

Significance
Inhibition of RIPK1 kinase has been shown to inhibit TNF-mediated apoptosis, necroptosis and inflammation. Exploring the mechanism that controls the activation of RIPK1 is key to understand its regulation and dysregulation in human diseases. Here, we explored the possibility of identifying small molecule activators of RIPK1-dependent cell death and identified R406/R788 that can directly bind and activate TAK1 to lead to sustained activation of RIPK1. Our study not only identified the mechanism by which the activation of TAK1 regulates RIPK1 but also a lead compound that can promote the cancer-killing activity of TRAIL, another member of TNF family. Our study suggests the possibility of developing small molecule activators of TAK1 as sensitizers for TRAIL-mediated anticancer therapies.
Keywords: TAK1, RIPK1, cell death
Abstract
TAK1 is a key modulator of both NF-κB signaling and RIPK1. In TNF signaling pathway, activation of TAK1 directly mediates the phosphorylation of IKK complex and RIPK1. In a search for small molecule activators of RIPK1-mediated necroptosis, we found R406/R788, two small molecule analogs that could promote sustained activation of TAK1. Treatment with R406 sensitized cells to TNF-mediated necroptosis and RIPK1-dependent apoptosis by promoting sustained RIPK1 activation. Using click chemistry and multiple biochemical binding assays, we showed that treatment with R406 promotes the activation of TAK1 by directly binding to TAK1, independent of its original target Syk kinase. Treatment with R406 promoted the ubiquitination of TAK1 and the interaction of activated TAK1 with ubiquitinated RIPK1. Finally, we showed that R406/R788 could promote the cancer-killing activities of TRAIL in vitro and in mouse models. Our studies demonstrate the possibility of developing small molecule TAK1 activators to potentiate the effect of TRAIL as anticancer therapies.
Stimulation of TNFR1 by TNFα can activate RIPK1-dependent apoptosis (RDA) or necroptosis in different cellular context (1–4). In TNFα stimulated cells, RIPK1 and TRADD, an adaptor protein, are rapidly recruited to bind with the intracellular domain of TNFR1 mediated by the respective death-domains (DD) to from TNF-signaling complex (TSC, also called complex I). The pattern of posttranslational modifications on RIPK1 in complex I, including ubiquitination and phosphorylation, decides if RIPK1 kinase activity is to be suppressed or activated. Ubiquitination of RIPK1 by cIAP1/2 in complex I is involved in mediating the activation of NF-κB by recruiting TAB1/2 to promote the activation of the TAK1 (Transforming growth factor- b-activated kinase 1, also called MAP3 K7) (5). TAK1 is an important regulator of cellular inflammatory pathways (6, 7). In the signaling pathways of TNF and TLRs ligands, TAK1, associated with TAB1, TAB2, and TAB3, is recruited by ubiquitination chain modified signaling complexes to promote its activation. Activated TAK1 phosphorylates the downstream components IKK complex as well as p38, JNK, ERK to mediate NF-κB and MAPK pathway activation (7–11). The transient phosphorylation of the intermediate domain of RIPK1 by TAK1 in complex I upon TNFα stimulation, such as Ser321 (S321), negatively regulates the activation of RIPK1, while the sustained TAK1-mediated phosphorylation of RIPK1 in the intermediate domain promotes its interaction with RIPK3 in complex IIb to mediate necroptosis (4). Thus, TAK1 is a dynamic regulator of RIPK1 activation. However, it remains unclear how sustained TAK1 activation promotes necroptosis.
TRAIL (Tumor necrosis factor-related apoptosis-inducing ligand), a member of TNF superfamily, is known to interact with its receptor DR4/5 to induce apoptosis (12, 13). TRAIL has been explored as an anticancer biotherapeutic drug candidate as it can induce apoptosis of cancer cells selectively without affecting normal cells and also with minimum systemic proinflammatory response (14). However, the outcome of TRAIL-R agonist human clinical trials suggests that additional sensitization strategies would need to be developed in order to overcome the apoptotic resistance of cancer cells to TRAIL-based therapy (15). The challenge is, however, if we can identify strategies that are safe and can sensitize cancer cells to TRAIL.
In this study, we explored the possibility of sensitizing cancer cells to necroptosis. Our study led to the identification of R406 and its pro-drug R788, an FDA-approved oral drug Fostamatinhib which is an inhibitor of the spleen tyrosine kinase (Syk) for the treatment of thrombocytopenia, as a sensitizer of TNF-induced necroptosis and RDA. We show that treatment with R406 activates TAK1 by directly binding with TAK1, independent of Syk, to promote the interaction and phosphorylation of RIPK1 with ubiquitinated TAK1. Furthermore, we demonstrate that the treatment with R406/R788 can potentiate the effect of TRAIL in promoting the death of cancer cells in vitro and in vivo. Our study suggests that promoting sustained activation of TAK1 may be considered as a potential anticancer strategy in combination with TRAIL.
Results
Identifying Small Molecule Sensitizers of Necroptosis.
We developed an assay based on TNFα-induced necroptosis of L929 cells for identifying sensitizers to necroptosis (SI Appendix, Fig. S1A). Using this assay, we screened 7,263 compounds in a known bioactive small molecule library for sensitizers to necroptosis. In this screen, we identified two closely related compounds, R406 [N4-(2,2-dimethyl-3-oxo-4H-pyrid[1,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine], an ATP-competitive inhibitor of Syk kinase (16), and its analog R788 which was designed to orally deliver the active metabolite R406, as the most effective sensitizers to necroptosis (SI Appendix, Fig. S1B and Fig. 1A). R788 (fostamatinib), sold under the brand names Tavalisse and Tavlesse, has been FDA-approved for the treatment of chronic immune thrombocytopenia (platelet loss). Treatment with R406 and R788 sensitized TNFα-induced cell death, either with TNFα alone or TNFα/zVAD.fmk, in multiple cell lines, including murine L929 cells, human FADD-deficient Jurkat cells, mouse embryonic fibroblasts (MEFs), murine 661W cells, human colorectal adenocarcinoma HT-29 cells, human lung cancer–derived NCI-H1975 cells and H1299 cells, with IC50s in single-digit micromolar concentrations (SI Appendix, Fig. S1 C–I).
Fig. 1.
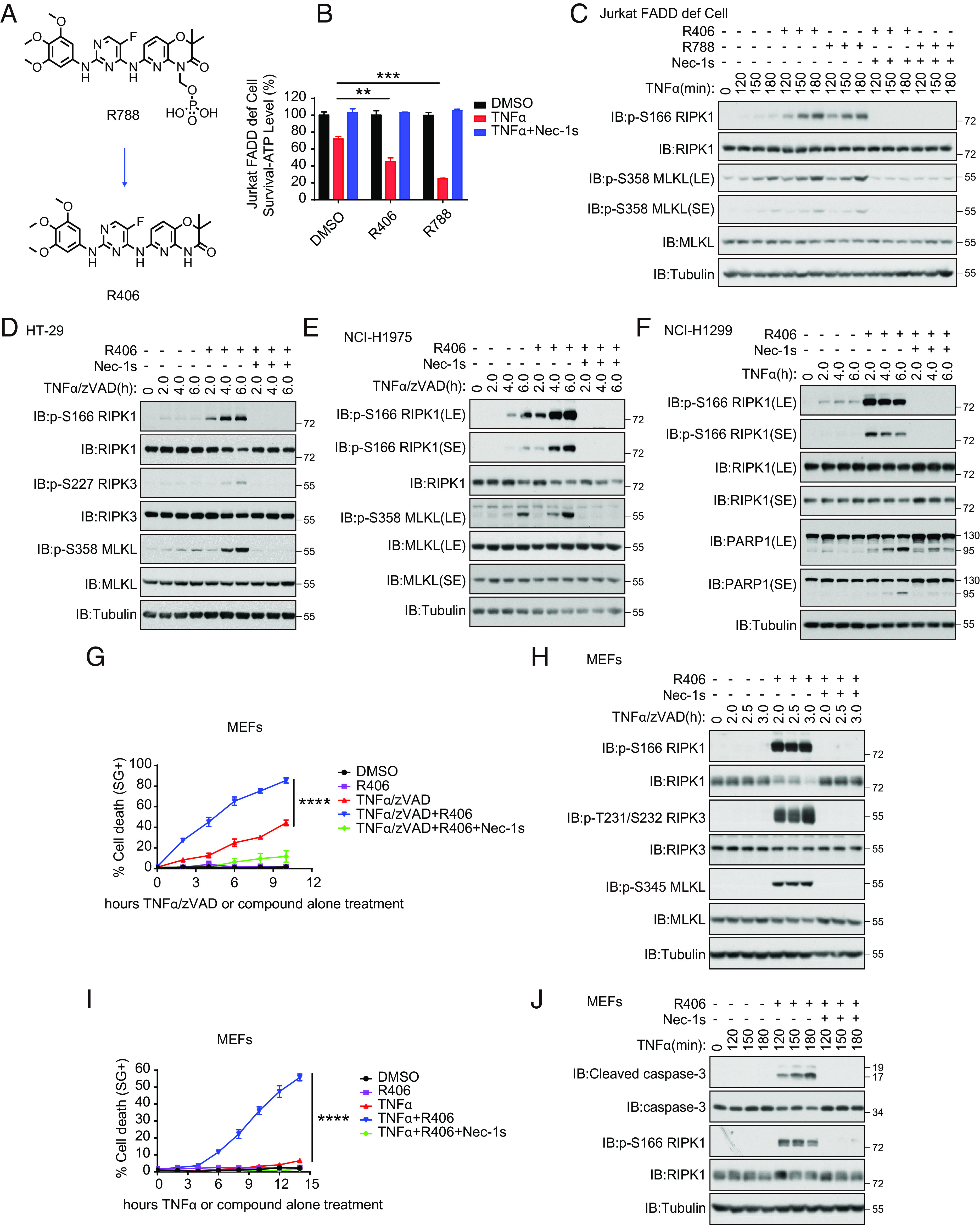
Identifying small molecule sensitizers of RIPK1-dependent cell death. (A) Chemical structure of R406 and R788. (B) FADD-deficient Jurkat cells were pretreated with or without 3 μM R406 or 3 μM R788 or 10 μM Nec-1s for 30 min as indicated, and then 2 ng/mL TNFα was added for 11 h. The cell viability was measured by CellTiter-Glo assay. Mean ± SEM of n= 3. **P< 0.01; ***P< 0.001; n.s. not significant. (C) FADD-deficient Jurkat cells were pretreated with or without R406 (3 μM) or R788 (3 μM) or Nec-1s (10 μM) for 30 min as indicated, and then 10 ng/mL TNFα was added for various time points. The cell lysates were analyzed by western blotting using antibodies for phosphorylated and total RIPK1, MLKL, and Tubulin as indicated. (D and E) HT-29 (D) and NCI-H1975 cells (E) were pretreated with or without 3 μM R406 or 10 μM. Nec-1s for 30 min as indicated and then 20 ng/mL TNFα with 20 μM zVAD.fmk were added for various time points, separately. The cell lysates were analyzed by phosphorylated and total RIPK1, RIPK3, MLKL, and Tubulin as indicated. (F) NCI-H1299 cells were pretreated with or without 3 μM R406 or 10 μM Nec-1s for 30 min as indicated, and then 20 ng/mL TNFα was added for various time points. The cell lysates were analyzed by phosphorylated and total RIPK1, PARP1 and Tubulin as indicated. (G) Necroptosis was induced in MEFs by the treatment with TNFα (20 ng/mL) and zVAD.fmk (20 μM) together with R406 (3 μM) at indicated times, and cell death was determined by STOX Green. Mean ± SEM of n= 4. ****P< 0.0001. (H) MEFs were pretreated with or without 3 μM R406 or 10 μM Nec-1s for 30 min as indicated, and then 20 ng/mL TNFα with 20 μM zVAD.fmk were added for various time points. The cell lysates were analyzed by phosphorylated and total RIPK1, RIPK3, MLKL, and Tubulin as indicated. (I) RDA was induced in MEFs by the treatment with TNFα (20 ng/mL) together with R406 (3 μM) at indicated times, and cell death was determined by SYTOX Green. Mean ± SEM of n= 4. ****P< 0.0001. (J) MEFs were pretreated with or without 3 μM R406 or 10 μM Nec-1s for 30 min as indicated, and then 20 ng/mL TNFα was added for various time points. The cell lysates were analyzed by phosphorylated-S166 and total RIPK1, Cleaved caspase-3, caspase-3 and Tubulin as indicated.
