

Abstract
Complex diseases are caused by a combination of genetic, lifestyle and environmental factors and comprise common non-communicable diseases, including allergies, cardiovascular disease, and psychiatric and metabolic disorders. More than 25% of Europeans suffer from a complex disease, and together these diseases account for 70% of all deaths. The use of genomic, molecular or imaging data to develop accurate diagnostic tools for treatment recommendations and preventive strategies, and for disease prognosis and prediction, is an important step towards precision medicine. However, for complex diseases, precision medicine is associated with several challenges. There is a significant heterogeneity between patients of a specific disease—both with regards to symptoms and underlying causal mechanisms—and the number of underlying genetic and non-genetic risk factors is often high. Here, we summarize precision medicine approaches for complex diseases and highlight the current breakthroughs as well as the challenges. We conclude that genomic-based precision medicine has been utilized mainly for patients with highly penetrant monogenic disease forms, such as cardiomyopathies. However, for most complex diseases—including psychiatric disorders and allergies—available polygenic risk scores are more probabilistic than deterministic and have not yet been validated for clinical utility. However, subclassifying patients of a specific disease into discrete homogenous subtypes based on molecular or phenotypic data is a promising strategy for improving diagnosis, prediction, treatment, prevention and prognosis. The availability of high-throughput molecular technologies, together with large collections of health data and novel data-driven approaches, offers promise towards improved individual health through precision medicine.
Keywords: precision medicine, complex diseases, genomic medicine, GWAS, polygenic risk score (PRS), genetic variations, molecular profiling, multi omics
Graphical Abstract
Precision medicine and health
In contemporary evidence-based medicine, people are frequently misdiagnosed and prescribed treatments fail, either because the wrong treatment is prescribed or because the patient does not respond adequately to the treatment. In many diseases, patients can also receive too much or too little treatment. There are many definitions of precision medicine and related terms—e.g. genomic medicine, personalized medicine, stratified medicine, and individualized medicine—some of which are complimentary. It is generally accepted that “precision” means a relative lack of random error. Thus, a pragmatic definition of “precision medicine” and “precision health” is to reduce error in medical decisions and health recommendations, respectively. The growing amount of genomic, molecular, and imaging data; electronic health records; and large-scale longitudinal cohorts—in combination with the development of novel analytical tools—suggests that precision medicine will readily complement contemporary evidence-based medicine in the not-so-distant future [1], and there has already been a rapid progression in several disease areas, including complex diseases.
Genetics of complex disease
Precision medicine approaches are already used in the clinic to diagnose rare conditions and to characterize cancers, aiming to provide the optimal treatment to each patient [2]. These approaches are mainly based on genomics, and the diagnostic tools have benefited from the rapid development of high-throughput sequencing technologies. Rare conditions are often caused by loss-of-function mutations or larger chromosomal rearrangements, and the identification of genetic causes for rare conditions has increased almost linearly during the last 30 years [3]. However, by definition rare genetic disorders only affect a minority of the population, and the advancements in diagnostics of rare conditions have no benefit for the majority of the population.
Complex diseases account for approximately 70% of global deaths, rendering them the main cost for the healthcare system [4] and a major burden to the society. For complex diseases, the heritability—i.e. the variation in disease risk that could be attributed to genetic factors—is often 40%−60%, and few cases are caused by mutations in single genes—so-called monogenic disease forms. Almost two decades ago, when the first genome-wide association studies (GWAS) were performed, the identification of genetic variants influencing the risk of common diseases began [5]. Since then, thousands of genetic loci associated with complex disease have been identified [6]. For example, the largest GWAS to date—with a sample size of 5.4 million individuals [7]—showed that 12,111 independent SNPs are associated with height, one of the most heritable human traits. For most complex diseases and traits, the genetic variants identified in GWAS explain a limited fraction of the heritability. For example, for systolic blood pressure (SBP) the 901 most significantly associated loci combined explain 5.7% of the variation in SBP [8]. However, all SNPs combined—even variants that are not genome-wide significant—explain as much as 21.3% of the variation in SBP [8]. During the last years, the use of polygenic risk scores (PRS) has therefore been widely discussed in the complex disease field. A PRS is an estimate that reflects an individual’s genetic liability to a complex disease or trait and often represents a combined effect of hundreds of thousands of genetic variants, of which a majority do not reach the threshold for significance in a GWAS (Figure 1). The PRS are constructed based on linear combinations of effect estimates for SNPs from a GWAS [9].
Figure 1.
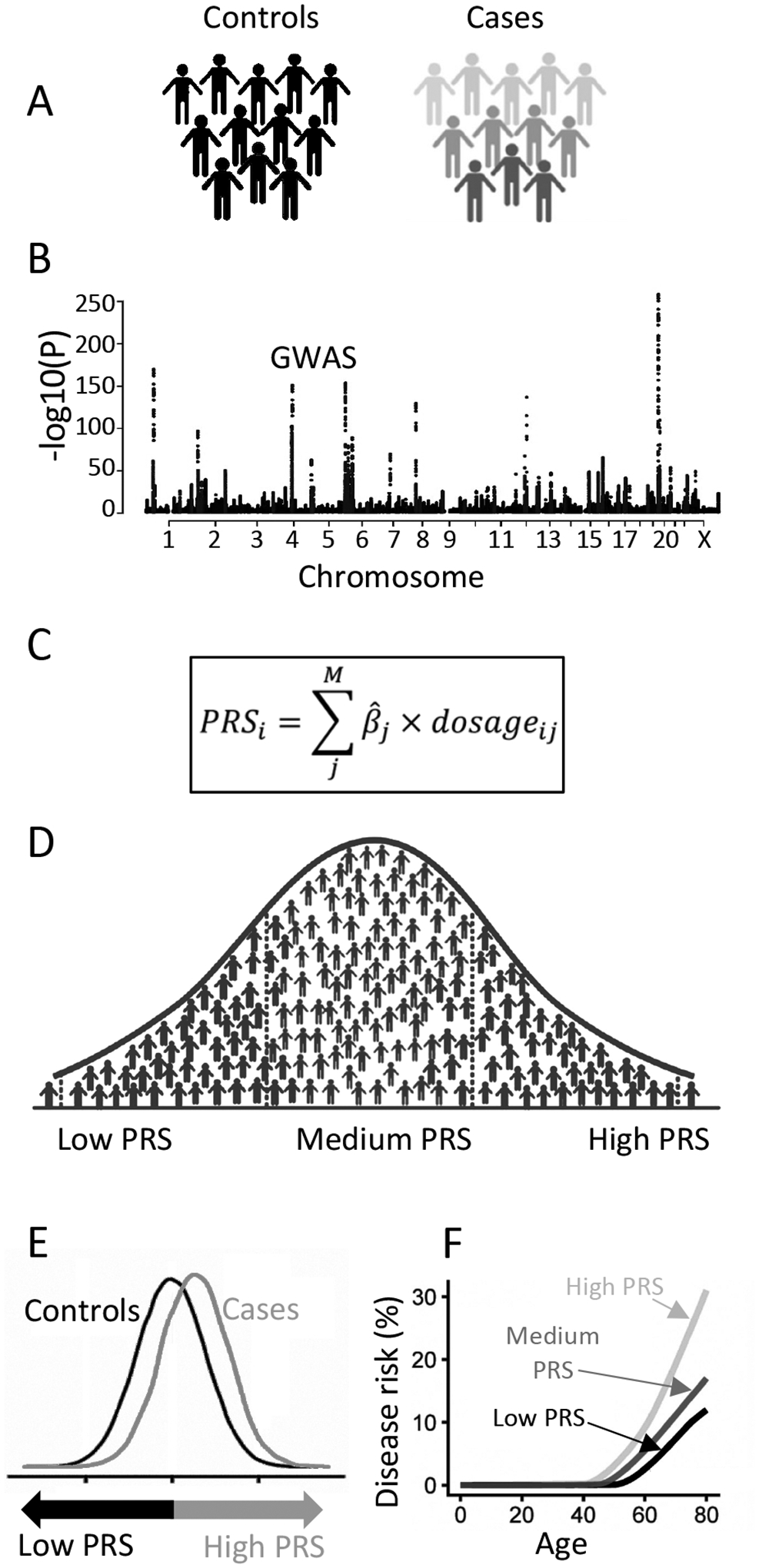
Construction and applications of a polygenic risk score (PRS). Cases and controls for a disease (A) are collected, and a GWAS is performed (B). The effect estimates are extracted from the GWAS and used, in combination with information on linkage disequilibrium, to construct weights for the PRS (C). PRSs are computed in an independent cohort, in which a majority of the participants will have intermediate PRSs, and a small fraction will have high vs. low PRSs respectively (D). The distribution of PRSs can be compared between cases and controls (E), or the disease incidence rate can be compared between participants with high, intermediate, or low PRSs (F) to evaluate the performance of the PRSs.
One successful example in which PRS have been evaluated in relation to risk assessment is among women with a suspected predisposition to breast cancer [10,11]. An associated web-based risk assessment tool, CanRisk, has been developed, which takes into account both PRS, family history, and rare pathogenic variants in known cancer susceptibility genes [12]. It supports integration of PRS into clinical risk calculators as a feasible approach to enhance disease prevention. However, for most complex diseases, the precision of the PRS is still too low to be clinically relevant, and there are yet no clear guidelines available on how to translate PRS to the benefit of people in general. The current days PRS are more probabilistic than deterministic [13] and are better suited for risk prediction modelling than for diagnostic purposes. Many international organizations recommend estimating the 10-year cardiovascular risk for adults between the ages of 40 to 75 years, and it has been shown that including PRS further increases the accuracy of the current risk prediction tools [14]. Studies have also shown that communicating risk-information—including both clinical risk factors and PRS—to middle-aged persons promotes positive lifestyle changes as well as the tendency to seek medical care [15]. This supports that PRS could be incorporated into clinical risk calculators to delay or prevent disease onset.
It is known that the majority of the heritability of complex diseases and traits is due to common genetic variants with small effects [16,17], and most individuals with a complex disease do not carry established single-gene mutations with large effects. In general, PRS could therefore represent an individual’s genetic liability to a disease, and people with high PRS have been shown to have a high disease risk equal to carriers of monogenic mutations [18]. As the accuracy of PRS depends on the power of the GWAS from which they are derived, the performance of PRS is likely to continue to improve, thanks to larger cohort sizes and better characterized samples together with further methodological developments [19–21]. However, for most complex diseases, monogenic forms have been identified. One well-known example is the melanocortin 4 receptor gene (MC4R), where common GWAS variants are associated with a small increase in the risk of obesity [22]. In contrast, rare deleterious mutations in MC4R represent the most common monogenic cause of severe early onset obesity [23]. Variants that have such considerable effects on risk of disease are often rare, specific to individual populations or families, or de novo mutations, and are therefore difficult to detect in GWAS. Such variants are often not in linkage disequilibrium with common variants [16], and therefore not captured by PRS. Thus, it is important to realize that persons with low PRS can still carry rare mutations that result in very high genetic liability to the disease.
