AAV2-Cre intact RGCs. Data are represented as mean ± S.E.M. Unpaired t test; ∗∗∗p value < 0.001; ns: not significant.
(E) Representative images and quantification of OPP incorporation in Ptenfl/fl+AAV2-Cre, Ptenfl/flHTTfl/fl+AAV2-Cre, and Ptenfl/flHTTfl/fl+AAV2-Plap intact RGCs.
(F) Representative images and quantification of OPP incorporation in Ptenfl/fl+AAV2-Cre, Ptenfl/flHTTfl/fl+AAV2-Cre, and Ptenfl/flHTTfl/fl+AAV2-Plap RGCs at 3dpc.
Data are represented as mean ± SEM. One-way ANOVA with Bonferroni multiple comparisons test; ∗∗∗p value < 0.001; ns, not significant.
Furthermore, we analyzed how HTT affected the outcome of ONC in terms of protein synthesis in the regenerative condition. When comparing Pten/HTT-deleted and Pten-deleted RGCs, we saw no difference in new protein synthesis in the intact condition and at 3dpc (Figures 4E and 4F). This was already the case not only at 12 h post crush (12hpc) and at 1dpc but also at a later time point post injury (7dpc) (Figures S4A–S4C). In contrast, the extent of protein synthesis was significantly lower upon ONC in the control condition (Ptenfl/flHTTfl/fl+AAV2-Plap) (Figures 4F and S4A–S4C). Altogether, these results show that HTT does not regulate the global rate of protein synthesis.
Nonetheless, the interaction between HTT and the translation machinery suggests a role for HTT in protein synthesis regulation. Thus, we examined whether HTT regulates the translation of specific mRNA subsets. To this end, we deleted HTT in MEFs (Figures 5A and 5B) and performed RNA sequencing of total cellular RNA and ribosome-associated RNA (Figure 5C). We then performed differential expression analysis by comparing HTT-deleted and control samples independently for both datasets (Figure 5D; Table S2). From this analysis, we identified 279 genes displaying differential transcriptional regulation and 131 genes displaying differential ribosome association upon HTT deletion (for each dataset: false discovery rate [FDR] p value < 0.05, |log2(fold-change)| > 1) (Figure 5E). Strikingly, out of the 131 translationally regulated genes, we found 115 genes for which total RNA expression level was unchanged (differential expressed gene [DEG] translatome only), meaning that their expression is not regulated at the transcriptional level (Figures 5E, S5A, and S5B). We found that the correlation between the transcriptional and translational regulations of all differentially regulated genes is only partial (Figure 5F). We then normalized the log fold-change of the translational regulation by that of the transcriptional regulation for all individual hits. The plot of log fold-changes of translatome versus transcriptome revealed target mRNAs whose translation is controlled by HTT (translational changes only) but whose total RNA level is not modified (Figure 5G). Very interestingly, from the list of transcripts with differential association with ribosomes, we found receptors such as the transient receptor potential ion channel Trpm6 and the Toll-like receptor TLR8, signal transducers such as Wnt2b, enzymes such as the 5-phosphatase Inpp5j, and transcription factors such as Tox2 (Figure 5G). Importantly, these results highlight the strong decoupling between translational and transcriptional regulations in cells. Thus, our results indicate a differential mRNA association with ribosomes at single transcript level, conditioned by HTT-ribosome interaction. Altogether, these data support the hypothesis that HTT regulates the translation of a specific subset of mRNAs.
Figure 5.

HTT regulates the association of a specific mRNA subset with ribosomes
(A) Schematic representation of total and ribosome-associated RNA analysis by RNA sequencing from MEF HTTfl/fl+LV-Cre versus control.
(B) Representative immunoblot of the total and purified ribosomal fractions of control and HTT-KO MEF lysate.
(C) Distribution of relative log expression (log-ratio of read count to median read count across samples) after normalization using residuals with RUVseq.
(D) Principal component analysis of the individual replicates of total and ribosome-associated RNA samples. EdgeR with RUVseq normalization based on residuals.
(E) Venn diagram of differentially expressed genes identified in total and ribosome-associated RNA samples (FDR-corrected p value < 0.05, |log2(fold-change)| > 1).
(F) Heatmap of log2(fold-change) values between HTT-deleted (KO) and control conditions in total and ribosome-associated RNA samples, for genes differentially expressed in both datasets.
(G) Scatterplot of the log2(fold-change) in ribosome-associated RNA (translatome) versus the log2(fold-change) in total RNA (transcriptome). Blue hits: FDR-corrected p value < 0.05 and |log2(fold-change)| > 1 in both datasets. Pink hits: FDR-corrected p value < 0.05 and |log2(fold-change)| > 1 in transcriptome only. Orange hits: FDR-corrected p value < 0.05 and |log2(fold-change)| > 1 in translatome only.
(H) Quantitative reverse-transcriptase PCR (RT-qPCR) analysis of Tox2 mRNA level normalized to Gapdh and Sdha levels in Ptenfl/fl+AAV2-Cre and Ptenfl/flHTTfl/fl+AAV2-Cre RGCs at 3dpc.
(I) RT-qPCR analysis of Tox2 and Gapdh mRNA levels normalized to 18S level in immunoprecipitated ribosome fraction in Ptenfl/fl+AAV2-Cre and Ptenfl/flHTTfl/fl+AAV2-Cre RGCs at 3dpc.
Data are represented as mean ± SEM. Unpaired t test; ∗∗p value < 0.01; ns, not significant.
HTT regulates Tox2 expression at the translational level
We then asked whether HTT modulates axon regeneration through the control of specific protein translation in RGCs. First, we used RT-qPCR on fluorescence-activated cell sorting (FACS)-isolated RGCs (Figure S5C) to compare the total level of target mRNAs between Pten/HTT-deleted and Pten-deleted conditions at 3dpc. In parallel, we analyzed the level of ribosome-associated target mRNAs in RGCs, using ribosome immunoprecipitation with RPL22-FLAG and RT-qPCR on ribosome-associated RNA (Figures S5D–S5G). Comparing Pten/HTT-deleted and Pten-deleted conditions at 3dpc, we validated several translational targets that are differentially associated with ribosomes in RGCs upon HTT deletion, whereas their total mRNA level is unchanged, e.g., Trarg1, Tox2, and Wnt2b (Figures 5H, 5I, and S5H). These results are consistent with HTT-deleted versus control MEF datasets. Conversely, no difference was found for several mRNAs that are regulated neither at the total nor at the ribosome-associated level, e.g., Tubb3 and Pex19 (Figure S5H). These results confirm the relevance of our in vitro dataset to injured RGCs.
Next, we focused on one translational target of HTT, Tox2 transcription factor (Figure 5G). Tox was previously shown to regulate corticogenesis by promoting the division of neural progenitors and neurite outgrowth via promotor binding of key genes such as Sox2 and Robo2 and members of the Shh, Wnt, and Notch signaling pathways.33 Tox family members participate in the transcriptional architecture of interneurons in the developing cortex.34 These features make Tox2 a relevant candidate in the context of neuronal circuit repair.
Our data reveal that Tox2 mRNA is differentially associated with ribosomes upon HTT deletion, but not transcriptionally regulated (Figure 5G). Tox2 mRNA is expressed in mature RGCs (Figure S5I) and is not regulated by HTT deletion (Figures S5J and S5K). Using RT-qPCR on FACS-isolated RGCs, we found no difference in Tox2 mRNA level between Pten/HTT-deleted and Pten-deleted conditions at 3dpc (Figure 5H). Conversely, using RT-qPCR on ribosome-associated mRNA, we found a differential association of Tox2 mRNA with ribosomes in the absence of HTT, comparing Pten/HTT-deleted and Pten-deleted conditions at 3dpc (Figure 5I), which was not the case for Gapdh. From these data, we concluded that HTT is a translational regulator of Tox2 mRNA target in RGCs.
To determine how HTT regulates Tox2 translation, we performed polysome profiling on MEF. Using ultracentrifugation on a sucrose gradient, mRNA species were separated according to the number of ribosomes that they carry. Lighter fractions contain untranslated or slowly translated mRNAs, whereas heavier fractions contain actively translated mRNAs (Figure 6A). We compared HTT-deleted versus control MEFs and assessed the distribution of Tox2 mRNA in monosomes (slow or no translation), light polysomes (medium translation), and heavy polysomes (fast, active translation).35,36 Interestingly, HTT deletion caused the distribution of Tox2 mRNA to shift from fast, active translation fractions toward slow to no-translation fractions, whereas no difference was found for the control mRNA Gapdh (Figure 6B). HTT deletion caused a significant increase in Tox2 mRNA in monosomal fractions. Conversely, we found a significant decrease in Tox2 mRNA in polysomal fractions, where mRNAs are actively translated (Figure 6C). Thus, this experiment shows that Tox2 is less actively translated in the absence of HTT and, hence, that HTT positively regulates Tox2 translation.
Figure 6.

HTT controls axon regeneration via the translational regulation of Tox2 mRNA
(A) Representative polysome profile of HTT-deleted (KO) MEF versus control on a 15%–50% sucrose gradient.
(B) Representative Tox2 and Gapdh mRNA distribution in fractions 4–14 of a polysome fractionation, in HTT-deleted (Cre) versus control MEFs.
(C) Tox2 and Gapdh mRNA distribution in monosomal (yellow) and polysomal (purple) fractions. Data are represented as min/max values with line at mean. Paired t test; ∗p value < 0.05; ns, not significant.
(D) Principle of puro-PLA experiment in vivo.
(E) PLA staining confocal images for puromycin-Tox2 or puromycin-Gapdh in intact RGCs in Ptenfl/fl+AAV2-Cre, Ptenfl/flHTTfl/fl+AAV2-Cre, and Ptenfl/flHTTfl/fl+AAV2-Plap.
(F) Quantification of puromycin-Tox2 PLA-positive events in Ptenfl/fl+AAV2-Cre, Ptenfl/flHTTfl/fl+AAV2-Cre, and Ptenfl/flHTTfl/fl+AAV2-Plap intact RGCs.
(G) Quantification of puromycin-Gapdh PLA-positive events in Ptenfl/fl+AAV2-Cre, Ptenfl/flHTTfl/fl+AAV2-Cre, and Ptenfl/flHTTfl/fl+AAV2-Plap intact RGCs.
(H) PLA staining confocal images for puromycin-Tox2 or puromycin-Gapdh in Ptenfl/fl+AAV2-Cre, Ptenfl/flHTTfl/fl+AAV2-Cre, and Ptenfl/flHTTfl/fl+AAV2-Plap RGCs at 3dpc.
(I) Quantification of puromycin-Tox2 PLA-positive events in Ptenfl/fl+AAV2-Cre, Ptenfl/flHTTfl/fl+AAV2-Cre, and Ptenfl/flHTTfl/fl+AAV2-Plap RGCs at 3dpc.
(J) Quantification of puromycin-Gapdh PLA-positive events in Ptenfl/fl+AAV2-Cre, Ptenfl/flHTTfl/fl+AAV2-Cre, and Ptenfl/flHTTfl/fl+AAV2-Plap RGCs at 3dpc.
Data are represented as mean ± SEM. Kruskal-Wallis with Dunn’s multiple comparisons test; ∗p value < 0.05, ∗∗p value < 0.01, and ∗∗∗p value < 0.001; ns: not significant.
To confirm this result in RGCs, we set up a puromycin-PLA (puro-PLA) in vivo, which allows the detection of nascent Tox2 protein (Figures 6D and S6A–S6C). In the intact condition, we found no significant difference in puromycin-Tox2 and puromycin-Gapdh PLA-positive events when comparing Pten-deleted and Pten/HTT-deleted RGCs (Figures 6E–6G). However, at 3dpc, we found that the number of new Tox2 protein synthesis events was significantly lower in RGCs in the absence of HTT, down to control level (Figures 6H and 6I). Conversely, the number of new Gapdh protein synthesis events was unchanged upon HTT deletion but was significantly lower in the control condition (Ptenfl/flHTTfl/fl + AAV2-Plap 3dpc) (Figure 6J). This was the case already at 1dpc (Figures S6D–S6F). Altogether, our data show that HTT deletion in RGCs decreases the level of new Tox2 protein synthesis, in contrast with the level of global translation, which is unchanged (Figure 4F). This effect is specific to the injured condition. This demonstrates that HTT selectively controls Tox2 protein synthesis in injured RGCs.
Translationally regulated target of HTT Tox2 controls axon regeneration
To investigate whether Tox2 is implicated in HTT-mediated axon regeneration, we modulated Tox2 expression and assessed axon regeneration at 14dpc. First, we used a short hairpin RNA (shRNA) directed against Tox2 (shTox2) (Figures S6B and S6C) and found that Tox2 knockdown suppresses axon regeneration in Pten-deleted RGCs at 14dpc (Figures 7A and 7B). This phenocopies HTT deletion in the same paradigm of regeneration (Figure 1E). Similarly to HTT deletion, the extent of RGC survival was unchanged (Figures S7A and S7B). pS6 protein levels were unchanged by Tox2 knockdown (Figures S7A and S7C), again showing that the suppression of the regenerative effect is not due to an alteration of the mTOR pathway.
Figure 7.