R406/R788 Sensitize to RIPK1-Dependent Necroptosis and Apoptosis.
We next characterized the effect of R406 and R788 on necroptosis. TNFα-induced necroptosis of FADD-deficient Jurkat cells, which was sensitized by the treatment with either R406 or R788, induced the activation of RIPK1 as marked by its biomarker p-S166 RIPK1 (1, 17) and p-S358 MLKL, a biomarker for necroptosis (18, 19) (Fig. 1 B and C). The cell death and phosphorylation of RIPK1 and MLKL were all inhibited by the presence of Nec-1s and thus, R406/R788 sensitized FADD-deficient Jurkat cells to necroptosis in RIPK1-dependent manner (Fig. 1 B and C). Similarly, treatment of HT-29 cells and NCI-H1975 human cancer cell lines with R406 sensitized the cell death and the activation of RIPK1 and necroptosis induced by TNFα/zVAD.fmk compared to that of TNFα/zVAD.fmk alone as indicated by the increased p-S227 RIPK3 and p-MLKL, which were inhibited by Nec-1s (Fig. 1 D and E and SI Appendix, Fig. S1 G and H). Treatment of human lung cancer cell line NCI-H1299 cells with TNFα and R406 also sensitized to RDA as shown by p-S166 RIPK1 and cleavage of PARP1 (SI Appendix, Fig. S1I and Fig. 1F).
Treatment with R406 also sensitized to necroptosis of murine cells, including MEFs, 661W cells, and BMDMs, treated with TNFα and zVAD.fmk, which was inhibited by Nec-1s (Fig. 1G and SI Appendix, Fig. S2 A and B). Increased levels of p-S166 RIPK1, p-T231/S232 RIPK3, and p-S345 MLKL in MEFs were detected upon the treatment of R406 with that of TNFα and zVAD.fmk (Fig. 1H). Similarly, treatment of 661W cells and BMDMs with R406 sensitized the activation of RIPK1 and necroptosis induced by TNFα/zVAD.fmk compared to that of TNFα/zVAD.fmk alone as indicated by the increased p-RIPK3 and p-MLKL, which were inhibited by Nec-1s (SI Appendix, Fig. S2 C and D).
Furthermore, treatment of MEFs, 661W cells and BMDMs with R406 sensitized the activation of RIPK1 and apoptosis induced by TNFα compared to that of TNFα alone as indicated by the cleavage of caspase-3, which were inhibited by Nec-1s (Fig. 1 I and J and SI Appendix, Fig. S2 E–H).
In contrast, RIPK1-independent apoptosis of MEFs, 661W cells, and BMDMs induced by the treatment with TNFα/CHX was not affected by R406 (SI Appendix, Fig. S2 I–K). Thus, treatment with R406/R788 can sensitize to RDA and necroptosis, but not TNFα-mediated apoptosis independent of RIPK1 kinase. Since R406 and R788 functioned equivalently in necroptosis and apoptosis assays, the subsequent cell-based experiments used primarily R406 only.
Sensitization of Cell Death by R406 Requires the Kinase Activity of RIPK1.
We next analyzed the requirement for R406 to sensitize RDA and necroptosis. While treatment with R406 and TNFα sensitized WT MEFs to apoptosis inhibitable by Nec-1s, the sensitization effect of R406 was lost in Ripk1−/− MEFs and restored upon the complementation of WT RIPK1 expression, but not D138N or K45M RIPK1 kinase-dead mutants (Fig. 2 A and B). The treatment of WT MEFs, but not D138N or K45M RIPK1 mutant expressing MEFs, with R406 and TNFα stimulated the activation of RIPK1, as marked by p-S166 RIPK1, was detected in MEFs expressing WT RIPK1, but not D138N or K45M RIPK1 (Fig. 2C). The addition of RIPK1 kinase inhibitor Nec-1s blocked the activation of RIPK1 induced by TNFα and R406 (Fig. 2D). The treatment of MEFs with R406 and TNFα induced apoptosis, as indicated by the cleavage of caspase-8 and PARP1, which was inhibited by genetic inactivation of RIPK1 with D138N or K45M mutations or by pharmacological inhibition of RIPK1 using Nec-1s (Fig. 2 C and D).
Fig. 2.

Sensitization of cell death by R406 requires the kinase activity of RIPK1. (A) Ripk1−/− MEFs were retrovirally transduced with vectors for empty control and HA-tagged mRIPK1, respectively. The cells stably selected with puromycin were treated as indicated for 15 h and cell death was assessed by CellTiter-Glo assay. Mean ± SEM of n = 3. **P < 0.01; ***P < 0.001; n.s. not significant (Left). The RIPK1 protein levels were analyzed by immunoblotting and equal loading was controlled by determining the tubulin levels (Right). (B) Ripk1−/− MEFs were retrovirally transduced with vectors for empty control, HA-tagged mRIPK1, and the catalytic dead mutants D138N or K45M, respectively. The cells stably selected with puromycin were treated as indicated for 11 h, and cell death was assessed by CellTiter-Glo assay. Mean ± SEM of n = 3. *P < 0.05; n.s. not significant (Left). The RIPK1 protein levels were analyzed by immunoblotting and equal loading was controlled by determining the tubulin levels (Right). (C) Ripk1−/− MEFs were retrovirally reconstituted with an empty vector (EV), HA-tagged mRIPK1, and catalytic dead mutants D138N or K45M, respectively. The cells were subsequently pretreated with or without R406 (3 μM) for 30 min and then stimulated with hTNFα (20 ng/mL) for 2 h. The cells were lysed with RIPA buffer. The cell lysates were analyzed by western blotting with the indicated antibodies. (D) MEFs were pretreated with or without 3 μM R406 or 10 μM Nec-1s for 30 min as indicated, and then 20 ng/mL TNFα was added for various time points. The cell lysates were analyzed by western blotting using antibodies for phosphorylated and total RIPK1, PARP1, Cleaved caspase-8, caspase-8, and Actin as indicated. (E) Ripk1-/- MEFs were retrovirally reconstituted with an empty vector (EV), HA-tagged mRIPK1, and catalytic dead mutants D138N or K45M, respectively. The cells were subsequently pretreated with or without R406 (3 μM) for 30 min, and then hTNFα (20 ng/mL) and zVAD.fmk (20 μM) were added for various time points. The cells were lysed with RIPA buffer. The cell lysates were analyzed by western blotting with the indicated antibodies. (F) MEFs were pretreated with or without 3 μM R406 or 10 μM Nec-1s for 30 min as indicated, and then 20 ng/mL TNFα and 20 μM zVAD.fmk were added for various time points. The cell lysates were analyzed by western blotting using antibodies for phosphorylated and total RIPK1, RIPK3, MLKL, and Tubulin as indicated. (G) HEK293T cells were transfected with NF-κB firefly luciferase plasmid and Renilla luciferase and subsequently cultured for 20 h. The cells were pretreated with or without 3 μM R406 or 0.5 μM 5Z-7, or 5 μM BAY11-7082 for 30 min and then 10 ng/mL TNFα were added for 60, 90, and 120 min. NF-κB luciferase activity was analyzed by the Dual-Luciferase Assay system.
