

Abstract
Chimeric antigen receptor (CAR) T cell therapy has shown remarkable success in the treatment of hematologic malignancies. Unfortunately, it has limited efficacy against solid tumors, even when the targeted antigens are well expressed. A better understanding of the underlying mechanisms of CAR T cell therapy resistance in solid tumors is necessary to develop strategies to improve efficacy. Here we report that solid tumors release small extracellular vesicles (sEVs) that carry both targeted tumor antigens and the immune checkpoint protein PD-L1. These sEVs acted as cell-free functional units to preferentially interact with cognate CAR T cells and efficiently inhibited their proliferation, migration, and function. In syngeneic mouse tumor models, blocking tumor sEV secretion not only boosted the infiltration and anti-tumor activity of CAR T cells but also improved endogenous anti-tumor immunity. These results suggest that solid tumors use sEVs as an active defense mechanism to resist CAR T cells and implicate tumor sEVs as a potential therapeutic target to optimize CAR T cell therapy against solid tumors.
Keywords: extracellular vesicles, CAR T cells, solid tumors, immune checkpoint, tumor antigens
Introduction
CAR T cell therapies have demonstrated remarkable efficacy in treating hematopoietic cancers. However, they are much less effective in treating solid tumors (1). There is an unmet need to understand the mechanisms by which solid tumors inhibit the infiltration and function of CAR T cells. To date, the proposed mechanisms for the resistance to CAR T cell therapy include new mutations in cancer cells and deficiencies in CAR T cells (2). With respect to cancer cells, tumor antigen loss is a major mechanisms for tumor to evade CAR T cells (1, 2). However, clinical studies showed that many patients with antigen-positive tumors were still resistant to the CAR T therapies (3–6), suggesting that additional mechanisms exist for the resistance.
The tumor microenvironment (TME) limits CAR T infiltration and function in solid tumors (1, 7, 8). It is important to identify immunosuppressive factors in the TME that inhibit CAR T cell functions. Extracellular vesicles (EVs), such as exosomes, are lipid-encapsulated vesicles released from cells to the extracellular milieu (9). EVs carry bioactive molecules and potently affect the pathophysiology of the recipient cells. Recent studies including our work have shown that tumor-derived small EVs (sEVs) inhibit anti-tumor immunity and T cell memory by immune checkpoint molecules including PD-L1 on their surface (9–17).
Here, we report that CAR T cell treatment promotes tumors to release sEVs that carry an increased level of PD-L1 on their surface, which suppresses the infiltration and function of CAR T cells. Importantly, tumor antigens are enriched on the sEVs together with PD-L1, which lead to enhanced interaction of sEVs with and potent inhibition of the CAR T cells. In a preclinical tumor model, inhibition of exosome secretion boosts CAR T therapy and improves endogenous anti-tumor immunity. Our results suggest a new mechanism by which solid tumors resist CAR T cells, and implicate tumor exosomes as a potential therapeutic target in CAR T cell therapy.
Materials and Methods
Cell culture and reagents
The murine PDAC cell line 4662 cell line was generated as previously described (18). 6419c5 cell line was obtained from Dr. Ben Stanger (University of Pennsylvania) (19). The cells were cultured at 37 °C with humidified 5% CO2. B16-F10 mouse melanoma cells (Cat#: CRL-6475, RRID: CVCL_0159) and 293T cells (Cat#: CRL-3216™, RRID: CVCL_0063) were purchased from ATCC, B16-F10-CD19 mouse melanoma cells were obtained from Dr. Andy Minn (University of Pennsylvania) (20). Murine oral cancer cell line MOC1 cell line was obtained from Dr. Steven Albelda (University of Pennsylvania). Human ovarian adenocarcinoma cell line SKOV3 and NIH-3T3-FAP cells were generated as previously described (21, 22). Authentication of these cell lines were performed using short tandem repeat (STR) profiling analysis. The cells were passaged for less than one month. All cells were regularly tested for mycoplasma using the Mycoplasma Detection Kit (InvivoGen, Cat#: rep-mys-50) before experiments.
Generation of CAR T cells
Mouse Meso-CAR was constructed by fusing anti-mesothelin scFv to a mouse CD3-ζ signaling domain and a mouse 4-1BB co-stimulatory domain along with a EGFP tag. The CAR was subcloned into the MSGV vector and packaged in the Phoenix packaging cells (Allele; Phoenix Eco Cells Line ABP-RVC-10002; purchased from and authenticated by ATCC, CRL-3214, RRID: CVCL_H717) to obtain the retrovirus. T cells were cultured in RPMI 1640 supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate, 1 mM pyruvate and 50 μM β-mercaptoethanol. Primary mouse T cells were isolated as suggested by the manufacturer (Miltenyi Biotec) from the spleens of mice and incubated in 12-well plates (2 × 106 cells per well), and Dynabeads Mouse T-Activator CD3/CD28 (Gibco, Cat#: 11453D) at a 1:1 ratio, as previously described (22, 23). To obtain CAR T cells with lower activation level, we also reduced the amount of Dynabeads Mouse T-Activator CD3/CD28 (from a 1:1 ratio to 0.5:1 ratio, Dynabeads vs. T cells). After 48 hrs, cells were plated in a 24-well plate coated with retrovirus and 5 μg/cm2 of retronectin (Takara, Cat#: T100B). Before plating T cells, a retronectin-coated 24-well plate was centrifuged without braking at room temperature for 1 hr at 1,000 × g with 1 ml per well crude viral supernatant. After overnight incubation, cells were expanded with 50 U/ml IL-2 for 48 hrs. The transduction efficiency with mouse Meso-CAR or MigR-GFP (control T cells) was assessed after transduction. MigR-CAR T cells and FAP-CAR T cells were generated as previously described (22). Human HER2-CAR was constructed by fusing anti-HER2 scFv to a human CD3-ζ signaling domain and a human 4-1BB costimulatory domain, and subcloned into pTRPE lentiviral vectors according to the previous study (24). Primary T cells were isolated from healthy donors provided by the University of Pennsylvania Human Immunology Core. Briefly, human CD3+ T cells were activated with Dynabeads CD3/CD28 CTSTM (Thermo Fisher Scientific) at a 3:1 ratio. After 24 hrs, T cells were transduced with lentivirus. Beads were removed from culture on Day 5. After CAR-expressing T cells were quantified and characterized by Anti-Trastuzumab (R&D Systems, Cat#: FAB95471R) and flow cytometry, CAR-T cells were cryopreserved for further studies. CD19-CAR T cells used in this study were produced as a previous description (20).
Isolation of sEVs from tumor tissues
To obtain tumor tissue derived EVs, tumor tissues were cut into small pieces (~2 mm diameter), and then dissociated enzymatically with 1 mg/ml type I collagenase, 50 U/ml RNase and DNase I for 30 mins at 37°C according a previous study (25). After being filtered through a 0.22-mm filter, the suspensions were centrifuged at 300 × g for 15 min to discard the cells, and 2,000 × g for 20 min to remove dead cells and debris (Beckman Coulter, Allegra X-14R). The supernatants were then centrifuged at 120,000 × g for 2 hrs at 4°C (Beckman Coulter, Optima XPN-100) to obtain sEVs. After discarding the supernatants, the sEV pellets were washed with a large volume of ice-cold PBS and centrifuged again at 120,000 × g for 2 hrs at 4 °C. The final sEV pellets were resuspended in PBS for further studies.
Isolation of mesothelin+ sEVs
Following our previous study (26), 500 μl of magnetic beads (MagniSort™ Streptavidin Positive Selection Beads, Invitrogen, Catalog Number: MSPB-6003) were incubated with biotinylated anti-mesothelin antibody (1:20, Biorbyt, Cat#: orb241037), or biotinylated anti-mouse IgG (1:20, Biolegend, Cat#: 02-6102, RRID: AB_2532938) on a shaker at room temperature for 2 hrs. The beads were then placed on the magnet and washed three times with PBS. 0.5 mg of tumor tissue-derived sEVs (in 0.5 ml PBS) were incubated with biotinylated anti-IgG or mesothelin antibody-bounded magnetic beads overnight at 4°C. The magnetic beads bound with mesothelin+ sEVs were collected, placed on the magnet, and washed 5 times with PBS to obtain enriched mesothelin+ sEVs. To obtain the rest of the sEVs after mesothelin+ sEV removal, the supernatant was transferred into a new tube for ultracentrifugation at 120,000 × g for 2 hrs. The same amounts of sEV proteins from each group were loaded for western blotting analysis.
Purification of EVs from cell culture media
EVs derived from the culture media of PDAC cell lines were isolated by ultracentrifugation as previously described (9, 26). In brief, bovine EVs were depleted by centrifugation at 100,000 × g overnight at 4 °C to obtain EV-free FBS. Cells were cultured in DMEM medium supplemented with 10% EV-depleted FBS. The culture medium was collected and centrifuged at 2,000 × g for 20 min at 4 °C to remove dead cells and cell debris (Beckman Coulter, Allegra X-14R). The supernatant was collected and further centrifuged at 16 500 × g for 40 min at 4 °C (Beckman Coulter, Allegra X-14R), followed by ultracentrifugation at 100 000 × g for 2 hrs at 4 °C to pellet sEVs (Beckman Coulter, Optima XPN-100). The purified sEVs were suspended in PBS and stored at 4 °C or −80 °C for subsequent studies.
The sEV-CAR T cell binding assay
To assess the interactions between exosomes and CAR T cells, sEVs were labeled with PKH26 in 100 μl PBS, and then washed with 30 ml PBS, and pelleted by ultracentrifugation. To block PD-L1 or mesothelin on sEVs, the sEVs were incubated with 10 μg/ml anti-PD-L1 or anti-mesothelin antibodies or IgG isotype antibodies overnight at 4°C. After washing with PBS, sEVs were collected by ultracentrifugation to remove the free antibodies. CAR T cells (2 × 105 cells/well in 96-well plates) with or without 20 ng/ml mouse mesothelin protein (R&D Systems, Cat#: 10516-MS-050) or IgG control (R&D Systems, Cat#: 4460-MG-100) treatments were incubated with 20 μg/ml PKH26 -labeled sEVs for 24 hrs, and then collected for flow cytometry analysis or imaged by confocal microscopy (9, 26).
