

Abstract
Protein N-glycosylation plays a critical role in a wide range of biological processes, and aberrant N-glycosylation is frequently associated with various pathological states. For global N-glycosylation analysis, N-glycans are typically released from glycoproteins mediated by endoglycosidases, primarily peptide N-glycosidase F (PNGase F). However, conventional N-glycan release by in-solution PNGase F is time-consuming and nonreusable. Although some immobilization methods can save time and reduce the enzyme dosage, including affinity interaction and covalent binding, the immobilized PNGase F by these traditional methods may compromises the immobilized enzyme's stability and biofunction. Therefore, a new approach is urgently needed to firmly and steadily immobilize PNGase F. To meet this demand, we have developed a spontaneous and site-specific way to immobilize PNGase F onto magnetic nanoparticles via Spy chemistry. The magnetic nanoparticles were synthesized and modified with SpyTag as a solid surface. The PNGase F fused with SpyCatcher can then be site-specifically and covalently immobilized onto this solid phase, forming a firm isopeptide bond via self-catalysis between the SpyTag peptide and SpyCatcher. Importantly, the immobilization process mediated by mild spy chemistry does not result in PNGase F inactivation; and allows immobilized PNGase F to rapidly release various types of glycans (high-mannose, sialylated, and hybrid) from glycoproteins. Moreover, the immobilized PNGase F exhibited good deglycosylation activity and facilitated good reusability in consecutive reactions. Deglycosylation of clinical samples was completed by the immobilized PNGase F as fast as several minutes.
Recombinant PNGase F was spontaneously, site-specifically, and covalently immobilized onto magnetic nanoparticles based on Spy chemistry. The immobilized PNGasae F exhibited excellent deglycosylation activity, thermostability, and reusability.
1. Introduction
N-Glycosylation is a major and conserved posttranslational modification in eukaryotes, with over half of all proteins undergoing N-glycosylation.1N-glycans can affect the function of modified proteins by influencing folding, half-life, and protein–protein interactions, thereby playing a role in various physiological events, including development, immunity, autophagy, and neurogenesis.2 Furthermore, aberrant glycosylation is involved in disease development and progression.3 Thus, efficient analysis of N-glycans and elucidation of N-glycan biological function is important to understand their role in physiological and pathological processes. To date, many analytical techniques, including NMR,4 capillary electrophoresis,5 liquid chromatography,6 and mass spectrometry,7 have been developed to efficiently analyze N-glycans. However, in the workflow of global N-glycosylation analysis, N-glycan removal by endoglycosidases, mainly peptide N-glycosidase F (PNGase F) with overnight incubation; prior to instrumental analysis is the main rate-limiting step. Traditional digestion methods require prolonged reaction times due to limited digestion efficiency.
There are many ways to shorten the reaction time, including microwave irradiation,8,9 high pressure-cycling technology (PCT),10 and enzyme immobilization.11,12 Among these strategies, immobilizing PNGase F onto a solid surface, especially the surface of magnetic particles, offers a straightforward and promising solution to shorten the incubation time, enhance protein stability and reduce enzyme consumption.12–15 Many method have been employed to immobilize enzymes onto particle surfaces, including absorption, affinity interactions, and covalent immobilization.16 To efficiently immobilize PNGase F, affinity interaction of the special tag coexpressed from fused protein and reactive groups modified on the surface of solid materials, including the hexahistidine – nickel,9 GST – GSH,17 and CBM – cellulose18 is the traditional way to immobilize PNGase F. However, the affinity interaction is derived from noncovalent forces such as hydrogen bonds, van der Waals forces, electrostatic bonds, and hydrophobic interactions, and the partial interactions may dissociate in the harsh deglycosylation environment. To construct more stable PNGase F conjugates, covalent linkages were reacted on functional groups from protein with modified reactive groups on the surface of solid phases, including amine,19 carboxyl,20,21 epoxy,22 cyanate ester,23 vinyl azlactone,24 and thiol–ene groups.25 Although the covalent immobilization of PNGase F can be achieved by the above reaction, the protein active domain and biofunction may be compromised and even inactivated after immobilization by these nonspecific reactions. Hence, site-selective and covalent protein conjugations are challenging since there are many free active groups in one protein chain. In a previous study, site-selective and covalent immobilization of PNGase F was mediated by an acyl transfer reaction between the glutamine tag of PNGase F and a free amine group on magnetic particles mediated by microbial transglutaminase.12 To further simplify the immobilization method without additional enzymes or chemical agents, spontaneous, covalent, and site-specific immobilization mediated by the SpyTag/SpyCatcher protein–ligase system was introduced to immobilize PNGase F.
The SpyTag/SpyCatcher protein–ligase system (spy chemistry), engineered from Streptococcus pyogenes, forms a covalent and stable isopeptide bond via self-catalysis between an aspartic acid residue of the SpyTag peptide (AHIVMVDAYKPTK) and a lysine residue of the SpyCatcher domain (PDB: 4MLI), withstanding harsh conditions such as a wide range of pH, temperatures, and detergents.26–28 Importantly, this spy chemistry is spontaneous, which can also minimize nonspecific protein immobilization as this reaction is highly efficient and specific.29,30 With these intriguing features, Spy chemistry, since it was reported in 2012,28 has been used in many applications, including protein ligation and protein immobilization in solid materials, including hydrogels,31 agarose,27 silica,32 gold nanoparticles,33 and magnetic nanoparticles.34,35 Therefore, Spy chemistry is an excellent approach to immobilize PNGase F, minimizing the risk of PNGase F inactivation in traditional covalent immobilization processes.
