

Abstract
Cell-specific microRNA (miRNA) expression estimates are important in characterizing the localization of miRNA signaling within tissues. Much of these data are obtained from cultured cells, a process known to significantly alter miRNA expression levels. Thus, our knowledge of in vivo cell miRNA expression estimates is poor. We previously demonstrated expression microdissection-miRNA-sequencing (xMD-miRNA-seq) to acquire in vivo estimates, directly from formalin-fixed tissues, albeit with limited yield. Here we optimized each step of the xMD process, including tissue retrieval, tissue transfer, film preparation, and RNA isolation, to increase RNA yields and ultimately show strong enrichment for in vivo miRNA expression by qPCR array. These method improvements, including the development of a non-crosslinked ethylene vinyl acetate (EVA) membrane, resulted in a 23–45 fold increase in miRNA yield, depending on cell type. By qPCR, miR-200a was increased 14-fold in xMD-derived small intestine epithelial cells, with a concurrent 336-fold reduction in miR-143 relative to the matched non-dissected duodenal tissue. xMD is now an optimized method to obtain robust in vivo miRNA expression estimates from cells. xMD will allow archival formalin-fixed tissues from surgical pathology archives to make theragnostic biomarker discoveries.
Keywords: tissue dissection, microRNA, methods
Background:
microRNAs (miRNAs), small noncoding RNAs, are essential regulators of mRNA translation into proteins. miRNAs are intrinsic regulators of cellular physiology and have been linked to multiple pathologies through expression level changes in tissues1–3. However, tissues are diverse landscapes of multiple cell types, all contributing to the miRNAome of that tissue4–8. The small intestine, for instance, has numerous discrete cell types of epithelial, endothelial, and inflammatory lineages9. It would be ideal for identifying the miRNA expression pattern of each cell type independently. Cell culture is frequently used for this task. However, culturing and passaging cells dramatically alter miRNA expression patterns10. Therefore, when obtainable from cell culture, the miRNA expression is not a perfect proxy to in vivo cellular expression patterns. Additionally, many cell types, such as cardiomyocytes and neurons, do not grow effectively in culture. There is currently a need for an effective and efficient method to isolate cells directly from tissues to best approximate in vivo miRNA expression patterns.
Two commonly used methods, laser capture microdissection, and flow cytometry, exist for quantifying expression levels of miRNAs within distinct cell subsets. Laser capture microdissection is perhaps the most global and accurate, but it is best performed with frozen tissues, which are limiting for human studies11. Also, it is laborious and requires technical expertise and expensive machinery. Flow cytometry with cell capture, like laser capture, requires expensive equipment and is somewhat limited by the available antibodies to mark different non-immune cell types12. The method to dissociate cells has long preparation times, during which cell stress increases expression alterations5. These concerns highlight the need for new microdissection techniques to retrieve the near in vivo miRNA expression signatures. Single-cell or single nuclei approaches exist for miRNAs (ex. Smart-seq-total, but even the best methods yield only ~15,000 miRNA reads/cell 13,14. This might work for the differentiation of cell types but not for deep discoveries of all miRNAs present in a given cell type.
Expression microdissection (xMD) is a method to rapidly and cost-effectively microdissect cells directly from formalin-fixed tissues on routine glass slides15,16. This method involves pigmenting a specific cell type by standard immunohistochemistry on formalin-fixed, paraffin-embedded tissues. An ethylene vinyl acetate (EVA) membrane is tightly affixed to the slide. A flash lamp is then used, where the pigmented cells convert the light to heat, focally melting and adhering the cells to the EVA membrane, which can be removed with the transferred material. We previously introduced xMD-miRNA-seq as an extension of the method to specifically obtain the miRNA signature of intestinal epithelial cells17. In our initial development of xMD-miRNA-seq, we noted an 80-fold reduction in RNA from an unprocessed slide to the final xMD membrane-obtained material during the xMD steps and a low percentage of miRNA reads from the sequencing library preparation. Here we present an optimized method of xMD to assay miRNAs where we have substantially optimized the RNA collection for the purpose of qPCR array. We demonstrate a step-by-step approach to evaluate each facet of the method to increase specificity and RNA yield. These improvements have increased the opportunities to utilize xMD on archival formalin-fixed tissues from surgical pathology files. This method can be used on both common and rare varieties of cell types to best understand the in vivo expression patterns of miRNAs in health and disease toward more accurate tissue-based biomarkers.
Methods
Procurement of Human Tissue.
Sections of the duodenum (small intestine) were procured from pancreatoduodenectomy specimens. Heart tissue was collected from an orthotopic heart transplant case in an expedited fashion in the surgical pathology suite at The Johns Hopkins Hospital. Johns Hopkins Investigational Review Board (IRB) approval was given (NA_00001584 and NA_00070313), and patient consent was obtained for the use of these tissues.
Experiments were performed in accordance with our guidelines, including anonymization upon receipt. Specimens were formalin-fixed for 24 hours, followed by standard processing and paraffin embedding. Four-micron (μm) sections were placed on Superfrost Plus slides (Fisherbrand, Cat No. 12–550-15, Waltham, MA, USA) and stored at −80°C until use.
Evaluating RNA loss at different steps in the IHC protocol.
Prior to optimizing the full protocol, we performed a general analysis of multiple steps of the original IHC protocol to determine key steps that caused significant RNA loss. Slides underwent one or more steps of baking, antigen retrieval by either high-temperature antigen retrieval (HTAR, in EDTA or citrate) or proteinase K (PK, 10 or 20 minutes [min]), primary antibody staining (AE1/AE3, Bio SB, Cat No: BSB 5432, Goleta, CA, USA), and or Poly linker (secondary antibody) staining. Following specific steps, heart tissue slides were scraped using a razor blade, and tissue was collected for RNA processing as described below. Variations including either 5 or 15 min of HTAR, either EDTA or citrate for the HTAR and the presence or absence of one of two RNAse inhibitors (1 – Millipore Sigma, Protector RNAse Inhibitor, Cat. No. 3335399001, Burlington, VT, USA; 2 – NEB, RNAse Inhibitor, M0314S, Ipswich, MA, USA). The RNA was then extracted from the tissue using the miRNeasy kit (Qiagen Cat. No. 217084, Germantown, MD, USA). The amount of RNA was evaluated using qPCR for hsa-miR-133 relative to a Cel-miR-39 spike-in.
Global Expression Microdissection Protocol.
The complete, final version of the protocol is given herein, with the experimentally modified steps noted in Fig. 1 and fully described in the supplementary methods(A–G). The slides were deparaffinized before staining by heating at 60°C for 20 min (Thermobrite StatSpin system, Des Plaines, IL, USA) and then washed in 3 xylene baths for 5 min each (Macron, ACS grade, Radnor, PA, USA), 2 ethanol baths for 3 min each (Pharmco, Cat No: 111000200, Shelbyville, KY, USA), followed by 3 min in 90%, and 3 min in 80% ethanol respectively. The slides were placed in a citrate solution (Bio SB, Cat No: BSB 0020), and antigen was retrieved using a high pressure high temperature (HTAR) method with a pressure cooker (Cuisinart, Stamford, CN, USA). The entire HTAR process included 20 min of ramp-up time, 1 min at full pressure and temperature, and 7 min of cool-down time (A). The slides were treated with peroxide blocker (Bio SB, PolyDetector Plus) for 5 min. For epithelial cell staining, the primary antibody was anti-AE1/AE3 (anti-pancytokeratin) (Bio SB, Cat No: BSB 5432) at a 1:100 dilution for 45 min. For endothelial cells, anti-CD31 (Bio SB, Cat No: BSB 5223) was used at a 1:75 dilution for 60 min. To each antibody solution, 15μl of RNASecure (Thermo Fisher, Cat No. AM7006) per ml was added (B). Primary antibodies were washed off with immuno-wash and treated with Poly linker and Poly HRP for 15 min each with washes in between. The slides were treated with the chromogen 3,3’-Diaminobenzidine (DAB) for 10 min (Biocare Medical, Cat No: DB801, Pachecho, CA, USA) (C). The slides were washed again with immuno-wash (Bio SB PolyDetector Plus) then dehydrated with serial ethanol baths of increasing concentrations followed by 3 xylene baths. No counterstaining or coverslipping was performed. After staining, slides were stored at −80°C until use.
