

Abstract
The members of the HNRNPF/H family of heterogeneous nuclear RNA proteins – HNRNPF, HNRNPH1, HNRNPH2, HNRNPH3, and GRSF1, are critical regulators of RNA maturation. Documented functions of these proteins include regulating splicing, particularly alternative splicing, 5’capping and 3’ polyadenylation of RNAs, and RNA export. The assignment of these proteins to the HNRNPF/H protein family members relates to differences in the amino acid composition of their RNA recognition motifs, which differ from those of other RNA binding proteins (RBPs). HNRNPF/H proteins typically bind RNA sequences enriched with guanine (G) residues, including sequences that, in the presence of a cation, have the potential to form higher-order G-quadruplex structures. The need to further investigate members of the HNRNPF/H family of RBPs has intensified with the recent descriptions of their involvement in several disease states, including the pediatric tumor Ewing sarcoma and the hematological malignancy mantle cell lymphoma; newly described groups of developmental syndromes; and neuronal-related disorders, including addictive behavior. Here, to foster the study of the HNRNPF/H family of RBPs, we discuss features of the genes encoding these proteins, their structures and functions, and emerging contributions to disease.
Keywords: HNRNPF/H RNA binding proteins, cancer, alternative splicing, neurodevelopment, HNRNPF, HNRNPH1, HNRNPH2, HNRNPH3, GRSF1
This article is categorized under: RNA Processing > Splicing Regulation/Alternative Splicing, RNA in Disease, RNA interactions with proteins and other Molecules > Protein-RNA Interactions: Functional Implications
Graphical Abstract
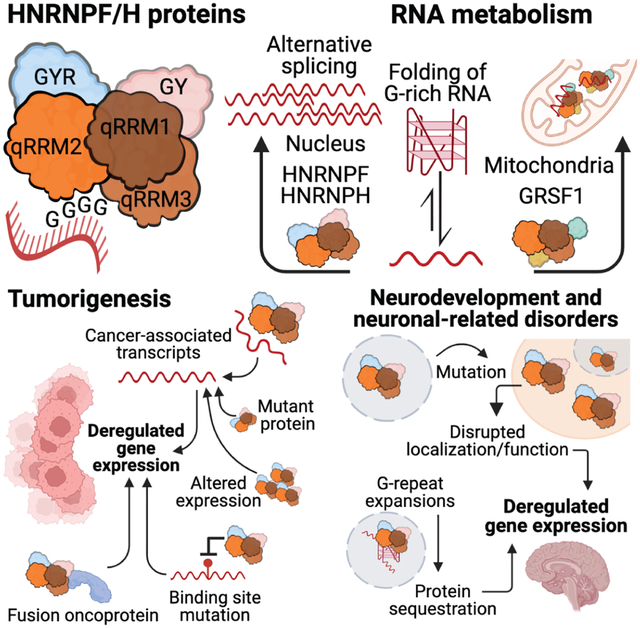
The HNRNPF/H RNA binding proteins are regulators of RNA maturation, particularly alternative splicing. Recent studies have highlighted the discovery of mutations in genes encoding members of the HNRNPF/H protein family in specific diseases, their function in regulating the expression of disease-specific transcripts, and the effect of sequence variants in their binding sites on the regulation of gene expression. Figure created with BioRender.com.
TWITTER:
The contributions of the HNRNPF/H family of RNA binding proteins to cancer and other diseases highlight their importance in regulating RNA maturation.
1. INTRODUCTION
The HNRNPF/H family of heterogeneous nuclear RNA proteins (hnRNPs) comprises five proteins, HNRNPF, HNRNPH1, HNRNPH2, HNRNPH3, and GRSF1. First identified and characterized in the late 1980s through the early to mid-1990s, these homologous RNA binding proteins (RBPs) lack the conserved aromatic amino acid residues in their RNA recognition motifs (RRMs) typical of most hnRNPs. This observation led to their designation as proteins containing quasi-RNA recognition motifs (qRRMs) (Honoré et al., 1995). Structural and functional studies have highlighted HNRNPF/H proteins binding of guanine (G)-rich sequences and how these interactions may alter RNA secondary structures. Studies of these RNA-protein interactions, through the generation of transcriptome-wide datasets and analysis of gene-specific events, have revealed the importance of the HNRNPF/H proteins’ function in regulating RNA processing, particularly alternative splicing. This review begins with summaries of key features of the HNRNPF/H genes and proteins. We then discuss the contributions of this family of RBPs to specific cancer sub-types and neurodevelopmental syndromes, other disease states, and the aging process. We aim to stimulate further study of the functions of the HNRNPF/H proteins to inform our understanding of the mechanism underlying these conditions and aid the development of new treatments based on these findings.
2. THE HNRNPF/H RNA BINDING PROTEINS
Dreyfuss and colleagues first described HNRNPF and what is now known as HNRNPH1 (formally HNRNPH) in a series of studies detailing their amino acid sequence and their binding of poly-G RNA (Matunis et al., 1994; Piñol-Roma et al., 1988; Swanson & Dreyfuss, 1988). In parallel and subsequently, Wilusz and co-workers identified GRSF-1, currently referred to as GRSF1 (Qian & Wilusz, 1994), and described a protein highly homologous to HNRNPH1 initially named DSEF-1 or HNRNPH’ and now designated as HNRNPH2 (Bagga et al., 1998; Bagga et al., 1995; Qian & Wilusz, 1991). HNRNPF, HNRNPH1, HNRNPH2, and GRSF1 have three qRRM domains but exhibit different cellular localizations. The HNRNPF, HNRNPH1, and HNRNPH2 proteins concentrate in the nucleoplasm, while the predominant isoform of GRSF1 (Isoform 1) localizes to mitochondria (Antonicka et al., 2013; Jourdain et al., 2013). The family member, HNRNPH3 (previously known as HNRNP2H9), has different N and C-termini; however, its two qRRM domains share significant sequence homology with the qRRM2 and qRRM3 domains of the other HNRNPF/H proteins (Mahé et al., 1997). Figure 1 summarizes the domain structure of the HNRNPF/H proteins (Figure 1A) and their sequence alignments (Figure 1B).
Figure 1. An overview of the HNRNPF/H proteins.
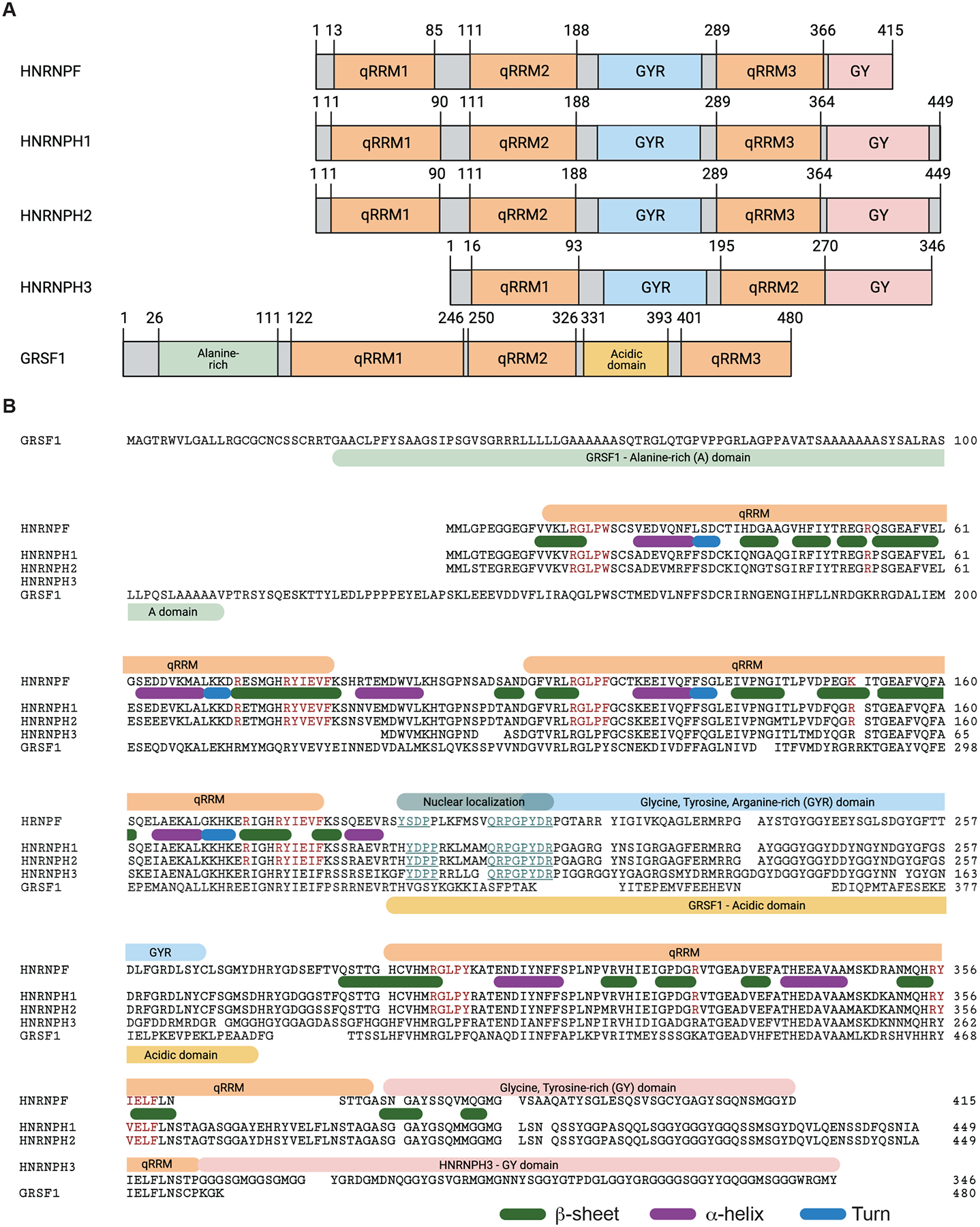
(A) Schematics of the HNRNPF/H protein family. The indicated domains are based on the UniProt reference human proteins as follows: HNRNPF - P52597, HNRNPH1 – P31943, HNRNPH2 – P55795, HNRNPH3 – P31942, GRSF1 – Q12849. (B) The amino acid sequence alignment of the reference HNRNPF/H proteins were generated using R-packages msa (Bodenhofer et al., 2015) and ggmsa (Zhou et al., 2022). The indicated qRRMs, related structural elements (β-sheet, α-helix, turns), and low complexity domains are based on those reported for HNRNPF (P52597), except for the C-terminus of HNRNPH3 (P31942) and the additional GRSF1 domains (Q12849). The amino acids in red indicate residues that contact RNA, and those in blue, indicate residues that form a putative nuclear localization signal. Figure panels created with BioRender.com.
2.1. The HNRNPF/H genes and gene expression
The human HNRNPF/H genes map to four human chromosomes (Banga et al., 1996; Honoré et al., 1995). The HNRNPF and HNRNPH3 genes map to chromosome 10, while HNRNPH1 maps to chromosome 5, HNRNPH2 to chromosome X, and GRSF1 to chromosome 4 (Figure 2A). Bulk RNA sequencing of various human tissues (GTEx Portal: https://gtexportal.org/home/) demonstrates the ubiquitous expression of the human HNRNPF/H gene family members with some variation in the expression levels detected in different tissues, particularly in brain-derived tissues (Figure 2B). The overall expression of HNRNPF and HNRNPH1 is higher than that of other family members, with GRSF1 RNA levels demonstrating the lowest abundance. Interestingly, the transcript variants expressed by the HNRNPF/H genes (Figure 3) are more distinctive and complex than the gene-level expression data, or the amino acid homology of these proteins (Figure 1B) may suggest. The HNRNPF gene (ENSG00000169813) expresses several protein-encoding transcript variants with different 5’ untranslated regions (Figure 3A), each of which encodes the same 415 amino acid protein (UniProt – P52597; ~55 kDa) (Figure 1B). In contrast, the Ensembl genome database reports ~50 annotated transcript variants expressed by the human HNRNPH1 gene (ENSG00000169045), including the subset of transcripts (coding and non-coding) detailed in Figure 3B. The dominant full-length HNRNPH1 transcripts ENST00000393432/NM_001257293 and ENST00000442819/NM_005520 and another variant (ENST00000356731) encode the same 449 amino acid protein (UniProt – P31943; ~50–55 kDa) while additional transcripts have the potential to encode small peptides (<200 amino acids), as well as putative protein isoforms of 212 to 472 amino acids. Interestingly, a substantial proportion of transcription from the HNRNPH1 locus results in the expression of transcripts that lack coding potential. The functional significance of the expression of non-dominant HNRNPH1 isoforms and HNRNPH1 non-coding transcripts is unclear. However, as discussed below (3.2), a study of cancer-associated mutations suggests that the ratio of the dominant protein-coding transcripts and other transcripts may regulate the expression of full-length HNRNPH1 protein (Pararajalingam et al., 2020). Interestingly, though the HNRNPH1 and HNRNPH2 proteins are over 90% homologous, the organization of the genes encoding these proteins differs substantially. Specifically, unlike the multi-exon HNRNPH1 gene, the HNRNPH2 locus expresses a single two-exon transcript (Figure 3C) that encodes the HNRNPH2 449 amino acid protein (UniProt – P55795; ~50–55 kDa). HNRNPH3 (Figure 3D) and GRSF1 (Figure 3E) express several transcript variants. Two HNRNPH3 variants (ENST00000265866 and ENST00000354695) encode proteins of 346 (UniProt – P31942; ~ 37 kDa) and 331 amino acids, respectively, while the longest GRSF1 transcript variant encodes the mitochondrial-localized GRSF1 isoform 1 (480 amino acids, UniProt – Q12849; ~55 kDa).
Figure 2. An overview of the HNRNPF/H genes.
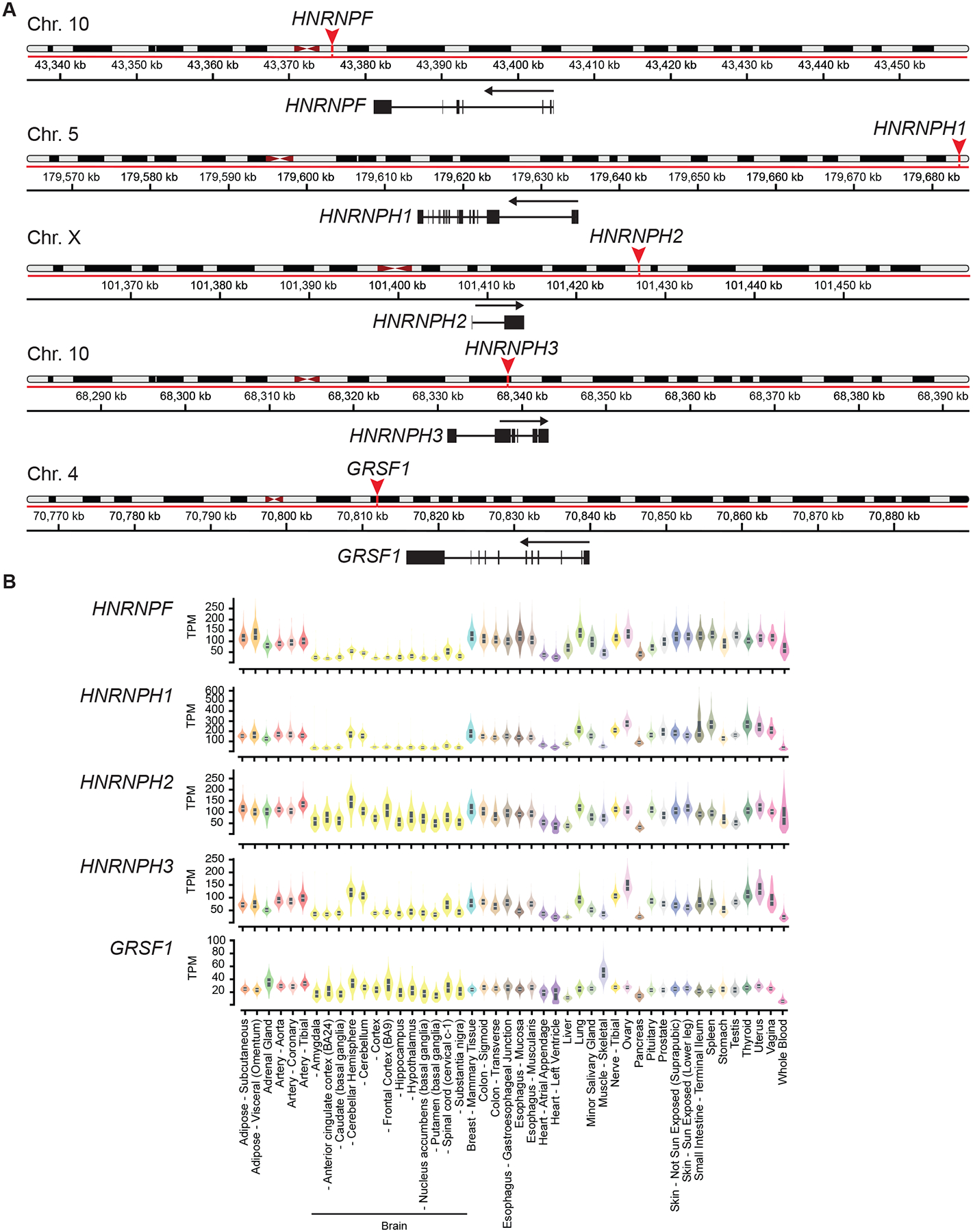
(A) Schematics of the location and organization of genes encoding members of the HNRNPF/H proteins (GTEx Integrated Genome Viewer). (B) An overview of gene level expression of each HNRNPF/H family member assessed using bulk RNA sequencing of selected human tissues. Data extracted from GTEx Analysis Release V8 (dbGaP Accession phs000424.v8.p2; please note the sampling biases in these datasets discussed in (Garsetti et al., 2022).
Figure 3. HNRNPF/H transcript variant expression.

The organization and expression of selected (A) HNRNPF, (B) HNRNPH1, (C) HNRNPH2, (D) HNRNPH3, and (E) GRSF1 transcript variants in selected tissues. Data extracted from GTEx Analysis Release V8 (dbGaP Accession phs000424.v8.p2; please note the sampling biases in these datasets discussed in (Garsetti et al., 2022). The arrow indicates the direction of expression.
2.2. HNRNPF/H protein stucture
The HNRNPF/H proteins’ structural similarities include their qRRM domains and two other domains, a glycine-tyrosine-arginine-rich (GYR) domain and in the case of HNRNPH1, HNRNPH2, and HNRNPH3, a C-terminal glycine-rich (GY) domain (Figure 1A). HNRNPH1 and HNRNPH2 are over 90% homologous, differing by only nine residues, with most in the qRRM1 domains of each protein (five amino acid differences) and other differences in the other qRRM domains (one in the qRRM2 and two in the qRRM3 domains) and one in the GYR region (Figure 1B). The similarity of the HNRNPH1 and HNRNPH2 proteins means that in some cases, experimentation cannot definitively determine whether one protein or the other, or both, are responsible for a given result. Therefore, in this review, when discussing studies that could not report HNRNPH1 or HNRNPH2-specific results, we will use the designation HNRNPH. HNRNPF and HNRNPH1 or HNRNPH2 are about 75% homologous, with amino acid differences found throughout the HNRNPF protein but particularly in the C-terminus region (Figure 1B). The family member, HNRNPH3, has different N and C-termini; however, its two qRRM domains share significant sequence homology with the qRRM 2 and 3 domains of the other HRNPF/H proteins (Figure 1B).