Although the immobilization of PNGase F has been widely studied, the spontaneous immobilization of this enzyme has yet to be conducted. In this trial, PNGase F was spontaneously, site-specifically, and covalently immobilized onto magnetic nanoparticles based on Spy chemistry. Fe3O4/SiO2 particles were synthesized and decorated with a reactive carboxyl group for SpyTag incorporation. Then, the modified magnetic particles can covalently and spontaneously immobilize the coexpression of SpyCatcher-PNGase F via Spy chemistry without additional enzymes or chemical agents. The immobilized SpyCatcher-PNGase F exhibited identical deglycosylation activity as its soluble counterpart, and showed good thermostability and reusability. Finally, immobilized SpyCatcher-PNGase F was employed in clinical samples to analyze its applications.
2. Materials and methods
2.1. Materials and reagents
Escherichia coli BL21 (DE3) competent cells, Escherichia coli DH5α competent cells, 2 × AceTaq Master Mix, and 2 × Phanta Max Master Mix were purchased from Vazyme (Nanjing, China). T4 liagese, Nde I, Nhe I, and corresponding buffers were purchased from Takara (Beijing, China). BCA Protein Assay Kit was bought from Beyotime Biotechnology (Shanghai, China). High Affinity Ni-charged resin FF and columns were obtained from GenScript (Nanjing, China). Ferric chloride hexahydrate (FeCl3·6H2O), sodium acetate (NaAc), ethylene glycol, ammonium hydroxide (NH4OH), carboxyethylsilanetriol sodium salt (CES), Tetraethyl orthosilicate (TEOS), isopropanol, N,N-dimethylformamide (DMF), succinic anhydride, IgG, Ribonuclease B (RNase B), transferrin, porous graphitic carbon (PGC), cellulose microcrystalline (MCC), imidazole, acetonitrile (ACN), dimethyl sulfoxide (DMSO), trifluoroacetic acid (TFA), N-methylmorpholine, methylamine hydrochloride, formic acid, acetic acid (HAc), 1-butanol, methyl alcohol, 2-aminobenzamide (2-AB), sodium cyanoborohydride (NaBH3CN), ethanol, and isopropyl-β-d-thiogalactopytanoside (IPTG) were purchased from Merck KgaA (Darmstadt, Germany). SpyTag peptide was purchased from Nanjing Peptide Biotech (Nanjing, China). The template DNA of PNGase F and SpyCatcher were obtained from laboratory storage. Human Serum was obtained from the school Hospital of Wuhan Sports University. All aqueous solutions were purified by a Milli-Q purification system (Merck, MA).
2.2. Preparation and characterization of magnetic nanoparticles
The magnetic nanoparticles modified with carboxyl groups were synthesized using a hydrothermal approach in the following steps.36 Firstly, FeCl3·6H2O (1.35 g) and NaAc (3.6 g) were dissolved in 40 mL ethylene glycol and mixed under vigorous stirring for 2 h. Then, the mixture was sealed in a Teflon-lined stainless-steel autoclave and heated at 200 °C for 8 h. After cooling to room temperature, the black products (magnetic nanoparticles, MNP) were washed alternately with ethanol and deionized water 5 times and dried at 60 °C for 12 h. Secondly, the black magnetic nanoparticles were dispersed into a mixture of ethanol, deionized water, NH4OH, and TEOS through ultrasonication, and the mixture was vigorously stirred at room temperature to produce the core–shell structured magnetic nanoparticles. After a 12 h reaction, the magnetic nanoparticles were separated with an external magnet and washed sequentially with deionized water and ethanol. Thirdly, 60 mg of magnetic nanoparticles was dispersed into isopropanol through ultrasonication and continuously stirred. The solution was then added dropwise with 300 μL of CES and constantly stirred for 24 h to obtain carboxyl-modified magnetic nanoparticles. Fourthly, the carboxyl-modified particles were stirred with 10% succinic anhydride in DMF solution under N2 protection for 3 h at room temperature, to introduce carboxyl groups to the surface. Finally, SpyTag peptides were further modified onto magnetic nanoparticles by EDC/NHS methods in previous studies.8,9 The corona structure of produced functionalized MNP was confirmed by transmission electron microscopy (TEM). Meanwhile, the MNP surface modification was confirmed by X-ray photoelectron spectroscopy (XPS) and dynamic light scattering (DLS).
2.3. Construction and expression of SpyCatcher-PNGase F
The gene encoding for SpyCatcher from Streptococcus pyogenes was used as the template and was amplified by PCR using specific primers (Table S1†). The PCR products were cloned into pET-28a-PNGase F vector between Nde I and Nhe I sites, resulting in plasmids encoding PNGase F proteins with an N-terminal SpyCatcher fusion. The constructed plasmids were transformed into E. coli DH5α competent cells and selected on Luria-Bertani (LB) agar plates containing 50 mg L−1 kanamycin, followed by bacterial culture and plasmid extraction. The recombinant plasmid was confirmed by sequencing and then transformed into E. coli BL21 (DE3) competent cells for the recombinant protein expression (SpyCatcher-PNGase F).