Figure 1.
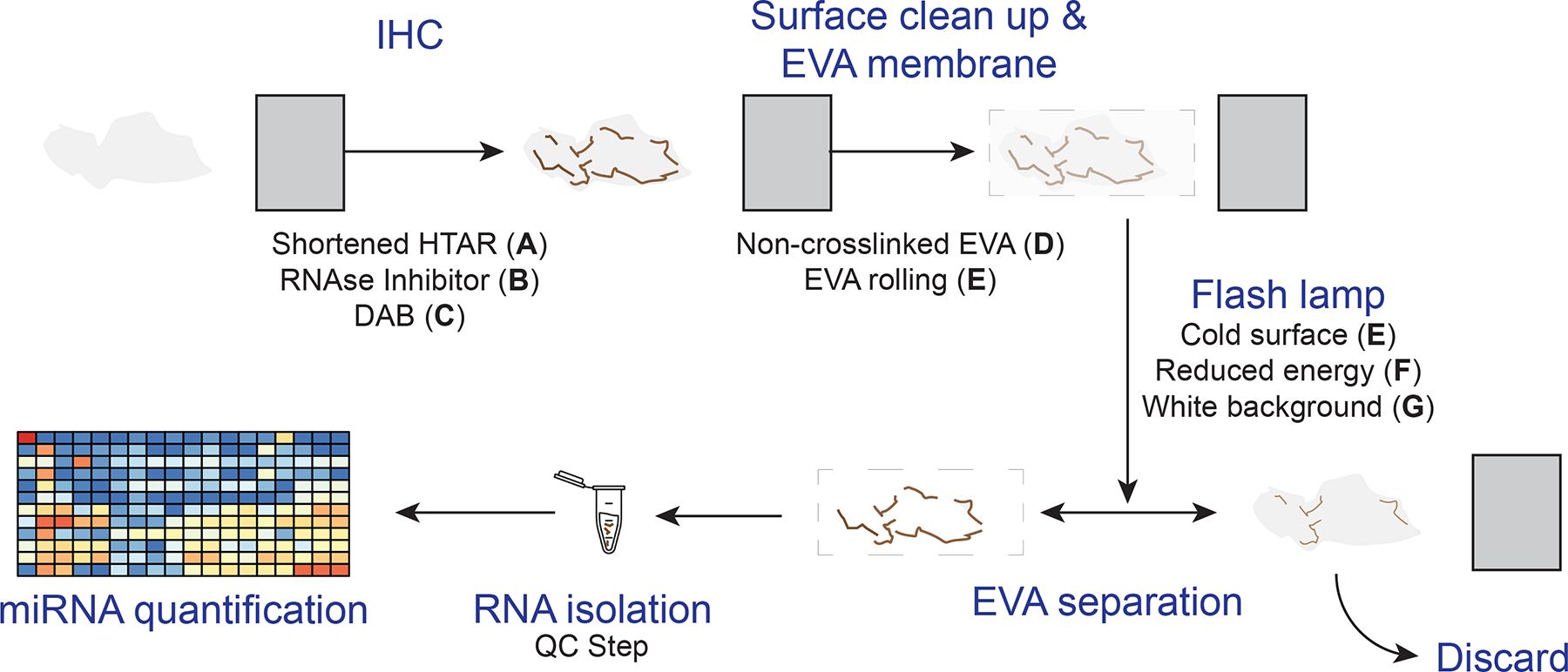
The general xMD method with improvements. A standard slide is generated from a formalin-fixed paraffin-embedded block on which IHC is performed for a cell type of interest. After IHC, the surface is cleared of ambient RNA, and an EVA membrane is tightly opposed. A flash lamp introduces light absorbed by pigmented cells that focally adhere to the EVA membrane. Then, the EVA membrane is dissolved to isolate RNA from the specific cell type for miRNA quantification. Improvements, described in this study in separate sections of the methods, were to shorten the HTAR time (A); add an RNAse inhibitor with the primary antibody (B); use DAB (C); create and use a noncrosslinked EVA membrane (D); and improve RNA isolation specificity by clearing the surface with EVA rolling (E) and using a cold surface (E); reducing the energy of the flash lamp (F), and using a white background for the flash (G). A quality control step before miRNA quantification was added. DAB. 3,3’-diaminobenzidine; EVA. ethylene vinyl acetate; HTAR, high-temperature antigen retrieval: IHC. immunohistochemistry; xMD. expression microdissection.
The xMD nucleic acid material isolations from tissues were performed using a SensEpil flash lamp (HomeSkinovations, AS101500A, Yokneam Illit, Israel), a food saver storage system (FoodSaver, v3835, Rye, NY, USA), and fullerene impregnated Ethylene Vinyl Acetate (EVA) (D). Stained slides were covered with an initial trimmed EVA membrane placed on the tissue and pressed down using a wooden dowel. A second EVA membrane was then sealed against the slide using the FoodSaver vacuum system to tightly oppose the two (E). Then a wet western blot sponge (Thermo Fisher, Cat No. EI9052) was placed on the vacuum bag. The flash lamp was placed on top of the sponge and flashed 5 times at the intensity 4 (of 5) over a shiny white background (F, G). The vacuum bag was opened, the slide/EVA removed, and the EVA membrane containing the transferred tissue was gently detached and placed in a low retention 1.5 ml microcentrifuge tube (Thermo Fisher, Cat No. 3446PK) for digestion.
After xMD, two membranes were placed in each sample tube and frozen at −80°C overnight, allowing a freeze/thaw to occur, which improved the dissolution of the membrane. 300 μl of Protein Kinase Digestion (PKD) buffer (Qiagen, Cat. No. 19131) was added to sample tubes to cover the membranes, more was added if the membranes were not covered. Membranes were incubated with 10 μl proteinase K (>600 mAU/ml) at 56°C for 30 min, followed by 15 min at 80°C to deactivate the enzyme. Afterwards, the samples were treated with DNase for 15 min at room temperature. Then, membranes were incubated for 5 min with one volume (generally 310 μl) of phenol:chloroform (Sigma, Cat. No. P3803). The membrane backers were removed from the tube with tweezers, leaving the mostly dissolved EVA membranes and tissue behind. The samples were incubated at room temperature for 1 hour, under aggressive agitation, then centrifuged for 30 min at max speed (16 000 rpm) before removing the chloroform layer. A 15 min incubation with 20 μl of proteinase K at 56°C was followed by a 5 min incubation on ice. Another volume of phenol:chloroform was added, and the samples were incubated for 5 min at room temperature. The samples were centrifuged for 30 min at max speed (16 000 rpm) in a tabletop centrifuge, and the supernatant (aqueous phase) was transferred to a new tube. Then one volume of isopropyl alcohol (VWR, Cat. No. 0918, Radnor, PA, USA) was added to the aqueous phase and incubated at −20°C for 3 hours to overnight. The samples were centrifuged for 30 min at 16 000 rpm. The supernatant was discarded, and the pellet washed with ethanol twice. The RNA was then resuspended in 20 μl of RNAse-free water (Invitrogen, 10977–015, Waltham, MA, USA). The samples, including the DNAse step, were cleaned with the Zymo Research Clean & Concentrator-5 Kit (Cat No. R1015, Irvine, CA, USA). The RNA (total) was stored at −80°C until use.
