

Significance
Chromosomal aberrations, such as dicentric or isochromosomes, are prevalent in cancer and developmental syndromes, yet mechanisms promoting their origin remain elusive. Here, we demonstrate that a key epigenetic mark, H3K36me3, and its regulator, the tumor suppressor SETD2, preserve genomic integrity by preventing isochromosome formation. Tumors with SETD2 loss exhibit increased intratumoral heterogeneity and are more resistant to chemotherapy. We propose that SETD2 loss causes the formation of isochromosomes, isodicentric and dicentric chromosomes that may be the source of this heterogeneity contributing to therapeutic resistance.
Keywords: chromosome, mitosis, isochromosome, intratumoral heterogeneity, epigenetics
Abstract
Isochromosomes are mirror-imaged chromosomes with simultaneous duplication and deletion of genetic material which may contain two centromeres to create isodicentric chromosomes. Although isochromosomes commonly occur in cancer and developmental disorders and promote genome instability, mechanisms that prevent isochromosomes are not well understood. We show here that the tumor suppressor and methyltransferase SETD2 is essential to prevent these errors. Using cellular and cytogenetic approaches, we demonstrate that loss of SETD2 or its epigenetic mark, histone H3 lysine 36 trimethylation (H3K36me3), results in the formation of isochromosomes as well as isodicentric and acentric chromosomes. These defects arise during DNA replication and are likely due to faulty homologous recombination by RAD52. These data provide a mechanism for isochromosome generation and demonstrate that SETD2 and H3K36me3 are essential to prevent the formation of this common mutable chromatin structure known to initiate a cascade of genomic instability in cancer.
Genome maintenance is one of the highest order functions of a cell. Numerous processes safeguard against errors in replication and segregation of chromosomes during mitosis. Genomic instability and aneuploidy are also hallmarks of cancer and can be initiated by chromosome instability (CIN), defined as a propensity to mis-segregate chromosomes during cell division (1). Mis-segregated chromosomes can become incorporated into micronuclei, the cytoplasmic DNA-containing structures that are subjected to double-stranded breaks (DSBs) and chromothripsis (2–5). Amplification or deletion of genomic material or altered chromosomal structures downstream of these events have potential to be deleterious and oncogenic.
One such structure is the class of abnormal chromosomes known as isochromosomes. These mirror-imaged chromosomes are commonly observed in hematopoietic and solid tumors and genetic disorders such as Turner syndrome (6, 7). Isochromosomes result in duplications of one chromosome arm contained in the isochromosome and deletion of the unincorporated arm. They may also contain two centromeres, generating a dicentric (8). Dicentric chromosomes form chromatin bridges during anaphase, as microtubules from opposite poles attach to each centromere. Following anaphase, dicentric chromosomes may break causing kataegis and chromothripsis, ultimately promoting subclonal heterogeneity within cell populations and tumors (9–11). Yet, while the high prevalence and importance of dicentric and isochromosomes are appreciated, mechanisms accounting for their origin in human disease are poorly understood.
SETD2 is a methyltransferase responsible for the trimethylation of lysine 36 on histone H3 (H3K36me3). SETD2 and this epigenetic mark regulate transcriptional fidelity, RNA splicing, and the DNA damage response (12). SETD2 is also widely recognized as a tumor suppressor that is frequently mutated or deleted in many tumor types, including clear cell renal cell carcinoma (ccRCC), pediatric acute lymphoblastic leukemia, lung, bladder, and uterine cancer (13). Intriguingly, recent work in ccRCC demonstrated that tumors with SETD2 mutations exhibit branched, clonal evolution and more intratumoral heterogeneity than tumors with other driver mutations (14). Consistent with a role in genome maintenance, SETD2 loss has been shown to disrupt mitotic spindle organization during mitosis to promote CIN, which may contribute to the tumor suppressive role for this enzyme (15, 16). However, a mechanism linking mitotic errors to the origin of branched evolution in cells or tumors with mutation or deletion of SETD2 remains unknown. As chromosome bridging can drive subclonal heterogeneity in cultured cells, we investigated the prevalence of bridging in cells lacking SETD2.
While investigating the origin of chromosome mis-segregation in cells lacking SETD2, we found that SETD2 and H3K36me3 prevent the generation of isochromosomes and isodicentric chromosomes, establishing a critical link between these epigenomic regulators and genomic instability.
Results
Deletion of SETD2 Leads to Bridging and Lagging Chromosomes in Mitosis.
To quantify the frequency of chromosome mis-segregation following Setd2 loss, mouse embryonic fibroblasts (MEFs) with wild-type (Wt/Wt), heterozygous (Fl/Wt), and homozygous (Fl/Fl) floxed alleles of Setd2 were treated with vehicle or 4-OHT to activate Cre-ER and acutely delete Setd2. Three days after treatment of 4-OHT, monoallelic deletion of Setd2 resulted in decreases in both Setd2 and H3K36me3, whereas biallelic loss resulted in complete loss of Setd2 and H3K36me3 (SI Appendix, Fig. S1A). H3K36me3 was also lost across chromosome arms and at subtelomeric and pericentric regions (SI Appendix, Fig. S1B). Mono- and biallelic loss of Setd2 increased the prevalence of mitotic cells with lagging, bridging, or both types of mis-segregated chromosomes during the late anaphase and early telophase (Fig. 1 A and B). We have previously posited that lagging chromosomes arise from alterations in chromosome-microtubule attachment, while bridges may be caused by dicentric chromosomes (SI Appendix, Fig. S1C) (1, 15, 16). To confirm whether acute deletion of SETD2 causes both lagging and bridging chromosomes in human cells, HeLa cells expressing tetracycline-inducible Cas9 (HeLa TetOn-Cas9) (17), and either nontargeting (NT) or SETD2-specific gRNAs, were treated with doxycycline for 3 d, and chromosome mis-segregation was quantified. Doxycycline treatment caused loss of SETD2 as well as H3K36me3 along chromosome arms and at subtelomeric and pericentric regions (SI Appendix, Fig. S1 D and E), promoting increases in both lagging and bridging chromosomes (Fig. 1 C and D).
Fig. 1.
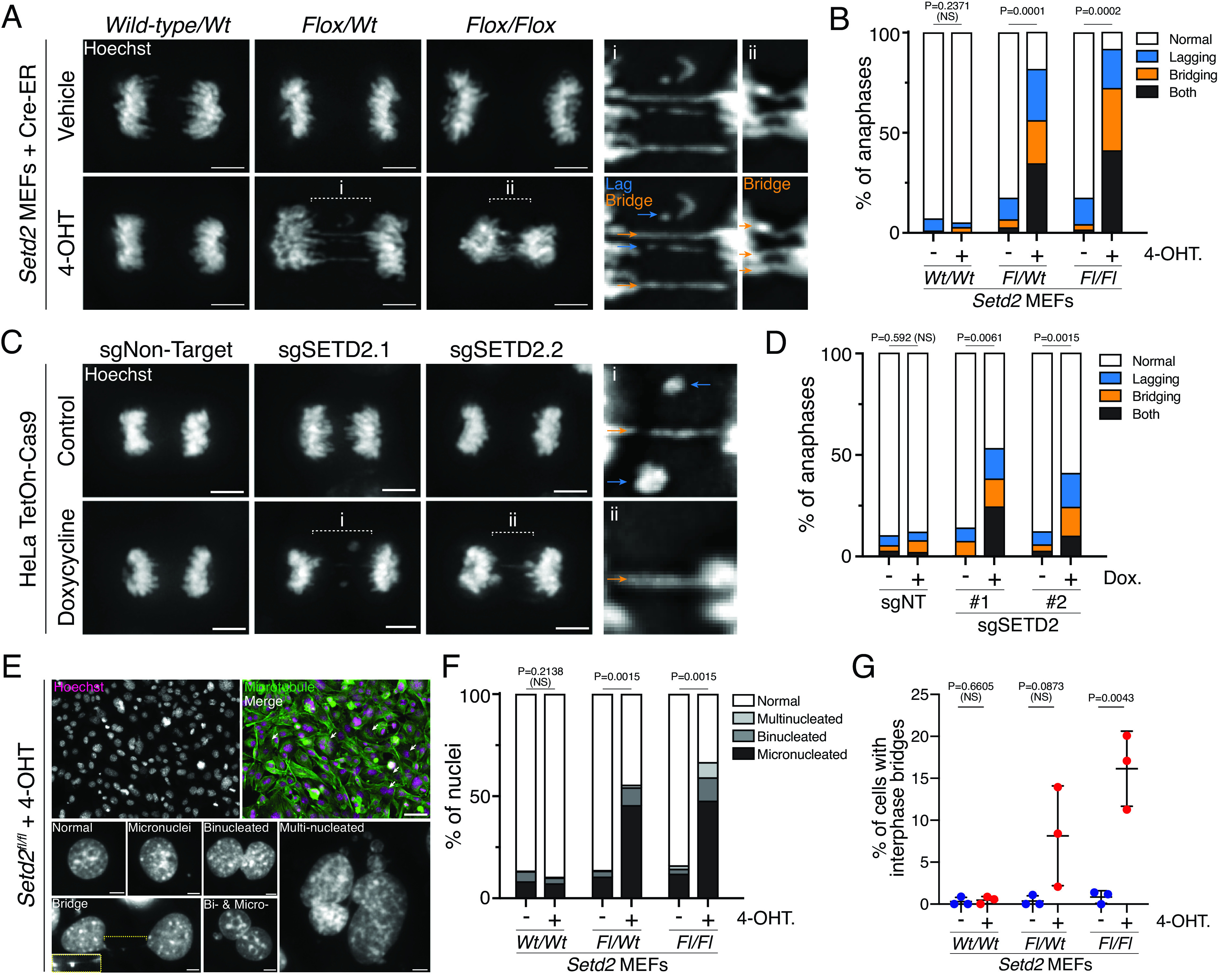