Treatment of CAR T cells with sEVs
CAR T cells were plated in a 96-well plate at a seeding density of 2 × 105 cells/well and incubated with sEVs with indicated treatment. 20 μg/ml sEVs were used for the treatment unless otherwise indicated. CAR T cells and their control T cells were incubated with sEVs for 48 hrs unless otherwise indicated. The treated CAR T cells were analyzed using flow cytometry. To block sEV PD-L1 or mesothelin, 200 μg sEVs in 100 μl PBS were incubated with 10 μg/ml of indicated blocking antibodies or IgG isotype antibodies as a control overnight at 4°C. To remove non-bound free antibodies, sEVs were then washed with PBS and collected by ultracentrifugation before incubation with cells.
Tumor cell killing assay
To study the effects of tumor cell-derived sEVs on the ability of CAR T cells to kill tumor cells, CAR T cells (4 × 105 cells/well in 48-well plate) were treated with PBS or tumor-derived sEVs (20 μg/ml for 48 hrs) with indicated treatment, and then co-cultured with tumor cells (4 × 105) in 6-well plates for 48 hrs with an effector to target (E:T) ratio of 1:1. Cells were then intracellularly stained with ITC-conjugated (BD Biosciences, Cat#: 560901, RRID: AB_10563896) or Alexa Fluor 647 (BD Biosciences, Cat#: 560626, RRID:AB_1727414)-conjugated antibody against cleaved caspase-3 (BD Biosciences) and prepared for flow cytometry. Relative cytotoxicity of CAR T cells to tumor cells was determined as previously described (9).
CAR T cell transmigration assay
CAR T cell transmigration assay was performed using a 96-well ChemoTx chemotaxis system with 3 μm pore size (Neuro Probe) following the manufacturer’s protocol. The bottom wells were filled with 30 μl of migration buffer with or without 100 ng/ml murine CXCL9 (Peprotech, Cat#: 250-18). CAR T cells incubated with or without sEVs (20 μg/ml) were applied to the top of filters. After 4 hrs of incubation at 37 °C, migrated cells collected from the bottom wells were quantified using a cell counter.
Nanoparticle tracking analysis (NTA)
To prepare EV sample for NTA, tumor cells were seeded into 6-well plates to reach a 50% confluence prior to culture medium collection. The culture medium was replaced with FBS-free medium the next day. After an additional 6 hrs of incubation, the medium was collected and centrifuged at 16,500 × g for 40 min at 4 °C to remove dead cells, cell debris, and large EVs. The supernatant was collected to analyze the concentration and size distribution of sEVs by an NTA system (Malvern Instruments, NanoSight NS300) (9, 26).
Iodixanol density gradient centrifugation
Iodixanol density gradient centrifugation was performed as described in our previous study (9, 26). To prepare a discontinuous iodixanol gradient (5%, 10%, 20%, and 40%), 60% OptiPrep aqueous iodixanol was diluted with 0.25 M sucrose in 10 mM Tris. Purified sEVs (200 μg) were loaded on top of the iodixanol gradient and centrifuged at 100,000 × g for 18 hrs at 4°C (Beckman Coulter, Optima XPN-100). Twelve fractions of density gradient layers were collected from the top of the gradients. The sEVs contained in each fraction were pelleted by additional ultracentrifugation at 100,000 × g for 2 hrs at 4°C and analyzed by western blotting (9).
CRISPR/Cas9 genome editing
The gRNA oligonucleotides against murine PD-L1 (sgRNA 1, 5’-GGTCCAGCTCCCGTTCTACA-3’, sgRNA 2, 5’-TGTAGAACGGGAGCTGGACC -3’), murine RAB27A (sgRNA 1, 5’-CCAAGGCCAAGAACTTGATG-3’, sgRNA 2, 5’-CATCAAGTTCTTGGCCTTGG-3’) (synthesized by Genewiz) were annealed and cloned into lentiCRISPR-v2-Puro vector (Addgene, Cat#: 52961, RRID: Addgene_52961) as previously described (26). These plasmids were co-transfected with lentiviral packaging plasmids into 293T cells. Lentiviral supernatants were harvested 48 – 72 hrs after transfection and then filtered before infecting cells. Stable transfecting cells were selected by 2 μg/ml puromycin. Monoclonal knockout cells were isolated using a limiting dilution method (26) and further identified by flow cytometry or western blotting.
Immunoelectron microscopy
Tumor-derived sEVs isolated by differential ultracentrifugation were loaded onto formvar carbon-coated nickel grids, fixed in 4% paraformaldehyde, and immunolabeled with rabbit anti-PD-L1 antibody (clone D4H1Z, Cell Signaling Technology, RRID: AB_2799672) or/and mouse anti-mesothelin antibody (clone 7E6A6, Thermo Fisher Scientific, RRID: AB_2898487) followed by incubation with an anti-rabbit secondary antibody conjugated with protein A-gold particles (10 nm) and an anti-mouse secondary antibody conjugated with protein A-gold particles (5 nm). Each step was followed by washing three times with PBS and ten times with ddH2O. The sEVs were then fixed in 2.5% glutaraldehyde, washed, contrasted with 2% uranyl acetate, dried in air, and then examined with a JEM-1011 transmission electron microscope.
Western blotting
Lysis buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM KCl, 5 mM MgCl2, 0.5% Triton X-100, 1 mM DTT, 1 mM PMSF, and protease inhibitor cocktail was used to lyse cells or EVs. The proteins were then separated by 10% sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes. The blots were blocked with 5% bovine serum albumin for 1 hr at room temperature and incubated with primary antibodies overnight at 4°C. The blots were probed with horseradish peroxidase-conjugated secondary antibodies for 1 hr at room temperature and then for incubated with ECL Western Blotting Substrate (Pierce) for developing.
Fluorescence microscopy
Cells cultured on glass coverslips were fixed in 4% paraformaldehyde for 15 min. After permeabilization by 0.1% Triton X-100 in PBS for 15 min, cells on glass coverslips were blocked with 5% BSA in PBS, incubated with primary antibodies overnight at 4°C and then fluorescence-labeled secondary antibodies (Life Technologies) for 1 hr at room temperature. After being enumerated by ProLong® Gold Antifade Reagent with DAPI (Cat#: 8961, Cell Signaling Technology), samples were visualized by Eclipse TE2000-U inverted microscope (Nikon). NIS-Elements Advanced Research software (Nikon, version 4.50) was used to analyze the images.
For immunofluorescence of tissue samples, fresh samples were fixed in 4% paraformaldehyde followed by dehydration, the samples were then embedded in paraffin wax. 5 μm paraffin sections were prepared and treated by microwave repair antigen retrieval. Primary antibodies in 2% BSA and related second antibodies were incubated on the same sections. After that, all sections were covered by coverslips with mounting medium containing DAPI. All slides were imaged under a Nikon fluorescence microscope as described above. The experiment was performed in a double-blind manner.
Flow cytometry
The staining, gating, and analysis strategy used in flow cytometry was described in our previous studies with some modifications (9, 26). Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll (catalog) density gradient centrifugation and analyzed by flow cytometry. Tumor tissues were dissociated enzymatically with 1 mg/ml type I collagenase in the presence of 50 U/ml RNase and DNase I at 37°C for 30 min to obtain single-cell suspension. Live/Dead Fixable Aqua Dead Cell Stain Kit (Life Technologies) was used to exclude dead cells. Cellular surface staining was performed for 40 min on ice. After fixation and permeabilization using Intracellular Fixation & Permeabilization Buffer Set (eBioscience), intracellular staining was performed for 45 min at room temperature. Stained cells were analyzed by flow cytometry (FACS LSR II). The complete information for antibodies used in this study is summarized in Supplementary Table 1.
RNA sequencing analysis
Meso-CAR T cells were treated with PBS, 4662 cell-derived sEVs with or without PD-L1 knockout for 48 hrs. CAR T cells were then collected and total RNA was extracted using a PureLink RNA Mini Kit (Invitrogen, Cat#: 12183018A). RNA sequencing was performed in Novogen Co.. The DEGSeq R package (1.20.0) was used to analyze the differentially expressed genes for downstream heatmap plotting. The data generated in this study are publicly available in Gene Expression Omnibus at GSE230000.
Animal studies
All mice used in this study were purchased from The Jackson Laboratory and bred and housed in the vivarium at the University of Pennsylvania under pathogen-free conditions. Animal experiments were carried out according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. Power analysis for the mouse number used in this study was performed as previous studies (9, 26). To study the response of solid tumors to CAR T cells in vivo, 4662 cells (3 × 105 cells in 100 μl medium) were subcutaneously injected into 8-week-old female NSG mice. Female mice were used in our study to be consistent with the literatures. Meso-CAR T cells or MigR T cells were intravenously injected when tumors reached a volume of ~150 mm3. After the treatment of transduced CAR T cells or MigR T cells for 3 days, tumor tissues were collected for sEV purification as previously described (25). To study the inhibitory effect of tumor cell-derived sEV mesothelin on the Meso-CAR T cells, 4662 wild type or RABA27KO cells (3 × 105 cells in 100 μl medium) were subcutaneously injected into 8-week-old female NSG mice. Mice were allocated randomly to each treatment group. Transduced CAR T cells were intravenously injected when tumors reached a volume of ~150 mm3. To block mesothelin on sEVs, purified sEVs (500 μg) were incubated with anti-mesothelin blocking antibodies (10 μg/ml) or IgG isotype control antibodies (10 μg/ml) in 500 μl PBS. The sEVs were then washed with 30 ml PBS and pelleted by ultracentrifugation to remove the free antibodies. 25 μg of 4662-derived sEVs with or without the IgG isotype antibodies, or anti-mesothelin antibodies (10 μg/ml) were injected into mouse tail vein twice a week. The dose of the sEVs used in this study was chosen according to previous reports (26, 27). To investigate the combined effect of exosome inhibitors and CAR T cells in immunocompetent C57BL/6 mice, 4662 cells (3 × 105 cells in 100 μl medium) or MOC1 cells (5 × 105 cells in 100 μl medium) were subcutaneously injected into 8-week-old female C57BL/6 mice and the mice were allocated randomly to each group. When tumors reached a volume of ~150 mm3, mice were intravenous injected with 2.5 × 106 Meso-CAR T cells or MigR T cells with or without intraperitoneal injection of 2.5 mg/kg GW4869 (Sigma-Aldrich, Cat#: D1692) or Nexinhib20 (Sigma-Aldrich, Cat#: SML1919) three times a week. The doses of these two inhibitors were based on previously published studies (13, 15, 28). Tumor volume was measured using a digital caliper and calculated by the formula: length × (width)2/2. Before the longest dimension of the tumors reached 2.0 cm, the mice were euthanized. For flow cytometry analysis, tumor tissues were dissociated with 1 mg/ml type I collagenase in the presence of 50 U/ml RNase and DNase I to obtain single-cell suspensions. For C57BL/6 mice, single-cell suspensions from lymphatic nodes and spleens were also prepared. Flow cytometry was performed in a double-blind manner.