The cells were induced with 0.1 mM IPTG in LB medium with 0.05 mg L−1 kanamycin at 200 rpm at 16 °C for 16 hours. The induced cells were harvested by centrifugation at 8000 rpm and disrupted by high-pressure homogenizers to release expression proteins. The induced proteins were purified by Ni-charged resin according to the manufacturer's instructions and stored at −20 °C.
2.4. Spontaneous immobilization of SpyCatcher-PNGase F
The MNP@SpyTag (1 g) was resuspended in 1 mL tris-buffered saline (TBS, 50 mM tris–HCl, 150 mM NaCl, pH 7.0) containing 0.5 mg purified SpyCatcher-PNGase F. The two components were incubated at 4 °C for overnight under continuously shaking. In parallel, control MNP lacking SpyTag modification were also incubated with SpyCatcher-PNGase F under identical conditions. To eliminate nonspecific protein adsorption onto MNP@SpyTag, the MNP@SpyTag were separated by a magnet and subsequently washed three times with PBS buffer containing 0.5% (v/v) Tween 20. Finally, the immobilized SpyCatcher-PNGase F was separated by the use of a magnetic field and resuspended in PBS buffer before further analysis. The amounts of SpyCatcher-PNGase F immobilized on MNP were calculated using the BCA method by measuring the initial and final amount of soluble proteins in the solution.
2.5. Enzymatic activity of PNGase F
According to reported methods,13 to ensure molar equivalence (66.7 μM) for further N-glycan release, the standard glycoproteins were dissolved in deionized water in 1 (RNase B), 10 (IgG), and 5.33 (transferrin) mg mL−1 final concentrations, respectively. 10 μL of each sample was diluted with 10 μL deionized water and heat-denatured with 3 μL denaturing buffer (5% SDS and 400 mM DTT) for 10 min. After cooling on ice, 3 μL 500 mM sodium phosphate and 3 μL 10% NP40 were added to the solution. The reaction was started by adding 4 μL free PNGase F or immobilized SpyCatcher-PNGase F (protein per mL, 0.4 mg mL−1, respectively) in the mixer at 1000 rpm incubated at their optimal temperature for 2, 5, 10, and 40 min. Finally, the reaction was stopped by heat-denaturation or separated by a magnet. The protein samples were separated in 8–16% sodium dodecyl sulfate-polyacrylamide gel at 120 V for approximately 2 h to 3 h. Gels were stained with Coomassie Brilliant Blue R250 and destained in 30% (v/v) ethanol and 10% (v/v) acetic acid overnight. The human serum was detected by BCA method to measure the concentration of the whole proteins and then treated with free PNGase F or immobilized PNGase F as mentioned above.
2.6. N-Glycans purification
The released N-glycans were purified using PGC Solid Phase Extraction (SPE). Briefly, PGC SPE was washed sequentially with 5 mL of ACN, 3 mL of 80% ACN in H2O with 0.1% TFA (v/v), and 3 mL of 5% ACN in H2O with 0.1% TFA (v/v). The samples were mixed with 1 mL of 5% ACN in H2O with 0.1% TFA (v/v) and loaded onto PGC SPE. The column samples were flowed through the column at a rate of no faster than 1 drop per second, followed by washing 3 mL of 5% ACN in H2O with 0.1% TFA (v/v). Finally, the N-glycans were eluted with 1 mL of 50% ACN in H2O with 0.1% TFA (v/v) into a clean tube and dried using Speed Vap.
2.7. Analysis of N-glycan with UPLC
To detect N-glycans with UPLC, 2-AB was employed to label N-glycan. 2-AB and NaBH4CN were dissolved in 20 μL of DMSO/acetic acid (7/3, v/v) at 0.7 and 2.0 M concentrations, respectively. The mixtures were added to the dried N-glycans and incubated at 65 °C for 2 h, following by purification with MCC cartridges. Briefly, MCC cartridges were washed sequentially with 5.0 mL of H2O, and 3.0 mL of 1-butanol/ethanol/H2O (4 : 1 : 1, v/v/v). The derivated mixture was mixed with 1 mL of 1-butanol/ethanol/H2O (4 : 1 : 1, v/v/v) and loaded onto the MCC column. The samples were slowly flowed through the column, followed by washing with 3.0 mL of 1-butanol/ethanol/H2O (4 : 1 : 1, v/v/v). The labeled N-glycans were eluted with 1.0 mL of ethanol/H2O (1 : 1, v/v), and dried using Speed Vap for further use. The 2-AB derivatives were analyzed using a UPLC analytical system (Shimadzu, Nakagyo-ku, Kyoto, Japan) equipped with RF-20A fluorescence detector. The 2-AB labeled samples were loaded onto an ACQUITY UPLC BEH Amide column (100 mm × 2.1 mm i.d., 1.7 μm particle size, Waters, Milford, USA) at 40 °C using a binary gradient. A linear decreasing gradient of solvent B (95% ACN with 50 mM ammonium formate, pH 4.4) from 70% to 50% was used to elute samples at a flow rate of 0.1 mL min−1, using 5% ACN with 50 mM ammonium formate, pH 4.4 as Solvent A. The excitation/emission wavelengths were 324/417 nm for 2-AB derivatives. The enzymatic activity in the experiment was evaluated by quantifying the peak areas of N-glycans.