Setd2 loss causes chromatin mis-segregation during mitosis. (A) Images of wild-type, heterozygous or homozygous deletion of Setd2 have lagging (blue arrows) and bridging (orange arrows) chromosomes during the anaphase. MEFs with no, one, or two floxed alleles of Setd2 (Wild-type/WtFlox/Wt, and Flox/Flox, respectively) were either treated with vehicle (EtOH, control in top panels) or 4-OHT (excise floxed alleles of Setd2, bottom panels) for 3 d, fixed, and counterstained with Hoechst (gray). Scale bars are 5 µm. (B) Quantification of chromosome segregation errors during the anaphase and early telophase in control (vehicle-treated) or 4-OHT-treated MEFs described in (A). n = 198 wt/wt vehicle, n = 215 wt/wt 4-OHT, n = 298 fl/wt vehicle, n = 227 fl/wt 4-OHT, n = 258 fl/fl Vehicle, n = 225 fl/fl 4-OHT cells across 2 (wt/wt), 4 (fl/wt), and 3 (fl/fl) biological replicates. P values derived from an unpaired t test of normal cell values between treatment groups for each genotype. (C) Images of anaphases in control (untreated) or doxycycline-treated HeLa cells expressing tetracycline-inducible Cas9 (TetOn-Cas9) and single-guide RNA (sgRNA) that are nontargeting (NT) or specific for SETD2. Cells are counterstained with Hoechst (gray). Lagging (blue) and bridging (orange) chromosomes occur in cells lacking SETD2. Scale bars are 5 µm. (D) Quantification of chromosome segregation errors during the anaphase and early telophase in cells described in (C). n = 183 sgNT control, n = 190 sgNT dox., n = 198 sgSETD2.1 control, n = 240 sgSETD2.1 dox., n = 187 sgSETD2.2 control, n = 246 sgSETD2.2 dox. cells across three biological replicates. P values derived from an unpaired t test of normal cell values between treatment groups for each sgRNA background. (E) Image of Setd2fl/fl MEFs treated with 4-OHT (at left), showing nuclear defects that occur in interphase after 3 d treatment. Scale bars are 50 µm (top images) and 5 µm (grayscale images at the bottom). (F) Quantifications of nuclear phenotypes from images described in (E) for Setd2 MEFs. n = 450 wt/wt vehicle, n = 617 wt/wt 4-OHT, n = 650 fl/wt vehicle, n = 509 fl/wt 4-OHT, n = 736 fl/fl Vehicle, n = 687 fl/fl 4-OHT cells across three biological replicates. P values derived from an unpaired t test of normal cell values between treatment groups of each genotype. (G) Quantification of interphase bridges observed in cell images described in (E). P values derived from an unpaired t test of normal cell values between treatment groups each genotype. Error bars are SD.
Increased frequency of lagging and bridging chromosomes should lead to an increase in micronuclei and chromatin bridges in interphase (10, 11). Both Setd2 heterozygous and homozygous-deleted MEFs, as well as HeLa cells acutely deleted of SETD2, exhibited increases in binucleation, micronucleation, and multinucleation (Fig. 1 E and F and SI Appendix, Fig. S2 A and B). Interphase bridges were identified by accumulation of RPA and cGAS (Fig. 1G and SI Appendix, Fig. S2C), markers of single-stranded DNA and cytoplasmic DNA, respectively (10, 18). Thus, either mono- or biallelic loss of Setd2 is sufficient to promote chromosome segregation defects, which can lead to micronucleation, binucleation, and interphase bridging.
Dicentric Chromosomes Are Prevalent in Cells Lacking SETD2.
Although lagging chromosomes and defects in spindle morphology could be explained by loss of SETD2-dependent tubulin methylation, the defect(s) underlying anaphase bridges was not clear (1, 19). Chromatin bridges are caused by structural changes in chromosomes and arise from alterations in chromosome decatenation, cohesion, and/or generation of dicentric chromosomes (11). To determine whether loss of Setd2 leads to aberrations in chromosomal structure, metaphase chromosomes from wild-type, mono- and biallelic Setd2 floxed MEFs were isolated 3 d after 4-OHT treatment, and chromosomal abnormalities were quantified.
Heterozygous or homozygous loss of Setd2 resulted in an increase in the percentage of cells that have dicentric chromosomes (Fig. 2 A and B), as observed by dense DAPI staining, which marks major satellite repeats at pericentric regions of murine chromosomes (20). To account for CIN in Setd2 deleted cells (15), the number of dicentric chromosome(s) was normalized to chromosome number for each cell, which was confirmed to be increased in Setd2-deleted cells (Fig. 2C). 1.1% and 2.0% of chromosomes were dicentric in Setd2fl/wt and Setd2fl/fl cells treated with 4-OHT or 0.43 and 0.81 chromosomes per diploid murine cell, respectively.
Fig. 2.
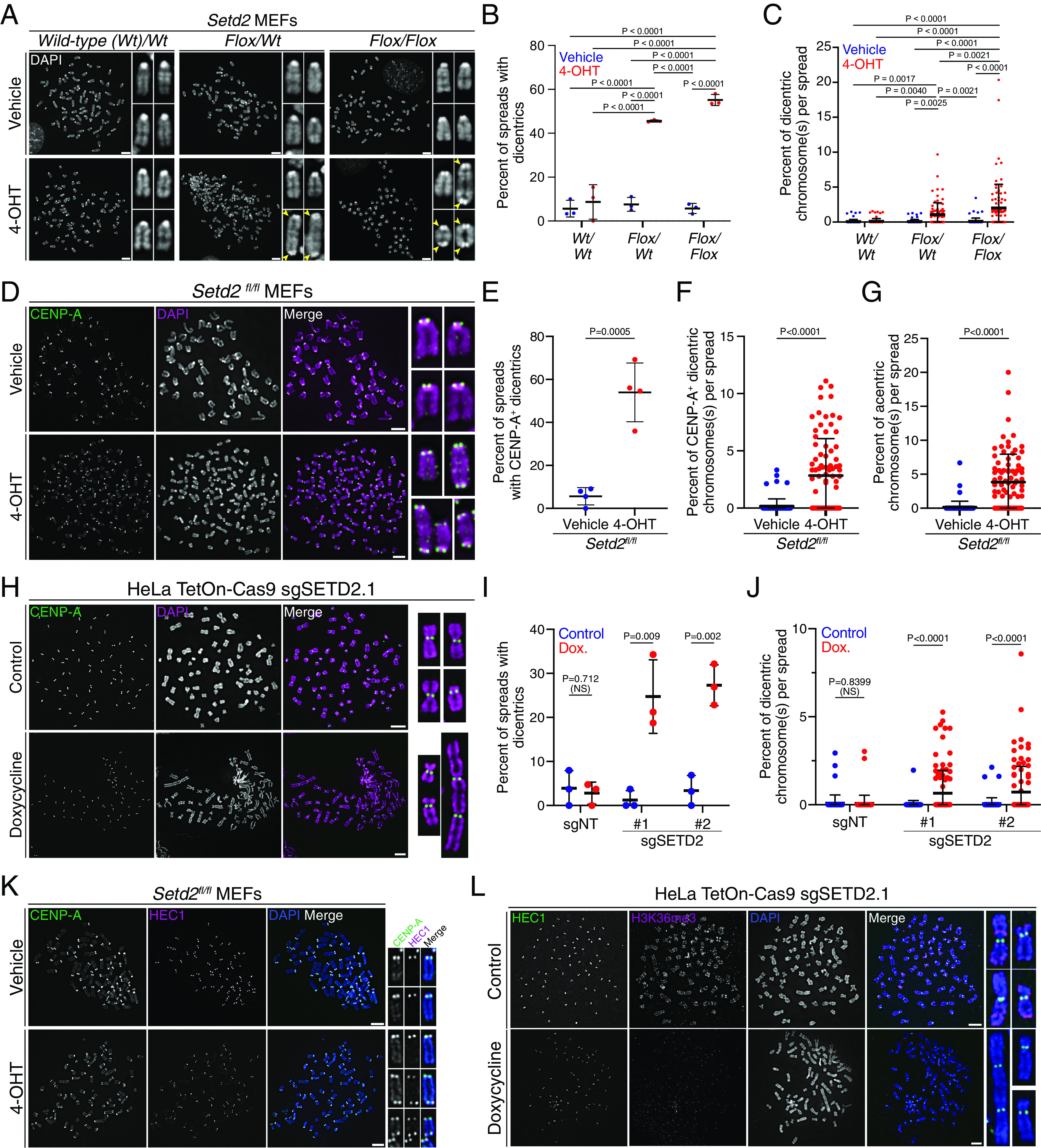
Dicentric chromosomes arise in cells lacking Setd2. (A) Metaphase spread from MEFs with no, one, or two floxed alleles of Setd2 (wt/wtfl/wt, and fl/fl, respectively) and treated with vehicle (control, in top panels) or 4-OHT (Bottom) for 3 d. Spreads are counterstained with DAPI (gray). Dicentric chromosomes (yellow arrowheads) are present in cells with homozygous and heterozygous Setd2 deletion. (B) Quantification of the percentage of metaphase spreads from MEFs described in (A) that have at least one dicentric chromosome from n = 3 biological replicates. P values derived from two-way ANOVA with Tukey’s multiple comparisons tests. (C) Quantification of the percentage of dicentric chromosome(s) per metaphase spread image from (B). Each point represents an individual image. n = 88 wt/wt vehicle, n = 81 wt/wt 4-OHT, n = 76 fl/wt vehicle, n = 79 fl/wt 4-OHT, n = 89 fl/fl vehicle, n = 85 fl/fl 4-OHT cells. P values derived from two-way ANOVA with Tukey’s multiple comparisons tests. (D) Metaphase spread from Setd2fl/fl MEFs treated with vehicle (control, top panels) or 4-OHT (Setd2 knockout, bottom panels). Spreads were immunostained for CENP-A (green) and counterstained for DAPI (magenta). Dicentric chromosomes in Setd2-knockout cells have two CENP-A foci. (E) Quantification of the percentage of metaphase spreads from Setd2fl/fl MEFs treated with vehicle (control) or 4-OHT (Setd2 knockout) that have dicentric chromosomes containing CENP-A from n = 4 biological replicates. P values from an unpaired t test between each condition. (F) Quantification of the percentage of chromosomes that are dicentric and contain two CENP-A foci on metaphase spreads from Setd2fl/fl MEFs from (E). n = 79 fl/fl vehicle, n = 85 fl/fl 4-OHT cells. P values from an unpaired t test between each condition. (G) Quantification of the percentage of chromosomes that are acentric from Setd2fl/fl MEFs treated with vehicle (n = 79 cells) or 4-OHT (n = 85 cells). P values from an unpaired t test between each condition. (H) Metaphase spread isolated from control (untreated) or doxycycline-treated HeLa TetOn-Cas9 cells expressing sgSETD2.1, immunostained for CENP-A (green) and counterstained for DAPI (magenta). Control cells (Top) have monocentric chromosomes, while SETD2-knockout cells (Bottom) have mono- and dicentric chromosomes. (I) Quantifications of the percentage of metaphase spreads that have at least one dicentric chromosome from HeLa TetOn-Cas9 cells expressing sgNT or sgSETD2 and were untreated (control) or treated with doxycycline and then stained for CENP-A and DAPI. SETD2-knockout caused increases in the frequency of cells that have dicentrics in n = 3 biological replicates. P values derived from an unpaired t test of normal cell values between treatment groups each genotype. (J) Quantifications of the percentage of dicentric chromosomes in each metaphase spread from cells described in (H). SETD2-knockout leads to an increase in the percentage of chromosomes that are dicentric in n = 75 sgNT control, n = 75 sgNT dox., n = 86 sgSETD2.1 control, n = 100 sgSETD2.1 dox., n = 85 sgSETD2.2 control, n = 89 sgSETD2.2 dox. cells. P values derived from an unpaired t test of normal cell values between treatment groups each genotype. (K) Metaphase spread from Setd2fl/fl MEFs treated with vehicle (control, top) or 4-OHT (Setd2 knockout, bottom), demonstrating that dicentric chromosomes assemble kinetochores (HEC1, magenta) at both centromeres (CENP-A, green). (L) Metaphase spread from HeLa TetOn-Cas9 cells with sgSETD2, control (Top) or treated with doxycycline (SETD2-knockout, Bottom), demonstrating that dicentric chromosomes assemble kinetochores (HEC1, green) at two locations. Scale bars are 5 µm. For each plot, error bars are SD.
