

Summary
Cell differentiation is achieved by acquiring a cell type-specific transcriptional program and epigenetic landscape. While the cell type-specific patterning of enhancers has been shown to precede cell fate decisions, it remains unclear how regulators of these enhancers are induced to initiate cell specification and how they appropriately restrict cells that differentiate. Here, using embryonic stem cell–derived hematopoietic cell differentiation cultures, we show the activation of some hematopoietic enhancers during arterialization of hemogenic endothelium, a prerequisite for hematopoiesis. We further reveal that ZEB2, a factor involved in the transcriptional regulation of arterial endothelial cells, and a hematopoietic regulator MEIS1 are independently required for activating these enhancers. Concomitantly, ZEB2 or MEIS1 deficiency impaired hematopoietic cell development. These results suggest that multiple regulators expressed from an earlier developmental stage non-redundantly contribute to the establishment of hematopoietic enhancer landscape, thereby restricting cell differentiation despite the unrestricted expression of these regulators to hematopoietic cells.
Subject areas: Natural sciences, Biological sciences, Developmental biology
Graphical abstract
Highlights
-
•
Hematopoietic enhancers are activated during hemogenic endothelium arterialization
-
•
ZEB2 regulates transcriptional program of arterial hemogenic endothelium
-
•
ZEB2 concomitantly activates hematopoietic enhancers
-
•
ZEB2 and MEIS1 are independently required for hematopoietic enhancer activation
Natural sciences; Biological sciences; Developmental biology
Introduction
The cell type-specific enhancer landscape facilitates the induction of transcriptional programs required for cell differentiation.1,2,3 The accumulation of transcription factors at the enhancers modulates chromatin looping and generates a chromatin architecture that enables efficient and stable gene transcription.1 Transcription factors expressed this way form transcription networks that make up the cellular identity.4 Thus, to determine how cell differentiation is initiated, it is necessary to understand how the cell type-specific enhancer landscape is established.
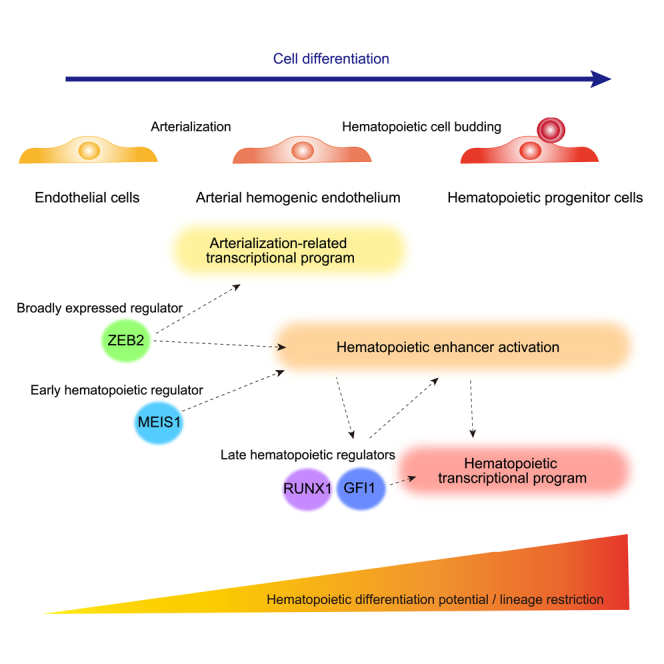
Initiators of cell type-specific enhancer activation can be specifically induced by environmental cues that drive cell differentiation or already exist from an earlier developmental stage. An example of the former is MyoD, which alone induces a substantial part of the muscle cell-specific transcriptional and epigenetic patterns.5 However, it has been difficult to find regulators with such pioneering activity for many other cell types. The latter, in contrast, depends on regulators that are not specific to the target cell type. How non-specific regulators contribute to lineage specification while tightly controlling the cells that can differentiate into the target cells is poorly understood. Stem cell-specific transcription factors have been shown to bind to and epigenetically regulate differentiated cell type-specific enhancers.6 Thus, cell type-specific enhancers may be regulated by nonspecific regulators throughout development long before lineage specification occurs. While such regulation could be extended to the initiation of enhancer activation that drives lineage specification, a mechanism that restricts cell differentiation to the target cells is also required. Given that cell differentiation requires the precise spatiotemporal presentation of multiple environmental cues, we hypothesized that multiple factors that are already expressed from an earlier developmental stage contribute to the initiation of enhancer activation required for the later developmental stage. Utilizing regulators from earlier developmental stages would allow the gradual accumulation of developmental potential, while the involvement of multiple factors could establish a multi-lock system to tightly restrict cell differentiation.
In definitive hematopoiesis, which generates all mature hematopoietic cell types, hematopoietic stem cells (HSCs) emerge from hemogenic endothelium (HE) of arterial tissue of the dorsal aorta.7 Since HSCs arise from arterial endothelium but not venous tissue, it has long been hypothesized that the arterialization of endothelial cells is a prerequisite for hematopoiesis.8,9 Although this idea had been controversial for decades due to the observations of arterialization-uncoupled hematopoiesis,10,11,12,13 recent studies using human pluripotent stem cell (PSC) differentiation cultures and single-cell transcriptomic technologies in human fetuses have revealed that the regulators required for hematopoiesis are up-regulated in arterial endothelial cells but not venous endothelial cells.14,15 These findings suggest that arterialization sets HE to a primed state for hematopoietic cell differentiation. However, the detailed molecular mechanisms underlying how arterialization contributes to hematopoiesis are unknown.
Here, we utilized a human embryonic stem cell (ESC)-derived two-dimensional hematopoietic cell differentiation system to assess the contribution of transcription factors at the earlier developmental stage to the establishment of the enhancer landscape required for the later development stage. We identified that ZEB2 participates in transcriptional regulation in arterial HE and concurrently contributes to the hematopoietic enhancer activation. While the involvement of broadly expressed ZEB2 raised the question of how cell differentiation is restricted, we showed that a hematopoietic regulator MEIS1 is additionally required for the hematopoietic enhancer activation and subsequent hematopoietic cell differentiation. Our results support a model in which cell differentiation is guided by the gradual accumulation of cell type-specific molecular features, with the non-redundant contribution of multiple regulators expressed from an earlier developmental stage.
Results
Hematopoietic molecular features gradually accumulate along development
In order to assess when and how hematopoietic transcriptional programs and the hematopoietic enhancer landscape are induced in the course of development, we utilized our human ESC-derived hematopoietic cell differentiation system.16,17 Using this system, we isolated ESCs on day 0, mesoderm on day 2, CD34+KDR+ endothelial cells on day 4 and 7, and CD34+CD45– endothelial cells and CD34+CD45+ hematopoietic progenitor cells (HPCs) on day 10 (Figure S1A). We confirmed that these HPCs could differentiate into multiple lineages (Figure S1B). Day 4 CD34+KDR+ cells, which mostly consisted of CD73–CD43– HE (Figure S1C), were isolated and re-plated to induce the endothelial-to-hematopoietic transition (EHT) by the addition of hematopoietic cytokines. On day 7, CD34+KDR+ cells contained a mixture of CD73–CD43– HE and CD73+CD43– non-HE committed to endothelial lineage, consistent with previous reports (Figure S1C).18,19,20 Similarly, day 10 CD34+CD45– cells included CD73– cells, which partially lost KDR but had not yet acquired CD43 expression, and CD73+KDR+ endothelial cells (Figure S1D). A global transcriptional analysis of these cells revealed the developmental trajectory in the principal component analysis (PCA) plot (Figure 1A). Of note, day 7 CD34+KDR+ cells and day 10 CD34+CD45– cells showed a gradual shift toward HPCs. Moreover, genes up-regulated in day 10 CD34+CD45– cells compared to day 7 CD34+KDR+ cells were enriched with lymphoid and myeloid cell differentiation gene sets (Figure 1B). These results validate that our ESC-derived hematopoietic cell differentiation system can be used to investigate the molecular events that trigger hematopoiesis.
Figure 1.

Activation of hematopoietic enhancers prior to induction of the hematopoietic transcriptional program
(A) PCA of the global gene expression during ESC-derived hematopoietic progenitor cell differentiation. RNA-seq of bulk cells from day 0 and 2, CD34+KDR+ cells from day 4 and 7, and CD34+CD45– cells and CD34+CD45+ cells from day 10 were analyzed.
(B) Association of differentially expressed genes with cell differentiation-related gene sets. Genes up-regulated along differentiation were subjected to a gene ontology analysis focused on cell differentiation and development.
(C) Heatmap showing the H3K27ac ChIP-seq signal along differentiation at hematopoietic enhancers (left), expression pattern of nearest genes (middle), and results of the gene ontology analysis (right). Hematopoietic enhancers were clustered into 3 clusters by the H3K27ac signal pattern along differentiation. The median is indicated by white dots in the violin plots (middle), and the top 5 results are shown for the gene ontology analysis.
(D) Examples of cluster 2 enhancers showing H3K27ac modification prior to transcription of nearby genes. The y axis shows the tag counts per 10 million mapped reads. Individual (A, C-left), average (B, C-middle, right), and representative (D) data from two independent experiments are shown.
Since the establishment of a cell type-specific enhancer landscape is the key determinant of cell lineage specification,2,3 we next examined when hematopoietic enhancers are activated. H3K27ac (histone H3 acetylated at Lys27) ChIP-seq was conducted to determine the active enhancer regions (Table S1). A developmental trajectory similar to that in the PCA of global transcription was observed with H3K27ac modification at hematopoietic enhancers (Figure S1E). To globally determine when hematopoietic enhancers are activated, we defined hematopoietic enhancers as active distal regulatory regions (excluding promoters) marked with H3K27ac modification in day 10 CD34+CD45+ cells (see STAR Methods for details). These enhancers were largely divided into three clusters depending on the timing of the H3K27ac signal acquisition in the course of hematopoietic cell development (Table S2): cluster 1 was activated almost exclusively in HPCs, cluster 2 was activated from day 7, and cluster 3 was active mainly in endothelial cells with a diminished signal in HPCs (Figures 1C, 1D, S1F, S1G, and S1H). Cluster 2 enhancers included the RUNX1 +23 enhancer, previously used as a marker of HE,21 and both CD34+KDR+ cells on day 7 and CD34+CD45– cells on day 10 showed the H3K27ac signal at this enhancer, confirming that these cells contained HE (Figure 1D). MEIS1 E2 enhancer22 was also included in cluster 2, while MEIS1 E3 enhancer at +140 kb22 was included in cluster 3, suggesting the gradual activation of multiple enhancers to ensure stable gene expression. While the timing of enhancer activation and that of nearest gene expression were generally correlated in clusters 1 and 3, enhancer activation preceded the expression of the nearest genes in cluster 2; despite the activation of enhancers from day 7, many of the nearest genes were up-regulated from the HPC stage (Figures 1C and S2A). Moreover, genes around cluster 1 enhancers were associated with immune responses, whereas those around cluster 2 enhancers were enriched with leukocyte-differentiation signatures. Collectively, these results suggest that enhancer activation guides the induction of key genes required for hematopoietic cell differentiation.
