

Abstract

Sulfonamides are one of the most important pharmacophores in medicinal chemistry, and sulfonamide analogues have gained substantial interest in recent years. However, the protein interactions of sulfonamides and especially of their analogues are underexplored. Using FKBP12 as a model system, we describe the synthesis of optically pure sulfenamide, sulfinamide, and sulfonimidamide analogues of a well characterized sulfonamide ligand. This allowed us to precisely determine the binding contributions of each sulfonamide oxygen atom and the consequences of nitrogen replacements. We also present high-resolution cocrystal structures of sulfonamide analogues buried in the pocket of a protein target. This revealed intimate contacts with the protein including an unprecedented hydrogen bond acceptor of sulfonimidamides. The use of sulfonamide analogues enabled new exit vectors that allowed remodeling of a subpocket in FKBP12. Our results illuminate the protein interaction potential of sulfonamides/sulfonamide analogues and will aid in their rational design.
Keywords: sulfonamides, drug design, protein–ligand-interactions, sulfonimidamides, FKBP
Introduction
Sulfonamides are indispensable in medicinal chemistry. Twenty-five percent of all FDA-approved new molecular entities from the first half of 2022 contained a sulfonamide motif.a Moreover, sulfonamide analogues, especially the =O to =NR exchanged sulfonimidamides (see Figure 1) and sulfoximines, have gained substantial momentum,1–3 and the first successful examples show the potential of these modalities.4–7 Notably, the new exit vector enabled by sulfonimidamides may open unprecedented opportunities for the structure-based design of sulfonamide analogues, but structurally well-documented examples for this are still lacking.
Figure 1.
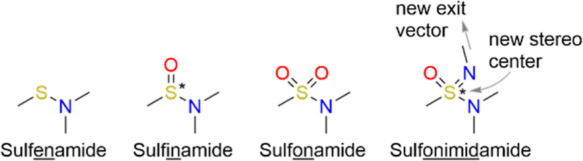
Structural motif of a sulfonamide and its analogues.
In line with the prevalence of sulfonamides, over 1000 cocrystal structures are available in the PDB harboring sulfonamide-containing ligands.b However, the contribution of the sulfonamide motif to affinity is usually poorly defined and little is known regarding the interaction energies resulting from the sulfonamide oxygen-protein contacts.
Results and Discussion
The FK506-binding protein 12 (FKBP12) is a widely used model system for ligand–protein interaction studies that binds to sulfonamides as one of its preferred recognition motifs.8–15 Notably, the sulfonamide oxygens are thought to be a key binding motif for these ligands and mimic the α-keto amide of FK506 (Supporting Information, Figure S5) and similarly engage in unusual but highly conserved CH···O=S interactions with the protein.16,17 Taking advantage of this highly conserved binding mode, we set out to explore the interactions of sulfonamides and their derivatives with FKBP12.
Toward this goal, we used the bicyclic sulfonamide 1 (known as compound 16a in Pomplun et al.11 and compound 16 in Kolos et al.,10 see Scheme 1 for the chemical structure) since it binds to FKBP12 with high affinity10 and the binding mode of this compound class is well established.
Scheme 1.

Reagents and conditions: (a) 3,5-dichlorobenzenethiol, HOAc, SO2Cl2, −40 °C to rt, then 2, DIPEA, MeCN, 19 h, rt, 55%; (b) KF, mCPBA, MeCN/H2O 5:1, 30 min, 0 °C, then 3, 5 h, 0 °C, 5% 4b and 7% 4a (separated diastereomers); (c) PhI(OAc)2, AcONH4, MeOH, 19 h, rt, 26% 5a and 35% 5b (separated diastereomers); (d) NaH, MeI, THF, 24 h, 0 °C to >rt, 82% 6a, 64% 6b; (e) NaH, allyl bromide, THF, 42 h, 0 °C to >rt, 100% 7a, 81% 7b; (f) NaH, 3-bromocyclohexene, 2–5 d 0 °C to >rt, 66% 8a, 85% 8b; (g) PhB(OH)2, Cu(OAc)2, TEA, MeCN, 18–42 h rt, 100% 9a, 87% 9b; (h) 2,6-lutidine, NMO, OsO4, acetone/water (10:1), 4 d, rt, 62%; (i) 3,5-dichlorobenzenesulfonyl chloride, DIPEA, dry MeCN, 19 h, rt, 48%; and (j) 2,6-lutidine, NMO, OsO4, acetone/water 7:1, 16 h, rt, 54%.
The synthesis of the sulfonamide analogues started with the known precursor 2,11 derived from the synthesis of sulfonamide-based FKBP ligands (Scheme 1). Here, amine 2 was coupled with freshly prepared sulfenyl chloride to give sulfenamide 3. The activation of 3,5-dichlorothiophenol was performed with sulfuryl chloride and acetic acid, as described by Martzel et al.18 The aim was to form the respective sulfinyl chloride and after the reaction with 2 the respective sulfinamide. Surprisingly, the only product found in this reaction was sulfenamide 3. The sulfenamide 3 was then oxidized to sulfinamides 4a/b. To prevent overoxidation, KF/mCPBA was used as oxidizing agent, as described by Datta and Buglass.19 The sulfinamides could be obtained as pure diastereomers by preparative HPLC. Oxidative imination of sulfenamide 3, following the approach of Zenzola et al.,20 provided sulfonimidamides 5a/b, which were readily separable by silica gel column chromatograpy. The isolated sulfonimidamides were then alkylated with methyl iodide (6a/b), allyl bromide (7a/b), and 3-bromocyclohexene (8a/b). A Chan–Lam reaction with phenylboronic acid, as described by Battula et al.,21 yielded products 9a/b. Compounds 8a/b were synthesized as respective mixtures of cyclohexenyl-C1-epimers, which were not preparatively separated.