Regulation of Tox2 translation by HTT controls axon regeneration in vivo
(A) Whole optic nerve confocal images showing CTB+ regenerating axons in Ptenfl/fl+AAV2-Cre+AAV2-shScrambled and Ptenfl/fl+AAV2-Cre+AAV2-shTox2 at 14dpc.
(B) Quantification of the integrated intensity of fluorescence along the optic nerve. Unpaired t test; ∗p value < 0.05 and ∗∗p value < 0.01.
(C) Whole optic nerve confocal images showing CTB+ regenerating axons in Ptenfl/fl mice injected with AAV2-Cre+AAV2-Plap, Ptenfl/flHTTfl/fl mice injected with AAV2-Cre+AAV2-Plap, and Ptenfl/flHTTfl/fl mice injected with AAV2-Cre+AAV2-Tox2 at 14dpc.
(D) Quantification of the integrated intensity of fluorescence along the optic nerve. One-way ANOVA with Bonferroni multiple comparisons test; ∗p value < 0.05 and ∗∗p value < 0.01; ns, not significant. The top graph gives the multiple comparisons test between Ptenfl/flHTTfl/fl+AAV2-Cre+AAV2-Plap and Ptenfl/flHTTfl/fl+AAV2-Cre+AAV2-Tox2 conditions.
(E) Proposed model showing the HTT-dependent customization of ribosomes, which specifically controls Tox2 translation and CNS axon regeneration.
The injury site is indicated by a red star. Data are represented as mean ± SEM.
Conversely, we overexpressed Tox2 in Pten/HTT-deleted RGCs by cloning Tox2 coding sequence in the AAV vector backbone under the CMV promoter. This way, we circumvented any contribution of regulatory untranslated regions (UTRs) in Tox2 mRNA. As controls, we injected AAV2-Cre + AAV2-Plap in Ptenfl/fl and Ptenfl/flHTTfl/fl mice. As expected, Pten/HTT-deleted RGCs showed very little regeneration compared with Pten-deleted RGCs (AAV2-Plap injection). On the other hand, Tox2 overexpression significantly rescued axon regeneration in Pten/HTT-deleted RGCs (Figures 7C and 7D). With an efficient overexpression of Tox2 under the CMV promoter, Tox2 protein synthesis is increased, regardless of the translation regulation process normally occurring in the cell, and thus produces robust regeneration even in the Pten/HTT-deleted condition. We also tested the effect of Tox2 overexpression in WT mice and found no difference in terms of regeneration (Figures S7D and S7E), survival, or pS6 expression (Figures S7F–S7H). This result suggests that Tox2 alone is not sufficient to promote axon regeneration but is necessary in the Pten-deleted paradigm. Analysis of Tox2 protein expression revealed a strong decrease in Pten/HTT-deleted RGCs in response to injury. Conversely, this drop is contained in Pten-deleted RGCs, where HTT expression is stable even at a late time point after injury (14dpc) (Figure S7I).
Altogether, our experiments provide evidence that HTT is a ribosome-associated factor that controls axon regeneration via the translational regulation of specific mRNAs such as Tox2. In the paradigm of mTOR-induced axon regeneration, the deletion of HTT or of its translationally regulated mRNA target abolishes the regenerative effect. Conversely, the overexpression of the translationally regulated target leads to increased protein synthesis of this specific target, regardless of its normal translation regulation, and rescues the regenerative phenotype after CNS injury (Figure 7E).
Discussion
CNS axon regeneration is a multimodal process that involves different layers of regulation. Gene expression, through epigenetic and transcriptional regulations, has been extensively studied in this context.3,37 Importantly, robust axon regeneration can be triggered by activating molecular pathways that are master regulators of several steps of protein synthesis, such as mTOR4,38 and c-myc, which is directly involved in the regulation of the translational complex and protein synthesis.7,39 Yet, to which extent protein translation is critical to achieve axonal growth and functional recovery remains largely unknown. In our study, we highlight for the first time that the process of mRNA-specific translation, coordinated by the functional interaction of specific factors with ribosomes, controls axon regeneration. We identify HTT as a key regulatory factor whose association with ribosomes conditions axon regeneration. The translational selectivity driven by HTT is specific to the regenerative condition following injury. Through the identification of this new regulatory mechanism, our study unravels candidate mRNAs whose selective translation coordinated by HTT is key for the axon regeneration of injured neurons. More generally, we show that specific translational control is as important as transcriptional control to promote axon regeneration.
Our data provide evidence of a strong, mRNA-independent interaction of HTT with ribosomes, which confirm previous screens of HTT interactants in the brain that revealed several ribosomal proteins.28,40 We show that WT HTT regulates mRNA-specific translation, but not global translation. This may involve binding partners that remain to be identified. Furthermore, our results are in line with findings related to HTT mutation in the context of Huntington’s disease. Indeed, the disease-related mutation of HTT may alter its role in protein translation, notably cap-dependent translation.41 Two recent studies showed that mutant HTT impairs global protein synthesis, either up by altering the activity of translation factors such as 4E-BP42 or down by stalling ribosomes on mRNA.43
Here, we show that HTT customizes ribosomes, which results in differential mRNA association with ribosomes and translation. More generally, ribosome-interacting factors stand as novel keys of translational regulation, directly influencing the functional outcome of mRNA translation depending on the physio-pathological context.44 It is now well described that the translational complex is not an invariant unit and that its composition, notably the differential association of translation factors with ribosomes, influences gene expression at the translational level.16 The partial correlation of mRNA and protein levels45,46,47 illustrates the necessity of a fine translational regulation to maintain cell homeostasis and adapt its response to stress or external stimuli. Translation-associated factors are critical regulators of gene expression, thereby building the developmental program for cell specification and tissue organization.48 In addition, the functional interactants of ribosomes, namely, the ribo-interactome, remain to be clearly characterized. So far, only few studies have explored the functional interaction between associated factors and ribosomes.49,50 Using endogenous tagging of ribosomal proteins, Simsek and colleagues provided an exhaustive description of the ribo-interactome in mouse embryonic stem cells.44 On top of the canonical components of the translation machinery, they identified more than 300 ribosome-associated proteins, as well as post-translational modifications that contribute to ribosome customization. Yet, this interactome that shapes ribosome diversity may well be specific to a particular developmental stage, cell type, or stress response.51,52,53 In the next years, the increasing sensitivity of proteomic techniques will allow the description of ribosome diversity in different neuronal subpopulations in the contexts of neuronal injury response, neuroprotection, and axon regeneration.
Furthermore, much effort is made to understand how variations in the composition of the translational complex lead to a switch in gene expression that causes pathological defects, for example, in the case of ribosomopathies54 and in cancer cells.55,56,57,58 In the context of CNS injury, our work highlights for the first time that translational regulation through the customization of the translational machinery is at play to control axon regeneration. Whether this selective translation process displays differential activity in subcellular domains (e.g., cell soma versus axon shaft versus growth cone) will be the next question to address. Indeed, how local protein synthesis influences CNS axon regeneration raises much attention,59 based on studies in embryonic and PNS neurons.60,61,62,63,64 In addition, the increasing resolution of imaging techniques has recently allowed the detection of the translational complex in the pre-synaptic compartments of intact adult CNS axons.65,66 Therefore, our work opens the path to explore the role of local translational control in CNS regeneration.
The selectivity of the translational complex for certain mRNA pools relies on the presence of cis-regulatory elements in the transcript sequence.67,68,69,70 These regulatory regions include internal ribosome entry site (IRES) elements in mRNA 5′ UTR. Initially described in viruses, IRESs have been identified in >100 mammalian mRNAs, e.g., c-myc71,72 and p53,73 which encode transcription factors implicated in axon regeneration.7,74 The translational complex composition is key to promote IRES-dependent mRNA translation, as reported for ribosomal-protein-RPL38-containing ribosomes that control Hox gene expression during development.75,76 Other regulatory elements of mRNA selectivity are upstream open reading frames (uORFs). Translation of uORFs has been shown to regulate tissue patterning77 and stress response.78 5′ terminal oligopyrimidine (5′ TOP) motifs also allow translational control in stress conditions and are found in mRNAs encoding ribosomal proteins and translation factors themselves.69 In addition, beside their role in target mRNA degradation, microRNAs can directly control protein synthesis through the inhibition of the initiation step by repressing cap-dependent translation or by preventing ribosomal subunit assembly.79 Such cis-regulatory elements mediate mRNA translation at the steps of mRNA identification by the ribosome, recruitment and initiation.
In our case, HTT may directly promote the recruitment of specific mRNAs by 5′ UTR- or IRES-mediated regulation, although this remains to be determined. Besides, HTT may participate in translation regulation at different steps, either directly or through an intermediate translational partner. Interestingly, HTT interacts with both small and large subunits of ribosomes. In addition, Tox2 mRNA relocates in the monosomal fraction in the absence of HTT. The ribosome density on a particular transcript correlates with its translation efficiency.80 Not only does this depend on the rate of translation initiation, but it also depends on how fast a firstly loaded ribosome will translate a transcript during elongation phase to allow the loading of more ribosomes. Moreover, a recent work in adult neurons has demonstrated an unexpected translational activity of monosomes, which translate synapse-associated mRNAs in physiological conditions.81 Beyond the translational regulatory state, specific subsets of mRNAs may be translated in monosomal fractions. Our results suggest a regulatory role of HTT in selective translation, with the two pieces of evidence: upon HTT deletion, (1) Tox2 mRNA relocates in the monosomal fraction, and (2) Tox2 protein is less synthesized in injured RGCs. The steps of translational regulation involved remain to be identified, as well as the precise mechanism by which HTT mediates mRNA-specific translation.
Enhancing selective translation rather than global translation may be more relevant to express efficiently pro-regenerative molecules in order to repair the neuronal circuit. Indeed, despite major advances in achieving long-distance regeneration in the past decade,7,25,82 no functional recovery has been obtained yet. Importantly, this massive extent of axon regeneration relies on the modulation of transcription- and translation-promoting pathways that may be oncogenic and, therefore, are currently inapplicable as therapies. Therefore, elucidating HTT mechanism of action as a pro-regenerative factor may allow better tackling of neuronal injury response and triggering of functional CNS axon regeneration.