To confirm that dicentric chromosomes have two functional centromeres, metaphase spreads from control or Setd2-deleted MEFs were immunostained for CENP-A, the histone H3-variant that specifies centromere identity and activity (21). CENP-A staining verified that 54% of Setd2-deleted cells have dicentric chromosomes with CENP-A at two distinct constriction sites on the same chromosome, compared to 6% of controls (Fig. 2 D and E). The percentage of chromosomes that are dicentric increased from 0.2% in control samples to 2.8% in Setd2-knockout cells (Fig. 2F, 0.08 to 1.12 chromosomes per cell). Additionally, the percentage of acentric chromosomes per metaphase was also increased in Setd2-deleted cells (Fig. 2G, 0.2 to 3.9%). We also examined dicentric formation in HeLa TetOn-Cas9 cells with SETD2 deletion, which also displayed an increase in the percentage of cells with dicentrics (1 to 25% for sgSETD2.1 and 3 to 27% for sgSETD2.2, Fig. 2 H and I). The percentage of dicentric chromosomes per spread, as determined by CENP-A staining, also increased in SETD2-knockout cells (0.02 to 0.65% for sgSETD2.1 and 0.06 to 0.72% for sgSETD2.2, Fig. 2J).
For dicentric chromosomes to bridge during mitosis, both centromeres must be active to assemble kinetochores. However, inactivation of either centromere decreases kinetochore assembly, causing the dicentric to segregate like a monocentric chromosome (8). To determine whether dicentric chromosomes assembled kinetochores at both centromeric locations, metaphase spreads from control or SETD2-deleted MEFs and HeLa cells were costained for CENP-A and HEC1, the outermost component of the kinetochore that binds microtubules. Additionally, metaphase chromosomes were stained for KNL1 or CENP-T, components of the outer kinetochore and constitutive centromere-associated network, respectively. Both centromeres of dicentric chromosomes contained HEC1, KNL1, and CENP-T and are likely active (Fig. 2 K and L and SI Appendix, Fig. S3A), indicating that these dicentrics contribute to the elevated frequency of anaphase bridges induced by SETD2 loss.
To determine how rapidly dicentric chromosomes arise following Setd2 deletion, metaphases were isolated from Setd2fl/fl cells, vehicle or 4-OHT treated, starting 1 d after initial treatment. Dicentric chromosome prevalence increased up to 3 d after treatment and then waned by 14 d but was still increased relative to controls (SI Appendix, Fig. S3 B and C). This result demonstrates that loss of Setd2 results in a robust generation of dicentric chromosomes during a discrete time window shortly following deletion. Long-term Setd2 deletion also resulted in a lower but still significant prevalence of dicentric chromosomes.
Interchromosomal Rearrangements, Telomere Fusion, and Neocentromere Formation Are Not Primary Drivers of Dicentric Chromosomes Following Setd2 Loss.
Dicentric chromosomes are a product of chromosome fusion or neocentromere formation and are known to occur in human diseases and promote genomic instability and tumor evolution (9, 11, 22, 23). A common driver of chromosome fusion is telomere dysfunction, and H3K36me3 is present near telomeric or subtelomeric regions (SI Appendix, Fig. S1 B and E) (10). To test whether telomere attrition occurs following acute Setd2 loss, quantitative fluorescence in situ hybridization (Q-FISH) was performed to determine telomere length in vehicle-and 4-OHT-treated Setd2fl/fl MEFs. There were no changes in telomere length associated with loss of Setd2 (SI Appendix, Fig. S3 D–F). Additionally, no telomeric FISH signal was observed between the two centromeres, indicating that telomere fusion was not occurring (SI Appendix, Fig. S3 G and H). Together, these results indicate that telomere dysfunction is not the primary driver of dicentric chromosome formation following acute loss of Setd2.
We next examined whether dicentric chromosomes arise due to neocentromere formation or telomere-independent chromosome fusion, which may be downstream of DSBs. If chromosome fusion drives dicentric formation, those chromosomes should harbor an interchromosomal rearrangement between two nonhomologous chromosomes. However, if dicentrics lack interchromosomal rearrangements, this may suggest neocentromere formation. To differentiate between these possibilities, multicolor FISH (m-FISH) was performed on metaphase spreads from vehicle- or 4-OHT-treated Setd2fl/fl MEFs to determine the genetic composition of the dicentric chromosomes (Fig. 3 A and B). m-FISH identified that 15.3% of dicentrics have interchromosomal rearrangements, generated from the fusion of either two or more different chromosomes. However, the majority (84.7%) of dicentric chromosomes do not have interchromosomal rearrangements and are composed of the same chromosome (Fig. 3C). Moreover, there is no preference for dicentric formation on specific chromosomes.
Fig. 3.

Dicentric chromosomes in Setd2-knockout cells lack interchromosomal rearrangements and neocentromeres. (A) m-FISH image on Setd2fl/fl MEFs treated with 4-OHT (Setd2-knockout). Arrow indicates a dicentric chromosome. (B) Representative images of dicentric chromosomes with interchromosomal (Top) or intrachromosomal rearrangements (Bottom). (C) Quantification of dicentric chromosomes (n = 189 across three biological replicates) with or without interchromosomal rearrangements. The majority of dicentrics lack interchromosomal rearrangements and are composed of genetic material from the same chromosome. (D) Schematic of dicentric chromosomes. CENP-B binds CENP-B box motif on centromeric DNA. Neocentromeres, which contain CENP-A nucleosomes, would lack CENP-B. (E) Representative immunofluorescent images of chromosomes from Setd2fl/fl MEFs treated with 4-OHT and stained for CENP-A (green), CENP-B (magenta), and DNA (DAPI, blue). CENP-A localizes with CENP-B at centromeres in monocentric cells and in some dicentrics. Chromosomes lacking CENP-B on one or both centromeres (yellow arrowheads) may be suggestive of neocentromeres. Scale bars are 1 µm. (F) Representative immunofluorescent and FISH images of chromosomes from Setd2fl/fl MEFs treated with 4-OHT and stained for CENP-A (green), CENP-B box FISH probe (magenta), and DNA (DAPI, blue). CENP-A localizes with CENP-B box DNA at centromeres in monocentric cells and in some dicentric cells. Some centromeres contain CENP-A but lack CENP-B box sequences (yellow arrowheads). The scale bar is 1 µm. (G) Quantifications of percent of dicentrics that have CENP-B colocalizing with CENP-A at 0,1, or 2 sites across n = 3 (IF) and n = 2 (FISH) biological replicates. Error bars are SD (H) Quantification of CENP-A colocalization with CENP-B from n = 192 dicentrics described in (G). Mean percentage (± SD) across five biological replicates (IF and FISH).