qPCR method for verifying optimization steps. According to manufacturer specifications, cDNA was synthesized using miScript II RT Kit (Qiagen, Cat No. 218161).
qPCR was performed using miScript assays (Qiagen, Cat No: 218075) The cDNA was diluted to approximately 40 ng/ul. A master mix was made for all wells with SYBR green master mix with universal primers. Then the mix was aliquoted for each primer, hsa-let-7a, hsa-miR-101 (or has-miR-222), hsa-miR-128, hsa-miR-133 and cel-miR-39 as appropriate for the experiment. The specific primers were added to the smaller master mixes. For each treatment, the qPCR was performed in quadruplicate. In each well, there was 160 ng cDNA at a concentration of 40 ng/μl. The reaction volume was 25 μl. The thermocycler used the following program: 40 cycles of 95°C for 15 seconds, 55°C for 30 seconds, and then 70°C for 30 seconds. The ΔΔ Ct value was calculated relative to the spike in and normalized to the slides that did not undergo IHC.
qPCR arrays for verifying overall optimization.
Human miRNA TLDA cards (Thermo Fisher, Cat No. 44913) were used for the global analysis of 384 miRNAs. The RNA extraction was performed as above. The RT was performed using Thermo Fisher Megaplex Primer Pools (Cat no. 4444750) and TaqMan MicroRNA Reverse Transcription Kit (Cat No. 4366596). The RT was followed by a preamplification step using Taqman PreAmp Master Mix (Cat. No. 4488593) and Megaplex PreAmp primers (Cat No. 444750) The array card was then prepared using TaqMan Universal Master Mix II (Cat. No. 444043) and run on a Quantstudio 21k, using the standard protocol (Thermo Fisher, publication 4399813).
Data were analyzed in Microsoft Excel. All controls were checked to assess the quality of work. The data were normalized using the average of three miRNAs known to have a steady expression in most cell types (miR-21, miR-22, and miR-103)8. All three were assessed to be expressed evenly in all cell types. The CT values for all three miRNAs were averaged for both the tissue scrape and xMD samples. This value was then used to calculate the ΔCT value for all 384 miRNAs in the plate, excluding miRNAs that did not have amplification on any of the sample plates. The 2−ΔΔCT value was then calculated between the tissue scrapes and xMD samples. The fold change was then graphed for each of the miRNAs. Statistical analysis was performed using a standard two-variable T-test between each miRNA or treatment individually.
Results
Identifying the IHC protocol steps with the largest impact on RNA degradation
There are a number of IHC steps in the xMD process which might be responsible for the loss of RNA through degradation. Therefore, we evaluated these steps by comparing levels of the miRNA let-7a normalized to a cel-miR-39 spike-in, by qPCR, stopping the method after different steps of the IHC process and with varying conditions. Three different antigen retrieval techniques were evaluated on cardiac tissues: proteinase K (PK) digestion, HTAR with EDTA buffer, and HTAR with citrate buffer. The method was stopped at points including just after HTAR, after primary antibody, or after the complete IHC protocol. At each stopping point, the slides were scraped of tissue material, RNA was isolated, and qPCR was performed. All miRNA Ct results were compared via fold change to the control unbaked slides. PK digestion for antigen retrieval showed a slight increase (1.4 fold) in let-7a retention (Fig. 2a). Experiments stopping after HTAR with EDTA or citrate showed between a 2- and 15-fold loss of let-7a depending on the length (5 or 15 min) of antigen retrieval. A full IHC protocol using EDTA HTAR resulted in an over 80-fold loss of let-7a. This was abrogated by using two different RNase inhibitors that reduced the loss to ~40–50 fold (Fig. 1a). Notably, the proteinase K method released more miRNA initially, but this miRNA appeared to be lost in later steps. This experiment indicated that the more significant loss of RNA occurred during the staining steps, rather than the antigen retrieval steps, but that both steps could be optimized to reduce RNase activity.
Figure 2.
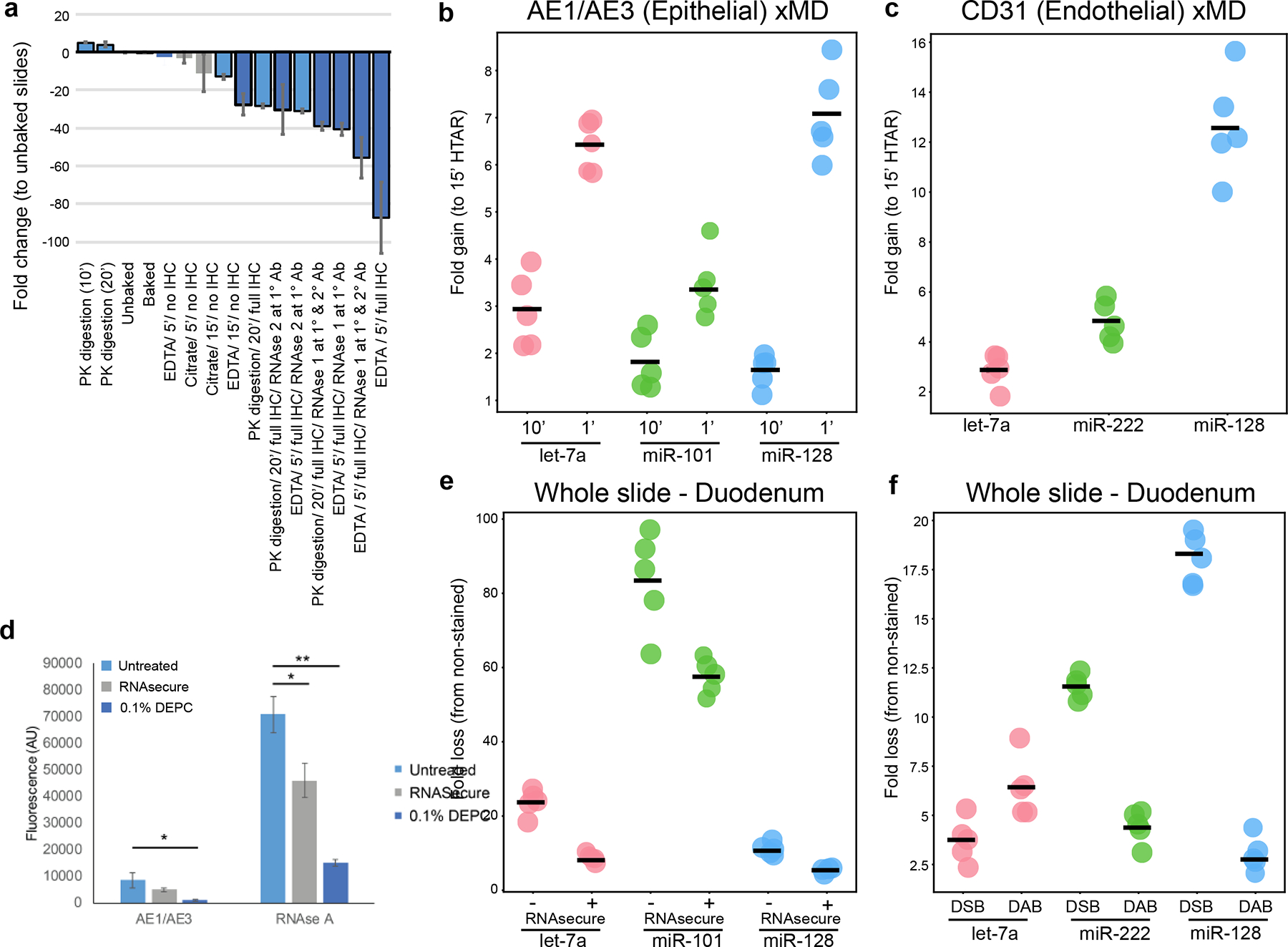
The RNA fold loss of multiple steps was compared. (A) Comparison of RNA fold loss between the steps of IHC. The fold change is a loss of let-7a expression relative to an unexperimented on tissue scrape from a matched slide. Antigen retrieval methods (Millipore Sigma) and RNAse inhibitors (NEB) were evaluated. RNA was collected after antigen retrieval steps, using a primary antibody, and at the end of the full IHC experiment. More than 80-fold RNA loss was observed with the standard protocol. (B) Increase of miRNA expression in xMD-obtained AE1/AE3* cells with shorter HTAR lengths (1 and 10 min vs 15 min). (C) Increased miRNA expression in xMD-obtained CD31+ cells with a shorter HTAR length (10 min vs 15 min). (D) RNAse A and 0.1% DEPC reduced RNAse activity (measured in arbitrary fluorescence units) in AEl/AE3-stained slide material or in material with an RNAse A spike-in. *P < .01. **P < .001. (E) Whole slides, either untreated (−) or treated (+) with RNAsecure during IHC. were compared with slides not treated. miRNA loss was greater for 3 representative miRNAs, Let-7a, miR-101, and miR-128, when RNAsecure was not used. (F) Slides treated with Deep Space Black (DSB) or DAB were compared with slides not treated with a chromogen. Overall. DSB had a more significant loss of miRNA. DAB. 3.3’-diaminobenzidine; HTAR, high-temperature antigen retrieval; IHC. immunohistochemistry; xMD. expression microdissection.