To conclude, we show that translational regulation, independent of transcriptional regulation, is a critical step to achieve CNS regeneration. Upon injury, ribosome customization through the association of factors such as HTT switches the translational landscape from a regeneration-primed to a regeneration-incompetent state. Altogether, our study allows (1) the identification of a new molecular mechanism of control of axon regeneration, (2) the highlighting of pro-regenerative targets of CNS axon regeneration regulated at the translational level, and (3) the development of novel therapeutic strategies based on the modulation of translation-associated factors to enhance the selective translation of such pro-regenerative targets.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea-pig polyclonal anti-RBPMS | Millipore | Cat# ABN1376; RRID: AB_2687403 |
| Mouse IgG isotype control | ThermoFisher Scientific | Cat# 31903; RRID: AB_10959891 |
| Mouse monoclonal anti-actin | Sigma-Aldrich | Cat# A1978; RRID: AB_476692 |
| Mouse monoclonal anti-HSP60 | Santa Cruz Biotechnology | Cat# sc-376240; RRID: AB_10986282 |
| Mouse monoclonal anti-HTT, clone 4c8 | Millipore | Cat# MAB2166; RRID: AB_11213141 |
| Mouse monoclonal anti-puromycin | Millipore | Cat# MABE343; RRID: AB_2566826 |
| Mouse monoclonal anti-RPL22 | Santa Cruz Biotechnology | Cat# sc-373993; RRID: AB_10918294 |
| Mouse monoclonal anti-rRNA, clone Y10b | Santa Cruz Biotechnology | Cat# sc-33678; RRID: AB_628226 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | Cat# 2118; RRID: AB_561053 |
| Rabbit monoclonal anti-pS6 | Cell Signaling Technology | Cat# 4857; RRID: AB_2181035 |
| Rabbit monoclonal anti-RPS6 | Cell Signaling Technology | Cat# 2217; RRID: AB_331355 |
| Rabbit polyclonal anti-RPL3 | Abcam | Cat# ab228638 |
| Rabbit monoclonal anti-HTT, clone d7F7 | Cell Signaling Technology | Cat# 5656; RRID: AB_10827977 |
| Rabbit polyclonal anti-eIF4G | Cell Signaling Technology | Cat# 2498; RRID:AB_2096025 |
| Rabbit polyclonal anti-Histone 3 | Cell Signaling Technology | Cat# 9715; RRID: AB_331563 |
| Rabbit polyclonal anti-Pabp1 | Cell Signaling Technology | Cat# 4992; RRID: AB_10693595 |
| Rabbit polyclonal anti-Tox2 | Sigma-Aldrich | Cat# HPA058396; RRID: AB_2683704 |
| Rabbit anti-SCG10 | Novus Biologicals | Cat# NPB1-49461 |
| Mouse anti-Tubulin β III (Tuj1) | Biolegend | Cat# 801202; RRID:AB_10063408 |
| Anti-digoxigenin antibody | Roche | Cat# 11093274910; RRID: AB_2313640 |
| Donkey HRP-conjugated anti-mouse | Thermo Fisher Scientific | Cat# A16011; RRID: AB_2534685 |
| Goat HRP-conjugated anti-rabbit | Proteintech | Cat# SA00001-2; RRID: AB_2722564 |
| Donkey anti-guinea pig Alexa Fluor 647 | Jackson Laboratory | Cat# 706-605-148; RRID: AB_2340476 |
| Donkey anti-mouse Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A21202; RRID: AB_141607 |
| Goat anti-mouse Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A11004; RRID: AB_2534072 |
| Donkey anti-mouse Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A31571; RRID: AB_162542 |
| Donkey anti-rabbit Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A21206; RRID: AB_2535792 |
| Donkey anti-rabbit Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A10042; RRID: AB_2534017 |
| Donkey anti-rabbit Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A31573; RRID: AB_2536183 |
| Bacterial and virus strains | ||
| AAV2-CMV-Cre | Belin et al.7 | N/A |
| AAV2-CMV-Placental alkalyne phosphatase (Plap) | Belin et al.7 | N/A |
| AAV2-SNCG-RPL22-flag | This study | N/A |
| AAV2-U6-shTox2 | This study | N/A |
| AAV2-U6-shScrambled | This study | N/A |
| AAV2-CMV-Tox2-HA | This study | N/A |
| AAV2-CMV-cRheb | Lim et al.25 | N/A |
| AAV2-CMV-BDNF-tdTomato | This study | N/A |
| AAV8-CMV-Plap | This study | N/A |
| AAV8-CMV-Cre | This study | N/A |
| H2B-eGFP | Addgene | Cat# 25999; RRID: Addgene_25999 |
| LV-Cre-GFP | Addgene | Cat# 86805; RRID: Addgene_86805 |
| Chemicals, peptides, and recombinant proteins | ||
| Alexa Fluor 555-conjugated-cholera toxin B subunit (CTB) | Thermo Fisher Scientific | Cat# C22843 |
| cOmplete EDTA-free Protease Inhibitor Cocktail | Roche | Cat#4693159001 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Thermo Fisher Scientific | Cat# 31966047 |
| Fetal bovine serum, FBS | Eurobio Scientific | Cat# CVFSVF00-01 |
| Penicillin-streptomycin | Thermo Fisher Scientific | Cat# 15140122 |
| L-Glutamin | Thermo Fisher Scientific | Cat# 25-005-CI |
| Trypsin-EDTA | Thermo Fisher Scientific | Cat# 25-050-CI |
| Mouse IgG | Thermo Fisher Scientific | Cat# 31903 |
| Puromycin | Sigma-Aldrich | Cat# P8833 |
| Emetine | Sigma-Aldrich | Cat# E2375 |
| Harringtonin | Santa Cruz Biotechnology | Cat# sc-204771 |
| RNase I | Invitrogen | Cat# AM2294 |
| SUPERase In | Invitrogen | Cat# AM2694 |
| NBT-BCIP | Roche | Cat# 11681451001 |
| Duolink blocking buffer | Sigma-Aldrich | Cat# DUO82007 |
| Wash buffer A Duolink | Sigma-Aldrich | Cat# DUO82046 |
| GlycoBlue coprecipitant | Thermo Fisher Scientific | Cat# AM9515 |
| TRIzol Reagent | Invitrogen | Cat# 15596-026 |
| Critical commercial assays | ||
| Protein G magnetic beads, SureBeads | Bio-Rad | Cat# 1614023 |
| SilverQuest silver staining kit | Thermo Fisher Scientific | Cat# 45-1001 |
| OPP labeling kit – Click-iT Plus OPP Alexa Fluor 647 Protein Synthesis Assay Kit | Thermo Fisher Scientific | Cat# C10458 |
| mRNA extraction kit PicoPure | Thermo Fisher Scientific | Cat# 12204-01 |
| mRNA extraction kit miRVana | Thermo Fisher Scientific | Cat# AM1560 |
| PicoPure RNA extraction kit | Thermo Fisher Scientific | Cat# KIT0204 |
| PicoChip | Agilent | Cat# 5067-1513 |
| RNeasy Micro kit | Qiagen | Cat # 74004 |
| SuperScript II kit | Invitrogen | Cat# 18064022 |
| SYBR Green Supermix | Bio-Rad | Cat# 1725271 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | Cat# 1725271 |
| Duolink PLA kit | Sigma-Aldrich | Cat# DUO92013 |
| Minus probe-conjugated anti-mouse antibody - Duolink | Sigma-Aldrich | Cat# DUO92004; RRID: AB_2713942 |
| Plus probe-conjugated anti-rabbit antibody - Duolink | Sigma-Aldrich | Cat# DUO92002; RRID: AB_2810940 |
| DIG RNA labelling kit | Roche | Cat# 11175025910 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Fast Red TR/Naphthol AS-MX Tablets | Sigma-Aldrich | Cat# F4648 |
| Deposited data | ||
| RNA sequencing data | GeneWiz/ This study |
GSE209946 |
| Proteomic data of HTT interactome in mouse brain | Shirasaki et al.28 | N/A |
| Experimental models: Cell lines | ||
| Mouse: MEF (Mouse Embryonic Fibroblasts) | This study | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: HTT floxed | Dragatsis et al.19 | N/A |
| Mouse: PTEN floxed | Jackson Laboratory | RRID:IMSR_JAX:004597 |
| Mouse: PTEN floxed HTT floxed | This study | N/A |
| Mouse: ROSA-tdTomato floxed | Jackson Laboratory | RRID:IMSR_JAX:007914 |
| Mouse: phosphorylated line S421D | Thion et al.24 | N/A |
| Mouse: phosphorylated line S1181D/S1201D | M’Barek et al.13 | N/A |
| Mouse: phosphorylated line S421A | Thion et al.24 | N/A |
| Mouse: phosphorylated line S1181A/S1201A | M’Barek et al.13 | N/A |
| Oligonucleotides | ||
| HTT probe for In situ hybridization: nucleotides 346-1614 of mouse HTT mRNA | NCBI | Accession number: NM_010414 |
| Tox2 probe for In situ hybridization: nucleotides 94-853 of mouse Tox2 mRNA | NCBI | Accession number: NM_001098799.2 |
| Primer for In situ hybridization: Tox2 forward: 5’-TCACGAGACACTGGTGCATT-3’ |
This study | N/A |
| Primer for In situ hybridization: Tox2 reverse: 5’-AAGGTGGCACTGGGATTCTG-3’ |
This study | N/A |
| Primer for shTox2: 5’-GGGCGAGAGTAACGAAGACTA-3’ |
This study | N/A |
| Primer for control shRNA: 5’-GCTTACTAACCTCGGCAGT-3’ |
This study | N/A |
| Primer for RT-qPCR: Tox2 forward: 5’-ATGGTGACAGTGCCTACGTG-3’ |
This study | N/A |
| Primer for RT-qPCR: Tox2 reverse: 5’-AGGGATGGCTCAGGGAGATT-3’ |
This study | N/A |
| Primer for RT-qPCR: GAPDH forward: 5’-GCATGGCCTTCCGTGTTC-3’, |
This study | N/A |
| Primer for RT-qPCR: GAPDH reverse: 5’-TGTCATCATACTTGGCAGGTTTCT-3’ |
This study | N/A |
| Primer for RT-qPCR: Sdha forward: 5’-TGTTCAGTTCCACCCCACA-3’ |
This study | N/A |
| Primer for RT-qPCR: Sdha reverse: 5’-TCTCCACGACACCCTTCTG-3’ |
This study | N/A |
| Primer for RT-qPCR: 18S forward: 5’-GCAATTATTCCCCATGAACG-3’ |
This study | N/A |
| Primer for RT-qPCR: 18S reverse: 5’-GGCCTCACTAAACCATCCAA-3’ |
This study | N/A |
| Primer for RT-qPCR: Firefly luciferase forward: 5’-GAGGCGAACTGTGTGTGAGA-3’ |
This study | N/A |
| Primer for RT-qPCR: Firefly luciferase reverse: 5’-GTGTTCGTCTTCGTCCCAGT-3’ |
This study | N/A |
| Primer for RT-qPCR: Gpr158 forward: 5’-AACACAGCCTAGATCCAGAAGAC-3’ | This study | N/A |
| Primer for RT-qPCR: Gpr158 reverse: 5’-GGGTTGTTTGTGATCATCTTTTTA-3’ | This study | N/A |
| Primer for RT-qPCR: Lpcat forward: 5’-GTGCACGAGCTGCGACT-3’ | This study | N/A |
| Primer for RT-qPCR: Lpcat reverse: 5’-GCTGCTCTGGCTCCTTATCA-3’ | This study | N/A |
| Primer for RT-qPCR: Pex19 forward: 5’-CAGCAGCACAGCGTCATGGTCA-3’ | This study | N/A |
| Primer for RT-qPCR: Pex19 reverse: 5’-GTTGAGGCCAGGAGGCATCT-3’ | This study | N/A |
| Primer for RT-qPCR: Rnf112 forward: 5’-CACTGCAGGAGACCTGTGC-3’ | This study | N/A |
| Primer for RT-qPCR: Rnf112 reverse: 5’-CCCAGGACAGCCAATAAGCA-3’ | This study | N/A |
| Primer for RT-qPCR: Surf4 forward: 5’-CTGTTGGCCTCATCCTTCGT-3’ | This study | N/A |
| Primer for RT-qPCR: Surf4 reverse: 5’-GGCAATTGTCTGCAGTGCG-3’ | This study | N/A |
| Primer for RT-qPCR: Tpt1 forward: 5’-GGAGCTGCAGAGCAGATTAAG-3’ | This study | N/A |
| Primer for RT-qPCR: Tpt1 reverse: 5’-GTCCAGGAGAGCAACCATACC-3’ | This study | N/A |
| Primer for RT-qPCR: Tubb3 forward: 5’-GGTGGACTTGGAACCTGGAA-3’ | This study | N/A |
| Primer for RT-qPCR: Tubb3 reverse: 5’-TAAAGTTGTCGGGCCTGAAT-3’ | This study | N/A |
| Primer for RT-qPCR: Trarg1 forward: 5’-GGTCCTTGCCATTGCCTCTT-3’ | This study | N/A |
| Primer for RT-qPCR: Trarg1 reverse: 5’-TGCTGCACACTACTTCGAGAC-3’ | This study | N/A |
| Primer for RT-qPCR: Wnt2b forward: 5’-AACGAGGGGACTTTGACTGG-3’ | This study | N/A |
| Primer for RT-qPCR: Wnt2b reverse: 5’-CCACTCACACCGTGACACTT-3’ | This study | N/A |
| Recombinant DNA | ||
| Plasmid Tox2 ISH probe in pGEMT | This study | N/A |
| Plasmid HTT ISH probe in pGEMT | This study | N/A |
| Plasmid for Firefly RNA synthesis in pGEMT | This study | N/A |
| Software and algorithms | ||
| Zen | Zeiss | RRID: SCR_013672 |
| Fiji | ImageJ | RRID: SCR_002285 |
| IMARIS | Bitplane | RRID: SCR_007370 |
| Metamorph | Molecular Devices | RRID: SCR_002368 |
| STRING version 11.5 | Elixir | RRID: SCR_005223 |
| Venny 2.1 | BioinfoGP | RRID: SCR_016561 |
| DAVID Bioinformatics Resources 6.8 | LHRI | RRID: SCR_001881 |
| STAR | A. Dobin, Github | RRID: SCR_004463 |
| HTSeq-Count | HTSeq | RRID: SCR_005514 |
| edgeR | Bioconductor | RRID: SCR_012802 |
| R | The R project | RRID:SCR_001905 |
| RUVSeq | Bioconductor | RRID:SCR_006263 |
| GraphPad Prism | GraphPad | RRID:SCR_002798 |
| Other | ||
| Polycarbonate tube | Beckman Coulter | Cat# 349622 |
| Ultracentrifuge tubes | Beckman Coulter | Cat# 344059 |
| Plate reader | BMG Labtech | PHERAstar FS |
| ChemiDoc | Bio-Rad | Chemidoc MP Imaging System |
| Gradient Master | BioComp Instruments | Gradient Master |
| Piston Gradient Fractionator | BioComp Instruments | PGFip |
| Fraction collector | Teledyne ISCO | FoxyR1 |
| Nanodrop | Thermo Fisher Scientific | Nanodrop One |
| Cell Sorter | BD Biosciences | Aria II |
| Tissue Homogenizer | Retsch | Mixer Mill MM400 |
| Confocal Microscope, spinning disk | Andor | Dragonfly |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Stephane Belin (stephane.belin@inserm.fr).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact without restriction.