Dicentrics may occur by establishing neocentromeres at an ectopic, noncentromeric site (24). To test whether neocentromeres form in Setd2-knockout cells, metaphase spreads from vehicle- and 4-OHT-treated Setd2fl/fl MEFs were immunostained for CENP-A and CENP-B (Fig. 3 D and E), the latter of which binds to CENP-B boxes, a 17-base-pair motif sequence located within native α-satellite-containing centromeres for all chromosomes (except the Y-chromosome) (25). Using a complementary approach, spreads were immunostained for CENP-A, followed by FISH using a CENP-B box probe (Fig. 3F). If centromeres form at the correct location, CENP-A should colocalize with CENP-B. If neocentromeres form at ectopic sites, CENP-A would be present at sites lacking CENP-B (Fig. 3D). The percentage of centromeres localizing with CENP-B or CENP-B box DNA was quantified, and, using either approach, both centromeres contained CENP-B in 88% of dicentrics, while 12% of dicentrics contained CENP-A at potential noncentromeric sites (Fig. 3 G and H). It should be noted that the Setd2fl/fl MEFs were derived from a male mouse, and the Y-chromosome lacks a CENP-B box. Thus, we cannot differentiate between Y-chromosome-derived dicentrics and potential neocentromeres. While these observations indicate that Setd2 deletion may possibly promote some degree of neocentromere formation, the vast majority of dicentric chromosomes are not formed by this mechanism.
Isodicentric Chromosomes Predominate in Setd2-Deleted Cells.
We examined the nature of the dicentrics in Setd2-deleted cells more carefully. One possibility was that the dicentrics were isodicentric, defined as a derivative chromosome composed of a mirror-imaged duplication that promotes gene amplification and deletion within an individual chromosome (26). Isodicentrics were suspected because centromeres were exclusively positioned at both chromosome poles in Setd2-deleted MEFs, suggesting symmetry (Fig. 4A). Dicentrics in SETD2-knockout HeLa cells also appeared symmetrical, often producing abnormally large chromosomes (Fig. 4B). To test whether isodicentric chromosomes occur in Setd2-mutant MEFs, metaphase spreads were subjected to Giemsa staining and G-banding analyses (Fig. 4C). This cytogenetic approach identified 75% of dicentrics as bona fide isodicentric chromosomes.
Fig. 4.
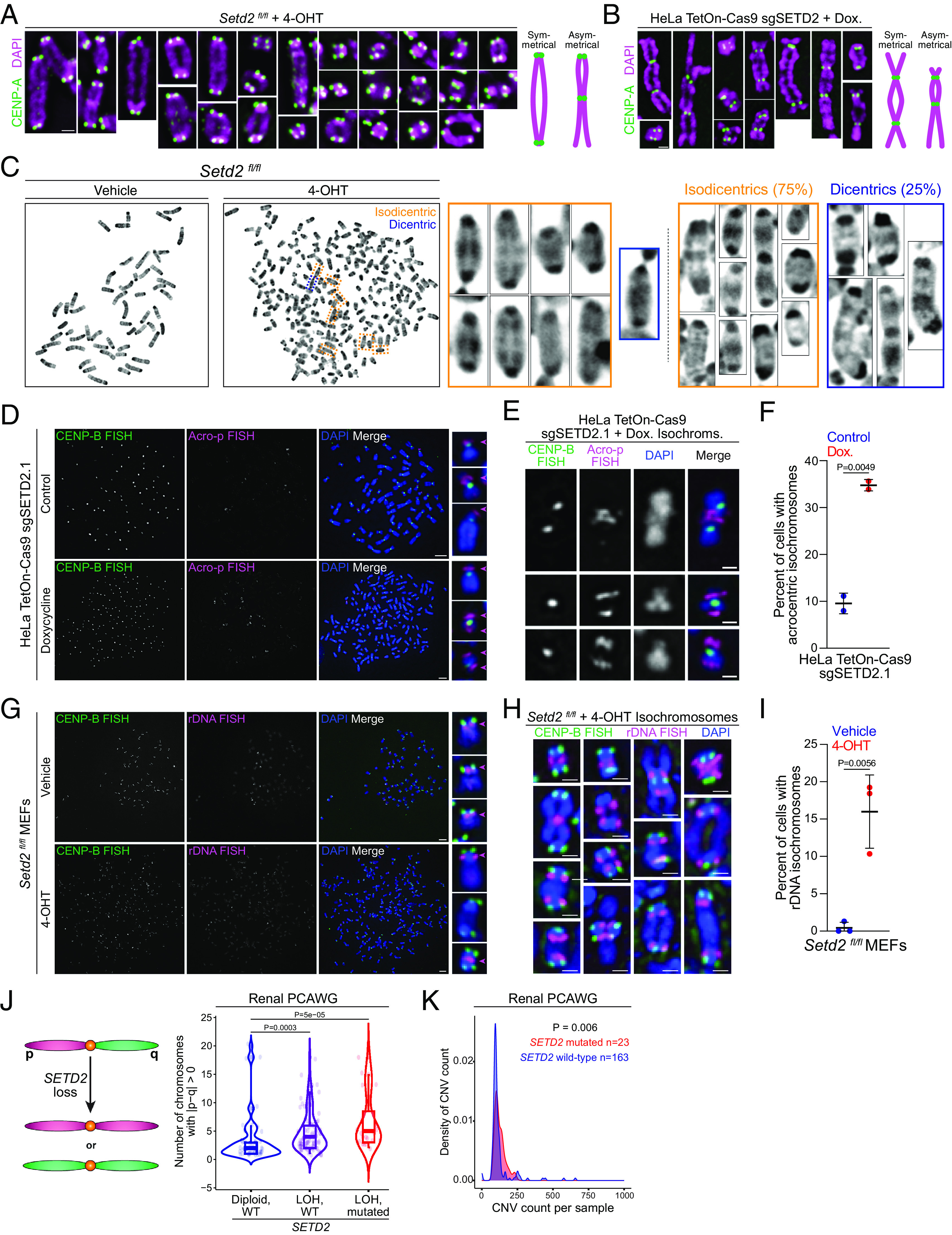
Setd2 knockout promotes formation of isodicentric chromosomes. (A) Immunofluorescent images of dicentric metaphase chromosomes from Setd2fl/fl MEFs treated with 4-OHT (Setd2-knockout) and stained for CENP-A (green) and DNA (DAPI, magenta). Nearly all dicentric chromosomes are symmetrical. The scale bar is 1 µm, and all images are set to the same scale. (B) Representative immunofluorescent images of dicentric metaphase chromosomes from HeLa TetOn-Cas9 cells expressing sgSETD2.1 and treated with doxycycline (Setd2-knockout). Chromosomes are stained for CENP-A (green) and DNA (DAPI, magenta). The scale bar is 1 µm. (C) Images of Giemsa-stained metaphase spreads from Setd2fl/fl MEFs treated with vehicle (control) or 4-OHT (Setd2 knockout). Cytogenetic analyses identified that vast majority of dicentrics are isochromosomes (isodicentric, orange boxes). Dicentric chromosomes (blue boxes) occur but the majority are isodicentric. n = 24 dicentrics across 18 spreads. (D) Images of FISH for CENP-B box and p-arms of acrocentric chromosomes (Acro-p) on metaphase spreads from control or doxycycline-treated HeLa TetOn-Cas9 cells expressing sgSETD2.1. Acro-p signal should be at short, p-arm (Top); however, SETD2-knockout cells have signal on both arms. Scale bars are 5 µm. (E) Representative images of isochromosomes, including isodicentric, from HeLa TetOn-Cas9 sgSETD2.1 cells treated with doxycycline. Scale bars are 1 µm. (F) Quantification of the percentage of cells with isochromosomes in control (79 spreads) or doxycycline-treated (132 spreads) HeLa TetOn-Cas9 sgSETD2. n = 2 biological replicates. (G) Images of FISH for CENP-B box and rDNA (BAC RP23-225M6) on metaphase spreads from control or 4-OHT-treated Setd2fl/fl MEFs. rDNA should be present at one location per chromosome, however Setd2-knockout leads to isodicentrics (Bottom Right). Scale bars are 5 µm. (H) Representative images of isochromosomes, including isodicentrics, from Setd2-knockout MEFs. Scale bars are 1 µm. (I) Quantification of the percentage of cells with isochromosomes in control (159 spreads) or 4-OHT-treated (241 spreads) Setd2fl/fl MEFs across n = 3 biological replicates. (J) Illustration of normal chromosome with p and q arm, however SETD2 loss results in isochromosomes with unequal chromosome arms. Quantification of number of chromosomes per sample with unequal p and q arms in Renal PCAWG data with diploid, WT SETD2 (blue, n = 47), heterozygous deletion of SETD2 (purple, n = 88), or deletion and mutation of SETD2 (red, n = 23). P values derived from the Wilcoxon test followed by Holm–Bonferroni to adjust for multiple comparisons. (K) Quantification of distribution of CNV count per sample in wild-type and mutated SETD2 (n = 163 and 23 respectively) from Renal PCAWG data. P values derived from the Wilcoxon signed-rank test. For each plot, error bars are SD and P values derived from an unpaired t test unless stated otherwise.