The sulfinamides, N-unsubstituted sulfonimidamides, and substituted sulfonimidamides (methyl, allyl, cyclohexenyl, and phenyl substituted) could be obtained as stable and separated diastereomers arising from the chiral sulfur. The configurations of the sulfur atoms of the alkylated sulfonimidamides 6a/b and 7a/b could be determined via NOESY-NMR (Supporting Information, Figures S1 and S2), which also defined the configuration of the respective precursors 5a/b and other derivatives thereof (8a/b and 9a/b). The configurations of the sulfinamides 4a/b were assigned indirectly by stereoselective conversion to 5a/b (Supporting Information, Figure S3). Cocrystal structures later confirmed the assigned configurations (Figure 2 and S6–12).
Figure 2.

Cocrystal structure of bicyclic sulfonamides and analogues thereof. 1 [(A) PDB: 8CHL], 4b [(C) PDB: 8CHM], and 6a [(E) PDB: 8CHI] in complex with FKBP12. In (A,C), the binding pose of ligand 1 has been superimposed as gray sticks. 2D-interaction networks for these compounds are shown on the right (B,D,F). Hydrogen bonds are shown in blue, halogen-π-interactions are shown in red, CH···O/N interactions are shown in green, and other polar contacts are shown in light blue. Distances of dashed lines are given in Å. Electron density of the ligand 1 is shown as a blue mesh in part A. The distance of the sulfonimidamide methyl group of ligand 6a to Phe36 is shown in pink.
The S-configured phenyl-substituted sulfonimidamide 9a and the reference sulfonamide 1 were dihydroxylated at their respective vinyl groups in the C5 position of the core to give 10 and 11 to increase aqueous solubility, which was required for protein NMR studies (see below).
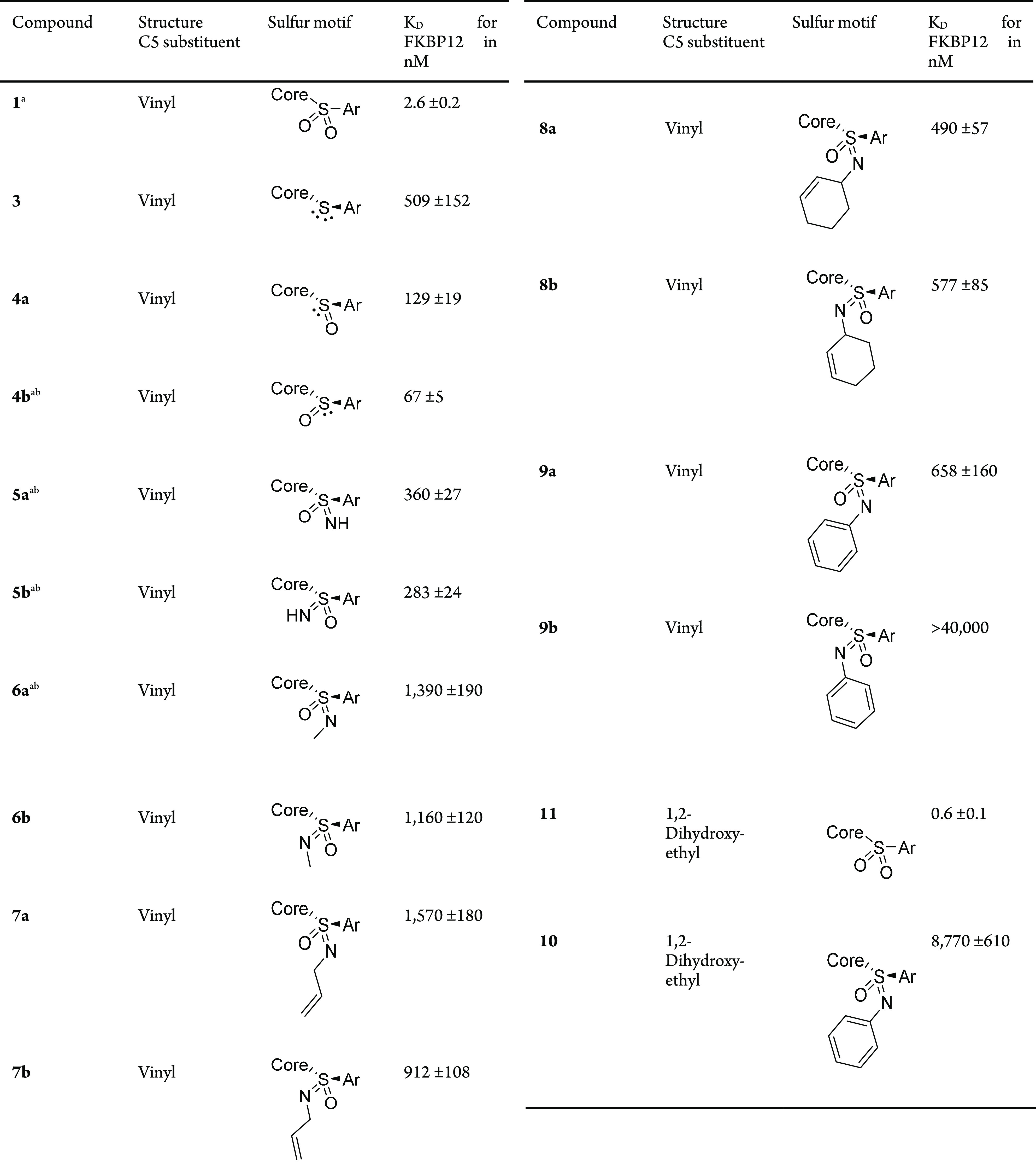
The binding affinities for FKBP12 of the sulfenamide, sulfinamide, and sulfonimidamide ligands were measured by a fluorescence polarization assay using sulfonamide 1 as a reference. For 1 and four analogues, we were also able to solve cocrystal structures with FKBP12 and the FK506-binding domain of FKBP5122 at high resolutions (1.0–1.7 Å, Figure 2 and Supporting Information Figures S6–S12).c
Additional binding affinity data for three other human FKBPs and two FKBP-like proteins from human pathogens are given in Supporting Information, Table S1. The same trends can be observed as for FKBP12, within an affinity offset characteristic for each specific FKBP homologue. Solely the preference for the sulfinamide stereochemistry (4a and 4b) changed from human FKBPs to FKBP-like proteins. This indicates that the findings obtained for FKBP12 are largely representative for the FKBP family.