Experimental model and study participant details
All animal care and surgeries were performed in line with institutional, French and European guidelines. Experimental procedures are deposited under the project numbers APAFIS # 20191202144779_v4 and APAFIS # 38155-202205021448189 v5. For all experimental procedures, mice aged 3 or 4 weeks regardless of their sex were used unless otherwise stated. Mice were anesthesized with intraperitoneal injection of ketamine (Clorketam, 60-100mg/kg) and xylazine (Rompun, 5-10mg/kg). Mice of the following mutant lines were used: HTT-floxed,19 Pten-floxed,83 Pten-floxed HTT-floxed, ROSA-tdTomato-floxed, HTT phosphorylation mutant lines (mimicking constitutive phosphorylation, S421D and S1181D/S1201D, or unphosphorylable, S421A and S1181A/S1201A).13,24
Method details
Intravitreal injection
Following anesthesia, the external edge of the eye was clamped using an artery clamp to display the conjunctiva. Using a glass micropipette connected to a 50μl Hamilton syringe (Hamilton, 80521), 1μl of vitreous humor was removed and 1μl of Adeno-associated type 2 virus (AAV2) was injected into the vitreous body of the eye. The following AAV2 viruses were used: AAV2-CMV-Cre-HA, AAV2-CMV-Placental alkaline phosphatase (Plap), AAV8-CMV-Cre-HA, AAV8-Plap, AAV2-SNCG-RPL22-flag, AAV2-U6-sh-Tox2-hSyn-mCherry, AAV2-U6-sh-scrambled-hSyn-mCherry, AAV2-CMV-Tox2-HA, AAV2-CMV-cRheb-HA, AAV2-CMV-BDNF-tdTomato. The shRNA targeting Tox2 (shTox2, 5’- GGGCGAGAGTAACGAAGACTA-3’) was cloned under the control of a U6 promoter in a pAAV-U6-hSyn-mCherry plasmid. The scrambled sequence for the control shRNA (shScrambled) was 5’-GCTTACTAACCTCGGCAGT-3’. For overexpression, full-length mouse Tox2 fused to HA tag was cloned under the control of a CMV promoter in a pAAV-CMV. For anterograde labeling of regenerating axons, 1μl of 1μg/μl Alexa Fluor 555-conjugated cholera toxin beta subunit (CTB) ((ThermoFisher Scientific, C22843) was injected intravitreally 2-3 days before sacrifice. For in vivo O-propargyl-puromycin (OPP) incorporation assay, 1μl of 20μM Click-iT OPP (ThermoFisher Scientific, C10458) diluted in sterile PBS was injected intravitreally 30min before intracardial perfusion. Mice with eye inflammation or damage were excluded from the study.
Optic nerve crush
Optic nerve crush was performed 4 weeks after intravitreal injection of the viral vector or in 6-week-old wild-type mice, unless otherwise stated. Following anesthesia, the conjunctiva was opened with fine scissors (Fine Science Tools, 91500-09) and dilating forceps were carefully slided in-between the two arteries behind the eye ball to expose the underlying optic nerve. The optic nerve was pinched for 5 seconds using Dumont #5 forceps (Fine Science Tools, 11251-20) 1-2mm behind the eye ball. Mice with unstoppable heavy bleeding were excluded from the study. Mice were treated with paracetamol (Doliprane, 4mg/ml in drinking water) as painkiller for 48h following optic nerve crush.
Intrathecal injection
Intrathecal injection was performed on 3 to 4 week-old mice, as described.84 Following anesthesia, the dorsal skin was shaved and the vertebral column exposed by a small incision of the skin at the L4-L6 level. Using a 30G needle connected to a 50μl Hamilton syringe (Hamilton, 702LT), 10μl of Adeno-associated type 8 (AAV8) virus was injected into the spinal canal at L5 level. The following AAV8 viruses were used: AAV8-CMV-Cre, AAV8-CMV-Placental alkaline phosphatase (Plap). Muscles and skin were stitched with surgical staples. Mice were treated with paracetamol (Doliprane, 4mg/ml in drinking water) as painkiller for 48h following intrathecal injection.
Sciatic nerve injury
Sciatic nerve injury was performed 4 weeks following intrathecal injection. Following anesthesia, the left sciatic nerve was exposed thanks to an incision of the skin and muscles of the left thigh. The sciatic nerve was pinched for 15 seconds using Dumont #SS forceps (Fine Science Tools, 11203-25). The sciatic nerve received a second pinch of 5 seconds to ensure full axotomy. Mice were treated with paracetamol (Doliprane, 4mg/ml in drinking water) as painkiller for 48h following sciatic nerve injury.
Tissue dissection and processing
At the time of sacrifice, following anesthesia, mice were intracardially perfused with 50-100ml ice-cold PBS followed by 50-100ml 4% formaldehyde in PBS. Eyes and optic nerves were dissected out of the skull with fine scissors and forceps (FST) and post-fixed in 4% formaldehyde overnight at 4°C. For sciatic nerve injury experiments, L3 to L5 DRG and sciatic nerve were dissected and post-fixed in 4% formaldehyde overnight at 4°C.
Optic nerve clearing
For analysis of axon regeneration, optic nerves were cleared as previously described.85 Briefly, optic nerves were carefully separated from the eye body and dehydrated in baths of 50%, 80% and 95% ethanol in PBS (20min each) and 100% absolute ethanol (overnight). Optic nerves were incubated for 2hours in hexane then cleared in benzyl benzoate:benzyl alcohol (2:1). Whole optic nerves were mounted in benzyl benzoate:benzyl alcohol (2:1) under a coverslip and imaged with confocal microscopy (Andor spinning disk). The entire volume of the optic nerve was imaged with a z-step of 2μm. The entire optic nerve was imaged using a custom stitching module in Metamorph Microscope imaging software. The extent of axon regeneration was determined from the maximum projection of the z-stack. Integrated fluorescence was determined along the optic nerve at defined distances from the injury site, as described.26
Whole-mount retina immunohistochemistry
Retinas were dissected out of the eye ball using fine scissors and forceps (FST). Whole retinas were cut radially in a flower shape to facilitate mounting. After several washes in PBS, whole retinas were incubated in blocking solution (PBS 0.5% Triton X-100 5% donkey serum) for 1h at room temperature. Whole retinas were incubated in primary antibody diluted in blocking solution overnight at 4°C with gentle shaking. Primary antibodies used were: guinea pig anti-RBPMS (Millipore, ABN1376) 1:300; rabbit anti-pS6 (Cell Signaling Technology, 4857) 1:400. After several washes in PBS 0.1% Triton X-100, whole retinas were incubated in secondary antibody diluted in blocking solution for 2h at room temperature. Secondary antibodies used were Alexa Fluor-conjugated secondary antibodies (Jackson ImmunoResearch) diluted 1:500. After several washes in PBS 0.1% Triton X-100, whole retinas were flat-mounted in mounting medium (Fluoromount-G, ThermoFisher Scientific, 00-4959-52). Whole-mount retinas were imaged with epifluorescence microscopy (Nikon Ti2 Eclipse). Four representative pictures per retina were taken for the intact conditions, and eight representative pictures per retina were taken for the 14dpc conditions. For RGC survival experiments, RBPMS-positive and pS6-positive RGC were manually quantified in regions of interest of each representative picture using the cell counter module in Fiji. Numbers were averaged per individual retina.
Tissue cryosectioning
Eye balls were incubated in 15% sucrose in PBS for 48h at 4°C, then embedded in tissue freezing medium (MM-France, F/TFM-C). DRG and sciatic nerves were incubated in 30% sucrose in PBS for 48h at 4°C, then embedded in tissue freezing medium (MM-France, F/TFM-C). Blocks were frozen at -80°C. 12μm longitudinal sections for sciatic nerves and 14μm sagittal sections for eyes were obtained with a cryostat device (Thermo Scientific CryoStar NX50 Cryostat) and mounted on SuperFrost Plus adhesion slides (Thermo Scientific, J1800AMNZ). All sections on slides were kept frozen at -20°C until use.
Immunohistochemistry
Sections on slides were left to defrost at room temperature before immunostaining. After several washes in PBS, sections were incubated in blocking solution (PBS 0.1% Triton X-100 5% donkey serum) for 1h at room temperature. For DRG and sciatic nerve tissues, sections were incubated in blocking solution (PBS 0.5% Triton X-100 5% bovine serum albumin (BSA)) for 1h at room temperature. Sections were incubated in primary antibody diluted in blocking solution overnight at 4°C. Primary antibodies used were: mouse anti-HTT (Sigma-Aldrich, MAB2166) 1:100; guinea pig anti-RBPMS (Millipore, ABN1376) 1:300; rabbit anti-pS6 (Cell Signaling Technology, 4857) 1:400; rabbit anti-Tox2 (Sigma-Aldrich, HPA058396) 1:100; mouse anti-puromycin (Sigma-Aldrich, MABE343) 1:250; mouse anti-Tubulin β III (Biolegend, 801213) 1:500; rabbit anti-SCG10 (Novus Biologicals, NBP1-49461) 1:1000. After several washes in PBS, sections were incubated in secondary antibody diluted in blocking solution for 2h at room temperature. Secondary antibodies used were Alexa Fluor-conjugated secondary antibodies (Jackson ImmunoResearch) 1:500. After several washes in PBS, sections were mounted in mounting medium with DAPI (Fluoromount-G, ThermoFisher Scientific, 00-4959-52). Retina sections and sciatic nerve sections were imaged with epifluorescence microscopy (Nikon Ti2 Eclipse) or with confocal microscopy (spinning disk Dragonfly from Andor). For eye sections, images were randomly taken along individual retina sections, so as to represent all eyes and all sections on the slide of interest. For fluorescence intensity experiments, the mean intensity was determined using Fiji in manually defined regions of interest corresponding to individual RGC on representative images. For sciatic nerve sections, all sections of an individual nerve were imaged and used for quantification, unless the section was altered. For fluorescence intensity quantification, the mean intensity was determined using Fiji in 100μm-wide regions of interest at defined steps from the injury site. For calculation of the regeneration index (RI50), an exponential regression was performed on the intensity data normalized to that of the crush site (distance 0). The RI50 index was calculated as the value of the distance at 50% of intensity.
OPP labeling
OPP was labeled on sections with Click-iT chemistry (Click-iT Plus OPP Alexa Fluor 647 Protein Synthesis Assay Kit, ThermoFisher Scientific, C10458). After several washes in PBS, sections were permeabilized in PBS 0.1% Triton X-100 for 15min at room temperature. Sections were incubated for 30min at room temperature in the reaction mix (containing Alexa Fluor dye picolyl azide, prepared according to manufacturer’s instructions), then incubated in rinsing buffer for 5min. After several washes in PBS, sections were immunostained, imaged and quantified as described above.
In situ hybridization
In situ hybridization was performed as previously described.86 Templates of antisense in situ hybridization probes for HTT and Tox2 were cloned in a pGEMT easy vector (Promega) and synthesized using digoxigenin (DIG) RNA labelling Kit (Roche) after linearization of the plasmid. The sequence for HTT probe corresponds to nucleotides 346-1614 of mouse HTT mRNA (accession number NM_010414). The sequence for Tox2 probe corresponds to nucleotides 94-853 of mouse Tox2 mRNA (accession number NM_001098799.2). After hybridization of the DIG-labelled probe on slides, the alkaline phosphatase-conjugated anti-digoxigenin antibody (Roche, 11093274910) was incubated overnight at room temperature. Alkaline phosphatase staining was probed with NBT-BCIP (Roche, 11681451001). Slides were washed and post-fixed after reaching the appropriate coloration intensity. For fluorescent in situ hybridization, signal detection of alkaline phosphatase was performed using Fast Red and naphthol (Sigma-Aldrich, F4648).
Proximity ligation assay
This assay is used to detect the proximity between target antigens with oligonucleotide-coupled secondary antibodies and signal amplification. To perform the proximity ligation assay, the Duolink PLA kit (Sigma-Aldrich, DUO92013) was used according to the manufacturer’s instructions. Sections on slides were first left to defrost at room temperature. After several washes in PBS, sections were permeabilized in PBS 0.5% Triton for 15min. Sections were blocked in Duolink blocking buffer (Duolink, Sigma-Aldrich, DUO82007) for 1h at 37°C. Sections were incubated overnight at 4°C with mouse anti-HTT (clone 4c8, Sigma-Aldrich, MAB2166) 1:100 and rabbit anti-RPS6 (Cell Signaling Technology, 2217) 1:250; or with rabbit anti-HTT (clone d7F7, Cell Signaling Technology, 5656) 1:100 and mouse anti-rRNA (clone Y10b, Santa Cruz Biotechnology, sc-33678) 1:100. After two 5min washes with wash buffer A (Duolink, Sigma-Aldrich, DUO82046), sections were incubated for 1h at 37°C with complementary minus probe-conjugated anti-mouse antibody and plus probe-conjugated anti-rabbit antibody (Duolink, Sigma-Aldrich, DUO92004 and DUO92002). After two 5min buffer A washes, sections were processed for ligation with ligase diluted 1:40 in ligation buffer for 30min at 37°C. After two 5min buffer A washes, sections were processed for amplification with polymerase diluted 1:80 in amplification buffer for 100min at 37°C. Sections were washed twice 10min with 1x wash buffer B, then 1min with 0.01x wash buffer B. Sections were post-fixed in 4% formaldehyde for 15min at room temperature, then processed for immunohistochemistry.