To confirm whether isochromosomes occur in SETD2-deleted cells, FISH against nucleolar organizer regions was performed to simultaneously mark the p-arms of the human acrocentric chromosomes (13, 14, 15, 21, and 22, Acro-p) or 45S rDNA of mouse chromosomes (11, 12, 15, 16, 18, and 19). While control HeLa samples showed stereotypic Acro-p FISH staining, SETD2-knockout chromosomes exhibited more cells with isochromosomes, including isodicentrics (Fig. 4 D–F). Formation of isochromosomes and isodicentrics were also observed in Setd2-knockout MEFs (Fig. 4 G–I).
The generation of isochromosomes, isodicentrics, and dicentrics should increase copy number variants (CNVs) as well as induce chromosome arm-level changes. To test whether these events occur in tumors with loss or mutation of SETD2, we counted the number of chromosomes in each sample with unequal p and q arms in the Renal Cell Carcinoma Analysis of Whole Genomes (PCAWG), excluding sex chromosomes (27). Renal tumors with loss of heterozygosity (LOH) and/or SETD2 mutation increase the number of chromosomes with unequal p:q arms (Fig. 4J). Concordantly, there is an increase in the number of CNVs in the tumors with SETD2 mutations (Fig. 4K). While this does not prove that isochromosomes, dicentrics, or isodicentrics drive CNVs in renal tumors, it is consistent with the consequence of those chromosomal aberrations.
To determine whether H3K36me3 or catalytic activity of SETD2 is required to suppress dicentric chromosome formation, HeLa or MEFs were treated with a pharmacologic inhibitor of SETD2 methyltransferase activity (EPZ-719, SETD2i) (28). SETD2i treatment led to a 96% decrease in H3K36me3 without perturbing SETD2 protein levels (SI Appendix, Fig. S4 A and B). Metaphase spreads were isolated from control, 4-OHT, or SETD2i Setd2fl/fl MEFs, and 33% of SETD2i-treated cells displayed dicentric chromosomes, compared to 44% of Setd2-knockouts or 1.7% of controls (Fig. 5A). These results demonstrate that the catalytic activity of SETD2 and/or the presence of H3K36me3 suppresses dicentric chromosome formation. To confirm this, rescue experiments were performed in HeLa TetOn-Cas9 sgSETD2.1 expressing cells. Full-length, wild-type or catalytically dead R1625C SETD2 or an empty vector control under a tetracycline-inducible promotor were introduced in these cells such that doxycycline treatment would simultaneously knock-out and rescue SETD2 expression (SI Appendix, Fig. S4C). We isolated metaphase spreads from these cells and observed that, while wild-type SETD2 rescues, R1625C mutated-SETD2 does not, confirming that methyltransferase activity is critical for suppressing dicentric and acentric chromosome formation (SI Appendix, Fig. S4D).
Fig. 5.

Loss of SETD2 catalytic activity during DNA replication promotes formation of dicentric and acentric chromosomes in a RAD52-dependent manner. (A) Quantification of prevalence of dicentric chromosomes in metaphase spreads from Setd2fl/fl MEFs treated with vehicle (DMSO, control), 4-OHT or 1 µM SETD2i, 3 d after treatment. Setd2-knockout or inhibition leads to an increase in dicentric chromosomes from n = 3 biological replicates. P values derived from one-way ANOVA with Tukey’s multiple comparisons test. (B) Schematic of cell synchronization and SETD2 inhibition or depletion during DNA replication. (C) Western blot of whole cell extract from HeLa SETD2-FKBP cells showing that dTAG treatment degrades SETD2 prior to S phase release. When arrested at mitosis, SETD2 is still depleted, and there is a decrease in H3K36me3 in both dTAG- and SETD2i-treated cells. (D) Quantification of the percentage of cells with dicentric and/or acentric chromosomes from cells described in (C), using n = 3 biological replicates. P values derived from one-way ANOVA with Tukey’s multiple comparisons test. (E) Quantification of the percentage of dicentric and acentric chromosomes per cell from cells described in (D). SETD2 depletion (dTAG, n = 71 cells) and SETD2i (n = 51) had more dicentric or acentric chromosomes than control (DMSO, n = 51 cells). P values derived from one-way ANOVA with Tukey’s multiple comparisons test. (F) Representative images of DMSO (control) or dTAG-treated HeLa SETD2-FKBP cells in S phase, stained for RAD52 and RAD51. Scale bars are 5 µm. (G) Quantification of the percentage of cells described in (F) with >5 RAD51 foci during the S phase. N = 10 technical replicates for each condition. P values derived from an unpaired t test. (H) Quantification of the percentage of cells described in (F) with >3 RAD52 foci during the S phase. N = 10 technical replicates for each condition. P values derived from an unpaired t test. (I) Quantification of average number of RAD52 foci of cells described in (F) during the S phase. N = 10 technical replicates for each condition. P values derived from an unpaired t test. (J) Quantification of the percentage of control or doxycycline-treated HeLa TetOn-Cas9 sgSETD2.1 cells with dicentric and/or acentric chromosomes with depletion of RAD52 using siRAD52 or siNon-Targeting control (NT) from n = 2 biological replicates. siRAD52 decreases the percentage of cells with dicentrics or acentrics. P values derived from one-way ANOVA with Tukey’s multiple comparisons test. (K) Quantification of the percentage of dicentric and/or acentric chromosomes per cell described in (J) n = 49, 48, 49, 62, and 58 cells across two biological replicates. P values derived from one-way ANOVA with Tukey’s multiple comparisons test. (L) Proposed model describing how SETD2 loss drives genetic heterogeneity. Isodicentric chromosomes initiate gene amplification and deletion, chromosome mis-segregation, and chromothripsis in subsequent divisions to increase clonality.
While dicentric chromosomes may arise from faulty DSB repair, telomere attrition, or neocentromere formation (22), mechanisms promoting isodicentrics are less well understood. In yeast, mirror-image dicentric and acentric chromosomes can arise through a DNA replication-dependent, DSB-independent manner called faulty template switching or replication template exchange (29, 30). During this process, stalled replication forks restart utilizing RAD52 instead of the recombinase RAD51 and anneal to a nearby inverted repeat, essentially performing “U-turns” to generate palindromic acentric and dicentric chromosomes. Loss of H3K36me3 decreases RAD51-dependent homologous recombination (31), so one hypothesis was that cells lacking SETD2 might preferentially utilize RAD52 during DNA replication.
To test whether SETD2 is required during DNA replication to prevent isochromosomes, we generated SETD2-FKBP knock-in HeLa cells and synchronized them at G1/S using a double thymidine block. For the final 2 h of the arrest, cells were treated with the SETD2i or dTAG47 to degrade SETD2 (Fig. 5 B and C). Cells were released into S phase, and then metaphase spreads were collected and analyzed for dicentric and acentric chromosomes. Degradation or inhibition of SETD2 methyltransferase activity increased the percentage of cells with dicentric or acentric chromosomes, as well as the percentage of chromosomes that are dicentric or acentric (Fig. 5 D and E). To test whether RAD51 and RAD52 function was altered following SETD2 loss, we synchronized cells at G1/S then released and processed for chromatin fractionation and immunofluorescence. While fractionation and western blot analyses did not reveal a change in loading of RAD51 or RAD52 onto chromatin during DNA replication (SI Appendix, Fig. S4E), SETD2-depleted cells displayed a decrease in RAD51 foci and a concordant increase in RAD52 foci (Fig. 5 F–I). To determine whether the increase in RAD52 utilization facilitates the increase in gross chromosomal rearrangements, RAD52 was knocked-down using siRNA; then, SETD2 was knocked out in HeLa TetOn-Cas9 cells (SI Appendix, Fig. S4F). Knockdown of RAD52 was sufficient to decrease the formation of acentric and dicentric chromosomes in SETD2-knockout cells, relative to nontargeting or untreated control cells (Fig. 5 J and K). These data suggest that cells lacking H3K36me3 and SETD2 may utilize a RAD52-dependent pathway that is more prone toward faulty template switching and isochromosome formation.
Discussion
SETD2 is the sole source of H3K36 trimethylation in somatic cells and a known tumor suppressor, with roles in mitotic spindle organization, DNA repair, and transcriptional fidelity. Yet, it has been unclear how SETD2 loss causes extensive intratumor heterogeneity and branched evolution. We propose that in addition to CIN, the formation of isochromosomes following SETD2 and H3K36me3 loss of function provides the mechanism to promote the genomic heterogeneity observed (Fig. 5L). This model is consistent with previous work demonstrating that a single chromatin bridge is sufficient to promote clonal heterogeneity (9, 11), suggesting that this structural intermediate may in fact be the major feature driving genomic instability.