3. Results and discussion
3.1. Synthesis of the magnetic nanoparticles
Immobilization of proteins on magnetic nanoparticles (MNP) is a convenient and effective approach to separate immobilized proteins from the reaction. Herein, the MNP was synthesized as a typical core–shell structure consisting of three domains: an Fe3O4 core, a SiO2 middle shell, and a surface-modified carboxyl corona. The carboxyl was further grafted with SpyTag to immobilize the spycatcher protein spontaneously. The surface elemental profile of the MNP was examined by XPS analysis, and Fe 2p, O 1s, Si 2p, C 1s, and N 1s at binding energies of approximately 710.5, 532.4, 103.3, 284.6, and 400.1 eV were obtained for each step of the fabrication processes, respectively (Fig. 1). Primordially, the obtained elements of iron and oxygen confirmed the presence of Fe3O4 at binding energies of 710.5 eV and 532.4 eV. Then, the survey scan confirms the presence of silicon and elevated contents of carbon and oxygen after TEOS and CES modification at binding energies of 103.3 eV, 284.6 eV, and 532.4 eV, respectively. Meanwhile, the binding of TEOS and CES was also supported by a drastic decrease in the intensity of the characteristic Fe 2p peaks, indicating that all MNP in the composite was fully decorated. The grafted peptides were significantly confirmed by the presence of nitrogen at binding energy of 400.1 eV. The overall obtained element analysis corroborated the successful synthesis of SpyTag-modified MNP. Moreover, the zeta potentials of bare MNP, MNP@COOH, and MNP@SpyTag were also measured (Table S2†). The zeta potential of bare MNP was found to be approximately 9.8 ± 0.8 mV, which is consistent with previous results.37 After coating TEOS and CES on the surface of MNP, they exhibited a decreased zeta potential of −22.1 ± 0.6 mV because of the existence of surface COOH and OH groups.37,38 Meanwhile, the zeta potential of MNP@SpyTag underwent significant changes due to the amino acids with a positive charge from SpyTag, which increased to −0.2 ± 0.2 mV. Based on the above XPS and zeta potential results, it is obviously demonstrated that the surface of MNP has been successfully modified with SpyTag. Characteristic TEM images of modified MNP are shown in Fig. 1C. The observed sizes of the MNP were approximately 871.5 ± 88.2 nm, consistent with the result of a hydrodynamic diameter (Dh) in dynamic light scattering (DLS) (Table S2†). Thus, a robust magnetic nanoparticle platform was established for spontaneous enzyme immobilization.
Fig. 1. Schematic illustrates steps of magnetic nanoparticle preparation (A) and analysis by XPS (B) and TEM (C).

3.2. Expression and activity of SpyCatcher-PNGase F
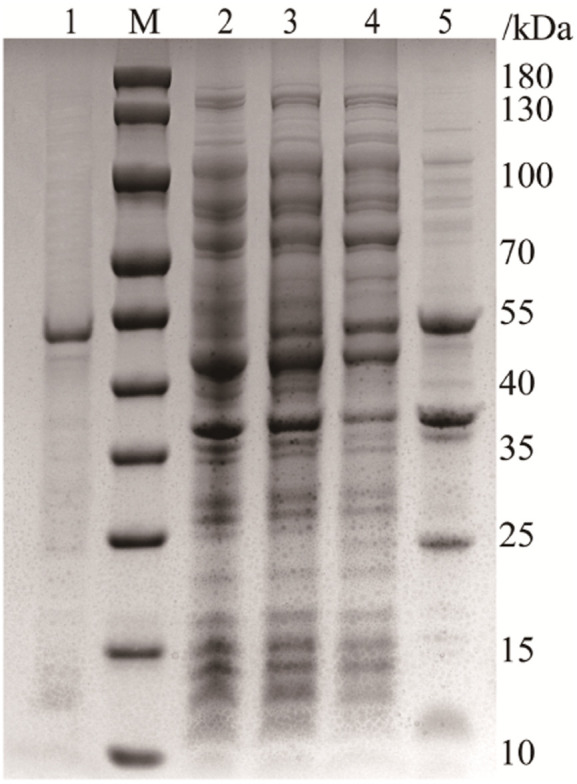
In this work, SpyCatcher was subcloned into the N-terminus of PNGase F for intracellular production of the fused proteins (Fig. S1†). The polypeptide chain at the N-terminus is comparably far from the active domain of PNGase F (PDB ID: 1PNF), and (Gly4Ser)3 was introduced as a flexible linker, allowing the catalytic centre to remain readily accessible.39 The successfully constructed plasmid was transformed into the expression strain BL21 (DE3), and the expression of the fused protein was induced by IPTG. The protein expression was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, Fig. 2), which showed an intense protein band at 52 kDa, consistent with the combined molecular mass of PNGase F (35 kDa), flexible linker (2 kDa), and SpyCatcher (15 kDa).40 In contrast to His-tagged PNGase F, which is mainly expressed in inclusion bodies,9 approximately 40% of SpyCatcher-PNGase F can be expressed in the supernatant and directly purified by affinity interaction, owing to the hydrophilic amino acids of SpyCatcher which can improve the solubility of the coexpressed proteins.
Fig. 2. SDS-PAGE analysis of the expression of SpyCatcher-PNGase F. Lanes M, 1–5: molecular weight standards, purified protein, total bacterial protein, bacterial protein induced with IPTG, soluble protein, and inclusion protein, respectively.