The qRRM domains of the HNRNPF/H proteins define their recognition and interactions with RNA (Honoré et al., 1995; Matunis et al., 1994) and their relative locations and sequences are detailed in Figure 1B. Honoré and colleagues first referred to the RNA recognition motifs of the HNRNPF/H family members as quasi-RRMs because their amino acid composition differed from those of the consensus RRM sequences, lacking positively charged and aromatic residues (Honoré et al., 1995). However, subsequently, studies showed that the qRRM domains adopt classical RRM folds. Specifically, analysis of the HNRNPF protein demonstrated that in the presence of RNA containing three consecutive guanines, each qRRM domain forms a β/α, β/β, α/β structural motif (Dominguez & Allain, 2006; Dominguez et al., 2010). These studies also defined the conserved amino acids within the qRRMs of the HNRNPF/H1/H2 proteins that contact RNA (Dominguez et al., 2010). Specifically, Dominguez and colleagues described a four amino acid motif (RGLP) followed by either W, F, or Y towards the beginning of the domain and an R or L in the middle as critical residues that contact RNA. Towards the end of the domain, conserved sequences in contact with RNA consist of an R or G residue followed by five amino acids and a motif consisting of A, T, I/V, E, V/L, and F (see Figure 1B for the location of each of these motifs). Structural studies of HNRNPH indicate a similar organization of their qRRM domains and that the first two qRRMs of HNRNPH1 can form compact and extended conformations (Penumutchu et al., 2018). Interestingly, a critical determinant of these conformational dynamics is a single amino acid between the qRRM1 and qRRM2 domains – an alanine in HNRNPF and a proline in the HNRNPH proteins (Penumutchu et al., 2018). The alanine residue present in HNRNPF favors the protein forming an extended state that has the potential to facilitate interactions with multiple G-tracts. In contrast, the proline present in HNRNPH confers a more compact state that facilitates interaction with a single G-tract (Penumutchu et al., 2018).
While the qRRM domains have defined structures, the GYR and GY portions of the HNRNPF/H proteins have the repetitive amino acid composition typical of low-complexity domains (LCDs). Many recent studies have LCDs facilitate the multivalent interactions regulating many dynamic processes, including gene expression (reviewed in Alberti & Hyman, 2021; Bhat et al., 2021; Sharp et al., 2022). One assayable property of proteins containing putative LCDs assesses their propensity to form a gel-like state. For example, recent in vitro studies demonstrated that the purified GYR domain region of HNRNPH1 can form a hydrogel detectable by confocal microscopy that transmission electron microscopy revealed consists of polymer-like fibers (Kim & Kwon, 2021). A subsequent analysis presented in the same study highlighted that the GYR domain could interact with other RBPs containing similar LCDs, including HNRNPF, and the importance of three tyrosine residues – Y236, Y240, and Y243 – within this domain as critical determinants of the interaction with other LCD containing proteins. Interestingly, Kim and Kwon observed that the GY domain of HNRNPH1 did not form hydrogel droplets. However, they did observe using a transgene reporter assay system that this domain can activate transcription, the potential significance of which we will discuss in sections 3.3 and 4.1.
Close to and overlapping with the GYR domains, HNRNPF, HNRNPH1, HNRNPH2, and HNRNPH3 are motifs associated with nuclear localization. Specifically, a region that includes a conserved motif recognized by the karyopherin receptor complex and associated with the nuclear transport of HNRNPF - YSDPP (Lee et al., 2006; Siomi et al., 1997) (YDPP in HNRNPH1, HNRNPH2, and HNRNPH3) and a region adjacent to this motif (amino acids 205 – 213 of HNRNPF, HNRNPH1, and HNRNPH2) (Van Dusen et al., 2010) (Figure 1B). Results supporting the importance of this latter region in the nuclear localization of HNRNPF/H proteins include studies showing that deletion or single amino acid substitutions affecting the highly conserved QRPGPYYDRP sequence results in a shift from a nuclear-only localization to nuclear and cytoplasmic localizations. As discussed below (3.2.2 and 4.1), this observation has proven particularly critical in assessing the potential effect of recurrent mutations in several HNRNPF/H genes.
GRSF1 is the only member of the HNRNPF/H family to contain an alanine-rich (A-rich) domain, which is present in the N-terminus of the protein’s longest isoform (480 amino acids), and it also has an acidic (glutamate and proline-rich) region between its qRRM2 and qRRM3 domains (Figure 1B). The mitochondrial location of GRSF1’s longest isoform, which contains the A-rich domain, suggests that this may contribute to this cellular localization (Antonicka et al., 2013; Jourdain et al., 2013). These and a subsequent study that observes a GRSF1 isoform lacking the A-rich domain concentrates in the cytoplasm support this hypothesis (Sofi et al., 2018). The function of the acidic region is undefined; however, one study has suggested that it may function as a negative regulator of RNA binding (Sofi et al., 2018).
2.3. HNRNPF/H protein functions
The regulation of many facets of RNA metabolism, including splice site selection and polyadenylation, involves the interaction of RNPs with G-tracts (3 or more consecutive guanines); (Huppert et al., 2008; McCullough & Berget, 1997; Wang et al., 2004; Xiao et al., 2009; Yeo et al., 2004; Zarudnaya et al., 2003). Multiple studies, including transcriptome-wide analysis, have demonstrated the enriched presence of G-tracts within the binding sites of the HNRNPF/H proteins (for example, (Caputi & Zahler, 2001; Huelga et al., 2012; Uren et al., 2016) and ENCODE RNA Bind-n-seq datasets – HNRNPF: ENCSR376SUZ and HNRNPH2 ENCSR328PGZ). Research related to the functions of the HNRNPF/H proteins has thus probed two broad themes. One theme focuses on the HNRNPF/H proteins’ regulation of RNA processing through their interaction with G-tracts adjacent to splice sites or polyadenylation signals. Complementing these efforts are studies focused on the interaction of the HNRNPF/H proteins with regions of RNA that can form complex G-quadruplex (G4) secondary structures. Here, we will first consider the functions of the nuclear-localized HNRNPF/H proteins, focusing on more general findings rather than reports of their regulation of gene-specific RNA processing, followed by a discussion of GRSF1’s functions in the mitochondria.
2.3.1. Nuclear-localized HNRNPF/H proteins
Before discussing findings related to the function of the nuclear HNRNPF/H proteins, it is crucial to note that due to their homology, assessing their protein-specific functions is particularly challenging. For example, sequence-based reagents (e.g., siRNAs) may target the transcripts expressed by more than one HNRNPF/H gene unless carefully designed. Also, antibodies against these proteins may detect more than one family member, particularly HNRNPH1 and HNRNPH2. It is also likely that the HNRNPF/H proteins regulate the expression of each other, and perturbation of one may alter the expression of another family member. Furthermore, much of our understanding of HNRNPF/H protein function comes from in vitro biochemical or cell-based studies, which will not capture the complexity of their protein-specific functions in vivo. For example, transgenic mice harboring disruptions of Hnrnph1 (Hnrnph1tm1b(KOMP)Wts1) or Hnrnph2 (Hnrnph2em1(IMPC)J) loci exhibit quite different phenotypes. Hnrnph2 hemizygous male and homozygous female mice are viable, though phenotypic analysis noted abnormal vocalization and coat/hair morphologies. In contrast, homozygous disruption of Hnrnph1 on a C57BL/6N background results in preweaning lethality, whereas heterozygous mice exhibit disruption of erythropoiesis. Of note, a recent study by Shan and co-workers highlighted this latter observation as part of their analysis of the differential binding of RBPs, including HNRNPF/H proteins, to RNAs that exhibit changes in expression during red blood cell differentiation (Shan et al., 2021). Generation of another line of Hnrnph1 knockout mice resulted in no post-13.5-week embryos, though its conditional deletion in germline cells produced viable offspring (Feng et al., 2022). However, assessment of the germline Hnrnph1-deleted mice at five months of age demonstrated both male and female infertility due to defects in spermatogenesis and a failure of oogenesis and folliculogenesis, respectively (Feng et al., 2022). Complete ablation of Hnrnpf (Hnrnpf−/− mice generated using B6.C-Tg(CMV-Cre)1Cgn/J x Hnrnpf-fl/fl mice) is embryonically lethal. However, the tissue-specific depletion of HNRNPF (principally in kidney tubules) achieved using a Pax8-Cre-derived mouse line did result in viable mice which exhibited hypertension and glycosuria (Lo et al., 2019). Notably, germline Hnrnph1-deleted mice and the Pax8-Cre-HNRNPF knockout mice exhibited evidence of altered maturation of specific transcripts, consistent with their functions as alternative splicing factors (Feng et al., 2022; Lo et al., 2019).
Multiple studies have described the functions of the HNRNPF/H proteins in alternative splicing, including studies of gene-specific splicing events (for example, (Buratti et al., 2004; Caputi & Zahler, 2001; Chen et al., 1999; Chou et al., 1999; Fisette et al., 2012; Garneau et al., 2005; Hastings et al., 2001; Královic̆ová & Vor̆echovský, 2006; Marcucci et al., 2007; Mauger et al., 2008; McCullough & Berget, 1997; Min et al., 1995). Complementing these studies are more global analyses that have used increasingly sophisticated approaches for determining and quantifying alternative splicing events (for example, (Huelga et al., 2012; Katz et al., 2010; Venables et al., 2008; Wang et al., 2012; Xiao et al., 2009). G-tracts, such as those bound by HNRNPF/H proteins, are over-represented near splice sites, particularly within regions about 70 nts downstream of 5’ splice sites that contribute to defining exon-intron boundaries (McCullough & Berget, 1997; Xiao et al., 2007; Yeo et al., 2004), though G-tracts are also present within other intronic and exonic regions. Thus, at least one mechanism by which HNRNPF/H proteins regulate alternative splicing involves their interaction with these sequences, which combined with 5’ splice sites of varying strengths, determines the frequency of an exon inclusion or exclusion event (Xiao et al., 2009). Additionally, the length and organization of other G-tracts and their intronic or exonic location, may contribute to the regulation of splicing events by the HNRNPF/H proteins. For example, HNRNPF/H proteins may inhibit or promote the recruitment of the spliceosome complex to specific sites in a precursor mRNA. HNRNPF/H proteins may also compete with other RBPs for binding if other cis-regulatory sequences are in proximity to target sites of the HNRNPF/H proteins. Collectively, these observations mean that the output of HNRNPF/H protein function can vary, resulting in exon inclusion in one context and exon exclusion in another.
Related to the interaction of HNRNPF/H proteins with G-rich sequences is the unusual property of nucleic acids containing multiple G-tracts. Specifically, in the presence of a monovalent cation (e.g., K+ or Na+), G-rich sequences can, via Hoosgteen-hydrogen bonds, form planer G-tetrads that can stack to generate a G-quadruplex (G4) structure (Varshney et al., 2020). Experimental studies have estimated that the human transcriptome contains thousands of G-rich sequences with the potential to form RNA parallel G4s stacking two or three guanine-tetrads (consensus motif: G≥2N1–7G≥2N1–7 G≥2N1–7G≥2) (Guo & Bartel, 2016; Kwok et al., 2016). However, it is unclear to what extent rG4s occur within cells, as many RBPs can suppress or resolve such structures. The recognition of G-tracts by HRNPHF/H proteins has meant these are prime candidates for interacting with regions of G-rich RNA with the potential to form rG4s and that their regulation of splicing includes altering rG4 formation as a result of the inhibition of folding (Dominguez et al., 2010; Samatanga et al., 2013), or as our recent in vitro studies have indicated the potential for destabilization of the folded G4 state (Vo et al., 2022).
Another important facet of the RNA processing functions mediated by members of the HNRNPF/H protein family includes their interaction with G-tracts present in the GU rich region 3’ of the AAUAAA polyadenylation (poly A) signal. In a series of studies employing viral or cellular RNAs, in vitro analysis demonstrated that HNRNPF/H proteins can bind G-rich sequences downstream of a poly A signal and as a consequence regulate the efficiency of 3’ end processing (Alkan et al., 2006; Arhin et al., 2002; Bagga et al., 1998; Bagga et al., 1995; Qian & Wilusz, 1991). Initial experiments demonstrated the promotion of cleavage stimulation factor (CSTF) complex assembly by HNRNPF/H proteins and thus 3’-end processing (Arhin et al., 2002; Bagga et al., 1998; Bagga et al., 1995). Subsequent experimentation showed that in other contexts, for example, the processing of B-cell nascent RNAs, HNRNPF/H proteins can also inhibit CSTF binding and RNA cleavage (Veraldi et al., 2001). More recent studies examining the human β-globin locus demonstrated that HNRNPH interacts with the polypyrimidine tract-binding protein PTBP1 and that, in this case, PTBP1 can recruit HNRNPH to a G-rich sequence 5’ of the poly A signal (Millevoi et al., 2009). In addition, analyses of the 3’-end processing of TP53 transcripts have linked the function of the HNRNPF/H proteins to G-rich sequences that can form G-quadruplexes. Specifically, Decorsiere et al. and Newman et al. showed that to overcome the inhibition of 3’ end processing following DNA damage, HNRNPF/H binds a G-rich sequence 3’ of the TP53 poly A signal that can form a quadruplex structure. This binding event together with the DHX36 resolution of the G4 structure maintains TP53 RNA processing and thus protein expression (Decorsiere et al., 2011; Newman et al., 2017). Finally, there is some evidence that the HNRNPF/H proteins may regulate the alternative splicing and polyadenylation of transcripts expressed by a single gene to mediate expression of different protein isoforms, for example isoforms of the acetylcholinesterase enzyme (Nazim et al., 2017).
2.3.2. GRSF1 and mitochondrial function
To date, the most detailed analyses of the cellular functions of GRSF1 have focused on the activity of its longest isoform – isoform 1 – in regulating the post-transcriptional processing of transcripts expressed in mitochondria (mt). As mentioned above (2.2), the 480 amino acid GRSF1-isoform 1 contains an N-terminus A-rich domain that functions as a mitochondrial localizing signal. In mitochondria, GRSF1 forms RNA-dependent foci (Antonicka et al., 2013; Jourdain et al., 2013). In the absence of GRSF1, studies have demonstrated significant disruption of the expression of several mitochondrial RNA (mtRNA) species. For example, Jourdain and co-workers and Antonicka and colleagues reported decreased levels of COX1, COX2, and MT-ND5 protein-encoding mRNAs following the silencing of GRSF1 (Antonicka et al., 2013; Jourdain et al., 2013). The same studies also noted alterations in the steady state levels of ribosomal RNAs, notably decreased detection of 16S ribosomal RNA (rRNA). Jourdain and co-workers also reported changes in the ratio of some mature and precursor tRNAs; for example, depletion of GRSF1 resulted in decreases in the levels of some mature transfer RNAs (tRNAPhe and tRNAleu) but increases in their respective precursor RNAs (Jourdain et al., 2013). Additional experimentation by Jourdain et al. demonstrated that GRSF1 interacts with subunits of RNase P, an enzyme required for processing many mtRNAs, and the RNase P-independent processing of other transcripts (Jourdain et al., 2013). Complementary findings of Antonicka and co-workers demonstrated GRSF1’s binding of RNAs encoded by the mitochondrial genome’s light strand (L-strand) (Antonicka et al., 2013). A subsequent study by Pietras and colleagues confirmed GRSF1’s binding of the mtRNA L-strand and showed GRSF1 functions as a critical component of a mtRNA degradation mechanism (Pietras et al., 2018). The nomenclature of the mtRNA L and heavy (H) strands refers to their asymmetric densities in a gradient, which reflect differences in their sequence distribution. Specifically, L-strand-transcripts are G-rich and thus predicted to have a greater likelihood of containing G-tracts that can form rG4s. In brief, Pietras et al. demonstrated that in mitochondria, GRSF1 could bind and disrupt G4 structures. A component of the mitochondrial degradosome – SUPVSL1 (previously SUV3) interacts with the GRSF1-bound G4 containing RNA to release GRSF1 and facilitate degradation of the RNA by the 3’ – 5’ ribonuclease PNPT1 (previously PNPase) (Pietras et al., 2018). This cooperative mechanism between GRSF1 and the mitochondrial degradosome is vital for the maintenance of the mitochondrial transcriptome. Interestingly, Pietras et al. speculate that the GC sequence bias of vertebrate mitochondrial genomes also required an adaption of a mechanism that limits the accumulation of transcripts prone to rG4 formation and that this may, at least in part, explain the late evolutionary emergence of GRSF1 (Pietras et al., 2018).
Other functions ascribed to GRSF1 include a report by Ufer and co-workers describing its interaction with a G-rich sequence in the 5’ untranslated region (UTR) of a transcript variant of glutathione peroxidase 4 (GPX4) that encodes a mitochondrial-specific isoform of the GPX4 protein (Ufer et al., 2008). Ufer and colleagues also showed that depletion of GRSF1 in embryos ex vivo by RNAi resulted in the impairment of mid and hind-brain development; effects rescued by the co-administration of a cDNA expressing the mitochondrial-specific isoform of GPX4 (Ufer et al., 2008). Noh, Kim, and colleagues noted the binding of GRSF1 to a similar sequence motif in a long non-coding RNA – RMRP (RNA component of mitochondrial RNA processing endoribonuclease) (Noh et al., 2016), as described for the interaction of GRSF1 and the transcript variant of GPX4. RMRP is a component of a complex required for the processing of mature 5.8S rRNAs, and subsequent experiments performed by Noh, Kim et al. showed that GRSF1 contributes to an accumulation of RMRP RNAs in the mitochondrial matrix (Noh et al., 2016).
3. THE HNRNPF/H RNA BINDING PROTEINS AND CANCER
Many proteins that regulate RNA maturation have emerged as contributing to the expression of cancer-specific oncogenic transcripts and alterations in the expression profiles of cancer cells, including core components of the spliceosome and hnRNPs. We discuss below recent studies that highlight the different mechanisms by which HNRNPF/H proteins can promote tumorigenesis (Figure 4A), including their function in regulating the expression of mRNAs encoding proteins that promote tumorigenesis or alter responses to treatment. We also discuss the effect of somatic mutations on the expression or function of members of this protein family or the effect of mutations that disrupt their binding sites and chromosomal rearrangements involving the HNRNPF/H genes.
Figure 4. HNRNPF/H proteins and the expression of cancer-specific transcripts.
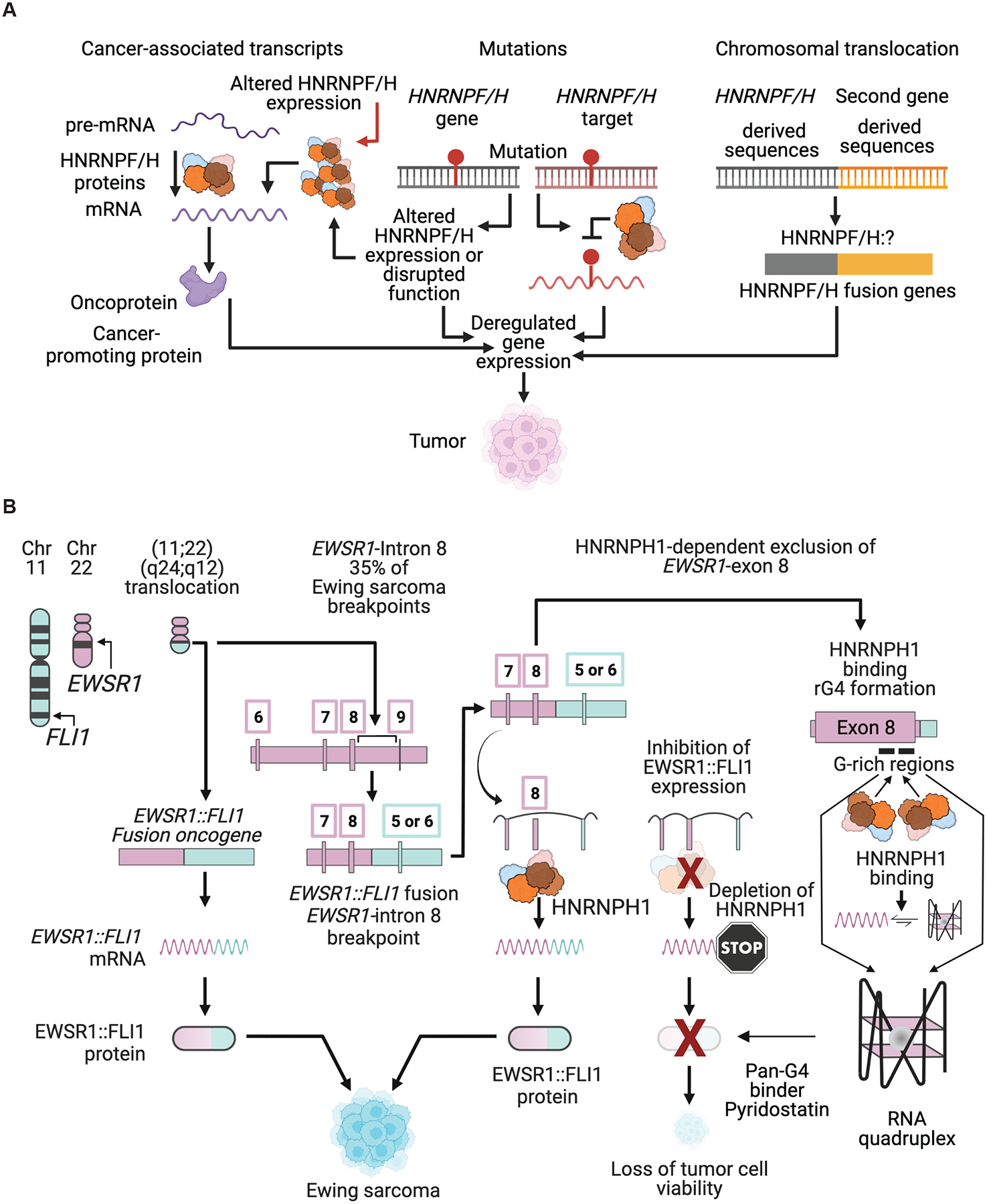
(A) Schematics of the mechanisms by which HNRNPF/H genes or proteins can contribute to tumorigenesis. Mechanisms shown are the HNRNPF/H-dependent processing of cancer-specific transcripts, the effect of mutations that disrupt HNRNPF/H protein expression or function or the binding sites of these proteins, and the involvement of HNRNPF/H genes in chromosomal translocations. (B) Schematic of the HNRNPH1-dependent exclusion of EWSR1-exon 8 present in the EWSR1::FLI1 transcripts expressed in a subset of the pediatric tumor Ewing sarcoma (see Grohar et al., 2016; Neckles et al., 2019; Vo et al., 2022) for further details). In vitro analysis has demonstrated that HNRNPH1 binds EWSR1-exon 8 G-rich sequences in non-G4 and G4 states, and that its binding favors accumulation of the bound RNA in an unfolded state (Vo et al., 2022). Figure panels created with BioRender.com.