Chromosome bridges are subject to cycles of breakage-fusion-bridge (BFBs), which initiate further gross chromosomal rearrangements and chromothripsis in subsequent cell divisions (11, 32). Genomic signatures of BFBs occur in myriad tumor types, and cellular transformation is driven by transient but not chronic formation of dicentric chromosomes (23, 33, 34). Importantly, we observed a robust appearance of dicentric chromosomes following acute SETD2 loss, with a subsequent waning over time. This suggests that a discrete temporal burst of isochromosomes and dicentric chromosomes following SETD2 or H3K36me3 loss may be sufficient to promote transformation, gross chromosomal rearrangements, and subclonal heterogeneity. The persistence of isochromosomes and isodicentrics may reflect a low level of ongoing rearrangements or silencing of one centromere such that chromosomes are functionally monocentric (8). Isochromosome persistence may be guided by selective pressures that favor the amplification of genes present in the duplicated chromosome arms.
Our findings establish that SETD2 and H3K36me3 prevent isochromosomes and isodicentric chromosomes, potentially by promoting RAD51 activity over the more error-prone RAD52 pathway (26, 35). While faulty template switching mediated by RAD52 has been shown to drive “U-turns” in yeast leading to copy number alterations and aneuploidy, this work shows that this pathway may also occur in mammalian cells (29, 30, 36). Nonallelic homologous recombination (HR) between sister chromatids, fork stalling and template switching (FoSTeS), microhomology-mediated break-induced replication or foldback inversions are potential mechanisms for isochromosome formation following SETD2 loss (6, 37, 38). Sequencing through these inverted repeats will be essential to differentiate between these mechanisms.
The ability to generate more clones provides tumors with a fitness advantage, and accordingly, a range of tumor types with SETD2 mutations are more resistant to a broad range of therapeutics (39–42). Notably, ccRCC, where SETD2 was initially established as a tumor suppressor, is among the most resistant cancers to chemotherapy (43). SETD2 mutations commonly occur in hepatosplenic T cell lymphoma (HSTL), a rare yet aggressive neoplasm that is also chemoresistant (44). The most common chromosomal abnormality in HSTL is isochromosome 7q [i(7q)], which promotes amplification of genes in 7q and deletion of 7p. Further analyses are needed to assess whether isochromosomes occur in other tumor types with SETD2 mutations. Additionally, our data demonstrate that MEFs and HeLa cells exhibit different frequencies of dicentric and isochromosome formation, suggesting that different genetic or phenotypic backgrounds may influence these rearrangements, which will be the focus of future work.
These data demonstrate that loss of the tumor suppressor SETD2 promotes the formation of isochromosomes and isodicentric chromosomes, leading to chromatin bridges during mitosis. While these structures have long been appreciated to promote intratumoral heterogeneity, their origin has been elusive. Our findings provide an unexpected and important link between an essential histone methyltransferase and genome maintenance, via an exciting and unique genomic structure that can simultaneously promote copy number alterations, and DNA strand breaks.
Materials and Methods
Cell Culture.
All cells were cultured at 37 °C and 5% CO2. Mouse embryonic fibroblasts (MEFs) were cultured as described previously (15). Briefly, cells were cultured in Dulbecco's Modified Eagle Medium (DMEM), phenol red-free (Gibco, Catalog number 21063-029) supplemented with 1 mM sodium pyruvate (Sigma, S8636-100 mL), GlutaMAX (Gibco, 35050-061), and 10% fetal bovine serum (FBS, GeminiBio, 100-106). HeLa TetOn-Cas9 and 293FTs cells were grown in DMEM (Gibco, 11965-092) supplemented with 1% penicillin/streptomycin (Gibco, 15150122) and 10% tetracycline-free FBS (GeminiBio, 100-800) (17). Setd2wt/wt, Setd2flox/wt, Setd2flox/flox MEFs expressing Cre-ER were treated with 3 µM (Z)-4-hydroxytamoxifen (4-OHT, Millipore Sigma, H7904) or vehicle (Ethanol, 0.1% final volume) for 3 d, unless stated otherwise, and then harvested for the experiment.
Cell Culture Methods, Treatments, and Cell Line Generation.
Single guides targeting SETD2 (sgSETD2.1 sense strand GTGCGGATCAGCCAATTGCCG and sgSETD2.2 sense strand GAATGAACTGGGATTCCGACG) were cloned into pLenti-sgRNA (Addgene, 71409) following previous methods (45). Lentivirus was generated using 293FT cells by cotransfection of guide plasmid with psPAX2 and pMD2.G (Addgene, 12260 and 12259, respectively). Conditioned media were harvested, filtered, and stored at -80. HeLa TetOn-Cas9 cells were transduced with 8 µg/mL polybrene (Millipore Sigma, TR-1003) and viral supernatant for guides targeting SETD2 or nontargeting control sgRNA (Addgene, 80189). Cells were selected using 0.5 µg/mL puromycin (Millipore Sigma, P9620). To activate expression of Cas9, cells were treated with 1 µg/mL doxycycline (Millipore Sigma, D5207) daily and harvested after 3 d unless stated otherwise. For SETD2i experiments, EPZ-719 (MedChemExpress, HY-139626) was resuspended in dimethyl sulfoxide (DMSO), and cells were treated and harvested after 72 h.
To generate a stable knock-in SETD2-FKBP, HeLa Kyoto cells were transduced with adeno-associated virus containing homology-directed repair templates (Vectorbuilder), designed using a previously published method (46). After 18 h, cells were nucleofected with SpCas9 2X NLS nuclease loaded with SETD2-targeting sgRNA (GACUCACGGUGUUAUGA with UAU-modified scaffold, Synthego and ThermoFisher Neon Transfection System). Cells were expanded and then selected using Hygromycin. Hygromycin-resistant cells were subjected to single-cell sorting (BD FACSAria) and then screened for homozygous insertion via PCR. The homozygous clone exhibited complete SETD2 depletion after 1.5 h of 1 µM dTag47 (Tocris).
siRAD52.1 and siRAD52.2 (Dharmacon, J-011760-05 and J-011760-06, respectively) and siNon-targeting (Silencer Select Negative Control No. 2, ThermoFisher, 4390846) were reverse transfected using Lipofectamine RNAiMAX using the manufacturer’s protocol. Twenty-four hours after transfection, cells were forward transfected again and treated with doxycycline daily. To generate SETD2 rescue cells, HeLa TetOn-Cas9 sgSETD2.1 cells were transfected with Super piggyBac Transposase (SBI, PB210PA-1) and codon-optimized SETD2 in a PiggyBac expression vector that coexpressed a blastidicin-resistance gene, which was used to select for a polyclonal population (Vectorbuilder).
Immunoblotting.
For immunoblots, whole cell extracts were generated by scraping and then collecting cells via centrifugation. Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed with RIPA buffer (Sigma), supplemented with Halt Phosphatase and Protease inhibitors (Pierce), 1 mM phenylmethylsulfonyl fluoride (PMSF, Sigma), and 25 U/mL Universal Nuclease (Pierce). Extracts were incubated on ice, and protein levels were quantified using the bicinchoninic acid (BCA) assay (Pierce) and supplemented with 4× Laemmli buffer with 2-mercaptoethanol (Bio-Rad). Samples were boiled and sonicated. To generate histone extracts, cells were scraped, centrifuged, and washed twice with ice-cold PBS. Cells were incubated with hypotonic lysis buffer (10 mM Tris–HCl, pH 8.0, 1 mM KCl, 1.5 mM MgCl2, 1 mM dithiothreitol (DTT), Halt, and 1 mM PMSF) for 30 min on rotator at 4 °C. Nuclei were pelleted at 10,000 g for 10 min at 4 °C, and histones were extracted from pelleted nuclei using 0.2N HCl at 4 °C overnight on a rocker. Nuclear debris was pelleted at 16,000 g for 10 min then discarded, and histone extract was quantified using the BCA assay, neutralized with an equal volume of 1M Tris–HCl, pH8.0, and supplemented with 4× Laemmli buffer and then boiled. For chromatin fractionations, cells were resuspended at 10M cells/mL of ice cold Buffer A (100 mM NaCl, 300 mM Sucrose, 3 mM MgCl2, 10 mM 1,4-Piperazinediethanesulfonic acid (PIPES), 1 mM EGTA, 0.2% TritonX-100, 1 mM PMSF, and Halt) and incubated on ice for 10 min. Lysate was centrifuged at 10,000 g for 10 min, saving supernatant and rinsing pellet in 1× volume of Buffer A. Lysate was pelleted again resuspended in an equal volume of Buffer B (50 mM Tris–HCl, pH7.5, 150 mM NaCl, 5 mM EDTA, 1% TritonX-100, 1 mM PMSF, Halt, and Universal Nuclease). Fractions were supplemented with 4× Laemmli buffer, boiled, and sonicated. Proteins were run out using 4 to 20% Criterion TGX Precast Gels (Bio-Rad) and then transferred using the Trans-blot Turbo Transfer System (Biorad) for semidry (histones and low-molecular-weight proteins) or Criterion Blotter for wet transfer (high-molecular-weight proteins). Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (ThermoFisher), blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline (TBS)–Tween 0.1% (blocking buffer), and then probed for antibodies in block buffer. SuperSignal West Pico PLUS or Femto Maximum chemiluminescent solution (Pierce, 34580 and 34095) was added, and blots were imaged using either film (HyBlot), Bio-Rad Gel Doc, or Amersham Imager 600. Primary antibodies used were anti-SETD2 (Sigma, HPA042451), H3K36me3 (Cell Signaling, 4909), β-actin (Cell Signaling, 4970), Histone 3 (Cell Signaling, 14269), Vinculin (Abcam, ab129002), β-tubulin (E7, gift of Kristen Verhey), RAD51 (Millipore Sigma, PC130), RAD52 (Santa Cruz Biotechnology, sc-365341), and Cas9 (Active Motif, 61978). Secondary antibodies used were anti-rabbit and anti-mouse horseradish peroxidase (HRP) conjugates (Pierce, W401B and W402B).