All sulfonamide analogues bound weaker compared to the reference sulfonamide 1 (KD = 2.6 nM). This was expected since the latter was highly optimized for FKBP12. The cocrystal structure of 1 in complex with FKBP12 revealed its interaction network (Figure 2A,B), confirming the expected binding mode.
The pipecolate core of 1 is buried in a hydrophobic pocket exactly like the pipecolate core of the natural ligand FK506 (see Supporting Information, Figure S5). 1 forms two hydrogen bonds with FKBP12 (amide-C=O···HN-Ile56 and pyridine-N···HO-Tyr82), one dipolar interaction [amide-C(=O)···HO-Tyr82], and one halogen-π-interaction (aryl-Cl···π-His87). In analogy to the carbonyl oxygens of FK506 (see Supporting Information, Figure S5), the sulfonamide oxygens form a network of S=O···HC interactions with aromatic chains of Tyr26, Phe36, Tyr82, and Phe99.
The availability of the sulfenamide (3), sulfonamide (1) and both sulfinamides (4a/b) allowed–for the first time–to dissect the energetic contributions of each sulfonamide oxygen atom to binding affinity (see Table 1 and Supporting Information, eq S1).24 Both oxygen atoms combined accounted for a binding energy of −13.1 kJ/mol. Surprisingly, the interactions of both oxygens were coupled, with the removal of the first oxygen (ΔΔG = 8.05–9.67 kJ/mol) being much more detrimental than the subsequent removal of the second oxygen (ΔΔG = 3.40–5.02 kJ/mol). This overproportional effect led to a 1.4-fold higher binding energy for the combined two oxygens than the sum of both oxygen atoms separately. In direct comparison, the R-configured sulfinamide 4b was slightly preferred over the S-configured sulfinamide 4a. This suggests that the oxygen predicted to be in vicinity to Tyr26 contributes more to the overall binding energy than the oxygen predicted to be in vicinity to Tyr82. This orientation was confirmed by the cocrystal structures of 4b (Figure 2C,D with FKBP12, Supporting Information, Figure S6 with FKBP51FK1), which also showed that the overall binding mode is only minimally perturbed by the removal of the pro-S oxygen and that all protein interactions typical for this class of ligands are maintained.
Table 1. Binding Affinities of all Sulfonamide Analogues for FKBP12, Measured in a Competitive Fluorescence Polarization Assay23a.
The interaction network of 4b is equivalent to the interactions of sulfonamide 1, apart from the missing oxygen. The remaining sulfinamide oxygen forms the same S=O···HC interactions with Tyr26, Phe36, and Phe99 as observed for 1. The protein did not adapt to the gap created by the removed oxygen atom in vicinity to Tyr82. The missing S=O···HC interactions of said oxygen obviously account for the sulfinamide’s lower binding affinity.
With the contributions of each sulfonamide oxygen defined, we explored the effect of stereospecific substitution by nitrogens. Replacing an oxygen with an unsubstituted nitrogen was more detrimental (KD = 360 and 283 nM) than removing an oxygen altogether, for either configuration. Cocrystal structures for both diastereomers (5a/b) revealed an overall conserved binding mode (Supporting Information, Figures S7–S10). The distances of the S=O···HC and S=NH···HC interactions did not significantly deviate from the corresponding S=O···HC distances in the sulfonamide 1, and the binding modes offer enough space to accommodate the additional hydrogen. For the diastereomer 5b, the dichlorophenyl ring adopts different poses compared to the sulfonamide 1 (Supporting Information, Figure S9E,G), likely reflecting an attempt to ameliorate unfavorable interactions of the buried free NH of the sulfonimidamide.
Taken together, the crystallographic analysis suggests that the desolvation cost for an unsubstituted S(=O)=NH in the free sulfonimidamides (5a/b) is much higher compared to the SO2 in sulfonamides and in this particular case is poorly compensated by new interactions with FKBP12.
To reduce the desolvation cost, we methylated the sulfonimidamides, as in 6a/b. However, this reduced the affinity even further (KD = 1390 and 1160 nM). The cocrystal structure of 6a showed that this can be attributed to a steric clash, which is reflected by a twist of the ligand’s dichlorophenyl ring (Supporting Information, Figures S11 and S12).
Surprisingly, in the complex of 6a with FKBP12, we observed a very short contact of the sulfonimidamide nitrogen [S(=O)=NMe] to the hydroxyl group of Tyr82 (2.7 Å, Figure 2E,F and Supporting Information Figure S11A). This is substantially below van-der-Waals distance. The geometry is consistent with a hydrogen bond between S(=O)=NMe and HO-Tyr82. To the best of our knowledge, this is the first direct experimental evidence that sulfonimidamides can act as hydrogen bond acceptors in protein complexes. It should be noted, however, that the results obtained by cocrystallography represent snapshots and the interaction pattern might be more dynamic in solution, including a competition between hydrogen bonding to S(=O)=NMe or the pyridine-N.
A key prospect of sulfonimidamides compared with sulfonamides is the additional exit vector available in the former. To explore this option, we further increased the size of the sulfonimidamide substituent as in 7a/b (allyl), 8a/b (cyclohexenyl), and 9a/b (phenyl). Surprisingly, this did not further compromise affinity, although all these modifications were expected to severely clash with the protein. With the exception of the R-configured phenyl-substituted sulfonimidamide 9b (KD > 40 μM), the affinities actually improved for the larger substituents, which is difficult to reconcile with the previously observed canonical binding mode.