Shortened High-Temperature Antigen Retrieval Improved the miRNA Yield:
In our initial evaluation, we noted differences in miRNA abundance at the HTAR step. Therefore, we varied the antigen retrieval time between 1 min, 10 min, and 15 min in an attempt to reduce the loss of RNA due to high-temperature degradation or miRNA diffusion. Each HTAR method had an additional 22 min of heat up/cool down time. Thus they represented 28, 32, and 42 min of full experimental time. The staining intensity for AE1/AE3 was equivocal at all HTAR lengths. The shortest HTAR had the most retained miRNA, with, on average 2.1-fold and 5.9-fold more at times 1 and 10 min respectively (Fig. 2b). Again, the different miRNAs had different fold increases. A second tested antibody, CD31, had insufficient staining with a HTAR of 1 and was evaluated by comparing 10 and 15 min of HTAR (Fig. 2c). The 10 min antigen retrieval had 6.9-fold more miRNAs compared to the 15 min using Let-7a, miR-222 and miR-128. Altogether, these data indicate that the shorter a feasible HTAR step can be, the higher the miRNA yield.
RNAse activity was highest among primary antibodies and partially mitigated by RNAsecure
Next, the RNase activity of the IHC reagents was tested using an RNase alert system and measuring arbitrary units (AU) of fluorescence intensity in a CLARIOstar microplate reader. The negative control had a value of 340 AU, and Millipore ddH20 water had a value of 325 AU. The highest value was for the AE1/AE3 antibody, 5,330 AU. Three additional antibodies (Podocin, Claudin 8, CD34) had values between 680–966 AU. This indicated a need to reduce RNAse activity in the primary antibody incubation step.
In order to address the RNAse activity, we tested the addition of either 0.1% DEPC or RNAsecure to AE1/AE3+ IHC material and compared this with a spike-in of RNAse A (N=3 each). Both RNAsecure and 0.1% DEPC significantly reduced RNAse activity after the spike-in (p≤0.01, t-test, Fig. 1d). Additionally, the RNAsecure-treated material had the highest concentration of RNA (65 ng/μl) followed by 0.1% DEPC (20 ng/μl) and the untreated slides (15 ng/μl) remaining after AE1/AE3 staining. Thus, we elected to use RNAsecure.
We then evaluated the levels of three miRNAs from slides that underwent the full IHC protocol with or without RNAsecure relative to control unbaked slides. On AE1/AE3 stained slides, RNAsecure-treated experiments had an average of 24.6-fold miRNA loss relative to unbaked slides, whereas unprotected slides had an average loss of 41.4-fold. The reduction in miRNA loss was variable across the three miRNAs for RNAsecure treated slides (let-7a 8.8-fold, miR-128 4.9-fold and miR-101, 60.2-fold) (Fig. 2e), but always significantly less than unprotected slides (let-7a 25.2-fold, p=0.00086; miR-128 10.9, p=0.0011; and miR-101 88.1, p=0.0068). Thus, the use of an RNAsecure reagent had a mild, measurable improvement in mitigating RNA loss.
RNAse activity in the chromogen also contributed to the loss of miRNA:
The initial xMD-miRNA-seq was performed with a DSB chromagen to provide a black stain versus a brown stain of DAB. It was reasoned the darker color would improve pigmented cell transfer. However, DSB contains nickel, and we became concerned this could have a deleterious effect on the RNA, due to the known effects of metal cations on DNA18. Therefore we compared DSB and DAB effects on RNA integrity. The fold changes in miRNA abundance were compared to slides not treated with chromogen. The DAB chromogen averaged a 4.7-fold loss, compared to an average 10.6-fold loss for the DSB in comparison to an unstained slide (Fig. 1f). However, this was not a universal effect across all three miRNAs. Since DAB is a more common chromagen, and the trend was toward better RNA amounts with DSB, we elected to use it going forward.
Non-cross-linked EVA membranes improved RNA yield
After the microdissection the cellular products must be extracted from the membrane, which is performed by dissolving the membrane in phenol:chloroform. The commercial 3M membrane is cross-linked, rendering it insoluble and yielding significantly lowering amounts of RNA. In order to improve miRNA yields after microdissection, a membrane using Elvax 410, a form of EVA, was generated and evaluated. We recognized that these Elvax 410 membranes were “stickier” than the crosslinked membranes, and impregnated fullerenes were used as a contaminant to break up the EVA polymer to provide greater specificity to the xMD method. To demonstrate this, heart tissue was stained with NCAD to highlight interacted disks (Fig. 3a) and xMD was performed. Fullerene-impregnated EVA allowed for more specific capture of intercalated disks (Fig. 3b, c). Several different thicknesses of Elvax 410 membranes were evaluated for specificity and sensitivity (Fig. 3d–f). A thinner membrane captures less pigmented material while a thicker membrane captures more non-specific tissue. The 8% EVA membrane resulted in the best tradeoff of sensitivity and specificity in capturing pigmented cells and was the most consistent membrane to generate. The membrane thickness was determined by the gravimetric method (average 6.9 μm) and the film method (average of 5.21 μm) (Table 1).
Figure 3.