There are several additional limitations when utilizing PRS in precision medicine. First, most GWAS have been performed in cohorts of European descent, and PRS computed to date are much less informative in other ethnicities [24]. Therefore, there is an urgent need for more diverse ancestries to be included in GWAS [25]. Second, for most diseases and traits, the PRS still explain a limited amount of the heritability [26]. This is mainly due to not having large enough sample sizes in GWAS, and even larger GWAS studies are needed [7]. Finally, individuals with high PRS will likely carry risk alleles in many—maybe hundreds or even thousands—of genes that influence different pathways in different tissues and matching an optimal treatment to patients’ genetic makeup will be more challenging compared to individuals with monogenic forms of the same disease. Therefore, using precision medicine approaches to identify subgroups of patients with similar features is believed to be a more promising strategy for complex diseases.
While most of the knowledge on the genetic contribution of complex disease has arisen from array-based SNP genotyping studies, followed by imputations and GWAS, whole genome sequencing (WGS) and whole exome sequencing (WES) will soon be state of the art in genetic studies [2]. Human genomics is—due to novel high-throughput technologies for genome sequencing—the fastest growing field with regards to big data production. The costs for WGS have dropped dramatically over the last decades, from $1,000,000,000 per genome—when the first human genome was sequenced twenty years ago—to soon reach $100 per genome. In 2013, Genomics England announced a flagship project for sequencing 100,000 whole genomes of patients with rare diseases as well as their family members, and tumor samples from patients with common cancers. Since then, large-scale WGS and WES studies have been performed in clinical trial participants [27] as well as in population-based cohorts [28–30], aiming to increase understanding of the genetic contribution also to complex diseases and to accelerate the development of genome-based precision medicine. Sequencing methods capture more than 10 times as many damaging genetic variants [28] compared to SNP genotyping and imputation, and the major parts of the genetic variants identified with WGS are rare [17,31]. However, the ability to identify genetic effects is proportional to the allele frequency, the allelic effect, and the sample size. Consequently, the contributions of large WGS and WES studies have been relatively limited to the complex disease genetics field [32], in comparison to the most recent GWAS studies where the sample size, instead of the number of variants, has increased dramatically [7]. This suggests that the genetic effects that are not captured by PRS or by known monogenic variants most likely comprise thousands of additional genetic variants with effect sizes too small to be captured in the current studies (Figure 2), ranging in frequency from very rare to common, and that even larger GWAS studies in the future will dramatically increase the accuracy of the PRS. However, it should be remembered that even with a highly accurate PRS that captures most of the heritability of a complex disease, many people will carry rare deleterious mutations that are not captured by the PRS, and there is high probability of those being incorrectly classified as being of low disease risk based on PRS only.
Figure 2.
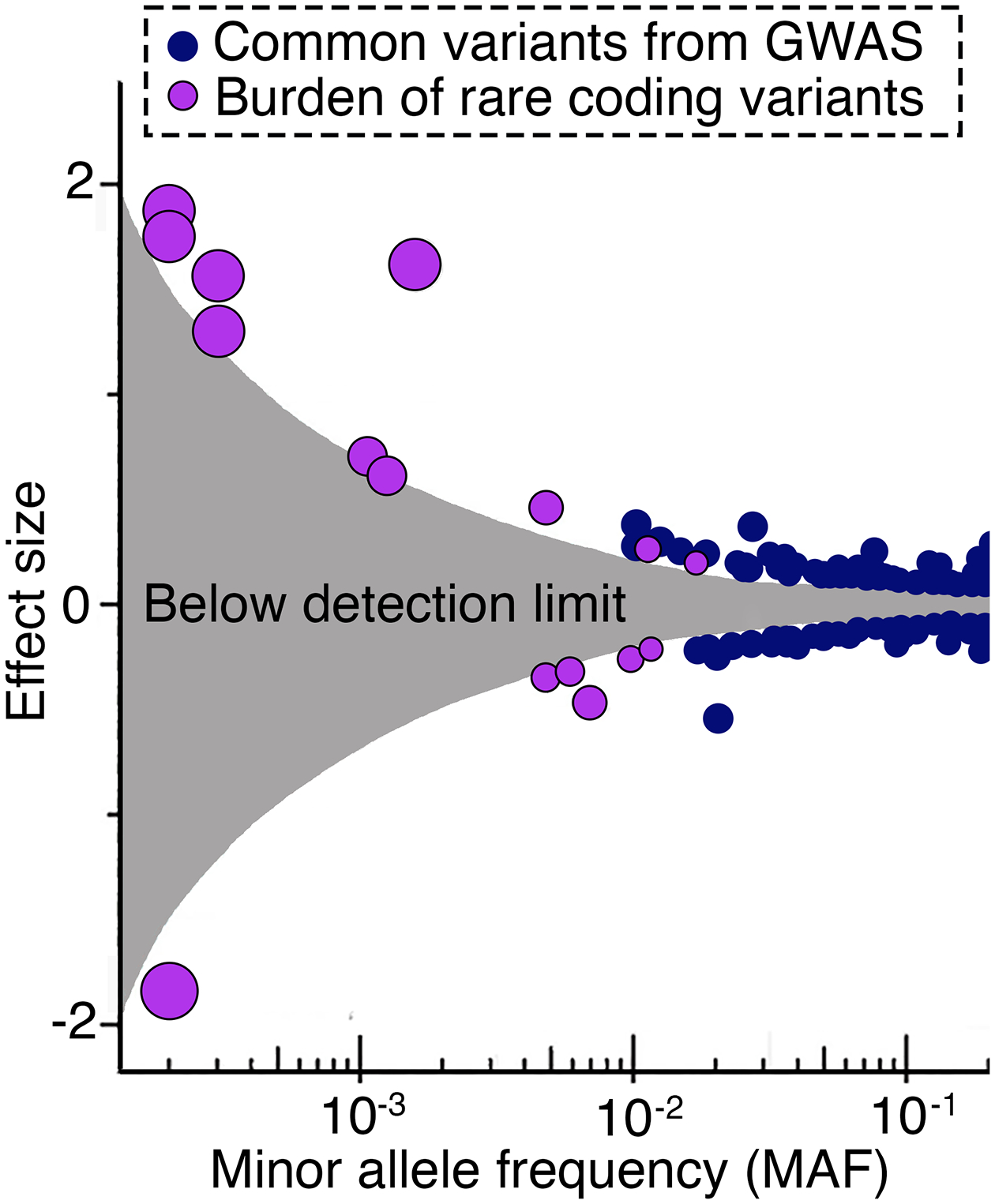
The effect size for genetic variants of different allele frequencies. The figure shows the effect on BMI in kg/m2 per copy of the minor allele for individual SNPs (blue circles), or for the burden of rare alleles (purple circles), of different allele frequencies. The blue circles represent the effect sizes from a genome-wide association study (GWAS), and the purple circles represent rare coding variants from gene-based tests performed in over 600,000 samples with whole-exome sequencing (WES) data. The grey area indicates the effect sizes for which the study was underpowered to detect any effects for different allele frequencies. The figure is adapted from Akbari et al [32]. Reprinted with permission from AAAS.
Precision medicine for cardiovascular disease
Genomic medicine approaches for clinical utility have been evaluated for several complex diseases, including cardiovascular diseases. In the cardiovascular field, there is a large interest in precision medicine approaches [33], and recent reviews include guidance for clinical implementation of PRS [14]. One determinant of success has been the fraction of patients who have a monogenic form of the disease, a factor that varies dramatically between different diseases. Cardiomyopathies are a heterogeneous group of disorders that affect mainly younger adults and can lead to severe outcomes and complications. Since up to 60% of cases are familial, genomics has been successfully used to enlighten the etiological basis and disease mechanisms [34]. This has recently resulted in many strategies to therapeutically target specific disease mechanisms. For example, cardiac amyloidosis—one of the leading causes of cardiomyopathy—has recently gained guideline class I recommendation for a first-in-class treatment using a protein stabilizer. Cardiac amyloidosis is caused by mutations in TTR, a gene that encodes the protein transthyretin, which causes a fibrillar structure in the myocardium. The protein structure stabilizer tafamidis was submitted for market approval already in 2010, for peripheral nerve disease caused by TTR amyloid deposits [35]. Following a landmark randomized clinical trial in cardiac amyloidosis, the FDA approved tafamidis for cardiac indication in 2019, 16 years after its first publication. Besides stabilizing the natural tetrameric structure of TTR, different approaches from blocking gene transcription to attacking deposited TTR-fibrils are under clinical investigation or are already approved. The latest success comes from a first-in-human clinical trial using CRISPR-Cas9 in hereditary amyloidosis [36]. Using intravenous infusions of lipid nanoparticles containing guide-RNA against the tafamidis-locus and mRNA coding for an optimized Cas9 enzyme, six patients have undergone experimental gene therapy. After a single-dose treatment, gene sequencing showed successful modification of TTR in liver cells, with the CRISPR-Cas9 editing leading to a premature stop codon and termination of translation, and consequently a dose-dependent decrease of serum TTR. Other indications have followed the stringent developmental pipeline, with mavacamten being the latest success in a randomized clinical phase 3 trial [37]. Aficamten, another myosin inhibitor, is currently in clinical investigation. First published in 2016 in a rat model of hypertrophic cardiomyopathy (HCM) [38], only 8 years later, the FDA approved the compound for treating symptomatic patients with obstructive HCM. This brings the first specifically developed drug to HCM and positions it as an alternative treatment to cardiac surgery or interventional septum ablation. In dilated cardiomyopathy (DCM), different trials in phase 1–3 investigate the use of small compounds, antibodies, non-coding RNAs, cellular therapies and gene therapy approaches. Only recently, a promising small compound trial in phase 3 against cardiac laminopathy was cancelled due to futility, leaving one of the most severe forms of DCM without the prospect of an immediate treatment option [39] besides primary prophylactic implantation of a defibrillator against sudden cardiac death.