Adult retina explant cultures and live imaging
For live imaging of BDNF-carrying vesicles, ex vivo cultures of adult retina explants were used as described.26 4 week-old WT or HTT phospho-mutant mice (S421 mutation site, HTT S421A/A or HTT S421D/D) were injected intravitreally with AAV2-cRheb, then with AAV2-BDNF-tdTomato one day after. Two weeks after injection, mice were sacrificed by cervical dislocation and retinas were dissected in ice-cold HBSS. Explants of 0.5mm in diameter were prepared and plated on glass-bottom dishes (MatTek, P35G-1.0-20-C) previously coated with poly-L-lysine (Sigma-Aldrich, P1399), laminin (Sigma-Aldrich, L2020) and coating medium (4μg/ml methylcellulose (Sigma-Aldrich, M0512) in Hibernate A (BrainBits, HA)). Explants were cultured in Neurobasal-A (Thermo Fisher Scientific, 10888022) supplemented with 1X B-27 (Thermo Fisher Scientific, 12587-010), 1% L-Glutamine (Thermo Fisher Scientific, 25-005-CI) and 1% penicillin-streptomycin (ThermoFisher Scientific, 15140122) for two weeks before imaging. Imaging was performed with confocal microscopy (Andor spinning disk). BDNF vesicles were imaged in individual axons, at a rate of 1 image per second for 2min.
Kymograph analysis
Kymograph analysis was performed using the KymoToolBox plugin in Fiji.87 Regions of interest were designed as segmented lines of 60-200μm along the axon, oriented towards the tip of the axon. Vescile trajectories were manually drawn from the kymogaph and analyzed with the Analyse Kymo tool. For each vesicle, the following parameters were calculated: Anterograde velocity = VMa (μm/s) = Anterograde distance (μm) / Time (s), Retrograde velocity = VMr (μm/s) = Retrograde distance (μm) / Time (s), Net directionality = Q (μm/s) = VMr × Relative number of retrograde vesicles + VMa × Relative number of anterograde vesicles.
STRING interactome analysis
The list of 747 binding partners of HTT in the mouse brain was retrieved from Shirasaki et al.28 The list was subjected to Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) network analysis and representation.29 Each node represents one protein. Network edges represent confidence with minimum interaction score of 0.9 (highest confidence). Disconnected nodes in the network are hidden from the representation. Network was clustered following a Markov cluster algorithm with inflation parameter of 3.
Culture of mouse embryonic fibroblasts (MEF)
Primary cultures of MEF were prepared from E13.5 embryos as described.88 E13.5 embryos were collected in PBS. Heart, liver and brain were dissected away and the remaining embryonic tissues were transferred to a culture dish containing 0.25% Trypsin-EDTA (Thermo Fisher Scientific). Tissue was cut in small pieces using a razor blade, then passed through a 10ml pipette several times to ensure dissociation. Tissue was incubated for 15min at 37°C and passed once more through a 5ml pipette.
Cell culture
MEF were maintained at 37°C 5% CO2 in 10cm dishes in cell culture medium: Dulbecco’s Modified Eagle Medium (DMEM, 31966047), high glucose, GlutaMAX Supplement, Thermo Fisher Scientific, 10% decomplemented fetal bovine serum (FBS, Eurobio Scientific, CVFSVF00-01, 56°C for 30min), 1% penicillin-streptomycin (ThermoFisher Scientific, 15140122), 1% L-Glutamine (Thermo Fisher Scientific, 25-005-CI). When reaching confluency, cells were detached by washing 1x in PBS then by incubating in 2ml Trypsin-EDTA (Thermo Fisher Scientific, 25-050-CI) at 37°C for 5-10min. Cells were collected by adding fresh cell culture medium and pelleted by centrifugating at 180rcf for 5min. Cells were resuspended in 1ml cell culture medium and replated 1:3 or 1:4 in fresh medium.
BCA protein assay
Protein concentration was quantified using a BCA protein assay (Pierce BCA Protein Assay Kit, Thermo Fisher Scientific, 23225) according to the manufacturer’s instructions. The standard curve was obtained by diluting bovine serum albumin (BSA) to concentrations ranging from 2mg/ml to 25μg/ml. 5μl of diluted sample or diluted BSA was mixed with 200μl of BCA working reagent (ratio Reagent A to Reagent B = 50:1) in a 96-well plate. Each sample or point of standard curve was quantified in duplicate. The reaction was incubated at 37°C for 30min. 562nm absorbance was measured with a plate reader (PHERAstar FS) and protein concentration was inferred from the standard curve.
Preparation of cell lysate
All steps were performed on ice or at 4°C unless otherwise stated. MEF were washed 1x in PBS then scraped in PBS and pelleted at 1,200rcf for 5min. Cells were lysed in 1ml RIPA buffer (10mM Tris-HCl pH7.5, TritonX-100 1%, 150mM NaCl, 1mM EDTA, 2.5mM sodium deoxycholate) supplemented with protease inhibitors (cOmplete EDTA-free Protease Inhibitor Cocktail, Roche, 04693159001). Proteins were extracted by 15min incubation on ice, followed by 15min centrifugation at 14,000rcf at 4°C. The supernatant was transferred to a new tube. Protein concentration was quantified with a BCA protein assay (Pierce BCA Protein Assay Kit, Thermo Fisher Scientific, 23225). Samples were mixed with Laemmli 6x and heated for 4min at 95°C.
Western blot
Polyacrylamide gels were precast (4-15%, Bio-Rad, #4568083) or handcast (6%, 10% or 12%). For handcast gels, the separating gel was obtained by polymerisation of acrylamide (Sigma, A3699) mixed with ammonium persulfate (APS) (Euromedex, EU0009-A) and TEMED (Euromedex, 50406-A) in SDS-containing buffer (375mM Tris-HCl, pH8.8, 0.1% SDS). The separating gel was overlaid with a 5% stacking gel. Handcast gels were casted in Bio-Rad polyacrylamide gel casting system. For Western blot analysis, 5-20μg protein were loaded in individual wells. Electrophoresis was run in SDS-containing running buffer (Tris-Glycine SDS, Euromedex, EU0510) for 10min at 90V, constant voltage, then for 45min-1h at 150V-220V, constant voltage, depending on polyacrylamide percentage. In at least one well, 10μl Precision Plus Protein Kaleidoscope Prestained Protein Standards (Bio-Rad, 1610375) were loaded as a molecular weight marker. Proteins separated by electrophoresis were transferred onto nitrocellulose membrane (ThermoFisher Scientific, 88018) in transfer buffer (Tris-Glycine, Euromedex, EU0550) containing 20% ethanol. Transfer was performed for 2hours 30min at 320mA, constant amperage. Membranes were stained with Ponceau to ensure correct transfer and even loading of proteins, then blocked for 1hour in Tris-buffered saline (TBS, Euromedex, ET220-B) 0.05% Tween-20 (Sigma, P9416) (TBS-T) 5% semi-skimmed powder milk. Membranes were incubated in primary antibody diluted in TBS-T 5 % milk overnight at 4°C with gentle agitation. Primary antibodies used were: mouse anti-HTT (Sigma-Aldrich, MAB2166) 1:500; mouse anti-puromycin (Millipore, MABE343) 1:1000; mouse anti-actin (Sigma-Aldrich, A1978) 1:5000; mouse anti-RPL22 (Santa Cruz Biotechnology, sc-373993) 1:1000; rabbit anti-RPL3 (Abcam, ab228638) 1:1000, mouse anti-Hsp60 (Santa Cruz Biotechnology, sc-376240) 1:1000; rabbit anti-Pabp1 (Cell Signaling Technology, 4992) 1:1000; rabbit anti-eIF4G (Cell Signaling Technology, 2498) 1:1000; rabbit anti-RPS6 (Cell Signaling Technology, 2217) 1:1000; rabbit anti-Gapdh (Cell Signaling Technology, 2118) 1:1000; rabbit anti-Histone 3 (His3) (Cell Signaling Technology, 9715) 1:1000. Membranes were washed 4 times 5min in TBS-T, then incubated in secondary antibody diluted in TBS-T 5% milk for 2hours at room temperature. Secondary antibodies used were: horseradish peroxidase (HRP)-conjugated anti-mouse (ThermoFisher Scientific, A16011), HRP-conjugated anti-rabbit (Proteintech, SA00001-2). Membranes were washed four times 5min in TBS-T, then probed for enhanced chemiluminescence (ECL) in 100mM Tris-HCl, pH8.5, 0.5% luminol, 0.5% coumaric acid and 0.01% H2O2. Protein detection was performed using ChemiDoc (ChemiDoc MP Imaging System).
Immunoprecipitation
All steps were performed at 4°C or on ice. WT MEF from 4 confluent 10cm dishes were washed once in PBS then harvested by scraping in PBS and pelleted at 1,200rcf for 5min. Cells were lysed in 1ml lysis buffer (20mM Tris-HCl, pH 7.5, 50mM NaCl, 2mM EDTA, 1% Triton) supplemented with protease inhibitors (cOmplete EDTA-free Protease Inhibitor Cocktail, Roche, 04693159001). Proteins were extracted by 20min incubation at 4°C with end-over-end agitation, followed by 20min centrifugation at 14,000rcf. The supernatant was transferred to a new tube and 50μl cell lysate was spared for analysis. 950μl cell lysate was pre-cleared for 1h with 95μl protein G magnetic beads (SureBeads, Bio-Rad, 1614023) pre-washed 3x in lysis buffer, with end-over-end agitation. Beads were separated on a magnetic rack and discarded. 50μl pre-cleared cell lysate was spared for analysis of input fraction. Half volume (450μl) of pre-cleared lysate was incubated with 5μl of mouse anti-HTT antibody (clone 4c8, Sigma-Aldrich, MAB2166) and the other half (450μl) with 5μl of mouse IgG (ThermoFisher Scientific, 31903) for 2h with end-over-end agitation. 45μl pre-washed protein G magnetic beads were added to each tube and incubated for 1h with end-over-end agitation. Beads were separated on a magnetic rack and washed 3x in lysis buffer. Beads were resuspended in 45μl Laemmli 1x, then vortexed for a few seconds and heated for 4min at 95°C. The immunoprecipitate was collected after separating beads on a magnetic rack and analysed by silver staining or Western blot.
Silver staining
Silver staining was performed according to the manufacturer’s instructions (SilverQuest Silver Staining Kit, ThermoFisher Scientific, 45-1001). Briefly, after electrophoresis, the gel was fixed in fixative solution (40% ethanol, 10% acetic acid) for 20 min. After 30% ethanol wash, the gel was incubated for 10 min in the sensitizing solution. After washes, the gel was incubated for 15min in the staining solution, followed by 4 to 8min incubation in the developing solution. When reaching the appropriate staining intensity, the reaction was stopped and the gel was imaged using ChemiDoc.
Cell transduction
MEF were transduced with a lentiviral vector encoding the Cre recombinase (LV-Cre-GFP, Addgene #86805) or a control lentiviral vector (H2B-eGFP, Addgene #25999). After 1 day in culture, medium was replaced. Cells were kept for 7 days after transduction before harvesting. Deletion of HTT was monitored using Western blot analysis.
Ribosome purification
Ribosome purification was performed as previously described.30 All steps were performed at 4°C or on ice, unless otherwise specified. WT MEF from 1 to 2 confluent 15cm dishes were washed in 1x PBS then scraped in PBS and pelleted at 500rcf for 5min. Cells were resuspended in 1ml buffer A (250mM sucrose, 250mM KCl, 5mM MgCl2, 50mM Tris-HCl, pH 7.4). 50μl cell suspension was spared and mixed 1:1 with Laemmli 2x (4% SDS, 60mM TrisHCl pH 6.8, 200mM DL-Dithiothreitol) to obtain the total fraction. NP-40 was added to the cell suspension to obtain a final concentration of 0.7% (v/v). Cells were lysed by 15min incubation on ice with pipetting every 5min. Cell lysate was centrifuged at 750rcf for 10min. The pellet (nuclear fraction) was resuspended in Laemmli 1x to obtain the nuclear fraction. The supernatant (cytoplasmic fraction) was centrifuged at 12,500rcf for 10min. The pellet (mitochondrial fraction) was resuspended in Laemmli 1x to obtain the mitochondrial fraction. 50μl of the supernatant was spared and mixed 1:1 with Laemmli 2x to obtain the post-mitochondrial fraction. The rest of the supernatant was carefully layed on a 1ml sucrose cushion (1M sucrose, 500mM KCl, 5mM MgCl2, 50mM Tris-HCl, pH 7.4) in a 3.5ml polycarbonate tube (Beckman Coulter). Tubes were balanced with buffer B (250mM sucrose, 500mM KCl, 5mM MgCl2, 50mM Tris-HCl, pH 7.4), then ultracentrifuged at 250,000rcf for 2h. 50μl of the top layer of the supernatant was collected and mixed with Laemmli 2x to obtain the post-ribosomal fraction. The pellet (ribosomal fraction) was resuspended in Laemmli 1x to obtain the ribosomal fraction. All fractions were quantified with a BCA protein assay (Pierce BCA Protein Assay Kit, Thermo Fisher Scientific, 23225). For Western blot analysis, 10-20μg proteins of each fraction were loaded, except the post-ribosomal fraction for which 30μl were loaded. The markers used for characterization of the different fractions are: Histone 3 (His3, nuclear), Hsp60 (mitochondrial), Gapdh (cytoplasmic), RPL22 or RPS6 (ribosomal).
Puromycin incorporation assay (SUnSET assay)
MEF were incubated with 25μg/ml puromycin (Sigma-Aldrich, P8833) for 15min at 37°C. MEF were washed once with fresh medium, then washed and scraped in ice-cold PBS for subsequent protein extraction and Western blot analysis.