Regulators of arterial HE are potential candidates of early hematopoietic enhancer activation
The precedent enhancer activation at key genes involved in hematopoietic cell differentiation suggested that the initial events of hematopoietic cell differentiation can be elucidated by addressing how these enhancers are activated. We thus examined when these enhancers become accessible to transcription factors. It is known that enhancers can be closed (marked by H3K27me3), poised (marked by H3K27me3 and H3K4me1), primed (marked by H3K4me1), or active (marked by H3K27ac and H3K4me1).23 While cell type-specific enhancers were expected to be closed or poised in ESCs and become primed and activated along development, only a small fraction of hematopoietic enhancers were marked with H3K27me3 in ESCs, which was lost along differentiation (Figure S2B). Moreover, some of the enhancers were already marked with H3K4me1 but not H3K27me3 or H3K27ac in ESCs, indicating the primed state; their numbers increased on day 2 and 4. Similar trends were observed for all three clusters of hematopoietic enhancers. These results suggest that the majority of hematopoietic enhancers are accessible by transcription factors before hematopoietic lineage specification occurs.
Since most hematopoietic enhancers became accessible before their activation, we sought to identify the factors involved in hematopoietic enhancer activation from transcription factor binding motifs within the enhancer regions. Differential enrichment of transcription factor motifs was observed among the three clusters of hematopoietic enhancers (Figures 2A and S2C). Cluster 1 enhancers, which are exclusively active in HPCs, contained binding motifs of hematopoietic transcription factors including RUNX1, MYB, and BCL11A. In contrast, cluster 2 and 3 enhancers, which are activated from the vascular endothelial cell stage, showed enriched motifs of transcription factors expressed in vascular endothelial cells (Figure 2B). Notably, some motifs, such as those of ZEB2 and SPI1 in group 2, were enriched in both cluster 1 and 2 enhancers, with the corresponding regulators expressed from the endothelial cell stage to the stage of HPC differentiation. Given that cluster 2 enhancers were activated prior to HPC development and found near hematopoietic cell differentiation-related genes, the transcription factors involved in the activation of cluster 2 enhancers were considered to play crucial roles in HPC differentiation (Groups 2, 3, and 4 in Figure 2A). These factors included hematopoietic transcription factors, such as MEIS1 and SPI1, and those known to be involved in the arterialization of HE, such as SOX17. These observations led us to hypothesize that an earlier transcriptional wave induced by the arterialization is involved in the activation of enhancers required for hematopoietic cell differentiation.
Figure 2.

Presence of arterialization-related regulator motifs in hematopoietic enhancers
(A) Motif analysis of the hematopoietic enhancers defined in Figure 1D and the expression pattern of enriched transcription factors (TFs). Left heatmap shows the enrichment score calculated as a percentage of motifs in the target sequence divided by the percentage of motifs in the background sequence, and the middle heatmap illustrates the significance of the enrichment. Results were clustered into 5 groups, based on the degree of the enrichment at the 3 clusters of the hematopoietic enhancers. Examples of enriched TFs are shown for each group.
(B) Violin plots showing the expression pattern of the enriched TFs in groups 1, 3 and 5, which correspond to TFs with their motifs specifically enriched in hematopoietic enhancer clusters 1, 2 and 3, respectively.
Notch signaling during arterialization is required for hematopoietic enhancer activation
We next explored whether the arterialization of HE contributes to hematopoietic enhancer activation. We confirmed that the induction of arterial endothelial cell-specific transcription factors in endothelial cells and their repression in HPCs are recapitulated in our ESC-derived hematopoietic cell differentiation system, consistent with previous findings20 (Figure S3A). Since Notch signaling plays central roles in arterialization, we inhibited Notch signaling to test whether arterialization is required for hematopoietic enhancer activation. The addition of a Notch inhibitor, DAPT, from day 4 prevented the activation of cluster 2 and 3 hematopoietic enhancers (Figure 3A). In line with this observation, HPC emergence was significantly impaired on day 10 in the presence of DAPT (Figure 3B). In contrast, although the enrichment of SMAD2, SMAD4, and ZEB2 (also known as Smad Interacting Protein 1, SIP1) motifs at hematopoietic enhancers (Figure 2A) suggested the involvement of TGF-β signaling,24 the inhibition of TGF-β signaling by SB431542 showed no effects on hematopoietic enhancer activation on day 7 or HPC development on day 10 (Figures S3B and S3C).
Figure 3.

Requirement of Notch signaling and hematopoietic cytokines for hematopoietic enhancer activation
(A) Heatmap and boxplots showing the effects of Notch signaling inhibition by DAPT and of the absence of hematopoietic cytokines on the H3K27ac ChIP-seq signal at the hematopoietic enhancers defined in Figure 1C. DAPT was added from day 4 at a concentration of 10 μM, and hematopoietic cytokines (SCF, TPO, FLT3L and FP6) were removed the same day. ns, p > 0.05; ∗∗∗∗P ≦ 0.0001 (repeated-measures one-way ANOVA followed by Dunnett’s multiple comparisons test).
(B) Flow cytometry of control and DAPT-treated live cells on day 10 for identifying HPCs by the expression of CD34 and CD45. DAPT was added from days 4–10 at a concentration of 10 μM. Representative (left) and summary data (right) are shown. ∗P ≦ 0.05 (paired t-test).
(C) Percentage of hematopoietic enhancers occupied by Notch1 on day 7. Hematopoietic enhancers are divided into 3 clusters, as shown in Figure 1C.
(D) Fold-change of the indicated gene expressions in day 7 CD34+ cells by DAPT treatment. Genes were selected from the transcription factors whose binding motifs were enriched at cluster 2 enhancers (see Figure 2A). ns, FDR >0.05; ∗∗FDR ≦ 0.01; ∗∗∗∗FDR ≦ 0.0001 (calculated by DESeq2).
(E) Binding of the indicated transcription factors to hematopoietic enhancers on day 7. The ChIP-seq signal distribution at enhancer regions (5′ to 3′ ends) +/− 1 kB (top) and the proportions of hematopoietic enhancers bound by the transcription factors (bottom) are shown. Hematopoietic enhancers were grouped according to the timing of their activation, as shown in Figure 1C.
(F) Expression of the indicated genes during hematopoietic cell differentiation. Genes were selected from candidate regulators of cluster 1 and 2 hematopoietic enhancers, as shown in Figure 2A.
(G) Expression patterns of ZEB2 and MEIS1 during in vitro hematopoietic cell differentiation. The averages and standard deviations are shown.
(H) Occupancy of the genomic loci of the genes shown in Figure 3E by the five indicated regulators. The average of two independent experiments (A, C, D, G), representative and summary of four independent experiments (B), individual data of two or three independent experiments (F), and representative data from one or two experiments (E, H) are shown.
We further examined whether hematopoietic enhancer activation is restricted to HE. Since the addition of hematopoietic cytokines from day 4 models the development of HE, we cultured day 4 CD34+CD73– cells in hematopoietic expansion medium, but replaced hematopoietic cytokines with VEGF. H3K27ac ChIP-seq analyses of CD34+ cells on day 7 revealed that some hematopoietic enhancers, particularly cluster 2 enhancers, showed reduced or failed activation (Figure 3A). In addition, we tested whether hematopoietic enhancer activation is observed specifically in arterial-type HE by dividing CD34+ endothelial cells on day 7 in hematopoietic cell differentiation culture into four subpopulations based on the expression of CD73, DLL4, and CXCR4 (Figure S3D).20 Although cluster 2 hematopoietic enhancers showed marginally higher activity in CD73– HE than in CD73+ non-HE, they were mostly activated in all examined fractions (Figure S3E), despite the differential expression of arterial endothelial cell-specific transcription factors (Figure S3F). This observation is likely due to the similar degree of Notch signaling activation (Figure S3G) and excessive availability of hematopoietic cytokines in the culture system. These results altogether suggest that Notch signaling, which induces arterialization, and hematopoietic cytokines facilitate hematopoietic enhancer activation.
Given the involvement of Notch signaling in hematopoietic enhancer activation, we tested whether Notch1 directly binds hematopoietic enhancers. Notch1 ChIP-seq indicated that Notch1 rarely occupied hematopoietic enhancer regions on day 7, suggesting that transcription factors downstream of Notch signaling are instead responsible for the activation (Figure 3C). Among the regulators whose binding motifs were enriched at cluster 2 hematopoietic enhancers (Figure 2A), some, such as SPI1, MEIS1, SOX17, and SOX6, were down-regulated by Notch signaling inhibition (Figure 3D), making them candidate downstream factors for hematopoietic enhancer activation. Notch signaling-independent regulators may also be necessary for enhancer activation to restrict cell differentiation, considering that not all cells receiving Notch signaling acquire the hematopoietic enhancer landscape.
To identify hematopoietic enhancer regulators, we next examined whether the regulators whose binding motifs were enriched at cluster 2 enhancers could bind hematopoietic enhancers. ChIP-seq of ZEB2, SPI1, MEIS1, SOX6 and SOX17 revealed that ZEB2 and MEIS1 occupied hematopoietic enhancers most frequently (Figure 3E). In contrast, SOX6, SOX17, and SPI1 hardly overlapped with hematopoietic enhancers, despite the detection of peaks at other H3K27ac-marked enhancer regions (Figure S3H). Moreover, ZEB2 and MEIS1 were expressed from day 4 before the activation of hematopoietic enhancers (Figures 3F and 3G) and able to bind the genomic loci of most transcription factors that came up as candidate regulators of cluster 1 and 2 hematopoietic enhancers by a motif enrichment analysis (Figure 3H). These results suggest the potential roles of ZEB2 and MEIS1 in the initiation of hematopoietic enhancer activation.