Precision medicine for allergic diseases
Allergic diseases are a group of complex diseases that represent very common conditions like asthma, eczema, hay fever, pet allergy and food allergy that affect large proportions of populations worldwide, from infants to the elderly. Both genetic and environmental factors are well recognized to influence disease occurrence, severity and progression. Therefore, precision medicine efforts need to encompass environmental, lifestyle and genomic domains to successfully improve diagnostics, treatment response or predictive scores in allergic disease [40]. Analyses of allergen-specific IgE in blood (or a skin prick test) today constitute a standard diagnostic test for any allergic condition. For some conditions, like peanut allergy, conventional peanut extract tests cannot distinguish true, potentially life-threatening peanut allergy from cross-reactivity with birch pollen associated with only mild symptoms. However, novel molecular allergy approaches—i.e. component-resolved analyses—may reveal IgE sensitization patterns at the protein level (e.g. towards the peanut protein Ara h2), which represents true peanut allergy rather than cross-reactivity [41]. Such tests are today available in most well-equipped laboratories and allow for targeted patient advice (e.g. strict avoidance), rescue treatment (prescription of adrenaline/epinephrine autoinjectors) or disease-modifying treatment recommendation (immunotherapy). In relation to allergy to airborne allergens—for example tree or grass pollens, or dust mites—diagnostics must reflect personal exposures that are often region- or country-specific (e.g. birch pollen very prevalent in Scandinavia). However, with climate change, allergen levels have changed in that peaks may occur outside the regular pollen season and new plant species with new allergens may appear in a given region [40]. As such, both the patient and the caregiver must be aware of these changes that will influence diagnostics (new allergens tested) and treatment (consider exposure outside traditional exposure periods), exemplifying that also environmental factors will have to be incorporated in precision medicine efforts.
While genetic factors strongly influence the risk of allergic and respiratory disease, no convincing examples of diagnostic tests applied in clinical settings have so far been seen. The predictive value of PRS is rather low, with AUCs around 0.65–0.70 [42]. However, genetics can improve clinical diagnostics, on top of disease history and routine clinical information, especially in the case of early onset severe asthma or an inherited respiratory or immunodeficiency disease. In such examples, whole-genome sequencing has predominantly been used to rule out monogenetic respiratory disease (e.g. cystic fibrosis, primary ciliary dyskinesia or surfactant dysfunction) and inform decisions about initiating adequate treatment.
Several precision medicine studies in allergic diseases have been performed to identify markers for treatment response. Local corticosteroid applications (e.g. in nasal spray, inhalation device or topical skin cream) are the primary anti-inflammatory treatments for all allergic diseases. In severe cases, highly potent and mechanism-specific biological treatments have, in the last years, become more broadly available. For the right patient, a dramatic treatment response can be observed with marked symptom reduction and increased quality of life. For severe asthma patients, there are today several biological drugs available, including omalizumab (anti-IgE), mepolizumab (anti-IL5), benralizumab (anti-IL5R), dupilumab (anti IL4/IL13) and tezepelumab (anti-TSLP) [43]. A clinical challenge is to predict which patient will respond to which treatment. The biomarkers—for example, blood eosinophil counts, total or specific IgEs, or fraction of exhaled NO—used to guide decision-making in the clinic today do not provide enough precision. Omics-based characterization of response, or prediction of response, holds great promise to allow for clinical precision medicine applications. Two examples using transcriptomics can be highlighted to illustrate this potential. In children with severe, eosinophilic asthma, treatment responses to mepolizumab were found to differ based on baseline nasal transcriptomic signatures; some children responded well, whereas others—depending on the signature—actually did worse on treatment [44]. The other example provides novel insights into oral immunotherapy effects in adolescents with peanut allergy, for which signatures of tolerance and immune response were captured in those who successfully completed the therapy [45].
Precision medicine in psychiatry
Psychiatric disorders—including depression and schizophrenia as well as bipolarity, anxiety, post-traumatic stress, and neurodevelopmental and eating disorders—affect our mental health. Personalized practice is an integrated part of clinical psychiatry. Psychiatric diagnoses are defined based on descriptive criteria [46], but the classical foundations of the diagnostic categories were based on detailed clinical phenotyping, including long-term outcome [47]. Recent evidence supports the notion that clinical characteristics can be helpful for personalized management of major depression and psychotic disorders [48,49]. The lack of knowledge about disease mechanisms and related treatments has so far limited the application of precision medicine in psychiatry.
Most psychiatric disorders have a high heritability, with estimates between 40% and 80% [50], and are highly polygenic [51]. Lately, there has been a large increase in discoveries of genetic variants associated with psychiatric disorders [50], followed by the development of PRS-based tools [51]. However, PRS for psychiatric disorders explain relatively small proportions of the liability of each disorder, and the current PRS are still not clinically useful [52]. In addition to genetics, several lines of research have focused on prediction modelling building on clinical, cognitive, and brain imaging approaches. With the emerging transformative knowledge in psychiatric genetics, neuroscience and brain imaging, there is a large potential for implementing precision psychiatry in several areas [53]. Despite this, psychiatry has been slow in implementing precision medicine [54], and potential applications have not yet been considered for real-world implementation [55], or even been demonstrated to be useful in the clinic [52]. Promising results have emerged for prediction models to support the diagnosis and predict treatment response in some specific clinical scenarios [56] with the most fruitful approaches being the prediction of diagnoses and disease onset, as well as treatment response and outcome [57]. Prediction algorithms applying machine learning methodology have reasonable levels of accuracy and precision when informed with clinical [58,59], brain MRI [55,58,59] and biomarker [58–60] data. For example, individuals at clinical high risk for psychosis could be classified with 78% sensitivity and 77% specificity [60], and integrating clinical information [56] further increases the prediction accuracy of transition to psychosis to 85%.
Precision medicine approaches for treatment stratification could have large clinical impact [61,62] due to the high rates of non-response to psychopharmacological agents. For example, approximately 30%−40% of schizophrenia patients are treatment-resistant [63]. Some recent findings in the prediction of treatment response including clinical data show reasonable accuracy [1]. However, the performance of these models will most likely increase when genetic, molecular or imaging data are also included. There are several ongoing efforts to develop PRS for treatment stratification. For example, for antipsychotic non-response, the PRS could predict non-response in independent samples [64], and the accuracy further improved when adding cognitive measures [65]. Genetic prediction may also be helpful for identifying individuals who are likely to develop specific side effects [66]. Combining genotyping with data available in large, prescription registers—such as those from the Nordic countries [67]—in addition to longitudinal clinical samples, will also provide new insight into the genetics of psychopharmacological treatment. In addition, including neurobiological deviances from normal trajectory during neurodevelopment [68] may help treatment development and stratification.
Precision diabetes medicine
In diabetes, very few people are ever cured, and most people at high risk will eventually develop the disease, even when aggressive interventions are used to mitigate risk. For example, the Diabetes Prevention Program [69] was a breakthrough trial that demonstrated that type 2 diabetes incidence can be substantially reduced through metformin treatment (31% risk reduction compared to placebo control) and intensive lifestyle intervention (58% risk reduction compared to standard-of-care control) in people at high risk of the disease. Nevertheless, the extent to which the metformin and lifestyle interventions delayed diabetes onset compared with the control intervention was only about 12 months with metformin and 36 months with intensive lifestyle intervention. Moreover, after more than 20 years of follow-up, the mortality rates were comparable in those initially randomized to metformin or lifestyle and in those randomized to the control interventions.
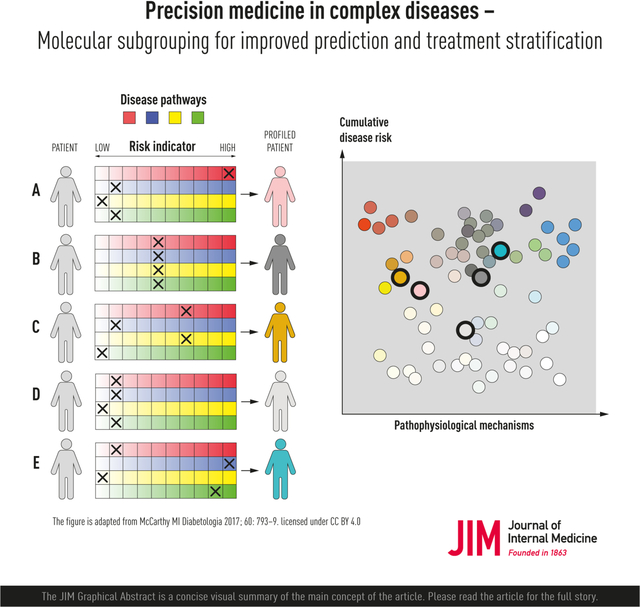
Precision medicine for diabetes is—as for many other complex diseases—very challenging, as both the common forms of the disease (type 1 and type 2) are remarkably complex in etiology, clinical presentation, pathogenesis and treatment requirements. Thus, no single solution will solve these challenges, and it is improbable that genomic medicine alone will play a major role in defeating these diseases, as so much of the etiology and pathogenesis is a consequence of environmental risk factors. Thus, precision diabetes medicine of the future is likely to involve a broad-scope approach, which will need to incorporate time-varying and environmentally sensitive markers, neither of which are characteristics of genetics. In an elegant commentary on the complex etiology of type 2 diabetes, McCarthy proposed the “palette” model [70]. This is a metaphor of the painter’s palette, where the bright colors around the edges represent the individual disease pathways, and the combinate in the middle—where the colors meet—represents the mixed combination of pathways that leads to disease development in each patient. The model infers that amongst the heterogenous group of people with type 2 diabetes, some have a relatively simple form caused by one or a few key factors, whereas most people with the disease cannot be so easily distinguished, owing to the complex interaction of many genetic and non-genetic factors that renders these cases unpredictable and difficult to accurately diagnose and treat.
In 2018, an important study describing the subclassification of diabetes into distinct subclasses was published [71]. A “hard clustering” machine-learning method called “K-means clustering” was utilized to stratify a dataset comprised of people of Swedish and Finnish ancestry into five subclasses, each representing a phenotypically distinct etiology. The research went beyond this to show that these subclasses can be used to predict differential treatment requirements and incidence of diabetes complications. Many subsequent papers showed that these subclasses are reproducible in other populations, some differing in ethnic origin to the original cohorts, and interpreted this work as evidence that people with diabetes can be assigned to one of five distinct sub-diagnoses of diabetes—which is, unfortunately, incorrect. Assignment of individuals to a subclass is a probabilistic phenomenon, where each person within the dataset has a certain probability to be assigned to each cluster. Thus, a given individual within the dataset may contribute to one or more of the five subclasses, similar to the palette model [70]. Subsequent analyses using “soft clustering” methods revealed that only around one-third of people with diabetes can confidently be assigned to a specific subclass, with the other two-thirds being of mixed etiology, aligning well with the palette model [72]. Moreover, while classification methods lead to easily interpretable results, stratification of initially continuous data usually results in a loss of predictive power. Indeed, Dennis et al. showed that when comparing a sub-classification method to a simple model that maintains the continuous nature of the clinical variables, the latter had superior predictive ability [73]. Building on this, the most recent advances in precision diagnostics for diabetes have used machine-learning methods that maintain the continuous distribution of data—for example, using UMAP and DDRTREE—to array continuous data distributions and to assign probabilities of diagnostic tendencies that might prove useful for clinical translation.