It is a traditional way to verify the activity of PNGase F by monitoring the pronounced mobility shift in gel shift assays. Three standard glycoproteins, RNase B, IgG, and transferrin, were used to analyze the deglycosylated activity of PNGase F. RNase B is a typical high-mannose glycoprotein that contains a single N-glycosylation site at Asn,34 with a molecular weight of approximately 15 kDa.41 IgG contained a single N-glycosylation site (its heavy chain, 50 kDa) occupied by complex N-glycans.42 Transferrin is attached to highly sialylated N-glycans with a molecular weight of approximately 80 kDa.43 The activities of SpyCatcher-PNGase F were identified with these three standard glycoproteins as substrates, using traditional overnight 37 °C digestion conditions.13 As shown in Fig. 3, the bands of deglycosylated substrates treated with similar amounts of enzymes were significantly shifted, indicating that SpyCatcher-PNGase F has the same deglycosylated activity as commercial PNGase F.
Fig. 3. SDS-PAGE analysis of RNase B (A), IgG (B), and transferrin (C) digested by PNGase F. Lane M: protein marker; lane N: glycoproteins; lanes P: digestion by commercial PNGase F, lane S: digestion by recombinant SpyCatcher-PNGase F; lane I: digestion by immobilized SpyCatcher-PNGase F.

3.3. Characterization of immobilized SpyCatcher-PNGase F
To verify the conjugation of SpyTag and SpyCatcher-PNGase F, mixing free SpyTag and SpyCatcher-PNGase F in TBS solution led to the spontaneous formation of a covalent complex that was stable against heat-denatured conditions in SDS solution.34 The molecular mass of this complex (53 kDa) was slightly larger than that of SpyCatcher-PNGase F (52 kDa), confirming that SpyTag was able to efficiently react with SpyCatcher-PNGase F under mild conditions (Fig. S2†). Furthermore, the conjugation of SpyTag/SpyCatcher manifested high selectivity and strong resistance against harsh washes or interference of nonfusion protein; conjugation of SpyCatcher-PNGase F from crude cell lysates could also be directly carried out by incubating MNP@SpyTag, bypassing protein purification processes. Due to these merits, SpyCatcher-PNGase F was spontaneously immobilized under mild conditions, avoiding the risk of PNGase F inactivation. In this study, we firstly performed simultaneous purification and immobilization of SpyCatcher-PNGase F from the culture supernatant. However, the protein concentration experienced minimal changes in this process (data not shown), rendering the quantification of the immobilized SpyCatcher-PNGase F challenging. To facilitate a more straightforward and efficient assessment of the amount of immobilized SpyCatcher-PNGase F, SpyCatcher-PNGase F was firstly purified using the affinity interaction between its co-expressed His-tag and Nickel resin. Subsequently, the purified protein was immobilized onto MNP@SpyTag. The spontaneously conjugated PNGase F amount was determined to be 61.5 ± 9.0 mg protein per g magnetic nanoparticles using the BCA method. To the best of our knowledge, this is the first platform for covalent, site-specific, and spontaneous immobilization of PNGase F.
The activity of immobilized SpyCatcher-PNGase F was also identified using the above three standard glycoproteins as substrates using traditional overnight 37 °C digestion conditions. As shown in Fig. 3, the band of deglycosylated substrates treated by similar amounts of enzymes was also significantly shifted, indicating that immobilized SpyCatcher-PNGase F has the same deglycosylated performance as in-solution SpyCatcher-PNGase F. Meanwhile, the released N-glycans from RNase B, IgG, and transferrin were labelled with 2-AB and separated by UPLC. The retention times and peak areas of the each glycan peaks, released by both SpyCatcher-PNGase F and immobilized SpyCatcher-PNGase F, were remarkably consistent (Fig. S3†), demonstrating that the immobilized enzyme possesses the capacity to release the completeness of N-glycan identical to the native enzyme without structural release bias. These results proved that the immobilized SpyCatcher-PNGase F have the performance to completely deglycosylate different kinds of glycoproteins as in-solution SpyCatcher-PNGase F.
Immobilization of SpyCatcher-PNGase F onto magnetic particles is a practical approach to improve its stability. The activities of the enzymes were evaluated by quantifying the peak areas of 2-AB-labelled Hex5HexNAc4Neu5Ac2 from transferrin. As shown in Fig. 4A, the optimal reaction temperature of immobilized SpyCatcher-PNGase F showed a higher activity at 52 °C compared to the optimal reaction temperature of in-solution SpyCatcher-PNGase F (42 °C). The thermal stability of the immobilized enzyme allows for greater yields at comparatively high temperatures. In addition, the deglycosylation kinetics of SpyCatcher-PNGase F and immobilized SpyCatcher-PNGase F were also investigated using a series of reaction times (2, 5, 10, and 40 min) with continuous stirring as previously described,13 and the results (Fig. 4B) showed somewhat different release characteristics for the two digestion techniques. With the use of immobilized SpyCatcher-PNGase F, a steeper increase in deglycosylation was obtained, and the release efficiency of immobilized SpyCatcher-PNGase F reached its maximum approximately 10 min incubation time, while the same level was reached at its optimal condition in only 40 min by in-solution digestion.
Fig. 4. The thermal stability (A) and release efficiency (B) of the relative fluorescent intensity of the average of the Hex5HexNAc4Neu5Ac2 from transferrin by SpyCatcher-PNGase F and immobilized SpyCatcher-PNGase F. The amount of N-glycan under optimal deglycosylation was set as 100%. Each result was repeated three times.