3.1. Cancer-associated transcripts and the HNRNPF/H proteins
One mechanism by which members of the HNRNPF/H proteins contribute to tumorigenesis is their promotion of the expression of specific transcript variants encoding proteins that tumor cells depend on for cancer initiation or progression or could modulate treatment efficacy. Here, we describe our recent studies highlighting the function of HNRNPH1 in processing the oncogenic fusion transcript EWSR1::FLI1. We also discuss studies performed by others that illustrate further examples of how the HNRNPF/H proteins regulate the expression of cancer-specific transcripts or gene expression more broadly.
3.1.1. HNRNPH1 and EWSR1::FLI1
Ewing sarcoma (EWS) is a pediatric bone and soft tissue cancer. In about 85% of Ewing sarcomas, a chromosomal translocation t(11:22)(q24:q12) generates the fusion oncogene EWSR1::FLI1 consisting of 5’ EWSR1-derived and 3’ FLI1-derived sequences. The resulting oncoprotein (EWSR1::FLI1) functions as an aberrant transcription factor that promotes malignant transformation and the proliferation and survival of EWS cells. Of the 85% of Ewing sarcomas that harbor an EWSR1::FLI1 fusion gene, a subset (approximately 35% of cases, see (Vo et al., 2022)) harbor EWSR1-intron 8 breakpoints. To express the EWSR1::FLI1 fusion oncoprotein, tumor cells that harbor EWSR1-intron 8 breakpoints must exclude EWSR1-exon 8 during RNA processing to generate an in-frame EWSR1::FLI1 mRNA that expresses the fusion oncoprotein. Failure to exclude EWSR1-exon 8 results in the expression of an EWSR1::FLI1 mRNA that includes a premature stop codon. The results of a genome-wide RNAi screen performed in an EWS cell line dependent on excluding EWSR1-exon 8 to express EWSR1::FLI1 and our subsequent studies determined that HNRNPH1 functions as the critical regulator of this splicing event (Grohar et al., 2016) (Figure 4B). In brief, silencing of HNRNPH1 in EWS cell lines dependent on the exclusion of EWSR1-exon 8 to express in-frame EWSR1::FLI1 fusion transcripts (e.g., the EWS cell lines TC-32 and SK-N-MC) results in the inclusion of EWSR1-exon 8, a decrease in the expression of the fusion oncoprotein, reversal of the well-characterized EWS gene expression signature, and loss of cell viability (Grohar et al., 2016). Interestingly, PCR analysis of EWSR1::FLI1 transcripts (Neckles et al., 2019) and minigene assays (Vo et al., 2022) demonstrated that neither HNRNPH2 nor HNRNPF function as regulators of EWSR1-exon 8 exclusion, indicating that this is an HNRNPH1-specific splicing event. Building on these observations, we showed that HNRNPH1 binds at least one G-rich sequence present at the 3’ end of EWSR1-exon 8 in vitro and that in the presence of cations, these RNAs can form G-quadruplex (G4) secondary structures (Neckles et al., 2019; Vo et al., 2022). Using this information, we assessed if TC-32 and SK-N-MC EWS cell lines are sensitive to the pan-G4-binder pyridostatin (PDS). We determined that these EWSR1-exon 8-positive EWS cell lines exhibit reduced cell viability at lower concentrations of PDS than other EWS cell lines or non-EWS cells. Furthermore, we observed that PDS generates a concentration-dependent increase in the inclusion of EWSR1-exon 8 and a concomitant decrease in the expression of the EWSR1::FLI1 protein and reverses the expression of established transcriptional targets of the fusion oncoprotein (activated and repressed genes) (Neckles et al., 2019). Collectively, these studies suggest a means for inhibiting EWSR1:FLI1 expression through disruption of the HNRNPH1-dependent processing of the pre-mRNA expressed in a subset of Ewing sarcomas.
3.1.2. HNRNPF/H and the regulation of cancer-associated transcripts
Tumorigenesis requires the disruption of many biological processes, including the deregulation of specific signaling pathways. One HNRNPF/H-dependent splicing event that could contribute to the activation of multiple signaling pathways involves the expression of the Δ165 isoform of the receptor tyrosine kinase, MST1R (macrophage stimulating 1 receptor, also known as RON) (see (Cazes et al., 2022) for discussion of MST1R/RON). The Δ165 isoform of MST1R requires no ligand binding for activation, resulting in the stimulation of multiple pathways associated with cancer, including the TGFß and PI3K/AKT pathways. First detected in a gastric carcinoma cell line (Collesi et al., 1996), tumor types expressing the MST1R-Δ165 transcript variant include colorectal and breast cancer (Ghigna et al., 2005; Zhou et al., 2003), glioblastoma multiforme (LeFave et al., 2011), and ovarian (Mayer et al., 2015) and pancreatic cancer (Chakedis et al., 2016). Expression of the MST1R-Δ165 transcript variant involves the exclusion of MST1R-exon 11 from the full-length transcript. Analysis by LeFave and colleagues highlighted the presence of two G-tracts (3Gs) at the 5’ end of MST1R-exon 11 (LeFave et al., 2011). Critically, depletion of HNRNPH1 in cells expressing the transcript encoding the MST1R-Δ165 isoform resulted in a significant shift from the exclusion of MST1R-exon 11 to the inclusion of this exon; findings confirmed using a minigene consisting of MST1R-exon 11 and flanking intronic sequences (LeFave et al., 2011). A subsequent study by Braun and co-workers confirmed and extended these findings. First, using a series of minigene constructs and random mutagenesis, this study reported the mapping of RBP sequence binding motifs to nucleotide (nt) changes that influence MST1R-exon 11 splicing (Braun et al., 2018). This analysis highlighted several RBPs, particularly HNRNPH1 and HNRNPH2, as putative regulators of the splicing of MST1R-exon 11. Consistent with earlier findings, the silencing of HNRNPH1 resulted in increased MST1R-exon 11 inclusion. Interestingly, further analysis indicated cooperativity of HNRNPH1/2’s binding of multiple sites in MST1R-exon 11, suggesting that even minor changes in HNRNPH1/2 expression could affect the splicing of this exon significantly (Braun et al., 2018). Furthermore, the authors speculate that the MST1R-exon 11 G-tracts could form G4 structures and that multiple binding events may be critical to altering the local secondary structure to favor exon skipping (Braun et al., 2018). Considering the relevance of these observations to cancer, analysis of data extracted from The Cancer Genome Atlas (TCGA) indicated a correlation of mutations within MST1R-exon 11 G-tracts, including those that HNRNPH1 or HNRNPH2 could bind and exclude this exon, particularly in carcinomas of the head and neck (squamous cell) or thyroid (Braun et al., 2018). We will discuss additional cases of the potential effect of sequence changes in the putative binding sites of HNRNPF/H proteins in 3.2.
As the Braun et al. study suggests, slight differences in the levels of HNRNPF/H these proteins in a pre-malignant or transformed cell, compared with a healthy cell, could have a profound effect on the transcriptome. Several studies have thus examined correlations between HNRNPF/H expression and the expression of cancer-associated transcripts. For example, in prostate cancer, two independent studies have implicated HNRNPF/H proteins in regulating the expression of androgen receptor (AR), a transcription factor involved in the initiation and progression of castration-resistant prostate cancer. First, Yang and colleagues demonstrated using multiple independent cohorts that prostate tumor samples express elevated HNRNPH1 transcript levels than matched healthy tissue (Yang et al., 2016). They also showed that prostate tumor samples from African American men exhibited further differential expression compared to similar tumor samples from Caucasian Americans (Yang et al., 2016). In addition, Yang and co-workers found a correlation between AR and HNRNPH1 expression in prostate tumors. Finally, they demonstrated that depleting HNRNPH1 expression via RNAi decreases the expression of full-length AR and the AR-V7 transcript variants (Yang et al., 2016). The generation of AR-V7 transcripts involves using a cryptic exon 3b present within AR-intron 3 and is associated with the resistance of prostate cancer cells to androgen deprivation therapy. Interestingly, a study by Fan and co-workers demonstrated that HNRNPF can bind a G-rich sequence within exon 3b of the AR gene and that HNRNPF promotes recruitment of U2AF65 to the AR-exon 3b 3’ splice site, which aids in the recognition and inclusion of this exon and thus the expression of AR-V7 (Fan et al., 2018).
Several reports have also noted deregulated expression of one or more HNRNPF/H proteins in the brain tumor glioblastoma multiforme (GBM). LeFave and colleagues first demonstrated elevated HNRNPH RNA and protein expression in GBM samples compared to healthy tissues using PCR-based and immunohistochemical analysis (LeFave et al., 2011). Critically, the authors speculated that HNRNPH’s elevated expression might explain the altered splicing of MST1R their study revealed, along with changes in the splicing of transcripts expressed that encode the MADD (MAP kinase activating death domain) gene, which functions in TNF-α signaling. More recently, Herviou et al. utilized the TCGA database to demonstrate increased expression of the HNRNPF and HNRNPF RNAs in tumors (n=154) compared with a small number of control samples (n=5) and confirmed elevated expression of the HNRNPH and HNRNPF proteins in three high-grade GBM samples versus low-grade glioma samples (Herviou et al., 2020). As part of the same study, Herviou and colleagues concluded that low levels of the HNRNPF/H proteins favor reduced translation of mRNAs encoding proteins involved in the DNA damage response. In contrast, higher levels of HNRNPF/H proteins promote the translation of these mRNAs. Evidence presented to support this conclusion included demonstrating the HNRNPF/H-DHX36 cooperative regulation of USP1, a protein that forms part of the Fanconi anemia DNA repair pathway (Herviou et al., 2020). Also, based on earlier studies that HNRNPH regulates the alternative splicing of transcripts encoding the serine/threonine kinase ARAF (Rauch et al., 2011; Rauch et al., 2010), Le Bras et al. recently confirmed that the depletion of HNRNPF/H increases the expression of an ARAF transcript variant that negatively regulates the MAPK pathway. (Le Bras et al., 2022). Additional analysis linked HNRNPF/H’s indirect regulation of the MAPK pathway to alterations in the expression of MAPK protein targets, including modification of the translation initiation factor EIF4E (eukaryotic translation initiation factor 4E) (Le Bras et al., 2022). The authors of this study thus propose that elevated levels of HNRNPF/H in GBM enhance expression of the full-length ARAF protein, resulting in an indirect increase in translation by promoting EIF4E phosphorylation. These changes in protein translation increase tumor growth. It is unclear what mechanism or mechanisms result in the elevated expression of HNRNPF/H in some tumor types. However, one report has indicated that the transcription factor MYC may regulate HNRNPH1 expression (Rauch et al., 2011).
3.2. Mutations in HNRNPF/H genes or the targets of HNRNPF/H proteins
Recurrent, loci-specific sequence changes are a feature of many cancer types, and their identification has proven critical to defining those genes that contribute to tumorigenesis. Here, we highlight recent studies reporting recurrent sequence changes in HNRNPF/H genes that may increase cancer risk or contribute directly to tumorigenesis. We will also feature one example of how sequence changes in sites bound by HNRNPF/H proteins can dramatically change the transcript variants expressed by a particular gene.
3.2.1. Ulcerative colitis, cancer risk, and HNRNPF
An increased risk of cancer is associated with the repetitive cycles of tissue damage and repair observed in clinical states involving chronic inflammation. One such inflammatory condition is ulcerative colitis, which affects the intestine and is associated with an increased risk of colorectal cancer. A recent study of sequence changes detected in epithelial cells isolated from the nondysplastic intestinal crypt structures of ulcerative colitis patients highlighted the selection for alterations in several genes, including HNRNPF (Kakiuchi et al., 2020). Observed in 18 samples, these sequence changes in HNRNPF are all predicted to affect the protein’s putative nuclear localization signal (nls) (p.R206Q, p.R206W, p.P209S, p.P209L, pP209T, pY210C, pY210D, p207–213del, p.209–214del). It is unclear whether these mutations contribute to the clinical course of ulcerative colitis and/or increased cancer risk, but, as we will discuss below (4.1), this cluster of mutations occurs in the same region as several germline mutations in other HNRNPF/H genes, particularly HNRNPH2, reinforcing that this portion of the protein is critical to its function.
3.2.2. Mantle-cell lymphoma
The B-cell malignancy, mantle-cell lymphoma (MCL), constitutes about 5 to 7% of lymphoma diagnoses per year (Armitage & Longo, 2022). The most frequently observed genetic event in MCL involves a chromosomal translocation that results in the constitutive expression of cyclin D1, a regulator of the cell cycle (reviewed in Navarro et al., 2020). Additional recurrent somatic sequence changes observed in MCL include those affecting genes that function in the DNA damage response, NOTCH or NFκB-signaling pathways, epigenetic regulation (reviewed in Silkenstedt et al., 2021) or, most recently, RNA processing, specifically, DAZAP1, EWSR1, and HNRNPH1 (Nadeu et al., 2020; Pararajalingam et al., 2020). Focusing on HNRNPH1, the sequence analysis of two independent cohorts of MCL samples (Nadeu et al., 2020; Pararajalingam et al., 2020) and retrospective analyses (Pararajalingam et al., 2020) of previous datasets (Agarwal et al., 2019; Beà et al., 2013; Khodadoust et al., 2017; Rule et al., 2018; Wu et al., 2016) revealed that as many as 10% of MCLs harbor sequence changes in HNRNPH1 with most mapping to intronic regions adjacent to or within HRNPNH1-exon 5 (exon 4 of the coding sequence) (Figure 5A). Interestingly, analysis of other lymphoma subtypes suggests that HNRNPH1 sequence changes represent an MCL-specific mutation (Pararajalingam et al., 2020).
Figure 5. Cancer-associated mutations in HNRNPF/H genes.
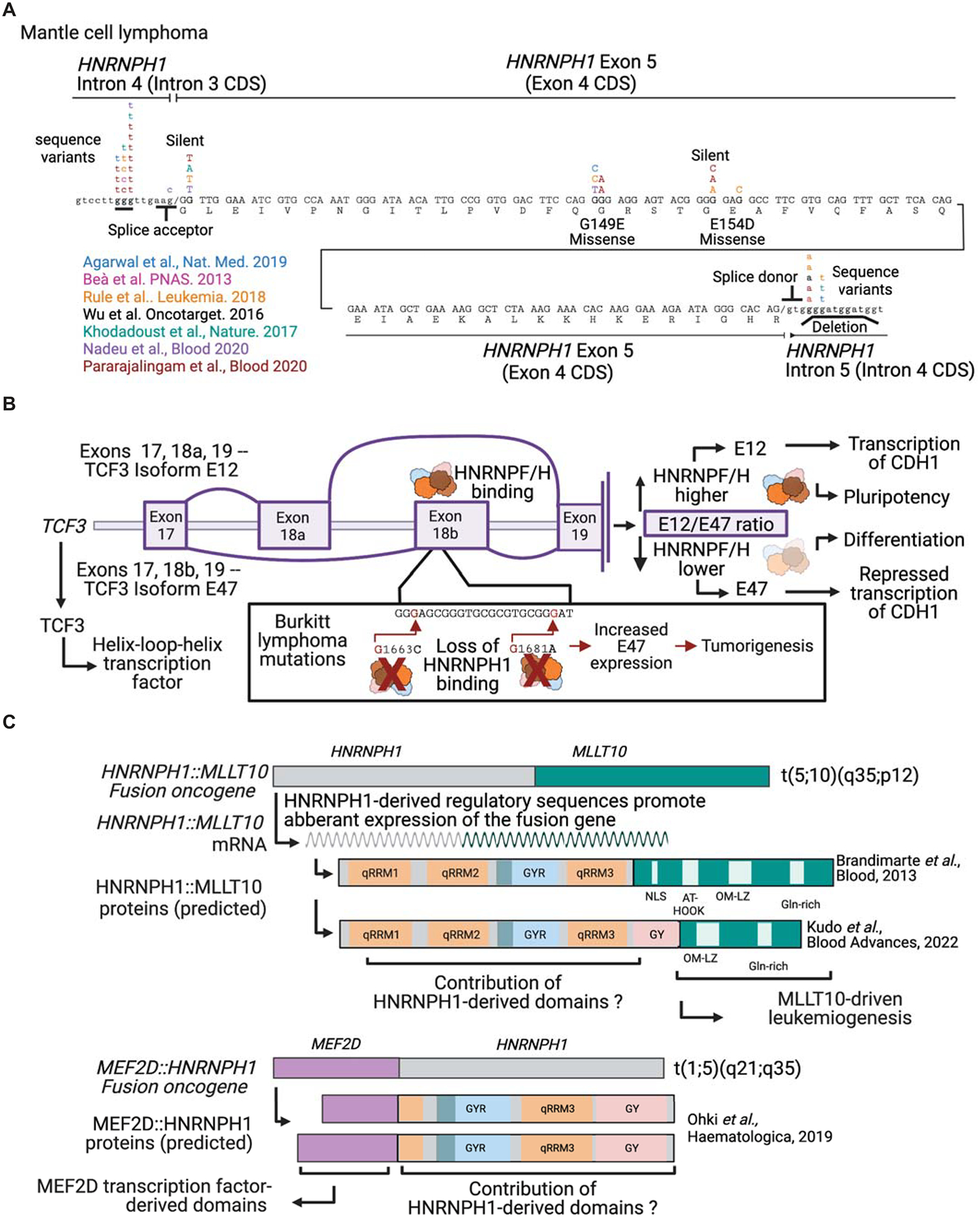
(A) A schematic of the position of mutations in the HNRNPH1 gene identified in mantle cell lymphomas reported by (Nadeu et al., 2020; Pararajalingam et al., 2020), with the latter study including reanalysis of the following studies (Agarwal et al., 2019; Beà et al., 2013; Khodadoust et al., 2017; Rule et al., 2018; Wu et al., 2016). (B) A schematic of the HNRNPF/H-dependent processing of TCF3 (transcription factor 3) transcript variants that express isoforms with different phenotypic outcomes and the effect of mutations that alter HNRNPF/H binding sites based on findings reported in Burkitt’s lymphoma(Yamazaki et al., 2020; Yamazaki et al., 2018; Yamazaki et al., 2019). (C) Schematics of MLLT10-HNRNPH1 fusion genes identified in pediatric T-acute lymphoblastic leukemia (Brandimarte et al., 2013) and pediatric acute myeloid leukemia (Kudo et al., 2022) and HNRNPH2-MEF2D identified in a case of pediatric B-cell precursor acute lymphoblastic leukemia (Ohki et al., 2019). MMT10 = MLLT10 histone lysine methyltransferase DOT1L cofactor; MEF2D = myocyte enhancer factor 2D. Figure panels created with BioRender.com.