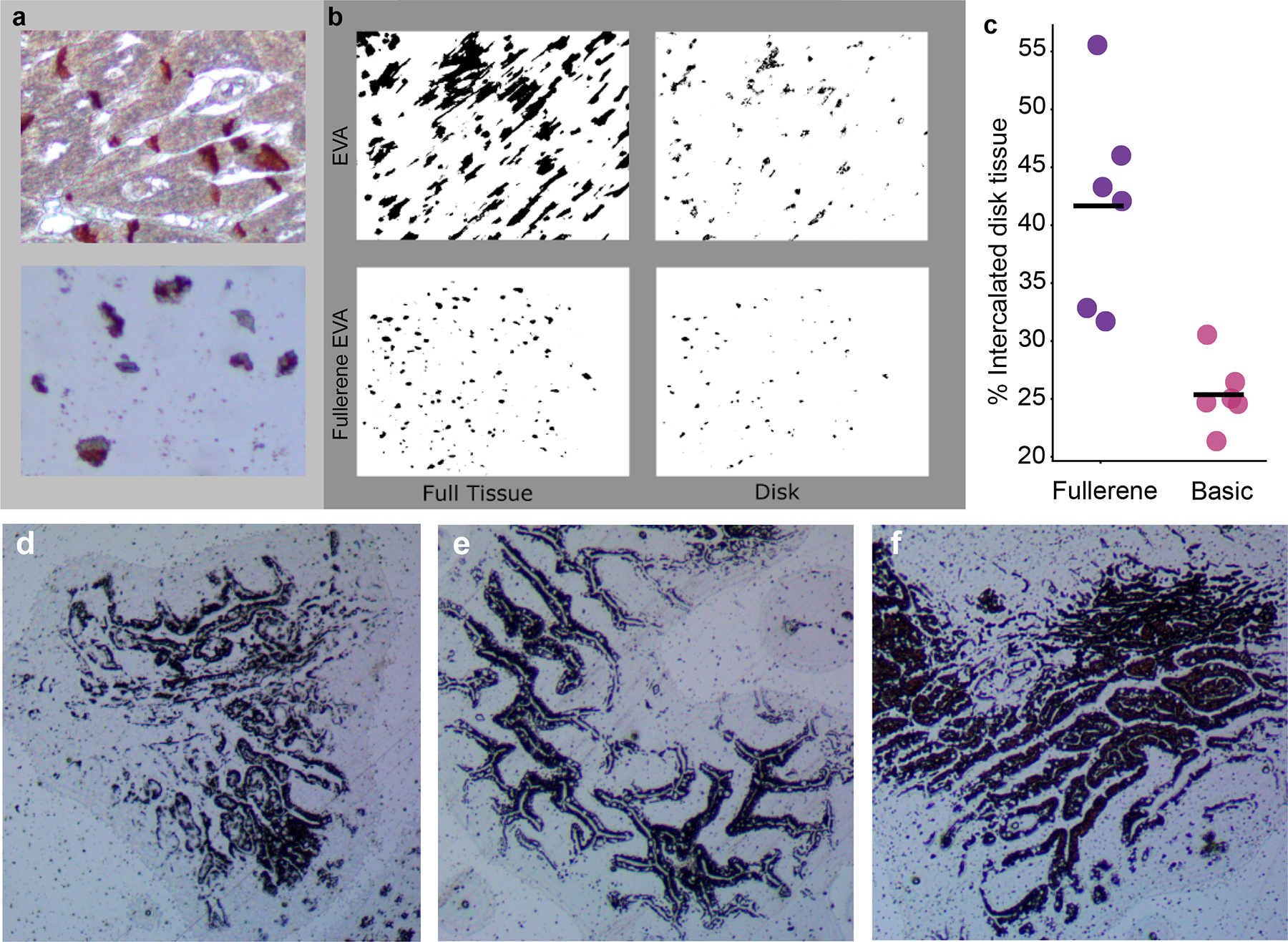
A comparison between fullerene-impregnated EVA membranes and unimpregnated membranes. (A) Human heart slides, stained for intercalated discs (N-cadherin) and cut at 4 μm. were used. Top panel demonstrates the staining pattern for intercalated discs and bottom panel an EVA membrane. (B) Binary color area images of a postdissected unimpregnated EVA membrane showing the full tissue captured (top left) vs the disk region within the collected tissue (top right). Similar images of a postdissected fullerene-impregnated EVA membrane (bottom left) demonstrating improved capture specificity with less full tissue relative to captured disk material (bottom right). (C) An ImageJ pixel count across multiple images (n = 12) demonstrated increased specificity in capturing intercalated disc tissue with fullerene-impregnated EVA membranes. (D-F) 6%. 8%. and 10% EVA membranes showing capture of AE1/AE3’ epithelial cells, with the cleanest pattern seen with 8% EVA. EVA. ethylene vinyl acetate
Table 1.
The thicknesses of membranes were tested gravimetrically and using a spectrometer.
| %6 EVA b/w | %8 EVA b/w | %10 EVA b/w | |
| Gravimetric (pm) | 3.84 | 6.90 | 18.17 |
| Spectrometer (pm) | 3.60 | 5.21 | 17.35 |
RNA yield was determined for this optimal EVA membrane. Nucleic acid material was captured and assayed from slides stained with AE1/AE3 using 3M membranes (N=5), Elvax 410 membranes (N=5), or Elvax 410 membranes impregnated with fullerenes (N=5). The 3M membrane yielded the least amount of RNA of the three membrane types (9.4 ng/μl on average, p=0.00043). The Elvax 410 produced the most (27.2 ng/μl on average, p=0.078) with the Elvax fullerene combination averaging 23.5 ng/μl, but with higher specificity, as discussed above (Fig. 3a, b, c).
Methods to optimize specific RNA transfer to EVA membranes:
Despite the optimizations, an initial small RNA sequencing experiment of xMD-obtained AE1/AE3 positive cells failed to demonstrate appropriate gain or loss of epithelial (miR-192) and mesenchymal-specific (miR-143) miRNAs, suggesting unnoticed non-specific capture of RNA from the FFPE slide. We then performed experiments to improve RNA transfer specificity. Removing superficial RNA from the slide tissue by pressing an extra EVA membrane over the tissue prior to the flash lamp step improved the miR-143/miR-192 Ct ratio by 4.93±0.41(N=5). Lowering the energy (kJ) in the flash lamp step improved the ratio by 3.51±0.55 (N=5). Combining these two steps further increased the miR-192 to miR-143 ratio, with a Ct ratio to 9.61±0.38 (N=5), suggesting a ~781-fold improvement (2^9.61) in enrichment by the combined method.
Tissue transfer is optimal with a glossy white background and high energy intensity.
The flash lamp produces a quick, high-intensity light which photothermally heats the pigmented areas to help affix them to the EVA membrane. Using the method described in the method sections, black, mirror, no background, white, and reflective white were tested. Both the black background (N=1) and no background (raised platform, N=1) qualitatively had a poor transfer to the EVA membrane. The mirror background averaged roughly 1 million pigmented pixels (N=3), the matte white averaged 1.4 million pigmented pixels (N=5), and the glossy white background had an average of 1.7 million pigmented pixels (N=3) (Suppl. Fig 1a). We concluded that a glossy white background would be superior for all future experiments.
We then tested a select number of options of the flash lamp for intensity and number of flashes used, again evaluating AE1/AE3 staining in the small intestine. The most tissue transfer occurred under the highest intensity (level 5) and was independent of the number of flashes (1, 3 or 5). Less efficient transfers of pigmented material occurred at the lowest intensity (level 1) and the medium intensity (level 3) (Suppl. Fig. 1b). All comparisons were qualitative in nature due to the clear differences observed.
The fully optimized method has higher yields of miRNA before and after xMD.