Beyond precision diagnostics, significant progress has also been made in precision nutrition. Several studies have shown success in individual-level prediction of post-prandial glycemic variation. The seminal paper on this topic was published in 2015 [73], where Zeevi et al. collected very detailed diet and phenotypic data in ~800 young adults. The phenotypic assessments included continuous glucose monitoring, physical activity, habitual diet and biological features including gut microbial diversity assessed using 16s rRNA sequencing of bacterial DNA obtained from participants’ stool. They derived machine-learning models that accurately predicted personalized postprandial glycemic response to real-life meals. The authors proceeded to conduct a small blinded randomized controlled dietary intervention and showed that the algorithmically personalized diets performed better compared to diets prescribed by a registered dietitian. Berry et al. undertook a slightly larger study [73] and broadened the scope to explore whether—in addition to postprandial glycaemia—triglyceride and c-peptide concentrations could also be predicted after randomized fixed-macronutrient meal assignment. They showed that while postprandial glycaemia could be adequately predicted, triglycerides and c-peptide could not. Importantly, glycaemia was assessed using flash monitors (a continuous measure of glucose), whereas triglycerides and c-peptide were assessed using dried blood spots manually obtained throughout the two-week study period. Thus, limitations of the method used to measure triglycerides and c-peptide may have undermined the ability to predict these phenotypes.
In early 2018, the American Diabetes Association (ADA) and European Association for the Study of Diabetes (EASD) launched the Precision Medicine in Diabetes Initiative (PMDI). The work undertaken within the ADA/EASD PMDI has focused on building robust networks and infrastructures through which research on precision diabetes medicine can be undertaken and translated into clinical practice [74]. In 2020, the first ADA/EASD Consensus Report on Precision Diabetes Medicine was published [75]. The report outlined the key pillars of precision diabetes medicine (prevention, diagnosis, treatment, prognosis and monitoring), outlined terminology, described clinical exemplars of precision diabetes medicine, and provided expert opinion on key steps to implementation—including opportunities and barriers. The motivation to establish the ADA/EASD PMDI came through recognition that research germane to precision diabetes medicine was advancing rapidly, yet there was little coordination of these processes, which stood in the way of clinical translation. Moreover, there has been little consensus about what precision and personalized medicine really are as research concepts and as practical clinical domains, with people often using the same words to mean different things and different words to mean the same thing. This is evident, not only within diabetes medicine, but also for other disease areas.
Genetic subtyping in obesity and metabolism
Obesity is a multifactorial disease, resulting from an intricate interplay between genetic and environmental factors. Obesity is one of the major risk factors for many complex diseases including type 2 diabetes, cardiovascular disease and cancer. At the population level, the genetic contribution to obesity (i.e. the heritability) has been estimated to range between 40%−70%, suggesting that a substantial proportion of the interindividual differences in obesity susceptibility is due to genetic factors. Indeed, GWAS have so far identified more than 1500 genetic variants that each are associated with body mass index (BMI), obesity risk or other adiposity outcomes [76]. At the individual level, people’s genetic susceptibility to obesity is quantified using PRS, and even though most individuals have an average score—and thus an average susceptibility to obesity—some individuals will have a very high genetic susceptibility, and others will have a very low genetic susceptibility (Figure 1). Increasingly, these scores are used in the context of precision health to predict which individuals are at high risk of gaining weight. However, so far the predictive ability of PRS has been limited, which may be due to how obesity is defined. Obesity is typically defined by a simple metric—i.e. a BMI of ≥30 kg/m2. However, two individuals with obesity, even if they have exactly the same BMI, may differ in the underlying (biological) causes of their weight gain. They may also differ in the presentation of the disease, the prognosis, the complications and the response to treatment. To account for this heterogeneity, there is a growing interest to subclassify obesity into smaller, more homogenous subtypes. So far, subclassifications have been predominantly performed based on clinical features present in individuals who have obesity (i.e. phenotypic subclassification). For example, individuals who have obesity, but do not demonstrate any comorbidities, have been subtyped as having “metabolically healthy obesity” [77]. Increasingly, more advanced methods—such as principal component analyses and machine learning based on anthropometric and clinical features—are being used for more refined subclassifications [78–80].
With the discovery of an increasing number of genetic variants associated with obesity, genetic subclassification has become possible. A key advantage of subclassifications that are based on genetic variants is that they may reveal new insights into the etiology of the disease and its subtypes. In addition, genetic subclassification can be done early on in life, long before the onset of disease, allowing for a timely prevention. There are various approaches that can be applied to use genetic information in the subclassification of obesity. For example, in a recent study, publicly available GWAS summary statistics were used to identify BMI-increasing variants that are associated with a lower risk of type 2 diabetes and cardiovascular disease, and with a favorable cardiometabolic profile [81]. With this multi-trait approach, 62 variants were identified, for which enrichment analyses point to a key role of adipose tissue and adipocyte biology in the uncoupling of obesity from its comorbidities, which is in stark contrast to the enrichment analyses performed for BMI-only associated variants, which point to the brain [22]. These subsets of variants help reveal new biological pathways that are not observed when all variants are considered together, as commonly done in PRS approaches. Importantly, these subsets of variants can contribute to the subtyping of obesity through using genetic risk scores that are based on them. The derived subtypes of obesity would allow for a more precise diagnosis of obesity—as compared to the simple “BMI≥30 kg/m2” metric—that will allow for more personalized prevention and treatment strategies that are tailored to the specific obesity subtype, and a more precise prognosis. Additional studies have confirmed the obesity–type 2 diabetes discordant genetic effects and the link to fat distribution and lipid metabolism, and genes that may represent targets for precision medicine approaches have been highlighted [82]. However, both fat distribution and the pathogenic effects by the adipose tissues are known to differ between sexes [83,84], and future studies stratifying by sex are therefore needed to increase the precision in prevention and treatment strategies for obesity [85].
Fetal precision medicine
During the last two decades, there has been considerable development in precision diagnostics within embryo and fetal precision medicine both before and during pregnancy. This has been brought about through huge advances within the field of genomics and also within the ultrasound and magnetic resonance imaging area. Before 2000 the genetic tests had low resolution and covered only a few conditions, but with the advancements within the genomic field, the tests now have a very high resolution and cover many genetic conditions [86]. The genomic advances cover both genomic screening techniques and diagnostic tools. For example, sampling the fetus’s genome has been revolutionized in recent years by the possibility of sampling the fetus DNA by testing the cell free DNA (cfDNA) in the mother’s blood [87]. Noninvasive diagnosis of fetal aneuploidy can be performed by shotgun sequencing DNA from maternal blood [88]. This non-invasive technique has substantially lowered the need for invasive diagnostic procedures that are associated with a risk of miscarriage. Non-invasive prenatal testing (NIPT) is built on discovering that fetal cfDNA is present extracellularly in the maternal plasma and serum during pregnancy. The fetal cfDNA comes mainly from apoptosis in the placenta, which is a fetal tissue. Of the total maternal cfDNA in maternal plasma, approximately 3%−10% is typically from the fetus in late first trimester [89]. Fetal cfDNA can be used for detecting fetal rhesus factor status, screening for fetal trisomies, and, more recently, for screening of some of the microdeletion syndromes [89–93]. The entire genome and mutational status have been sequenced from fetal cfDNA, and some fetal single-gene mutations have already been diagnosed using fetal cfDNA [94]. The advancements in assisted reproductive techniques have now made it possible to check the genetics of a 5-day old embryo before the embryo is transferred to the mother—preimplantation genetic diagnosis (PGD) [95]. Many more applications for non-invasive fetal screening, including targeting monogenic forms of complex disease, are expected in the future.
Complex disease, multi-morbidities and precision medicine
Due to aging populations, complex diseases in a single patient do unfortunately not typically manifest in a mutually exclusive manner. They co-exist and provoke each other over time, complicating work-up and treatmentin particular in a precision medicine context [96]. As explained above, a given complex disease can have multiple etiologies and in fact represent different routes or mechanisms that interact towards similar conditions in a single patient. One can consider this a “special case” of the general multi-morbidity problem in which several diseases that for long have been recognized as distinct, separate diseases interact—for example, via shared genes and pathways [97]. In some cases, a “driver disease” provoked by genetic risk, exposures or both may lead to longitudinal disease development in which the order of events is quite well understood (even if it in the individual case may be hard to predict), while in other cases it may be less clear what represents a risk factor and what represents a complication in a life-course perspective.
The concept of “promiscuous” diseases has been introduced to reflect the difficulties in understanding the mechanistic aspects of temporal disease development [98]. Atrial fibrillation is one such example in which this condition may contribute to increasing the risk of other cardiac events, but it may also be a complication to other diseases without prior manifestation. Sex differences in disease trajectories across men and women add to the complexity of the problem [99]. The way diseases have been defined over the history of medicine has been influenced by symptomatology, anatomy and tissues, not by detailed knowledge at the mechanistic, molecular level that now increasingly can be revealed by deep multi-omics technologies [100]. When such technologies are used to produce readouts that over several time points reflect disease progression, it becomes possible to better decipher mechanisms over time and to include exposure information along the way.
Drugs are designed to change cellular behavior, which today also is characterized by multiple omics technologies. When multi-omics data are produced in large cohorts, they have the potential to reveal patient-level disease characteristics and individualized response to treatment. In a recent study [101], a deep-learning-based framework (variational autoencoders) was used to integrate multi-omics data from a cohort of newly diagnosed diabetes patients (genomics, transcriptomics, proteomics, metabolomics, and microbiomes). In addition to the molecular level data, the autoencoder also received data on medication, diet questionnaires describing lifestyle and clinical measurements. This type of approach can then be used to quantify and assess how different interventions are likely to change the multi-omics readouts and hence how exposures change cellular behavior. Exposures can be everything from lifestyle interventions to drugs given to patients. The authors focused on drug-omics associations and found many new such associations. Of relevance in the precision medicine context, the methodology can be used to perform “thought experiments” on how a drug that a patient did not yet receive would likely change the omics readouts for that patient. The variational autoencoder uses correlations learned from other patients who received the drug to make this estimate. It is often said in healthcare that one cannot know what would have happened if drug B were given before drug A, when A was the initial choice. However, with this kind of data reconstruction scheme one can make such estimates, even in the context of multi-morbidities where polypharmacy is a significant problem.