HPC development is impaired by ZEB2 deletion
Our observations that ZEB2 binds hematopoietic enhancers prior to HPC development prompted us to determine whether ZEB2 is functionally involved in HPC development. We therefore deleted ZEB2 in ESCs using the CRISPR-Cas9 system (Figure S4A). We confirmed the loss of ZEB2 protein expression in two lines (Figure S4B) in which we induced hematopoietic cell differentiation. While both CD34+CD45+ HPCs and CD34–CD45+ mature hematopoietic cells were observed in wild-type (WT) cells on day 10, neither fractions were present in ZEB2-deficient lines (Figures 4A and 4B). The hematopoietic cells remained absent on day 14, indicating that ZEB2 deletion did not delay but impaired hematopoietic cell differentiation (Figure S4C). Moreover, defective HPC differentiation was similarly observed in ZEB2-deficient induced pluripotent stem cells (iPSCs; 201B2 background) (Figure S4D). The effects of ZEB2 deletion were cell-intrinsic, as co-culture with GFP-labelled WT CD34+ cells from day 4 did not rescue defective HPC differentiation in the ZEB2-deficient cells (Figure S4E). We also observed a significant reduction in EHT using time-lapse imaging (Figure S4F). The few floating cells that emerged even in the absence of ZEB2 were prone to cell death (Figure S4F). These results suggested that ZEB2 is required for HPC development.
Figure 4.

Complete impairment of HPC differentiation in the absence of ZEB2
(A) Flow cytometry of WT and two lines of ZEB2-deficient live cells on day 10 for identifying HPCs and mature hematopoietic cells by the expression of CD34 and CD45.
(B) Percentages (left) and numbers (right) of WT and ZEB2-deficient CD34+CD45+ and CD34–CD45+ cells on day 10. Individual results, mean and standard deviation are shown. ∗P ≦ 0.05; ∗∗P ≦ 0.01; ∗∗∗P ≦ 0.001; ∗∗∗∗P ≦ 0.0001 (two-way ANOVA followed by the Holm–Sidak multiple comparisons test).
(C) PCA of the global gene expression during WT and ZEB2-deficient HPC differentiation.
(D) Gene ontology analysis of genes down-regulated in ZEB2-deficient day 10 CD34+CD45– cells compared to WT counterparts. The top 10 gene sets are shown.
(E) The expression of RUNX1 splice variants in WT and ZEB2-deficient day 7 and 10 endothelial cells, shown as transcripts per million (TPM) of RNA-seq data. ns, p > 0.05; ∗P ≦ 0.05; ∗∗P ≦ 0.01 (one-way ANOVA followed by Tukey’s multiple comparisons test).
(F) Heatmap showing the H3K27ac ChIP-seq signal at the hematopoietic enhancers along the hematopoietic cell differentiation of WT and ZEB2-deficient ESCs. Hematopoietic enhancers are grouped by the timing of activation in WT, as shown in Figure 1C ns, p > 0.05; ∗∗P ≦ 0.01; ∗∗∗∗P ≦ 0.0001 (repeated-measures one-way ANOVA followed by Dunnett’s multiple comparisons test).
(G) Gene ontology analysis of genes nearby cluster 2 enhancers whose activity was reduced by ZEB2 deletion, as shown in Figure 4F. The top 10 gene sets are shown. Representative (A) and summary (B) data from five independent experiments, and individual (C, E) and average (F) data from two or three independent experiments are shown.
We next examined whether ZEB2 deletion caused dysregulation at the transcriptional level in the course of hematopoietic cell differentiation. Transcriptional dysregulation was most prominent in CD34+CD45– cells on day 10, as indicated by the PCA and the number of differentially expressed genes between WT and ZEB2-deficient cells (Figures 4C and S5A). Notably, the global gene expression pattern of ZEB2-deficient CD34+CD45– cells on day 10 was closer to day 7 WT CD34+CD45– cells rather than day 10 WT CD34+CD45– cells, suggesting that ZEB2 deletion impaired the acquisition of hematopoietic potential in endothelial cells (Figures 4C and S5B). Indeed, a gene ontology analysis of differentially expressed genes in day 10 CD34+CD45– cells revealed the down-regulation of immune response-related genes (Figure 4D). In addition, the expression of RUNX1c isoform, which has been reported to mark the emergence of definitive HPCs,25 was detectable from day 10 in WT CD34+CD45– cells, whereas ZEB2-deficient counterparts showed no expression of RUNX1c and also a reduced expression of other RUNX1 isoforms (Figure 4E).
To address whether the impaired hematopoiesis by ZEB2 deficiency is associated with the failure to activate hematopoietic enhancers, we conducted H3K27ac ChIP-seq of ZEB2-deficient cells. H3K27ac modification was reduced in more than half of the cluster 2 enhancers; the difference was modest on day 7 but became more apparent on day 10, when these enhancers became more activated in WT (Figure 4F). Moreover, whereas cluster 1 enhancers showed a slight increase in the H3K27ac signal in WT CD34+CD45– cells on day 10, this increase was not observed in the ZEB2-deficient counterparts (Figure 4F). In contrast, H3K4me1 modification, an indicator of primed enhancers, was comparable at hematopoietic enhancers between WT and ZEB2-deficient CD34+ cells on day 7 (Figure S5C). These findings suggest that ZEB2 deletion altered the composition of trans-regulatory elements that bind primed enhancers, thereby impairing hematopoietic enhancer activation.
HPC differentiation is reduced in the absence of MEIS1
To further elucidate how ZEB2 regulates hematopoietic enhancer activation, we divided cluster 2 enhancers into two sub-clusters by k-means clustering, which showed the differential effects of ZEB2 deletion, and examined differences in their characteristics (reduced and unaltered/increased, see cluster 2 in Figure 4F). Enhancers whose activity was reduced by ZEB2 deletion were found near genes associated with leukocyte differentiation, whereas those with unaltered or increased activity showed no enrichment of gene sets (Figure 4G). While both sub-clusters contained similar portions of enhancers bound by ZEB2 (Figure S5D), they differed in the enrichment of transcription factor binding motifs (Figure 5A). Of particular note was the preferential enrichment of MEIS1 motifs at the enhancers affected by ZEB2 deletion. MEIS1 was another factor expressed from before hematopoietic enhancer activation and showed binding to hematopoietic enhancers (Figures 3E and 3G). Moreover, the MEIS1 gene locus contained cluster 2 enhancers bound by ZEB2, and ZEB2 deletion reduced H3K27ac signal at these enhancers and MEIS1 gene expression level (Figures 5B and 5C). These observations collectively suggest that in addition to ZEB2-dependent hematopoietic enhancer activation, the subsequent up-regulation of factors such as MEIS1 further orchestrates generation of the hematopoietic enhancer landscape. Alternatively, ZEB2 and MEIS1 may independently contribute to the enhancer activation.
Figure 5.

Severe reduction of HPC differentiation in the absence of MEIS1
(A) The enrichment of transcription factor motifs at cluster 2 hematopoietic enhancers, whose activity was affected or unaffected by ZEB2 deletion (left). The top 15 differentially enriched transcription factors and ZEB2 are shown. The expression of the indicated transcription factors in ZEB2-deficient cells compared to WT counterparts on day 7 (middle) and occupation of their gene loci by ZEB2 in WT (right) are shown by the color code.
(B) H3K27ac modification, binding of ZEB2, and mRNA transcription at the MEIS1 gene locus in WT and ZEB2-deficient CD34+CD45– cells on day 7 and 10. Location of cluster 2 enhancer regions are also indicated.
(C) Expression pattern of MEIS1 during in vitro hematopoietic cell differentiation in WT and ZEB2-deficient lines. The averages and standard deviations are shown. ∗∗∗∗FDR ≦ 0.0001 (calculated by DESeq2).
(D) Flow cytometry of WT and two lines of MEIS1-deficient live cells on day 10 for identifying HPCs and mature hematopoietic cells by the expression of CD34 and CD45.
(E) Percentages (left) and numbers (right) of WT and MEIS1-deficient CD34+CD45+ and CD34–CD45+ cells on day 10. Individual results, mean and standard deviation are shown. Ns, p > 0.05; ∗∗∗∗P ≦ 0.0001 (two-way ANOVA followed by Holm–Sidak multiple comparisons test).
(F) PCA of the global gene expression during WT and MEIS1-deficient hematopoietic progenitor cell differentiation.
(G) The expression of RUNX1 splice variants in WT and MEIS1-deficient CD34+CD45– cells on day 7 and 10 endothelial cells, shown as transcripts per million (TPM) in the RNA-seq data. ns, p > 0.05; ∗P ≦ 0.05; ∗∗P ≦ 0.01 (one-way ANOVA followed by Tukey’s multiple comparisons test). Representative (B) and individual (F, G) data from two or three independent experiments, average of two independent experiments (C), and representative (D) and summary (E) data from four to five independent experiments are shown.
To clarify the relationship between ZEB2 and MEIS1, we generated MEIS1-deficient ESCs (Figures S5E and S5F). Upon hematopoietic cell differentiation, the number and percentage of CD34+CD45+ HPCs were significantly reduced in the absence of MEIS1, in concordance with a previous report26 (Figures 5D and 5E). Transcriptional analyses revealed that acquisition of the hematopoietic transcriptional program was impaired in MEIS1-deficient CD34+KDR+CD45– cells on day 10, similar to ZEB2-deficient cells (Figures 5F and S5G). Defective hematopoiesis was also indicated by the failed induction of RUNX1c isoform in MEIS1-deficient lines (Figure 5G). In contrast, the transcriptional profile of HPCs that developed in the absence of MEIS1 was similar to WT counterparts (Figure 5F). These results suggest that MEIS1 deletion substantially reduces the efficiency of hematopoietic cell differentiation while allowing a minor population to differentiate into HPCs.
Involvement of ZEB2 in transcriptional regulation of arterial HE
Since multipotent HPCs are known to emerge from arterial HE,20 we examined whether ZEB2 and MEIS1 deletion also affects their formation. The number of CD34+CD73–DLL4+CXCR4+ cells was comparable among the WT, ZEB2-deficient, and MEIS1-deficient lines on day 7 (Figures 6A and 6B). However, ZEB2-deficient arterial HE showed dysregulated transcriptional profiles, whereas MEIS1-deficient counterparts were similar to the WT (Figure 6C). Notably, transcriptional dysregulation was observed in several Notch signaling targets, including HES4 and DLL4 in the absence of ZEB2 (Figures 6D, S6A, and S6B). Mechanistically, ZEB2 deletion reduced the binding of Notch1 (Figures 6E and S6A).
Figure 6.

Transcriptional dysregulation by ZEB2 deletion in arterial HE
(A) Flow cytometry of WT and two lines of ZEB2-deficient and MEIS1-deficient CD34+CD73–CD43– cells on day 7 for identifying arterial HE by the expression of DLL4 and CXCR4.
(B) Number of WT, ZEB2-deficient, and MEIS1-deficient CD34+CD73–CD43–DLL4+CXCR4+ arterial HE cells. The individual results, means, and standard deviations are shown. ns, p > 0.05 (one-way ANOVA followed by Holm–Sidak multiple comparisons test).
(C) PCA of global gene expression in WT, ZEB2-deficient, MEIS1-deficient, and ZEB2/MEIS1 double-deficient CD34+CD73–CD43–DLL4+CXCR4+ arterial HE, and CD34+KDR+CD73+CD43–DLL4+CXCR4+ arterial non-HE on day 7.