While treatment with R406 also sensitized to necroptosis of WT MEFs treated with TNFα and zVAD.fmk, which was inhibited by Nec-1s, the sensitization effect of R406 was lost in Ripk1−/− MEFs and restored upon the complementation of WT RIPK1 expression, but not D138N or K45M RIPK1 kinase-dead mutants (Fig. 2 A and B). The treatment of WT MEFs, but not D138N or K45M mutant RIPK1 expressing MEFs, with R406, TNFα and zVAD.fmk increased levels of p-S166 RIPK1 and p-S345 MLKL (Fig. 2E). The addition of RIPK1 kinase inhibitor Nec-1s blocked the activation of RIPK1, RIPK3, and MLKL induced by TNFα, zVAD.fmk and R406 (Fig. 2F).
Since the scaffold of RIPK1 is involved in mediating NF-κB signaling induced by TNFα (20), we next measured the effect of R406 on the NF-κB signaling in cells stimulated by TNFα using Firefly/Renilla dual luciferase assay. We found that treatment with R406 increased TNFα-stimulated NF-κB promoter driven transcriptional activity, which was inhibited by TAK1 5z-7-oxozeaenol and NF-κB inhibitor BAY11-7082, suggesting that treatment with R406 promotes the activation of NF-κB (Fig. 2G). Consistently, treatment with TNFα and R406 together, compared to that of TNFα alone, led to further increases in the transcription of proinflammatory cytokines such as TNFα, Cxcl1, Cxcl2, Ccl2, and Ccl5, that are inhibited by RIPK1 inhibitor Nec-1s (SI Appendix, Fig. S3 A–E).
Taken together, these results suggest that treatment with R406 sensitized to both TNFα-mediated apoptosis and necroptosis in RIPK1-kinase-dependent manner and R406 also promotes TNFα-induced NF-κB activation and RIPK1-dependent inflammatory cytokine production.
R406 Promotes the Activation of TAK1 Kinase.
Since R406/R788 are known as inhibitors for SYK (21), we next tested the effect of SYK knockdown on TNFα-induced cell death. We found that the knockdown of SYK had no effect on the survival of MEFs, nor did it sensitize cell death in the presence of TNFα (SI Appendix, Fig. S4 A and B). In addition, treatment of PRT, another SYK inhibitor, did not sensitize cells to apoptosis or necroptosis (SI Appendix, Fig. S4C). These results suggest that the ability of R406 to sensitize RIPK1-mediated cell death is independent of SYK.
We next analyzed the effect of R406 on RIPK1 activation in complex I. We found that the increased RIPK1 activation (p-S166 RIPK1) could be detected after 15 min TNFα/R406 stimulation in Ripk1−/− MEFs complemented with WT Flag-RIPK1 (Fig. 3A). Increased activation of RIPK1 was detected in complex I associated with TNFR1 in WT MEFs stimulated with TNFα in the presence of R406 (Fig. 3 A and B). In TNFα- and R406-stimulated WT MEFs, we also detected increased and sustained p-T187 TAK1, p-S321 RIPK1, and p-IKKα/β (Fig. 3 B–D). Similarly, treatment of 661W cells and BMDMs with TNFα and R406 strikingly increased sustained p-T187 TAK1, p-S321 RIPK1, and p-IKKα/β, which were all known to be mediated by TAK1 kinase activity, compared to that of TNFα as indicated (SI Appendix, Fig. S5 A and B). Notably, the phosphorylation of IκBα, MK2 and p65, which were the cytoplasmic substrates of IKKα/β, were also increased in the presence of R406 (Fig. 3C and SI Appendix, Fig. S5 A and B). These phosphorylation events, including p-T187 TAK1, p-S321 RIPK1, p-IKKα/β, and p-IκBα, were strongly inhibited by TAK1 inhibitor (5Z)-7-oxozeaenol (5Z-7). Since p-T187 is an activation biomarker for TAK1 (22) and activated TAK1 can mediate the phosphorylation of IKKα/β and S321 RIPK1 (4), these results suggest that TAK1 is overactivated in cells treated with both TNFα and R406 compared to that of TNFα alone. Furthermore, overactivated TAK1 leads to sustained phosphorylation of its substrates associated with complex I, including IKKα/β and RIPK1, as well as its downstream cytoplasmic substrate, such as IκBα, p65, and MK2.
Fig. 3.

R406 promotes the activation of TAK1 kinase. (A) Flag-RIPK1 MEFs were pretreated with or without 3 μM R406 for 30 min and then 20 ng/mL TNFα were added for 5, 15, 30, 60 min. The cells were lysed with 0.5% Nonidet P-40 buffer. The cell lysates were collected and sequentially immunoprecipitated with anti-TNFR1 and anti-Flag antibody. First immunoprecipitation (IP): The TNFR1 complex I was immunoprecipitated using anti-TNFR1 antibody. Second IP: the supernatants after the first IP were then immunoprecipitated with anti-Flag antibody. The immunoprecipitated complexes and whole-cell lysates were analyzed by western blotting with the indicated antibodies. (B) MEFs were pretreated with or without 3 μM R406 for 30 min and then 20 ng/mL TNFα were added for 5, 15, 30, 60 min. The cells were lysed with 0.5% Nonidet P-40 buffer. The cell lysates were collected and immunoprecipitated with anti-TNFR1 antibody. The immunoprecipitated complexes and whole-cell lysates were analyzed by western blotting with the indicated antibodies. (C and D) MEFs were treated for 30 min with R406 (3 μM) or 1 h with 5Z-7 (0.5 μM), then stimulated with 20 ng/mL TNFα for indicated time points. The cells were lysed with RIPA buffer. The cell lysates were analyzed by western blotting with the indicated antibodies. (E) MEFs were pretreated with or without R406 (3 μM) and Nec-1s (10 μM) for 30 min, then 20 ng/mL TNFα and 20 μM zVAD.fmk were added for indicated time points. The cells were lysed with 0.5% Nonidet P-40 buffer and cell lysates were immunoprecipitated with anti-RIPK3 antibody. All immunoprecipitated complexes and whole-cell lysates were analyzed by western blotting with the indicated antibodies. (F) MEFs were pretreated with or without 3 μM R406 for 30 min, then 20 ng/mL TNFα and 20 μM. zVAD.fmk were added for indicated time points. The cells were lysed with 0.5% Nonidet P-40 buffer, and cell lysates were immunoprecipitated with anti-FADD antibody. All immunoprecipitated complexes and whole-cell lysates were analyzed by western blotting with the indicated antibodies.
Since overactivation of TAK1 is known to promote necroptosis (23) and we indeed detected increased phosphorylation of RIPK3 and MLKL in MEFs treated with TNFα/zVAD.fmk and R406, we next characterized the levels of complex II by immunoprecipitating FADD, RIPK1, and caspase-8 (Fig. 3 E and F and SI Appendix, Fig. S5 C and D). Supporting the ability of R406 to promote necroptosis, we detected increased amount of complex II as indicated by the coimmunoprecipitation of FADD with RIPK1 and caspase-8 as well as that of RIPK1 and RIPK3 in MEFs treated with TNFα/zVAD.fmk and R406 (Fig. 3 E and F). Furthermore, we found that the presence of Nec-1s inhibited p-S166 RIPK1, p-T231/S232 RIPK3, and p-S345 MLKL, which are the biomarkers for the activated RIPK1, RIPK3, and MLKL, respectively, as well as the interaction of RIPK1 and RIPK3 in MEFs treated with TNFα/zVAD.fmk and R406 (Fig. 3E). Similar increased formation of complex IIb was also found in 661W cells treated with TNFα/zVAD.fmk and R406 (SI Appendix, Fig. S5C).
Taken together, treatment with R406 increased the phosphorylation of TAK1 and its downstream targets and sensitized cells to TNFα-mediated necroptosis in RIPK1 kinase-dependent manner.
R406 Promotes the Interaction of TAK1 with Ubiquitinated RIPK1.
Since the treatment of R406 promoted the activation of RIPK1 in complex I upon TNFα stimulation, we hypothesized that R406 might target a component of the complex I to promote the activation of RIPK1. We examined the requirement for R406 to sensitize RIPK1 activation in MEFs, which had been made genetically missing a specific key component of the complex I, including A20, HOIP, TAK1, and ABIN-1. We found that knockout of TAK1, but not A20, HOIP, or ABIN-1, eliminated the ability of R406 to sensitize to cell death (SI Appendix, Fig. S6A). Treatment with R406 sensitized Tak1+/+ MEFs to TNFα-induced cell death, but could not further sensitize Tak1−/− MEFs to TNFα-induced cell death (SI Appendix, Fig. S6 A and B). We also measured the biochemical requirement of TAK1 for R406 to activate RIPK1. TAK1 knockout is known to sensitize the activation of RIPK1 to promote RDA (4). In Tak1-/- MEFs, treatment with R406 could not further enhance the activation of RIPK1 (SI Appendix, Fig. S6C). Inhibition of TAK1 kinase by 5z-7-oxozeaenol or expressing kinase dead TAK1-K63W mutant blocked the ability of R406 to promote the activation of TAK1 and the binding of TAK1 with RIPK1 (SI Appendix, Fig. S6D). Consistently, R406 could sensitize TNFα-treated Tak1−/− MEFs expressing WT TAK1, but not K63W mutant TAK1 (SI Appendix, Fig. S6E). Biochemically, increased levels of p-T187 TAK1, p-IKKα/β, and p-S166 RIPK1 were found in R406 and TNFα-treated Tak1−/− MEFs complemented with WT, but not K63W TAK1 mutant, compared to that of TNFα alone (Fig. 4A).
Fig. 4.