Cell and Metaphase Spread Immunofluorescence and Imaging.
Cells were cultured on cleaned glass coverslips (No. 1.5) in multiwell plates or glass-bottom multiwell plates (CellVis). Cells were fixed either by ice-cold 100% methanol for 10 min or 4% paraformaldehyde in Cytoskeleton Buffer (100 mM NaCl, 300 mM Sucrose, 10 mM PIPES pH6.8, 3 mM MgCl2, and 0.5% TritonX100) for images in Fig. 1 and SI Appendix, Fig. S2C. For chromatin extractions (Fig. 5F), cells were incubated in 20 mM N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES), 50 mM NaCl, 3 mM MgCl2, 300 mM Sucrose, and 0.5% TritonX-100 for 5 min on ice and then fixed with PFA at 4% final concentration in extraction buffer. Cells were then rinsed three times with PBS and incubated with PBS with 0.1% TritonX100 (PBSTx) for 10 min. Cells were blocked with 1% BSA in PBSTx and then stained with primary and secondary antibodies in blocking buffer. Cells were washed with PBS, counterstained with Hoechst (2.5 µg/mL), and then mounted with ProLong Antifade Gold or stored in PBS. Primary antibodies used were anti-α Tubulin (12G10, gift of K. Verhey), cGAS (Cell Signaling, 31659), RPA (Cell Signaling, 2208), RAD51 (Millipore Sigma, PC130), and RAD52 (Santa Cruz Biotechnology, sc-365341).
Metaphase spreads were stained as described (47). Briefly, cells were treated with 150 ng/mL of colcemid (KaryoMAX, ThermoFisher) for 1 to 2 h, harvested via shake-off, and then pelleted via centrifugation. Cells were gently resuspended and swelled with a 1:1 ratio of 75 mM KCl:0.9% sodium citrate (MEFs) or 75 mM KCl (HeLa) and prewarmed to 37 °C. Cells were incubated for 5 to 15 min and then cytospun onto poly-lysine coated slides at 1,300 rpm for 10 min. Cells were extracted using KCl chromosome media (KCM) (10 mM Tris pH8.0, 120 mM KCl, 20 mM NaCl, 0.5 mM EDTA, and 0.1% TritonX100) for 10 min. Samples were blocked using 1% BSA in KCM, stained with antibodies in 1% BSA in KCM, and then fixed with 4% PFA in KCM for 10 min. Chromosomes were rinsed with PBS, counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (1 µg/mL) in PBS, and then mounted with VectaShield (H100) and sealed. Primary antibodies used were CENP-A (Cell Signaling, 2048), CENP-A (Abcam, ab13939), HEC1 (Santa Cruz Biotechnology, sc-135934), HEC1 (GeneTex, GTX70268), H3K36me3 (Abcam, ab9050), CENP-B (Santa Cruz, sc-32285), KNL1 (Abcam, ab222055), CENP-T (Abcam, ab220280), and Centromere Protein Antibody (ACA, Antibodies Inc., 15-234). Secondary antibodies used were anti-mouse, anti-rabbit, or anti-human AlexaFluor conjugates (ThermoFisher, A21121, A11001, A21236, A11032, A32733, A11034, A11037, A21135, and A21445).
Images of mitotic cells and metaphase spreads were acquired on Deltavision Elite imaging system (GE Healthcare) equipped with a Cool SnapHQ2 charge-coupled device camera (Roper), using a 60× 1.4 numerical aperture (NA) or 100× 1.4 NA objectives (Olympus) or Nikon MultiExcitation TIRF imaging system, using widefield settings, Andor Zyla 4.2 Plus sCMOS monochrome camera, and 60× 1.5NA objective (Olympus). Images were acquired using preset Alexa filter settings, and optical sections were collected at 200-nm intervals and processed using ratio deconvolution in softWoRx (GE Healthcare, version 6.5.2) or NIS Elements. Images were prepared for publication using FIJI (Version 2.1.0/1.53o). Images of mitotic and interphase cells are maximum-intensity projects, and metaphase spreads are single z-slices. Images of interphase cells for quantifying nuclear phenotypes were acquired using Cytation 5 (Biotek) using 20× 0.45 NA objective (Olympus) with standard filter sets or Nikon MultiExcitation TIRF imaging system, using widefield settings. Nuclear images and metaphase spreads were manually quantified. Chromatin bridges were defined as DNA structures that were parallel to the axis of poleward elongation and spanned most of the length between the segregated anaphase chromosomes. Lagging chromosomes were scored as DNA that remained in the midzone during anaphase and lacked obvious orientation along the axis of elongation. To count RAD51 and RAD52 foci, nuclei (Hoechst) were thresholded, and foci were identified by finding local maxima using FIJI.
FISH.
Cells were treated with KaryoMAX and then harvested via trypsinization. Cells were centrifuged and then gently resuspended in a 1:1 ratio of 75 mM KCl: 0.9% sodium citrate (MEFs) or 75 mM KCl (HeLa) for 5 to 15 min. Cells were partially fixed by addition of 1/10 volume of ice-cold Carnoy’s fixative (3:1 methanol:acetic acid). Cells were centrifuged and gently resuspended; then, fixative was added, dropwise while vortexing. Cells were incubated at least 30 min on ice, spun down, and resuspended in ~0.5 mL fixative and then dropped onto ice-cold slides. Slides were air-dried at room temperature overnight and then stored at 4 °C. Slides were rehydrated in 2× SSC for 2 min, dehydrated in ethanol series (2 min each in 75%, 90%, 100% EtOH), and then air dried. Centromere (PNA Bio, CENPB-AF488 or -647, 1:100), telomere (PNA, TelC-A647, 1:500), Acro-P (Cytocell, LPE NOR, undiluted), or rDNA (Empire Genomics, RPCI23-225M6, 1:5) FISH probes were diluted in hybridization solution (10 mM Tris–HCl pH7.2, 70% formamide, and 0.5% blocking reagent, Roche 11096176001) and then added to the slide. Slides were denatured at 78 °C for 2 to 5 min and then incubated for overnight at 37 °C. Slides were washed in 0.4× SSC and then 2× SSC with 0.5% Tween20 for 2 min each. Slides were washed three times with PBS adding DAPI (1 µg/mL) to second wash. Slides were dehydrated using ethanol series and then mounted with ProLong Antifade Gold (Invitrogen). If spreads were subjected to FISH following immunofluorescence, the immunofluorescence protocol was used, and spreads were dehydrated using an ethanol series prior to FISH protocol. If spreads were stained only with DAPI, the denaturing and hybridization steps were omitted. For G-banding and Q-FISH, samples were prepared, imaged, and analyzed by Creative Bioarray.
Multicolor FISH.
Setd2flox/flox MEFs were treated with vehicle or 3 µM 4-OHT for 3 d, and mitotic cells were enriched by incubation in 100 ng/mL colcemid (KaryoMAX) for ~5 h. To prepare metaphase chromosome spreads, cells were collected by trypsinization, swollen with prewarmed 0.075 M KCl at 37 °C for 6 min, and fixed by washing and resuspending in ice-cold Carnoy fixative. Metaphase spreads were then dropped onto slides and allowed to air dry. For multicolor FISH, 3 µL of 21XMouse probes (MetaSystems) was applied to the sample and sealed with a coverslip using rubber cement. Samples and probes were codenatured at 75 °C for 2 min followed by incubation at 37 °C overnight in a humidified chamber using the ThermoBrite System (Leica). Slides were then washed for 2 min in 0.4× SSC at 75 °C followed by a 30-s wash in 2× SSC at room temperature. Slides were counterstained with DAPI and mounted with an antifade solution. Metaphases were imaged using a Metafer Slide Scanning Platform (MetaSystems). Slides were first scanned to search for metaphase spreads using MSearch and then automatically captured using Autocapt at 63× magnification. Multicolor karyotypes were generated using Isis (v5.8.12, MetaSystems) and adjusted for imaging threshold. Dicentric chromosomes were identified as chromosomes harboring two DAPI-bright staining regions, and interchromosomal rearrangements were assessed by the appearance of different colored FISH probes hybridizing to the same dicentric chromosome.
CNV Association Analysis with SETD2 Mutated Patient Samples.
Patient metadata, mutation annotations, and gene-level copy number annotations were obtained from the Pan-Cancer Analysis of Whole Genomes (PCAWG) (27), provided by the cBioPortal (48, 49). Segment-level copy number annotations for the PCAWG-11 dataset were obtained from the UCSC Xena data browser. The data from all sources was processed in R and joined by sample ID and/or Tumor Sample Barcode.