After all optimizations across the entire protocol, two comparisons were made. One compared the original and optimized protocols after all steps of the IHC method but prior to the flash lamp capture of material. The second compared all steps through the full xMD protocol between the original and optimized protocols from material on the ELVAX 410 EVA membrane. For the pre-flash lamp experiment (scraped slide) on AE1/AE3 stained slides, the optimized method yielded an average of 39.9-fold more miRNA than the non-optimized method (Fig. 4a). The individual miRNAs again showed a difference in miRNA increase (Let-7a. 100.5 fold; miR-222, 6.2-fold; and miR-128, 11.4-fold). For the ELVAX 410 EVA membrane comparison of AE1/AE3+ material, there was, on average, a 16.45-fold greater yield in the optimized method relative to the original method, with some variation by miRNA tested (let-7a, 25.8-fold; miR-222, 13.7-fold; and miR-128, 9.9-fold) (Fig. 4b). The same experiments were performed with a CD31+ IHC to capture endothelial cells (Fig. 4c, d). Here the increases were individually 8.4, 14.4, and 29-fold improved for let-7a, miR-222, and miR-128, respectively, in scraped tissue and 21.3-, 15- and 25-fold improved for let-7a, miR-222 and miR-128 respectively in xMD captured material.
Figure 4.
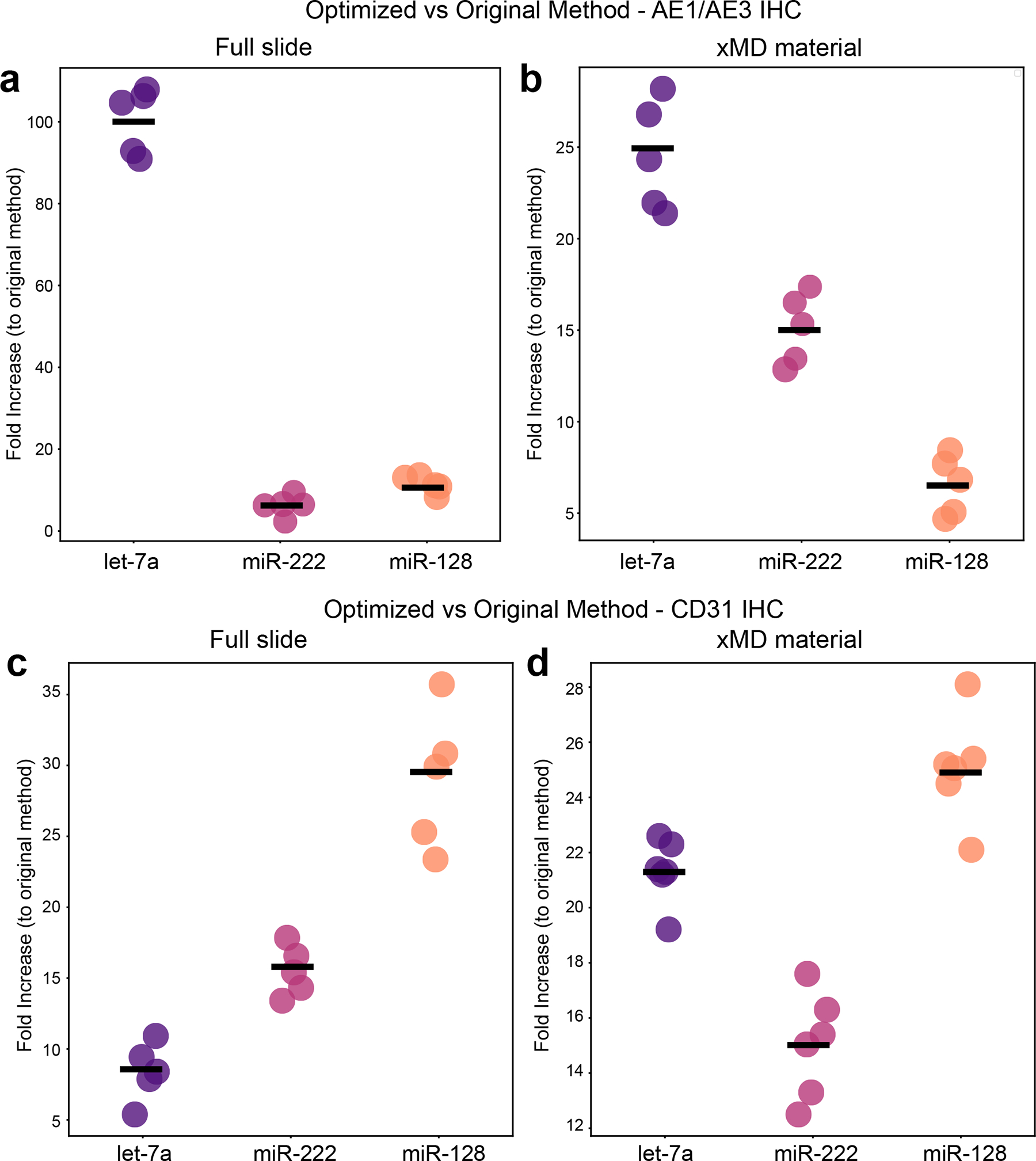
A comparison of miRNA yield from optimized IHC and xMD with original methods. (A) miRNA yield directly from slides that underwent AE1/AE3 1HC, showing the fold improvement of the optimized protocol to the original protocol (n = 5).(B) miRNA yield after xMD capture of AE1/AE3 material, showing the fold improvement of the optimized protocol to the original protocol (n = 5). (C) miRNA yield directly from slides that underwent CD31 IHC, showing the fold improvement of the optimized protocol to the original protocol (n − 5). (D) miRNA yield after xMD capture of CD31+ material, showing the fold improvement of the optimized protocol to the original protocol (n = 6). EVA, ethylene vinyl acetate; IHC, immunohistochemistry; xMD, expression microdissection.
A TLDA qPCR array demonstrates the enrichment of AE1/AE3+ cells
After all optimizations, TLDA miRNA array was performed on duodenal tissue scraped from the slide or from AE1/AE3 positive xMD material to document global miRNA changes in the epithelial cell-specific population. The TLDA miRNA array contains 370 miRNAs, of which 258 miRNAs had expression that could be analyzed. TLDA was chosen as it could be combined with a QC step directly on the same RNA sample due to the low input RNA requirement. Thus, prior to the TLDA, QC was performed to compare the ratio of miR-192 to miR-143 between AE1/AE3+ cells and a tissue scrape using standard qPCR. This method demonstrated a 29.9-fold relative increase in miR-192 to miR-143 between AE1/AE3+ epithelial cells and the total tissue scrape, indicating enrichment was made, and the TLDA proceeded. The TLDA qPCR Ct data were normalized, and fold changes between the whole tissue and epithelial cells were calculated. Epithelial cells represented ~15% of the cellular material in the duodenal slide, which indicates potential maximal gains and losses in miRNA relative to the full tissue sample. Globally, 54 miRNAs had >4-fold decreased expression in epithelial cells, while only 12 were > 4-fold enriched. Two main mesenchymal markers, miR-143 and miR-145, were 336- and 279-fold reduced in the epithelial cell sample (Fig. 5a, Suppl. Table 1). Conversely, the miR-200 family of epithelial markers increased between 1.7-fold and 14-fold in the epithelial cell material (Fig. 5b). Two additional miRNAs increased in the epithelial cells were miR-488 and miR-302c (9.6- and 7.1-fold respectively).
Figur 5.