R406 promotes the interaction of TAK1 with ubiquitinated RIPK1. (A) Tak1−/− MEFs were retrovirally reconstituted with the Flag-tagged TAK1 (Flag-TAK1) and the catalytic dead mutant Flag-TAK1(K63W). The cells were subsequently pretreated with or without 3 μM R406 for 30 min and then stimulated with 20 ng ml−1 TNFα for indicated time points. The cells were lysed with 0.5% Nonidet P-40 buffer and cell lysates were immunoprecipitated with anti-Flag antibody-conjugated agarose. All immunoprecipitated complexes, and whole-cell lysates were analyzed by western blotting with the indicated antibodies. (B) MEFs were pretreated with or without 3 μM R406 for 30 min, and then 20 ng/mL TNFα was added for indicated time points. The cells were lysed with 0.5% Nonidet P-40 buffer and cell lysates were immunoprecipitated with anti-TAK1 antibody. All immunoprecipitated complexes and whole-cell lysates were analyzed by western blotting with the indicated antibodies. (C) Flag-RIPK1 MEFs were treated with or without 3 μM R406 for 30 min, and then 20 ng/mL TNFα was added for 30 min, DMSO as control. The cell lysates were immunoprecipitated with anti-Flag beads, the RIPK1 immunocomplexes were eluted with glycine followed by trypsin digestion, phosphor-peptides were enriched and analyzed by mass spectrometry.
We next characterized the effect of R406 on the interaction of TAK1 and RIPK1. TAK1 and RIPK1 are known to interact (11). We found that treatment with R406 increased p-T187 TAK1, an activation biomarker for TAK1, suggesting that R406 promotes the activation of TAK1. Strikingly even though treatment with R406 did not increase the overall ubiquitination levels of RIPK1 in complex I (Fig. 3A), it did increase the binding of TAK1 and ubiquitinated RIPK1 within 15 min of treatment while p-S166 RIPK1 was only detected at 30 min (Fig. 4 A and B), suggesting that the interaction of activated TAK1 with RIPK1 occurred before the activation of RIPK1. Furthermore, we found that inhibition of TAK1 by 5Z-7 inhibited its binding with RIPK1 (SI Appendix, Fig. S6D). The treatment with R406 promoted the binding of activated TAK1, but not inactivated K63W TAK1, with RIPK1 (Fig. 4A).
Since treatment with R406 promoted the activation of TAK1 and binding of TAK1 with RIPK1, we next determined the changes in the phosphorylation pattern of RIPK1 by mass spectrometry analysis. We found that the treatment with R406 increased p-S166 RIPK1 by 25-fold (Fig. 4C), confirming the western blot data on the increased p-S166 RIPK1 in cells treated with R406 and TNFα (Figs. 2D, 3 C and D, and 4A and SI Appendix, Fig. S6 C and F). In addition, our mass spec analysis identified increased phosphorylation of S321 RIPK1 (Fig. 4C), which was known to be phosphorylated by TAK1 (4). Murine RIPK1 Ser321 and Ser332/334/336 are conserved across mammals and are located in the intermediate domain close to the caspase-8 cleavage site Asp325 (SI Appendix, Fig. S6G). Since S332, S334, and S336 residues in RIPK1 were all known to be phosphorylated by TAK1 and we have generated anti-p-S332/S334 RIPK1 ab (4), we next used this anti-p-S332/S334 RIPK1 ab to characterize the effect of R406. We found that the treatment with R406 led to increased phosphorylation of not only S321, but also that of S332/S334, which was blocked by the presence of TAK1 inhibitor 5Z-7 (SI Appendix, Fig. S6F).
Taken together, these cellular data suggest that R406 targets TAK1 to stimulate its interaction with ubiquitinated RIPK1 to promote the activation of RIPK1.
R406 Promotes the Ubiquitination of TAK1 in the Kinase-Dependent Manner.
Since the activation of TAK1 is strongly modulated by ubiquitination and the ubiquitination is required for TAK1 to phosphorylate IKK complex (11, 24), we next analyzed the effect of R406 on TAK1 ubiquitination. Since we noted that increased TAK1 in complex I was associated with an upshifted tail (Fig. 3B), which is indicative of increased ubiquitination, we next determined the effect of R406 on K63, M1, and K48 ubiquitination of TAK1. We found that TAK1 was modulated by K63, M1, and K48 ubiquitinations, which were all inhibited by TAK1 inhibitor 5Z-7, suggesting that TAK1 is modified by different types of ubiquitin chains in the activity-dependent manner (Fig. 5A). Interestingly, treatment with R406 specifically increased K63 ubiquitination of TAK1 (Fig. 5A and SI Appendix, Fig. S7 A and B). Furthermore, inhibition of TAK1 by 5Z-7 strongly inhibited the basal as well as R406-activated TAK1 ubiquitination (Fig. 5B), suggesting that the kinase activity of TAK1 is required for the basal and R406-activated TAK1 ubiquitination.
Fig. 5.

R406 promotes the ubiquitination of TAK1 in the kinase-dependent manner. (A) HEK293T cells were co-transfected with the indicated expression vectors (Flag-TAK1, 0.5 μg; His-K63, 0.5 μg; His-M1, 0.5 μg; His-K48, 0.5 μg) for 24 h. 3 μM R406 and 500 nM 5Z-7 were added at 18 h after transfection. Cell lysates were then subjected to pull-down via Ni-NTA and analyzed by western blotting with antibodies as indicated. (B) HEK293T cells were cotransfected with expression vectors of Flag-TAK1 (0.5 μg) and His-Ub. (0.5 μg) or His-K63 (0.5 μg) for 24 h. 3 μM R406 and 500 nM 5Z-7 were added at 18 h after transfection. Cell lysates were then subjected to pull-down via Ni-NTA and analyzed by western blotting with antibodies as indicated. (C) Expression vectors harboring His-Ub (0.5 μg) and Flag-tagged TAK1 (0.5 μg), activated mutant Flag-TAK1-T178/184E (0.5 μg) or the catalytic dead mutant Flag-TAK1-K63W (0.5 μg) were transfected into HEK293T cells, 3 μM R406 and 500 nM 5Z-7 were added at 18 h after transfection. Cell lysates were then subjected to pull-down via Ni-NTA and analyzed by western blotting with antibodies as indicated. (D) Expression vectors harboring His-Ub (0.5 μg) and Flag-tagged TAK1 (0.5 μg) or various ubiquitin lysine mutants (K158R, K54R, K227R, K532R, and K447R) (0.5 μg) were transfected into HEK293T cells, cell lysates were then subjected to pull-down via Ni-NTA and analyzed by western blotting with antibodies as indicated. (E) HEK293T cells were cotransfected with expression vectors of His-Ub (0.5 μg) and Flag-TAK1. (0.5 μg) or the mutant Flag-TAK1(K158R) (0.5 μg) for 24 h. Cells were pretreated with or without 5Z-7 (250 nM) for 6 h. Cell lysates were then subjected to pull-down via Ni-NTA and analyzed by western blotting with antibodies as indicated. (F) Expression vectors harboring His-Ub (0.5 μg) and Flag-tagged TAK1 (0.5 μg) or the mutant Flag-TAK1(K158R) (0.5 μg) were transfected into HEK293T cells, 3 μM R406 and 500 nM 5Z-7 were added at 18 h after transfection. Cell lysates were then subjected to pull-down via Ni-NTA and analyzed by western blotting with antibodies as indicated. (G) Tak1−/− MEFs were retrovirally reconstituted with the Flag-tagged TAK1 (Flag-TAK1) and the mutant Flag-TAK1(K158R). The cells were subsequently pretreated with or without 3 μM R406 for 30 min and then stimulated with TNFα for indicated time points. The cell survival was measured by CellTiter-Glo assay. Mean ± SEM of n= 3. **P< 0.01; n.s. not significant. (H) Tak1−/− MEFs were retrovirally reconstituted with the Flag-tagged TAK1 (Flag-TAK1) and the mutant Flag-TAK1(K158R). The cells were subsequently pretreated with or without 3 μM R406 for 30 min and then stimulated with 20 ng ml−1 TNFα for indicated time points. The cells were lysed with 0.5% Nonidet P-40 buffer, and cell lysates were immunoprecipitated with anti-Flag antibody-conjugated agarose. All immunoprecipitated complexes and whole-cell lysates were analyzed by western blotting with the indicated antibodies.
To further investigate the role of TAK1 activity in its ubiquitination, we generated a construct expressing T178E/184E-activated TAK1 (25). We found that the ubiquitination levels of phosphomimetic T178E/184E TAK1 mutant were higher than those of WT TAK1; furthermore, the effect of treatment with R406 on TAK1 ubiquitination was reduced with this T178/184E-activated TAK1 mutant compared to that of WT (Fig. 5C). In contrast, treatment with R406 could not increase ubiquitination of TAK1-K63W kinase-dead mutant (Fig. 5C). Finally, the ubiquitination of T178E/184E-activated TAK1 was also inhibited by the treatment with TAK1 inhibitor 5Z-7. Taken together, these data suggest that the activity of TAK1 kinase promotes its own ubiquitination and is required for R406 to promote the K63 ubiquitination of TAK1.