Polysome gradient
MEF were incubated with 25μg/ml Emetine (Sigma-Aldrich, E2375) for 20min at 37°C. Cells were washed with PBS then incubated with 0.25% Trypsin-EDTA and collected by centrifugation. The cell pellet was washed twice with ice-cold PBS and centrifuged at 500rcf for 5min at 4°C. Cells were resuspended in 3x cell pellet volumes of buffer A30 (250mM sucrose, 250mM KCl, 5mM MgCl2, 50mM Tris-HCl, pH7.4) containing protease inhibitors (cOmplete EDTA-free Protease Inhibitor Cocktail, Roche). For cell lysis, NP-40 was added to a final concentration of 1% and the cell suspension was homogenized on ice for 10min, with multiple pipetting. The cell lysate was centrifuged at 750rcf for 10min at 4°C to isolate the nuclei, then the supernatant was centrifuged at 12,500rcf for 10min at 4°C to isolate the mitochondria. The supernatant was transferred to a new tube and protein concentration was quantified using a BCA protein assay. Protein samples (1-3mg) were loaded on a 15-50% sucrose gradient in 2.5mM KCl, 0.5mM MgCl2, 5mM Tris-HCl, pH 7.4. Gradients were prepared in 13.2ml ultracentrifuge tubes (Beckman Coulter, 344059) using a Gradient Master (BioComp Instruments). After balancing the tubes, gradients were ultracentrifuged in SW41-Ti swinging buckets at 250,000rcf for 1h45 at 4°C with slow brake speed. 254nm absorbance was read with a fractionator (Piston Gradient Fractionator PGFip, BioComp Instruments) and 700μl successive fractions were collected using the FoxR1 fraction collector (Teledyne ISCO). For RNA extraction, fractions were immediately frozen on dry ice and kept at -80°C until further processing. For protein extraction, 175μl trichloroacetic acid (Sigma-Aldrich, T0699) was added to each fraction and proteins were left to precipitate overnight at -20°C.
Cell treatment
For analysis of HTT distribution in polysomes, MEF were first incubated with 25μg/ml puromycin (Sigma-Aldrich, P8833) for 30min at 37°C, or with 2μg/ml harringtonin (Santa Cruz Biotechnology, sc-204771) for 5min at 37°C. For RNase I treatment, MEF lysate was obtained from WT MEF and incubated with 1μl RNase I (100U/μl) (Invitrogen, AM2294) for 10min on ice, then with 2μl SuperaseIn RNase inhibitor (20U/μl) (Invitrogen, AM2694) for 10min on ice. For EDTA treatment, MEF lysate was obtained from WT MEF and incubated with 30mM EDTA for 20min on ice.
Protein extraction from polysomal fractions
Fractions were centrifuged at 20,000rcf for 20min at 4°C. Each supernatant was carefully removed and discarded, and each pellet was washed with 500μl ice-cold acetone then centrifuged again at 20,000rcf for 5min at 4°C. The acetone washing step was repeated twice. After the third wash, the residual acetone was removed by heating the samples at 95°C for 5min. Each pellet was rsuspended in 50μl Laemmli 2x, then vortexed for a few seconds and heated for 5min at 95°C. For Western blot analysis, the same volume of each fraction was loaded on a polyacrylamide gel.
RNA extraction and RT-qPCR from polysomal fractions
Polysomal fractions were defrosted on ice and half of each fraction was used for RNA extraction. 100pg of Firefly luciferase RNA was added in each fraction for spike-in normalization. Fractions of interest were pooled according to their distribution (fractions 4 and 5 corresponding to monosomes, fractions 6 to 9 corresponding to light polysomes and fractions 10 to 14 corresponding to heavy polysomes). RNA was precipitated overnight at -20°C using 11.25μg GlycoBlue coprecipitant (ThermoFisher Scientific, AM9515), 30μl of 7.5M LiCl (ThermoFisher Scientific, 10498254) and 1ml of 100% ethanol per 350μl fraction volume. RNA was pelleted by centrifugation at 16,000rcf for 30min at 4°C. The supernatant was decanted and the pellet was resuspended in 350μl buffer RLT (RNeasy Micro kit, Qiagen, 74004) containing β-mercaptoethanol according to manufacturer’s instructions. RNA was subsequently purified with the RNeasy Micro kit for RNA isolation (Qiagen), with an in-column step of DNase I treatment. RNA was finally eluted in 14μl H2O per column and RNA concentration was determined using Bioanalyzer analysis on a PicoChip (Agilent, 5067-1513). Reverse transcription was performed using SuperScript II kit (Invitrogen, 18064022) with at least 2ng starting amount of RNA per fraction in a final reaction volume of 40μl. qPCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, 1725271). The following primers were used: Tox2 forward 5’-ATGGTGACAGTGCCTACGTG-3’, Tox2 reverse 5’-AGGGATGGCTCAGGGAGATT-3’, Gapdh forward 5’-GCATGGCCTTCCGTGTTC-3’, Gapdh reverse 5’-TGTCATCATACTTGGCAGGTTTCT-3’, Firefly luciferase forward 5’-GAGGCGAACTGTGTGTGAGA-3’, Firefly luciferase reverse 5’-GTGTTCGTCTTCGTCCCAGT-3’. The experiment was performed three times.
MEF ribosome-associated RNA extraction
For transcriptome and translatome analysis, HTTfl/fl MEF were transduced with Cre-expressing lentivirus or control lentivirus for 7 days. Cells were processed for ribosome purification. A fraction (1/15) of the cell suspension was kept for total RNA analysis (transcriptome), pelleted and resuspended in 1ml TRIzol Reagent (Invitrogen, 15596-026). After ribosome purification, the ribosome pellet was washed twice with ice-cold H2O, resuspended in 1ml TRIzol Reagent and homogenized very well by vortexing. Total RNA and ribosome-associated RNA was extracted with Trizol/chloroform extraction and isopropanol precipitation as per the manufacturer’s instructions. After washing once with 75% ethanol and drying, the RNA pellet was resuspended in 50μl H2O. 8μl RNA was treated with DNase I (Sigma-Aldrich, AMPD1) as per the manufacturer’s instructions. The final RNA concentration was determined using the Nanodrop (ThermoFisher Scientific, Nanodrop One).
Library preparation
Libraries were prepared from n=3 independent replicates for total RNA and n=3 independent replicates for translated RNA. Quality control was performed by Genewiz using RNA Screentape on Agilent TapeStation. All samples had a RNA quality number (RQN) > 9. Library preparation and RNA-sequencing were performed by Genewiz. Libraries (stranded, reverse) were prepared with NEBNext Ultra II Directional RNA Library preparation kit with 250ng RNA as starting amount. RNA-sequencing analysis was performed on an Illumina NovaSeq 6000 platform, with paired-end reading (2x150bp). Adapters were trimmed by Genewiz before data processing. FastQC control was performed by Genewiz. Mean quality score was > 35 for all samples. The percentage of bases with a quality score ≥ 30 was > 92% for all samples. Read depth was > 19M reads for all samples.
RNA-seq data analysis
Fastq files were processed for alignment and read count. The mouse genome (Mus_musculus.GRCm38.dna.toplevel.fa) was indexed using STAR (version 2.7.2b), using 149bp overhangs and gene annotation (Mus_musculus.GRCm38.97.chr.gtf). Reads were aligned to the genome using STAR. The percentage of uniquely mapped reads was > 88% for all samples. Reads were counted with HTSeq-count (version 0.11.1). Differential gene expression analysis was performed separately for the transcriptome and for the translatome using edgeR.89 Read counts were first normalized using RUVSeq90 with the RUVr method based on the residuals from the GLM fit after upper quartile normalization (number of residuals k=2). RNA-sequencing data are deposited in the NCBI Gene Expression Omnibus (GEO) database under accession code GSE209946.
Retina dissociation for cell sorting
Ptenfl/fl or Ptenfl/flHTTfl/fl were previously injected bilaterally with AAV2-Cre. Four weeks later, both optic nerves were crushed. 3 days after injury (3dpc) mice were sacrificed by cervical dislocation. All steps were performed on ice, unless otherwise stated. Eyes were enucleated and retinas were dissected in ice-cold HBSS and transferred to a new tube containing 1ml HBSS (35 retinas per tube). The tissue was incubated in 500μl of dissociation buffer (HBSS containing 1mg/ml papain (Sigma-Aldrich), 1mM L-cystein, 0.5mM EDTA, DNase I 1:500) at 37°C for 10min. Digestion was deactivated using 750μl trypsing inhibitor solution (1mg/ml in HBSS containing DNase I 1:500). The tissue was collected by 150rcf centrifugation for 1min30s. The tissue was resuspended in 200μl of RGC solution (L-glutamin 1x in Hibernate A, A12475-01) and cells were mechanically dissociated on ice with gentle pipetting through a 200μl pipette tip. Staining was performed by adding 200μl staining solution (PE-Cyanine 5 anti-mouse CD90.2 (Thy1.2, eBioscience) to a final concentration of 1:400, and DAPI to a final concentration of 10μg/ml). Cells were stained 20min on ice in the dark. Cells were washed twice with centrifugation at 150rcf for 5min and resuspension in 500μl RGC solution. Final volume was adjusted to 1ml with RGC solution and samples were processed for cell sorting.
RGC cell sorting and RNA extraction
Fluorescence-activated cell sorting (FACS) was performed on the FACS and microscopy facility of Institute of Advanced Biology (IAB), Grenoble, France. Cells are isolated by flow cytometry on a Aria II (BD Biosciences) cell sorter through a 100μm nozzle at 20psi. Dissociated retinal cells are gated based on scattering characteristics (FSC/SSC), doublet exclusion with width and height parameters and live/dead discrimination. DAPI and PE-Cy5 fluorescence were excited respectively with 405nm and 561nm lasers and collected through 405/50nm and 670/30nm band pass filters. WT unstained retinal cells are used as a control for autofluorescence assessment. PE-Cy5-Thy1-positive DAPI-negative population was retained for the sorting. 100,000 PE-Cy5-positive DAPI-negative cells were collected per individual replicate. Cells were collected in a tube containing 400μl of lysis buffer of PicoPure RNA extraction kit (KIT0204). The lysate was incubated 30min at 42°C, then processed for RNA extraction according to the manufacturer’s instructions, with a DNase I in-column digestion step. Finally, RNA was eluted in 25μl water.
Ribosome immunoprecipitation and RNA extraction
3-week-old Ptenfl/fl or Ptenfl/flHTTfl/fl were injected bilaterally with AAV2-RPL22-flag. 1 week later, mice were injected bilaterally with AAV2-Cre. Four weeks later, optic nerve crush was performed bilaterally. 3 days after injury, mice were sacrificed by cervical dislocation. All subsequent steps were performed on ice or at 4°C, unless otherwise stated. Eyes were enucleated and retinas were dissected in ice-cold PBS and transferred to a new tube containing 200μl lysis buffer (10mM Tris-HCl pH 7.5, 150mM NaCl, 0.5mM EDTA, 1% Triton X-100) supplemented with protease inhibitors (cOmplete EDTA-free Protease Inhibitor Cocktail, Roche, 04693159001). For each biological replicate, 4-5 retinas were pooled. The tissue was mechanically lysed using stainless steel grinding beads and a tissue homogenizer (Retsch, Mixer Mill MM400, 30 pulsations per second for 30sec). The lysate was transferred to a new tube and centrifuged at 750rcf for 10min. The supernatant was transferred to a new tube and centrifuged at 12,500rcf for 10min. The supernatant was transferred to a new tube and protein concentration was determined using a BCA protein assay (Pierce, 23225). 1mg protein was prepared in 400μl lysis buffer and 600μl wash buffer (10mM Tris-HCl pH 7.5, 150mM NaCl, 0.5mM EDTA). Protein lysates were pre-cleared with 100μl protein G beads (Bio-Rad, 1614023) (previously washed with wash buffer) for 2hours at 4°C with end-over-end agitation. Beads were separated on a magnetic rack and discarded. Half volume (500μl) of pre-cleared lysate was incubated with 5μl of mouse anti-flag antibody (1mg/ml, Sigma-Aldrich, F1804) and the other half (500μl) with 5μl of 1mg/ml mouse IgG (ThermoFisher Scientific, 31903) overnight with end-over-end agitation. 45μl pre-washed protein G magnetic beads were added to each tube and incubated for 2h with end-over-end agitation. Beads were separated on a magnetic rack and washed 4x in lysis buffer. Beads were resuspended in 350μl RLT buffer (Qiagen RNeasy kit, 74004) containing β-mercaptoethanol, then vortexed for a few seconds. Ribosome-associated RNA was purified according to manufacturer’s instructions, with a DNase I in-column digestion step. Finally, the RNA was eluted in 14μl water.