To clarify this, we analyzed the complexes by native MS and protein NMR experiments. For solubility reasons, we used the dihydroxylated analogues 11 and 10 since this modification is known to maintain the canonical binding mode for sulfonamide ligands.15
Native mass spectrometry further confirmed the direct binding of 11 and 10 to FKBP12. Complementary ion mobility measurements indicated no major conformational change for either ligand (Supporting Information, Figure S13). The 11-FKBP12 complex was found to be more stable toward collisional activation than the 10-FKBP12 complex (Supporting Information, Figure S14), in line with the higher affinity of the former. HDX analyses revealed that similar to the sulfonamide ligands, 10 induced a more shielded conformation in parts of FKBP12. In contrast, some regions such as the 30s loop preceding F36 showed enhanced accessibility, similar to the F36-displacing FKBP ligand SAFit125,26 (Supporting Information, Figure S15).
When investigating the 11- and 10-FKBP12 complexes by NMR spectroscopy (Supporting Information, Figures S16 and S17), four sets of signals were particularly informative: (i) NOESY experiments showed the pyridines of 10 and 11 to be in close proximity to the Tyr82 side chain (Figure 3 and Supporting Information, Figure S17), consistent with a key hydrogen bond (see Figure 2). (ii) The amide signal of the indole side chain of Trp59 was shifted downfield in the presence of 10 and 11 (Supporting Information, Figure S16C), which is highly characteristic for Trp59 being pushed deeper into the binding pocket. (iii) The HSQC signals of Ile56(NH) were strongly shifted by either 10 and 11 (Supporting Information, Figure S16D). This is consistent with a hydrogen bond to the carbonyl group of 10 and 11, which is a key interaction of FKBP ligands, including all structures presented in this work (Figure 2). (iv) The only HSQC signal missing for the 10-FKBP12 complex was for Phe36 and Tyr82 (Supporting Information, Figure S16E), which are predicted to be close to the N-substitution site of 10 (Figure 2E,F).
Figure 3.

2D NOESY spectra of [u-13C/15N]-labeled FKBP12 with 11 (A,C) and 10 (B,D) recorded at 700 MHz, showing the Tyr82 side chain being close to the ligand pyridine rings. Spectra in panels A and B were acquired at a sample temperature of 272 K using 0.5 mM FKBP sample in 25 mM phosphate buffer, pH 7, containing 5% D2O with a twofold excess of inhibitors. Samples for recording spectra in panels (C,D) contained 0.1 mM of [u-13C/15N]-FKBP12 + 0.36 mM 11 and 0.04 mM of [u-13C/15N]-FKBP12 + 0.2 mM 10, respectively, in 95–98% D2O. The latter spectra were recorded at a temperature of 298 K without 13C decoupling such that peaks involving the labeled protein exhibit a large doublet splitting due to 1JCH, whereas inhibitor peaks do not.
Collectively, these data show that 10 binds with the overall typical FKBP ligand binding mode where the N-phenyl substituent of the sulfonimidamide is accommodated by adaptions of F36 and Y82.
Conclusions
Our set of optically pure sulfenamide, sulfinamide, and sulfonimidamide analogues, together with the experimentally confirmed highly conserved binding mode, allowed us to precisely investigate the binding contributions of each sulfonamide oxygen as well as nitrogen substitutions thereof. For one ligand (6a), we provide clear evidence that sulfonimidamide nitrogen atoms can act as hydrogen bond acceptors in protein complexes. Finally, we show that substituents on sulfonimidamides enable new ways to engage proteins, which, in this case, revealed a dynamic rearrangement of a subpocket in FKBP12. Taken together, our findings provide a better understanding of how sulfonamides and sulfonimidamide analogues interact with proteins, which will help in the rational design of sulfon(imid)amide-containing drugs.
Methods
FP Assay
For most pipetting steps of the FP Assay, a Beckman Coulter FXP Laboratory Automation Workstation was used. As tracer, the fluorescent ligand 16g developed by Pomplun et al.11 was used. The compound was diluted in a 1:2 serial dilution in DMSO and then mixed with protein and tracer in buffer (20 mM HEPES, pH 8, 0.002% v/v Triton X-100, 150 mM NaCl) in a black, nonbinding 384-well plate, and then incubated in the dark for 30 min. Polarization was measured on a Tecan Spark at room temperature with an excitation wavelength of 535 nm and an emission wavelength of 595 nm. The assay was performed in triplicates. The competition curves were visualized using GraphPad Prism 6.0, KD values were calculated from the fitting according to Kozany et al.23 The final parameters are listed in Supporting Information, Table S2.
Crystallization
Complexes were prepared by mixing FKBP12 C22 V at 25.6 mg/mL (5a, 5b, and 6a) or 31.7 mg/mL (1 and 4b) or FKBP51FK1 A19T, C103A, C107I (14–140) at 28 mg/mL formulated in 20 mM HEPES-NaOH pH 8.0 and 20 mM NaCl with a slight molar excess of ligand previously dissolved at 20 mM in DMSO. For complexes of FKBP12 and 1 and 4b crystallization was performed at room temperature using the sitting drop vapor-diffusion method, equilibrating mixtures of 1 μL of protein complex and 1 μL of reservoir against 30 μL of reservoir solution. For 1, crystals were obtained from reservoir solutions containing 2.2 M ammonium sulfate and 0.2 M cadmium sulfate. For 4b, crystals were obtained from reservoir solutions containing 2.1 M ammonium sulfate, 0.2 M cadmium chloride, and 0.1 M HEPES-NaOH pH 7.5. For complexes of FKBP12 and 5a, 5b, and 6a crystallization was performed at room temperature using the hanging drop vapor-diffusion method, equilibrating mixtures of 1 μL of protein complex and 1 μL of reservoir against 500 μL of reservoir solution. Crystals were obtained from reservoir solutions containing 1.32–1.4 M Na/K tartrate, 0.2 M ammonium citrate, and 0.1 M MES pH 6.5. Crystals were fished, cryoprotected with LV CryoOil (Jena Bioscience), and flash frozen in liquid nitrogen. For complexes with FKBP51FK1 crystallization was performed at room temperature using the hanging drop vapor-diffusion method, equilibrating mixtures of 1 μL protein complex and 1 μL reservoir against 500 μL reservoir solution. Crystals were obtained from reservoir solutions containing 30–32% PEG-3350, 0.2 M NH4-acetate, and 0.1 M HEPES-NaOH pH 7.5. Crystals were fished and flash frozen in liquid nitrogen.