Further observations made by Pararajalingam and colleagues included highlighting that the recurrent HNRNPH1 intronic sequence changes predominately affect G-tracts (Figure 5A) and that at least one of these had the potential for binding by HNRNPH1. They also noted that the ratio of HNRNPH1 transcripts that include or exclude the exon these sequences flank differed when they examined the HNRNPH1 mRNAs expressed in mutated and unmutated tumor samples (Pararajalingam et al., 2020). These observations led Pararajalingam and co-workers to speculate that the non-coding mutations identified in the MCL samples disrupt the expression of one or more of its transcript variants (coding and/or non-coding), resulting in the increased expression of full-length HNRNPH1 mRNAs and HNRNPH1 protein (Pararajalingam et al., 2020). Indeed, subsequent assessment of protein expression in MCL tissue samples demonstrated a correlation between mutation status and a shift in the splicing ratio that favors the generation of full-length transcripts with the intensity of anti-HNRNPH staining (Pararajalingam et al., 2020). Further studies are needed to determine how the altered expression of HNRNPH1 contributes to MCL. However, this study does highlight the need to consider how disruption of the autoregulation of HNRNPH1 expression may represent an important mechanism for deregulating its expression and potentially other protein family members and RNA binding proteins, a finding that warrants further investigation.
3.2.3. Burkitt’s lymphoma
Cancer-specific sequence changes that alter the binding site for a member of the HNRNPF/H protein family can also influence the expression of a protein that contributes to tumorigenesis. One example is the regulation of two isoforms of the E-box helix-loop-helix transcription factor, TCF3. In a series of studies conducted by Yamazaki and colleagues, they demonstrated that HNRNPF/H proteins are critical regulators of the expression of two TCF3 isoforms, E12 and E47, by mediating an alternative splicing event involving TCF3-Exon 18a and TCF3-Exon 18b, respectively (Figure 5B) (Yamazaki et al., 2018; Yamazaki et al., 2019). HNRNPF/H proteins bind sites in TCF3-Exon 18b, suppressing the inclusion of this exon and favoring the expression of TCF3-Exon 18a containing E12 isoform (Yamazaki et al., 2018). Regulation of the expression of the relative levels of each TCF3 isoform is critical for defining human embryonal stem cell fate as the TCF3-E47 isoform represses the transcription of CDH1, resulting in reduced E-cadherin expression and thus cell differentiation. In contrast, the expression of the E12 isoform favors CDH1 expression and the maintenance of a pluripotent state (Yamazaki et al., 2018). A follow-up study identified a distal intronic conserved region (ICR) between theTCF3 Exons 18a and 18b as an additional cis-regulatory element that regulates the usage of these exons (Yamazaki et al., 2019). Specifically, Yamazaki et al. determined that a second RBP, PTBP1, binds this ICR and functions cooperatively with HNRNPH1 bound to TCF3-Exon 18b to regulate the alternative splicing of TCF3. In brief, low levels of HNRNPH1 and PTBP1’s binding of the ICR favors the inclusion of TCF3-exon 18b and TCF3-exon 18a exclusion and, thus, expression of the TCF3-E47 isoform. Contrastingly, elevated HNRNPH1 levels result in the expression of the TCF3-E12 isoform (Yamazaki et al., 2019). Interestingly, a study of recurrent mutations observed in the MYC-driven B-cell lymphoma, Burkitt’s lymphoma, has shown significant enrichment of alterations that map to TCF3-Exon 18b (Schmitz et al., 2012). Furthermore, employing minigene reporters and gel shift assays, Yamazaki et al. demonstrated that two of these mutations – G1663C and G1681A – alter the ratio of E12 and E47 transcripts due to the loss of HNRNPH1 binding (Figure 5B) (Yamazaki et al., 2020). These results suggest that disruption of HNRNPF/H’s regulation of TCF3 splicing may contribute to the pathology of a subset of Burkitt’s lymphoma.
3.3. Fusion oncogenes
Many tumors harbor genomic rearrangements that result in the fusion of sequences from two different genes, a proportion of which initiate or contribute to tumorigenesis. Fusion oncogenic events can include the juxtaposition of the regulatory regions from one gene such that these sequences deregulate the expression of another gene. In other cases, the fusion of the 5’ regulatory region and a portion of the coding sequence from one gene adjacent to coding sequences derived from a second gene results in the expression of a protein with aberrant function. In recent years, the detection of fusion events in tumor samples involving the HNRNPF/H loci has increased primarily because of the availability of computational tools that enable the detection of fusion transcripts within RNA-seq data. The genomic rearrangement that underlies the expression of several of these HNRNPF/H-fusion transcripts remains to be determined. However, particularly in the case of two or more reports of a similar fusion event, these could indicate the generation of a bona fide fusion oncogene. If a HNRNPF/H family member is the 5’ partner gene, the regulatory elements responsible for the ubiquitous constitutive expression of these genes could deregulate the expression of the 3’ partner gene’s protein product. Alternatively, or in addition, the HNRNPF/H protein domains could contribute directly to the function of a derived fusion oncoprotein by altering protein-nucleic acid or protein-protein interactions.
Cases of fusion oncogenes in which HNRNPH1 is the 5’ partner gene include t(5;10)(q35;p12) translocations found in pediatric leukemias, including T-cell acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) (Brady et al., 2022; Brandimarte et al., 2013; Chen et al., 2018; Kudo et al., 2022) (Figure 5C). Analysis of the fusion of 5’ HNRNPH1-derived and MLLT10-derived 3’ sequences in one case of early T-cell precursor-ALL identified transcripts that joined either HNRNPH1 nt 1324 (exon 11) or HNRNPH1 nt 6701 (intron 10) to nt 2097 of MLLT10 (exon 15) (Brandimarte et al., 2013). These transcripts are in-frame and predicted to express a fusion protein that retains most of the three qRRM domains of the RNA binding protein, along with the MLLT10-derived OM-LZ domain required for leukemogenesis. Sequencing of another HNRNPH1-MLLT10 fusion expressed in a case of pediatric AML indicated fusion of HNRNPH1-intron 12 sequences to MLLT10-intron 14 sequences, which, following splicing, generates an in-frame transcript predicted to express a fusion protein including the three qRRM domains of HNRNPH1 (Kudo et al., 2022). Currently, it is unclear if the presence of the HNRNPH1 RNA binding domains contributes to the fusion oncoproteins function or whether it is only the utilization of HNRNPH1’s regulatory sequences to mediate the deregulated expression of the transcription factor MTTL10 that is critical to inducing tumorigenesis. The same applies to HNRNPH1::ERG fusions described in a small number of acute myeloid leukemia (AML) cases (Bolouri et al., 2018; Jiang et al., 2022; Kerbs et al., 2022; Liu et al., 2022; Smith et al., 2021) in which the chromosomal rearrangement results in the 5’ HNRNPH1-derived regulatory sequences likely activating the expression of the ETS transcription factor ERG, which, deregulates gene expression to favor carcinogenesis. However, the study of other fusion proteins, such as EWSR1::FLI1, that include domains similar to those included within the HNRNPH1-fusions, suggest that the incorporation of protein regions, particularly those with low sequence complexity, could promote protein-protein interactions, inducing, for example, fusion protein multimerization, that enhances aspects of the aberrant function of these oncogenic proteins. (Boulay et al., 2017).
In addition to the rare but multiple reports of HNRNPH1::MTTL10 and HNRNPH1::ERG fusions, several studies have reported single cases of HNRNPF/H-fusion transcripts, including an HNRNPH3::ALK rearrangement identified in a single case of a salivary duct carcinoma (Dogan et al., 2019) and an HNRNPH3::TFE3 fusion detected in case of alveolar sort part sarcoma (Dickson et al., 2019). Also, as part of analyses of The Cancer Genome Atlas (TCGA) resource, several groups have used RNA-seq data to detect potential fusion transcripts and have noted cases involving HNRNPF/H genes (Gao et al., 2018; Hu et al., 2018; Yoshihara et al., 2015). HNRNPF/H fusion transcripts reported in TCGA datasets include HNRNPF::ATAD1 and HNRNPH3::ATAD1 fusions reported in breast cancer and lung adenocarcinoma, respectively. Other fusions include HNRNPF::ZNF33B and HNRNPF::ZNF569 fusions in ovarian adenocarcinoma; an HNRNPH1::RNF130 fusion in breast adenocarcinoma; an HNRNPH2::CLTRN fusion in a prostate adenocarcinoma; an HNRNPH3::KLK1 fusion in a kidney adenocarcinoma; an HNRNPH3::SLC29A3 in a stomach adenocarcinoma; and a GRSF1::ELFN2 fusion in adenocarcinoma of the large intestine.
Interestingly, one recent study reported a fusion in which HNRNPH1 sequences form the C-terminus of the derived protein, specifically a MEF2D::HNRNPH1 fusion identified in a case of pediatric B-cell precursor acute lymphoblastic leukemia (ALL) (Ohki et al., 2019) (Figure 5C). In the report by Ohki and co-workers, they describe two in-frame transcripts that fused exons 4 or 5 of MEF2D to exon 5 of HNRNPH1. Modeling of fusion protein predicts it will contain part of the HNRNPH1 qRRM3 domain and its GY region. Other MEF2D-fusions (e.g., MEF2D::BCL9) function as aberrant transcription factors, which led Kim and Kwon to assess if the qRRM3/GY region of HNRNPH1 can stimulate transcription (Kim & Kwon, 2021). Using a luciferase reporter system, Kim and Kwon demonstrated that the qRRM3/GY or the GY domain of HNRNPH1 can enhance transcriptional activity (Kim & Kwon, 2021); however, it remains to be determined how these domains contribute to the tumorigenic activity of the MEF2D::HNRNPH1 fusion protein.
4. THE HNRNPF/H RNA BINDING PROTEINS AND OTHER DISEASE STATES
4.1. Neurodevelopment
The functions of many RBPs are critical to ensuring the precise spatial and temporal regulation of gene expression required for normal neurodevelopment and neurological function. For example, the regulation of alternative splicing by specific RBPs during neuronal development can result in the expression of specific protein isoforms that exert different functional outcomes (Su et al., 2018). The HNRNPH-dependent splicing of TERF2 (or TRF2) (telomeric repeat-binding factor 2) transcripts illustrates such a result. TRF2 forms part of the telomeric shelterin complex that also functions as a regulator of neuronal differentiation via its interaction with REST (repressor element-1 silencing transcription factor). In brief, studies conducted using rat cells demonstrated that HNRNPH promotes the expression of the TRF2 full-length transcript and inhibits the expression of a TRF2 transcript variant (TRF2-S) encoding an isoform lacking TRF2’s DNA-binding domain and nls (Zhang et al., 2011). In the presence of HNRNPH, the nuclear expression of full-length TRF2 protein enhances the repression of neuronal genes by REST and the maintenance of a stem cell state (Grammatikakis et al., 2016). In contrast, lower levels of HNRNPH expression results in an accumulation of the TRF2-S isoform and sequestration of REST in the cytoplasm and the de-repression of neuronal genes silenced by REST and neuronal differentiation (Grammatikakis et al., 2016). Given these types of observations, it is unsurprising that deregulated RBP function is the underlying cause of several neurodevelopmental disorders (LaForce et al., 2022; Parra & Johnston, 2022; Prashad & Gopal, 2021), including Fragile X syndrome (FXS), which develops because of disrupted expression of the fragile X mental retardation protein (FMRP), a regulator of mRNA translation, RNA stability and subcellular localization (Protic et al., 2022). Added to this list in recent years are reports of neurodevelopmental disease-associated sequence changes in genes encoding hnRNPs, including the HNRNPF/H genes. Here, we discuss the genetic analyses that have identified multiple mutations in HNRNPF/H genes in individuals with clinical features characteristic of a neurodevelopmental disorder. Currently, there is limited research investigating the effect of these mutations on the organization of the HNRNPF/H proteins and their functions. However, by delineating these alterations in HNRNPF/H genes in this review, we aim to stimulate future research that will address these questions.
4.1.1. Neurodevelopmental disease-associated sequence changes in HNRNPH2
The first evidence of sequence changes in HNRNPH2 emerged through an assessment of six unrelated females (2 to 34 years) who presented with developmental delay and/or intellectual disability (Bain et al., 2016). The report by Bain and colleagues described de novo heterozygous sequence changes at nucleotides 616 (c.616C>T, four cases), 617 (c.617G>A, one case), and 626 (c.626C>T, one case) of HNRNPH2. The first of these nucleotide variants changes the HNRNPH2 amino acid 206 from arginine to tryptophan – p.R206W, and the second, the same amino acid to glutamine – p.(R206Q). The sixth case alters the HNRNPH2 amino acid 209 from proline to leucine – p.P209L. Since the publication of this study, multiple reports have documented additional cases of de novo pathogenic HNRNPH2 variants present in individuals with similar clinical phenotypes (Bain et al., 2021; Gillentine et al., 2021; Harmsen et al., 2019; Jepsen et al., 2019; Kreienkamp et al., 2022; Peron et al., 2020; Somashekar et al., 2020; White-Brown et al., 2021). Termed Bain-type X-linked Intellectual Development Disorder (MRXSB; OMIM #300986), clinical observations associated with heterozygous or hemizygous sequence variants in HNRNPH2 include developmental delay, intellectual disability, deficits in speech and motor function, musculoskeletal abnormalities, and an increased diagnosis of autism spectrum disorders (Bain et al., 2021; Madhok & Bain, 2022; Salazar et al., 2021).
The distribution of changes in the HNRNPH2 sequence is striking, with most altered nucleotides within the conserved region encoding the putative nls (Van Dusen et al., 2010) and the beginning of the GYR region between qRRM2 and qRRM3 (Figure 6A). Specifically, multiple reports have confirmed and extended the initial study by Bain and co-workers to show the presence of de novo heterozygous mutations in females and hemizygous mutations in males affecting nucleotides 616, 617, 626, 629, 634, 635, or 638 of HNRNPH2 (c.616C>T, p.R206W or c616C>G, p.R206G; c.617G>A, p.R206Q or c617G>T, p.R206L; c626C>T, p.P209L; c629A>G p.Y210C; c634A>G, p.R212G; c635G>C p.R212T; c638C>T p.P213L (Bain et al., 2021; Gillentine et al., 2021; Harmsen et al., 2019; Jepsen et al., 2019; Peron et al., 2020; Somashekar et al., 2020; White-Brown et al., 2021). Though rarer, reports of de novo mutations predicted to alter the HNRNPH2 qRRM domains include the following: qRRM1 – c85C>T, p.R29C, qRRM2 – c340C>T, p.R114W, and c422T>A, p.M141K, or qRRM3 c1019A>T, p.D340V (Bain et al., 2021; Jepsen et al., 2019; Kreienkamp et al., 2022). Interestingly, an individual harboring a c85C>T mutation (p.R29C), which maps to the region that encodes the first qRRM domain, exhibited milder symptoms (Kreienkamp et al., 2022) in comparison with those that map to a region encoding the qRRM2 domain (Bain et al., 2021; Jepsen et al., 2019; Kreienkamp et al., 2022; Somashekar et al., 2020). This observation suggests that the HNRNPH2 qRRM2 domain may contribute more significantly to the protein’s function in mediating splicing events needed for neurodevelopment, though this concept will require experimental confirmation.
Figure 6. Mutations in HNRNPF/H genes and neurodevelopment disorders.
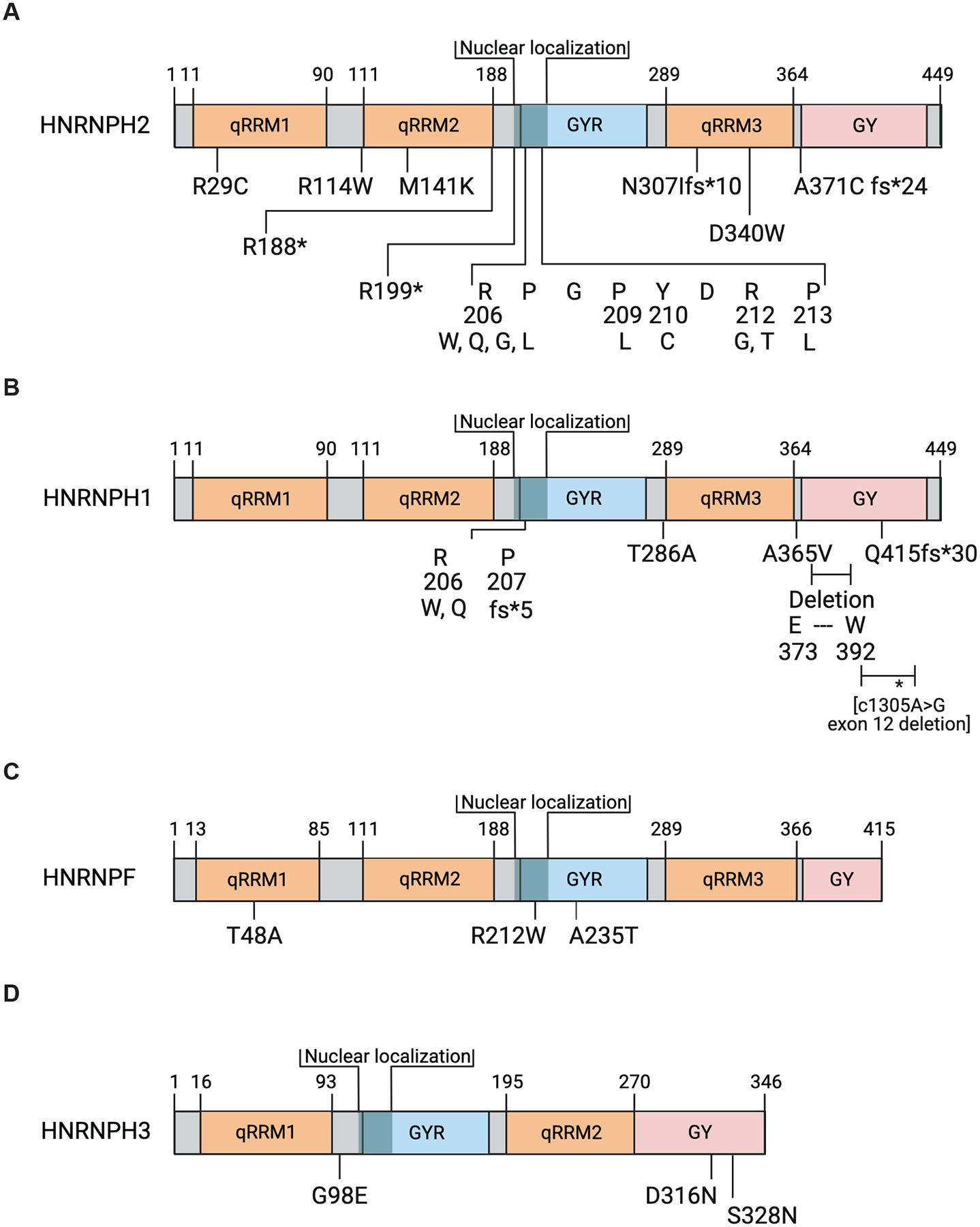
Schematics of (A) HNRNPH2, (B) HNRNPH1, (C) HNRNPF, and (D) HNRNPH3 proteins indicating the locations of amino acid changes predicted based on mutations identified in individuals diagnosed with a neurodevelopmental disorder and reported in the following studies: Bain et al., 2016; Bain et al., 2021; Epi et al., 2013; Gillentine et al., 2021; Harmsen et al., 2019; Jepsen et al., 2019; Kreienkamp et al., 2022; Peron et al., 2020; Pilch et al., 2018; Reichert et al., 2020; Somashekar et al., 2020; White-Brown et al., 2021. Figure panels created with BioRender.com.