(D) Heatmap and boxplots showing the expression patterns of genes associated with Notch signaling (GO:0007219) in WT, ZEB2-deficient, MEIS1-deficient, and ZEB2/MEIS1 double-deficient CD34+CD73–CD43–DLL4+CXCR4+ arterial He. ns, p > 0.05; ∗∗∗P ≦ 0.001; ∗∗∗∗P ≦ 0.0001 (repeated-measures one-way ANOVA followed by Dunnett’s multiple comparisons test).
(E) Average Notch1 ChIP-seq signal at the Notch1 binding sites in WT and ZEB2-deficient CD34+ cells on day 7. Notch1 binding sites were defined in WT CD34+ cells on day 7.
(F) Number of differentially expressed genes by ZEB2 deletion, MEIS1 deletion, or ZEB2/MEIS1 double deletion compared to WT on day 7. Representative (A) and summary (B) of four independent experiments and individual (C) or average (D, E, F) data from two independent experiments are shown.
As MEIS1 deletion increased ZEB2 expression level (Figure S6C), we generated ZEB2/MEIS1 double-deficient lines to assess their redundancy (Figures S6D and S6E). Double deficiency impaired HPC differentiation, similar to ZEB2 deletion (Figures S6F and S6G). Transcriptional profiles of double deficient cells also resembled those of ZEB2-deficient cells, although the dysregulation was more pronounced in the absence of two regulators, indicating partial redundancy (Figures 6C, 6D, and 6F). Taken together, these results suggest that ZEB2 is broadly involved in transcriptional regulation in arterial HE and during HPC differentiation, whereas MEIS1 functions more specifically in hematopoietic cell differentiation.
Independent contribution of ZEB2 and MEIS1 to hematopoietic enhancer activation
Next, we compared the contributions of ZEB2 and MEIS1 to hematopoietic enhancer activation. While cluster 1 enhancers showed reduced activity by both ZEB2 and MEIS1 deletion, divergent effects of their deletion were observed at the cluster 2 and 3 enhancers (Figures 7A and S7B). Thus, cluster 2 and 3 enhancers were further divided into four groups each (cluster 2a-2d and cluster 3a-3d, as shown in the right panel of Figure 7A) based on the effects of ZEB2 and MEIS1 deletion. Certain groups (cluster 2b-2d and cluster 3b-2d) showed reduced H3K27ac signals upon both ZEB2 and MEIS1 deletion on day 7 or 10. For example, enhancer activity was reduced at the RUNX1, GFI1B, SPI1, and MED4 loci in both ZEB2-deficient and MEIS1-deficient cells (Figures 7B and S7A). In contrast, there were also groups of enhancers whose activity was reduced by MEIS1 deletion, but rather increased by ZEB2 deletion (cluster 2a and cluster 3a). Notably, some cluster 2 enhancers (cluster 2b and 2c) showed down-regulated activity by MEIS1 deletion on day 7, but became active on day 10, while the activity of cluster 1 enhancers remained lower than WT (Figure 7A). These observations suggest that hematopoietic enhancer activation was delayed in the absence of MEIS1. Indeed, the hematopoietic enhancer profile of day 10 MEIS1-deficient CD34+CD45– cells showed a higher correlation with their WT counterparts on day 7 than on day 10 (Figure S7C). These results altogether indicate that both ZEB2 and MEIS1 are required to appropriately activate the majority of hematopoietic enhancers, while their differential function is also observed at some enhancers.
Figure 7.

Cooperative activation of hematopoietic enhancers by ZEB2 and MEIS1
(A) Heatmap and boxplots showing the z-score-normalized H3K27ac ChIP-seq signal at hematopoietic enhancers in WT, ZEB2-deficient, and MEIS1-deficient day 7 CD34+KDR+, day 10 CD34+CD45–, and day 10 CD34+CD45+ (HPC) cells. Hematopoietic enhancers were grouped by the timing of activation in WT, as shown in Figure 1C, and clusters 2 and 3 were further clustered by the effects of ZEB2 and MEIS1 deletion. ns, p > 0.05; ∗∗P ≦ 0.01; ∗∗∗∗P ≦ 0.0001 (repeated-measures one-way ANOVA followed by Dunnett’s multiple comparisons test).
(B) H3K27ac modification and binding of ZEB2 and MEIS1 at RUNX1 and SPI1 gene loci in WT, ZEB2-deficient, and MEIS1-deficient CD34+CD45– cells on day 7 and 10.
(C) The overlap of ZEB2 and MEIS1 genomic occupancy outside and inside hematopoietic enhancers.
(D) Heatmap showing the MEIS1 ChIP-seq signal at ZEB2 binding sites (5′ to 3′ ends) +/− 1 kB within hematopoietic enhancers in WT and ZEB2-deficient CD34+ cells on day 7.
(E) Expression patterns of genes down-regulated and up-regulated by ZEB2 deficiency in WT, ZEB2-deficient, MEIS1-deficient, MEIS1-induced ZEB2-deficient, and ZEB2-overexpressed ZEB2-deficient CD34+KDR+ cells on day 7. Black circles indicate medians. ns, p > 0.05; ∗P ≦ 0.05; ∗∗∗∗P ≦ 0.0001 (repeated-measures one-way ANOVA followed by Dunnett’s multiple comparisons test).
(F) Histograms showing the H3K27ac ChIP-seq signal at the hematopoietic enhancers defined in Figure 4F in WT, ZEB2-deficient, MEIS1-induced ZEB2-deficient, and ZEB2-overexpressed ZEB2-deficient CD34+KDR+ cells on day 7.
(G) Heatmap and violin plots showing the z-score-normalized H3K27ac ChIP-seq signal at the subfraction of cluster 2 hematopoietic enhancers, whose activity is reduced by ZEB2 deletion, in cells shown in E. Black circles indicate medians. ∗∗P ≦ 0.01; ∗∗∗∗P ≦ 0.0001 (repeated-measures one-way ANOVA followed by Dunnett’s multiple comparisons test).
(H) Flow cytometry of WT, ZEB2-deficient, and MEIS1-induced ZEB2-deficient, and ZEB2-overexpressed ZEB2-deficient live cells on day 10 for identifying HPCs and mature hematopoietic cells by the expression of CD34 and CD45. Both MEIS1 and ZEB2 were induced by the addition of doxycycline (dox) from day 4. Representative (top) and summary data (bottom) are shown. ns, p > 0.05; ∗∗∗∗P ≦ 0.0001 (two-way ANOVA followed by Sidak’s multiple comparisons test). Average (A, C, E, F, G) and representative (B, D) from two independent experiments, and representative and summary results of four independent experiments (H) are shown.
The partially overlapping effects of ZEB2 and MEIS1 deletion on hematopoietic enhancer activation and HPC differentiation prompted us to clarify whether MEIS1 acts as a downstream factor of ZEB2 or whether they are independently required. ZEB2 deletion down-regulated the expression of MEIS1 to a certain extent, whereas MEIS1 deletion up-regulated ZEB2 expression (Figures 5C and S6A). Their genomic binding regions were largely different outside of hematopoietic enhancers and, albeit to a lesser extent, within hematopoietic enhancers (Figure 7C). Moreover, ZEB2 deficiency did not alter MEIS1 binding at or near WT ZEB2 binding sites within hematopoietic enhancers (Figure 7D). To further validate the differential roles of ZEB2 and MEIS1, we tested whether MEIS1 or ZEB2 expression could rescue the abnormality in ZEB2-deficient cells. MEIS1 expression was enhanced to a comparable level as WT by introducing dCas9-VPR and MEIS1-targeted gRNAs from day 4 in ZEB2-deficient HE, and the ZEB2 transgene was overexpressed from day 4 (Figures S7D and S7E). Both ZEB2 and MEIS1 supplementation at this timing resulted in limited correction of transcriptional abnormality on day 7 (Figure 7E). However, while the forced expression of ZEB2 partially rescued the defects in hematopoietic enhancer activation on day 7 and HPC differentiation on day 10, MEIS1 expression failed to do so (Figures 7F, 7G, and 7H). Taken together, these results suggest that ZEB2 activates key hematopoietic enhancers required for hematopoietic cell development, independently of transcriptional dysregulation in HE, including MEIS1 down-regulation.
Lastly, given that both ZEB2 and MEIS1 are required but not sufficient for hematopoietic enhancer activation, we sought to address how they could control hematopoietic developmental potential at the single-cell level. Since ZEB2 and MEIS1 are expressed in endothelial cells but further up-regulated in HPCs, unlike typical transcription factors specifically expressed in endothelial cells (Figure 3G), we hypothesized that a higher expression of ZEB2 and MEIS1 could promote hematopoiesis. Indeed, reanalyzing single-cell RNA-seq data of arterial endothelial cells from human fetuses15 revealed that the expression of ZEB2 or MEIS1 showed a higher correlation with that of RUNX1 compared with endothelial genes, such as ERG and SOX6, or arterialization-related genes, such as DLL4 and HEY1 (Figure S7F). In contrast, ZEB2 and MEIS1 showed a lower correlation with each other (Figure S7G). However, when arterial endothelial cells were grouped by the expression level of ZEB2 and MEIS1, those highly expressing both ZEB2 and MEIS1 exhibited a higher expression of RUNX1 compared to cells expressing only one of ZEB2 or MEIS1 (Figure S7H). These observations suggest that cells expressing high levels of both ZEB2 and MEIS1 are favored to differentiate into HPCs. Collectively, the independent requirement of ZEB2 and MEIS1 in hematopoietic enhancer activation is suggested to contribute to the tight restriction of cell differentiation (Figure S7I).
Discussion
Cell differentiation is orchestrated by the coordinated regulation of a transcriptional network and epigenetic landscape that induces and stabilizes gene expressions. In this study, the stage-specific analysis of enhancer activity revealed that two transcription factors, ZEB2 and MEIS1, independently contribute to the initiation of the hematopoietic enhancer landscape in arterial HE. Deletion of either factor impaired hematopoietic enhancer activation and subsequently blocked hematopoietic cell development.