We next characterized the ubiquitination of TAK1 in cells treated with TAK1 inhibitor 5Z-7 or R406 by mass spectrometry. Our analysis identified 11 lysine residues in TAK1 that are ubiquitinated (SI Appendix, Table S1). Interestingly, the ubiquitination of all lysine residues in TAK1 was reduced by 5Z-7 treatment and increased by R406 treatment. We generated multiple Lys(K)-to-Arg(R) TAK1 mutants to analyze the function of TAK1 ubiquitination on its activation. K63-polyubiquitination of TAK1 at Lys158 can be induced by TNFα and plays an important role in regulating its kinase activation and downstream signal transduction (26, 27). We found that K158R, but not K54R, K227R, K532R, and K447R, completely eliminated the ubiquitination of TAK1 (Fig. 5D). Importantly, K158R mutation also eliminated the autophosphorylation of p-T187 TAK1, an activation biomarker of TAK1, suggesting that ubiquitination on K158 TAK1 is important for its kinase activity (Fig. 5E and SI Appendix, Fig. S7C). Furthermore, treatment with R406 could not increase the ubiquitination or activation of TAK1 K158R mutant as indicated by p-T187 TAK1 (Fig. 5F and SI Appendix, Fig. S7D). To determine whether mutation of K158 to Arg can further abolish the kinase activity in TAK1, we tested the catalytic activity of TAK1-K158R-TAB1 protein in vitro using an ADP-Glo Kinase Assay. We found that the production of ADP catalyzed by TAK1-K158R-TAB1 was much lower than that of WT TAK1-TAB1 but nevertheless increased in a time-dependent manner, while that catalyzed by kinase dead mutant TAK1-K63W-TAB1 remained at a minimum level. The result indicates that K158R mutant retains certain levels of kinase activity (SI Appendix, Fig. S7E). Thus, K158 TAK1 is important for R406 to promote the ubiquitination and activation of TAK1.
To determine the functional role of K158 ubiquitination in cell death induced by TNFα and R406, we compared the sensitivity of TAK1−/− MEFs expressing WT or K158R mutant. We found that the expression of K158R TAK1 sensitized to TNFα-mediated RIPK1-dependent cell death compared to that of cells expressing WT TAK1; but treatment with R406 could no longer sensitize TAK1-K158R MEFs to TNFα-mediated cell death (Fig. 5G). Biochemically, treatment with R406 and TNFα increased the levels of p-T187 TAK1, p-IKKα/β, and p-S166 RIPK1 in Tak1−/− MEFs complemented with WT, but not K158R TAK1 mutant, compared to that of TNFα alone (Fig. 5H).
Taken together, these results demonstrate that K158 residue is important for TAK1 activity and for R406 to promote TNFα-mediated cell death. Future investigation on K158 may elucidate the contribution of K158 or its ubiquitination in mediating TAK1 activation.
R406 Directly Interacts with TAK1.
The data described above suggest that R406 promotes TAK1 activation in the ubiquitination-dependent manner in cells. We next characterized the effect of R406 on recombinant TAK1. Since TAB1 is essential for mediating the activation of TAK1 (22), we used a TAK1-TAB1 fusion protein known to be constitutively active (28). Flag-IKKβ (K44A) or Flag-RIPK1 was expressed and purified from HEK293T cells and incubated with recombinant TAK1-TAB1 (Fig. 6 A and B). Treatment with R406 increased the phosphorylation of p-S176/177 IKKα/β and p-S321 RIPK1 in vitro, which was blocked upon inhibition of TAK1 with 5Z-7. Treatment with R406 also increased the autophosphorylation of p-T187 TAK1, supporting the activation of TAK1 kinase activity by R406 (Fig. 6C).
Fig. 6.
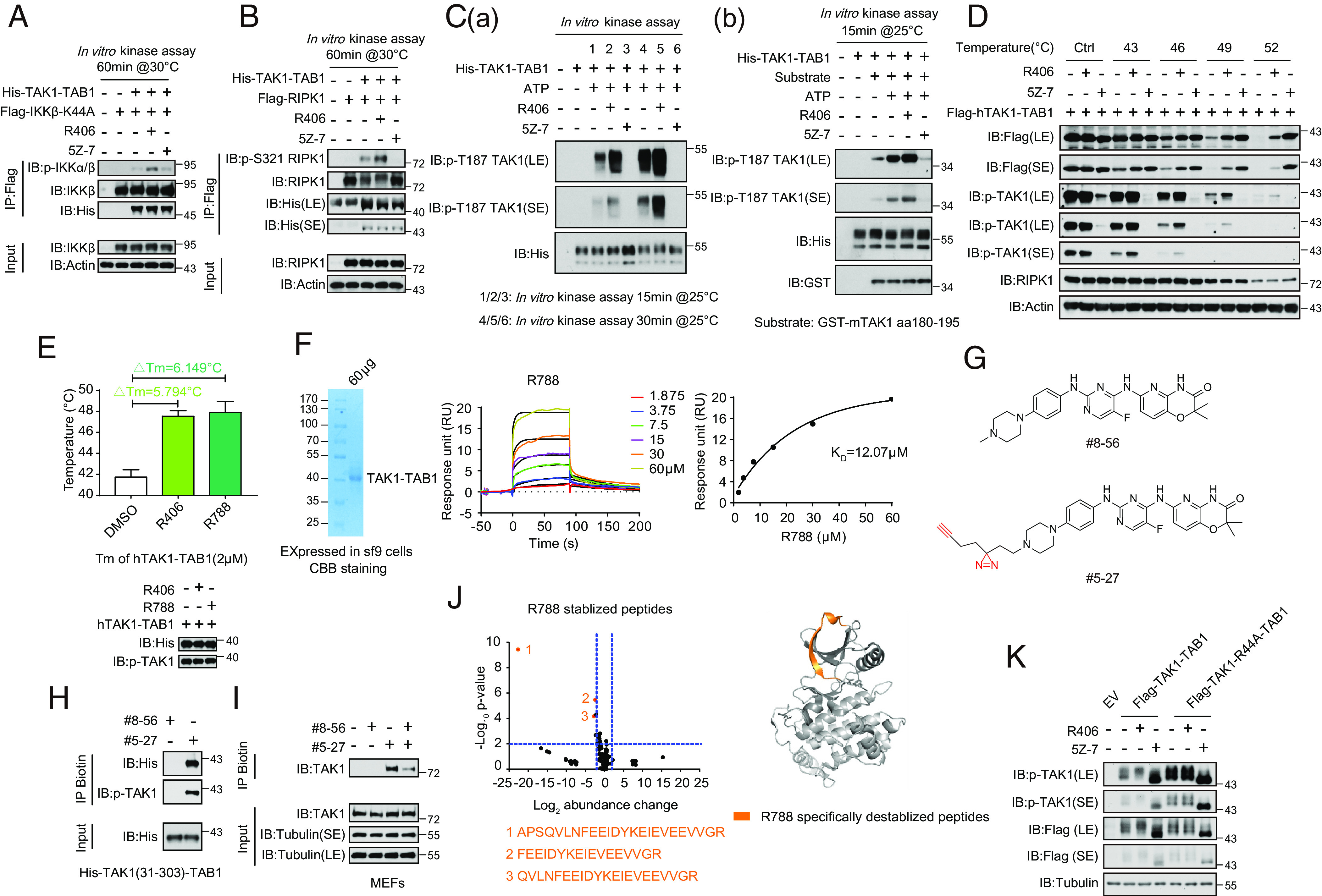
R406/R788 directly interacts with TAK1. (A) Flag-IKKβ (K44A) was expressed and purified from HEK293T cells and incubated with recombinant TAK1-TAB1 (2 μM) or R406 (20 μM), 5Z-7 (1 μM) as control. The products were analyzed by western blotting using indicated antibodies. (B) Flag-RIPK1 was expressed and purified from HEK293T cells and incubated with recombinant TAK1-TAB1 (2 μM) or R406 (20 μM), 5Z-7 (1 μM) as control. The products were analyzed by western blotting using indicated antibodies. (C, a) R406 promotes TAK1 autophosphorylation at Thr187. The TAK1 kinase activity was measured by incubating His-TAK1-TAB1 protein (2 μM) with R406 (20 μM) or 5Z-7 (1 μM) at 25 °C for 15 min and 30 min in the presence of ATP, respectively. The samples were analyzed by immunoblotting with p-TAK1 (Thr187). (b) The TAK1 kinase activity was measured by incubating His-TAK1-TAB1 protein with 4 μg GST fused mouse TAK1 peptide (aa180-195) in vitro in the presence of ATP or R406 or 5Z-7 at 25 °C for 15 min. The samples were analyzed by immunoblotting with p-TAK1(Thr187). (D). Cellular thermal stability. HEK293T cells were transfected with expression vectors for Flag-tagged hTAK1(15-303)-TAB1 fusion expression plasmids for 24 h. 3μM R406 and 500 nM 5Z-7 were added at 20 h after transfection. The HEK293T cells were harvested and resuspended with PBS and then were incubated in 0, 43, 46, 49, 52 °C for 3 min, and then frozen in liquid nitrogen quickly. The cells were subject to repeated freeze–thaw three times, and then centrifuged at 15000rpm for 15min. The products were analyzed by western blot using indicated antibodies. (E) Thermal stability profiles of protein thermal shift assay. The recombinant hTAK1(15-303)-TAB1 fusion protein (2 μM) were purified from the Bac-to-BacTM Baculovirus Expression System and treated with 160 μM R406, R788 for 2 h. The protein thermal stability was analyzed using differential scanning calorimetry by real-time PCR, and the melting temperatures were calculated by Protein Thermal Shift Software. Four replicates for each reaction were performed. (F) Kinetic profile of binding of R788 to TAK1-TAB1 from SPR analysis. (G) The chemical structures of the photoaffinity probe compound #5-27 for R406 and the photostable control compound #8-56. (H) Purified TAK1-TAB1(IIe31-Gln303, His468-Pro504) were incubated with #8-56 (200 μM) or #5-27 (200 μM) for 60 min. Samples were subjected to photo-cross-linking assay and enriched by streptavidin-coupled beads. All isolated complexes were analyzed by western blotting with indicated antibodies. (I) MEFs were pretreated with #8-56 (20 μM) or #5-27 (20 μM) or #8-56 with #5-27 for 1 h, and the cells were lysed with Nonidet P-40 buffer. Samples were subjected to photo-cross-linking assay and enriched by streptavidin-coupled beads. All immunoprecipitated complexes and whole-cell lysates were analyzed by western blotting with indicated antibodies. (J) Volcano plot showing peptides identified by LiP-MS. Recombinant hTAK1(15-303)-TAB1(468-504) purified from Sf-9 cells was treated with vehicle, R788 (200 μM), respectively, for 2 h at room temperature and then subjected to LiP-MS analysis. Abundance change of peptides in R788 treatment group compared to control group was presented as volcano plots. The x axis shows Log2 fold changes and the y axis shows −Log10 P values. Destabilized LiP-peptides were cherry picked in R788 treatment group with P-value<0.05 and abundance change>2 and further categorized into different subgroups. R788 destabilized peptides were colored orange (Left). R788 destabilized peptides were mapped to aligned structure of R788 with TAK1-TAB1 in right. R788 specifically destabilized peptides were colored orange. (K) HEK293T cells were transfected with expression vectors for Flag-tagged TAK1-TAB1 and Flag-TAK1-R44A-TAB1 fusion expression plasmid for 24 h, respectively. 3 μM R406 and 500 nM 5Z-7 were added at 20 h after transfection. The HEK293T cells were lysed with RIPA buffer and analyzed by western blotting with indicated antibodies.