A recent study by Kreienkamp and colleagues also described several male individuals harboring hemizygous sequence variants predicted to result in premature termination of translation. Specifically, monozygotic twins harboring a de novo c.562C>T, p.(R188*) variant; one case of a c.595C>T, p.R199* variant; and two sequence changes that result in frameshifts – c918_919dupTA, p.N307Ifs*10, and c1110dupT, p.A371Cfs*24 (Kreienkamp et al., 2022). In interpreting the effect of the predicted premature termination events, it is critical to consider HNRNPH2’s exonic organization, which consists of only two exons (Figure 3C). This organization suggests that the mutant transcripts are less likely to be the subject of nonsense-mediated mRNA decay (Kurosaki & Maquat, 2016). Instead, it is probable that the c918_919dupTA mutation results in a transcript that expresses a truncated protein lacking part of the qRRM3 domain and the whole of the GY C-terminus, while the c1110dupT mutation will result in a protein lacking most of the GY C-terminus. Interestingly, individuals harboring sequence changes predicted to express a truncated protein exhibited less severe developmental abnormalities, though several subsequently developed symptoms of a neuropsychiatric disorder (Kreienkamp et al., 2022). It is currently unclear if these milder symptoms are due to the truncated protein retaining its sequence-specific RNA binding and functions in RNA processing while disrupting other aspects of its functionality. For example, as discussed above (2.2), the inclusion of the HNRNPH1-GY domain in a reporter construct enhances gene expression (Kim & Kwon, 2021), which suggests that a truncated protein without this domain may lack this activity.
While most of the identified HNRNPH2 sequence changes are de novo mutations, it is also crucial to consider that the location of the HNRNPH2 locus on the X-chromosome means the potential for maternal inheritance of a disrupted allele. For example, the individual expressing the p.(N307Ifs*10) truncated proteins inherited this allele from a mosaic, unaffected mother (Kreienkamp et al., 2022). An earlier study also suggested that maternal germline mosaicism explains the inheritance of the c.616C>T p.R206W mutation by siblings (one male, one female) (Somashekar et al., 2020), though the authors did not confir this assertion experimentally. However, a study by White-Brown and colleagues showed inheritance of the same mutation c616C>T, p.R206W from an affected individual’s biological mother, though she exhibited no symptoms because of skewed X-chromosome inactivation (by a ratio of 1:99) of the disease-causing allele. (White-Brown et al., 2021).
4.1.2. Neurodevelopmental disease-associated sequence changes in HNRNPH1 and other family members
The identification of HNRNPH2 mutations prompted the investigation of other family members, and in 2018 Pilch et al. described the first HNRNPH1 sequence variant in a child who exhibited similarities in the clinical presentation of the first cases of Bain-type X-linked Intellectual Development Disorder. Specifically, Pilch and colleagues detected a de novo HNRNPH1 c.616C>T, p.R206W mutation in a 13-year-old boy diagnosed with significant intellectual disability, dysmorphic features, and other developmental abnormalities (Pilch et al., 2018). A subsequent study by Reichert and co-workers described an additional seven individuals (five male, two female) harboring HNRNPH1 mutations who exhibited similar clinical characteristics to the first case (Reichert et al., 2020). The HNRNPH1 sequence changes observed in these seven cases included single nucleotide alterations at residues that are part of the GYR region associated with nuclear localization (c.616C>T, p.R206W) – two cases and c.617G>A (p.R206Q) one case), and a duplication that is predicted to generate a frameshift (c.618dupG, p.P207fs*5) resulting in either expression of a truncated protein or reduced protein expression as a result of nonsense-mediated decay of the aberrant transcript. Another duplication (c1240_1243dup) also generates a frameshift (p.Q415fs*30); however, as the mutation affects a residue in the C-terminus of the protein (within the GY region), this may result in truncated protein expression. Of note, the individual harboring this mutation had a less severe phenotype than the individuals reported in this study with mutations that affect the GYR region. Interestingly, an individual with an in-frame deletion (c.1116–1175del152, p.E373-W392del) that removes 19 amino acids within the HNRNPH1 GY domain also had a less severe phenotype. The study by Reichert and colleagues also described a large duplication encompassing most of the HNRNPH1 locus and an adjacent gene, RUFY1 (Chr 5:178977572–179050134 GRCh37; Chr 5: 179550571–170623133 GRCh38). The functional effect of this duplication event is unknown; however, it is probable that disruption of HNRNPH1 function contributes to the clinical features of the individual harboring this mutation. A recent study also described two additional de novo HNRNPH1 mutations, c.856A>G (p.(T286A; C-terminus end of GYR region) and c.1094C>T (p.(A365V; GY region), in individuals with a neurodevelopmental disorder (Gillentine et al., 2021). The study by Gillentine and colleagues also highlighted an earlier cohort study of individuals with a severe pediatric epilepsy disorder (Epi et al., 2013) that noted a c.1305A>G synonymous mutation, which cell-based studies showed results in the expression of a transcript missing HNRNPH1-exon 12. This HNRNPH1-exon 12 exclusion event has the potential to express a variant HNRNPH1 transcript that expresses a protein lacking part of the GY domain, though to establish if this is the case, there is a need for further studies. See Figure 6B.
The study by Gillentine and co-workers also reported cases of mutations in HNRNPF (c.142A>G; c.634C>T; c.703G>A) in individuals with a neurodevelopmental disorder. The sequence changes in HNRNPF are located within the coding sequence for the first qRRM of the RBP (p.T48A), the second, the GYR region associated with nuclear localization (p.R212W)), and the third, the GYR region (p.A235T) (Figure 6C). Finally, the same study described alterations in the coding sequence of HNRNPH3 (c.293G>A, p.G98Q; c.946G>A, p.D316N; c.983G>A, p.s328N), which will alter a site between the first qRRM and the GYR region of HNRNPH3 and the other sites in its C-terminus region (Figure 6D).
4.1.3. Experimental analysis of HNRNPH2 mutants
As mentioned above, the interrogation of the functional effect of changes in the HNRNPH2 sequence is in its infancy, but one study has reported some initial findings (Kreienkamp et al., 2022). In the study by Kreienkamp and colleagues, the authors report the analysis of ectopically expressed wild-type HNRNPH2 protein and a panel of protein variants, HNRNPH2-R114W, HNRNPH2-R206Q, and HNRNPH2-P209L. First, they examined the cellular localization of these proteins, finding that the HNRNPH2 wild-type and the HNRNPH2-R114W protein that has an altered qRRM2 domain localized to the nucleoplasm only, while the HNRNPH2-R206Q and HNRNPH2-P209L proteins exhibited localization to both the nucleus and cytoplasm. These observations are consistent with mutations that cluster in a region around amino acids 206 – 212 of HNRNPH2, altering protein localization. Kreienkamp and colleagues next examined the interaction of the wild-type and mutant HNRNPH2 proteins with other RBPs with which it interacts. For example, the authors report reduced interaction of the HNRNPH2-R114W protein with DDX5 compared with the interaction of the wild-type protein and DDX5. This finding suggests that changes in the sequence of a qRRM domain could alter the protein-protein interactions needed to execute HNRNPH2-regulated splicing events. Critically, this study also examined the expression profiles of fibroblasts derived from an individual harboring a mutation that generates an HNRNPH2-R114W protein compared with cells derived from four unaffected males. Differential gene expression noted that the fibroblasts expressing the mutant protein exhibited deregulation of ~200 genes, while differential alternative splicing analysis indicated an alteration in the processing of RNA transcripts, particularly a substantial change in the number of skipped exon events. These studies suggest that defects in splicing are a critical outcome of de novo or inherited mutation of HNRNPF/H genes. Hopefully, future studies, including the generation of mouse models, will address how such splicing changes result in the clinical phenotypes observed in individuals harboring mutations in these genes.
4.2. HNRNPF/H genes and other neuro-related disease states
4.2.1. Neurodegenerative disorders
The genetic basis of a substantial proportion of the neurodegenerative disorders amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) is the expansion of a GGGGCC (G4C2) repeat located in intron 1 of C9ORF72 from an average of two repeats in healthy cells to several hundred or even over a thousand repeats in affected cells (Zampatti et al., 2022). The mechanisms by which expanded G4C2 repeats contribute to disease development remain unclear, but one focuses on the secondary conformations of transcripts containing expanded G4C2 repeats could form, including stable rG4s and the recruitment of RNA-binding proteins to these G-rich RNA molecules (reviewed in Malik et al., 2021). Collectively, the intramolecular RNA-RNA and intermolecular RNA-protein interactions associated with expanded G4C2 repeats appear to promote the appearance of nuclear-retained RNA foci. For example, fluorescent in situ hybridization (FISH) analysis showed, on average, the presence of six RNA foci per nucleus following the expression of transcripts containing 38 G4C2 repeats and twelve RNA foci per nucleus when the expressed transcripts contained 72 G4C2 repeats (Lee et al., 2013). Importantly, analysis of the RNA foci formed by transcripts expressed from constructs containing 38 or 72 G4C2 repeats demonstrated colocalization with HNRNPH, and subsequent RNA-immunoprecipitation studies confirmed HNRNPH’s binding of RNA containing the expanded G4C2 repeats (Lee et al., 2013). Furthermore, the analysis of an HNRNPH-dependent splicing event – inclusion of TARBP2-exon 7 – demonstrated slightly reduced retention of this exon in the presence of transcripts containing 72 G4C2 repeats (Lee et al., 2013). Critically, the analysis of ALS and FTD brain tissues also indicated the colocalization of G4C2-RNA foci and HNRNPH in the cerebellum (~70% of foci) (Lee et al., 2013). A complementary study of C9ORF72-G4C2-ALS-derived brain tissues confirmed the localization of HRNPF/H proteins (immunofluorescence) and C9ORF72-RNA foci (in situ hybridization) in cerebellar granule cells and motor neuron cells (Cooper-Knock et al., 2014).
The results reported by Lee and colleagues prompted them to speculate that expanded G4C2 repeats sequester HNRNPH and that disruption of HNRNPH function could contribute to the pathology of ALS and FTD. A subsequent study by Conlon et al. built upon this concept (Conlon et al., 2016). First, they confirmed HNRNPH’s interaction with RNAs containing C9ORF72-G4C2 repeats and that G4C2 repeats could form stable rG4 structures. To address the clinical relevance of their initial in vitro and cell line-based studies, Conlon and colleagues next examined the nuclear localization of HNRNPH proteins and rG4s in cells and tissue obtained from individuals with C9ORF72-G4C2-ALS compared with control samples. Among their reported findings, they detected many more HNRNPH-positive RNA foci in spinal cord sections from ALS patients than in non-ALS control samples. To assess the potential functional consequences of the observed altered localization of HNRNPH, Conlon et al. next compared the outcome of putative and established HNRNPH-dependent alternative splicing events using RNA purified from postmortem-cerebellum. PCR analysis demonstrated differences in the percent inclusion of specific exons when compared to control and C9ORF72-G4C2-ALS-derived samples, a finding they confirmed and extended through analysis of RNA-seq data. Critically, this study determined that the motor cortex of C9ORF72-G4C2-ALS-derived tissue contained more insoluble HNRNPH protein (1.9 x) than control samples. Supporting data suggested that this increase in insoluble HNRNPH is related to the presence of rG4s formed by the expanded G4C2 repeats. However, it is still unclear to what extent alterations in the function of HNRNPF/H proteins contribute to the pathology of C9ORF72-G4C2-ALS and FTD; a more recent study offers evidence that sequestration of HNRNPH proteins does have transcriptome-wide effects (Wang et al., 2020). Specifically, Wang et al. demonstrated that elevated levels of the HNRNPH proteins in the insoluble fraction of C9ORF72-G4C2-ALS-derived samples are associated with increased intron retention events compared to control samples (healthy individuals and ALS-derived samples with less HNRNPH protein in the insoluble fraction). The function of the genes encoding the transcripts that exhibited enhanced intron retention indicated enrichment for those that function in many pathways implicated in ALS, including those coding for proteasome subunits. This study also reported enrichment for the presence of at least one HNRNPH binding site close to the 5’ splice site of introns retained in C9ORF72-G4C2-ALS-derived samples (Wang et al., 2020).
4.2.2. Congenital myasthenic syndromes and other neuromuscular disorders
Congenital myasthenic syndromes (CMS) encompasses a heterogeneous group of disorders characterized by muscle weakness that arise because of mutations in genes encoding proteins involved in neuromuscular transmission, including subunits of the acetylcholine receptor (AChR) and COLQ, a protein that anchors acetylcholinesterase on the surface of muscle fibers (Finsterer, 2019). The CHRNA1 gene expresses a transcript variant ENST00000348749, consisting of nine exons, which encodes the full-length alpha subunit of AChR. Another variant, ENST00000261007, includes an additional 75 nt in-frame exon between CHRNA1-exons 3 and 4. Frequently referred to as CHRNA1-exon P3A, the inclusion of this exon results in a transcript that encodes a nonfunctional AChR protein that does not localize to the cell surface. Early studies (Beeson et al., 1990) and more recent RNA-seq analysis show that skeletal muscle expresses both variants (e.g., GTEx resources: ENST00000348749 – 28.8 TPM and ENST00000261007 – 12.1 TPM). In 2008, Masuda and colleagues demonstrated that an individual with severe myasthenic symptoms harbored two heterozygous CHRNA1 mutations, one of which they identified as a G >A mutation within the intron preceding CHRNA1-exon P3A (Masuda et al., 2008). Subsequent analysis demonstrated that this mutation disrupts an intron splicing silencer (ISS) that HNRNPH can bind. Furthermore, using minigenes to model the relevant region of CHRNA1, Masuda and co-workers demonstrated that depletion of HNRNPH results in an increased inclusion of the CHRNA1-exon P3A (Masuda et al., 2008). In their discussion, the authors speculate that the HNRNPH-dependent exclusion of CHRNA1-exon P3A contributes to the synapse-specific expression of AChR, though this hypothesis still needs evaluation. Mutations in COLQ, which result in acetylcholinesterase deficiency at neuromuscular junctions, are also associated with CMS. Interestingly, one COLQ mutation involves the HNRNPH1/HNRNPH2’s regulation of splicing. Specifically, a missense mutation in COLQ, (c.1244A>G (p.(E415G) (Kimbell et al., 2004), results in a COLQ-exon 16 exclusion event (Rahman et al., 2015) that likely generates a transcript encoding a truncated protein lacking its C-terminal domain. Further analysis of the consequence of the c.1244A>G mutation focused on its alteration of a G-rich motif from GGAGG to GGGGG and a shift from the binding of SRSF1, which favors exon inclusion, to the binding of HNRNPH, which in this context, promotes exon exclusion (Rahman et al., 2015).
4.2.3. Addictive behavior
Addictive behaviors and addiction develop because of the complex interaction of many genetic and environmental risk factors, including the changes in gene expression promoted by a particular stimulus. The isolation of regulators of gene expression that may contribute to addictive behaviors and addiction has proven challenging; however, an increasing number of reports have described the critical function of RNA binding proteins in regulating the expression of proteins that influence an addiction-related phenotype, including members of the HNRNPF/H protein family (Bryant & Yazdani, 2016). Insights into the potential function of HNRNPH1 in altering the response to addictive stimuli, such as methamphetamine, emerged from quantitative trait locus (QTL) mapping in mice (Bryant et al., 2009; Parker et al., 2012). Subsequent fine mapping of one mouse chromosome 11 QTL highlighted a genomic region containing the Hnrnph1 and Rufy1 genes (Yazdani et al., 2015). Employing TALEN-mediated gene editing to introduce frameshift deletions into each gene, Yazdani et al. next generated Hnrnph1+/− and Rufy1+/− mice lines, assessed the expression of the targeted gene, and the sensitivity of the mice to methamphetamine compared to wild-type mice. Molecular analysis of Hnrnph1+/− mice confirmed the disrupted expression of Hnrnph1, and compared to their wild-type litter mates, a reduced increase in locomotor activity following the administration of methamphetamine. Disruption of the Rufy1 locus generated no phenotypic differences.
In a follow-up study that further characterized the phenotype of Hnrnph1+/− mice, Ruan et al. demonstrated that the disrupted allele (a frameshift 16 bp deletion of the first coding exon of Hnrnph1) expresses an mRNA containing a premature stop codon (Ruan, Yazdani, Blum, et al., 2020). Using complementary assays of behavior, assessment of brain metabolism, and protein expression, Ruan and colleagues confirmed that Hnrnph1+/− mice exhibit reduced behavioral responses to methamphetamine and a decrease in the methamphetamine-induced release of dopamine in the nucleus accumbens region of the brain (Ruan, Yazdani, Blum, et al., 2020). Interestingly, the authors also linked alterations in the synaptic localization of the Hnrnph protein to a difference in mitochondrial function in the brain of Hnrnph1+/−mice following exposure to methamphetamine (Ruan, Yazdani, Blum, et al., 2020). Further analysis of the murine Hnrnph1 locus (specifically, its 5’ UTR) determined that alterations in the levels of Hnrnph protein expression are sufficient to result in quantifiable differences in the behavioral response of mice to methamphetamine (Ruan, Yazdani, Reed, et al., 2020). Of note, an assessment of the sensitivity of Hnrnph1+/−mice to other stimuli associated with addictive behavior showed that male mice consumed less alcohol than wild-type controls or female heterozygous mice (Fultz et al., 2021). Furthermore, Bryant et al. observed sex-independent and sex-specific effects when they examined the response of Hnrnph1+/− mice to the opioid fentanyl, nevertheless some effects, e.g., alterations in locomotor activity, were less pronounced than that observed following exposure to methamphetamine (Bryant et al., 2021). The mechanistic basis for these observations remains to be determined, but one clue may come from a study of the OPRM1 gene. OPRM1 codes for the mu-opioid receptor. Two studies have implicated the HNRNPF/H proteins in regulating the expression of OPRM1 transcripts (Song et al., 2012), including the sequence-specific binding of an intron splice enhancer sequence by HNRNPH1 that increases the likelihood of a specific exon exclusion event and differences in opioid dependency (Xu et al., 2014).
5. GRSF1, mitochondrial homeostasis, and cellular senescence
Cellular senescence, a state in which cells remain metabolically active but exhibit permanent cell cycle arrest, is a critical feature of aging, age-related disease, and other disease states, including cancer (Hernandez-Segura et al., 2018; McHugh & Gil, 2018; Ou et al., 2021). One feature of senescent cells is alterations in mitochondrial function and the accumulation of dysfunctional mitochondria (Ghosh-Choudhary et al., 2021). As discussed in sections 2.2 and 2.3, GRSF1 is the only HNRNPF/H protein that localizes to the cytoplasm, specifically mitochondria, where it is a component of the RNA granules that process precursor polycistronic mitochondrial RNAs and facilitates the loading of mature mRNA onto mitochondrial ribosomes. These observations have prompted several studies focused on exploring the function of GRSF1 in the maintenance of mitochondrial function and cellular senescence. As a starting point, Noh, Kim, and colleagues demonstrated that GRSF1 was one of only two RBPs that exhibited reduced expression in senescent fibroblasts and confirmed using a cell-based model of replicative senescence that GRSF1 protein levels decrease following the induction of senescence (Noh et al., 2018). A complementary report by Kim and colleagues confirmed the decrease in GRSF1 expression following the induction of senescence in a cell-based model and extended this observation to the study of liver, muscle, and heart tissues harvested from mice at 4, 18, and 32 months (Kim et al., 2019). PCR-based analysis of GRSF1 mRNA levels in murine muscle and liver showed substantial decreases at the later time-points, and analysis of GRSF1 protein levels in muscle and heart showed similar declines, though the results for liver proved variable. It is unclear which mechanism or mechanisms result in the senescence-associated decrease in GRSF1 expression. However, the study by Noh, Kim, and co-workers reported evidence that in senescent fibroblasts, GRSF1 protein exhibits reduced stability (Noh et al., 2018), while Kim and colleagues describe an increase in the methylation of the GRSF1 promoter in senescent cells compared to control cells (Kim et al., 2019).