Log2 normalization of segment-level CNV data was performed. We excluded sex chromosomes and only kept chromosomes 1:22. Segments with an initial copy number of 0 were set to -2 in the log2 transformation space. Additionally, we normalized the log2 CNVs by subtracting the mean across each of the patients. This is to normalize samples with whole genome doublings and to look for changes in CNV. To identify samples with SETD2 LOH, we separated samples by SETD2 copy number. Samples with a SETD2 copy number of 0 were neutral, and samples with a copy number of -1 were classified as LOH. Segment level copy number was plotted from our normalized CNV data using the cnSpec() function of the R package GenVisR. P and Q arm annotations were derived from the UCSC hg19 cytoband annotation. Association analysis between SETD2 mutation status and CNV was performed by computing different metrics from the CNV data: 1) Count of CNV segments: computing the number of segments with a copy number not equal to zero. 2) Count of chromosomes with a difference between in CNV for P and Q arms greater than 0.
Statistics.
Statistical analyses were performed in GraphPad Prism 10.0.0, except for PCAWG analyses, which were performed R version 4. All statistical tests are listed in figure legends, along with the number of biological or technical replicates. Statistical significance was considered to be P < 0.05, and all significant P values are listed, while nonsignificant values are listed if space permits. All data shown are mean with SD unless otherwise noted.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Jared Nordman, Rahul Bhowmick, David Cortez, Jeffrey Rathmell, and members of the W.K.R. laboratory for their constructive input; I. Cheeseman (Massachusetts Institute of Technology), K. Verhey (University of Michigan), and J. Rathmell (Vanderbilt University Medical Center) laboratories for reagent and resource sharing; the Cell and Developmental Biology Equipment Resource (Vanderbilt University, VU), Cell Imaging Shared Resource (VU), and Vanderbilt Ingram Cancer Center for research and technical support. Bioinformatic analyses of PCAWG data were performed by Vindhya Data Science. SI Appendix, Fig. S1C and Fig. 4K were modified from “Mitosis” by Servier Medical Art, licensed under a Creative Commons Attribution 3.0 unported license. Figs. 3D and 5L are modified from illustrations on Bioicons. Funding for this project is provided by Kidney Cancer Association (F.M.M.), US DOD W81XWH2110786 (F.M.M.), NIH 5T32CA009592 (L.V.), CPRIT RP220332 (R. Dere), NIH R01CA275082 (R. Dere and W.K.R.), US DOD W81XWH221076 (P.L.), and NIH R01CA20301 (C.L.W. and W.K.R.).
Author contributions
F.M.M., R. Dahiya, C.A.L., R.O., P.L., and W.K.R. designed research; F.M.M., E.S.K., A.T.T., R. Dahiya, and L.V. performed research; F.M.M., E.S.K., A.T.T., A.K., L.V., S.R.N., R. Dere, B.D.S., and C.L.W. contributed new reagents/analytic tools; F.M.M., E.S.K., A.T.T., R. Dere, T.G., C.A.L., P.L., and W.K.R. analyzed data; and F.M.M. and W.K.R. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Frank M. Mason, Email: frank.mason@vumc.org.
W. Kimryn Rathmell, Email: kimryn.rathmell@vumc.org.
Data, Materials, and Software Availability
Data will be made available upon request.
Supporting Information
References
- 1.Levine M. S., Holland A. J., The impact of mitotic errors on cell proliferation and tumorigenesis. Gene. Dev. 32, 620–638 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang C.-Z., et al. , Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crasta K., et al. , DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ly P., et al. , Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol. 19, 68–75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ly P., et al. , Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet. 51, 705–715 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lange J., et al. , Isodicentric Y chromosomes and sex disorders as byproducts of homologous recombination that maintains palindromes. Cell 138, 855–869 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tuna M., Amos C. I., Mills G. B., Whole-chromosome arm acquired uniparental disomy in cancer development is a consequence of isochromosome formation. Neoplasia 25, 9–17 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stimpson K. M., Matheny J. E., Sullivan B. A., Dicentric chromosomes: Unique models to study centromere function and inactivation. Chromosome Res. 20, 595–605 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gisselsson D., et al. , Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc. Natl. Acad. Sci. U.S.A. 97, 5357–5362 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maciejowski J., Li Y., Bosco N., Campbell P. J., de Lange T., Chromothripsis and kataegis induced by telomere crisis. Cell 163, 1641–1654 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Umbreit N. T., et al. , Mechanisms generating cancer genome complexity from a single cell division error. Sci. New York N Y 368, eaba0712 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharda A., Humphrey T. C., The role of histone H3K36me3 writers, readers and erasers in maintaining genome stability. Dna Repair 119, 103407 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Fahey C. C., Davis I. J., SETting the stage for cancer development: SETD2 and the consequences of lost methylation. Csh Perspect. Med. 7, a026468 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turajlic S., et al. , Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell 173, 595–610.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park I. Y., et al. , Dual chromatin and cytoskeletal remodeling by SETD2. Cell 166, 950–962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiang Y.-C., et al. , SETD2 haploinsufficiency for microtubule methylation is an early driver of genomic instability in renal cell carcinoma. Cancer Res. 78, 3135–3146 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKinley K. L., Cheeseman I. M., Large-scale analysis of CRISPR/Cas9 cell-cycle knockouts reveals the diversity of p53-dependent responses to cell-cycle defects. Dev. Cell 40, 405–420.e2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flynn P. J., Koch P. D., Mitchison T. J., Chromatin bridges, not micronuclei, activate cGAS after drug-induced mitotic errors in human cells. Proc. Natl. Acad. Sci. U.S.A. 118, e2103585118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiang Y. C., et al. , SETD2 haploinsufficiency for microtubule methylation is an early driver of genomic instability in renal cell carcinoma. Cancer Res. 78, canres.3460.2017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guenatri M., Bailly D., Maison C., Almouzni G., Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol. 166, 493–505 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKinley K. L., Cheeseman I. M., The molecular basis for centromere identity and function. Nat. Rev. Mol. Cell Bio. 17, 16–29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barra V., Fachinetti D., The dark side of centromeres: Types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 9, 4340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y., et al. , Patterns of somatic structural variation in human cancer genomes. Nature 578, 112–121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeBose-Scarlett E. M., Sullivan B. A., Genomic and epigenetic foundations of neocentromere formation. Annu. Rev. Genet. 55, 1–18 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Fachinetti D., et al. , DNA sequence-specific binding of CENP-B enhances the fidelity of human centromere function. Dev. Cell 33, 314–327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mizuno K., Miyabe I., Schalbetter S. A., Carr A. M., Murray J. M., Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature 493, 246–249 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.T. I. P.-C. A. of W. G. Consortium et al. , Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lampe J. W., et al. , Discovery of a first-in-class inhibitor of the histone methyltransferase SETD2 suitable for preclinical studies. Acs Med. Chem. Lett. 12, 1539–1545 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizuno K., Lambert S., Baldacci G., Murray J. M., Carr A. M., Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Gene. Dev. 23, 2876–2886 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paek A. L., et al. , Fusion of nearby inverted repeats by a replication-based mechanism leads to formation of dicentric and acentric chromosomes that cause genome instability in budding yeast. Genes Dev. 23, 2861–2875 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfister S. X., et al. , SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 7, 2006–2018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shoshani O., et al. , Chromothripsis drives the evolution of gene amplification in cancer. Nature 591, 137–141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cortés-Ciriano I., et al. , Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 52, 331–341 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gascoigne K. E., Cheeseman I. M., Induced dicentric chromosome formation promotes genomic rearrangements and tumorigenesis. Chromosome Res. 21, 407–418 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu L., et al. , Two replication fork maintenance pathways fuse inverted repeats to rearrange chromosomes. Nature 501, 569–572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costantino L., et al. , Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hermetz K. E., et al. , Large inverted duplications in the human genome form via a fold-back mechanism. PLoS Genet. 10, e1004139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koumbaris G., et al. , FoSTeS, MMBIR and NAHR at the human proximal Xp region and the mechanisms of human Xq isochromosome formation. Hum. Mol. Genet. 20, 1925–1936 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mar B. G., et al. , SETD2 alterations impair DNA damage recognition and lead to resistance to chemotherapy in leukemia. Blood 130, 2631–2641 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kadara H., et al. , Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann. Oncol. 28, 75–82 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim I.-K., et al. , Acquired SETD2 mutation and impaired CREB1 activation confer cisplatin resistance in metastatic non-small cell lung cancer. Oncogene 38, 180–193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dong Y., et al. , SETD2 mutations confer chemoresistance in acute myeloid leukemia partly through altered cell cycle checkpoints. Leukemia 33, 2585–2598 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rini B. I., Campbell S. C., Escudier B., Renal cell carcinoma. Lancet 373, 1119–1132 (2009). [DOI] [PubMed] [Google Scholar]
- 44.McKinney M., et al. , The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov. 7, 369–379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKinley K. L., Chapter 4 employing CRISPR/Cas9 genome engineering to dissect the molecular requirements for mitosis. Methods Cell Biol. 144, 75–105 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Layden H. M., Eleuteri N. A., Hiebert S. W., Stengel K. R., A protocol for rapid degradation of endogenous transcription factors in mammalian cells and identification of direct regulatory targets. STAR Protoc. 2, 100530 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergmann J. H., et al. , Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. Embo J. 30, 328–340 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cerami E., et al. , The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao J., et al. , Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Data will be made available upon request.