Structure Solution and Refinement
The crystallographic experiments were performed on the BL14.1 and BL14.2 beamlines at the Helmholtz-Zentrum BESSY II synchrotron, Berlin, Germany.27 Diffraction data were integrated with XDS and further processed with the implemented programs of the CCP4i and CCP4i2 interface.28–31 The data reduction was conducted with Aimless.31–33 Crystal structures were solved by molecular replacement using Phaser.34 Iterative model improvement and refinement were performed with Coot and Refmac5.35–40 The dictionaries for the compounds were generated wit PRODRG implemented in CCP4i.41 Residues facing solvent channels without detectable side chain density were truncated. Refinement data are shown in Supporting Information, Tables S3–S5
Ion Mobility Mass Spectroscopy
An FKBP12 stock solution of 44 mg/mL in 20 mM sodium chloride and 20 mM HEPES was rebuffered. For the rebuffering, a 3k Amicon Ultra Centrifugal Filter from Merck was first precleared with 500 μL of a 200 mM ammonium acetate buffer (pH 7, LC–MS grade) at 14g for 30 min. The ammonium acetate buffer is a native volatile buffer.42 Then, 100 μL of protein stock solution was added, filled up to 500 μL with ammonium acetate buffer, and rebuffered at 14g for 30 min. The previous step was performed five times in total. The rebuffered protein solution was then diluted with an ammonium acetate buffer to an initial concentration of 44 mg/mL (∼3.7 mM). Of the ligands, a stock solution of 20 mM in DSMO was diluted to 2 mM with DMSO. For the final solution, 0.5 μL of the protein dilution and 1 μL of ligand dilution or DMSO (for Apo-FKBP12) were combined and filled to 50 μL with 200 mM ammonium acetate buffer. This resulted in a final concentration of a 40 μM protein–ligand solution.
All sample solutions were measured using direct-infusion nanoESI on a Synapt G2-S instrument from Waters. Capillary voltage was between 1 and 2 kV; sampling cone voltage was at 10 V, and source temperature was 30 °C. For the ion mobility measurements and collision-induced unfolding experiments, a Trap DC Bias of 35 V was used in combination with an IMS Wave Velocity of 1000 m/s. The drift gas was nitrogen, and the collision gas was argon. The collision-induced unfolding experiments started at a trap collision energy of 4 V, continuing at 10 V in 5 V steps to 70 V and then in 10 V steps to a maximum of 160 V. The evaluation of collision-induced unfolding experiments was done with CIUSuite 2.43 CCS calibration was performed with ubiquitin from bovine44 (charge state 4+ with 949 Å2), cytochrome c from horse44 (charge state 7+ with 1536 Å2) and holomyoglobin from horse44 (charge state 7+ with 1863 Å2, 9+ with 2085 Å2). A logarithmic regression model was chosen for calibration.45 Regression coefficient R2 was 0.98964.
HDX
An FKBP12 stock solution of 4.24 mM in 20 mM sodium chloride and 20 mM HEPES was diluted with 200 mM ammonium acetate buffer (pH 7, LC–MS grade) to a concentration of 424 μM. The ligand stock solutions of 20 mM in DMSO were diluted to a concentration of 2.4 mM with DMSO. For the final solution, 9.44 μL of protein dilution and 2 μL of ligand dilution or DMSO (for Apo-FKBP12) were combined and filled up to 400 μL with 200 mM ammonium acetate buffer.
Labeling and measurements were performed by using an HDX setup from Waters. This includes a PAL RTC Autosampler from LEAP Technologies, a UHPLC with μBinary Pump and Auxiliary Pump from Waters, the HDX Manager of separate column ovens for the pepsin column and analytical column from Waters, and a Synapt XS from Waters. Protein solutions were stored in the quench tray at 1 °C under nitrogen. Three μL of protein solution was injected into the label vial. Then, 57 μL of label buffer (pH 7, 5 mM K2HPO4 and 5 mM KH2PO4 in D2O) or equilibration buffer (pH 7, 5 mM K2HPO4 and 5 mM KH2PO4 in H2O) was added and allowed to react for the set time at 20 °C. A 50 μL portion of the reaction solution was transferred to the quench vial in the quench tray. In this process, 50 μL of the quench buffer (pH 2.3, 50 mM K2HPO4 and 50 mM KH2PO4 in H2O) had already been placed at 1 °C at least 30 min earlier.
50 μL of quenched sample was injected and flowed for 3 min with a flow of 200 μL/min with 0.2% formic acid in H2O over the BEH pepsin column, 5 μm, 30 × 2.1 mm, 300 Å from Waters at 20 °C to the trap column ACQUITY UPLC BEH C18 VanGuard Precolumn, 1.7 μm, 5 × 2.1 mm, 130 Å from Waters at 1 °C. Thereafter, it started a gradient at 95% eluent A (0.2% formic acid inside H2O) and 5% eluent B (acetonitrile with 0.2% formic acid) over the trap column to the analytical column ACQUITY UPLC BEH C18, 1.7 μm, 150 × 1 mm, 130 Å from Waters at 1 °C. The gradient continued to 40% eluent A and 60% eluent B over 10 min and then to 100% eluent B over 3 min which was held for 2 min, all with a flow of 45 μL/min. ESI was used as ionization and as MS method, an HDMSe with nitrogen as the collision gas was used. As LockSpray for recalibration, a solution of 2 ng/μL leucine enkephalin inside 50:50 acetonitrile/water with 0.1% formic acid was infused.