Decreased expression of GRSF1, mediated using RNAi or CRISPR-Cas9 gene editing of HEK-293T cells, induces DNA damage, suppresses cell proliferation, and accelerates senescence following its induction (Noh et al., 2018; Noh et al., 2019). Associated with the acceleration of senescence was the enhanced secretion of interleukin 6 (IL6), a component of the senescence-associated secretory phenotype (SASP) (Noh et al., 2018; Noh et al., 2019). Analysis of the GRSF1-HEK-293T knockout cell lines also demonstrated reduced levels of mitochondrial protein complex components, increases in glycolysis, and changes in the redox balance of cells (Noh et al., 2019). Collectively, these results suggest that GRSF1 function is critical for maintaining mitochondrial activity. Furthermore, an examination of the mechanism of IL6 induction following GRSF1 loss revealed a signaling cascade that results in the activation of mTOR and NFκB and the presence of a feedback mechanism that promotes the secretion of pro-inflammatory factors and SASP (Noh et al., 2019). These findings have implications for GRSF1 as a regulator of inflammation and warrant further studies to determine if its decreased expression contributes to the elevated levels of IL6 observed in chronic inflammation and cancer. However, one part of such efforts will need to consider the in vivo consequences of altered GRSF1 expression. As a first step in assessing GRSF1’s function in a whole organism, Driscoll, Krasniewski, and colleagues generated tissue-specific knockout mice in which they ablated expression of Grsf1 in skeletal muscle (Grsf1cKO) (Driscoll et al., 2021). Interestingly, the Grsf1cKO mice exhibited no overt phenotype until 16 to 18 months of age, when their endurance in a treadmill exercise test reduced significantly. However, this observation was not associated with any histological changes in skeletal muscle or detection of oxidative stress. It is unclear at this time if differences between results obtained using cell lines and those observed in vivo reflect the presence of compensatory mechanisms or if other tissue types may prove more sensitive to loss of GRSF1. For example, investigation of hematopoiesis may prove informative as homozygous Grsf1 knockout mice (Grsf1tm1b(EUCOMM)Wts1) exhibit decreased leukocyte cell numbers compared to control animals.
6. Conclusion
The need to understand both the shared and distinguishing functions of members of the HNRNPF/H family of RBPs in the regulation of gene expression has increased with the recent identification of several protein-specific disease-related phenotypes. Critical to addressing this need is the generation of resources that will aid the selective detection or perturbation of each family member. For example, the homology of these proteins has limited the utility of antibodies to detect family members, particularly HNRNPH1 and HNRNPH2, selectively. However, at least in cell-based systems, the specific tagging of these proteins through the employment of gene editing strategies should enhance our ability to distinguish, for example, the transcripts they bind and the analysis of their functions. Accompanying such efforts is the need for additional whole organism models, particularly mouse models that assess the effect of the recurrent mutations identified in the disease states discussed in this review. Critical questions that the generation of novel resources should focus on addressing include an enhanced understanding of the effect of the mutations that cluster between the qRRM2 and the GYR regions of the HNRNPF/H proteins. Specifically, do these mutations alter the cellular localization of the HNRNPF/H, as some early studies may suggest, or could they also alter the RNA binding functions of these proteins or have other consequences? To inform our understanding of how this protein family contributes to transcript diversity, we also need further analysis of the molecular, biophysical, and structural characteristics of the interaction of each protein with G-tracts of varied sizes and sequence configurations (e.g., single G-tracts, interspersed G-tracts of diverse sizes), RNA conformations (e.g., G4 or non-G-states), and context (e.g., within exons or introns). Such efforts will be critical to further delineating how members of the HNRNPF/H protein family contribute to pathology and, in time, improve the diagnosis and treatment of diseases involving these proteins.
Acknowledgments
We apologize to colleagues whose work we could not cite due to space restrictions. We thank Tamara Jones, Daniel Zelmonovich, and Patricio Pichling, Genetics Branch, CCR, NCI for helpful discussion.
Funding Information
The Intramural Research Program of the National Cancer Institute (NCI), Center for Cancer Research (CCR supported this work – project numbers ZIA BC 011704 (NJC)
Footnotes
Conflict of Interest
The authors have declared no conflicts of interest for this article.
Contributor Information
Tayvia Brownmiller, Functional Genetics Section, Genetics Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, DHHS.
Natasha J. Caplen, Functional Genetics Section, Genetics Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, DHHS
Data Availability Statement
The Common Fund of the Office of the Director of the National Institutes of Health (NIH) and NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS support The Genotype-Tissue Expression (GTEx) Project. We downloaded the data used for the analyses described in this manuscript from the GTEx Portal June – July 2022.
References
- Agarwal R, Chan YC, Tam CS, Hunter T, Vassiliadis D, Teh CE, Thijssen R, Yeh P, Wong SQ, Ftouni S, Lam EYN, Anderson MA, Pott C, Gilan O, Bell CC, Knezevic K, Blombery P, Rayeroux K, Zordan A, Li J, Huang DCS, Wall M, Seymour JF, Gray DHD, Roberts AW, Dawson MA, & Dawson SJ (2019, Jan). Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med, 25(1), 119–129. 10.1038/s41591-018-0243-z [DOI] [PubMed] [Google Scholar]
- Alberti S, & Hyman AA (2021, Mar). Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat Rev Mol Cell Biol, 22(3), 196–213. 10.1038/s41580-020-00326-6 [DOI] [PubMed] [Google Scholar]
- Alkan SA, Martincic K, & Milcarek C (2006, Jan 1). The hnRNPs F and H2 bind to similar sequences to influence gene expression. Biochem J, 393(Pt 1), 361–371. 10.1042/BJ20050538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonicka H, Sasarman F, Nishimura T, Paupe V, & Shoubridge EA (2013, Mar 5). The mitochondrial RNA-binding protein GRSF1 localizes to RNA granules and is required for posttranscriptional mitochondrial gene expression. Cell Metab, 17(3), 386–398. 10.1016/j.cmet.2013.02.006 [DOI] [PubMed] [Google Scholar]
- Arhin GK, Boots M, Bagga PS, Milcarek C, & Wilusz J (2002, Apr 15). Downstream sequence elements with different affinities for the hnRNP H/H’ protein influence the processing efficiency of mammalian polyadenylation signals. Nucleic Acids Res, 30(8), 1842–1850. 10.1093/nar/30.8.1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage JO, & Longo DL (2022, Jun 30). Mantle-Cell Lymphoma. N Engl J Med, 386(26), 2495–2506. 10.1056/NEJMra2202672 [DOI] [PubMed] [Google Scholar]
- Bagga PS, Arhin GK, & Wilusz J (1998, Dec 1). DSEF-1 is a member of the hnRNP H family of RNA-binding proteins and stimulates pre-mRNA cleavage and polyadenylation in vitro. Nucleic Acids Res, 26(23), 5343–5350. 10.1093/nar/26.23.5343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagga PS, Ford LP, Chen F, & Wilusz J (1995, May 11). The G-rich auxiliary downstream element has distinct sequence and position requirements and mediates efficient 3’ end pre-mRNA processing through a trans-acting factor. Nucleic Acids Res, 23(9), 1625–1631. 10.1093/nar/23.9.1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain JM, Cho MT, Telegrafi A, Wilson A, Brooks S, Botti C, Gowans G, Autullo LA, Krishnamurthy V, Willing MC, Toler TL, Ben-Zev B, Elpeleg O, Shen Y, Retterer K, Monaghan KG, & Chung WK (2016, Sep 1). Variants in HNRNPH2 on the X Chromosome Are Associated with a Neurodevelopmental Disorder in Females. Am J Hum Genet, 99(3), 728–734. 10.1016/j.ajhg.2016.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain JM, Thornburg O, Pan C, Rome-Martin D, Boyle L, Fan X, Devinsky O, Frye R, Hamp S, Keator CG, LaMarca NM, Maddocks ABR, Madruga-Garrido M, Niederhoffer KY, Novara F, Peron A, Poole-Di Salvo E, Salazar R, Skinner SA, Soares G, Goldman S, & Chung WK (2021, Feb). Detailed Clinical and Psychological Phenotype of the X-linked HNRNPH2-Related Neurodevelopmental Disorder. Neurol Genet, 7(1), e551. 10.1212/NXG.0000000000000551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banga SS, Ozer HL, & Wilusz J (1996). Assignment of GRSF1 encoding a poly(A)+ mRNA binding protein to human chromosome 4q13. Cytogenet Cell Genet, 73(4), 295–296. 10.1159/000134359 [DOI] [PubMed] [Google Scholar]
- Beà S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, Gine E, Pinyol M, Royo C, Nadeu F, Conde L, Juan M, Clot G, Vizan P, Di Croce L, Puente DA, Lopez-Guerra M, Moros A, Roue G, Aymerich M, Villamor N, Colomo L, Martinez A, Valera A, Martin-Subero JI, Amador V, Hernandez L, Rozman M, Enjuanes A, Forcada P, Muntanola A, Hartmann EM, Calasanz MJ, Rosenwald A, Ott G, Hernandez-Rivas JM, Klapper W, Siebert R, Wiestner A, Wilson WH, Colomer D, Lopez-Guillermo A, Lopez-Otin C, Puente XS, & Campo E (2013, Nov 5). Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A, 110(45), 18250–18255. 10.1073/pnas.1314608110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeson D, Morris A, Vincent A, & Newsom-Davis J (1990, Jul). The human muscle nicotinic acetylcholine receptor alpha-subunit exist as two isoforms: a novel exon. EMBO J, 9(7), 2101–2106. 10.1002/j.1460-2075.1990.tb07378.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat P, Honson D, & Guttman M (2021, Oct). Nuclear compartmentalization as a mechanism of quantitative control of gene expression. Nat Rev Mol Cell Biol, 22(10), 653–670. 10.1038/s41580-021-00387-1 [DOI] [PubMed] [Google Scholar]
- Bodenhofer U, Bonatesta E, Horejs-Kainrath C, & Hochreiter S (2015, Dec 15). msa: an R package for multiple sequence alignment. Bioinformatics, 31(24), 3997–3999. 10.1093/bioinformatics/btv494 [DOI] [PubMed] [Google Scholar]
- Bolouri H, Farrar JE, Triche T Jr., Ries RE, Lim EL, Alonzo TA, Ma Y, Moore R, Mungall AJ, Marra MA, Zhang J, Ma X, Liu Y, Liu Y, Auvil JMG, Davidsen TM, Gesuwan P, Hermida LC, Salhia B, Capone S, Ramsingh G, Zwaan CM, Noort S, Piccolo SR, Kolb EA, Gamis AS, Smith MA, Gerhard DS, & Meshinchi S (2018, Jan). The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med, 24(1), 103–112. 10.1038/nm.4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Sandoval GJ, Riggi N, Iyer S, Buisson R, Naigles B, Awad ME, Rengarajan S, Volorio A, McBride MJ, Broye LC, Zou L, Stamenkovic I, Kadoch C, & Rivera MN (2017, Sep 21). Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell, 171(1), 163–178 e119. 10.1016/j.cell.2017.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, Cheng C, Dai Y, Devidas M, Qu C, Hill AN, Payne-Turner D, Ma X, Iacobucci I, Baviskar P, Wei L, Arunachalam S, Hagiwara K, Liu Y, Flasch DA, Liu Y, Parker M, Chen X, Elsayed AH, Pathak O, Li Y, Fan Y, Michael JR, Rusch M, Wilkinson MR, Foy S, Hedges DJ, Newman S, Zhou X, Wang J, Reilly C, Sioson E, Rice SV, Pastor Loyola V, Wu G, Rampersaud E, Reshmi SC, Gastier-Foster J, Guidry Auvil JM, Gesuwan P, Smith MA, Winick N, Carroll AJ, Heerema NA, Harvey RC, Willman CL, Larsen E, Raetz EA, Borowitz MJ, Wood BL, Carroll WL, Zweidler-McKay PA, Rabin KR, Mattano LA, Maloney KW, Winter SS, Burke MJ, Salzer W, Dunsmore KP, Angiolillo AL, Crews KR, Downing JR, Jeha S, Pui CH, Evans WE, Yang JJ, Relling MV, Gerhard DS, Loh ML, Hunger SP, Zhang J, & Mullighan CG (2022, Sep). The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet, 54(9), 1376–1389. 10.1038/s41588-022-01159-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandimarte L, Pierini V, Di Giacomo D, Borga C, Nozza F, Gorello P, Giordan M, Cazzaniga G, Te Kronnie G, La Starza R, & Mecucci C (2013, Jun 20). New MLLT10 gene recombinations in pediatric T-acute lymphoblastic leukemia. Blood, 121(25), 5064–5067. 10.1182/blood-2013-02-487256 [DOI] [PubMed] [Google Scholar]
- Braun S, Enculescu M, Setty ST, Cortes-Lopez M, de Almeida BP, Sutandy FXR, Schulz L, Busch A, Seiler M, Ebersberger S, Barbosa-Morais NL, Legewie S, Konig J, & Zarnack K (2018, Aug 17). Decoding a cancer-relevant splicing decision in the RON proto-oncogene using high-throughput mutagenesis. Nat Commun, 9(1), 3315. 10.1038/s41467-018-05748-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant CD, Chang HP, Zhang J, Wiltshire T, Tarantino LM, & Palmer AA (2009, Nov). A major QTL on chromosome 11 influences psychostimulant and opioid sensitivity in mice. Genes Brain Behav, 8(8), 795–805. 10.1111/j.1601-183X.2009.00525.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant CD, Healy AF, Ruan QT, Coehlo MA, Lustig E, Yazdani N, Luttik KP, Tran T, Swancy I, Brewin LW, Chen MM, & Szumlinski KK (2021, Mar). Sex-dependent effects of an Hnrnph1 mutation on fentanyl addiction-relevant behaviors but not antinociception in mice. Genes Brain Behav, 20(3), e12711. 10.1111/gbb.12711 [DOI] [PubMed] [Google Scholar]
- Bryant CD, & Yazdani N (2016, Jan). RNA-binding proteins, neural development and the addictions. Genes Brain Behav, 15(1), 169–186. 10.1111/gbb.12273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle M, De Conti L, Baralle D, Romano M, Ayala YM, & Baralle FE (2004). hnRNP H binding at the 5’ splice site correlates with the pathological effect of two intronic mutations in the NF-1 and TSHbeta genes. Nucleic Acids Res, 32(14), 4224–4236. 10.1093/nar/gkh752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputi M, & Zahler AM (2001, Nov 23). Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H’/F/2H9 family. J Biol Chem, 276(47), 43850–43859. 10.1074/jbc.M102861200 [DOI] [PubMed] [Google Scholar]
- Cazes A, Childers BG, Esparza E, & Lowy AM (2022, Apr 18). The MST1R/RON Tyrosine Kinase in Cancer: Oncogenic Functions and Therapeutic Strategies. Cancers (Basel), 14(8). 10.3390/cancers14082037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakedis J, French R, Babicky M, Jaquish D, Mose E, Cheng P, Holman P, Howard H, Miyamoto J, Porras P, Walterscheid Z, Schultz-Fademrecht C, Esdar C, Schadt O, Eickhoff J, & Lowy AM (2016, Jul 19). Characterization of RON protein isoforms in pancreatic cancer: implications for biology and therapeutics. Oncotarget, 7(29), 45959–45975. 10.18632/oncotarget.10009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Jiang L, Zhong ML, Li JF, Li BS, Peng LJ, Dai YT, Cui BW, Yan TQ, Zhang WN, Weng XQ, Xie YY, Lu J, Ren RB, Chen SN, Hu JD, Wu DP, Chen Z, Tang JY, Huang JY, Mi JQ, & Chen SJ (2018, Jan 9). Identification of fusion genes and characterization of transcriptome features in T-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A, 115(2), 373–378. 10.1073/pnas.1717125115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Kobayashi R, & Helfman DM (1999, Mar 01). Binding of hnRNP H to an exonic splicing silencer is involved in the regulation of alternative splicing of the rat beta-tropomyosin gene. Genes Dev, 13(5), 593–606. https://www.ncbi.nlm.nih.gov/pubmed/10072387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou MY, Rooke N, Turck CW, & Black DL (1999, Jan). hnRNP H is a component of a splicing enhancer complex that activates a c-src alternative exon in neuronal cells. Mol Cell Biol, 19(1), 69–77. 10.1128/MCB.19.1.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collesi C, Santoro MM, Gaudino G, & Comoglio PM (1996, Oct). A splicing variant of the RON transcript induces constitutive tyrosine kinase activity and an invasive phenotype. Mol Cell Biol, 16(10), 5518–5526. 10.1128/MCB.16.10.5518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA, & Manley JL (2016, Sep 13). The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife, 5. 10.7554/eLife.17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D, Ince PG, Wharton SB, Wilson SA, Kirby J, Hautbergue GM, & Shaw PJ (2014, Jul). Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain, 137(Pt 7), 2040–2051. 10.1093/brain/awu120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decorsiere A, Cayrel A, Vagner S, & Millevoi S (2011, Feb 1). Essential role for the interaction between hnRNP H/F and a G quadruplex in maintaining p53 pre-mRNA 3’-end processing and function during DNA damage. Genes Dev, 25(3), 220–225. 10.1101/gad.607011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BC, Chung CT, Hurlbut DJ, Marrano P, Shago M, Sung YS, Swanson D, Zhang L, & Antonescu CR (2019, Aug 21). Genetic diversity in alveolar soft part sarcoma: A subset contain variant fusion genes, highlighting broader molecular kinship with other MiT family tumors. Genes Chromosomes Cancer. 10.1002/gcc.22803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan S, Ng CKY, Xu B, Kumar R, Wang L, Edelweiss M, Scott SN, Zehir A, Drilon A, Morris LGT, Lee NY, Antonescu CR, Ho AL, Katabi N, Berger MF, & Reis-Filho JS (2019, Jun). The repertoire of genetic alterations in salivary duct carcinoma including a novel HNRNPH3-ALK rearrangement. Hum Pathol, 88, 66–77. 10.1016/j.humpath.2019.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C, & Allain FH (2006). NMR structure of the three quasi RNA recognition motifs (qRRMs) of human hnRNP F and interaction studies with Bcl-x G-tract RNA: a novel mode of RNA recognition. Nucleic Acids Res, 34(13), 3634–3645. 10.1093/nar/gkl488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C, Fisette JF, Chabot B, & Allain FH (2010, Jul). Structural basis of G-tract recognition and encaging by hnRNP F quasi-RRMs. Nat Struct Mol Biol, 17(7), 853–861. 10.1038/nsmb.1814 [DOI] [PubMed] [Google Scholar]