Statistical analysis
All the statistical analyses were performed using GraphPad Prism v.8.0 software. Each experiment was performed with three replications. For equal variance data, the significance of mean differences was determined using unpaired Student’s t-test (two groups) or one-way ANOVA with appropriate post-hoc tests (more than two groups); for data that differed in variance, unpaired t-test with Welch’s correction (two groups) or Welch’s ANOVA with appropriate post-hoc tests (more than two groups) was carried out. Two-way ANOVA was used to analyze mouse data among different groups. Error bars of data in this study represent mean ± s.d. A two-tailed value of P < 0.05 was considered statistically significant.
Data availability
The data generated in this study are publicly available in Gene Expression Omnibus at GSE230000. All other raw data are available upon request from the corresponding author.
Results
Increased level of PD-L1 on sEVs released from PDAC cells in response to Meso-CAR T cells
Using mouse splenic T cells, we generated Meso-CAR T cells that target mesothelin, a tumor-associated antigen expressed at high levels on various tumor cells such as pancreatic cancers (8, 23). T cells expressing an empty MigR vector were also generated as a control as described previously (22) (Supplementary Fig. 1A and B). We then established a mouse tumor model in NSG mice using PDAC 4662 cells known to express mesothelin (18). Meso-CAR T cells or MigR T cells were intravenously injected when tumors reached a volume of ~150 mm3. Tumor tissues were collected 3 days post-administration of Meso-CAR T cells or MigR T cells. We then purified sEVs from tumor tissues as previously described (25). Transmission electron microscopy (TEM) and nanoparticle tracking analysis (NTA) showed that tumor tissue sEVs had sizes ranging from 30 nm to 150 nm in diameter, which is characteristic of exosomes (Fig. 1A and B). We further isolated 4662 tumor cell-derived sEVs using Dynabeads conjugated with anti-mesothelin antibodies (Fig. 1C). Western blot analysis showed that Dynabeads bound to a significant amount of mesothelin+ sEVs (Fig. 1D). Next, we analyzed proteins in these mesothelin+ sEVs using Reverse Phase Protein Array (RPPA), an antibody-based quantitative proteomics technology (9, 29, 30). The RPPA analysis showed that PD-L1 was present on mesothelin+ sEVs isolated from the tumors from both MigR T cell-treated and Meso-CAR T cell-treated mice. However, the levels of PD-L1 were significantly higher in the Meso-CAR T cell-treated group (Fig. 1E). Western blot analysis further confirmed the upregulation of PD-L1 in the sEVs, which expressed the same amounts of mesothelin and exosome marker proteins (Hrs, CD63 and TSG101) (Fig. 1F). Collectively, these data showed that PDAC cells released sEVs carrying a higher level of PD-L1 in response to Meso-CAR T cell administration.
Figure 1. PDAC cells release sEVs that carry high levels of PD-L1 in response to Meso-CAR T treatment.
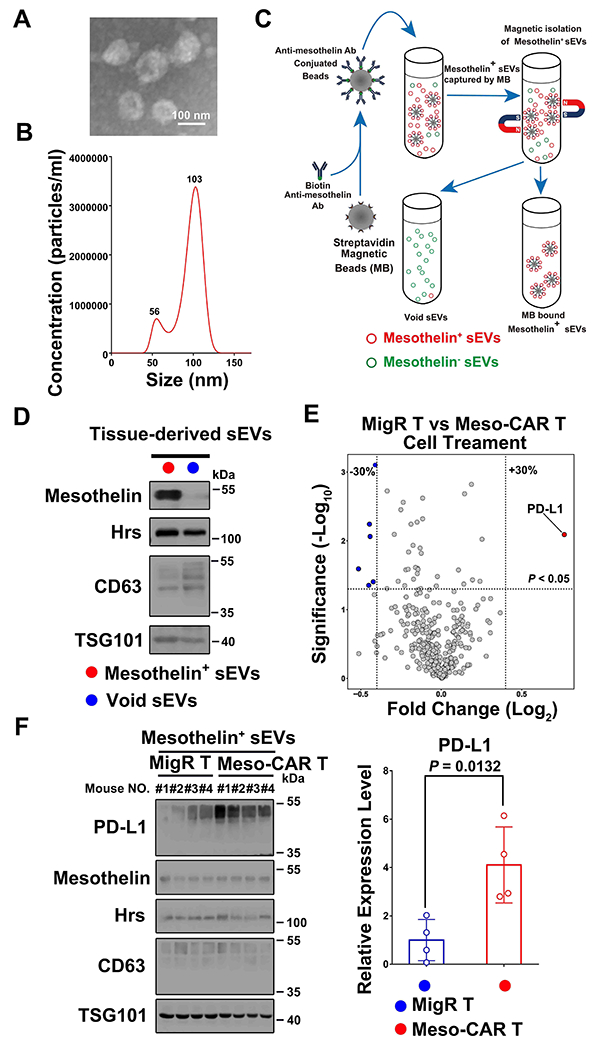
A, Representative electron microscopic image of sEVs isolated from tumor tissues. Scale bar, 100 nm. B, Characterization of sEVs purified from PDAC tumor tissues by nanoparticle tracking. The X-axis represents the diameters of the isolated vesicles; the Y-axis represents the concentration of isolated vesicles (particles/ml). C, Isolation of Mesothelin+ sEVs from PDAC tumor tissue-derived sEVs by magnetic beads. See Materials and Methods for details. D, Characterization of Mesothelin+ sEVs and remaining (“Void sEVs”) sEVs by western blotting. The expression levels of mesothelin and exosome marker proteins (Hrs, CD63 and TSG101) in indicated sEVs are shown. An equal amount of sEV proteins from the different fractions was loaded on the gel. E, Volcano plot of RPPA data displaying the pattern of protein expression comparing sEVs derived from PDAC tumors treated with MigR T cells or Meso-CAR T cells. The dotted horizontal line represents a significance level of P < 0.05, while dotted vertical lines represent differential expression differences of ± 30% (n=3). Each point represents the difference in the expression of each protein in the indicated sEVs. PD-L1 level was significantly higher in the sEVs from tumors with Meso-CAR T treatment than those with MigR T cell treatment. F, Western blot analysis of PD-L1, mesothelin and exosome marker proteins (Hrs, CD63 and TSG101) in purified tumor cell-derived Mesothelin+ sEVs from PDAC tumors with MigR T cell or Meso-CAR T cell treatment. All lanes were loaded with equal amounts of proteins. Quantification of the levels of PD-L1 in the sEVs is shown to the right (n=4). Data represent mean ± s.d. (n=3 or as indicated). Statistical analysis was performed using two-sided unpaired multiple t-test (E) or two-sided unpaired t-test (F).
PDAC cell-derived sEVs suppress Meso-CAR T cells
The induction of PD-L1 on PDAC tumor derived sEVs in response to Meso-CAR T cells led us to investigate whether these sEVs inhibit the function of CAR T cells. The sEVs from 4662 cells were purified by differential centrifugation and verified by TEM and NTA (Fig. 2A and B). Western blotting analysis showed that both mesothelin and PD-L1 were present on sEVs (Fig. 2C). Iodixanol density gradient centrifugation further showed that mesothelin and PD-L1 were presented in the same sEV fractions that contain exosome marker proteins CD63, Hrs and TSG101 (Fig. 2D). Immuno-electron microscopy (IEM) with antibodies against the extracellular domains of mesothelin and PD-L1 demonstrates that sEV PD-L1 and mesothelin have the same membrane topology as their cell surface counterparts, with their extracellular domains exposed on the surface of the same sEV (Fig. 2E). To examine the relationship between mesothelin and PD-L1 on sEVs, we isolated mesothelin+ sEVs from tumor cells using Dynabeads conjugated with anti-mesothelin antibodies. Western blot analysis showed that these beads enriched the mesothelin+ sEVs, and PD-L1 co-enriched on mesothelin+ sEVs (Fig. 2F and G). Even though it was impossible to remove all the mesothelin+ sEVs using these beads, PD-L1 level was significantly decreased in the remaining fraction (Fig. 2F and G), indicating a close correlation between PD-L1 and mesothelin in sEVs. Together with the immuno-EM analysis (Fig. 2E), the data strongly suggest that the majority of mesothelin+ sEVs carry PD-L1.
Figure 2. PDAC cell-derived sEVs inhibit Meso CAR T cells.