Beyond genomics
While genetic subtyping is a promising approach, genetic variation explains only part of the susceptibility to complex diseases. Therefore, genetics alone will not be sufficient to generate precise discriminatory subtypes. Similarly, genomic diagnostic approaches for complex disease have been most successful for monogenic disease forms—as in the example for cardiomyopathy above—and the use of PRS for risk prediction has still not been shown to be useful in the clinic for most diseases. There is a need to include other omics and non-genetic factors to further refine the precision medicine approaches. The genomic component of complex diseases captures the heritable effects, but importantly, the risk accumulation throughout a lifetime is not captured. The incidence rate of most complex diseases increases with age, and it is therefore important to consider other types of clinical and biological information that can serve as markers for disease risk and progression, markers that can be used for diagnostic purposes, prognosis or for clinical decision support. Such data could be based on, for example, molecular profiling, imaging or electronic health records. This provides an opportunity to identify, for example, molecular signatures that can be linked to disease development or increased disease risk. While many omics technologies—including proteomics, epigenomics, lipidomics and metabolomics—are useful for molecular profiling and for discovery of novel biomarkers, there are some important limitations that are less pronounced in genomics. It should, for example, be considered that genetic variation influences an individual’s baseline levels of many molecules, and genetic variants can also change the amino acid sequence of proteins with dramatic effect on the ability to measure it in some individuals [102–105]. Combining genomic information with other molecular measurements is therefore important to improving the predictive value of molecular profiling. In addition, high-throughput technologies for genome-wide assessment of genetic variation have high reproducibility at a reasonable cost. In complex disease genetics, genetic variations that are inherited from the parents are investigated, and therefore a blood sample—or any other tissue sample—taken at any timepoint is suited for the experiment. The technologies for RNA and DNA methylation analyses are in some aspects similar to DNA sequencing; however, both RNA expression and DNA methylation differ between cells, tissues and change over time. Epigenetic factors have been suggested to play an important role in the regulation of gene expression [106] and in the pathogenesis of complex diseases [107]. The epigenetic pattern is linked to cell-specific gene expression and transcription factor binding [108], but it is also associated with many environmental factors such as, for example, smoking [109]. Epigenome-wide association studies (EWAS) seeking to identify associations between epigenetic modifications and disease traits [110,111] have mainly been performed using large-scale DNA methylation data. DNA methylation is the attachment of a methyl group to a CpG (cytosine-phosphate-guanine) dinucleotide, which can introduce stable changes in gene expression [112,113]. High-throughput arrays for interrogating the DNA methylation at hundreds of thousands of positions in the genome have been developed, including the Infinium Human Methylation 450K and MethylationEPIC BeadChip (Illumina, San Diego, USA). Numerous studies have, for example, linked epigenetic changes in blood or nasal epithelium / bronchial wash samples with allergic disease [40] and other complex respiratory diseases like chronic obstructive pulmonary disease [114]. Epigenetics-based prediction models using a handful of markers are now appearing in the literature [115]. This is a very rapidly expanding field with huge potential for both diagnostics and novel treatment approaches. However, one major limitation with RNA sequencing and DNA methylation studies in complex diseases is that access to the relevant cells and tissues might not always be feasible—for example, in psychiatric disorders—and most studies have used blood samples as proxies for the tissue of interest.
In contrast to DNA methylation and RNA, proteins are often released or leaked out to the plasma, and for that reason, plasma proteome profiling has been suggested to better capture disease processes that occur in tissues other than blood. The development of proteomic technologies has taken a rapid step forward during the last decade, since the first proteomic analyses in larger cohorts were performed [104,105]. Untargeted proteomic analyses can detect and quantify thousands of proteins and are a powerful approach for discovering novel biomarkers. The first step in a biomarker discovery study should ideally be untargeted, similarly to a GWAS, aiming to identify novel protein biomarkers. However, due to high costs and relatively low throughput, most untargeted biomarker discovery studies are performed in a very limited number of samples [116]. Targeted proteomic approaches have been developed, including immunoassays based on nucleic acid proximity-based methods [117] or aptamer-based proteomic assays [118]. These methods provide an improvement in the dynamic range and sensitivity, as well as the intra-assay precision, and over 1000 articles have been published using these assays. However, similarly to genomics, most biomarker studies focus on the discovery phase, and very few new biomarkers have been taken forward into clinical use. This is mainly because most biomarkers show predictive value that it too low to enhance a clinical decision. In addition, the process of developing and validating biomarker assays for clinical utility is time consuming and requires validation in real-life clinical settings through clinical trials.
The omics cascade proceeds from the genome to the epigenome and transcriptome, followed by the proteome, and culminates in the metabolome, which is the end-scale integrative product. Accordingly, the metabolome has been described as being closest to the patient phenotype and therefore particularly useful for molecular phenotyping [119]. Metabolomics is defined as the comprehensive analysis of the small molecule component (conventionally defined as compounds <1500 Da). The sensitivity with which small molecules respond to physiological fluxes has led to them being termed the “canaries of the genome” [119], because dysregulation of the metabolome may be detected earlier than, or even without, alterations observed at the genome or transcriptome level. Estimates of the size of the metabolome vary widely from thousands to hundreds of thousands of small molecules. However, to place these estimates within context, in silico libraries of molecular lipids alone reported >250,000 different species [120]. In addition, if the metabolome is expanded to include exogenous compounds (e.g., dietary components, xenobiotics); conjugation products (e.g., glucuronides, sulfides); and peptides as well as isomers (e.g., regio- and stereoisomers), then the metabolome starts to approach that of the million-molecule size that has been proposed [121]. Metabolomics provides a real-time biochemical profile that reflects exogenous (e.g., xenobiotics, dietary, occupational, microbial) and endogenous (e.g., inflammation, oxidative stress, infection, microbiota) factors—the metabotype. This comprehensive chemical signature makes the metabolome particularly useful for investigating the etiology of complex diseases, which incorporates genetic and environmental factors and their interactions. However, the complete analysis of the metabolome remains challenging, and limits in the technology have been a significant bottleneck in performing large-scale metabolomic epidemiology or metabolome-wide association analyses (MWAS) [122]. To date, no single method is capable of accurately capturing the high level of physicochemical diversity and concentration ranges that span the several orders of magnitude of the metabolome. Accordingly, multiple assays across multiple instruments are necessary to acquire a comprehensive metabolite profile. Mass spectrometry coupled to separation techniques and nuclear magnetic resonance spectroscopy (NMR) are the most used analytical techniques for metabolomic analysis. NMR has multiple benefits including rapidity, minimal sample preparation, quantification and reproducibility across laboratories. However, NMR suffers from low sensitivity, limiting the detection of some metabolites. The high-throughput and relatively low-cost of NMR-based metabolomics have led to its use in large-scale precision medicine studies, most notably its selection for acquiring a metabolic profile of all 500,000 individuals in the UK Biobank [123]. The application of NMR is being actively pursued for in vitro diagnostic applications for precision medicine by multiple commercial entities [124]. However, mass spectrometry is the most common technology for measuring the metabolome, due to its high specificity and sensitivity. Mass spectrometry-based metabolomics data acquisition is generally broadly divided into targeted and untargeted methods. In a targeted approach, a selected panel of known or predefined metabolites is measured and identified based upon a match to known chemical standards. This is the most common approach employed by most commercial metabolomics laboratories. Untargeted metabolomics approaches involve the acquisition of thousands to tens-of-thousands of unidentified metabolite features. While consisting of true discovery-based approaches to biomarker analysis, the data analysis is complex and not readily applicable to large-scale studies or commercial services.
The metabolomics biomarker discovery pipeline is demonstrated in work by Wigger et al [125]. Using general profiling methods, they identified a group of sphingolipids (3 ceramides, 2 lactosylceramides and 1 dihydroceramide) that correlated with glucose intolerance and altered insulin secretion in 6 differential genetic mice strains fed a high-fat and high-sucrose diet. The findings were validated in two separate prospective human cohorts, a targeted quantitative method was developed, and a subset of sphingolipids (3 ceramides and 1 dihydroceramide) were linked to a predisposition to develop Type 2 Diabetes up to 9 years before disease diagnosis. These findings were proposed to aid in designing improved clinical trials to assess disease onset and provide alternative avenues for treatment. Laaksonen et al. [126] employed lipidomics profiling methods to identify a panel of four ceramides (Ceramide [d18:1/16:0]; Ceramide [d18:1/18:0]; Ceramide [d18:1/24:1] and Ceramide [d18:1/24:0]) that predicted cardiovascular mortality. These markers have been independently validated by other laboratories [127–129] and are currently offered as a clinical assay by, for example, the Mayo Clinic. These examples demonstrate how applications of metabolomics profiling approaches lead to the development of targeted biomarker assays for application in the clinic. There is great promise for metabolomics for precision medicine applications, but there is an increased need for method development to enable large-scale robust methods.
In the complex disease field, integrating different types of omics data has shed light into the functionality of GWAS findings, by statistical colocalization [123] with quantitative trait loci (QTLs) for intermediate omics phenotypes, such as transcriptomics, proteomics or metabolomics. Genetic data has also been used to identify causal effects by, for example, proteins [130] and metabolites [131] through Mendelian randomization analyses. In addition, multi-omics approaches have been suggested to be useful for precision medicine. One such example is the suggested integrative personalized omics profile (iPOP), which combines wearable and omics technology for diagnostic purposes [132]. By measuring the subject’s complete molecular profile (genomic, transcriptomic, proteomic, metabolomic and autoantibody), the authors were able to detect the onset of T2D following two viral infections, prior to being detected by conventional clinical-based measurements. To do this, the iPOP of a single subject was created over a 14-month period combining orthogonal approaches that resulted in >3 billion measurements taken over 20 time points [132]. This study illustrates the benefit of longitudinal monitoring via the identification of a dysregulated phenotype to provide actionable measures that can be taken by study participants.
Beside the molecular precision medicine approaches, data from electronic health records can provide important information for disease prediction and for selecting optimal treatments. In 2014, an important study was published describing the temporal disease progression patterns (trajectories) in the whole Danish population, using data from national electronic health registries [133]. The Nordic countries provide a unique opportunity to use electronic health records due to the personal identity number and the possibility to link the whole population to, for example, registers for total population, tax, education, birth, patient, cancer, prescription, and causes of death [134]. A tool—the Danish Disease Trajectory Browser (DTB)—has been developed to explore data from the Danish National Patient Register. The dataset comprises almost 25 years of data for 7.2 million Danes and 122 million admissions, and allows for exploring disease trajectories that are associated with, for example, specific diagnoses [98]. The prescribed drug registers have been used to construct prescription trajectories that can be used to provide information on suboptimal or futile prescriptions but also to suggest initial treatments [135].