Furthermore, the bond between SpyCatcher-PNGase F and magnetic nanoparticles mediated by Spy chemistry enables the immobilized SpyCatcher-PNGase F to be reused multiple times. As depicted in Fig. 4B, the release efficiency of immobilized SpyCatcher-PNGase F reached half of its maximum value after a 5 minute incubation at 52 °C. Consequently, this specific condition was selected to assess the enzyme's reusability. As shown in Fig. 5, the immobilized SpyCatcher-PNGase F exhibiting a relative 78.5% deglycosylated activity after the fifth reuse (Fig. 5). This indicates the high reusability of immobilized SpyCatcher-PNGase F. The negligible decrease in released N-glycan in each experiment may be due to the inevitable loss of magnetic particles or aggregation during the reapplication process.12,13
Fig. 5. The reusability of immobilized SpyCatcher-PNGase F. Each result was repeated three times.
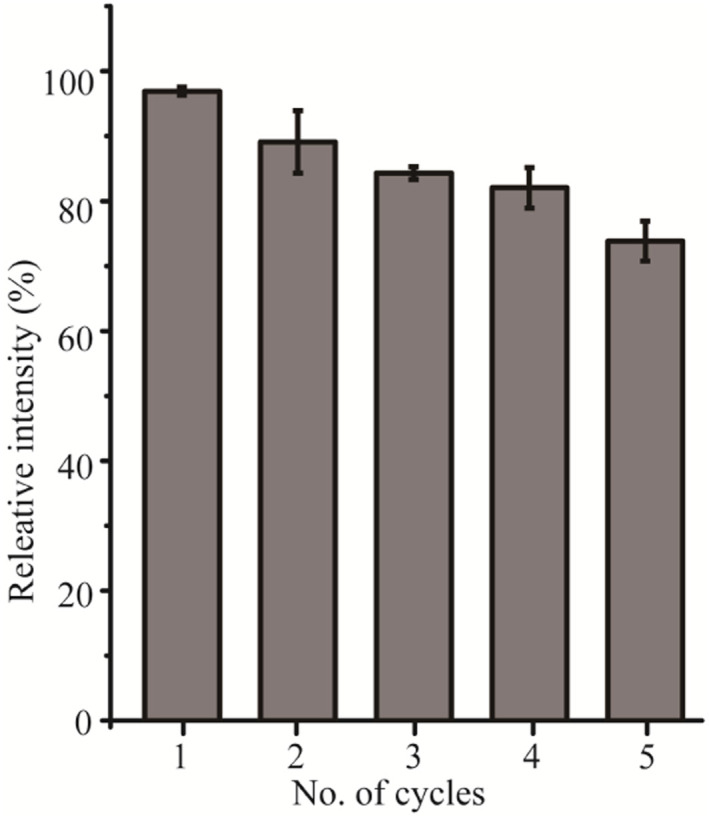
It is concluded that immobilized SpyCatcher-PNGase F possesses effective thermostability and reusability. Moreover, the natural attribution of immobilized PNGase F on magnetic nanoparticles can be easily removed from the reaction mixture assisted by magnetic partitioning, without affecting the rest of the workflow process.
3.4. The application of immobilized SpyCatcher-PNGase F
While the deglycosylation of standard glycoproteins by immobilized SpyCatcher-PNGase F appeared easy to achieve, clinical samples, human serum, were employed to determine its practical application. Human serum N-glycans are considered specific markers, and alteration in the serum N-glycan profile are related to ageing or various diseases.44,45 Therefore, N-glycan profiles of clinical samples were used to evaluate the activity of immobilized SpyCatcher-PNGase F. As shown Fig. 6, the retention time and relative intensities of each N-glycan released by immobilized SpyCatcher-PNGase F were almost the same as those of free SpyCatcher-PNGase F, indicating that the immobilized SpyCatcher-PNGase F mediated by Spy chemistry possesses the same deglycosylation activity as its free counterpart. Therefore, these findings suggest that immobilized SpyCatcher-PNGase F can be practically implemented to release N-glycans from numerous clinical samples, exhibiting comparable performance to the free enzyme.
Fig. 6. UPLC analysis of N-glycans released from human serum in-solution SpyCatcher-PNGase F and immobilized SpyCatcher-PNGase F. N-Acetyl glucosamine (HexNAc), blue ■; fucose (DeoxyHex), red ▼, mannose (Hex), green ●; galactose (Hex), yellow ●, and sialic acid (Neu5Ac), purple ◆.

4. Conclusion
This study presented a novel approach for immobilizing PNGase F onto magnetic nanoparticles via spy chemistry. This approach allows for spontaneous, site-specific, and covalent immobilization of SpyCatcher-PNGase F onto magnetic nanoparticles modified with SpyTag. The presence of hydrophilic amino acids in SpyCatcher facilitates the expression of PNGase F in the supernatant and its oriented immobilization onto the solid phase containing SpyTag, resulting in the immobilization of 61.5 ± 9.0 mg of SpyCatcher-PNGase F onto 1 g of SpyTag-modified magnetic nanoparticles. Notably, the immobilization process using Spy chemistry did not cause inactivation of the enzyme, allowing the immobilized SpyCatcher-PNGase F to exhibit good thermal stability and enzymatic activity. Furthermore, after five use cycles, the immobilized enzyme retained 78.5% activity. In summary, the results suggest that the spontaneous, covalent, and site-specific immobilization of SpyCatcher-PNGase F via Spy chemistry can be applied to many solid materials and has promising practical applications in N-glycan analysis.