Enhancer activation has been shown to precede the associated gene transcription.27,28 In concordance with these reports, we observed the activation of a fraction of hematopoietic enhancers prior to the associated gene expression or HPC differentiation. These enhancers included the RUNX1 +23 enhancer, whose activity was previously reported in DLL4+ arterial HE and has been used as a marker of cells undergoing hematopoiesis.20,21 Our findings further revealed that early hematopoietic enhancer activation extends to multiple enhancers spread globally, indicating the initiation of the hematopoietic epigenetic landscape establishment, rather than mere regulation of a gene. Further tracing back the differentiation, these enhancers lost the repressive H3K27me3 mark and gained the permissive H3K4me1 mark on days 2 and 4, demonstrating the gradual regulation of enhancers began much earlier than the associated gene expression. This idea is consistent with a previous report showing that some cell type-restricted enhancers are bound by transcription factors in ESCs and epigenetically modified.6 Thus, in order to induce cell type-specific gene expression, associated enhancers are likely bound and regulated by non-cell type-specific transcription factors expressed along cell differentiation. In addition to such stepwise regulation, a hierarchical order existed among hematopoietic enhancers, as exemplified by the failed activation of cluster 1 enhancers when the earlier activation of cluster 2 enhancers was impaired or delayed (Figure 7A). Collectively, our observations demonstrate the presence of multi-layered regulation to establish the hematopoietic enhancer landscape.
While master regulators of cell differentiation have been identified for a number of cell types, how these factors are induced during cell differentiation has remained a question, particularly because their induction must be tightly restricted to the differentiating cells but is presumably driven by factors not specific to the target cell type at the beginning of the differentiation process. We found that ZEB2 and MEIS1 are required for hematopoietic enhancer activation and HPC differentiation. These two factors appeared to be upstream of RUNX1c, which is regarded as a master regulator of hematopoiesis.29,30 Since ZEB2 was expressed in endothelial cells and regulated their gene expression, its direct binding to hematopoietic enhancers and requirement for enhancer activation suggest that hematopoiesis utilizes a regulator of the earlier developmental stage. In contrast, MEIS1 was found to be dispensable for gene regulation in endothelial cells despite being expressed at this stage of development (Figures 5C, 6C, and 6D), and acts as a hematopoietic regulator involved in the early hematopoietic enhancer activation and subsequent hematopoietic gene regulation. These observations suggest that initiators of cell differentiation could include regulators of the earlier developmental stage, and those that are expressed from the earlier stage, but function specifically toward the target cell differentiation. Importantly, ZEB2 and MEIS1 were independently required for activating overlapping sets of enhancers. Moreover, since not all cells expressing ZEB2 and MEIS1 become HPCs, the accumulation of additional factors at the hematopoietic enhancers may be necessary. For example, RUNX1 activates hematopoietic enhancers and drives EHT.31 Although the RUNX1 motif was not enriched in cluster 2 enhancers, it may bind to these enhancers via other transcription factors to further amplify enhancer activity, in addition to the de novo activation of cluster 1 enhancers. Such cooperative functions of multiple regulators are considered the key mechanism for restricting cell differentiation when utilizing regulators expressed in a non-cell type-specific manner.
The arterialization of HE by Notch signaling is considered a prerequisite for multipotent hematopoiesis.32 Although we did not confirm the lymphoid potential of our HPCs, we observed that HPC differentiation and hematopoietic enhancer activation were largely dependent on Notch signaling. Despite previous observations that definitive hematopoiesis only occurs from arterial but not venous endothelium, the molecular contribution of arterialization to hematopoiesis has long been unclear. In addition to the recent findings showing that Notch signaling—which plays a central role in arterialization—is required for definitive hematopoiesis via the induction of hematopoietic transcription factors such as HOXA genes,14,20 our findings further support the molecular link between arterialization and hematopoiesis, as we revealed hematopoietic enhancer activation is facilitated by Notch signaling and hematopoietic cytokines. Although ZEB2 regulates the expression of some Notch signaling targets, which may partly account for the impaired hematopoietic enhancer activation and HPC development, the enrichment of ZEB2 motifs at hematopoietic enhancers and the binding of ZEB2, but not Notch1, to these regions suggest the direct contribution of ZEB2 to hematopoietic enhancer activation. In contrast, Notch signaling up-regulated MEIS1, which could serve as a downstream regulator to facilitate hematopoietic enhancer activation. However, hematopoietic enhancer activation was more drastically impaired by Notch signaling inhibition compared to when MEIS1 was deleted, suggesting the involvement of additional downstream regulators in hematopoietic enhancer activation upon Notch signaling. Therefore, the pre-existence of factors, such as ZEB2 and MEIS1, and the induction of additional regulators downstream of Notch signaling are required to drive hematopoietic enhancer activation. Although detailed analysis demonstrating the precise order of regulator accumulation at the enhancers needs further investigation, our findings suggest that environmental cues, such as arterialization-inducing signals, are translated into the composition of the enhanceosome, which eventually determines the potential to develop into hematopoietic cells.
The development of HPCs has been mainly studied in experimental animals, such as mice and zebrafish, and in humans using PSC-derived in vitro culture. Particularly, the PSC-derived culture system has been a valuable tool for the analysis of human developmental processes prior to HPC emergence due to the difficulty in obtaining human specimens. The involvement of ZEB2 in embryonic hematopoiesis was previously studied in conditional knock-out mouse, in which HSCs emerged from ZEB2-deleted endothelial cells but were found defective and failed to differentiate into HPCs.33 That observation appears to contradict our results, which show the absence of ZEB2-deficient hematopoietic cells using flow cytometry. However, we observed EHT microscopically, albeit at a reduced frequency, along with an increase in the number of dead cells. Thus, a small number of HPCs are suggested to emerge even without ZEB2 but are prone to death. In addition, CD34+CD45+ cells in PSC-derived in vivo hematopoiesis are equivalent to in vivo HPCs, rather than HSCs.34 Thus, while more study is needed to clarify whether ZEB2 is indispensable for the development of HSCs in humans, our results concordantly indicate the requirement of ZEB2 in human HPC development. In contrast, the requirement of MEIS1 in primitive and definitive hematopoiesis has been demonstrated in zebrafish, mouse embryo, and human PSC-derived cultures, consistent with our results.26,35,36 However, the roles of neither ZEB2 nor MEIS1 in transcriptional and epigenetic regulation, particularly with regard to how they confer hematopoietic potential on arterial HE, have been uncovered beyond the effects of their deletion on the expression of some hematopoietic transcription factors. By focusing on stage-specific changes in the hematopoietic enhancer landscape, we show novel roles of these regulators prior to HPC emergence. Notably, there are single nucleotide polymorphisms (SNPs) associated with red and/or white blood cell counts around ZEB2 and MEIS1 genes (according to GWAS Catalog, accessed on 29 September 2021). These SNPs could potentially alter the efficiency of hematopoietic enhancer activation, thereby affecting the frequency of cells with the potential to develop into hematopoietic cells.
Lastly, our findings have important implications for artificially generating target cells from iPSCs. iPSC-derived cells are promising tools for regenerative medicine and drug screening. While the forced expression of transcription factors in iPSCs enables an accelerated differentiation process with reduced cost, the differentiation often results in insufficient acquisition of the desired cellular identity, which makes the generated cells inappropriate for clinical use.37 As our study revealed the gradual establishment of cell type-specific molecular features, understanding the order of transcription factors to be introduced could help unleash the potential of each transcription factor. In fact, the induction of RUNX1b in PSCs, but not in HE, has been shown to block HPC differentiation, demonstrating that transcription factors act divergently in a context-dependent manner.38 While MEIS1 was previously identified as a factor supporting the reprogramming of mature blood cells to HSCs,39 ZEB2 was never identified in such screens, most likely due to the broad expression of ZEB2. Since multiple factors are required to activate and potentiate the full spectrum of hematopoietic enhancers, the forced expression of ZEB2 and MEIS1 is still considered insufficient for HPC generation. However, our findings suggest that their contribution to establishing an appropriate epigenetic landscape prior to the introduction of key transcription factors is critical for stably inducing cell differentiation.
In conclusion, the present study suggests that cooperative enhancer activation by multiple regulators expressed from an earlier developmental stage controls the acquisition of developmental potential into hematopoietic cells. The absence of one factor causes defective enhancer activation, which sequentially leads to failed activation of later activated enhancers, finally blocking development. Further study of how these regulators cooperate to activate enhancers should aid in understanding the molecular control of cell lineage specification.
Limitations of the study
Our study demonstrates the contribution of multiple regulators from the earlier developmental stages to the generation of hematopoietic enhancer landscape. Although the roles of ZEB2 and MEIS1 were explored using in vitro human ES-derived hematopoietic cell differentiation system, their roles during in vivo hematopoietic cell differentiation in human fetus and adults need to be elucidated in future studies.