We next characterized the effect of R406 on the stability of TAK1 in cells. Treatment of R406 significantly increased the thermal stability of activated TAK1 including full-length TAK1 or TAK1-TAB1 expressed in 293T cells (Fig. 6D and SI Appendix, Fig. S8A). To study if the effect of R406 on the thermal stability of TAK1 is direct, we expressed and purified the recombinant hTAK1(15-303)-TAB1 fusion protein from the Bac-to-BacTM Baculovirus Expression System (SI Appendix, Fig. S8B). Treatment with R406 in vitro increased the phosphorylation of full length TAK1 and TAK1-KD-TAB1 fusion, but not TAK1-KD alone (SI Appendix, Fig. S8C). The protein thermal stability was analyzed using differential scanning calorimetry by real-time PCR and the melting temperatures were calculated by Protein Thermal Shift Software. We found that treatment with either R406 or R788 could increase the melting temperatures of activated TAK1 (Fig. 6E and SI Appendix, Fig. S8 D and E). Thus, both R406 and R788 can increase the thermostability of activated TAK1.
We then used saturation transfer difference (STD) NMR spectroscopy to detect the interaction of compounds with TAK1-TAB1 fusion protein. The strong STD NMR signals of R406/R788 were observed in the presence of TAK1-TAB1 fusion protein but absent in the presence of GST protein as a negative control (SI Appendix, Fig. S8F). The results indicate specific direct binding between R406/R788 and TAK1-TAB1.
The binding affinity between R788 and TAK1-TAB1 was quantified by surface plasmon resonance (SPR) assay. His-tagged TAK1-TAB1 protein purified from Sf-9 cells was immobilized on the chip to detect its association with R788. The equilibrium binding constant (KD) for the interaction was determined to be 12.07 μM (Fig. 6F).
We designed and synthesized a photoclickable probe compound #5-27 for R406 and a photostable compound #8-56 as a control (Fig. 6G). Notably, only the photoreactive probe #5-27, but not the photostable control #8-56, was able to efficiently pulldown endogenous TAK1 from MEFs and recombinant TAK1-TAB1 (TAK1 residues 31-303) purified from Sf-9 cells (Fig. 6 H and I). MEFs were pretreated with #8-56 or #5-27 individually or #8-56 with #5-27 together. The cells were lysed and the cell lysates were photo cross-linked by exposing to u.v. The products of probe labeling was detected by biotin-pulldown after click reaction. We found that TAK1 was pulled down after incubating with compound #5-27, but not #8-56 (Fig. 6I). Furthermore, the presence of compound #8-56 was able to reduce the binding of TAK1 to #5-27, demonstrating the competitive nature of the interaction (Fig. 6I).
We next determined the binding site of #5-27 in TAK1 using mass spectrometry. TAK1-TAB1 (TAK1 residues 31-303, 50 μg) was cross-linked with photo-affinity probe #5-27 by UV and click-chemistry reaction. Biotin–streptavidin affinity purification enriched peptides of TAK1 were identified by mass spectrometry. MS/MS of a TAK1-TAB1 peptide 35-EIEVEEVVGR-44 cross-linked with biotinylated compound #5-27 (SI Appendix, Fig. S9A).
Limited Proteolysis-Mass Spectrometry (Lip-MS) and Hydrogen deuterium exchange-Mass Spectrometry (HDX-MS) were also performed to examine protein conformational changes affected by protein interaction and provide structural information directly or indirectly involved in protein-compound binding. As shown in Fig. 6J, compared with LiP patterns in the nonbound control group, three peptides including 21-APSQVLNFEEID YKEIEVEEVVGR-44, 28-FEEIDYKEIEVEEVVGR-44, and 24-QVLNFEEIDYKEIEVEEVVGR-44 were observed destabilized in the treatment of R788, which locate in the same region identified by the photo-affinity probe. Consistently, the hydrogen–deuterium exchange rate in this region measured by HDX-MS was also decreased in the presence of R788 (SI Appendix, Fig. S9B). Interestingly, the hydrogen–deuterium exchange rate in DFG-hinge region was significantly increased with the treatment of R788 (SI Appendix, Fig. S9B). The compound R788 can be docked into the cleft between N- and C-lobe of TAK1 kinase domain, and occupies a region with some overlap with the ATP pocket, which is consistent with the mass-spectrometry evidence (SI Appendix, Fig. S9C). Collectively, these results suggest that R788 interacts with the N-terminal β1 strand region to induce conformation change in the DFG-hinge, which promotes TAK1 kinase activation. Future structural studies may further verify this modeling study.
We analyzed our structural model of the binding between R406/R788 and TAK1 (Fig. 6L and SI Appendix, Fig. S9B). Our analysis predicts that R44 in TAK1 was important for R406 to bind and promote TAK1 kinase activity. To experimentally analyze this prediction, we overexpressed WT- and R44A-TAK1-TAB1 in HEK293T cells and found that R44A TAK1 mutant exhibited higher levels of pT187 compared to that of WT. Interestingly, treatment with R406 was not able to enhance the kinase activity of R44A-TAK1 anymore, while 5Z-7 inhibited TAK1 kinase in both WT- and R44A-TAK1 (Fig. 6K). These data support our model of R406/TAK1 interaction.
R406/R788 Promote the Cancer-Killing Activity of TRAIL.
We next examined the effect of R406/R788 on TRAIL-induced cell death. Triple-negative mammary tumor–derived E0771 cells (29) were treated with different concentrations of TRAIL and R406. We found that the treatment with R406 dose–dependently sensitized E0771 cells to TRAIL-induced killing from IC50 ~34.21 nM to 1 nM (Fig. 7A). Treatment with R406 in the presence of TRAIL also increased the binding of TAK1 with RIPK1, p-S166, RIPK1 and the cleavage of caspase-3 and PARP-1, as that of TRAIL-treated cells (Fig. 7B). In order to test the effect of TRAIL and R788 combination in vivo, we established ZsGreen-luciferase-expressing E0771 cells and found that E0771-ZsGreen cells were equally sensitive to TRAIL/R788 combination as that of parental E0771 cells (SI Appendix, Fig. S10A).
Fig. 7.
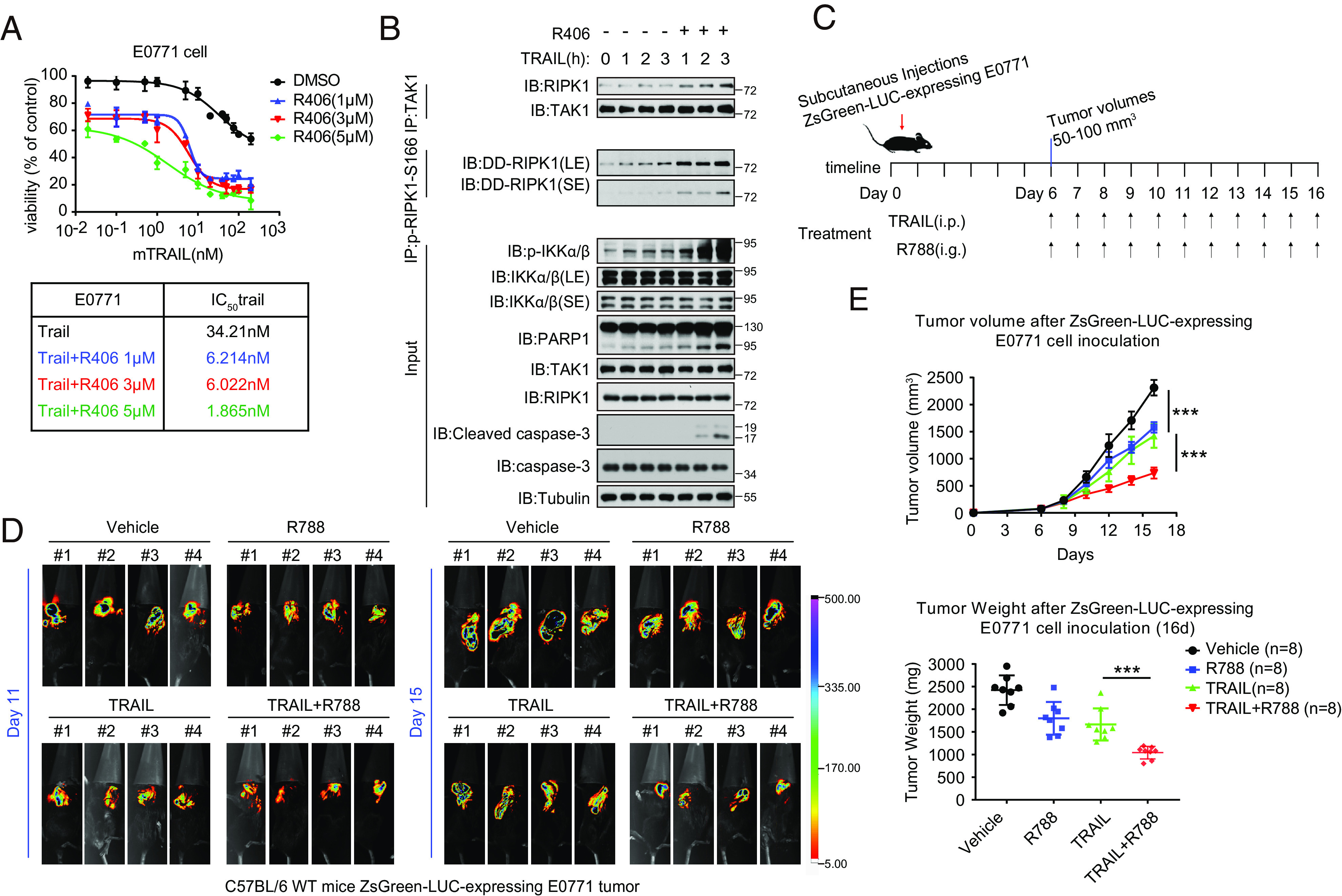
R406/R788 promote the cancer-killing activity of TRAIL. (A) E0771 cells treated with TRAIL, with or without three different concentrations of R406, for 24 h. Representative dose–response curves, tabular listing of IC50 values for trail with or without increasing concentrations of R406. (B) E0771 tumor cells were treated with 3 μM R406 for 30 min and then 10 ng/mL mTRAIL was added for various time points. The cell lysates were analyzed by western blotting. (C) Schematic diagram of the experimental schedule. E0771 cells were engineered to express. ZsGreen luciferase (ZsGreen-LUC-expressing E0771). After SC injection 11 and 15 d, C57BL/6 (6 to 8 wk old, female), were injected with D-Luciferin (150 mg/kg) in situ, respectively. Progression of breast tumor burden and treatment effects in mice were monitored in vivo and ex vivo by bioluminescent imaging (BLI). (D) ZsGreen-LUC-expressing E0771 tumor cells were implanted in 6-wk-old C57BL/6 WT female mice. After 16 d, collected the tumor tissues of female mice. (E) ZsGreen-LUC-expressing E0771 tumor parameters in C57BL/6 mice during indicated treatment. Mean ± SEM of n = 8. ***P < 0.001.