Concluding remarks
In order to make use of all types of health data for developing novel precision medicine approaches, there is a need for better frameworks for big data analytical approaches, and studies need to be scaled up dramatically. This will enable development of mathematical modelling approaches that can leverage high-dimensional, longitudinal and multimodal data. The combination of the high computing power of modern computer facilities and the ever-increasing ability to gather and store extremely large amounts of data have also paved the way for artificial intelligence (AI) as a major tool for big data analysis. In addition to prediction modelling, such models may also increase etiological insights and define the roadmap towards precision medicine—for example, in psychiatry [1], where multimodal data integration has been suggested to provide the fastest route to clinical practice for genomic precision psychiatry [60]. Prediction models integrating clinical, genetic and neuroimaging data, have, for example, been developed to accurately predict age of onset in neuropsychiatric diseases [136–138]. Still, even when the prediction tools have obtained clinically relevant accuracy, there are some bottlenecks before they can be implemented in real-world healthcare. This includes an initial lack of cost-effectiveness, inadequate levels of education and training of health care personnel as well as patients, and structural barriers of healthcare providers [53]. Still, the recent findings are encouraging, and it is expected that precision medicine will lead to a paradigm change in healthcare with transformative potential for individuals who are at high risk or are diagnosed with a complex disease. However, there are several additional challenges including the heterogeneity within each complex disease and the need to incorporate many layers of molecular, clinical and imaging data in combination with advanced mathematical modelling. Establishing networks, such as the ADA/EASD PMDI for diabetes, but also collaborations across disease fields will be crucial to meeting the rapid advancements in precision medicine.
Figure 3.
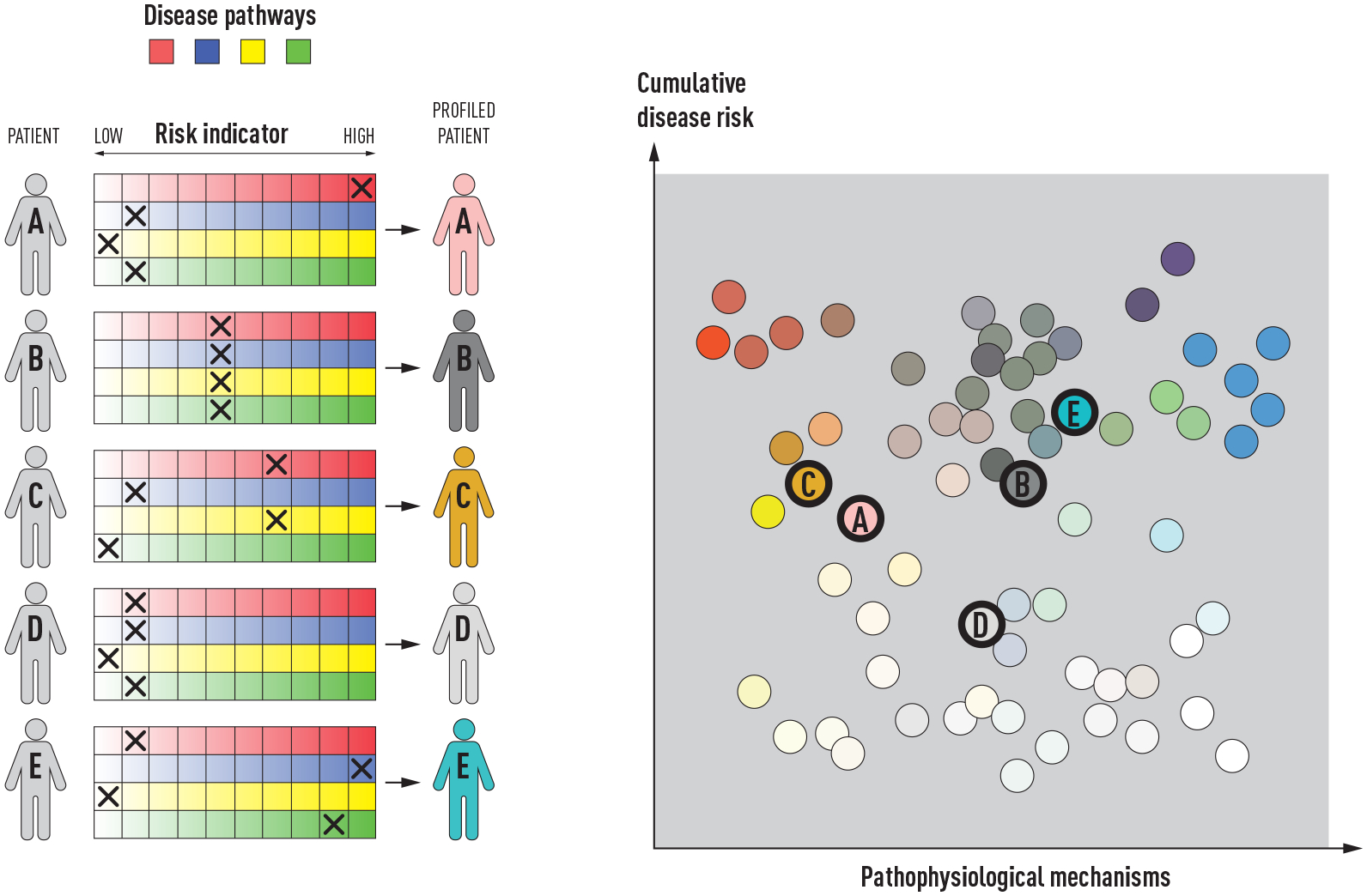
The “palette” model of the multifactorial etiology of a complex disease. The figure is adapted from the figure by McCarthy [70] licensed under CC BY 4.0 [139], where the model was proposed for type 2 diabetes. The different colors to the left represent four tentative disease pathways or pathophysiological mechanisms. Each of the five individuals (A-F) have different contributions of the colors representing the various pathways that can contribute to disease. The contribution of each pathway is illustrated by an “X”, and individuals with the same disease can have very different underlying pathophysiological mechanisms, as illustrated by the difference in colors of the individuals to the right.
Acknowledgments
This work has been supported by the Swedish Research Council (AJ, 2019-01497. EM, 2016-0386, 2018-05619 PERMEABLE), the Swedish Heart-Lung foundation (AJ, 20200687. EM, 20210546, CEW 20210519, 20200693), The Swedish Brain Foundation (AJ), and the Swedish Cancer Society (AJ, 22 2222 Pj), Research Council of Norway (OAA, 223273, 300309, 324252, 3244999). SB acknowledges the Novo Nordisk Foundation (grants NNF17OC0027594 and NNF14CC0001) and RJFL is supported by grants from the National Institutes of Health (R01DK110113, R01DK124097) and the Novo Nordisk Foundation (NNF20OC0059313) and a Danish National Research Foundation Chair (DNRF161). BM thanks for third party funding from Informatics for Life (Klaus Tschira Foundation), the German Center for Cardiovascular Research (DZHK) and the German Research Foundation (DFG: CRC1550).
Footnotes
Conflicts of Interests
OAA is a consultant to CorTechs.ai, Biogen, Milken and received speaker’s honoraria from Lundbeck, Janssen, and Sunovion.
EM has received consultant honoraria from ALK, AstraZeneca, Chiesi, Sanofi and Viatris outside the submitted work.
SB has ownerships in Intomics A/S, Hoba Therapeutics Aps, Novo Nordisk A/S, Lundbeck A/S, ALK A/S and has had managing board memberships in Proscion A/S and Intomics A/S.
BJ has performed NIPT (fetal diagnostics) clinical diagnostic trials with Ariosa, Vanadis and Natera, with expenditures reimbursed per patient.
PWF is head of the Department of Translational Medicine at the Novo Nordisk Foundation, a non-profit philanthropic organization based in Denmark.
ÅJ, RJFL, CEW, HH, and BM have nothing to declare.