Author contributions
Liang Zhang: investigation, methodology, validation, writing – original draft, funding acquisition. Wenhui Wang: methodology, software. Yueqin Yang: investigation, validation. Xiang Liu: data curation. Wenjie Zhu: formal analysis. Lingrui Pi: resources. Xin Liu: conceptualization, project administration, writing – review & editing, resources. Song Wang: supervision, project administration, funding acquisition.
Conflicts of interest
The authors state that there is no conflict of interest.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81827901), the 14th Five-Year-Plan Advantageous and Characteristic Disciplines (Groups) of Colleges and Universities in Hubei Province for Exercise and Brain Science from Hubei Provincial Department of Education in China, Natural Science Foundation of Hubei province (2022CFC069), Educational Commission of Hubei Province of China (Q20224102), the China Postdoctoral Science Foundation (2022M712486 and 2023T160500).
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04591a
References
- Hirata T. Kizuka Y. Adv. Exp. Med. Biol. 2021;1325:3–24. doi: 10.1007/978-3-030-70115-4_1. [DOI] [PubMed] [Google Scholar]
- Fahie K. Zachara N. E. J. Mol. Biol. 2016;428:3305–3324. doi: 10.1016/j.jmb.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmail S. Manolson M. F. Eur. J. Cell Biol. 2021;100:151186. doi: 10.1016/j.ejcb.2021.151186. [DOI] [PubMed] [Google Scholar]
- Noel M. Chasman D. I. Mora S. Otvos J. D. Palmer C. D. Parsons P. J. Smoller J. W. Cummings R. D. Mealer R. G. Clin. Chem. 2023;69:80–87. doi: 10.1093/clinchem/hvac160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagt S. de Haan N. Wang W. Zhang T. Wuhrer M. Lageveen-Kammeijer G. S. M. Anal. Chem. 2022;94:12954–12959. doi: 10.1021/acs.analchem.2c02840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C. Yu L. Yan J. Fu D. Yuan J. Liang X. Talanta. 2021;221:121382. doi: 10.1016/j.talanta.2020.121382. [DOI] [PubMed] [Google Scholar]
- Wu Y. Zhang Y. Li W. Xu Y. Liu Y. Liu X. Xu Y. Liu W. Talanta. 2022;249:123652. doi: 10.1016/j.talanta.2022.123652. [DOI] [PubMed] [Google Scholar]
- Liu X. Chan K. Chu I. K. Li J. J. Carbohydr. Res. 2008;343:2870–2877. doi: 10.1016/j.carres.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Zhang L. Wang C. Wu Y. Sha Q. Liu B. F. Lin Y. Liu X. J. Chromatogr. A. 2020;1619:460934. doi: 10.1016/j.chroma.2020.460934. [DOI] [PubMed] [Google Scholar]
- Szabo Z. Guttman A. Karger B. L. Anal. Chem. 2010;82:2588–2593. doi: 10.1021/ac100098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Wang P. Wang C. Wu Y. K. Feng X. J. Huang H. Ren L. J. Liu B. F. Gao S. Liu X. Sci. Rep. 2019;9:4865. doi: 10.1038/s41598-019-41378-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Wang W. Yang Y. Zhu W. Li P. Wang S. Liu X. Anal. Chim. Acta. 2023;1250:340972. doi: 10.1016/j.aca.2023.340972. [DOI] [PubMed] [Google Scholar]
- Bodnar J. Szekrenyes A. Szigeti M. Jarvas G. Krenkova J. Foret F. Guttman A. Electrophoresis. 2016;37:1264–1269. doi: 10.1002/elps.201500575. [DOI] [PubMed] [Google Scholar]
- Donohoo K. B. Wang J. Goli M. Yu A. Peng W. Hakim M. A. Mechref Y. Electrophoresis. 2022;43:119–142. doi: 10.1002/elps.202100199. [DOI] [PubMed] [Google Scholar]
- Matveeva V. G. Bronstein L. M. Nanomaterials. 2021;11:2257. doi: 10.3390/nano11092257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghraby Y. R. El-Shabasy R. M. Ibrahim A. H. Azzazy H. M. E. ACS Omega. 2023;8:5184–5196. doi: 10.1021/acsomega.2c07560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenkova J. Szekrenyes A. Keresztessy Z. Foret F. Guttman A. J. Chromatogr. A. 2013;1322:54–61. doi: 10.1016/j.chroma.2013.10.087. [DOI] [PubMed] [Google Scholar]
- Kwan E. M. Boraston A. B. McLean B. W. Kilburn D. G. Warren R. A. Protein Eng., Des. Sel. 2005;18:497–501. doi: 10.1093/protein/gzi055. [DOI] [PubMed] [Google Scholar]
- Qu Y. Xia S. Yuan H. Wu Q. Li M. Zou L. Zhang L. Liang Z. Zhang Y. Anal. Chem. 2011;83:7457–7463. doi: 10.1021/ac201665e. [DOI] [PubMed] [Google Scholar]