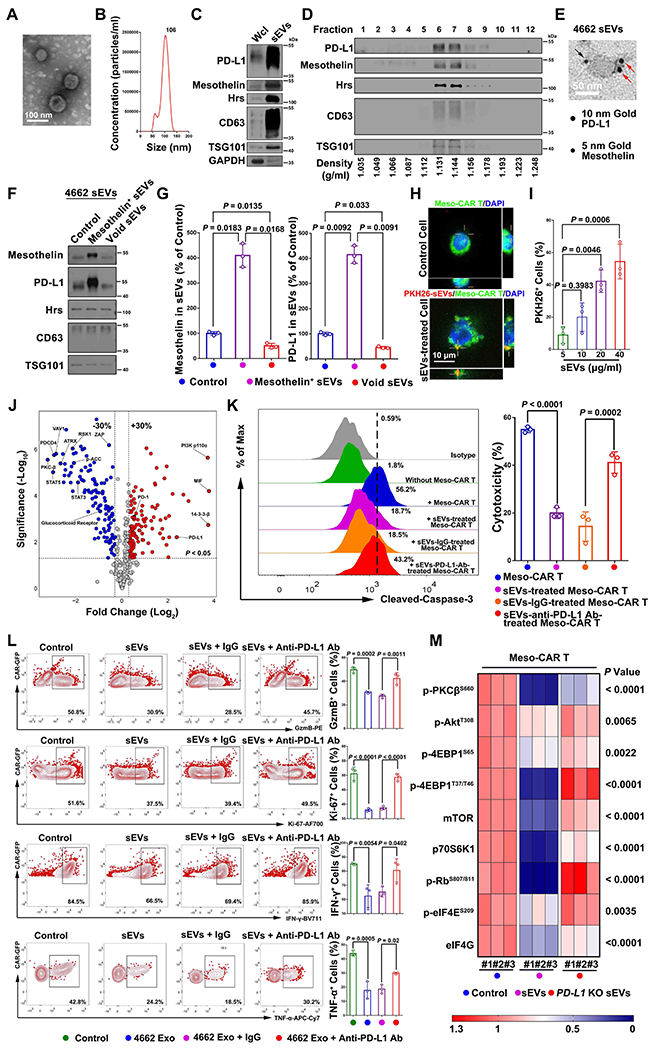
A, A representative EM image of purified sEVs from 4662 cells. Scale bar, 100 nm. B, Nanoparticle tracking analysis of sEVs purified from 4662 cells. The X-axis represents the diameters of the particles, and the Y-axis represents the concentration (particles/ml) of the sEVs. C, Western blot analysis of PD-L1, mesothelin and exosome marker proteins (Hrs, CD63 and TSG101) in the whole cell lysate (“Wcl”) and purified sEVs from 4662 cells. All lanes were loaded with an equal amount of proteins. D, PD-L1 and mesothelin co-fractionated with exosome marker proteins (Hrs, CD63 and TSG101) on iodixanol density gradients for 4662 cell-derived sEVs. E, A representative immuno-EM image of 4662 cell-derived sEV co-stained with anti-PD-L1 antibodies (conjugated with 10 nm gold particles, indicated by the red arrows) and anti-mesothelin antibodies (conjugated with 5 nm gold particles, indicated by the black arrows). Scale bar, 50 nm. F, Co-enrichment of PD-L1 on mesothelin+ sEVs. Proteins on mesothelin+ sEVs isolated by magnetic beads and the remaining sEVs (“Void sEVs”) were analyzed by western blotting. An equal amount of sEV proteins from the different fractions was loaded. G, Quantification of sEV mesothelin (left) and PD-L1 (right) expression in 4662 cell-derived sEVs. H, Confocal microscopy imaging showing the association of Meso-CAR T cells (green) with 4662 cell-derived sEVs labeled with PKH26 (red). Nuclei were stained with DAPI (blue). The interaction of sEVs on Meso-CAR T cells was indicated by crosses. Scale bar, 10 μm. I, The percentages of sEV-bound Meso-CAR T cells. Different amounts of sEVs were used in the binding assay. J, Volcano plot of RPPA data displaying the pattern of protein expression comparing Meso-CAR T cells with or without tumor sEVs treatment. Dotted vertical lines represent expression differences of 30%. Dotted horizontal lines represent a significance level of P < 0.05. K, Meso-CAR T cells treated with PBS, 4662 cell-derived sEVs with or without IgG isotype or PD-L1 antibody blocking, were co-cultured with 4662 cells for 48 hrs. Apoptosis of 4662 cells was assayed by flow cytometry using cleaved caspase-3. The relative cytotoxicity is shown to the right. L, Representative flow cytometric images of Meso-CAR T cells examined for the expression of GzmB, Ki-67, interferon gamma (IFN-γ) and tumor necrosis factor-α (TNF-α) after indicated treatments. Quantification of cells with GzmB, Ki-67, IFN-γ and TNF-α expression in Meso-CAR T cells after indicated treatments is shown to the right. M, Heatmap showing the expression levels of the representative proteins in Meso-CAR T cells with or with indicated sEVs treatment. Data represent mean ± s.d. (n=3). Statistical analysis is performed using one-way ANOVA analysis with Dunnett’s multiple comparison tests (G), two-sided unpaired multiple t-test (J), Welch ANOVA with Sidak’s T3 multiple comparison tests (I, K, L) or two-way ANOVA analysis with Tukey’s multiple comparison tests (M).
Next, we examined the effect of 4664 cell-derived sEVs on CAR T cells. First, using confocal fluorescence microscopy, we visualized a dose-dependent interaction of PKH26-labeled sEVs with Meso-CAR T cells (Fig. 2H and I). We then treated the mesothelin CAR T cells with sEVs and analyzed the profiles of signaling proteins in CAR T cells by RPPA. A number of proteins including those related to T cell activation (e.g. VAV1, PKC-β, RSK1, ZAP) and T cell survival (e.g. glucocorticoid receptor, STAT3, STAT5) were significantly down-regulated, whereas proteins involved in T cell exhaustion (e.g. PD-1, PD-L1, MIF), and regulatory T cell expression (e.g. PI3K p100α) (31) were up-regulated (Fig. 2J), suggesting that the sEVs had an inhibitory effect on CAR T cells. Treating Meso-CAR T cells with sEVs inhibited the cytotoxic effect of Meso-CAR T cells on 4662 cells in a dose- and time-dependent manner (Fig. 2K, Supplementary Fig. 1C and D), and the inhibitory effect was attenuated when the sEVs were pre-treated with an anti-PD-L1 blocking antibody (Fig. 2K). Moreover, sEVs inhibited the proliferation and function of Meso-CAR T cells in a dose- and time-dependent manner, as shown by the decreased expression of Ki-67, granzyme B (GzmB), interferon-γ (IFN-γ) and tumor necrosis factor (TNF-α) (Fig. 2L, Supplementary Fig. 1E and 1F). Pre-treatment of the sEVs with anti-PD-L1 antibodies, or sEVs derived from PD-L1 knockout (KO) cells, attenuated these effects (Fig. 2L, Supplementary Fig. 2A–C). The same inhibition pattern was observed when the CAR T cells were treated with a smaller amount of CD3/28 beads for their activation at lower levels (see Materials and Methods) (Supplementary Fig. 3A and B). In addition to the expression of Ki-67, GzmB, IFN-γ, and TNF-α, we also observed the inhibitory effect of sEVs by monitoring the expression of CD69 and CD25 on the CAR T cells (Supplementary Fig. 3C and D). Data from RNA-seq analysis further show that a series of genes including those related to T cell migration (CCL4), T cell growth (POLR1β, DKC1, RRS, etc.) were significantly down-regulated while other genes including T cell exhaustion (TGF1β1, TGF1β2, etc.), and regulatory T cell expression (SBSN, SDC1) were up-regulated, while PD-L1 KO attenuated these effects (Supplementary Fig. 3E). Similar observations were made for sEVs derived from another pancreatic cell line, 6419c5 cells (Supplementary Fig. 4, Supplementary Fig. 5A–D). Furthermore, we found that pancreatic cancer cells derived sEVs significantly inhibited the migration of Meso-CAR T cells (Supplementary Fig. 5E–H). Together, these data suggest that sEVs from pancreatic cancer cells inhibit the function of Meso-CAR T cells.
From the RPPA analysis, we found that the levels of proteins in the Akt-mTOR signaling pathway, including phospho-PKCβS660, phospho-AktT308, phospho-4EBP1S65, phospho-4EBP1T37/T46, mTOR, p70S6K and p-RbS807/811, were down-regulated in Meso-CAR T cells after treatment with sEVs, and this pattern was not observed in cells treated with sEVs from PD-L1 KO 4662 cells (Fig. 2M). Together, the data suggest that sEVs suppress the proliferation of the Meso-CAR T cells.
Tumor antigens promote the interaction between sEVs and CAR T cells
Next, we investigated whether tumor antigens on tumor derived sEVs are involved in the association of TEVs with CAR T cells. sEVs collected from PDAC cells (both 4662 cells and 6419c5 cells) were labeled with PKH26 and incubated with Meso-CAR T or the MigR T cells. The association of the sEVs with the cells was analyzed using flow cytometry. The level of sEVs association with Meso-CAR T cells was significantly higher than with the MigR T cells (Fig. 3A and B). Pre-treatment of sEVs with anti-mesothelin antibodies markedly inhibited Meso-CAR T cell-sEV association (Fig. 3C and D). Also, pre-treatment of Meso-CAR T cells with recombinant mesothelin significantly reduced the binding of sEVs to Meso-CAR T cells (Fig. 3E and F). In addition to PDAC cells, we also find that mesothelin is involved in the binding of murine oral squamous cell carcinoma MOC1 cell-derived sEVs to Meso-CAR T cells (Supplementary Fig. 6).
Figure 3. Mesothelin mediates the interaction between TEVs and CAR T cells.
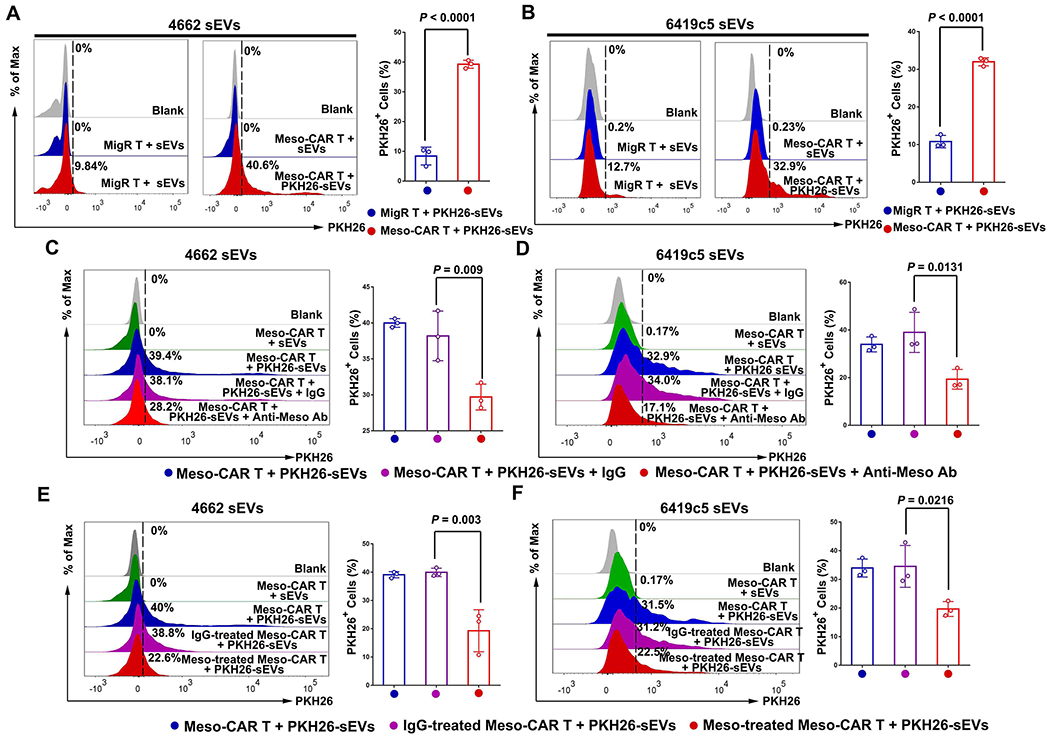
A and B, Representative images of flow cytometry of MigR and Meso-CAR T cells after incubation with PKH26-labeled 4662 cell-derived sEVs (A) and 6419c5 cell-derived sEVs (B). The percentages of sEV-bound MigR and Meso-CAR T cells are shown at the right. C and D, Representative images of flow cytometry of Meso-CAR T cells bound to PKH26-labeled-4662 cell-derived sEVs (C) and 6419c5 cell-derived sEVs (D) that were pre-treated with or without IgG or anti-mesothelin antibodies. The percentages of PKH26 positive Meso-CAR T cells are shown at the right. E and F, Representative images of flow cytometry of Meso-CAR T cells that bound to PKH26-labeled sEVs. Meso-CAR T cells were pre-treated with recombinant mesothelin and then incubated with PKH26-labeled sEVs derived from 4662 cells (E) and 6419c5 cells (F). The percentages of PKH26-positive Meso-CAR T cells are shown at the right. Data represent mean ± s.d. (n=3). Statistical analysis is performed using two-sided unpaired t-test (A, B) or one-way ANOVA analysis with Dunnett’s multiple comparison tests (C-F).