Through the analyses of hematopoietic cell differentiation, we suggest the cooperative roles of multiple factors from the earlier developmental stage in the enhancer activation required for the later developmental stage as a means to restrict cell differentiation. However, whether such mechanisms are also applicable for the development of other cell types awaits future examination.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-H3K27ac | GeneTex | GTX60815; RRID: AB_2888004 |
| Anti-H3K4me1 | ActiveMotif | 39297; RRID: AB_2615075 |
| Anti-H3K27me3 | Millipore | 07-449; RRID: AB_310624 |
| Anti-SIP1 (ZEB2) | Bethyl Laboratories | A302474A; RRID: AB_1944271 |
| Anti-MEIS1 | Abcam | ab19867; RRID: AB_776272 |
| Anti-NOTCH1 | Cell Signaling Technology | 3608S; RRID: AB_2153354 |
| BV421 anti-CD34 | BD Biosicneces | 744904; RRID: AB_2742572 |
| APC anti-CD309 | BIOLEGEND | 359916; RRID: AB_2565928 |
| PECy7 anti-CD45 | BIOLEGEND | 304016; RRID: AB_314404 |
| FITC anti-CD73 | BIOLEGEND | 344015; RRID: AB_2561808 |
| PE anti-DLL4 | Miltenyi Biotec | 130-096-567; RRID: AB_10831209 |
| APC anti-CD184 (CXCR4) | BD Biosicneces | 555976; RRID: AB_398616 |
| Anti-ꞵ-Actin | Cell Signaling Technology | 5125S; RRID: AB_1903890 |
| Anti-rabbit-HRP | Cell Signaling Technology | 7074S; RRID: AB_2099233 |
| Chemicals, peptides, and recombinant proteins | ||
| BMP-8 | R&D | 314-BP-010 |
| VEGF | R&D | 293-VE-010 |
| CHIR99021 | Wako | 038-23101 |
| SCF | R&D | 255-SC/CF |
| bFGF | Wako | 064-05381 |
| SB431542 | Wako | 031-24291 |
| TPO | R&D | 288-TPN |
| FLT3L | R&D | 308-GMP |
| FP6 | R&D | 8954-SR |
| iMatrix-511 | Matrixome | 892 012 |
| iMatrix-511 silk | Matrixome | 892 021 |
| RetroNectin | TAKARA | T100B |
| mTeSR1 | STEMCELL Technologies | ST-85850 |
| Essential 8 | Thermo Fisher Scientific | A1517001 |
| Essential 6 | Thermo Fisher Scientific | A1516401 |
| StemLine II | Sigma-Aldrich | S0192 |
| TrypLE Select | Gibco | A1217702 |
| Puromycin | InvivoGen | ant-pr-1 |
| G418 | Nacalai Tesque | 09380-44 |
| Critical commercial assays | ||
| SMART-seq v4 Ultra Low Input RNA Kit for Sequencing | Takara | Z4891N |
| SMARTer ThruPLEX DNA-seq 48S Kit | Takara | RB4427 |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | 20024906 |
| CD34 MicroBead Kit, human | Miltenyi Biotec | 130-046-702 |
| ChIP DNA Clean & Concentrator | Zymo Research | D5205 |
| Deposited data | ||
| RNA-seq and ChIP-seq data | This study | PRJNA783033 |
| Oligonucleotides | ||
| gRNA for CRISPR, see method details for oligonucleotide sequences | This study | N/A |
| Recombinant DNA | ||
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Cong et al.41 | Addgene #42230 |
| AAVS1-idCas9-vpr | Guo et al.51 | Addgene #89985 |
| Software and algorithms | ||
| Cutadapt (v1.15) | EMBnet.Journal42 | https://cutadapt.readthedocs.io/en/stable/index.html |
| Bowtie2 (v2.2.5) | Langmead and Salzberg47 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Hisat2 (v2.1.0) | Kim et al.43 | http://daehwankimlab.github.io/hisat2/ |
| BEDTools (v.2.27.1) | Bedtools | https://github.com/arq5x/bedtools2 |
| MACS2 (v2.1.1.20160309) | Zhang et al.48 | https://github.com/taoliu/MACS |
| samtools (v1.7) | Samtools | http://www.htslib.org/ |
| featureCounts (v1.6.0) | Liao et al.44 | http://subread.sourceforge.net/ |
| R (v3.6.1) | The R Project | https://www.r-project.org/ |
| R: DESeq2 (v1.24.0) | Bioconductor | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R: ClusterProfiler (v3.12.0) | Bioconductor | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| R: ComplexHeatmap (v2.0.0) | Bioconductor | https://bioconductor.riken.jp/packages/3.2/bioc/html/ComplexHeatmap.html |
| R: ggplot2 (v3.2.1) | CRAN | https://cran.r-project.org/web/packages/ggplot2/index.html |
| GenomeJack (v3.1) | Mitsubishi Space Software | http://genomejack.net/japanese/index.html |
| Homer | Heinz et al.49 | http://homer.ucsd.edu/homer/ |
| TPMCalculator | Vera Alvarez et al.45 | https://github.com/ncbi/TPMCalculator |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo | BD Biosicneces | https://www.flowjo.com/ |
| Other | ||
| CountBright Absolute Counting Beads | Thermo Fisher Scientific | C36950 |
| Dynabeads M-280 Sheep anti-Rabbit IgG | Thermo Fisher Scientific | 11203D |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Megumu K. Saito (msaito@cira.kyoto-u.ac.jp).
Materials availability
All requests for resources and reagents should be directed to the lead contact author. All reagents will be made available on request after completion of a Materials Transfer Agreement.
Experimental model and subject details
Cell lines
The human ESC line KhES-1 and iPSC line 201B2 were kindly provided by Norio Nakatsuji (Kyoto University, Kyoto, Japan) and Shinya Yamanaka (Kyoto University, Kyoto, Japan), respectively. Both lines were generated from female donors. All cells were regularly karyotyped and tested for the presence of mycoplasma. The use of human embryonic stem cells (ESCs) in Kyoto University was approved by the Ministry of Education Culture, Sports, Science and Technology of Japan (MEXT). The study plan for recombinant DNA research was approved by the recombinant DNA experiments safety committee of Kyoto University.
Method details
Monolayer hematopoietic cell differentiation via HE
PSCs spheroids were formed on an EZSPHERE SP microplate (IWAKI) to tightly control colony numbers and speed up the time required to obtain colonies of sufficient size. They were transferred to iMatrix-511-coated plates the next day and maintained in mTeSR1 (STEMCELL Technologies) for three more days. When undifferentiated colonies reached 750–1000 μm in diameter (day 0), hematopoietic cell differentiation was initiated based on previously described protocols.16,17,40 Briefly, the culture media was replaced with Essential-8 medium (#A1517001, Thermo Fisher Scientific) containing 80 ng/mL BMP4 (#314-BP-010, R&D), 80 ng/mL VEGF 165 (#293-VE-010, R&D), and 4 μM GSK-3 inhibitor CHIR99021 (#038-23101, Wako). On day 2, the media was changed to Essential-6 medium (#A1516401, Thermo Fisher Scientific) with 80 ng/mL VEGF, 25 ng/mL bFGF (#064-05381, Wako), 2 μM ALK5 inhibitor SB431542 (#031-24291, Wako), and 50 ng/mL SCF (#255-SC/CF, R&D). On day 4, CD34+ cells were isolated using a human CD34 Microbead Kit (Miltenyi Biotec) and re-plated on Retronectin-coated plates at a density of 2.5 x 104 cells/cm2 in hematopoietic cell differentiation medium, which contains Stemline-II medium (#S0192, Sigma-Aldrich), 50 ng/mL SCF, 50 ng/mL Flt-3 Ligand (#308-GMP, R&D), 5 ng/mL TPO (#288-TPN, R&D) and 20 ng/mL FP6 (#8954-SR, R&D). The media was thereafter changed every 3 days. 10 μM DAPT or 2 μM SB431542 were added on day 4 and/or 7 for the inhibition of Notch and TGF-β signaling, respectively, in Figures 3 and S3.
Flow cytometric analysis and cell sorting
For the isolation of cells from day 0 and 2, adherent cells were treated with 50% TrypLE Select (Gibco) in PBS supplemented with 0.5 mM EDTA (Gibco) and harvested by pipetting. Viability of cells was ensured to be > 90% for the RNA-seq and ChIP-seq experiments. On days 4 and 7, the supernatant was removed, and adherent cells were collected with 50% TrypLE Select. After staining with DAPI and antibodies, CD34+KDR+ cells were sorted or analyzed using BD FACSAriaII (BD Biosciences). To analyze both CD34+CD45– and CD34+CD45+ cells on day 10, the supernatant and adherent cells were collected. Dead cells were removed by DAPI staining, and the gating was set so that the purity of the sorted cells was over 90%. The following antibodies were used: BV421 anti-CD34 (744904, BD Biosciences), APC anti-CD309 (3559916, Biolegend), PECy7 anti-CD45 (304016, Biolegend), FITC anti-CD73 (344015, Biolegend), PE anti-DLL4 (130-096-567, Miltenyi Biotec), and APC anti-CD184 (555976, BD Biosciences). Flow cytometric data were analyzed using FlowJo (version 10.6.1, BD Biosciences).
Generation of ZEB2-deficient and MEIS1-deficient PSCs
A pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid41 (Addgene #42230) was modified to replace Cas9 with Cas9-T2A-puromycin. This plasmid was digested by BbsI, and the following oligonucleotides were annealed and inserted.
ZEB2: (forward) CACCGATCCAGACCGCAATTAACAA
(reverse) AAACTTGTTAATTGCGGTCTGGATC
MEIS1: (forward) CACCG TACTTGTACCCCCCGCGAGC
(reverse) AAACGCTCGCGGGGGGTACAAGTAC
The plasmids were transfected into human ESCs or human iPSCs using a NEPA21 electroporator (NEPAGENE) and cuvettes of 2-mm gap. The transfected cells were transiently selected by adding 1 μg/mL puromycin from 24 to 48 hours after the transfection. Single colonies were harvested for genotyping by Sanger sequencing. At least two knockout colonies were expanded for further analysis. To generate ZEB2/MEIS1 double-deficient ESCs, ZEB2-deficient ESCs were transfected with the pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid containing MEIS1-targeting gRNA.
Loss of the target protein was confirmed by western blotting. Briefly, CD34+ cells on day 4 were isolated using the human CD34 Microbead Kit (Miltenyi Biotec) and lysed with RIPA buffer (Wako) for 30 minutes on ice. After removing cell debris by centrifugation, the lysate was mixed with 2× Laemmli Sample Buffer (Bio-Rad Laboratories) containing 5% total volume 2-mercaptoethanol (Nacalai tesque) and boiled for 5 minutes at 95°C. Polyacrylamide gel electrophoresis was performed on SDS-polyacrylamide gels, and proteins were transferred to a nitrocellulose membrane (Merck Millipore). The membrane was then incubated with 5% BSA in Tris-buffered saline with tween 20 (Santa Cruz Biotechnology, lnc.) for blocking. The primary antibody reaction was performed at 4°C overnight. The secondary antibody incubation was performed for 90 minutes at room temperature. The target protein was detected using ECL chemiluminescence reagents (Thermo Fisher Scientific). An antibody against ꞵ-Actin was reacted for 60 minutes at room temperature. The following primary and secondary antibodies were used: anti-ZEB2 (1:1000, 61095, Active Motif), anti-MEIS1 (1:1000, abcam ab19867), anti-ꞵ-Actin (1:5000, #5125S, Cell Signaling Technology), and anti-rabbit-HRP (1:2500, #7074S, Cell Signaling Technology).
RNA-seq
FACS-sorted cells were lysed in RLT buffer (Qiagen), and RNA was extracted using RNAClean XP beads (Beckman Coulter). Reverse transcription was performed using a SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara). cDNA was then fragmented using a Covaris Focused-ultrasonicator M220 (M&S Instruments Inc.). The library was constructed using a SMARTer ThruPLEX DNA-seq 48S Kit (Takara) and sequenced on a NextSeq 500 System (Illumina) with 75-bp single-end reads. No technical replicates were generated.
Reads were trimmed by Cutadapt (version 1.15)42 and mapped to the human genome hg19 by Hisat2 (version 2.1.0).43 Tags were counted by featureCounts (version 1.6.0)44 and normalized using the DESeq2 package (version 1.24.0) in R. Differentially expressed genes were defined as those with FDR < 0.01 and log2 fold-change > 2. The expression of RUNX1 splice variants was determined by TPMCalculator (version 0.0.3).45 Heatmaps showing gene expression patterns were generated using the ComplexHeatmap package (version 2.0.0)46 in R. Data from individual samples were treated independently in all PCA plots and heatmaps.