RT-qPCR of RGC transcriptome and translatome
RNA concentration and integrity were determined using Bioanalyzer analysis on a PicoChip (Agilent, 5067-1513). Reverse transcription was performed using SuperScript II kit (Invitrogen, 18064022) with equal starting amounts of RNA (4ng) in a final reaction volume of 40μl. qPCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, 1725271). The following primers were used: Tox2 forward 5’-ATGGTGACAGTGCCTACGTG-3’, Tox2 reverse 5’-AGGGATGGCTCAGGGAGATT-3’, Gpr158 forward 5’-AACACAGCCTAGATCCAGAAGAC-3’, Gpr158 reverse 5’-GGGTTGTTTGTGATCATCTTTTTA-3’, Lpcat forward 5’-GTGCACGAGCTGCGACT-3’, Lpcat reverse 5’-GCTGCTCTGGCTCCTTATCA-3’, Pex19 forward 5’-CAGCAGCACAGCGTCATGGTCA-3’, Pex19 reverse 5’-GTTGAGGCCAGGAGGCATCT-3’, Rnf112 forward 5’-CACTGCAGGAGACCTGTGC-3’, Rnf112 reverse 5’-CCCAGGACAGCCAATAAGCA-3’, Surf4 forward 5’-CTGTTGGCCTCATCCTTCGT-3’, Surf4 reverse 5’-GGCAATTGTCTGCAGTGCG-3’, Tpt1 forward 5’-GGAGCTGCAGAGCAGATTAAG-3’, Tpt1 reverse 5’-GTCCAGGAGAGCAACCATACC-3’, Tubb3 forward 5’-GGTGGACTTGGAACCTGGAA-3’, Tubb3 reverse 5’-TAAAGTTGTCGGGCCTGAAT-3’, Trarg1 forward 5’-GGTCCTTGCCATTGCCTCTT-3’, Trarg1 reverse 5’-TGCTGCACACTACTTCGAGAC-3’, Wnt2b forward 5’-AACGAGGGGACTTTGACTGG-3’, Wnt2b reverse 5’-CCACTCACACCGTGACACTT-3’, Gapdh forward 5’-GCATGGCCTTCCGTGTTC-3’, Gapdh reverse 5’-TGTCATCATACTTGGCAGGTTTCT-3’, Sdha forward 5’-TGTTCAGTTCCACCCCACA-3’, Sdha reverse 5’-TCTCCACGACACCCTTCTG-3’, 18S forward 5’-GCAATTATTCCCCATGAACG-3’, 18S reverse 5’-GGCCTCACTAAACCATCCAA-3’.
Puro-PLA
For puromycin-PLA, the Duolink PLA kit (Sigma-Aldrich, DUO92013) was used according to manufacturer’s instructions and as described above. Sections of mouse retinas previously injected with OPP were first left to defrost at room temperature. Sections were blocked with Duolink Blocking Solution at 37°C for 1h. Primary antibodies used were mouse anti-puromycin (Sigma-Aldrich, MABE343) 1:250, and rabbit anti-Tox2 (Sigma-Aldrich, HPA058396) 1:100 or rabbit anti-Gapdh (Cell Signaling Technology, 2118) 1:250 diluted in Duolink Antibody Diluent and left overnight at 4°C. Secondary antibodies used were minus probe-conjugated anti-mouse (Sigma-Aldrich, DUO92004) and plus probe-conjugated anti-rabbit (Sigma-Aldrich, DUO92002). After PLA reaction, sections were post-fixed for 15min in 4% formaldehyde at room temperature, then processed for immunohistochemistry. Retina sections were imaged with confocal microscopy (Dragonfly spinning disk from Andor). For PLA experiments, the maximum projection of a z-stack of the retina in the region of interest corresponding to the RGC layer was used in each representative image. Regions of interest corresponding to individual RBPMS-positive cells were randomly defined using Fiji. The number of PLA-positive spots was automatically quantified in these individual cells using Fiji.
Quantification and statistical analysis
All quantitative data are expressed as mean +/- standard error of the mean, except polysome data represented as a boxplot with min/max values and line at mean. Unless otherwise specified, in all quantitative data, n represents the number of individual events and N the number of independent mouse eyes. All datasets were subjected to the Shapiro-Wilk normality test before statistical comparison. For quantification of OPP flurorescence intensity on sections, datasets were subjected to a log-transform for representation and statistical analysis. When applicable, two or more conditions were compared with unpaired t-test or one-way ANOVA with Bonferroni multiple comparisons test, respectively. To compare directionality of BDNF vesicle transport, a one-sample Wilcoxon signed-rank test was applied, with comparison of median to theoretical value of 0. To compare mRNA levels in mono- and polysomal fractions, a paired t-test was applied. To compare the number of puro-PLA-positive events, a Kruskal-Wallis with Dunn’s multiple comparisons test was applied.
Acknowledgments
The authors would like to thank Yohann Couté, Anne-Marie Hesse, Jan Warnking, Emmanuel Barbier, Deniz Dalkara, Mylène Pezet, and Moein Sarhadi for technical assistance, critical reading, and resource sharing. This work was supported by French National Research Agency (Agence Nationale de la Recherche [ANR]) grants to H.N. (C7H-ANR16C49 and ANR-21-CE16-0008-01) and S.B. (ANR-18-CE16-0007 and ANR-21-CE16-0008-02) and a European Research Council grant to H.N. (ERC-St17-759089). This work was supported by ANR under “Investissements d’avenir” programme (ANR-17-EURE-0003). H.N. is supported by NRJ Foundation. S.B. was supported by Fondation pour la Recherche Médicale (FRM) (ARF20150934123). N.V. is supported by FRM (FDT202204014716). J.S. is supported by UNADEV/AVISAN (2017 call for proposals “Maladies de la vision, origine et traitement” to F.S. and H.N.) and by FRM (SPF201909009106). This work was supported by the Photonic Imaging Center of Grenoble Institute Neuroscience (Univ Grenoble Alpes – Inserm U1216), which is part of the IsdV core facility and certified by the IbiSA label.
Author contributions
H.N. and S.B. designed the project, with input from F.S. and S.H. J.S., N.V., H.N., and S.B. wrote the manuscript, with input from all co-authors. J.S. was involved in all experiments and analyses. N.V. performed histology experiments and imaging. J.S. and E.P. performed mouse surgeries and perfusions. C.D. performed mouse surgeries and histology for sciatic nerve injury procedures. B.B., N.E., C.D., B.E., and A.P. performed molecular cloning of plasmid constructs, viral vector production, and biochemical validation. F.C. and J.-J.D. provided training and equipment for polysome profiling experiments. S.H. and F.S. provided HTT mutant mouse lines and tools for molecular biology.
Declaration of interests
The authors declare no competing interests.
Published: July 12, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.neuron.2023.06.005.
Contributor Information
Homaira Nawabi, Email: homaira.nawabi@inserm.fr.
Stephane Belin, Email: stephane.belin@inserm.fr.
Supplemental information
The HTT binding partners were identified by Shirasaki et al.28
Data and code availability
RNA-sequencing data are deposited in the NCBI Gene Expression Omnibus (GEO) database under accession code GSE209946 and are publicly available as of the date of publication. Data that support the findings of this study are either provided as supplemental information or are available from the lead contact upon request.
References
- 1.Filbin M.T. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat. Rev. Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- 2.Silver J., Miller J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 3.He Z., Jin Y. Intrinsic control of axon regeneration. Neuron. 2016;90:437–451. doi: 10.1016/j.neuron.2016.04.022. [DOI] [PubMed] [Google Scholar]
- 4.Park K.K., Liu K., Hu Y., Smith P.D., Wang C., Cai B., Xu B., Connolly L., Kramvis I., Sahin M., et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore D.L., Blackmore M.G., Hu Y., Kaestner K.H., Bixby J.L., Lemmon V.P., Goldberg J.L. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326:298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith P.D., Sun F., Park K.K., Cai B., Wang C., Kuwako K., Martinez-Carrasco I., Connolly L., He Z. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron. 2009;64:617–623. doi: 10.1016/j.neuron.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belin S., Nawabi H., Wang C., Tang S., Latremoliere A., Warren P., Schorle H., Uncu C., Woolf C.J., He Z., et al. Injury-induced decline of intrinsic regenerative ability revealed by quantitative proteomics. Neuron. 2015;86:1000–1014. doi: 10.1016/j.neuron.2015.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobi A., Tran N.M., Yan W., Benhar I., Tian F., Schaffer R., He Z., Sanes J.R. Overlapping transcriptional programs promote survival and axonal regeneration of injured retinal ganglion cells. Neuron. 2022;110:2625–2645.e7. doi: 10.1016/j.neuron.2022.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tian F., Cheng Y., Zhou S., Wang Q., Monavarfeshani A., Gao K., Jiang W., Kawaguchi R., Wang Q., Tang M., et al. Core transcription programs controlling injury-induced neurodegeneration of retinal ganglion cells. Neuron. 2022;110:2607–2624.e8. doi: 10.1016/j.neuron.2022.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun F., Park K.K., Belin S., Wang D., Lu T., Chen G., Zhang K., Yeung C., Feng G., Yankner B.A., et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011;480:372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poplawski G.H.D., Kawaguchi R., Van Niekerk E., Lu P., Mehta N., Canete P., Lie R., Dragatsis I., Meves J.M., Zheng B., et al. Injured adult neurons regress to an embryonic transcriptional growth state. Nature. 2020;581:77–82. doi: 10.1038/s41586-020-2200-5. [DOI] [PubMed] [Google Scholar]
- 12.Saudou F., Humbert S. The biology of huntingtin. Neuron. 2016;89:910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Ben M’Barek K.B., Pla P., Orvoen S., Benstaali C., Godin J.D., Gardier A.M., Saudou F., David D.J., Humbert S. Huntingtin mediates anxiety/depression-related behaviors and hippocampal neurogenesis. J. Neurosci. 2013;33:8608–8620. doi: 10.1523/JNEUROSCI.5110-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liot G., Zala D., Pla P., Mottet G., Piel M., Saudou F. Mutant huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J. Neurosci. 2013;33:6298–6309. doi: 10.1523/JNEUROSCI.2033-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawai H., Clarke D.B., Kittlerova P., Bray G.M., Aguayo A.J. Brain-derived neurotrophic factor and Neurotrophin-4/5 stimulate growth of axonal branches from regenerating retinal ganglion cells. J. Neurosci. 1996;16:3887–3894. doi: 10.1523/JNEUROSCI.16-12-03887.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue S., Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 2012;13:355–369. doi: 10.1038/nrm3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maier T., Güell M., Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–3973. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 18.Marques Sousa C., Humbert S. Huntingtin: here, there, everywhere! J. Huntingtons Dis. 2013;2:395–403. doi: 10.3233/JHD-130082. [DOI] [PubMed] [Google Scholar]
- 19.Dragatsis I., Levine M.S., Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez A.R., de Sevilla Müller L.P., Brecha N.C. The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. J. Comp. Neurol. 2014;522:1411–1443. doi: 10.1002/cne.23521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarbassov D.D., Ali S.M., Sabatini D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Colin E., Zala D., Liot G., Rangone H., Borrell-Pagès M., Li X.-J., Saudou F., Humbert S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008;27:2124–2134. doi: 10.1038/emboj.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gauthier L.R., Charrin B.C., Borrell-Pagès M., Dompierre J.P., Rangone H., Cordelières F.P., De Mey J., MacDonald M.E., Lessmann V., Humbert S., et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 24.Thion M.S., McGuire J.R., Sousa C.M., Fuhrmann L., Fitamant J., Leboucher S., Vacher S., du Montcel S.T., Bièche I., Bernet A., et al. Unraveling the role of huntingtin in breast cancer metastasis. J. Natl. Cancer Inst. 2015;107 doi: 10.1093/jnci/djv208. [DOI] [PubMed] [Google Scholar]
- 25.Lim J.H., Stafford B.K., Nguyen P.L., Lien B.V., Wang C., Zukor K., He Z., Huberman A.D. Neural activity promotes long distance, target-specific regeneration of adult retinal axons. Nat. Neurosci. 2016;19:1073–1084. doi: 10.1038/nn.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaeffer J., Delpech C., Albert F., Belin S., Nawabi H. Adult Mouse retina Explants: from ex vivo to in vivo Model of central nervous system Injuries. Front. Mol. Neurosci. 2020;13 doi: 10.3389/fnmol.2020.599948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anne S.L., Saudou F., Humbert S. Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J. Neurosci. 2007;27:7318–7328. doi: 10.1523/JNEUROSCI.1831-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shirasaki D.I., Greiner E.R., Al-Ramahi I., Gray M., Boontheung P., Geschwind D.H., Botas J., Coppola G., Horvath S., Loo J.A., et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 2012;75:41–57. doi: 10.1016/j.neuron.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belin S., Hacot S., Daudignon L., Therizols G., Pourpe S., Mertani H.C., Rosa-Calatrava M., Diaz J.J. Purification of ribosomes from human cell lines. Curr. Protoc. Cell Biol. 2010;Chapter 3 doi: 10.1002/0471143030.cb0340s49. Unit 3.40. [DOI] [PubMed] [Google Scholar]