In addition to mesothelin-expressing tumors, we have also examined sEVs derived from human ovarian adenocarcinoma SKOV3 cells expressing HER2, mouse B16-F10 melanoma cells expressing CD19 (20), and NIH-3T3 cells expressing fibroblast activation protein (FAP) (22). HER2, CD19 and FAP regulated the adhesion of sEVs on their cognate HER2-CAR T, CD19-CAR T and FAP-CAR T cells, respectively (Supplementary Fig. 7 and Supplementary Fig. 8). These data suggest that CAR T-targeted antigens on sEVs interact with their matching CARs, which enable the preferential binding of their sEVs with CAR T cells.
Tumor antigens on sEVs mediate the preferential inhibition of CAR T cells
We next investigated the role of mesothelin on sEV-mediated CAR T cell inhibition. sEVs from 4662 cells had an inhibitory effect on both MigR T cells and Meso-CAR T cells, as shown by the decreased expression of Ki-67, GzmB, IFN-γ and TNF-α. However, a significantly stronger inhibitory effect of sEVs was observed on Meso-CAR T cells than on MigR T cells (Fig. 4A). Similarly, sEVs from 6419c5 cells and MOC1 cells also showed a stronger inhibitory effect on Meso-CAR T cells than MigR T cells (Supplementary Fig. 9). Anti-mesothelin antibodies significantly attenuated the inhibitory effect of sEVs on tumor killing (Fig. 4B, Supplementary Fig. 10), proliferation, and function (Fig. 4C–F, Supplementary Fig. 11 and Supplementary Fig. 12) of the Meso-CAR T cells. As controls, anti-mesothelin antibodies did not block the inhibitory effect of sEVs on MigR T cells (Supplementary Fig. 13). A similar pattern was observed for CD19-CAR T cells (20) in solid tumors formed by human CD19-expressing B16-F10 melanoma cells (Supplementary Fig. 14). Collectively, these data showed that CAR T-targeted antigen-mediated interaction enhanced the inhibitory effect of sEVs on the CAR T cells in solid tumors (Fig. 4G).
Figure 4. Mesothelin contributes to the preferential inhibition of CAR T cells by tumor sEVs.
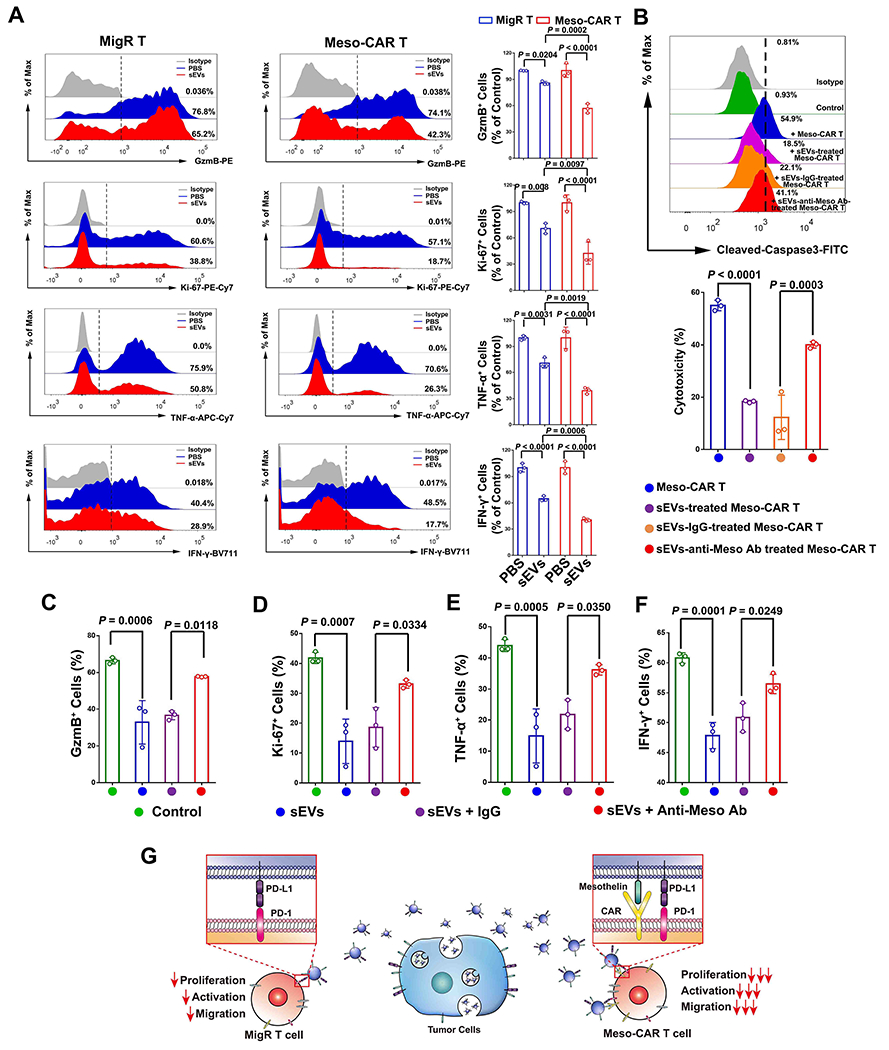
A, Representative flow cytometry analysis of MigR and Meso-CAR T cells examined for the expression of GzmB, Ki-67, IFN-γ and TNF-α after indicated treatments. Quantification of cells with positive GzmB, Ki-67, IFN-γ and TNF-α expression in Meso-CAR T cells after the indicated treatment is shown at the right. B, Meso-CAR T cells treated with PBS, sEVs, sEVs with or without IgG isotype or anti-mesothelin antibody blocking, were then co-cultured with 4662 cells for 48 hrs. Apoptosis of tumor cells detected by flow cytometry analysis of cleaved caspase-3 (upper panel), and the relative cytotoxicity is calculated (lower panel). C-F, Quantification of cells with positive GzmB (C), Ki-67 (D), TNF-α (E), IFN-γ (F) expression in Meso-CAR T cells after indicated treatments. G, Schema showing that CAR T-targeted tumor antigens on sEVs interact with CARs on CAR T cells, leading to the preferential inhibitory effect of sEVs on CAR T cells. Statistical analysis is performed using Welch ANOVA with Sidak’s T3 multiple comparison tests (A-F).
Mesothelin is required for sEV-induced CAR T cell immunosuppression in mice
Previous studies have established that Rab27a in tumor cells controls the secretion of exosomes, and is involved in the suppression of cytotoxic T cells (13, 32). To test the role of sEV mesothelin on Meso-CAR T cell suppression in vivo, we established an NSG mouse model using 4662 cells with or without RAB27A KO using the CRISPR/Cas9 system (Fig. 5A, Supplementary Fig. 15A). RAB27A KO markedly decreased the secretion of sEVs from 4662 cells without affecting the cell surface expression of mesothelin (Supplementary Fig. 15B and C). There was no significant difference in tumor growth between RAB27A KO and control 4662 tumor-bearing NSG mice treated with MigR T cells (Supplementary Fig. 15D). Next, Meso-CAR T cells were intravenously injected into NSG mice. Meso-CAR T cells had no significant effect on the growth of the control tumors, but significantly inhibited the growth of RAB27 KO tumors (Fig. 5B, Supplementary Fig. 15E). To further confirm the effect of Meso-CAR T cells on RAB27A KO tumors was dependent on the block of sEV secretion, purified sEVs were administrated to the mice by tail vein injection. Purified sEVs offset the effect of Meso-CAR T cells on RAB27A KO tumors in a dose-dependent manner (Supplementary Fig. 15F and G). Infusion of purified 25 μg sEVs derived from 4662 cells per mice twice a week completely offset the effect of Meso-CAR T cells on RAB27A KO tumors (Fig. 5B). Pre-treatment of sEVs with anti-mesothelin antibody, but not isotype control IgG, attenuated the inhibitory effect (Fig. 5B). Analysis of the weights of tumors harvested 5 days post tail vein injection of Meso-CAR T cells showed the same effect (Fig. 5C). RAB27A KO also significantly prolonged the survival of mice bearing tumors compared to the control group (Fig. 5D, Supplementary Fig. 15H), which was offset by sEVs injection. The anti-mesothelin antibodies, but not the IgG isotype control antibodies, attenuated the effect of sEVs on the survival (Fig. 5D).
Figure 5. Rab27 knockout attenuated the inhibitory effect of TEVs on Meso-CAR T cells.