References
- 1.Denny JC, Collins FS. Precision medicine in 2030—seven ways to transform healthcare. Cell 2021; 184: 1415–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fioretos T, Wirta V, Cavelier L, et al. Implementing precision medicine in a regionally organized healthcare system in Sweden. Nat Med 2022; 28: 1980–2. [DOI] [PubMed] [Google Scholar]
- 3.Claussnitzer M, Cho JH, Collins R, et al. A brief history of human disease genetics. Nature 2020; 577: 179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.James SL, Abate D, Abate KH, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 Diseases and Injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018; 392: 1789–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burton PR, Clayton DG, Cardon LR, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007; 447: 661–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catalog GWAS. GWAS Catalog. The NHGRI-EBI Catalog of human genome-wide association studies. GWAS Cat 2022. [Google Scholar]
- 7.Yengo L, Vedantam S, Marouli E, et al. A saturated map of common genetic variants associated with human height. Nature 2022; 610: 704–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evangelou E, Warren HR, Mosen-Ansorena D, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet 2018; 50: 1412–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi SW, Mak TSH, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc 2020; 15: 2759–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lakeman IMM, Rodríguez-Girondo M, Lee A, et al. Validation of the BOADICEA model and a 313-variant polygenic risk score for breast cancer risk prediction in a Dutch prospective cohort. Genet Med 2020; 22: 1803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mavaddat N, Michailidou K, Dennis J, et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am J Hum Genet 2019; 104: 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carver T, Hartley S, Lee A, et al. Canrisk tool—A web interface for the prediction of breast and ovarian cancer risk and the likelihood of carrying genetic pathogenic variants. Cancer Epidemiol Biomarkers Prev 2021; 30: 469–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franks PW, Melén E, Friedman M, et al. Technological readiness and implementation of genomic-driven precision medicine for complex diseases. J Intern Med 2021; 290: 602–20. [DOI] [PubMed] [Google Scholar]
- 14.O’Sullivan JW, Raghavan S, Marquez-Luna C, et al. Polygenic Risk Scores for Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2022; 146: E93–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Widén E, Junna N, Ruotsalainen S, et al. How Communicating Polygenic and Clinical Risk for Atherosclerotic Cardiovascular Disease Impacts Health Behavior: an Observational Follow-up Study. Circ Genomic Precis Med 2022; 15: E003459. [DOI] [PubMed] [Google Scholar]
- 16.Wainschtein P, Jain D, Zheng Z, et al. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nat Genet 2022; 54: 263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kierczak M, Rafati N, Höglund J, et al. Contribution of rare whole-genome sequencing variants to plasma protein levels and the missing heritability. Nat Commun 2022; 13: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018; 50: 1219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai N, Revez JA, Adams MJ, et al. Minimal phenotyping yields genome-wide association signals of low specificity for major depression. Nat Genet 2020; 52: 437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis ACF, Green RC, Vassy JL. Polygenic risk scores in the clinic: Translating risk into action. Hum Genet Genomics Adv 2021; 2: 100047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wand H, Lambert SA, Tamburro C, et al. Improving reporting standards for polygenic scores in risk prediction studies. Nature 2021; 591: 211–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015; 518: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farooqi IS, Keogh JM, Yeo GSH, Lank EJ, Cheetham T, O’Rahilly S. Clinical Spectrum of Obesity and Mutations in the Melanocortin 4 Receptor Gene. N Engl J Med 2003; 348: 1085–95. [DOI] [PubMed] [Google Scholar]
- 24.Ruan Y, Lin YF, Feng YCA, et al. Improving polygenic prediction in ancestrally diverse populations. Nat Genet 2022; 54: 573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 2019; 51: 584–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanigawa Y, Qian J, Venkataraman G, et al. Significant sparse polygenic risk scores across 813 traits in UK Biobank. PLoS Genet 2022; 18: e1010105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ledford H Drug firm seeks genome bounty. Nature 2016; 532: 427. [DOI] [PubMed] [Google Scholar]
- 28.Van Hout CV, Tachmazidou I, Backman JD, et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 2020; 586: 749–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Backman JD, Li AH, Marcketta A, et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 2021; 599: 628–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halldorsson BV, Eggertsson HP, Moore KHS, et al. The sequences of 150,119 genomes in the UK Biobank. Nature 2022; 607: 732–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ameur A, Dahlberg J, Olason P, et al. SweGen: A whole-genome data resource of genetic variability in a cross-section of the Swedish population. Eur J Hum Genet 2017; 25: 1253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akbari P, Gilani A, Sosina O, et al. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science (80-) 2021; 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antman EM, Loscalzo J. Precision medicine in cardiology. Nat Rev Cardiol 2016; 13: 591–602. [DOI] [PubMed] [Google Scholar]
- 34.Haas J, Frese KS, Peil B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J 2015; 36: 1123–35. [DOI] [PubMed] [Google Scholar]
- 35.Jones D Modifying protein misfolding. Nat Rev Drug Discov 2010; 9: 825–7. [DOI] [PubMed] [Google Scholar]
- 36.Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med 2021; 385: 493–502. [DOI] [PubMed] [Google Scholar]
- 37.Spertus JA, Fine JT, Elliott P, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): health status analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2021; 397: 2467–75. [DOI] [PubMed] [Google Scholar]
- 38.Green EM, Wakimoto H, Anderson RL, et al. Heart disease: A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science (80-) 2016; 351: 617–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Judge DP, Lakdawala NK, Taylor MRG, et al. Long-Term Efficacy and Safety of ARRY-371797 (PF-07265803) in Patients With Lamin A/C–Related Dilated Cardiomyopathy. Am J Cardiol 2022; 183: 93–8. [DOI] [PubMed] [Google Scholar]
- 40.Melén E, Koppelman GH, Vicedo-Cabrera AM, Andersen ZJ, Bunyavanich S. Allergies to food and airborne allergens in children and adolescents: role of epigenetics in a changing environment. Lancet Child Adolesc Heal 2022; 6: 810–9. [DOI] [PubMed] [Google Scholar]
- 41.Tedner SG, Klevebro S, Bergström A, et al. Development of sensitization to peanut and storage proteins and relation to markers of airway and systemic inflammation: A 24-year follow-up. Allergy Eur J Allergy Clin Immunol 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Namjou B, Lape M, Malolepsza E, et al. Multiancestral polygenic risk score for pediatric asthma. J Allergy Clin Immunol 2022; 150: 1086–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Busse WW, Melén E, Menzies-Gow AN. Holy Grail: the journey towards disease modification in asthma. Eur Respir Rev 2022; 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackson DJ, Bacharier LB, Gergen PJ, et al. Mepolizumab for urban children with exacerbation-prone eosinophilic asthma in the USA (MUPPITS-2): a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet 2022; 400: 502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Björkander S, Merid SK, Brodin D, et al. Transcriptome changes during peanut oral immunotherapy and omalizumab treatment. Pediatr Allergy Immunol 2022; 33. [DOI] [PubMed] [Google Scholar]
- 46.Häfner H Descriptive psychopathology, phenomenology, and the legacy of Karl Jaspers. Dialogues Clin Neurosci 2015; 17: 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kendler KS. The Development of Kraepelin’s Mature Diagnostic Concepts of Paranoia (Die Verrücktheit) and Paranoid Dementia Praecox (Dementia Paranoides): A Close Reading of His Textbooks from 1887 to 1899. JAMA Psychiatry 2018; 75: 1252–60. [DOI] [PubMed] [Google Scholar]
- 48.Maj M, Stein DJ, Parker G, et al. The clinical characterization of the adult patient with depression aimed at personalization of management. World Psychiatry 2020; 19: 269–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maj M, van Os J, De Hert M, et al. The clinical characterization of the patient with primary psychosis aimed at personalization of management. World Psychiatry 2021; 20: 4–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sullivan PF, Geschwind DH. Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders. Cell 2019; 177: 162–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andreassen OA, Hindley GFL, Frei O, Smeland OB. New insights from the last decade of research in psychiatric genetics: discoveries, challenges and clinical implications. World Psychiatry 2023; 22: 4–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smeland OB, Andreassen OA. Polygenic risk scores in psychiatry – Large potential but still limited clinical utility. Eur Neuropsychopharmacol 2021; 51: 68–70. [DOI] [PubMed] [Google Scholar]
- 53.Fusar-Poli P, Manchia M, Koutsouleris N, et al. Ethical considerations for precision psychiatry: A roadmap for research and clinical practice. Eur Neuropsychopharmacol 2022; 63: 17–34. [DOI] [PubMed] [Google Scholar]
- 54.Manchia M, Pisanu C, Squassina A, Carpiniello B. Challenges and future prospects of precision medicine in psychiatry. Pharmgenomics Pers Med 2020; 13: 127–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salazar De Pablo G, Studerus E, Vaquerizo-Serrano J, et al. Implementing Precision Psychiatry: A Systematic Review of Individualized Prediction Models for Clinical Practice. Schizophr Bull 2021; 47: 284–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koutsouleris N, Dwyer DB, Degenhardt F, et al. Multimodal Machine Learning Workflows for Prediction of Psychosis in Patients with Clinical High-Risk Syndromes and Recent-Onset Depression. JAMA Psychiatry 2021; 78: 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fernandes BS, Williams LM, Steiner J, Leboyer M, Carvalho AF, Berk M. The new field of “precision psychiatry”. BMC Med 2017; 15: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nunes A, Ardau R, Berghöfer A, et al. Prediction of lithium response using clinical data. Acta Psychiatr Scand 2020; 141: 131–41. [DOI] [PubMed] [Google Scholar]
- 59.Sanfelici R, Dwyer DB, Antonucci LA, Koutsouleris N. Individualized Diagnostic and Prognostic Models for Patients With Psychosis Risk Syndromes: A Meta-analytic View on the State of the Art. Biol Psychiatry 2020; 88: 349–60. [DOI] [PubMed] [Google Scholar]
- 60.Murray GK, Lin T, Austin J, McGrath JJ, Hickie IB, Wray NR. Could Polygenic Risk Scores Be Useful in Psychiatry?: A Review. JAMA Psychiatry 2021; 78: 210–9. [DOI] [PubMed] [Google Scholar]
- 61.El-Hage W, Leman S, Camus V, Belzung C. Mechanisms of antidepressant resistance. Front Pharmacol 2013; 4 NOV: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Howes OD, Thase ME, Pillinger T. Treatment resistance in psychiatry: state of the art and new directions. Mol Psychiatry 2022; 27: 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lally J, MacCabe JH. Antipsychotic medication in schizophrenia: A review. Br Med Bull 2015; 114: 169–79. [DOI] [PubMed] [Google Scholar]
- 64.Pardiñas AF, Smart SE, Willcocks IR, et al. Interaction Testing and Polygenic Risk Scoring to Estimate the Association of Common Genetic Variants with Treatment Resistance in Schizophrenia. JAMA Psychiatry 2022; 79: 260–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smart SE, Agbedjro D, Pardiñas AF, et al. Clinical predictors of antipsychotic treatment resistance: Development and internal validation of a prognostic prediction model by the STRATA-G consortium. Schizophr Res 2022; 250: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Campos AI, Mulcahy A, Thorp JG, et al. Understanding genetic risk factors for common side effects of antidepressant medications. Commun Med 2021; 1: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kurki MI, Karjalainen J, Palta P, et al. FinnGen: Unique genetic insights from combining isolated population and national health register data. medRxiv 2022: 2022.03.03.22271360. [Google Scholar]
- 68.Millan MJ, Andrieux A, Bartzokis G, et al. Altering the course of schizophrenia: Progress and perspectives. Nat Rev Drug Discov 2016; 15: 485–515. [DOI] [PubMed] [Google Scholar]
- 69.Diabetes Prevention Program Research Group. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009; 374: 1677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McCarthy MI. Painting a new picture of personalised medicine for diabetes. Diabetologia 2017; 60: 793–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ahlqvist E, Storm P, Käräjämäki A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 2018; 6: 361–9. [DOI] [PubMed] [Google Scholar]