E0771-ZsGreen cells were implanted subcutaneously to C57BL/6 mice to generate triple negative medullary breast adenocarcinoma model to be treated with vehicle alone, R788 alone, TRAIL alone, or TRAIL plus R788 (Fig. 7C). The progression of breast tumor burden and treatment effects in mice were monitored in vivo and ex vivo by bioluminescent imaging (BLI). We found that the combination of R788 and TRAIL treatment significantly reduced the tumor burden compared to that of TRAIL alone, R788 alone or vehicle (Fig. 7 D and E and SI Appendix, Fig. S10B). We analyzed the tumor samples for histological biomarkers including cleaved caspase-3 for apoptosis, Ki-67 for proliferation and CD31 for angiogenesis (SI Appendix, Fig. S10 C–E). We found that the combination treatment of TRAIL and R788 increased levels of cleaved caspase-3 while reduced the levels of Ki-67 and CD31 compared to that of TRAIL. Thus, R788 can promote the effect of TRAIL on promoting apoptosis of cancer cells while inhibiting proliferation and angiogenesis in the tumors.
We chose to test the combination of R788 and TRAIL on another cancer model, primary Lewis lung carcinoma-derived LLC cells. The treatment with R406 also dose-dependently sensitized LLC cells to TRAIL-induced killing from IC50 ~37.77 nM to 3.56 nM (SI Appendix, Fig. S11A). Treatment of LLC cells with R406 in the presence of TRAIL also increased the binding of TAK1 with RIPK1, p-S166 RIPK1, and the cleavage of PARP-1, as that of TRAIL-treated cells (SI Appendix, Fig. S11B). LLC cells were implanted subcutaneously to C57BL/6 mice to generate xenograft lung carcinoma model to be treated with vehicle alone, R788 alone, TRAIL alone, or TRAIL plus R788 (SI Appendix, Fig. S11C). We found that the combination treatment of R788 and TRAIL significantly reduced the tumor burden compared to that of TRAIL alone, R788 alone, or vehicle (SI Appendix, Fig. S11 C and D). We found that the levels of p-S166 RIPK1 were also increased in LLC tumor samples (SI Appendix, Fig. S11E). Similar to that of E0771 xenograft tumors, the combination treatment of R788 and TRAIL reduced the levels of Ki-67 and CD31 but increased cleaved caspase-3 (SI Appendix, Fig. S11F).
Taken together, our data suggest that the combination of TRAIL and R788 can promote the cancer-killing activity of TRAIL.
Discussion
In this study, we isolated R406/R788 as small molecule activators of TAK1 that promote the activation of TAK1 in K63 ubiquitination-dependent manner. The activation of TAK1 is strongly modulated by K63 ubiquitin chains (11, 24). Our results suggest that ubiquitinated RIPK1 might provide important source of such ubiquitin chains in the local environment of complex I where TAK1 is recruited and activated. Upon recruitment into complex I, TRADD organized complex ubiquitin modification on RIPK1, including M1, K63, and K48 ubiquitination, which dictates if RIPK1 is to be activated (30). Activated TAK1 in R406-stimulated cells preferentially binds with ubiquitinated RIPK1 in complex I within 15 min of stimulation by TNFα, even though R406 treatment does not increase the ubiquitination levels of RIPK1 per se. Binding of activated TAK1 with ubiquitinated RIPK1 in R406-treated cells may have three major consequences. The first is the ubiquitination of TAK1 at K158 residue and other sites, which may be mediated by one or more E3 ubiquitinating enzymes that are recruited into complex I that can also mediate RIPK1 ubiquitination. The identity of such E3(s) needs to be worked out in future. The second is the stabilization of activated TAK1 by its ubiquitination. In the absence of R406, TNFα stimulation can only lead to transient activation of TAK1 for ~15 min as marked by p-T187 TAK1, downstream p-S176/177 IKKα/β and p-S321 RIPK1. Treatment with R406 leads to sustained activation of TAK1 upto an hour. The third is the activation of RIPK1 mediated by TAK1 phosphorylation. Activated RIPK1 in turn mediates apoptosis and necroptosis (Fig. 8).
Fig. 8.

A schematic model of R406/R788 promotes TAK1 activity-dependent ubiquitination and TNFα-mediated cell death. Activated TAK1 in R406/R788-stimulated cells preferentially binds with ubiquitinated RIPK1 in complex I within 15 min of stimulation by TNFα. Binding of activated TAK1 with ubiquitinated RIPK1 in R406/R788 treated cells may have three major consequences. The first is the ubiquitination of TAK1 at K158 residue and other sites, which may be mediated by one or more E3 ubiquitinating enzymes that are recruited into complex I that can mediate RIPK1 ubiquitination. The second is the stabilization of activated TAK1 by its ubiquitination. Treatment with R406/R788 leads to sustained activation of TAK1 upto an hour. The third is the activation of RIPK1 mediated by TAK1 phosphorylation. Activated RIPK1 in turn mediates apoptosis and necroptosis.
NF-κB is a double-edged sword regulating both proinflammatory and prosurvival genes. A well-balanced and precisely controlled response is essential to maintain the homeostasis. A rapid inflammatory response to stimuli followed by a rapid drop of the signal may ensure effective clearance of pathogens and avoid excessive inflammation. In our study, we identify that treatment with R406 promotes sustained activation of TAK1-RIPK1, which directly leads to downstream caspase cleavage (apoptosis), RIPK3 activation (necroptosis), and prolonged NF-κB activation, Therefore, treatment with R406/R788 promotes a sustained procell death signaling, which ultimately leads to cell death. Thus, sustained activation of TAK1 leads to upregulation of immune response by promoting the ubiquitination of TAK1 and activation of RIPK1-mediated cell death. It may be interesting in future to test if the activation of TAK1 by R788 not only can sensitize TRAIL-mediated cancer cell death but also to be considered to potentiate PD1/PD-L1-based cancer checkpoint immune therapies.
The possibility of TRAIL as a proapoptotic member of TNF family to promote cancer killing independent of p53 without inducing systemic inflammatory response has raised much excitement as a new anticancer therapy in late 1990s (31). However, the subsequent studies suggest that new strategies must be developed to overcome the inherent and acquired resistance of cancer cells to TRAIL (32). Here we show that the sensitivity of cancer cells to TRAIL-induced apoptosis is regulated by TAK1, a key signaling kinase that regulates both the activation of NF-κB pathway as well as the activation of RIPK1. TRAIL can activate apoptosis via two death-domain-containing membrane receptors, TRAIL-R1 (DR4) and TRAIL-R2 (DR5) (31). The binding of TRAIL with DR4 and DR5 leads to the formation of DISC to the intracellular DD of DR4 and DR5 by the direct recruitment of FADD, a death-domain-containing adaptor, and caspase-8 via the DD-mediated interaction. Alternatively, the activation of DR4/DR5 can also lead to the formation of intracellular signaling complexes that may be similar to that of complex I, involving RIPK1 and TRADD, as well as TAK1, IKKs, and MAPKs, as that in TNFR1 signaling (33). The ability of R406/R788 to sensitize cancer cells to both TNFα and TRAIL-mediated RIPK1-dependent cell death suggests that the intracellular signaling pathways activated by TNFα and TRAIL may be very similar.