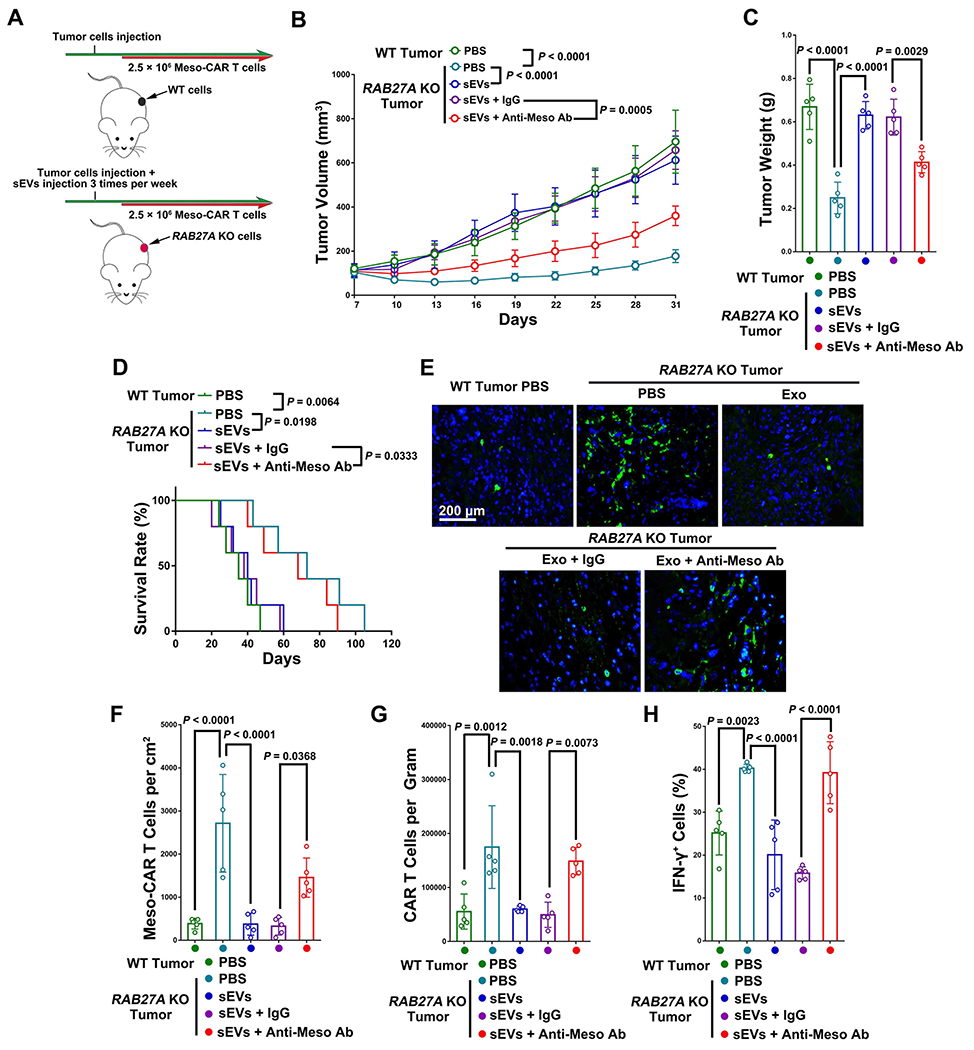
A, Schema of NSG mice with 4662 tumors or RAB27A KO 4662 tumors were treated with Meso-CAR T cells and injected with 4662 cell-derived sEVs with indicated treatments. See Materials and Methods for details. B, Growth curves of tumors in mice with indicated treatment. C, Tumor weights with indicated treatment. D, Survival curves of mice with indicated treatment. E, Representative immunofluorescence images of Meso-CAR T cells in tumor tissues. Scale bar, 200 μm. F, The number of Meso-CAR T cells for each group of mice quantified from (E). G, The number of Meso-CAR T cells in tumors for each group of mice quantified from flow cytometry analysis. H, The percentage of IFN-γ+ Meso-CAR T cells quantified by flow cytometry. Data represent mean ± s.d. (n=5). Statistical analysis is performed using Welch ANOVA with Dunnett’s T3 multiple comparison tests (B) or Welch ANOVA with Dunnett’s T3 multiple comparison tests (C, F-H) or log-rank test (D).
We next examined the infiltration of Meso-CAR T cells in tumors using immunofluorescence microscopy (Fig. 5E) and flow cytometry (Supplementary Fig. 15I). The level of infiltration was significantly higher in RAB27A KO tumors than in control tumors, and was significantly decreased with sEV infusion (Fig. 5E–G). Infusion of sEVs pre-treated with anti-mesothelin antibody attenuated this inhibitory effect. RAB27A KO also led to increased expression of IFN-γ of the infiltrating Meso-CAR T cells in the tumors, consistent with our in vitro studies. Tumor cell-derived sEVs inhibited IFN-γ expression in Meso-CAR T cells in the tumor site, which was also attenuated by anti-mesothelin antibody (Fig. 5H). Taken together, the results suggest that mesothelin is necessary for tumor sEV-mediated suppression of Meso-CAR T cells in vivo.
Inhibitors of exosome secretion improve the efficacy of CAR T treatment
To investigate the potential pharmacological inhibition of exosomes in CAR T therapy against solid tumors, two chemical inhibitors, GW4869 and Nexinhib20, which have been previously used for exosome inhibition in vitro and in vivo (13, 15, 28, 33, 34), were used in the treatment with Meso-CAR T cells in immunocompetent C57BL/6 mice with 4662 tumors (Fig. 6A). Both GW4869 and Nexinhib20 significantly reduced sEV secretion from 4662 cells without affecting mesothelin expression and mouse body weights (Supplementary Fig. 16). Meso-CAR T cells alone had no significant effect on the growth of tumors compared to the MigR T cells. The combination of Meso-CAR T cells and GW4869 showed the strongest inhibitory effect on tumor growth (Fig. 6B and C), and significantly prolonged the survival of the mice (Fig. 6D). We also examined the infiltration of Meso-CAR T cells in tumors using immunofluorescence microscopy (Fig. 6E) and flow cytometry (Supplementary Fig. 17A). The combination of Meso-CAR T cells and GW4869 significantly increased the infiltration of Meso-CAR T cells in tumors (Fig. 6E and F). In addition, GW4869 enhanced the activation of Meso-CAR T cells in the tumor site, as demonstrated by increased expression of IFN-γ (Fig. 6G). Similar results were obtained for Nexinhib20 (Supplementary Fig. 17B–F). In addition to PDAC cells, we also found that inhibition of exosome secretion by GW4869 or Nexinhib20 improved the efficacy of CAR T treatment in C57BL/6 mice with MOC1 tumors (Supplementary Fig. 18). GW4869 or Nexinhib20 also improved the efficacy of CAR T treatment in NSG mice with 4662 tumors (Supplementary Fig. 19). Collectively, our data suggest that inhibitors of exosome production have translational potential to enhance the efficacy of CAR T cell therapy in solid tumors.
Figure 6. GW4869 boosted the CAR T therapy in mouse PDAC model.
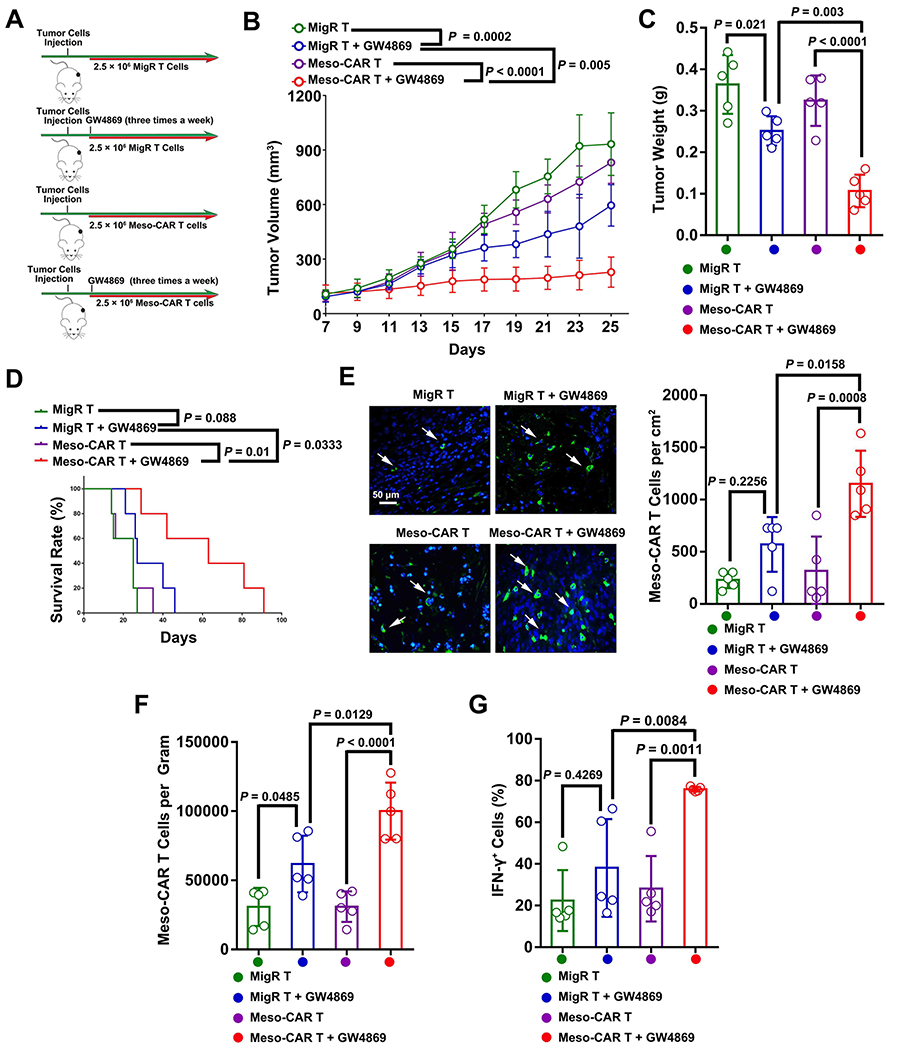
A, Schema of C57BL/6 mice with 4662 tumors were treated with MigR or Meso-CAR T cells with or without GW4869. See Materials and Methods for details. B, Growth curves of tumors in mice with indicated treatments. C, Tumor weights with indicated treatments. D, Survival rates of mice with indicated treatments. E, Representative IHC images of Meso-CAR T cells (green) in tumor tissues. Nuclei were stained with DAPI (blue). Scale bar, 50 μm. The number of Meso-CAR T cells for each group of mice quantified from IHC analysis is shown to the right. F, The number of Meso-CAR T cells in tumors for each group of mice quantified based on flow cytometry analysis. G, The percentage of IFN-γ+ Meso-CAR T cells quantified by flow cytometry. Data represent mean ± s.d. (n=5). Statistical analysis is performed using Welch ANOVA with Dunnett’s T3 multiple comparison tests (B, C, E-G) or log-rank test (D).