ChIP-seq
The following number of cells were used for the ChIP-seq: 1 x 105 for H3K27ac, 2 x 105 for H3K4me1 and H3K27me3, and 1-5 x 106 for transcription factor ChIP-seq. The antibodies used were anti-H3K27ac (GeneTex, GTX60815), anti-H3K4me1 (Active Motif, 39297), anti-H3K27me3 (Millipore, 07-449), anti-ZEB2 (Bethyl Laboratories, A302474A), anti-MEIS1 (Abcam, ab19867), anti-Notch1 (Cell Signaling Technology, 3608S), anti-SOX6 (Abcam, ab30455), anti-SOX17 (R&D Systems, AF1924-SP), and anti-SPI1 (Cell Signaling Technology, 2266). Cells were cross-linked in 1% (w/v) formaldehyde solution for 5 min (histone ChIP-seq) or 30 min (transcription factor ChIP-seq) and lysed. Cross-linked DNA was fragmented by sonication using a Digital Sonifier (Branson) and incubated overnight at 4°C with 50 μL DynaBeads IgG magnetic beads (Thermo Fisher) conjugated with 2.5 μg antibodies in the presence of 1% FBS and 10-30 μg salmon sperm DNA. Samples were then washed, eluted, reverse cross-linked at 65°C overnight, and purified using ChIP DNA Clean & Concentrator (Zymo Research). For transcription factor ChIP-seq, purified ChIP DNA was fragmented using a Covaris Focused-ultrasonicator M220 (M&S Instruments Inc.). The library was prepared using a SMARTer ThruPLEX DNA-seq 48S Kit (Takara) and sequenced on a NextSeq 500 System (Illumina) with 75-bp single-end reads.
ChIP-seq reads were trimmed by Cutadapt and mapped by bowtie247 to hg19 after removing the reads mapped to salmon. For visualization, ChIP peaks were called and normalized by the number of mapped reads (-SPMR option) using MACS248 with input reads as a control and are presented in GenomeJACK Browser (version 3.1, Mitsubishi Space Software). The y-axis of a ChIP-seq track indicates the count per million (CPM). To identify ChIP peaks, the findPeaks program in the Homer package49 was used. For H3K27ac ChIP-seq, the program was used with -region option and 20-fold enrichment over input as a cutoff. For transcription factor ChIP-seq, both the peak size and minimum distance of peaks were set to 500 bp, the local fold change cutoff was disabled, and the default settings were applied for all other parameters. ChIP-seq tag counts at the target regions were determined by featureCounts (version 1.6.0) and normalized by the number of uniquely mapped reads (tag counts per region ÷ uniquely mapped reads × 1,000,000). Heatmaps showing relative signal intensities were generated using the ComplexHeatmap package (version 2.0.0) in R.
Definition of hematopoietic enhancers
First, regions with H3K27ac marks in day 10 CD34+CD45+ cells were identified by the findPeaks program in the Homer package. Regions overlapping promoter regions (defined as up to 2.5 kb upstream of the transcription start site indicated in NCBI RefSeq) were removed. Regions with H3K27ac marks were similarly identified in day 0 and day 2 cells and H3K27ac-marked regions in day 10 CD34+CD45+ cells which overlap with H3K27ac-marked regions in day 0 and day 2 cells were also removed to define hematopoietic enhancers.
Gene ontology analysis
A gene ontology analysis of defined gene sets was conducted using ClusterProfiler (version 3.12.0)50 in R. All gene sets in the Gene Ontology database were used, except for the data in Figure 1B, where the gene sets whose name contain ‘development’ or ‘differentiation’ were selected.
Motif analysis
A motif analysis was performed using the findMotifsGenome program in the Homer package with default settings. For the background sequence, proximal enhancers [ENCFF036NSJ] and distal enhancers [ENCFF535MKS] from candidate cis-Regulatory Elements predicted in the ENCODE project (https://www.encodeproject.org/) were combined, and the genome coordinates were converted from hg38 to hg19 using UCSC LiftOver. Results with q-value < 0.0001, log P-value < -30 and the ratio of % target sequences with motifs to % background sequences with motifs > 1.5 in at least one cluster of hematopoietic enhancers were selected for the data presentation.
Colony forming unit assay
Kh1 ESCs were differentiated into HPCs, and 3,000 CD34+CD45+ cells were collected on day 10 using BD FACSAria II (BD Biosciences). These cells were incubated in MethoCult (Stem Cell Technologies, H4435) for 2 weeks.
Mixed culture of wild-type and ZEB2-deficient cells
GFP+ Kh1 ESCs were generated by introducing the EF1α-AcGFP-IRES-PuroR cassette using the PiggyBac transposon. GFP+ WT and GFP– ZEB2-deficient CD34+ cells were isolated using MACS beads on day 4 and mixed at a 1:1 ratio for further differentiation into HPCs. The development of HPCs was examined using flow cytometry on day 10.
Time-lapse imaging of endothelial-to-hematopoietic transition
CD34+ cells were isolated on day 4, cultured on a Retronectin-coated 12 well plate, and incubated in a hematopoietic cell differentiation medium (see monolayer hematopoietic cell differentiation via HE section). The medium was replaced on days 7 and 9 just before the initiation of imaging. Green fluorescent YO-PRO-1 (Thermo Fisher Scientific #Y3603) was added at 500 nM to stain dead cells on day 9. Time-series images were captured using BioStation CT (Nikon) at 15 min intervals for 24 hours, and a 20X objective lens was used. We defined round, floating, and green fluorescent-negative cells as hematopoietic cells and green fluorescent-positive cells as dead cells.
Forced expression of MEIS1 and ZEB2 in ZEB2-deficient cells
An inducible dCas9-VPR cassette was obtained from AAVS1-idCas9-vpr plasmid (Addgene #89985)51 and inserted in a PiggyBac transposon vector for the doxycycline-dependent induction of dCas9-VPR expression. The following gRNAs targeting the MEIS1 promoter were inserted under the U6 promoter in a separate PiggyBac transposon vector.
-110bp from TSS: (forward) CACCGTGCATTGGGCTGCAGCAAGT
(reverse) AAACACTTGCTGCAGCCCAATGCAC
-124bp from TSS: (forward) CACCGGCAAGTAGGCTCCTCGGCAG
(reverse) AAACCTGCCGAGGAGCCTACTTGCC
These plasmids, together with the vector containing PiggyBac transposase, were electroporated into ESCs using the NEPA21 electroporator (NEPAGENE). Cells with successful incorporation of the dCas9-VPR cassette and gRNAs were selected by adding G418 and puromycin, respectively. Doxycycline was added at 2 μg/mL on days 4 and 7.
For ZEB2 overexpression in ZEB2-deficient cells, ZEB2 cDNA was cloned and inserted into a PiggyBac transposon vector under the control of a doxycycline-dependent promoter. This plasmid was transfected into day 4 CD34+ cells by Fugene HD (Promega) according to the manufacturer’s instructions. Doxycycline was added at 2 μg/mL on days 4 and 7.
Single-cell transcriptional analysis of hematopoietic cell development in human fetuses
Data from GSE135202 were reanalyzed using Seurat52 in R. Data from Carnegie stages 13, 14 and 15 were combined and clustered into 3 groups, representing arterial endothelial cells, venous endothelial cells and HPCs. Markers of each fraction were defined, and genes with DNA-binding transcription factor activity (GO:0003700 in QuickGO Database53) were selected as cell type-specific transcription factors.
Quantification and statistical analysis
Experiments were independently replicated at least twice, and representative and/or summary data are shown. Flow cytometric analysis was independently repeated three or more times. The number of experiments conducted is stated in each figure legend. Statistical differences were determined by the statistical tests stated in each figure legend using GraphPad Prism (version 7). P < 0.05 was accepted for statistical significance.
Acknowledgments
We thank Ms. Maasa Matsuura for technical support, Ms. Harumi Watanabe for providing administrative assistance, and Dr. Peter Karagiannis for proofreading the paper. Bioinformatics analyses were partially performed on the NIG supercomputer at ROIS National Institute of Genetics. This work was supported by Grants-in-Aid for Japan Society for the Promotion of Science (JSPS) Fellows (18J02122) to Y.K., JSPS Grant-in-Aid for Early-Career Scientists (18K14713 and 20K15800) to Y.K., the grant for the Core Center for Regenerative Medicine and Cell and Gene Therapy (JP23bm1323001) and Core Center for iPS Cell Research of Research Center Network for Realization of Regenerative Medicine (JP21bm0104001) from the Japan Agency for Medical Research and Development (AMED) to M.K.S., the Program for Intractable Diseases Research utilizing Disease-specific iPS cells of AMED (21bm0804004) to M.K.S., grant from Terumo Life Science Foundation to M.K.S., and a grant from iPS Cell Research Fund to M.K.S.
Author contributions
Y.K. designed and performed most of the experiments. A.I. conducted the western blotting and time-lapse imaging experiments. R.S. and A.N. provided crucial advice. Y.K. and M.K.S. wrote the manuscript. All authors reviewed and approved the manuscript.
Declaration of interests
The authors declare no conflict of interest.
Published: September 9, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107893.
Contributor Information
Yohko Kitagawa, Email: y.kitagawa@cira.kyoto-u.ac.jp.
Megumu K. Saito, Email: msaito@cira.kyoto-u.ac.jp.