The labeled protein samples were measured n = 5 times for each time point at 1, 10, and 100 min, and the protein samples with equilibration buffer with time point 0 min were measured with n = 2 for each protein conformation. All measurements with a time point of 0 min were combined as reference. The evaluation was performed using ProteinLynx Global Server and DynamX, both software from Waters. Based on the peptide fragmentation pattern, a score threshold of 7.75 was chosen for the identified peptides and an intensity threshold of 10,000. This resulted in a sequence coverage of 100% with 30 peptides, of which the most important ones are presented.
Nuclear Magnetic Resonance
For each FKBP-inhibitor complex, two different samples were employed to collect NOESY data. The first set of samples contained 1 mM inhibitor and 0.5 mM [u-13C/15N]-labeled FKBP in 25 mM phosphate buffer at pH7 with 5% D2O and 5% DMSO and had volumes of 0.6 mL in standard 5 mm NMR tubes (“H2O samples”). For the second set, stock solutions of inhibitors 10 and 11 in DMSO-d6 were dissolved in 99% D2O and titrated with [u-13C/15N]-labeled FKBP in a nondeuterated phosphate buffer. Final concentrations were 0.2 mM 10 with 0.04 mM FKBP and 0.36 mM 11 with 0.1 mM FKBP, respectively, with sample volumes of 0.3 mL placed in 5 mm Shigemi tubes (“D2O samples”).
Spectra were recorded at a Bruker AvIII HD 700 MHz spectrometer, equipped with a cryogenic 1H{31P/13C/15N} QCI probe and Bruker AvIII HD 800 and AvIII 950 MHz spectrometers, both equipped with cryogenic 1H{13C/15N} TCI probes.
For detection of NOEs involving the FKBP Tyr82 OH group, 2D NOESY spectra were recorded with the H2O samples at a temperature of 272 K. To avoid saturation of the exchanging hydroxyl protons, a 1–1 echo water suppression scheme was employed with the excitation maximum adjusted to 12 ppm, whereas the strong DMSO signal was suppressed by presaturation. Carbon-13 decoupling was applied in the F1 dimension. Mixing times were 30 ms for the complex with 10 and 50 ms for the complex with 11. The same set of samples was employed for the detection of NOEs with Tyr82 CεH protons, but measurements were carried out at 298 K. A 3D F1-13C/15N-filtered NOESY-[13C,1H]-HMQC (100 ms mixing time) optimized for aromatic CH groups was recorded for the complex with 11. For sensitivity reasons, only a 2D 1H–1H plane from a simple (nonfiltered) aromatic 3D NOESY-[13C,1H]-HMQC (50 ms mixing time) was recorded for the complex with 10. Here, 13C decoupling was omitted in the indirect 1H dimension to enable the identification of intermolecular NOEs from the lack of a 1JCH splitting of the cross peaks. Standard 2D NOESY spectra (10: 100 ms mixing time; 11: 70 ms mixing time) were recorded with the D2O-samples to allow for a clean detection of inhibitor peaks without a background of FKBP amide signals. The residual water signal was suppressed using excitation sculpting. Again, no 13C decoupling was applied, such that intermolecular NOEs involving the 13C labeled FKBP can be identified by the 1JCH splitting (ca. 160 Hz in the case of Tyr Hε).
NMR Titration experiments were recorded on the spectrometers described above. Sample temperatures were either 298 K (Supporting Information, Figure 16A) or 313 K (Supporting Information, Figure 16B). 15N-BEST-TROSY spectra were recorded with 384 points in t1 and 40 ppm spectral width. Protein starting concentration was 50 μM and titration steps were conducted by the addition of the corresponding volume of compounds (20 mM stock concentration).
Acknowledgments
Diffraction data have been collected on BL14.1 at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin, Germany.27 We thank HZB for the allocation of synchrotron radiation beamtime and we would particularly like to acknowledge the help and support of Manfred Weiss and the whole MX team during the experiment. We acknowledge support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) and the Open Access Publishing Fund of Technical University of Darmstadt.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00241.
NMR assignments of sulfonimidamide diastereomers, binding affinities of all sulfonamide analogues, cocrystal structures, ion mobility mass spectra, collision-induced unfolding and dissociation results, HDX measurements, NOESY spectra of FKBP12 complexes, parameters of the performed FP assays, refinement data of the cocrystal structures, synthetic procedures, and 1H- and 13C NMR spectra (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. CRediT: Thomas Nehls investigation, methodology.
This work was supported by the BMBF grant iMIP (16GW0211K) and the LOEWE cluster TRABITA. The HDX-MS instrument was funded through a grant by the German Research Foundation (DFG, project number 461372424).
The authors declare no competing financial interest.
Footnotes
https://www.rcsb.org/ → advanced search → Chemical Similarity → Query Type “Descriptor”; Descriptor Type “SMILES”; Match Type “Substructure (including Stereoisomers)” → Search “CS(N)(=O)=O”.
To the best of our knowledge, the only PDB entries for sulfonamide analogues are 2CI1, which harbors a N-thiosulfinamide, which is generated enzymatically in situ from a S-nitroso-thiol, and 7JTC and 7T36, where the geometry of the ligands needs to be confirmed and the associated publication remains to be published.