Combining Meso-CAR T cell treatment and exosome inhibitors improves the infiltration and activation of endogenous CD8 T cells
CD8 T cells play a central role in anti-tumor immunity. Activation of the anti-tumor function of endogenous CD8 T cells was shown to contribute to effective CAR T cell therapies (20). In the same PDAC mouse model as used above, GW4869 significantly enhanced the infiltration of CD8 T cells into the tumor sites in mice treated with Meso-CAR T cells (Fig. 7A). Flow cytometry analysis showed that GW4869 increased the infiltration of endogenous CD8 T cells (CD45+ CD3+ GFP− CD8+) into the tumor sites (Supplementary Fig. 20A, Fig. 7B). The combination of Meso-CAR T and GW4869 significantly increased IFN-γ expression in CD8 T cells compared to Meso-CAR T or GW4869 alone (Fig. 7C). The combination of Meso-CAR T and GW4869 also led to the strongest activation of endogenous CD8 T cells in spleen and sentinel lymph nodes, as demonstrated by the high levels of expression of IFN-γ (Fig. 7D and E). A similar effect was observed for the combination of Meso-CAR T and Nexinhib20 (Supplementary Fig. 20B–E). In C57BL/6 mice bearing MOC1 tumors, we also found that GW4869 or Nexinhib20 improved the infiltration and activation of endogenous CD8 T cells (Supplementary Fig. S21). Together, these results suggest that the combination of Meso-CAR T and exosome inhibitors improves the anti-tumor activity of endogenous CD8 T cells.
Figure 7. The combination of Meso-CAR T cells and GW4869 enhanced the activation of endogenous CD8 T cells in immunocompetent mice.
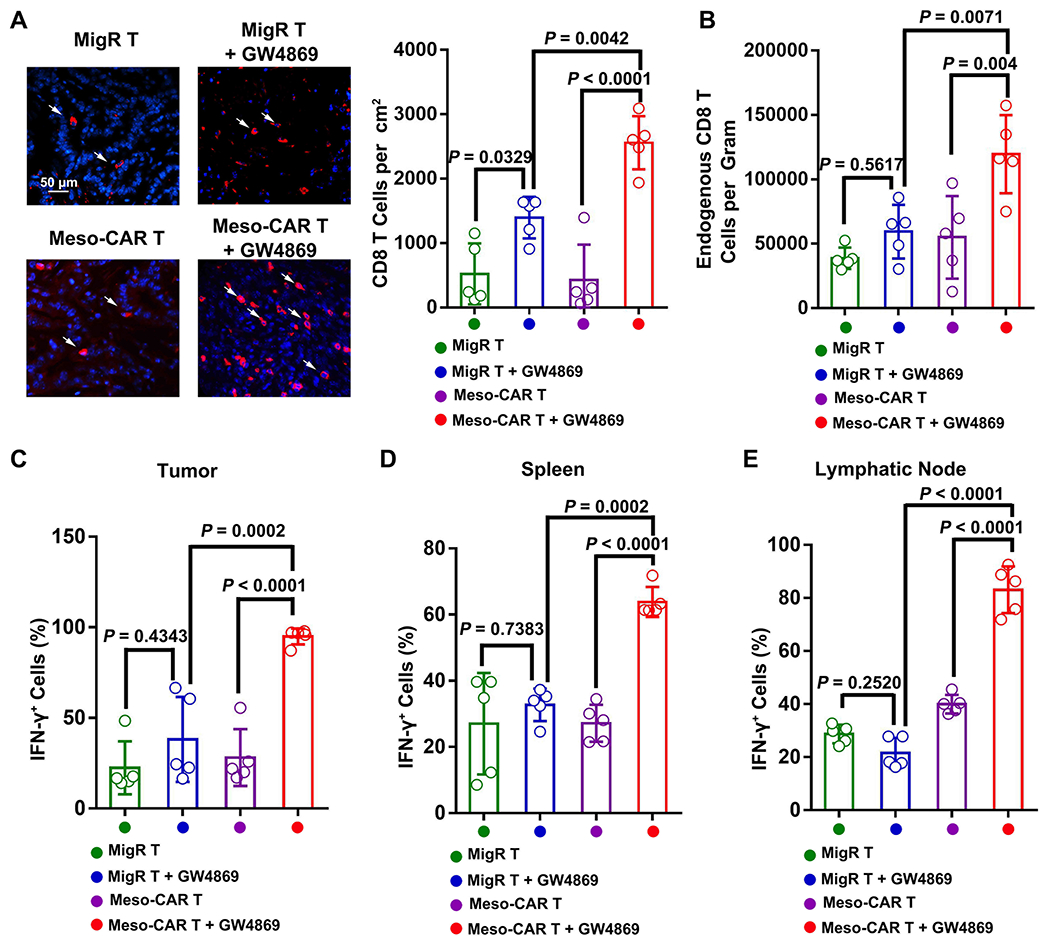
A, Representative immunofluorescence images of endogenous CD8 T cells in tumor tissues. Scale bar, 50 μm. The number of CD8 cells for each group of mice quantified from immunofluorescence analysis is shown at the right. B, The number of CD8 T cells in tumors for each group of mice quantified from flow cytometry analysis. C-E, The percentage of IFN-γ+ CD8 T cells in tumors (C), spleen (D), and lymphatic nodes (E) quantified from flow cytometry. Data represent mean ± s.d. (n=5). Statistical analysis is performed using Welch ANOVA with Dunnett’s T3 multiple comparison tests (A-E).
Discussion
Limited tumor infiltration and functional deficiency are two major issues in CAR T cell treatment of solid tumors. To develop effective new strategies to improve treatment efficacy, it is imperative to understand the molecular mechanisms by which tumors inhibit CAR T cells. Our current study led to the following findings: (1) Administration of CAR T cells induces higher levels of PD-L1 expression in sEVs released from tumor cells. (2) Tumor sEVs inhibited the migration, proliferation, and function of CAR T-cells in vitro and in vivo. (3) The tumor antigens targeted by CAR T cells are expressed on the surface of sEVs, contributing to the binding of immunosuppressive sEVs to the CAR T cells, and leading to the preferential inhibitory effect of sEVs on CAR T cells. (4) Blocking exosome secretion by RAB27A KO or chemical inhibitors boosted CAR T therapy in mouse models. (5) The combination of Meso-CAR T cells and exosome inhibitors improved the anti-tumor profile of endogenous CD8 T cells. Our study demonstrates a special vulnerability of CAR T cells to tumor-derived sEVs and suggests the potential application of exosome inhibitors for CAR T therapy against solid tumors.
In hematological tumors, EVs were implicated in cytokine release and CAR T cell activation and exhaustion (35, 36). In our study of solid tumors, we did not observe any stimulatory effect on CAR T cells. A recent study also observed that sEVs derived from neuroblastoma cells inhibited CAR T cells in vitro (37), consistent with our observations. Hematological tumors that are present in blood and the lymphatic system differ very much from solid tumors in respect to their extracellular matrix (ECM) (38–40), which has been speculated to contribute to the differences in CAR T treatments (1, 37, 41, 42). Tumor ECM contributes to the establishment of the TME that contains a high concentration of tumor-derived sEVs (38–40). The high concentration of sEVs in the TME would make a major difference in immune suppression and T cell infiltration, and thus different efficacy in treating solid tumors versus lymphoma or other blood tumors. Indeed, our study demonstrates that sEVs effectively inhibit CAR T cell infiltration and function in tumor tissues.
Using an antibody-based quantitative proteomics technology (RPPA), we found that PD-L1 was significantly up-regulated in tumor sEVs in a PDAC mouse model in response to Meso-CAR T cell treatment, suggesting an active defense mechanism for solid tumors to resist CAR T cells. CAR T cells release IFN-γ in response to tumors, which in turn leads to the increase of sEV PD-L1 levels (43, 44). Tumor sEVs pre-treated with anti-PD-L1 blocking antibodies or sEVs derived from PD-L1 KO cells show much attenuated suppressive effect, suggesting that PD-L1 on mesothelin+ sEVs induced by CAR T cell administration is crucial for the observed CAR T cell suppression. We show that the majority of PD-L1 was co-enriched with mesothelin on the same sEVs fractions. On the other hand, we do not rule out the possibility that there is a small mesothelin+ PD-L1− subpopulation of sEVs that is also involved in the suppression of CAR T cells.
Our proteomics analysis further suggests the inhibition of Akt-mTOR signaling in Meso-CAR T cells treated with sEVs. Akt/mTOR signaling pathway plays key roles in T cell differentiation, migration, metabolism, and survival (45, 46). Studies have also implicated that activated Akt is required for T cells to resist tumor-associated inhibitions (47, 48). The PD-L1-PD-1 interaction was shown to block the Akt activity in T cells (49, 50). Our results suggest that the interaction between sEV PD-L1 with PD-1 leads to the inhibition of the Akt-mTOR signaling pathway in CAR T cells, resulting in their decreased proliferation, activation, migration.
Our data suggest opposing roles that tumor antigens play in CAR T therapy. In principle, a tumor antigen should be expressed on tumor cell surface and CARs bind to the targeted tumor antigen with high affinity, which lays the foundation for a CAR T therapy (4, 8, 49, (51). However, when tumor antigens are loaded on sEVs together with PD-L1, an inhibitory effect is delivered. The expression of both tumor antigens and PD-L1 on the same sEVs allows these sEVs to bind efficiently and specifically bind to CAR T cells and inhibit their proliferation and function. In this sense, the antigens on sEVs actually confer a vulnerability to CAR T cells.