- 72.Wesolowska-Andersen A, Brorsson CA, Bizzotto R, et al. Four groups of type 2 diabetes contribute to the etiological and clinical heterogeneity in newly diagnosed individuals: An IMI DIRECT study. Cell Reports Med 2022; 3: 100477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dennis JM, Shields BM, Henley WE, Jones AG, Hattersley AT. Disease progression and treatment response in data-driven subgroups of type 2 diabetes compared with models based on simple clinical features: an analysis using clinical trial data. Lancet Diabetes Endocrinol 2019; 7: 442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nolan JJ, Kahkoska AR, Semnani-Azad Z, et al. ADA/EASD Precision Medicine in Diabetes Initiative: An International Perspective and Future Vision for Precision Medicine in Diabetes. Diabetes Care 2022; 45: 261–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chung WK, Erion K, Florez JC, et al. Precision medicine in diabetes: a Consensus Report from the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2020; 63: 1671–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loos RJF, Yeo GSH. The genetics of obesity: from discovery to biology. Nat Rev Genet 2022; 23: 120–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stefan N Metabolically Healthy and Unhealthy Normal Weight and Obesity. Endocrinol Metab 2020; 35: 487–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Acosta A, Camilleri M, Shin A, et al. Quantitative gastrointestinal and psychological traits associated with obesity and response to weight-loss therapy. Gastroenterology 2015; 148: 537–546.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fagherazzi G, Zhang L, Aguayo G, et al. Towards precision cardiometabolic prevention: results from a machine learning, semi-supervised clustering approach in the nationwide population-based ORISCAV-LUX 2 study. Sci Rep 2021; 11: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin Z, Feng W, Liu Y, et al. Machine Learning to Identify Metabolic Subtypes of Obesity: A Multi-Center Study. Front Endocrinol (Lausanne) 2021; 12: 843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang LO, Rauch A, Mazzaferro E, et al. Genome-wide discovery of genetic loci that uncouple excess adiposity from its comorbidities. Nat Metab 2021; 3: 228–43. [DOI] [PubMed] [Google Scholar]
- 82.Coral DE, Fernandez-Tajes J, Tsereteli N, et al. A phenome-wide comparative analysis of genetic discordance between obesity and type 2 diabetes. Nat Metab 2023: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rask-Andersen M, Karlsson T, Ek WE, Johansson Å. Genome-wide association study of body fat distribution identifies adiposity loci and sex-specific genetic effects. Nat Commun 2019; 10: 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Karlsson T, Rask-Andersen M, Pan G, et al. Contribution of genetics to visceral adiposity and its relation to cardiovascular and metabolic disease. Nat Med 2019; 25: 1390–5. [DOI] [PubMed] [Google Scholar]
- 85.Rask-Andersen M, Johansson Å. Illuminating the ‘healthy obese’ phenotype. Nat Metab 2023: 1–2. [DOI] [PubMed] [Google Scholar]
- 86.Sabbagh R, Van den Veyver IB. The current and future impact of genome-wide sequencing on fetal precision medicine. Hum Genet 2020; 139: 1121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci U S A 2008; 105: 16266–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dennis Lo YM, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. Lancet 1997; 350: 485–7. [DOI] [PubMed] [Google Scholar]
- 89.Bianchi DW. From prenatal genomic diagnosis to fetal personalized medicine: Progress and challenges. Nat Med 2012; 18: 1041–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dar P, Jacobsson B, MacPherson C, et al. Cell-free DNA screening for trisomies 21, 18, and 13 in pregnancies at low and high risk for aneuploidy with genetic confirmation. Am J Obstet Gynecol 2022; 227: 259.e1–259.e14. [DOI] [PubMed] [Google Scholar]
- 91.Iwarsson E, Jacobsson B, Dagerhamn J, Davidson T, Bernabé E, Heibert Arnlind M. Analysis of cell-free fetal DNA in maternal blood for detection of trisomy 21, 18 and 13 in a general pregnant population and in a high risk population – a systematic review and meta-analysis. Acta Obstet Gynecol Scand 2017; 96: 7–18. [DOI] [PubMed] [Google Scholar]
- 92.Dar P, Jacobsson B, Clifton R, et al. Cell-free DNA screening for prenatal detection of 22q11.2 deletion syndrome. Am J Obstet Gynecol 2022; 227: 79.e1–79.e11. [DOI] [PubMed] [Google Scholar]
- 93.Norton ME, Jacobsson B, Swamy GK, et al. Cell-free DNA Analysis for Noninvasive Examination of Trisomy. N Engl J Med 2015; 372: 1589–97. [DOI] [PubMed] [Google Scholar]
- 94.Lo YMD, Chan KCA, Sun H, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010; 2. [DOI] [PubMed] [Google Scholar]
- 95.Ingerslev HJ, Kesmodel US, Jacobsson B, Vogel I. Personalized medicine for the embryo and the fetus – Options in modern genetics influence preconception and prenatal choices. Acta Obstet Gynecol Scand 2020; 99: 689–91. [DOI] [PubMed] [Google Scholar]
- 96.Hu JX, Thomas CE, Brunak S. Network biology concepts in complex disease comorbidities. Nat Rev Genet 2016; 17: 615–29. [DOI] [PubMed] [Google Scholar]
- 97.Jørgensen IF, Aguayo-Orozco A, Lademann M, Brunak S. Age-stratified longitudinal study of Alzheimer’s and vascular dementia patients. Alzheimer’s Dement 2020; 16: 908–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Siggaard T, Reguant R, Jørgensen IF, et al. Disease trajectory browser for exploring temporal, population-wide disease progression patterns in 7.2 million Danish patients. Nat Commun 2020; 11: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Westergaard D, Moseley P, Sørup FKH, Baldi P, Brunak S. Population-wide analysis of differences in disease progression patterns in men and women. Nat Commun 2019; 10: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haendel MA, Chute CG, Robinson PN. Classification, Ontology, and Precision Medicine. N Engl J Med 2018; 379: 1452–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Allesøe RL, Lundgaard AT, Hernández Medina R, et al. Discovery of drug–omics associations in type 2 diabetes with generative deep-learning models. Nat Biotechnol 2023: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hicks AA, Pramstaller PP, Johansson Å, et al. Genetic determinants of circulating sphingolipid concentrations in European populations. Gibson G, ed. PLoS Genet 2009; 5: e1000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Demirkan A, van Duijn CM, Ugocsai P, et al. Genome-wide association study identifies novel loci associated with circulating phospho- and sphingolipid concentrations. Gibson G, ed. PLoS Genet 2012; 8: e1002490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Enroth S, Johansson Å, Enroth SB, Gyllensten U. Strong effects of genetic and lifestyle factors on biomarker variation and use of personalized cutoffs. Nat Commun 2014; 5: 4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Johansson Å, Enroth S, Palmblad M, Deelder AM, Bergquist J, Gyllensten U. Identification of genetic variants influencing the human plasma proteome. Proc Natl Acad Sci U S A 2013; 110: 4673–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jaenisch R, Bird A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33: 245–54. [DOI] [PubMed] [Google Scholar]
- 107.Javierre BM, Fernandez AF, Richter J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res 2010; 20: 170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schübeler D Function and information content of DNA methylation. Nature 2015; 517: 321–6. [DOI] [PubMed] [Google Scholar]
- 109.Besingi W, Johansson Å. Smoke-related DNA methylation changes in the etiology of human disease. Hum Mol Genet 2014; 23: 2290–7. [DOI] [PubMed] [Google Scholar]
- 110.Liang L, Willis-Owen SAG, Laprise C, et al. An epigenome-wide association study of total serum immunoglobulin e concentration. Nature 2015; 520: 670–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dick KJ, Nelson CP, Tsaprouni L, et al. DNA methylation and body-mass index: A genome-wide analysis. Lancet 2014; 383: 1990–8. [DOI] [PubMed] [Google Scholar]
- 112.Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science (80-) 2001; 293: 1068–70. [DOI] [PubMed] [Google Scholar]
- 113.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007; 447: 425–32. [DOI] [PubMed] [Google Scholar]
- 114.Strom JE, Merid SK, Pourazar J, et al. Chronic Obstructive Pulmonary Disease Is Associated with Epigenome-Wide Differential Methylation in BAL Lung Cells. Am J Respir Cell Mol Biol 2022; 66: 638–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van Breugel M, Qi C, Xu Z, et al. Nasal DNA methylation at three CpG sites predicts childhood allergic disease. Nat Commun 2022; 13: 7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nakayasu ES, Gritsenko M, Piehowski PD, et al. Tutorial: best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat Protoc 2021; 16: 3737–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Assarsson E, Lundberg M, Holmquist G, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One 2014; 9: e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Candia J, Cheung F, Kotliarov Y, et al. Assessment of Variability in the SOMAscan Assay. Sci Rep 2017; 7: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wishart DS. Metabolomics for investigating physiological and pathophysiological processes. Physiol Rev 2019; 99: 1819–75. [DOI] [PubMed] [Google Scholar]
- 120.Koelmel JP, Kroeger NM, Ulmer CZ, et al. LipidMatch: An automated workflow for rule-based lipid identification using untargeted high-resolution tandem mass spectrometry data. BMC Bioinformatics 2017; 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Uppal K, Walker DI, Liu K, Li S, Go YM, Jones DP. Computational Metabolomics: A Framework for the Million Metabolome. Chem Res Toxicol 2016; 29: 1956–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chaleckis R, Meister I, Zhang P, Wheelock CE. Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Curr Opin Biotechnol 2019; 55: 44–50. [DOI] [PubMed] [Google Scholar]
- 123.Bešević J, Lacey B, Conroy M, et al. New Horizons: the value of UK Biobank to research on endocrine and metabolic disorders. J Clin Endocrinol Metab 2022; 107: 2403–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hoyt L, O’Day EM. Perspective: A potential role for NUS in metabolite-based in vitro diagnostics. Magn Reson Chem 2021; 59: 257–63. [DOI] [PubMed] [Google Scholar]
- 125.Wigger L, Cruciani-Guglielmacci C, Nicolas A, et al. Plasma Dihydroceramides Are Diabetes Susceptibility Biomarker Candidates in Mice and Humans. Cell Rep 2017; 18: 2269–79. [DOI] [PubMed] [Google Scholar]
- 126.Laaksonen R, Ekroos K, Sysi-Aho M, et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur Heart J 2016; 37: 1967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Akhiyat N, Vasile V, Ahmad A, et al. Plasma Ceramide Levels Are Elevated in Patients With Early Coronary Atherosclerosis and Endothelial Dysfunction. J Am Heart Assoc 2022; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fretts AM, Jensen PN, Hoofnagle AN, et al. Circulating Ceramides and Sphingomyelins and Risk of Mortality: The Cardiovascular Health Study. Clin Chem 2021; 67: 1650–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Akawi N, Checa A, Antonopoulos AS, et al. Fat-Secreted Ceramides Regulate Vascular Redox State and Influence Outcomes in Patients With Cardiovascular Disease. J Am Coll Cardiol 2021; 77: 2494–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ek WE, Karlsson T, Höglund J, Rask-Andersen M, Johansson Å. Causal effects of inflammatory protein biomarkers on inflammatory diseases. Sci Adv 2021; 7: eabl4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Holmes MV, Asselbergs FW, Palmer TM, et al. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J 2015; 36: 539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen R, Mias GI, Li-Pook-Than J, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 2012; 148: 1293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jensen AB, Moseley PL, Oprea TI, et al. Temporal disease trajectories condensed from population-wide registry data covering 6.2 million patients. Nat Commun 2014; 5: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Laugesen K, Ludvigsson JF, Schmidt M, et al. Nordic health registry-based research: A review of health care systems and key registries. Clin Epidemiol 2021; 13: 533–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Aguayo-Orozco A, Haue AD, Jørgensen IF, et al. Optimizing drug selection from a prescription trajectory of one patient. npj Digit Med 2021; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Desikan RS, Fan CC, Wang Y, et al. Genetic assessment of age-associated Alzheimer disease risk: Development and validation of a polygenic hazard score. PLoS Med 2017; 14: e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kauppi K, Fan CC, McEvoy LK, et al. Combining polygenic hazard score with volumetric MRI and cognitive measures improves prediction of progression from mild cognitive impairment to Alzheimer’s disease. Front Neurosci 2018; 12: 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pihlstrøm L, Fan CC, Frei O, et al. Genetic Stratification of Age-Dependent Parkinson’s Disease Risk by Polygenic Hazard Score. Mov Disord 2022; 37: 62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.CC BY 4.0. https://creativecommons.org/licenses/by/4.0/