Supplementary Material
References
- Frings M.; Bolm C.; Blum A.; Gnamm C. Sulfoximines from a Medicinal Chemist’s Perspective: Physicochemical and in vitro Parameters Relevant for Drug Discovery. Eur. J. Med. Chem. 2017, 126, 225–245. 10.1016/j.ejmech.2016.09.091. [DOI] [PubMed] [Google Scholar]
- Chinthakindi P. K.; Naicker T.; Thota N.; Govender T.; Kruger H. G.; Arvidsson P. I. Sulfonimidamides in Medicinal and Agricultural Chemistry. Angew. Chem., Int. Ed. Engl. 2017, 56 (15), 4100–4109. 10.1002/anie.201610456. [DOI] [PubMed] [Google Scholar]
- Lücking U. Sulfoximines: a neglected opportunity in medicinal chemistry. Angew. Chem., Int. Ed. Engl. 2013, 52 (36), 9399–9408. 10.1002/anie.201302209. [DOI] [PubMed] [Google Scholar]
- Davies T. Q.; Hall A.; Willis M. C. One-Pot, Three-Component Sulfonimidamide Synthesis Exploiting the Sulfinylamine Reagent N-Sulfinyltritylamine, TrNSO. Angew. Chem., Int. Ed. Engl. 2017, 56 (47), 14937–14941. 10.1002/anie.201708590. [DOI] [PubMed] [Google Scholar]
- Steinkamp A.-D.; Schmitt L.; Chen X.; Fietkau K.; Heise R.; Baron J. M.; Bolm C. Synthesis of a Sulfonimidamide-Based Analog of Tasisulam and Its Biological Evaluation in the Melanoma Cell Lines SKMel23 and A375. Skin Pharmacol. Physiol. 2016, 29 (6), 281–290. 10.1159/000453042. [DOI] [PubMed] [Google Scholar]
- Thota N.; Makam P.; Rajbongshi K. K.; Nagiah S.; Abdul N. S.; Chuturgoon A. A.; Kaushik A.; Lamichhane G.; Somboro A. M.; Kruger H. G.; Govender T.; Naicker T.; Arvidsson P. I. N-Trifluoromethylthiolated Sulfonimidamides and Sulfoximines: Anti-microbial, Anti-mycobacterial, and Cytotoxic Activity. ACS Med. Chem. Lett. 2019, 10 (10), 1457–1461. 10.1021/acsmedchemlett.9b00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücking U. New Opportunities for the Utilization of the Sulfoximine Group in Medicinal Chemistry from the Drug Designer’s Perspective. Chem.—Eur. J. 2022, 28 (56), e202201993 10.1002/chem.202201993. [DOI] [PubMed] [Google Scholar]
- Bischoff M.; Sippel C.; Bracher A.; Hausch F. Stereoselective construction of the 5-hydroxy diazabicyclo4.3.1decane-2-one scaffold, a privileged motif for FK506-binding proteins. Org. Lett. 2014, 16 (20), 5254–5257. 10.1021/ol5023195. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan R.; Kozany C.; Wang Y.; Schneider S.; Hoogeland B.; Bracher A.; Hausch F. Exploration of pipecolate sulfonamides as binders of the FK506-binding proteins 51 and 52. J. Med. Chem. 2012, 55 (9), 4123–4131. 10.1021/jm201747c. [DOI] [PubMed] [Google Scholar]
- Kolos J. M.; Pomplun S.; Jung S.; Rieß B.; Purder P. L.; Voll A. M.; Merz S.; Gnatzy M.; Geiger T. M.; Quist-Løkken I.; Jatzlau J.; Knaus P.; Holien T.; Bracher A.; Meyners C.; Czodrowski P.; Krewald V.; Hausch F. Picomolar FKBP inhibitors enabled by a single water-displacing methyl group in bicyclic 4.3.1 aza-amides. Chem. Sci. 2021, 12 (44), 14758–14765. 10.1039/D1SC04638A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomplun S.; Sippel C.; Hähle A.; Tay D.; Shima K.; Klages A.; Ünal C. M.; Rieß B.; Toh H. T.; Hansen G.; Yoon H. S.; Bracher A.; Preiser P.; Rupp J.; Steinert M.; Hausch F. Chemogenomic Profiling of Human and Microbial FK506-Binding Proteins. J. Med. Chem. 2018, 61 (8), 3660–3673. 10.1021/acs.jmedchem.8b00137. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Kirschner A.; Fabian A.-K.; Gopalakrishnan R.; Kress C.; Hoogeland B.; Koch U.; Kozany C.; Bracher A.; Hausch F. Increasing the efficiency of ligands for FK506-binding protein 51 by conformational control. J. Med. Chem. 2013, 56 (10), 3922–3935. 10.1021/jm400087k. [DOI] [PubMed] [Google Scholar]
- Sun F.; Li P.; Ding Y.; Wang L.; Bartlam M.; Shu C.; Shen B.; Jiang H.; Li S.; Rao Z. Design and Structure-Based Study of New Potential FKBP12 Inhibitors. Biophys. J. 2003, 85 (5), 3194–3201. 10.1016/S0006-3495(03)74737-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.; Augelli S. C. E.; Barta N. S.; Bender S. L.; Bigge C. F.; Caprathe B. W.; Chatterjee A.; Deal J.; Dong L.; Fay L. K.; Hou X.; Hudack R. A. JR.; Warner L. C.. Heterobicycles fkbp-Ligands. WO 2002089806 A1, 2002.