- Driscoll RK, Krasniewski LK, Cockey SG, Yang JH, Piao Y, Lehrmann E, Zhang Y, Michel M, Noh JH, Cui CY, & Gorospe M (2021, Jun 2). GRSF1 deficiency in skeletal muscle reduces endurance in aged mice. Aging (Albany NY), 13(11), 14557–14570. 10.18632/aging.203151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi KC, Epilepsy Phenome/Genome, P., Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu YF, Madou MR, Marson AG, Mefford HC, Esmaeeli Nieh S, O’Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin Thio L, Venkat A, Vining EP, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, & Winawer MR (2013, Sep 12). De novo mutations in epileptic encephalopathies. Nature, 501(7466), 217–221. 10.1038/nature12439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Zhang F, Xu S, Cui X, Hussain A, Fazli L, Gleave M, Dong X, & Qi J (2018, May 15). Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc Natl Acad Sci U S A, 115(20), E4584–E4593. 10.1073/pnas.1802415115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Li J, Wen H, Liu K, Gui Y, Wen Y, Wang X, & Yuan S (2022, Jun 23). hnRNPH1 recruits PTBP2 and SRSF3 to modulate alternative splicing in germ cells. Nat Commun, 13(1), 3588. 10.1038/s41467-022-31364-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J (2019, Feb 26). Congenital myasthenic syndromes. Orphanet J Rare Dis, 14(1), 57. 10.1186/s13023-019-1025-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisette JF, Montagna DR, Mihailescu MR, & Wolfe MS (2012, Jun). A G-rich element forms a G-quadruplex and regulates BACE1 mRNA alternative splicing. J Neurochem, 121(5), 763–773. 10.1111/j.1471-4159.2012.07680.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fultz EK, Coelho MA, Lieberman D, Jimenez-Chavez CL, Bryant CD, & Szumlinski KK (2021, Mar 1). Hnrnph1 is a novel regulator of alcohol reward. Drug Alcohol Depend, 220, 108518. 10.1016/j.drugalcdep.2021.108518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, Liao WW, Reynolds SM, Wyczalkowski MA, Yao L, Yu L, Sun SQ, Fusion Analysis Working G, Cancer Genome Atlas Research, N., Chen K, Lazar AJ, Fields RC, Wendl MC, Van Tine BA, Vij R, Chen F, Nykter M, Shmulevich I, & Ding L (2018, Apr 3). Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep, 23(1), 227–238 e223. 10.1016/j.celrep.2018.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau D, Revil T, Fisette JF, & Chabot B (2005, Jun 17). Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator Bcl-x. J Biol Chem, 280(24), 22641–22650. 10.1074/jbc.M501070200 [DOI] [PubMed] [Google Scholar]
- Garsetti DE, Sahay K, Wang Y, & Rogers MB (2022, Oct 4). Sex and the basal mRNA synthesis machinery. Wiley Interdiscip Rev RNA, e1765. 10.1002/wrna.1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, Green MR, Riva S, & Biamonti G (2005, Dec 22). Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell, 20(6), 881–890. 10.1016/j.molcel.2005.10.026 [DOI] [PubMed] [Google Scholar]
- Ghosh-Choudhary SK, Liu J, & Finkel T (2021, Dec). The role of mitochondria in cellular senescence. FASEB J, 35(12), e21991. 10.1096/fj.202101462R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillentine MA, Wang T, Hoekzema K, Rosenfeld J, Liu P, Guo H, Kim CN, De Vries BBA, Vissers L, Nordenskjold M, Kvarnung M, Lindstrand A, Nordgren A, Gecz J, Iascone M, Cereda A, Scatigno A, Maitz S, Zanni G, Bertini E, Zweier C, Schuhmann S, Wiesener A, Pepper M, Panjwani H, Torti E, Abid F, Anselm I, Srivastava S, Atwal P, Bacino CA, Bhat G, Cobian K, Bird LM, Friedman J, Wright MS, Callewaert B, Petit F, Mathieu S, Afenjar A, Christensen CK, White KM, Elpeleg O, Berger I, Espineli EJ, Fagerberg C, Brasch-Andersen C, Hansen LK, Feyma T, Hughes S, Thiffault I, Sullivan B, Yan S, Keller K, Keren B, Mignot C, Kooy F, Meuwissen M, Basinger A, Kukolich M, Philips M, Ortega L, Drummond-Borg M, Lauridsen M, Sorensen K, Lehman A, Study C, Lopez-Rangel E, Levy P, Lessel D, Lotze T, Madan-Khetarpal S, Sebastian J, Vento J, Vats D, Benman LM, McKee S, Mirzaa GM, Muss C, Pappas J, Peeters H, Romano C, Elia M, Galesi O, Simon MEH, van Gassen KLI, Simpson K, Stratton R, Syed S, Thevenon J, Palafoll IV, Vitobello A, Bournez M, Faivre L, Xia K, Consortium S, Earl RK, Nowakowski T, Bernier RA, & Eichler EE (2021, Apr 19). Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders. Genome Med, 13(1), 63. 10.1186/s13073-021-00870-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammatikakis I, Zhang P, Panda AC, Kim J, Maudsley S, Abdelmohsen K, Yang X, Martindale JL, Motino O, Hutchison ER, Mattson MP, & Gorospe M (2016, May 3). Alternative Splicing of Neuronal Differentiation Factor TRF2 Regulated by HNRNPH1/H2. Cell Rep, 15(5), 926–934. 10.1016/j.celrep.2016.03.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohar PJ, Kim S, Rangel Rivera GO, Sen N, Haddock S, Harlow ML, Maloney NK, Zhu J, O’Neill M, Jones TL, Huppi K, Grandin M, Gehlhaus K, Klumpp-Thomas CA, Buehler E, Helman LJ, Martin SE, & Caplen NJ (2016, Jan 26). Functional Genomic Screening Reveals Splicing of the EWS-FLI1 Fusion Transcript as a Vulnerability in Ewing Sarcoma. Cell Rep, 14(3), 598–610. 10.1016/j.celrep.2015.12.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, & Bartel DP (2016, Sep 23). RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science, 353(6306), aaf5371. 10.1126/science.aaf5371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen S, Buchert R, Mayatepek E, Haack TB, & Distelmaier F (2019, Jun). Bain type of X-linked syndromic mental retardation in boys. Clin Genet, 95(6), 734–735. 10.1111/cge.13524 [DOI] [PubMed] [Google Scholar]
- Hastings ML, Wilson CM, & Munroe SH (2001, Jun). A purine-rich intronic element enhances alternative splicing of thyroid hormone receptor mRNA. RNA, 7(6), 859–874. 10.1017/s1355838201002084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Segura A, Nehme J, & Demaria M (2018, Jun). Hallmarks of Cellular Senescence. Trends Cell Biol, 28(6), 436–453. 10.1016/j.tcb.2018.02.001 [DOI] [PubMed] [Google Scholar]
- Herviou P, Le Bras M, Dumas L, Hieblot C, Gilhodes J, Cioci G, Hugnot JP, Ameadan A, Guillonneau F, Dassi E, Cammas A, & Millevoi S (2020, May 27). hnRNP H/F drive RNA G-quadruplex-mediated translation linked to genomic instability and therapy resistance in glioblastoma. Nat Commun, 11(1), 2661. 10.1038/s41467-020-16168-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honoré B, Rasmussen HH, Vorum H, Dejgaard K, Liu X, Gromov P, Madsen P, Gesser B, Tommerup N, & Celis JE (1995, Dec 1). Heterogeneous nuclear ribonucleoproteins H, H’, and F are members of a ubiquitously expressed subfamily of related but distinct proteins encoded by genes mapping to different chromosomes. J Biol Chem, 270(48), 28780–28789. 10.1074/jbc.270.48.28780 [DOI] [PubMed] [Google Scholar]
- Hu X, Wang Q, Tang M, Barthel F, Amin S, Yoshihara K, Lang FM, Martinez-Ledesma E, Lee SH, Zheng S, & Verhaak RGW (2018, Jan 4). TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucleic Acids Res, 46(D1), D1144–D1149. 10.1093/nar/gkx1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, Donohue JP, Shiue L, Hoon S, Brenner S, Ares M Jr., & Yeo GW (2012, Feb 23). Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep, 1(2), 167–178. 10.1016/j.celrep.2012.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert JL, Bugaut A, Kumari S, & Balasubramanian S (2008, Nov). G-quadruplexes: the beginning and end of UTRs. Nucleic Acids Res, 36(19), 6260–6268. 10.1093/nar/gkn511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepsen WM, Ramsey K, Szelinger S, Llaci L, Balak C, Belnap N, Bilagody C, De Both M, Gupta R, Naymik M, Pandey R, Piras IS, Sanchez-Castillo M, Rangasamy S, Narayanan V, & Huentelman MJ (2019, Aug). Two additional males with X-linked, syndromic mental retardation carry de novo mutations in HNRNPH2. Clin Genet, 96(2), 183–185. 10.1111/cge.13580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Lang X, Chen N, Jin L, Liu L, Wei X, Pan J, Yu F, Blake A, & Xiao S (2022, Aug). A novel HNRNPH1::ERG rearrangement in aggressive acute myeloid leukemia. Genes Chromosomes Cancer, 61(8), 503–508. 10.1002/gcc.23051 [DOI] [PubMed] [Google Scholar]
- Jourdain AA, Koppen M, Wydro M, Rodley CD, Lightowlers RN, Chrzanowska-Lightowlers ZM, & Martinou JC (2013, Mar 5). GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab, 17(3), 399–410. 10.1016/j.cmet.2013.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakiuchi N, Yoshida K, Uchino M, Kihara T, Akaki K, Inoue Y, Kawada K, Nagayama S, Yokoyama A, Yamamoto S, Matsuura M, Horimatsu T, Hirano T, Goto N, Takeuchi Y, Ochi Y, Shiozawa Y, Kogure Y, Watatani Y, Fujii Y, Kim SK, Kon A, Kataoka K, Yoshizato T, Nakagawa MM, Yoda A, Nanya Y, Makishima H, Shiraishi Y, Chiba K, Tanaka H, Sanada M, Sugihara E, Sato TA, Maruyama T, Miyoshi H, Taketo MM, Oishi J, Inagaki R, Ueda Y, Okamoto S, Okajima H, Sakai Y, Sakurai T, Haga H, Hirota S, Ikeuchi H, Nakase H, Marusawa H, Chiba T, Takeuchi O, Miyano S, Seno H, & Ogawa S (2020, Jan). Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature, 577(7789), 260–265. 10.1038/s41586-019-1856-1 [DOI] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, & Burge CB (2010, Dec). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods, 7(12), 1009–1015. 10.1038/nmeth.1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbs P, Vosberg S, Krebs S, Graf A, Blum H, Swoboda A, Batcha AMN, Mansmann U, Metzler D, Heckman CA, Herold T, & Greif PA (2022, Jan 1). Fusion gene detection by RNA-sequencing complements diagnostics of acute myeloid leukemia and identifies recurring NRIP1-MIR99AHG rearrangements. Haematologica, 107(1), 100–111. 10.3324/haematol.2021.278436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodadoust MS, Olsson N, Wagar LE, Haabeth OA, Chen B, Swaminathan K, Rawson K, Liu CL, Steiner D, Lund P, Rao S, Zhang L, Marceau C, Stehr H, Newman AM, Czerwinski DK, Carlton VE, Moorhead M, Faham M, Kohrt HE, Carette J, Green MR, Davis MM, Levy R, Elias JE, & Alizadeh AA (2017, Mar 30). Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature, 543(7647), 723–727. 10.1038/nature21433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GH, & Kwon I (2021, Dec 14). Distinct roles of hnRNPH1 low-complexity domains in splicing and transcription. Proc Natl Acad Sci U S A, 118(50). 10.1073/pnas.2109668118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Chun M, Wan J, Lee C, Yen K, & Cohen P (2019, Apr 3). GRSF1 is an age-related regulator of senescence. Sci Rep, 9(1), 5546. 10.1038/s41598-019-42064-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbell LM, Ohno K, Engel AG, & Rotundo RL (2004, Mar 19). C-terminal and heparin-binding domains of collagenic tail subunit are both essential for anchoring acetylcholinesterase at the synapse. J Biol Chem, 279(12), 10997–11005. 10.1074/jbc.M305462200 [DOI] [PubMed] [Google Scholar]
- Královic̆ová J, & Vor̆echovský I (2006, Feb 15). Position-dependent repression and promotion of DQB1 intron 3 splicing by GGGG motifs. J Immunol, 176(4), 2381–2388. 10.4049/jimmunol.176.4.2381 [DOI] [PubMed] [Google Scholar]
- Kreienkamp HJ, Wagner M, Weigand H, McConkie-Rossell A, McDonald M, Keren B, Mignot C, Gauthier J, Soucy JF, Michaud JL, Dumas M, Smith R, Lobel U, Hempel M, Kubisch C, Denecke J, Campeau PM, Bain JM, & Lessel D (2022, Feb). Variant-specific effects define the phenotypic spectrum of HNRNPH2-associated neurodevelopmental disorders in males. Hum Genet, 141(2), 257–272. 10.1007/s00439-021-02412-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo K, Kubota Y, Toki T, Kanezaki R, Kobayashi A, Sato T, Kamio T, Sasaki S, Shiba N, Tomizawa D, Adachi S, Yoshida K, Ogawa S, Seki M, Takita J, Ito E, & Terui K (2022, May 24). Childhood acute myeloid leukemia with 5q deletion and HNRNPH1-MLLT10 fusion: the first case report. Blood Adv, 6(10), 3162–3166. 10.1182/bloodadvances.2021006383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T, & Maquat LE (2016, Feb 1). Nonsense-mediated mRNA decay in humans at a glance. J Cell Sci, 129(3), 461–467. 10.1242/jcs.181008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok CK, Marsico G, Sahakyan AB, Chambers VS, & Balasubramanian S (2016, Oct). rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat Methods, 13(10), 841–844. 10.1038/nmeth.3965 [DOI] [PubMed] [Google Scholar]
- LaForce GR, Philippidou P, & Schaffer AE (2022, Sep 19). mRNA isoform balance in neuronal development and disease. Wiley Interdiscip Rev RNA, e1762. 10.1002/wrna.1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bras M, Gorelick N, Pautet S, Tyler B, Manenti S, Skuli N, Millevoi S, & Cammas A (2022, Mar 2). Translational Regulation by hnRNP H/F Is Essential for the Proliferation and Survival of Glioblastoma. Cancers (Basel), 14(5). 10.3390/cancers14051283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BJ, Cansizoglu AE, Suel KE, Louis TH, Zhang Z, & Chook YM (2006, Aug 11). Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell, 126(3), 543–558. 10.1016/j.cell.2006.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, Adachi Y, Sardone V, Miller JW, Smith BN, Gallo JM, Ule J, Hirth F, Rogelj B, Houart C, & Shaw CE (2013, Dec 12). Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep, 5(5), 1178–1186. 10.1016/j.celrep.2013.10.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeFave CV, Squatrito M, Vorlova S, Rocco GL, Brennan CW, Holland EC, Pan YX, & Cartegni L (2011, Oct 5). Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. Embo Journal, 30(19), 4084–4097. 10.1038/emboj.2011.259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Rao J, Hu W, Cui B, Cai J, Liu Y, Sun H, Chen X, Tang Y, Chen J, Wang X, Wang H, Qian W, Mao B, Guo S, Wang R, Liu Y, & Shen S (2022, Mar 28). Distinct genomic landscape of Chinese pediatric acute myeloid leukemia impacts clinical risk classification. Nat Commun, 13(1), 1640. 10.1038/s41467-022-29336-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CS, Miyata KN, Zhao S, Ghosh A, Chang SY, Chenier I, Filep JG, Ingelfinger JR, Zhang SL, & Chan JSD (2019, Oct 31). Tubular Deficiency of Heterogeneous Nuclear Ribonucleoprotein F Elevates Systolic Blood Pressure and Induces Glycosuria in Mice. Sci Rep, 9(1), 15765. 10.1038/s41598-019-52323-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhok S, & Bain J (2022). HNRNPH2-Related Neurodevelopmental Disorder. In Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, & Amemiya A (Eds.), GeneReviews((R)). https://www.ncbi.nlm.nih.gov/pubmed/36108116 [PubMed] [Google Scholar]
- Mahé D, Mahl P, Gattoni R, Fischer N, Mattei MG, Stevenin J, & Fuchs JP (1997, Jan 17). Cloning of human 2H9 heterogeneous nuclear ribonucleoproteins. Relation with splicing and early heat shock-induced splicing arrest. J Biol Chem, 272(3), 1827–1836. 10.1074/jbc.272.3.1827 [DOI] [PubMed] [Google Scholar]
- Malik I, Kelley CP, Wang ET, & Todd PK (2021, Sep). Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat Rev Mol Cell Biol, 22(9), 589–607. 10.1038/s41580-021-00382-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci R, Baralle FE, & Romano M (2007). Complex splicing control of the human Thrombopoietin gene by intronic G runs. Nucleic Acids Res, 35(1), 132–142. 10.1093/nar/gkl965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda A, Shen XM, Ito M, Matsuura T, Engel AG, & Ohno K (2008, Dec 15). hnRNP H enhances skipping of a nonfunctional exon P3A in CHRNA1 and a mutation disrupting its binding causes congenital myasthenic syndrome. Hum Mol Genet, 17(24), 4022–4035. 10.1093/hmg/ddn305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matunis MJ, Xing J, & Dreyfuss G (1994, Mar 25). The hnRNP F protein: unique primary structure, nucleic acid-binding properties, and subcellular localization. Nucleic Acids Res, 22(6), 1059–1067. 10.1093/nar/22.6.1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauger DM, Lin C, & Garcia-Blanco MA (2008, Sep). hnRNP H and hnRNP F complex with Fox2 to silence fibroblast growth factor receptor 2 exon IIIc. Mol Cell Biol, 28(17), 5403–5419. 10.1128/MCB.00739-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer S, Hirschfeld M, Jaeger M, Pies S, Iborra S, Erbes T, & Stickeler E (2015, Jul). RON alternative splicing regulation in primary ovarian cancer. Oncol Rep, 34(1), 423–430. 10.3892/or.2015.3995 [DOI] [PubMed] [Google Scholar]
- McCullough AJ, & Berget SM (1997, Aug). G triplets located throughout a class of small vertebrate introns enforce intron borders and regulate splice site selection. Mol Cell Biol, 17(8), 4562–4571. 10.1128/MCB.17.8.4562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, & Gil J (2018, Jan 2). Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol, 217(1), 65–77. 10.1083/jcb.201708092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millevoi S, Decorsiere A, Loulergue C, Iacovoni J, Bernat S, Antoniou M, & Vagner S (2009, Aug). A physical and functional link between splicing factors promotes pre-mRNA 3’ end processing. Nucleic Acids Res, 37(14), 4672–4683. 10.1093/nar/gkp470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min H, Chan RC, & Black DL (1995, Nov 1). The generally expressed hnRNP F is involved in a neural-specific pre-mRNA splicing event. Genes Dev, 9(21), 2659–2671. 10.1101/gad.9.21.2659 [DOI] [PubMed] [Google Scholar]
- Nadeu F, Martin-Garcia D, Clot G, Diaz-Navarro A, Duran-Ferrer M, Navarro A, Vilarrasa-Blasi R, Kulis M, Royo R, Gutierrez-Abril J, Valdes-Mas R, Lopez C, Chapaprieta V, Puiggros M, Castellano G, Costa D, Aymerich M, Jares P, Espinet B, Muntanola A, Ribera-Cortada I, Siebert R, Colomer D, Torrents D, Gine E, Lopez-Guillermo A, Kuppers R, Martin-Subero I, Puente XS, Bea S, & Campo E (2020, Jun 25). Genomic and epigenomic insights into the origin, pathogenesis and clinical behavior of mantle cell lymphoma subtypes. Blood. 10.1182/blood.2020005289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Bea S, Jares P, & Campo E (2020, Oct). Molecular Pathogenesis of Mantle Cell Lymphoma. Hematol Oncol Clin North Am, 34(5), 795–807. 10.1016/j.hoc.2020.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazim M, Masuda A, Rahman MA, Nasrin F, Takeda JI, Ohe K, Ohkawara B, Ito M, & Ohno K (2017, Feb 17). Competitive regulation of alternative splicing and alternative polyadenylation by hnRNP H and CstF64 determines acetylcholinesterase isoforms. Nucleic Acids Res, 45(3), 1455–1468. 10.1093/nar/gkw823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckles C, Boer RE, Aboreden N, Cross AM, Walker RL, Kim BH, Kim S, Schneekloth JS Jr., & Caplen NJ (2019, Dec). HNRNPH1-dependent splicing of a fusion oncogene reveals a targetable RNA G-quadruplex interaction. RNA, 25(12), 1731–1750. 10.1261/rna.072454.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M, Sfaxi R, Saha A, Monchaud D, Teulade-Fichou MP, & Vagner S (2017, Oct 27). The G-Quadruplex-Specific RNA Helicase DHX36 Regulates p53 Pre-mRNA 3’-End Processing Following UV-Induced DNA Damage. J Mol Biol, 429(21), 3121–3131. 10.1016/j.jmb.2016.11.033 [DOI] [PubMed] [Google Scholar]