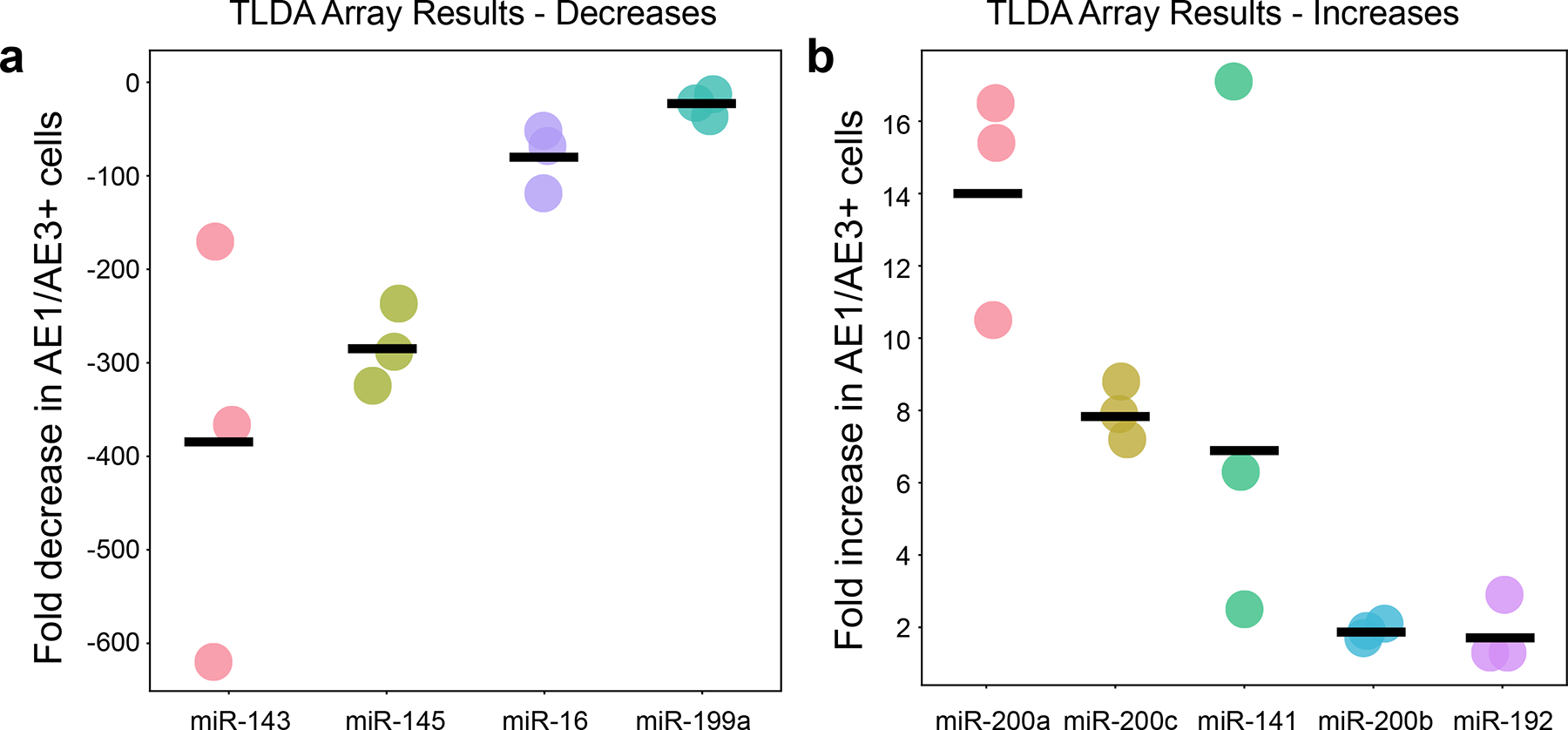
TLDA array results: (A) Selected miRNAs that had notably lower expression in AE1/AE3+ epithelial cells relative to the total tissue. (B) Selected miRNAs that had notably higher expression in AE1/AE3 cells relative to total tissue.
Discussion
The original publication of the xMD-miRNA-seq method demonstrated the potential utility of a method to capture near in vivo miRNA expression patterns of specific cells17. However, the method was inefficient, with notable RNA loss during the xMD steps and a low yield of miRNAs in the sequencing step. Thus, it could have been more practical for widespread usage. Here we assessed each step of the xMD-miRNA-seq method to reduce RNA loss and increase the specificity of miRNAs transferred onto the EVA membrane. The key improvements were realized by adding an RNAse inhibitor, shortening the HTAR time, using a nickel-free chromogen, generating a non-crosslinked fullerene-impregnated EVA membrane, and using a phenol:chloroform extraction method. These changes increased RNA yield, of all three examined miRNAs, individually and combined. The qPCR fold changes showed an increase in miRNA collection, averaging 16.5-fold when all changes were combined. Through these improvements, we were able to obtain ~200 ng of total RNA per slide, and ~1 μg when five slides are combined. As newer small RNA sequencing methods can be performed with as little as 10 ng of starting RNA, these improvements greatly expand the opportunity of using this method across rarer and more specific cell populations.
These optimizations, however, did not result in improved specificity of the collected RNA. Thus, a separate set of optimizations resulted in a marked reduction of ambient RNA from the surface of the slides and reduced non-target transfer of material. These steps were needed due to the stronger transfer properties of the non-crosslinked EVA membranes compared to previously used crosslinked EVA membranes.
A limitation of the method was our inability to execute the full assay with small RNA sequencing. Two attempts ably provided a robust miRNA dataset (>580 bona fide miRNAs). Still, neither had the appropriate enrichment expected19,20 The first was performed before the corrections to the protocol (E – xMD specificity optimization) and had no meaningful enrichment. The second was performed after developing a QC step, but the QC was not done concurrently. This sample had a ~50% enrichment in epithelial cells based on gains/losses of expression of epithelial and mesenchymal markers. Neither assay was performed with a concurrent qPCR QC step due to the relatively high need of RNA for both routine qPCR and sequencing library preparation protocols. Utilizing a QC step prior to performing the global miRNA array ensured the quality of the xMD, which we now recognize as critical prior to genomic level assaying and must be included in future iterations of this method that employ sequencing. Nonetheless, using all of the optimized approaches, we noted a strong enrichment for epithelial cells by the qPCR array-based method. Future developments in the assay, with improved technologies, will allow us to return to a sequencing-based system, which we believe is ultimately superior to array-based data.
We demonstrated the utility of the xMD method with a 4–13 fold increase in epithelial-specific markers (miR-200 family) and more importantly, a >300-fold decrease in mesenchymal miRNA (miR-143/145) expression compared to the entire duodenal tissue4,21. Of note, AE1/AE3 is a pan-cytokeratin marker of all epithelial cells of duodenum from the stem cells at the crypt base, through progenitor cells, to the mature enterocytes at the top of the crypt. This may explain why a couple of miRNAs known for their specificity to stem cells (miR-488, miR-302c) were also enriched8. Future uses of xMD will employ IHC antibodies selected for more specific labeling sub-groups of the epithelium based on single cell and proteomic expression data9,22–25.
The IHC and miRNA extraction were optimized, and the EVA membranes used for xMD were optimized. We developed non-polymerized EVA membranes with lattice disruption, which were able to partially dissolve in phenol:chloroform allowing for easier extraction of cellular products and higher yield.
While there is much excitement for single-cell RNA sequencing methods to assay miRNAs, challenges also remain in that domain. First, miRNAs represent a very small fraction of total RNA in a given cell. Thus the yield per cell is low, and obtaining that limited information on a cell-by-cell basis is cost-ineffective. Secondly, at the low yield per cell, the ability to identify the full repertoire of miRNAs (low to modestly expressed) in a cell type is challenging to impossible by that approach13. Finally, the great strength of single-cell RNA sequencing is to define populations of cells; however, based on our experiences with the limited set of miRNAs that exist (relative to genes), miRNAs will be inferior to genes for this purpose8.