Treatment of R788/TRAIL combination showed a strong effect in inhibiting angiogenesis in xenograft cancer models. Angiogenesis is a vital process for promoting the growth and metastasis of solid cancers. Angiogenic or proangiogenic responses in cancers have been shown to underlie cell adaption to facilitate TRAIL resistance and cancer progression (34). TRAIL is known to modulate angiogenesis as endothelial cells from tumor vasculature can also express TRAIL receptors and thus, a part of antitumor effect of TRAIL/R788 combination may be due to its targeting of tumor endothelial cells. Such adaptive mechanism that promotes TRAIL resistance is known to be associated with activation of self-renewing pathways by mitogen-activated protein kinases (MAPKs) and NF-κB pathway, which can regulate the downstream target genes such as c-FLIP, which can directly control the activation of caspase-8 to mediate apoptosis.
Taken together, our study suggests that sustained activation of NF-κB and of RIPK1 kinase by promoting the activation of TAK1 may be considered as a strategy to potentiate existing anticancer therapies.
Materials and Methods
Reagents and Antibodies.
The following commercial antibodies and reagents were used in this study: RIPK1 (Cell Signaling Technology, #3493); p-S166 RIPK1 (Cell Signaling Technology, #31122, mouse specific); p-S166 RIPK1 (Cell Signaling Technology, #65746, human specific); p-RIPK3 (Cell Signaling Technology, #93654, human specific); p-RIPK3 (Cell Signaling Technology, #91702, mouse specific); FADD (Abcam, #ab124812; Santa Cruz, #6036); α-Tubulin (Sigma-Aldrich, #T9026); β-actin (TransGen Biotech HC201); Flag (Cell Signaling Technology, #2368); Caspase-3 (Cell Signaling Technology, #9662); cl-Caspase-3 (Cell Signaling Technology, #9661); Caspase-8 (Cell Signaling Technology, #4927); cl-Caspase-8 (Cell Signaling Technology, #8592); TAK1 (Cell Signaling Technology, #5206; Santa Cruz, sc166562); p-TAK1 (Cell Signaling Technology, #4536); Syk (Cell Signaling Technology, #13198); TNFR1 (R&D Systems, AF-425-PB); Recombinant hTNFα (Novoprotein, #C008); Flag antibody-conjugated agarose (Sigma, #A2220); Ni-NTA His-Bind Resin (EMD Millipore 70666-4); p-S345 MLKL (Cell Signaling Technology, #37333, mouse specific); p-S358 MLKL (Cell Signaling Technology, #91689, human specific); MLKL (Abcam, #ab172868); ABIN-1 (Ubiquigent, #68-0001-100); A20 (Cell Signaling Technology, #5630); TRAF2 (Cell Signaling Technology, #4712); HOIP (Abcam, #ab46322); anti-sheep (SouthernBiotech, #2018-02); anti-rabbit (SouthernBiotech, #4050-05); anti-mouse (SouthernBiotech, #1031-05); IκBα (Cell Signaling Technology, #4814); p-IκBα (Cell Signaling Technology, #2859); IKKα (Cell Signaling Technology, #2682); IKKβ (Cell Signaling Technology, #8943); p-IKKα/β (Cell Signaling Technology, #2078); p38 (Cell Signaling Technology, #9212); p-p38 (Cell Signaling Technology, #9211); p65 (Cell Signaling Technology, #8242); p-p65 (Cell Signaling Technology, #3033); MK2 (Cell Signaling Technology, #3042); p-MK2 (Cell Signaling Technology, #3041); His (Cell Signaling Technology, #12698); GST (Santa Cruz, #sc-138); PARP (Cell Signaling Technology, #9532); SYRO Orange Protein gel stain (Life Technologies, #s6651); R406 (Selleck, S1533); R788 (Selleck, S2625); PRT (Selleck, S7738); 5Z-7 (Sigma-Aldrich, #O9890); zVAD.fmk (MedChemExpress, HY-16658); Cycloheximide (MCE, HY-12320); Sf-9 cell medium ESF-921 (ExpressionSystems, #96-001-01); ADP-GloTM kinase assay (Promega, #V6930).
Quantification and Statistical Analysis.
All cell death data are presented as mean ± SEM of one representative experiment. Each experiment was repeated at least three times. Mouse tumor data are presented as mean ± SEM of the indicated n values. All immunoblots were repeated at least three times independently with similar results. Curve fitting and statistical analyses were performed using GraphPad Prism 8.0 or Microsoft Excel 2016 software, using either unpaired two-tailed Student’s t test for comparison between two groups. Differences were considered statistically significant if P < 0.05. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Dr. Henning Walczak for kindly providing Hoip−/− MEF cells, Dr. Zhenggang Liu for Traf2−/− MEFs, Dr. Averil Ma of UCSF for Abin-1−/− MEFs and A20−/− MEFs. This work was supported in part by the National Natural Science Foundation of China (82188101, 21837004, 92049303, 32170755 and 32071297), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB39030200 and XDPB1003), the Shanghai Municipal Science and Technology Major Projects (2019SHZDZX02, 22JC410400 and E1D0211A19). A patent has been filed on the use of TAK1 activators for sensitization of TRAIL-induced cancer cell death.
Author contributions
J.Y. conceived the idea; W.S., Jianping Liu, Y.L., and J.Y. designed research; W.S., G.W., Xinyu Tian, C.Q., Jingli Liu, Y.T., M.Z., J.G., Z.C., Y.Z., Z.L., Xiaoxu Tian, P.W., C.P., J. Li, L.T., and B.S. performed research; W.S., Jianping Liu, Y.L., and J.Y. analyzed data; and W.S., Jianping Liu, Y.L., and J.Y. wrote the manuscript.
Competing interests
A patent has been filed on small molecule activators of TAK1 as sensitizers for TRAIL-mediated anti-cancer therapies.
Footnotes
Reviewers: J.N.-T., North Carolina State University; and E.W., Rutgers University.
Contributor Information
Jianping Liu, Email: jpliu@sioc.ac.cn.
Ying Li, Email: liying@sioc.ac.cn.
Junying Yuan, Email: junying_yuan@sioc.ac.cn.
Data, Materials, and Software Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD044897 (35). All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Degterev A., et al. , Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Degterev A., et al. , Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Yuan J., Amin P., Ofengeim D., Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geng J., et al. , Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 8, 359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanayama A., et al. , TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol. Cell 15, 535–548 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Omori E., et al. , TAK1 is a master regulator of epidermal homeostasis involving skin inflammation and apoptosis. J. Biol. Chem. 281, 19610–19617 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato S., et al. , Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 6, 1087–1095 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Adhikari A., Xu M., Chen Z. J., Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 26, 3214–3226 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Ajibade A. A., Wang H. Y., Wang R. F., Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 34, 307–316 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Ninomiya-Tsuji J., et al. , The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398, 252–256 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Wang C., et al. , TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Wiley S. R., et al. , Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3, 673–682 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Pitti R. M., et al. , Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 271, 12687–12690 (1996). [DOI] [PubMed] [Google Scholar]
- 14.Walczak H., et al. , Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 5, 157–163 (1999). [DOI] [PubMed] [Google Scholar]
- 15.von Karstedt S., Montinaro A., Walczak H., Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 17, 352–366 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Braselmann S., et al. , R406, an orally available spleen tyrosine kinase inhibitor blocks fc. receptor signaling and reduces immune complex-mediated inflammation. J. Pharmacol. Exp. Ther. 319, 998–1008 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Ofengeim D., et al. , Activation of necroptosis in multiple sclerosis. Cell Rep. 10, 1836–1849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun L., et al. , Mixed lineage kinase domain-like protein mediates necrosis signaling. downstream of RIP3 kinase. Cell 148, 213–227 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Chen W., et al. , Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 288, 16247–16261 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelliher M. A., et al. , The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8, 297–303 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Suljagic M., et al. , The Syk inhibitor fostamatinib disodium (R788) inhibits tumor growth in the Emu- TCL1 transgenic mouse model of CLL by blocking antigen-dependent B-cell receptor signaling. Blood 116, 4894–4905 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Shibuya H., et al. , TAB1: An activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science 272, 1179–1182 (1996). [DOI] [PubMed] [Google Scholar]
- 23.Mihaly S. R., Ninomiya-Tsuji J., Morioka S., TAK1 control of cell death. Cell Death Differ 21, 1667–1676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia Z. P., et al. , Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 461, 114–119 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu Y., et al. , Phosphorylation of Thr-178 and Thr-184 in the TAK1 T-loop is required for interleukin (IL)-1-mediated optimal NFkappaB and AP-1 activation as well as IL-6 gene expression. J. Biol. Chem. 283, 24497–24505 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan Y., et al. , Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor. necrosis factor alpha- and interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J. Biol. Chem. 285, 5347–5360 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang L., et al. , TAK1 ubiquitination regulates doxorubicin-induced NF-κB activation. Cell Signal 25, 247–254 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakurai H., et al. , TAK1-TAB1 fusion protein: A novel constitutively active mitogen-activated protein kinase kinase kinase that stimulates AP-1 and NF-kappaB signaling pathways. Biochem. Biophys. Res. Commun. 297, 1277–1281 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Yelek C., et al. , Tumor metabolism is affected by obesity in preclinical models of triple-negative breast cancer. Cancers (Basel) 14, 562 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ofengeim D., Yuan J., Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 14, 727–736 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Pan G., et al. , The receptor for the cytotoxic ligand TRAIL. Science 276, 111–113 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Deng D., Shah K., TRAIL of hope meeting resistance in cancer. Trends Cancer 6, 989–1001 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Azijli K., Weyhenmeyer B., Peters G. J., de Jong S., Kruyt F. A., Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: Discord in the death receptor family. Cell Death Differ 20, 858–868 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quiroz-Reyes A. G., et al. , Behind the adaptive and resistance mechanisms of cancer stem cells to TRAIL. Pharmaceutics 13, 1062 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan J., Shan B., R406 binding site on TAK1 identified by MS. ProteomeXchange. https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD044897. Deposited 27 August 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD044897 (35). All study data are included in the article and/or SI Appendix.