- Pomplun S.; Wang Y.; Kirschner A.; Kozany C.; Bracher A.; Hausch F. Rational design and asymmetric synthesis of potent and neurotrophic ligands for FK506-binding proteins (FKBPs). Angew. Chem., Int. Ed. Engl. 2015, 54 (1), 345–348. 10.1002/anie.201408776. [DOI] [PubMed] [Google Scholar]
- van Duyne G. D.; Standaert R. F.; Karplus P. A.; Schreiber S. L.; Clardy J. Atomic structure of FKBP-FK506, an immunophilin-immunosuppressant complex. Science 1991, 252 (5007), 839–842. 10.1126/science.1709302. [DOI] [PubMed] [Google Scholar]
- van Duyne G. D.; Standaert R. F.; Karplus P. A.; Schreiber S. L.; Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 1993, 229 (1), 105–124. 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- Martzel T.; Lohier J.-F.; Gaumont A.-C.; Brière J. F.; Perrio S. Sulfinate-Organocatalyzed (3 + 2) Annulation Reaction of Propargyl or Allenyl Sulfones with Activated Imines. Eur. J. Org. Chem. 2018, 2018 (36), 5069–5073. 10.1002/ejoc.201800749. [DOI] [Google Scholar]
- Datta M.; Buglass A. J. Fast and Efficient Synthesis of Sulfinamides by the Oxidation of Sulfenamides Using Potassium Fluoride and m -Chloroperoxybenzoic Acid. Synth. Commun. 2012, 42 (12), 1760–1769. 10.1080/00397911.2010.543748. [DOI] [Google Scholar]
- Zenzola M.; Doran R.; Degennaro L.; Luisi R.; Bull J. A. Transfer of Electrophilic NH Using Convenient Sources of Ammonia: Direct Synthesis of NH Sulfoximines from Sulfoxides. Angew. Chem., Int. Ed. Engl. 2016, 55 (25), 7203–7207. 10.1002/anie.201602320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battula S.; Subbareddy G. V.; Chakravarthy I. E. A mild and efficient copper-catalyzed N-arylation of unprotected sulfonimidamides using boronic acids. Tetrahedron Lett. 2014, 55 (2), 517–520. 10.1016/j.tetlet.2013.11.084. [DOI] [Google Scholar]
- Bracher A.; Kozany C.; Thost A. K.; Hausch F. Structural characterization of the PPIase domain of FKBP51, a cochaperone of human Hsp90. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (6), 549–559. 10.1107/S0907444911013862. [DOI] [PubMed] [Google Scholar]
- Kozany C.; März A.; Kress C.; Hausch F. Fluorescent probes to characterise FK506-binding proteins. ChemBioChem 2009, 10 (8), 1402–1410. 10.1002/cbic.200800806. [DOI] [PubMed] [Google Scholar]
- Du X.; Li Y.; Xia Y.-L.; Ai S.-M.; Liang J.; Sang P.; Ji X.-L.; Liu S.-Q. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Math. Stat. 2016, 17 (2), 144. 10.3390/ijms17020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaali S.; Kirschner A.; Cuboni S.; Hartmann J.; Kozany C.; Balsevich G.; Namendorf C.; Fernandez-Vizarra P.; Sippel C.; Zannas A. S.; Draenert R.; Binder E. B.; Almeida O. F. X.; Rühter G.; Uhr M.; Schmidt M. V.; Touma C.; Bracher A.; Hausch F. Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat. Chem. Biol. 2015, 11 (1), 33–37. 10.1038/nchembio.1699. [DOI] [PubMed] [Google Scholar]
- Jagtap P. K. A.; Asami S.; Sippel C.; Kaila V. R. I.; Hausch F.; Sattler M. Selective Inhibitors of FKBP51 Employ Conformational Selection of Dynamic Invisible States. Angew. Chem., Int. Ed. Engl. 2019, 58 (28), 9429–9433. 10.1002/anie.201902994. [DOI] [PubMed] [Google Scholar]
- Gerlach M.; Mueller U.; Weiss M. S. The MX Beamlines BL14.1–3 at BESSY II. JLSRF 2016, 2, A47. 10.17815/jlsrf-2-64. [DOI] [Google Scholar]
- The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50 (5), 760–763. 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Potterton E.; Briggs P.; Turkenburg M.; Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2003, 59 (7), 1131–1137. 10.1107/S0907444903008126. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (4), 235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potterton L.; Agirre J.; Ballard C.; Cowtan K.; Dodson E.; Evans P. R.; Jenkins H. T.; Keegan R.; Krissinel E.; Stevenson K.; Lebedev A.; McNicholas S. J.; Nicholls R. A.; Noble M.; Pannu N. S.; Roth C.; Sheldrick G.; Skubak P.; Turkenburg J.; Uski V.; von Delft F.; Waterman D.; Wilson K.; Winn M.; Wojdyr M. CCP4i2: the new graphical user interface to the CCP4 program suite. Acta Crystallogr., Sect. D: Struct. Biol. 2018, 74 (2), 68–84. 10.1107/s2059798317016035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (4), 282–292. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69 (7), 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40 (4), 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. A.; Steiner R. A.; Lebedev A. A.; Potterton L.; McNicholas S.; Long F.; Murshudov G. N. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60 (12), 2184–2195. 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (4), 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (4), 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls R. A.; Long F.; Murshudov G. N. Low-resolution refinement tools in REFMAC5. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68 (4), 404–417. 10.1107/S090744491105606X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53 (3), 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Murshudov G. N.; Papiz M. Z. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003, 374, 300–321. 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- van Aalten D. M.; Bywater R.; Findlay J. B.; Hendlich M.; Hooft R. W.; Vriend G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Aided Mol. Des. 1996, 10 (3), 255–262. 10.1007/BF00355047. [DOI] [PubMed] [Google Scholar]
- Konermann L. Addressing a Common Misconception: Ammonium Acetate as Neutral pH ″Buffer″ for Native Electrospray Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28 (9), 1827–1835. 10.1007/s13361-017-1739-3. [DOI] [PubMed] [Google Scholar]
- Polasky D. A.; Dixit S. M.; Fantin S. M.; Ruotolo B. T. CIUSuite 2: Next-Generation Software for the Analysis of Gas-Phase Protein Unfolding Data. Anal. Chem. 2019, 91 (4), 3147–3155. 10.1021/acs.analchem.8b05762. [DOI] [PubMed] [Google Scholar]
- May J. C.; Jurneczko E.; Stow S. M.; Kratochvil I.; Kalkhof S.; McLean J. A. Conformational Landscapes of Ubiquitin, Cytochrome c, and Myoglobin: Uniform Field Ion Mobility Measurements in Helium and Nitrogen Drift Gas. Int. J. Mass Spectrom. 2018, 427, 79–90. 10.1016/j.ijms.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruotolo B. T.; Benesch J. L. P.; Sandercock A. M.; Hyung S.-J.; Robinson C. V. Ion mobility-mass spectrometry analysis of large protein complexes. Nat. Protoc. 2008, 3 (7), 1139–1152. 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.