- 31.Sonenberg N., Hinnebusch A.G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerashchenko M.V., Gladyshev V.N. Ribonuclease selection for ribosome profiling. Nucleic Acids Res. 2017;45:e6. doi: 10.1093/nar/gkw822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Artegiani B., de Jesus Domingues A.M., Bragado Alonso S., Brandl E., Massalini S., Dahl A., Calegari F. Tox: a multifunctional transcription factor and novel regulator of mammalian corticogenesis. EMBO J. 2015;34:896–910. doi: 10.15252/embj.201490061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paul A., Crow M., Raudales R., He M., Gillis J., Huang Z.J. Transcriptional architecture of synaptic communication delineates GABAergic neuron identity. Cell. 2017;171:522–539.e20. doi: 10.1016/j.cell.2017.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gandin V., Sikström K., Alain T., Morita M., McLaughlan S., Larsson O., Topisirovic I. Polysome fractionation and analysis of mammalian translatomes on a genome-wide scale. J. Vis. Exp. 2014 doi: 10.3791/51455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warner J.R., Knopf P.M., Rich A. A multiple ribosomal structure in protein synthesis. Proc. Natl. Acad. Sci. USA. 1963;49:122–129. doi: 10.1073/pnas.49.1.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahar M., Cavalli V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018;19:323–337. doi: 10.1038/s41583-018-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu G.Y., Sabatini D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020;21:183–203. doi: 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Riggelen J., Yetil A., Felsher D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer. 2010;10:301–309. doi: 10.1038/nrc2819. [DOI] [PubMed] [Google Scholar]
- 40.Sap K.A., Guler A.T., Bury A., Dekkers D., Demmers J.A.A., Reits E.A. Identification of full-length wild-type and mutant huntingtin interacting proteins by crosslinking immunoprecipitation in mice brain cortex. J. Huntingtons. Dis. 2021;10:335–347. doi: 10.3233/JHD-210476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Culver B.P., Savas J.N., Park S.K., Choi J.H., Zheng S., Zeitlin S.O., Yates J.R., Tanese N. Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J. Biol. Chem. 2012;287:21599–21614. doi: 10.1074/jbc.M112.359307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Creus-Muncunill J., Badillos-Rodríguez R., Garcia-Forn M., Masana M., Garcia-Díaz Barriga G., Guisado-Corcoll A., Alberch J., Malagelada C., Delgado-García J.M., Gruart A., et al. Increased translation as a novel pathogenic mechanism in Huntington’s disease. Brain. 2019;142:3158–3175. doi: 10.1093/brain/awz230. [DOI] [PubMed] [Google Scholar]
- 43.Eshraghi M., Karunadharma P.P., Blin J., Shahani N., Ricci E.P., Michel A., Urban N.T., Galli N., Sharma M., Ramírez-Jarquín U.N., et al. Mutant huntingtin stalls ribosomes and represses protein synthesis in a cellular model of Huntington disease. Nat. Commun. 2021;12 doi: 10.1038/s41467-021-21637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simsek D., Tiu G.C., Flynn R.A., Byeon G.W., Leppek K., Xu A.F., Chang H.Y., Barna M. The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell. 2017;169:1051–1065.e18. doi: 10.1016/j.cell.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y., Beyer A., Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell. 2016;165:535–550. doi: 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 46.Komili S., Silver P.A. Coupling and coordination in gene expression processes: a systems biology view. Nat. Rev. Genet. 2008;9:38–48. doi: 10.1038/nrg2223. [DOI] [PubMed] [Google Scholar]
- 47.Vogel C., Marcotte E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujii K., Zhulyn O., Byeon G.W., Genuth N.R., Kerr C.H., Walsh E.M., Barna M. Controlling tissue patterning by translational regulation of signaling transcripts through the core translation factor eIF3c. Dev. Cell. 2021;56:2928–2937.e9. doi: 10.1016/j.devcel.2021.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Darnell J.C., Van Driesche S.J., Zhang C., Hung K.Y.S., Mele A., Fraser C.E., Stone E.F., Chen C., Fak J.J., Chi S.W., et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim H.D., Kong E., Kim Y., Chang J.-S., Kim J. RACK1 depletion in the ribosome induces selective translation for non-canonical autophagy. Cell Death Dis. 2017;8 doi: 10.1038/cddis.2017.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Genuth N.R., Barna M. The discovery of ribosome heterogeneity and its implications for gene regulation and organismal life. Mol. Cell. 2018;71:364–374. doi: 10.1016/j.molcel.2018.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo H. Specialized ribosomes and the control of translation. Biochem. Soc. Trans. 2018;46:855–869. doi: 10.1042/BST20160426. [DOI] [PubMed] [Google Scholar]
- 53.Shi Z., Barna M. Translating the genome in time and space: specialized ribosomes, RNA regulons, and RNA-binding proteins. Annu. Rev. Cell Dev. Biol. 2015;31:31–54. doi: 10.1146/annurev-cellbio-100814-125346. [DOI] [PubMed] [Google Scholar]
- 54.Gay D.M., Lund A.H., Jansson M.D. Translational control through ribosome heterogeneity and functional specialization. Trends Biochem. Sci. 2021;47:66–81. doi: 10.1016/j.tibs.2021.07.001. [DOI] [PubMed] [Google Scholar]
- 55.Lee L.J., Papadopoli D., Jewer M., del Rincon S., Topisirovic I., Lawrence M.G., Postovit L.-M. Cancer plasticity: the role of mRNA translation. Trends Cancer. 2021;7:134–145. doi: 10.1016/j.trecan.2020.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de la Parra C., Walters B.A., Geter P., Schneider R.J. Translation initiation factors and their relevance in cancer. Curr. Opin. Genet. Dev. 2018;48:82–88. doi: 10.1016/j.gde.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robichaud N., Sonenberg N., Ruggero D., Schneider R.J. Translational control in cancer. Cold Spring Harb. Perspect. Biol. 2019;11 doi: 10.1101/cshperspect.a032896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ho J.J.D., Cunningham T.A., Manara P., Coughlin C.A., Arumov A., Roberts E.R., Osteen A., Kumar P., Bilbao D., Krieger J.R., et al. Proteomics reveal cap-dependent translation inhibitors remodel the translation machinery and translatome. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.109806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schaeffer J., Belin S. Axonal protein synthesis in central nervous system regeneration: is building an axon a local matter? Neural Regen. Res. 2022;17:987–988. doi: 10.4103/1673-5374.324835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jung H., Yoon B.C., Holt C.E. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat. Rev. Neurosci. 2012;13:308–324. doi: 10.1038/nrn3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verma P., Chierzi S., Codd A.M., Campbell D.S., Meyer R.L., Holt C.E., Fawcett J.W. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J. Neurosci. 2005;25:331–342. doi: 10.1523/JNEUROSCI.3073-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Donnelly C.J., Willis D.E., Xu M., Tep C., Jiang C., Yoo S., Schanen N.C., Kirn-Safran C.B., van Minnen J., English A., et al. Limited availability of ZBP1 restricts axonal mRNA localization and nerve regeneration capacity. EMBO J. 2011;30:4665–4677. doi: 10.1038/emboj.2011.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sahoo P.K., Lee S.J., Jaiswal P.B., Alber S., Kar A.N., Miller-Randolph S., Taylor E.E., Smith T., Singh B., Ho T.S.-Y., et al. Axonal G3BP1 stress granule protein limits axonal mRNA translation and nerve regeneration. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-05647-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koley S., Rozenbaum M., Fainzilber M., Terenzio M. Translating regeneration: local protein synthesis in the neuronal injury response. Neurosci. Res. 2019;139:26–36. doi: 10.1016/j.neures.2018.10.003. [DOI] [PubMed] [Google Scholar]
- 65.Younts T.J., Monday H.R., Dudok B., Klein M.E., Jordan B.A., Katona I., Castillo P.E. Presynaptic protein synthesis is required for long-term plasticity of GABA release. Neuron. 2016;92:479–492. doi: 10.1016/j.neuron.2016.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hafner A.S., Donlin-Asp P.G., Leitch B., Herzog E., Schuman E.M. Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science. 2019;364 doi: 10.1126/science.aau3644. [DOI] [PubMed] [Google Scholar]
- 67.Chen H.-H., Tarn W.-Y. uORF-mediated translational control: recently elucidated mechanisms and implications in cancer. RNA Biol. 2019;16:1327–1338. doi: 10.1080/15476286.2019.1632634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xue S., Barna M. Cis-regulatory RNA elements that regulate specialized ribosome activity. RNA Biol. 2015;12:1083–1087. doi: 10.1080/15476286.2015.1085149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cockman E., Anderson P., Ivanov P. TOP mRNPs: molecular mechanisms and principles of regulation. Biomolecules. 2020;10 doi: 10.3390/biom10070969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oliveto S., Mancino M., Manfrini N., Biffo S. Role of microRNAs in translation regulation and cancer. World J. Biol. Chem. 2017;8:45–56. doi: 10.4331/wjbc.v8.i1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nanbru C., Lafon I., Audigier S., Gensac M.C., Vagner S., Huez G., Prats A.C. Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. J. Biol. Chem. 1997;272:32061–32066. doi: 10.1074/jbc.272.51.32061. [DOI] [PubMed] [Google Scholar]
- 72.Stoneley M., Paulin F.E., Le Quesne J.P., Chappell S.A., Willis A.E. C-Myc 5′ untranslated region contains an internal ribosome entry segment. Oncogene. 1998;16:423–428. doi: 10.1038/sj.onc.1201763. [DOI] [PubMed] [Google Scholar]
- 73.Ray P.S., Grover R., Das S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006;7:404–410. doi: 10.1038/sj.embor.7400623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Di Giovanni S., Knights C.D., Rao M., Yakovlev A., Beers J., Catania J., Avantaggiati M.L., Faden A.I. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006;25:4084–4096. doi: 10.1038/sj.emboj.7601292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kondrashov N., Pusic A., Stumpf C.R., Shimizu K., Hsieh A.C., Ishijima J., Shiroishi T., Barna M. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell. 2011;145:383–397. doi: 10.1016/j.cell.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xue S., Tian S., Fujii K., Kladwang W., Das R., Barna M. RNA regulons in Hox 5′UTRs confer ribosome specificity to gene regulation. Nature. 2015;517:33–38. doi: 10.1038/nature14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alghoul F., Laure S., Eriani G., Martin F. Translation inhibitory elements from Hoxa3 and Hoxa11 mRNAs use uORFs for translation inhibition. eLife. 2021;10 doi: 10.7554/eLife.66369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou J., Wan J., Shu X.E., Mao Y., Liu X.-M., Yuan X., Zhang X., Hess M.E., Brüning J.C., Qian S.-B. N6-methyladenosine guides mRNA alternative translation during integrated stress response. Mol. Cell. 2018;69:636–647.e7. doi: 10.1016/j.molcel.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Filipowicz W., Bhattacharyya S.N., Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 80.Li G.-W., Burkhardt D., Gross C., Weissman J.S. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Biever A., Glock C., Tushev G., Ciirdaeva E., Dalmay T., Langer J.D., Schuman E.M. Monosomes actively translate synaptic mRNAs in neuronal processes. Science. 2020;367 doi: 10.1126/science.aay4991. [DOI] [PubMed] [Google Scholar]
- 82.de Lima S., Koriyama Y., Kurimoto T., Oliveira J.T., Yin Y., Li Y., Gilbert H.-Y., Fagiolini M., Martinez A.M.B., Benowitz L. Full-length axon regeneration in the adult mouse optic nerve and partial recovery of simple visual behaviors. Proc. Natl. Acad. Sci. USA. 2012;109:9149–9154. doi: 10.1073/pnas.1119449109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Groszer M., Erickson R., Scripture-Adams D.D., Lesche R., Trumpp A., Zack J.A., Kornblum H.I., Liu X., Wu H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- 84.Parikh P., Hao Y., Hosseinkhani M., Patil S.B., Huntley G.W., Tessier-Lavigne M., Zou H. Regeneration of axons in injured spinal cord by activation of bone morphogenetic protein/Smad1 signaling pathway in adult neurons. Proc. Natl. Acad. Sci. USA. 2011;108:E99–E107. doi: 10.1073/pnas.1100426108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dodt H.-U., Leischner U., Schierloh A., Jährling N., Mauch C.P., Deininger K., Deussing J.M., Eder M., Zieglgänsberger W., Becker K. Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain. Nat. Methods. 2007;4:331–336. doi: 10.1038/nmeth1036. [DOI] [PubMed] [Google Scholar]
- 86.Moret F., Renaudot C., Bozon M., Castellani V. Semaphorin and neuropilin co-expression in motoneurons sets axon sensitivity to environmental semaphorin sources during motor axon pathfinding. Development. 2007;134:4491–4501. doi: 10.1242/dev.011452. [DOI] [PubMed] [Google Scholar]
- 87.Zala D., Hinckelmann M.-V., Yu H., Lyra da Cunha M.M., Liot G., Cordelières F.P., Marco S., Saudou F. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 2013;152:479–491. doi: 10.1016/j.cell.2012.12.029. [DOI] [PubMed] [Google Scholar]
- 88.Durkin M.E., Qian X., Popescu N.C., Lowy D.R. Isolation of mouse embryo fibroblasts. Bio Protoc. 2013;3 doi: 10.21769/bioprotoc.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Risso D., Ngai J., Speed T.P., Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014;32:896–902. doi: 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The HTT binding partners were identified by Shirasaki et al.28
Data Availability Statement
RNA-sequencing data are deposited in the NCBI Gene Expression Omnibus (GEO) database under accession code GSE209946 and are publicly available as of the date of publication. Data that support the findings of this study are either provided as supplemental information or are available from the lead contact upon request.