The optimized xMD method, described here, can allow any researcher access to in vivo miRNA expression patterns from cells of interest from FFPE tissues. This opens the recent surgical pathology archive of an institution for inquiry towards novel tissue-based biomarkers related to diagnosis and therapeutic responses in pathologic tissues. Additionally, between single-cell human cell atlas projects and the Human Protein Atlas, many cell-type defining genes/proteins are being realized that can further refine the selectivity of this xMD method9,22,23,26,27. Most materials used in this optimized version of xMD are widely available and cost-effective. Those that are not, require little training to make. Other methods of microdissection, such as laser capture, are limited by the ability of users to obtain enough specific material and/or access to expensive machinery. The smaller expense and experience required make xMD ideal for a new method of microdissection28.
This new optimized method will allow for more near in vivo miRNA expression patterns of cells to be determined. Previous work has shown miRNA expression in vitro differs from in vivo cells, highlighting the importance of the in vivo environment in FFPE tissues. miRNAs are effective in the cells in which they are expressed, and this population of miRNAs varies from cell type to cell type. The expression profiles of individual cells are lost when whole tissue expression is performed, limiting our current understanding of miRNA expression in cell types.
In conclusion, we demonstrate a fully optimized xMD method that obtains significant amounts of highly specific miRNA from cells obtained from FFPE tissues. This method showed robust isolation of epithelial cells from duodenal tissue, establishing an in vivo expression pattern of this cell type. Future developments in reducing RNA requirements for QC and sequencing steps will allow a full xMD-miRNA-Seq approach to be realized.
Supplementary Material
Acknowledgements
We thank the James Segars laboratory for the use of the ClarioSTAR fluorometer, the Ben Larman lab for the use of the qPCR thermocycler, the Oncology Tissue and Imaging Services Core for slide preparation, Margaret Weinstein and Mohammad Dbouk for sample acquisition, Arun Patil for figure assistance, Anandita Rajpurohit and Joo Heon Shin for library preparation assistance, and Roxane Ashworth and the GRCF DNA services staff for TLDA plate discussion and assistance.
Funding:
This work was supported by the National Institute of General Medical Sciences, R01GM130564, and the National Heart, Lung, and Blood Institute, R01HL137811 to M Halushka.
Footnotes
Conflicts of Interest:
The authors declare no conflicts of interest.
Competing Interests statement: The authors declare no competing interests. This work was supported by the National Institute of General Medical Sciences, R01GM130564, and the National Heart, Lung, and Blood Institute, R01HL137811, to M Halushka.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability
TLDA array data are available at the Gene Expression Omnibus, study GSE205216.
References
- 1.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148(6):1172–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. [DOI] [PubMed] [Google Scholar]
- 3.Jiang Q, Wang Y, Hao Y, et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37(Database issue):D98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kent OA, McCall MN, Cornish TC, Halushka MK. Lessons from miR-143/145: the importance of cell-type localization of miRNAs. Nucleic Acids Res. 2014;42(12):7528–7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCall MN, Kim MS, Adil M, et al. Toward the human cellular microRNAome. Genome Res. 2017;27(10):1769–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Rie D, Abugessaisa I, Alam T, et al. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nature biotechnology. 2017;35(9):872–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halushka MK, Fromm B, Peterson KJ, McCall MN. Big Strides in Cellular MicroRNA Expression. Trends in genetics : TIG. 2018;34(3):165–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patil AH, Baran A, Brehm ZP, McCall MN, Halushka MK. A curated human cellular microRNAome based on 196 primary cell types. Gigascience. 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haber AL, Biton M, Rogel N, et al. A single-cell survey of the small intestinal epithelium. Nature. 2017;551(7680):333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuosmanen SM, Kansanen E, Sihvola V, Levonen AL. MicroRNA Profiling Reveals Distinct Profiles for Tissue-Derived and Cultured Endothelial Cells. Scientific reports. 2017;7(1):10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray GI. An overview of laser microdissection technologies. Acta Histochem. 2007;109(3):171–176. [DOI] [PubMed] [Google Scholar]
- 12.Porichis F, Hart MG, Griesbeck M, et al. High-throughput detection of miRNAs and gene-specific mRNA at the single-cell level by flow cytometry. Nature communications. 2014;5:5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hucker SM, Fehlmann T, Werno C, et al. Single-cell microRNA sequencing method comparison and application to cell lines and circulating lung tumor cells. Nature communications. 2021;12(1):4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isakova A, Neff N, Quake SR. Single cell profiling of total RNA using Smart-seq-total. BioRxiv. 2020( 10.1101/2020.06.02.131060). [DOI] [Google Scholar]
- 15.Rosenberg AZ, Armani MD, Fetsch PA, et al. High-Throughput Microdissection for Next-Generation Sequencing. PLoS One. 2016;11(3):e0151775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tangrea MA, Chuaqui RF, Gillespie JW, et al. Expression microdissection: operator-independent retrieval of cells for molecular profiling. Diagnostic molecular pathology : the American journal of surgical pathology, part B. 2004;13(4):207–212. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg AZ, Wright C, Fox-Talbot K, et al. xMD-miRNA-seq to generate near in vivo miRNA expression estimates in colon epithelial cells. Scientific reports. 2018;8(1):9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLachlan GA, Muller JG, Rokita SE, Burrows CJ. Metal-mediated oxidation of guanines in DNA and RNA: a comparison of cobalt(II), nickel(II) and copper(II) complexes. Inorganica Chimica Acta. 1996;251(1–2):193–199. [Google Scholar]
- 19.Fromm B, Hoye E, Domanska D, et al. MirGeneDB 2.1: toward a complete sampling of all major animal phyla. Nucleic Acids Res. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patil AH, Halushka MK. miRge3.0: a comprehensive microRNA and tRF sequencing analysis pipeline. NAR Genom Bioinform. 2021;3(3):lqab068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chivukula RR, Shi G, Acharya A, et al. An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration. Cell. 2014;157(5):1104–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cornish TC, Chakravarti A, Kapoor A, Halushka MK. HPASubC: A suite of tools for user subclassification of human protein atlas tissue images. Journal of pathology informatics. 2015;6:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uhlen M, Oksvold P, Fagerberg L, et al. Towards a knowledge-based Human Protein Atlas. Nature biotechnology. 2010;28(12):1248–1250. [DOI] [PubMed] [Google Scholar]
- 24.Regev A, Teichmann SA, Lander ES, et al. The Human Cell Atlas. eLife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shanahan MT, Kanke M, Oyesola OO, et al. Multiomic analysis defines the first microRNA atlas across all small intestinal epithelial lineages and reveals novel markers of almost all major cell types. American journal of physiology Gastrointestinal and liver physiology. 2021;321(6):G668–G681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rozenblatt-Rosen O, Stubbington MJT, Regev A, Teichmann SA. The Human Cell Atlas: from vision to reality. Nature. 2017;550(7677):451–453. [DOI] [PubMed] [Google Scholar]
- 27.Burclaff J, Bliton RJ, Breau KA, et al. A Proximal-to-Distal Survey of Healthy Adult Human Small Intestine and Colon Epithelium by Single-Cell Transcriptomics. Cellular and molecular gastroenterology and hepatology. 2022;13(5):1554–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walsh EM, Halushka MK. A Comparison of Tissue Dissection Techniques for Diagnostic, Prognostic, and Theragnostic Analysis of Human Disease. Pathobiology. 2022:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
TLDA array data are available at the Gene Expression Omnibus, study GSE205216.