- Noh JH, Kim KM, Abdelmohsen K, Yoon JH, Panda AC, Munk R, Kim J, Curtis J, Moad CA, Wohler CM, Indig FE, de Paula W, Dudekula DB, De S, Piao Y, Yang X, Martindale JL, de Cabo R, & Gorospe M (2016, May 15). HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes Dev, 30(10), 1224–1239. 10.1101/gad.276022.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh JH, Kim KM, Idda ML, Martindale JL, Yang X, Abdelmohsen K, & Gorospe M (2018, Aug 7). GRSF1 suppresses cell senescence. Aging (Albany NY), 10(8), 1856–1866. 10.18632/aging.101516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh JH, Kim KM, Pandey PR, Noren Hooten N, Munk R, Kundu G, De S, Martindale JL, Yang X, Evans MK, Abdelmohsen K, & Gorospe M (2019, Mar 18). Loss of RNA-binding protein GRSF1 activates mTOR to elicit a proinflammatory transcriptional program. Nucleic Acids Res, 47(5), 2472–2486. 10.1093/nar/gkz082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohki K, Kiyokawa N, Saito Y, Hirabayashi S, Nakabayashi K, Ichikawa H, Momozawa Y, Okamura K, Yoshimi A, Ogata-Kawata H, Sakamoto H, Kato M, Fukushima K, Hasegawa D, Fukushima H, Imai M, Kajiwara R, Koike T, Komori I, Matsui A, Mori M, Moriwaki K, Noguchi Y, Park MJ, Ueda T, Yamamoto S, Matsuda K, Yoshida T, Matsumoto K, Hata K, Kubo M, Matsubara Y, Takahashi H, Fukushima T, Hayashi Y, Koh K, Manabe A, Ohara A, & Tokyo Children’s Cancer Study, G. (2019, Jan). Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica, 104(1), 128–137. 10.3324/haematol.2017.186320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou HL, Hoffmann R, Gonzalez-Lopez C, Doherty GJ, Korkola JE, & Munoz-Espin D (2021, Oct). Cellular senescence in cancer: from mechanisms to detection. Mol Oncol, 15(10), 2634–2671. 10.1002/1878-0261.12807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pararajalingam P, Coyle KM, Arthur SE, Thomas N, Alcaide M, Meissner B, Boyle M, Qureshi Q, Grande BM, Rushton C, Slack GW, Mungall AJ, Tam CS, Agarwal R, Dawson SJ, Lenz G, Balasubramanian S, Gascoyne RD, Steidl C, Connors J, Villa D, Audas TE, Marra MA, Johnson NA, Scott DW, & Morin RD (2020, Jul 30). Coding and noncoding drivers of mantle cell lymphoma identified through exome and genome sequencing. Blood, 136(5), 572–584. 10.1182/blood.2019002385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CC, Cheng R, Sokoloff G, & Palmer AA (2012, Feb). Genome-wide association for methamphetamine sensitivity in an advanced intercross mouse line. Genes Brain Behav, 11(1), 52–61. 10.1111/j.1601-183X.2011.00747.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra AS, & Johnston CA (2022, Jun 10). Emerging Roles of RNA-Binding Proteins in Neurodevelopment. J Dev Biol, 10(2). 10.3390/jdb10020023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penumutchu SR, Chiu LY, Meagher JL, Hansen AL, Stuckey JA, & Tolbert BS (2018, Sep 19). Differential Conformational Dynamics Encoded by the Linker between Quasi RNA Recognition Motifs of Heterogeneous Nuclear Ribonucleoprotein H. J Am Chem Soc, 140(37), 11661–11673. 10.1021/jacs.8b05366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peron A, Novara F, La Briola F, Merati E, Giannusa E, Segalini E, Anniballi G, Vignoli A, Ciccone R, & Canevini MP (2020, Apr). Missense variants in the Arg206 residue of HNRNPH2: Further evidence of causality and expansion of the phenotype. Am J Med Genet A, 182(4), 823–828. 10.1002/ajmg.a.61486 [DOI] [PubMed] [Google Scholar]
- Pietras Z, Wojcik MA, Borowski LS, Szewczyk M, Kulinski TM, Cysewski D, Stepien PP, Dziembowski A, & Szczesny RJ (2018, Jul 2). Dedicated surveillance mechanism controls G-quadruplex forming non-coding RNAs in human mitochondria. Nat Commun, 9(1), 2558. 10.1038/s41467-018-05007-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilch J, Koppolu AA, Walczak A, Murcia Pienkowski VA, Biernacka A, Skiba P, Machnik-Broncel J, Gasperowicz P, Kosinska J, Rydzanicz M, Emich-Widera E, & Ploski R (2018, Oct). Evidence for HNRNPH1 being another gene for Bain type syndromic mental retardation. Clin Genet, 94(3–4), 381–385. 10.1111/cge.13410 [DOI] [PubMed] [Google Scholar]
- Piñol-Roma S, Choi YD, Matunis MJ, & Dreyfuss G (1988, Feb). Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev, 2(2), 215–227. 10.1101/gad.2.2.215 [DOI] [PubMed] [Google Scholar]
- Prashad S, & Gopal PP (2021, Jul). RNA-binding proteins in neurological development and disease. RNA Biol, 18(7), 972–987. 10.1080/15476286.2020.1809186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protic DD, Aishworiya R, Salcedo-Arellano MJ, Tang SJ, Milisavljevic J, Mitrovic F, Hagerman RJ, & Budimirovic DB (2022, Feb 9). Fragile X Syndrome: From Molecular Aspect to Clinical Treatment. Int J Mol Sci, 23(4). 10.3390/ijms23041935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, & Wilusz J (1994, Jun 25). GRSF-1: a poly(A)+ mRNA binding protein which interacts with a conserved G-rich element. Nucleic Acids Res, 22(12), 2334–2343. 10.1093/nar/22.12.2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian ZW, & Wilusz J (1991, Oct). An RNA-binding protein specifically interacts with a functionally important domain of the downstream element of the simian virus 40 late polyadenylation signal. Mol Cell Biol, 11(10), 5312–5320. 10.1128/mcb.11.10.5312-5320.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MA, Azuma Y, Nasrin F, Takeda J, Nazim M, Bin Ahsan K, Masuda A, Engel AG, & Ohno K (2015, Aug 18). SRSF1 and hnRNP H antagonistically regulate splicing of COLQ exon 16 in a congenital myasthenic syndrome. Sci Rep, 5, 13208. 10.1038/srep13208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch J, Moran-Jones K, Albrecht V, Schwarzl T, Hunter K, Gires O, & Kolch W (2011, Jul 1). c-Myc regulates RNA splicing of the A-Raf kinase and its activation of the ERK pathway. Cancer Res, 71(13), 4664–4674. 10.1158/0008-5472.CAN-10-4447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch J, O’Neill E, Mack B, Matthias C, Munz M, Kolch W, & Gires O (2010, Feb 15). Heterogeneous nuclear ribonucleoprotein H blocks MST2-mediated apoptosis in cancer cells by regulating A-Raf transcription. Cancer Res, 70(4), 1679–1688. 10.1158/0008-5472.CAN-09-2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert SC, Li R, S AT, van Jaarsveld RH, Massink MPG, van den Boogaard MH, Del Toro M, Rodriguez-Palmero A, Fourcade S, Schluter A, Planas-Serra L, Pujol A, Iascone M, Maitz S, Loong L, Stewart H, De Franco E, Ellard S, Frank J, & Lewandowski R (2020, Jul). HNRNPH1-related syndromic intellectual disability: Seven additional cases suggestive of a distinct syndromic neurodevelopmental syndrome. Clin Genet, 98(1), 91–98. 10.1111/cge.13765 [DOI] [PubMed] [Google Scholar]
- Ruan QT, Yazdani N, Blum BC, Beierle JA, Lin W, Coelho MA, Fultz EK, Healy AF, Shahin JR, Kandola AK, Luttik KP, Zheng K, Smith NJ, Cheung J, Mortazavi F, Apicco DJ, Ragu Varman D, Ramamoorthy S, Ash PEA, Rosene DL, Emili A, Wolozin B, Szumlinski KK, & Bryant CD (2020, Jan 2). A Mutation in Hnrnph1 That Decreases Methamphetamine-Induced Reinforcement, Reward, and Dopamine Release and Increases Synaptosomal hnRNP H and Mitochondrial Proteins. J Neurosci, 40(1), 107–130. 10.1523/JNEUROSCI.1808-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan QT, Yazdani N, Reed ER, Beierle JA, Peterson LP, Luttik KP, Szumlinski KK, Johnson WE, Ash PEA, Wolozin B, & Bryant CD (2020, May 13). 5’ UTR variants in the quantitative trait gene Hnrnph1 support reduced 5’ UTR usage and hnRNP H protein as a molecular mechanism underlying reduced methamphetamine sensitivity. FASEB J. 10.1096/fj.202000092R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rule S, Jurczak W, Jerkeman M, Rusconi C, Trneny M, Offner F, Caballero D, Joao C, Witzens-Harig M, Hess G, Bence-Bruckler I, Cho SG, Thieblemont C, Zhou W, Henninger T, Goldberg J, Vermeulen J, & Dreyling M (2018, Aug). Ibrutinib versus temsirolimus: 3-year follow-up of patients with previously treated mantle cell lymphoma from the phase 3, international, randomized, open-label RAY study. Leukemia, 32(8), 1799–1803. 10.1038/s41375-018-0023-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar R, Beenders S, LaMarca NM, Thornburg O, Rubin-Thompson L, Snow A, Goldman S, Chung WK, & Bain JM (2021, Dec). Cross-sectional, quantitative analysis of motor function in females with HNRNPH2-related disorder. Res Dev Disabil, 119, 104110. 10.1016/j.ridd.2021.104110 [DOI] [PubMed] [Google Scholar]
- Samatanga B, Dominguez C, Jelesarov I, & Allain FH (2013, Feb 1). The high kinetic stability of a G-quadruplex limits hnRNP F qRRM3 binding to G-tract RNA. Nucleic Acids Res, 41(4), 2505–2516. 10.1093/nar/gks1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, Liu X, Powell J, Yang Y, Xu W, Zhao H, Kohlhammer H, Rosenwald A, Kluin P, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Ogwang MD, Reynolds SJ, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Pittaluga S, Wilson W, Waldmann TA, Rowe M, Mbulaiteye SM, Rickinson AB, & Staudt LM (2012, Oct 4). Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature, 490(7418), 116–120. 10.1038/nature11378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan M, Ji X, Janssen K, Silverman IM, Humenik J, Garcia BA, Liebhaber SA, & Gregory BD (2021, Sep). Dynamic changes in RNA-protein interactions and RNA secondary structure in mammalian erythropoiesis. Life Sci Alliance, 4(9). 10.26508/lsa.202000659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PA, Chakraborty AK, Henninger JE, & Young RA (2022, Jan). RNA in formation and regulation of transcriptional condensates. RNA, 28(1), 52–57. 10.1261/rna.078997.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkenstedt E, Linton K, & Dreyling M (2021, Oct). Mantle cell lymphoma - advances in molecular biology, prognostication and treatment approaches. Br J Haematol, 195(2), 162–173. 10.1111/bjh.17419 [DOI] [PubMed] [Google Scholar]
- Siomi MC, Eder PS, Kataoka N, Wan L, Liu Q, & Dreyfuss G (1997, Sep 22). Transportin-mediated nuclear import of heterogeneous nuclear RNP proteins. J Cell Biol, 138(6), 1181–1192. 10.1083/jcb.138.6.1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JL, Ries RE, Wang Y-C, Leonti AR, Alonzo TA, Gamis AS, Aplenc R, Kolb AE, Huang BJ, Ma X, Shaw TI, & Meshinchi S (2021, 2021/11/23/). ETS Family Transcription Factor Fusions in Childhood AML: Distinct Expression Networks and Clinical Implications. Blood, 138, 2356. 10.1182/blood-2021-148894 [DOI] [Google Scholar]
- Sofi S, Stehling S, Niewienda A, Janek K, Kuhn H, & Ufer C (2018, Apr). Functional characterization of isolated RNA-binding domains of the GRSF1 protein. Biochim Biophys Acta Gen Subj, 1862(4), 946–957. 10.1016/j.bbagen.2017.12.009 [DOI] [PubMed] [Google Scholar]
- Somashekar PH, Narayanan DL, Jagadeesh S, Suresh B, Vaishnavi RD, Bielas S, Girisha KM, & Shukla A (2020, Jan). Bain type of X-linked syndromic mental retardation in a male with a pathogenic variant in HNRNPH2. Am J Med Genet A, 182(1), 183–188. 10.1002/ajmg.a.61388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song KY, Choi HS, Law PY, Wei LN, & Loh HH (2012, Feb). Post-transcriptional regulation of mu-opioid receptor: role of the RNA-binding proteins heterogeneous nuclear ribonucleoprotein H1 and F. Cell Mol Life Sci, 69(4), 599–610. 10.1007/s00018-011-0761-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CH, D D, & Tarn WY (2018). Alternative Splicing in Neurogenesis and Brain Development. Front Mol Biosci, 5, 12. 10.3389/fmolb.2018.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, & Dreyfuss G (1988, May). Classification and purification of proteins of heterogeneous nuclear ribonucleoprotein particles by RNA-binding specificities. Mol Cell Biol, 8(5), 2237–2241. 10.1128/mcb.8.5.2237-2241.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ufer C, Wang CC, Fahling M, Schiebel H, Thiele BJ, Billett EE, Kuhn H, & Borchert A (2008, Jul 1). Translational regulation of glutathione peroxidase 4 expression through guanine-rich sequence-binding factor 1 is essential for embryonic brain development. Genes Dev, 22(13), 1838–1850. 10.1101/gad.466308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren PJ, Bahrami-Samani E, de Araujo PR, Vogel C, Qiao M, Burns SC, Smith AD, & Penalva LO (2016). High-throughput analyses of hnRNP H1 dissects its multi-functional aspect. RNA Biol, 13(4), 400–411. 10.1080/15476286.2015.1138030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dusen CM, Yee L, McNally LM, & McNally MT (2010, May). A glycine-rich domain of hnRNP H/F promotes nucleocytoplasmic shuttling and nuclear import through an interaction with transportin 1. Mol Cell Biol, 30(10), 2552–2562. 10.1128/MCB.00230-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney D, Spiegel J, Zyner K, Tannahill D, & Balasubramanian S (2020, Aug). The regulation and functions of DNA and RNA G-quadruplexes. Nat Rev Mol Cell Biol, 21(8), 459–474. 10.1038/s41580-020-0236-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP, Koh CS, Froehlich U, Lapointe E, Couture S, Inkel L, Bramard A, Paquet ER, Watier V, Durand M, Lucier JF, Gervais-Bird J, Tremblay K, Prinos P, Klinck R, Elela SA, & Chabot B (2008, Oct). Multiple and specific mRNA processing targets for the major human hnRNP proteins. Mol Cell Biol, 28(19), 6033–6043. 10.1128/MCB.00726-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veraldi KL, Arhin GK, Martincic K, Chung-Ganster LH, Wilusz J, & Milcarek C (2001, Feb). hnRNP F influences binding of a 64-kilodalton subunit of cleavage stimulation factor to mRNA precursors in mouse B cells. Mol Cell Biol, 21(4), 1228–1238. 10.1128/MCB.21.4.1228-1238.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo T, Brownmiller T, Hall K, Jones TL, Choudhari S, Grammatikakis I, Ludwig KR, & Caplen NJ (2022, Jun 24). HNRNPH1 destabilizes the G-quadruplex structures formed by G-rich RNA sequences that regulate the alternative splicing of an oncogenic fusion transcript. Nucleic Acids Res, 50(11), 6474–6496. 10.1093/nar/gkac409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Aslanzadeh V, Papa F, Zhu H, de la Grange P, & Cambi F (2012). Global profiling of alternative splicing events and gene expression regulated by hnRNPH/F. PLoS One, 7(12), e51266. 10.1371/journal.pone.0051266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Conlon EG, Manley JL, & Rio DC (2020, Dec). Widespread intron retention impairs protein homeostasis in C9orf72 ALS brains. Genome Res, 30(12), 1705–1715. 10.1101/gr.265298.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, & Burge CB (2004, Dec 17). Systematic identification and analysis of exonic splicing silencers. Cell, 119(6), 831–845. 10.1016/j.cell.2004.11.010 [DOI] [PubMed] [Google Scholar]
- White-Brown AM, Lemire G, Ito YA, Thornburg O, Bain JM, & Dyment DA (2021, Oct 31). A disease-causing variant in HNRNPH2 inherited from an unaffected mother with skewed X-inactivation. Am J Med Genet A. 10.1002/ajmg.a.62549 [DOI] [PubMed] [Google Scholar]
- Wu C, de Miranda NF, Chen L, Wasik AM, Mansouri L, Jurczak W, Galazka K, Dlugosz-Danecka M, Machaczka M, Zhang H, Peng R, Morin RD, Rosenquist R, Sander B, & Pan-Hammarstrom Q (2016, Jun 21). Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget, 7(25), 38180–38190. 10.18632/oncotarget.9500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Wang Z, Jang M, & Burge CB (2007, Nov 20). Coevolutionary networks of splicing cis-regulatory elements. Proc Natl Acad Sci U S A, 104(47), 18583–18588. 10.1073/pnas.0707349104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Wang Z, Jang M, Nutiu R, Wang ET, & Burge CB (2009, Oct). Splice site strength-dependent activity and genetic buffering by poly-G runs. Nat Struct Mol Biol, 16(10), 1094–1100. 10.1038/nsmb.1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lu Z, Xu M, Pan L, Deng Y, Xie X, Liu H, Ding S, Hurd YL, Pasternak GW, Klein RJ, Cartegni L, Zhou W, & Pan YX (2014, Aug 13). A heroin addiction severity-associated intronic single nucleotide polymorphism modulates alternative pre-mRNA splicing of the mu opioid receptor gene OPRM1 via hnRNPH interactions. J Neurosci, 34(33), 11048–11066. 10.1523/JNEUROSCI.3986-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Liu L, Conlon EG, & Manley JL (2020, Oct). Burkitt lymphoma-related TCF3 mutations alter TCF3 alternative splicing by disrupting hnRNPH1 binding. RNA Biol, 17(10), 1383–1390. 10.1080/15476286.2020.1772559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Liu L, Lazarev D, Al-Zain A, Fomin V, Yeung PL, Chambers SM, Lu CW, Studer L, & Manley JL (2018, Sep 1). TCF3 alternative splicing controlled by hnRNP H/F regulates E-cadherin expression and hESC pluripotency. Genes Dev, 32(17–18), 1161–1174. 10.1101/gad.316984.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Liu L, & Manley JL (2019, Nov). TCF3 mutually exclusive alternative splicing is controlled by long-range cooperative actions between hnRNPH1 and PTBP1. RNA, 25(11), 1497–1508. 10.1261/rna.072298.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Jia D, Kim H, Abd Elmageed ZY, Datta A, Davis R, Srivastav S, Moroz K, Crawford BE, Moparty K, Thomas R, Hudson RS, Ambs S, & Abdel-Mageed AB (2016, Apr 1). Dysregulation of miR-212 Promotes Castration Resistance through hnRNPH1-Mediated Regulation of AR and AR-V7: Implications for Racial Disparity of Prostate Cancer. Clin Cancer Res, 22(7), 1744–1756. 10.1158/1078-0432.CCR-15-1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani N, Parker CC, Shen Y, Reed ER, Guido MA, Kole LA, Kirkpatrick SL, Lim JE, Sokoloff G, Cheng R, Johnson WE, Palmer AA, & Bryant CD (2015, Dec). Hnrnph1 Is A Quantitative Trait Gene for Methamphetamine Sensitivity. PLoS Genet, 11(12), e1005713. 10.1371/journal.pgen.1005713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Hoon S, Venkatesh B, & Burge CB (2004, Nov 2). Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc Natl Acad Sci U S A, 101(44), 15700–15705. 10.1073/pnas.0404901101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K, Wang Q, Torres-Garcia W, Zheng S, Vegesna R, Kim H, & Verhaak RG (2015, Sep 10). The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene, 34(37), 4845–4854. 10.1038/onc.2014.406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampatti S, Peconi C, Campopiano R, Gambardella S, Caltagirone C, & Giardina E (2022). C9orf72-Related Neurodegenerative Diseases: From Clinical Diagnosis to Therapeutic Strategies. Front Aging Neurosci, 14, 907122. 10.3389/fnagi.2022.907122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarudnaya MI, Kolomiets IM, Potyahaylo AL, & Hovorun DM (2003, Mar 1). Downstream elements of mammalian pre-mRNA polyadenylation signals: primary, secondary and higher-order structures. Nucleic Acids Res, 31(5), 1375–1386. 10.1093/nar/gkg241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Casaday-Potts R, Precht P, Jiang H, Liu Y, Pazin MJ, & Mattson MP (2011, Sep 27). Nontelomeric splice variant of telomere repeat-binding factor 2 maintains neuronal traits by sequestering repressor element 1-silencing transcription factor. Proc Natl Acad Sci U S A, 108(39), 16434–16439. 10.1073/pnas.1106906108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Feng T, Xu S, Gao F, Lam TT, Wang Q, Wu T, Huang H, Zhan L, Li L, Guan Y, Dai Z, & Yu G (2022, Jul 18). ggmsa: a visual exploration tool for multiple sequence alignment and associated data. Brief Bioinform, 23(4). 10.1093/bib/bbac222 [DOI] [PubMed] [Google Scholar]
- Zhou YQ, He C, Chen YQ, Wang D, & Wang MH (2003, Jan 16). Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: generation of different splicing RON variants and their oncogenic potential. Oncogene, 22(2), 186–197. 10.1038/sj.onc.1206075 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The Common Fund of the Office of the Director of the National Institutes of Health (NIH) and NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS support The Genotype-Tissue Expression (GTEx) Project. We downloaded the data used for the analyses described in this manuscript from the GTEx Portal June – July 2022.