Over the past decade, strategies to enhance the effect of CAR T cells, including targeting a different tumor antigen, targeting of multiple antigens, and combination of CAR T cells with checkpoint blockade, have been developed (7, 20, 23, 41, 52). These strategies mostly focus on strengthening the activation and function of CAR T cells through the antigens. While promising, these strategies have shown limited efficacy or severe side effects (6, 36, 41, 42, 53). The benefits of blocking PD-L1 pathway for CAR T cell therapy have been shown in treating solid tumors (54, 55). However, only short-term clinical remission was observed (55, 56). Furthermore, combining CAR T treatment with PD-1/PD-L1 blockade has the risk of increased toxicity as the PD-1/PD-L1 signaling is also required to contain autoimmunity (57). A combination of exosome secretion blockers with CAR T therapy has not been tested. Our study showed that the application of exosome inhibitors, GW4869 and Nexinhib20, significantly improved the function of CAR T cells in the pancreatic cancer model. Solid tumor microenvironment is characterized by low levels of infiltration of lymphocytes, and an optimal CAR T-cell therapy should have the capability to activate host immune cells (20, 58). The combination of CAR T cells with exosome inhibitors systematically drives the infiltration and activation of endogenous CD8 T cells in our mouse model. Our study suggests that inhibition of immunosuppressive exosome production enhances the efficacy of CAR T cell therapy in solid tumors.
In summary, our study demonstrates that solid tumors produce sEVs carrying tumor antigens as well as immune checkpoint molecules that act as cell-free units to suppress the anti-tumor function of CAR T cells. Targeting sEV secretion may provide a new strategy to improve CAR T cell therapy in solid tumors. Further study of the molecular composition of the sEVs and their relationship with CAR T cells is important for a better understanding of CAR T cell function in solid tumors and guide our effort to develop new strategies that reshape the TME and anti-tumor immunity for effective CAR T cell therapy in cancer patients.
Supplementary Material
SIGNIFICANCE.
Small extracellular vesicles secreted by solid tumors inhibit CAR T cells, which provides a molecular explanation for CAR T cell resistance and suggests that strategies targeting exosome secretion may enhance CAR T-cell efficacy.
Acknowledgements
We are grateful to Dr. John Scholler (University of Pennsylvania) for providing the GFP-Meso-CAR plasmid, Dr. Steven Albelda (University of Pennsylvania) for the MOC1 cells, and Dr. Andy Minn (University of Pennsylvania) for B16-F10-CD19 mouse melanoma cells. This work was supported by NIH R35 GM141832 to Wei Guo, NCI P50 CA261608 grant to Wei Guo, Meenhard Herlyn and Xiaowei Xu, and NCI P01 CA217805 and a sponsored research agreement from TMUNITY Therapeutics to Ellen Puré.
Footnotes
Declaration of interests: The authors declare no conflicts of interest.
References
- 1.Hou AJ, Chen LC, Chen YY. Navigating CAR-T cells through the solid-tumour microenvironment. Nat Rev Drug Discov. 2021;20:531–50. [DOI] [PubMed] [Google Scholar]
- 2.Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21:145–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568:112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. 2018;378:449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ko AH, Jordan AC, Tooker E, Lacey SF, Chang RB, Li Y, et al. Dual Targeting of Mesothelin and CD19 with Chimeric Antigen Receptor-Modified T Cells in Patients with Metastatic Pancreatic Cancer. Mol Ther. 2020;28:2367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morello A, Sadelain M, Adusumilli PS. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer discovery. 2016;6:133–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan Y, Che X, Qu J, Hou K, Wen T, Li Z, et al. Exosomal PD-L1 Retains Immunosuppressive Activity and is Associated with Gastric Cancer Prognosis. Ann Surg Oncol. 2019;26:3745–55. [DOI] [PubMed] [Google Scholar]
- 11.Kim DH, Kim H, Choi YJ, Kim SY, Lee JE, Sung KJ, et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Experimental & molecular medicine. 2019;51:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monypenny J, Milewicz H, Flores-Borja F, Weitsman G, Cheung A, Chowdhury R, et al. ALIX Regulates Tumor-Mediated Immunosuppression by Controlling EGFR Activity and PD-L1 Presentation. Cell Rep. 2018;24:630–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poggio M, Hu T, Pai CC, Chu B, Belair CD, Chang A, et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell. 2019;177:414–27 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ricklefs FL, Alayo Q, Krenzlin H, Mahmoud AB, Speranza MC, Nakashima H, et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Science advances. 2018;4:eaar2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Li CW, Chan LC, Wei Y, Hsu JM, Xia W, et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018;28:862–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gargiulo E, Viry E, Morande PE, Largeot A, Gonder S, Xian F, et al. Extracellular Vesicle Secretion by Leukemia Cells In Vivo Promotes CLL Progression by Hampering Antitumor T-cell Responses. Blood cancer discovery. 2023;4:54–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong W, Guo W. Immunosuppressive Extracellular Vesicles in CLL. Blood cancer discovery. 2023;4:5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015;75:2800–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity. 2018;49:178–93 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson LR, Lee DY, Eacret JS, Ye D, June CH, Minn AJ. The immunostimulatory RNA RN7SL1 enables CAR-T cells to enhance autonomous and endogenous immune function. Cell. 2021;184:4981–95 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castellarin M, Sands C, Da T, Scholler J, Graham K, Buza E, et al. A rational mouse model to detect on-target, off-tumor CAR T cell toxicity. JCI insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer immunology research. 2014;2:154–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Good CR, Aznar MA, Kuramitsu S, Samareh P, Agarwal S, Donahue G, et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell. 2021;184:6081–100 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015;75:3596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cianciaruso C, Beltraminelli T, Duval F, Nassiri S, Hamelin R, Mozes A, et al. Molecular Profiling and Functional Analysis of Macrophage-Derived Tumor Extracellular Vesicles. Cell Rep. 2019;27:3062–80 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W, Zhong W, Wang B, Yang J, Yang J, Yu Z, et al. ICAM-1-mediated adhesion is a prerequisite for exosome-induced T cell suppression. Dev Cell. 2022;57:329–43 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madeo M, Colbert PL, Vermeer DW, Lucido CT, Cain JT, Vichaya EG, et al. Cancer exosomes induce tumor innervation. Nat Commun. 2018;9:4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hennessy BT, Lu Y, Gonzalez-Angulo AM, Carey MS, Myhre S, Ju Z, et al. A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clinical proteomics. 2010;6:129–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu H, Liu S, Zhang G, Bin W, Zhu Y, Frederick DT, et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature. 2017;550:133–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aragoneses-Fenoll L, Ojeda G, Montes-Casado M, Acosta-Ampudia Y, Dianzani U, Portoles P, et al. T-Cell-Specific Loss of the PI-3-Kinase p110alpha Catalytic Subunit Results in Enhanced Cytokine Production and Antitumor Response. Front Immunol. 2018;9:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12:19-30; sup pp 1–13. [DOI] [PubMed] [Google Scholar]
- 33.Ding H, Li LX, Harris PC, Yang J, Li X. Extracellular vesicles and exosomes generated from cystic renal epithelial cells promote cyst growth in autosomal dominant polycystic kidney disease. Nat Commun. 2021;12:4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen L, Charrier A, Zhou Y, Chen R, Yu B, Agarwal K, et al. Epigenetic regulation of connective tissue growth factor by MicroRNA-214 delivery in exosomes from mouse or human hepatic stellate cells. Hepatology. 2014;59:1118–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cox MJ, Lucien F, Sakemura R, Boysen JC, Kim Y, Horvei P, et al. Leukemic extracellular vesicles induce chimeric antigen receptor T cell dysfunction in chronic lymphocytic leukemia. Mol Ther. 2021;29:1529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu X, Hu H, Xiao Y, Li Q, Zhong Z, Yang J, et al. Tumor-derived extracellular vesicles induce invalid cytokine release and exhaustion of CD19 CAR-T Cells. Cancer Lett. 2022;536:215668. [DOI] [PubMed] [Google Scholar]
- 37.Ali S, Toews K, Schwiebert S, Klaus A, Winkler A, Grunewald L, et al. Tumor-Derived Extracellular Vesicles Impair CD171-Specific CD4(+) CAR T Cell Efficacy. Front Immunol. 2020;11:531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huleihel L, Hussey GS, Naranjo JD, Zhang L, Dziki JL, Turner NJ, et al. Matrix-bound nanovesicles within ECM bioscaffolds. Science advances. 2016;2:e1600502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Y, Deng W, McGinley EC, Klinke DJ, 2nd. Melanoma exosomes deliver a complex biological payload that upregulates PTPN11 to suppress T lymphocyte function. Pigment cell & melanoma research. 2017;30:203–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sung BH, Ketova T, Hoshino D, Zijlstra A, Weaver AM. Directional cell movement through tissues is controlled by exosome secretion. Nat Commun. 2015;6:7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown CE, Mackall CL. CAR T cell therapy: inroads to response and resistance. Nat Rev Immunol. 2019;19:73–4. [DOI] [PubMed] [Google Scholar]
- 42.June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med. 2018;379:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017;19:1189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, et al. CAR T cell killing requires the IFNgammaR pathway in solid but not liquid tumours. Nature. 2022;604:563–70. [DOI] [PubMed] [Google Scholar]
- 45.Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol. 2011;11:109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun J, Dotti G, Huye LE, Foster AE, Savoldo B, Gramatges MM, et al. T cells expressing constitutively active Akt resist multiple tumor-associated inhibitory mechanisms. Mol Ther. 2010;18:2006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Newick K, O’Brien S, Sun J, Kapoor V, Maceyko S, Lo A, et al. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer immunology research. 2016;4:541–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol. 2013;33:3091–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gudipati V, Rydzek J, Doel-Perez I, Goncalves VDR, Scharf L, Konigsberger S, et al. Inefficient CAR-proximal signaling blunts antigen sensitivity. Nat Immunol. 2020;21:848–56. [DOI] [PubMed] [Google Scholar]
- 52.Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. 2020;367:446–53. [DOI] [PubMed] [Google Scholar]
- 53.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–5. [DOI] [PubMed] [Google Scholar]
- 54.Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nature biotechnology. 2018;36:847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther. 2017;25:2214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell. 2019;36:471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alizadeh D, Wong RA, Gholamin S, Maker M, Aftabizadeh M, Yang X, et al. IFNgamma Is Critical for CAR T Cell-Mediated Myeloid Activation and Induction of Endogenous Immunity. Cancer discovery. 2021;11:2248–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are publicly available in Gene Expression Omnibus at GSE230000. All other raw data are available upon request from the corresponding author.