- Ren X. J. Bai H. H. Pan Y. T. Tong W. Qin P. B. Yan H. Deng S. S. Zhong R. G. Qin W. J. Qian X. H. Anal. Methods. 2014;6:2518–2525. [Google Scholar]
- Wei L. Zhang W. Lu H. Yang P. Talanta. 2010;80:1298–1304. doi: 10.1016/j.talanta.2009.09.029. [DOI] [PubMed] [Google Scholar]
- Bynum M. A. Yin H. Felts K. Lee Y. M. Monell C. R. Killeen K. Anal. Chem. 2009;81:8818–8825. doi: 10.1021/ac901326u. [DOI] [PubMed] [Google Scholar]
- Bidondo L. Festari F. Freire T. Giacomini C. Biotechnol. Appl. Biochem. 2022;69:209–220. doi: 10.1002/bab.2099. [DOI] [PubMed] [Google Scholar]
- Krenkova J. Lacher N. A. Svec F. J. Chromatogr. A. 2009;1216:3252–3259. doi: 10.1016/j.chroma.2009.02.036. [DOI] [PubMed] [Google Scholar]
- Lafleur J. P. Senkbeil S. Novotny J. Nys G. Bogelund N. Rand K. D. Foret F. Kutter J. P. Lab Chip. 2015;15:2162–2172. doi: 10.1039/c5lc00224a. [DOI] [PubMed] [Google Scholar]
- Li L. Fierer J. O. Rapoport T. A. Howarth M. J. Mol. Biol. 2014;426:309–317. doi: 10.1016/j.jmb.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J. Jia R. Wenge D. Sun H. Wang Y. Chang Y. Luo H. Biotechnol. Lett. 2021;43:1075–1087. doi: 10.1007/s10529-021-03098-x. [DOI] [PubMed] [Google Scholar]
- Zakeri B. Fierer J. O. Celik E. Chittock E. C. Schwarz-Linek U. Moy V. T. Howarth M. Proc. Natl. Acad. Sci. U. S. A. 2012;109:E690–E697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeble A. H. Howarth M. Chem. Sci. 2020;11:7281–7291. doi: 10.1039/d0sc01878c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakeri B. Howarth M. J. Am. Chem. Soc. 2010;132:4526. doi: 10.1021/ja910795a. [DOI] [PubMed] [Google Scholar]
- Sun F. Zhang W. B. Mahdavi A. Arnold F. H. Tirrell D. A. Proc. Natl. Acad. Sci. U. S. A. 2014;111:11269–11274. doi: 10.1073/pnas.1401291111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. Q. Zhang G. Y. ACS Appl. Nano Mater. 2020;3:44–48. [Google Scholar]
- Ma W. Saccardo A. Roccatano D. Aboagye-Mensah D. Alkaseem M. Jewkes M. Di Nezza F. Baron M. Soloviev M. Ferrari E. Nat. Commun. 2018;9:1489. doi: 10.1038/s41467-018-03931-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X. Ye Q. Wang C. W. Wu Y. Ma K. Yu S. Wei N. Gao H. ACS Appl. Mater. Interfaces. 2021;13:44147–44156. doi: 10.1021/acsami.1c14670. [DOI] [PubMed] [Google Scholar]
- Ye Q. H. Jin X. Y. Gao H. F. Wei N. ACS Appl. Bio Mater. 2022;5:5665–5674. doi: 10.1021/acsabm.2c00709. [DOI] [PubMed] [Google Scholar]
- Deng H. Li X. Peng Q. Wang X. Chen J. Li Y. Angew Chem. Int. Ed. Engl. 2005;44:2782–2785. doi: 10.1002/anie.200462551. [DOI] [PubMed] [Google Scholar]
- Ulu A. Noma S. A. A. Koytepe S. Ates B. Appl. Biochem. Biotechnol. 2019;187:938–956. doi: 10.1007/s12010-018-2853-9. [DOI] [PubMed] [Google Scholar]
- Li K. X. Zeng Z. X. Xiong J. J. Yan L. S. Guo H. Q. Liu S. F. Dai Y. H. Chen T. Colloids Surf., A. 2015;465:113–123. [Google Scholar]
- Kovacs N. Farsang R. Szigeti M. Vonderviszt F. Jankovics H. Mol. Biotechnol. 2022;64:914–918. doi: 10.1007/s12033-022-00464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer J. A. Ruta A. Therien A. M. West J. L. Biomacromolecules. 2019;20:2486–2493. doi: 10.1021/acs.biomac.9b00183. [DOI] [PubMed] [Google Scholar]
- Wu Y. Wang C. Luo J. Liu Y. Zhang L. Xia Y. Feng X. Liu B. F. Lin Y. Liu X. Anal. Bioanal. Chem. 2017;409:4027–4036. doi: 10.1007/s00216-017-0346-2. [DOI] [PubMed] [Google Scholar]
- Liu S. Liu X. Adv. Clin. Chem. 2021;105:1–47. doi: 10.1016/bs.acc.2021.02.001. [DOI] [PubMed] [Google Scholar]
- Wang C. Wu Y. Liu S. Zhang L. Liu B. F. Liu X. Anal. Chim. Acta. 2020;1131:56–67. doi: 10.1016/j.aca.2020.07.044. [DOI] [PubMed] [Google Scholar]
- Vilaj M. Lauc G. Trbojevic-Akmacic I. Glycobiology. 2021;31:2–7. doi: 10.1093/glycob/cwaa047. [DOI] [PubMed] [Google Scholar]
- Yoshida Y. Furukawa J. I. Naito S. Higashino K. Numata Y. Shinohara Y. Proteomics. 2016;16:2747–2758. doi: 10.1002/pmic.201500550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.