Supplemental information
Data and code availability
-
•
RNA-seq and ChIP-seq data have been deposited at NCBI SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
All original code is available in this paper’s supplemental information (Methods S1).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Bulger M., Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heinz S., Romanoski C.E., Benner C., Glass C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015;16:144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spitz F., Furlong E.E.M. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 5.Davis R.L., Weintraub H., Lassar A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-X. [DOI] [PubMed] [Google Scholar]
- 6.Kim H.S., Tan Y., Ma W., Merkurjev D., Destici E., Ma Q., Suter T., Ohgi K., Friedman M., Skowronska-Krawczyk D., Rosenfeld M.G. Pluripotency factors functionally premark cell-type-restricted enhancers in ES cells. Nature. 2018;556:510–514. doi: 10.1038/s41586-018-0048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Medvinsky A., Dzierzak E. Definitive Hematopoiesis Is Autonomously Initiated by the AGM Region. Cell. 1996;86:897–906. doi: 10.1016/S0092-8674(00)80165-8. [DOI] [PubMed] [Google Scholar]
- 8.de Bruijn M.F., Speck N.A., Peeters M.C., Dzierzak E. Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. EMBO J. 2000;19:2465–2474. doi: 10.1093/emboj/19.11.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clements W.K., Traver D. Signalling pathways that control vertebrate haematopoietic stem cell specification. Nat. Rev. Immunol. 2013;13:336–348. doi: 10.1038/nri3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakagawa M., Ichikawa M., Kumano K., Goyama S., Kawazu M., Asai T., Ogawa S., Kurokawa M., Chiba S. AML1/Runx1 rescues Notch1-null mutation-induced deficiency of para-aortic splanchnopleural hematopoiesis. Blood. 2006;108:3329–3334. doi: 10.1182/blood-2006-04-019570. [DOI] [PubMed] [Google Scholar]
- 11.Robert-Moreno A., Guiu J., Ruiz-Herguido C., López M.E., Inglés-Esteve J., Riera L., Tipping A., Enver T., Dzierzak E., Gridley T., et al. Impaired embryonic haematopoiesis yet normal arterial development in the absence of the Notch ligand Jagged1. EMBO J. 2008;27:1886–1895. doi: 10.1038/emboj.2008.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burns C.E., Galloway J.L., Smith A.C.H., Keefe M.D., Cashman T.J., Paik E.J., Mayhall E.A., Amsterdam A.H., Zon L.I. A genetic screen in zebrafish defines a hierarchical network of pathways required for hematopoietic stem cell emergence. Blood. 2009;113:5776–5782. doi: 10.1182/blood-2008-12-193607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monteiro R., Pinheiro P., Joseph N., Peterkin T., Koth J., Repapi E., Bonkhofer F., Kirmizitas A., Patient R. Transforming Growth Factor β Drives Hemogenic Endothelium Programming and the Transition to Hematopoietic Stem Cells. Dev. Cell. 2016;38:358–370. doi: 10.1016/j.devcel.2016.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung H.S., Uenishi G., Park M.A., Liu P., Suknuntha K., Raymond M., Choi Y.J., Thomson J.A., Ong I.M., Slukvin I.I. SOX17 integrates HOXA and arterial programs in hemogenic endothelium to drive definitive lympho-myeloid hematopoiesis. Cell Rep. 2021;34 doi: 10.1016/j.celrep.2021.108758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeng Y., He J., Bai Z., Li Z., Gong Y., Liu C., Ni Y., Du J., Ma C., Bian L., et al. Tracing the first hematopoietic stem cell generation in human embryo by single-cell RNA sequencing. Cell Res. 2019;29:881–894. doi: 10.1038/s41422-019-0228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niwa A., Heike T., Umeda K., Oshima K., Kato I., Sakai H., Suemori H., Nakahata T., Saito M.K. A novel serum-free monolayer culture for orderly hematopoietic differentiation of human pluripotent cells via mesodermal progenitors. PLoS One. 2011;6 doi: 10.1371/journal.pone.0022261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohta R., Sugimura R., Niwa A., Saito M.K. Hemogenic Endothelium Differentiation from Human Pluripotent Stem Cells in A Feeder- and Xeno-free Defined Condition. J. Vis. Exp. 2019 doi: 10.3791/59823. [DOI] [PubMed] [Google Scholar]
- 18.Choi K.-D., Vodyanik M.A., Togarrati P.P., Suknuntha K., Kumar A., Samarjeet F., Probasco M.D., Tian S., Stewart R., Thomson J.A., Slukvin I.I. Identification of the hemogenic endothelial progenitor and its direct precursor in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:553–567. doi: 10.1016/j.celrep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ditadi A., Sturgeon C.M., Tober J., Awong G., Kennedy M., Yzaguirre A.D., Azzola L., Ng E.S., Stanley E.G., French D.L., et al. HUMAN DEFINITIVE HAEMOGENIC ENDOTHELIUM AND ARTERIAL VASCULAR ENDOTHELIUM REPRESENT DISTINCT LINEAGES. Nat. Cell Biol. 2015;17:580–591. doi: 10.1038/ncb3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uenishi G.I., Jung H.S., Kumar A., Park M.A., Hadland B.K., McLeod E., Raymond M., Moskvin O., Zimmerman C.E., Theisen D.J., et al. NOTCH signaling specifies arterial-type definitive hemogenic endothelium from human pluripotent stem cells. Nat. Commun. 2018;9:1828. doi: 10.1038/s41467-018-04134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nottingham W.T., Jarratt A., Burgess M., Speck C.L., Cheng J.-F., Prabhakar S., Rubin E.M., Li P.-S., Sloane-Stanley J., Kong-a-San J., de Bruijn M.F.T.R. Runx1-mediated hematopoietic stem-cell emergence is controlled by a Gata/Ets/SCL-regulated enhancer. Blood. 2007;110:4188–4197. doi: 10.1182/blood-2007-07-100883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang P., Wei W., Lo C., Rosten P., Hou J., Hoodless P.A., Bilenky M., Bonifer C., Cockerill P.N., Kirkpatrick A., et al. Delineating MEIS1 cis-regulatory elements active in hematopoietic cells. Leukemia. 2014;28:433–436. doi: 10.1038/leu.2013.287. [DOI] [PubMed] [Google Scholar]
- 23.Shlyueva D., Stampfel G., Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 2014;15:272–286. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- 24.Verschueren K., Remacle J.E., Collart C., Kraft H., Baker B.S., Tylzanowski P., Nelles L., Wuytens G., Su M.-T., Bodmer R., et al. SIP1, a Novel Zinc Finger/Homeodomain Repressor, Interacts with Smad Proteins and Binds to 5′-CACCT Sequences in Candidate Target Genes. J. Biol. Chem. 1999;274:20489–20498. doi: 10.1074/jbc.274.29.20489. [DOI] [PubMed] [Google Scholar]
- 25.Challen G.A., Goodell M.A. Runx1 isoforms show differential expression patterns during hematopoietic development but have similar functional effects in adult hematopoietic stem cells. Exp. Hematol. 2010;38:403–416. doi: 10.1016/j.exphem.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H., Liu C., Liu X., Wang M., Wu D., Gao J., Su P., Nakahata T., Zhou W., Xu Y., et al. MEIS1 Regulates Hemogenic Endothelial Generation, Megakaryopoiesis, and Thrombopoiesis in Human Pluripotent Stem Cells by Targeting TAL1 and FLI1. Stem Cell Rep. 2018;10:447–460. doi: 10.1016/j.stemcr.2017.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arner E., Daub C.O., Vitting-Seerup K., Andersson R., Lilje B., Drabløs F., Lennartsson A., Rönnerblad M., Hrydziuszko O., Vitezic M., et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science. 2015;347:1010–1014. doi: 10.1126/science.1259418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim Y.W., Lee S., Yun J., Kim A. Chromatin looping and eRNA transcription precede the transcriptional activation of gene in the β-globin locus. Biosci. Rep. 2015;35 doi: 10.1042/BSR20140126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Menendez P., Navarro-Montero O., Ramos-Mejia V., Ayllón V., Stanley E., Elefanty A., Real P. RUNX1C regulates hematopoietic specification of human embryonic stem cells. Exp. Hematol. 2016;44:S89. doi: 10.1016/j.exphem.2016.06.186. [DOI] [Google Scholar]
- 30.Navarro-Montero O., Ayllon V., Lamolda M., López-Onieva L., Montes R., Bueno C., Ng E., Guerrero-Carreno X., Romero T., Romero-Moya D., et al. RUNX1c Regulates Hematopoietic Differentiation of Human Pluripotent Stem Cells Possibly in Cooperation with Proinflammatory Signaling. Stem Cell. 2017;35:2253–2266. doi: 10.1002/stem.2700. [DOI] [PubMed] [Google Scholar]
- 31.Lichtinger M., Ingram R., Hannah R., Müller D., Clarke D., Assi S.A., Lie-A-Ling M., Noailles L., Vijayabaskar M.S., Wu M., et al. RUNX1 reshapes the epigenetic landscape at the onset of haematopoiesis. EMBO J. 2012;31:4318–4333. doi: 10.1038/emboj.2012.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park M.A., Kumar A., Jung H.S., Uenishi G., Moskvin O.V., Thomson J.A., Slukvin I.I. Activation of Arterial Program Drives Development of Definitive Hemogenic Endothelium with Lymphoid Potential. Cell Rep. 2018;23:2467–2481. doi: 10.1016/j.celrep.2018.04.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goossens S., Janzen V., Bartunkova S., Yokomizo T., Drogat B., Crisan M., Haigh K., Seuntjens E., Umans L., Riedt T., et al. The EMT regulator Zeb2/Sip1 is essential for murine embryonic hematopoietic stem/progenitor cell differentiation and mobilization. Blood. 2011;117:5620–5630. doi: 10.1182/blood-2010-08-300236. [DOI] [PubMed] [Google Scholar]
- 34.Wang L., Menendez P., Shojaei F., Li L., Mazurier F., Dick J.E., Cerdan C., Levac K., Bhatia M. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J. Exp. Med. 2005;201:1603–1614. doi: 10.1084/jem.20041888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azcoitia V., Aracil M., Martínez-A C., Torres M. The homeodomain protein Meis1 is essential for definitive hematopoiesis and vascular patterning in the mouse embryo. Dev. Biol. 2005;280:307–320. doi: 10.1016/j.ydbio.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 36.Cvejic A., Serbanovic-Canic J., Stemple D.L., Ouwehand W.H. The role of meis1 in primitive and definitive hematopoiesis during zebrafish development. Haematologica. 2011;96:190–198. doi: 10.3324/haematol.2010.027698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doss M.X., Sachinidis A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells. 2019;8:403. doi: 10.3390/cells8050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen B., Teng J., Liu H., Pan X., Zhou Y., Huang S., Lai M., Bian G., Mao B., Sun W., et al. Inducible overexpression of RUNX1b/c in human embryonic stem cells blocks early hematopoiesis from mesoderm. J. Mol. Cell Biol. 2017;9:262–273. doi: 10.1093/jmcb/mjx032. [DOI] [PubMed] [Google Scholar]
- 39.Riddell J., Gazit R., Garrison B.S., Guo G., Saadatpour A., Mandal P.K., Ebina W., Volchkov P., Yuan G.-C., Orkin S.H., Rossi D.J. Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell. 2014;157:549–564. doi: 10.1016/j.cell.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yanagimachi M.D., Niwa A., Tanaka T., Honda-Ozaki F., Nishimoto S., Murata Y., Yasumi T., Ito J., Tomida S., Oshima K., et al. Robust and highly-efficient differentiation of functional monocytic cells from human pluripotent stem cells under serum- and feeder cell-free conditions. PLoS One. 2013;8 doi: 10.1371/journal.pone.0059243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 43.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 45.Vera Alvarez R., Pongor L.S., Mariño-Ramírez L., Landsman D. TPMCalculator: one-step software to quantify mRNA abundance of genomic features. Bioinformatics. 2019;35:1960–1962. doi: 10.1093/bioinformatics/bty896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 47.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based Analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo J., Ma D., Huang R., Ming J., Ye M., Kee K., Xie Z., Na J. An inducible CRISPR-ON system for controllable gene activation in human pluripotent stem cells. Protein Cell. 2017;8:379–393. doi: 10.1007/s13238-016-0360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Binns D., Dimmer E., Huntley R., Barrell D., O’Donovan C., Apweiler R. QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics. 2009;25:3045–3046. doi: 10.1093/bioinformatics/btp536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-seq and ChIP-seq data have been deposited at NCBI SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
All original code is available in this paper’s supplemental information (Methods S1).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.