

Abstract
Cellular machineries that drive and regulate gene expression often rely on the coordinated assembly and interaction of a multitude of proteins and RNA together called ribonucleoprotein complexes (RNPs). As such, it is challenging to fully reconstitute these cellular machines recombinantly and gain mechanistic understanding of how they operate and are regulated within the complex environment that is the cell. One strategy for overcoming this challenge is to perform single molecule fluorescence microscopy studies within crude or recombinantly supplemented cell extracts. This strategy enables elucidation of the interaction and kinetic behavior of specific fluorescently labeled biomolecules within RNPs under conditions that approximate native cellular environments. In this review, we describe single molecule fluorescence microcopy approaches that dissect RNP-driven processes within cellular extracts, highlighting general strategies used in these methods. We further survey biological advances in the areas of pre-mRNA splicing and transcription regulation that have been facilitated through this approach. Finally, we conclude with a summary of practical considerations for the implementation of the featured approaches to facilitate their broader future implementation in dissecting the mechanisms of RNP-driven cellular processes.
Graphical Abstract:
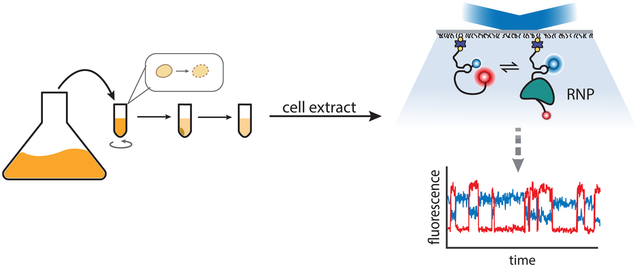
The thermodynamic and kinetic behavior of ribonucleoprotein complexes (RNPs) can be probed using cell extracts and single-molecule fluorescence microscopy techniques to approximate the cellular environment.
1. INTRODUCTION
The transfer of genome-encoded information into functional proteins and the complex regulatory mechanisms that control gene expression across all domains of life rely on the activity of ribonucleoprotein complexes (RNPs) with RNA-binding proteins (RBPs) at their core. For example, prokaryotic clustered regularly interspersed short palindromic repeat (CRISPR) based adaptive immunity mechanisms and eukaryotic gene regulatory mechanisms such as RNA splicing and RNA interference are each RNP-driven processes. In the CRISPR-mediated gene silencing mechanisms of bacteria and archaea, RNPs composed of a guide RNA and a CRISPR-associated endonuclease can target specific DNA cleavage sites, thereby blocking gene expression at the DNA level (1, 2). In eukaryotes, precursor mRNA (pre-mRNA) splicing relies on the formation of large RNP complexes that assemble into a sequence of stable spliceosome assemblies to mediate the identification of splice sites, excision of introns, and ligation of exons leading to mature mRNA synthesis (3). In the RNA silencing mechanisms of eukaryotes, RNPs known as RNA-induced silencing complexes (RISC), formed through the association of microRNAs (miRNAs) or small interfering (si)RNAs with Argonaute (AGO) proteins, target RNA transcripts for translation regulation and/or degradation (4). In addition, PIWI-interacting (pi)RNAs form RNP complexes with PIWI proteins of the Argonaute family and maintain germline genomic integrity by repressing transposon activity (5). Decades of detailed genetic, biochemical, and structural studies have largely elucidated the composition and molecular events carried out by these and other gene regulatory mechanisms. Yet what remains less clear is how RNP-mediated gene regulatory and gene expression mechanisms are influenced by the complex cellular environment.
Short-lived, often non-specific interactions between RBPs and potential targets dominate molecular interactions in the complex, crowded intracellular environment. Furthermore, RNPs rely on a network of specifically interacting regulatory components. RNP activity will thus be sensitive to small changes in their concentration and local cellular properties, especially those resulting from processes such as macromolecular phase separation (6, 7). All these factors may significantly perturb RNP-mediated mechanisms in cells. In eukaryotic gene silencing mechanisms, for example, it remains unclear how the activity of miRNAs or RISC-associated RBPs is modulated by competitive binding to untranslated regions (UTRs) of non-target RNAs (8). The accessibility of RBP target sites is also influenced by the conformational dynamics inherent to RNAs and RBPs. In pre-mRNA splicing for example, local RNA secondary structural perturbations can either enhance or inhibit the interactions with splicing factors, leading to direct effects on the splicing efficiency or identity of spliced products, with associated downstream effects (9). Thus, one important aspect of studying native biochemical behavior of molecular RNP complexes is designing assays that approximate the complex environment of the cell and allow for characterization of the role that RNA conformational dynamics plays on their mechanisms.
Recent advances in single molecule fluorescence microscopy techniques such as fluorescence co-localization and single-molecule Förster resonance energy transfer (smFRET) have made it possible to follow coupled and non-equilibrium processes in gene expression (10, 11). Studies with these techniques often follow two experimental designs: in vitro experiments with fully reconstituted, recombinant systems or experiments using live cells. Yet, each of these approaches impose significant constraints on studying complex RNP-driven mechanisms. On the one hand, in vitro experiments using purified proteins and mixtures of purified biomolecules offer a large degree of control and reproducibility. This allows one to address very targeted aspects of assembly dynamics and molecular rates. Yet, the large number of components and complex assemblies of much of the cellular expression machinery often precludes them from purification of single components and their functional reconstitution. Single molecule microscopy studies in cells, on the other hand, offer an alternative to fully reconstituted systems. These studies can elucidate the timing and localization of particular components within larger RNP assemblies, as in the case of tracking miRNA assembly within RNA-induced silencing complexes (12). However, due to the immense complexity of cells, biochemical RNP interactions cannot be well controlled in composition or environmental conditions, making it difficult to perturb these systems in a way that leads to direct insights about the molecular interactions underlying biological functions. Imaging within cells also raises significant measurement challenges such as limited fluorophore longevity, making it very difficult to address targeted mechanistic questions about RNP assembly and dynamics.
An alternative approach for approximating complex cellular environments that has proven effective for characterization of complex RNP biology is the reconstitution of the desired RNP system within cell extracts. Cell extracts have enabled seminal discoveries in RNA biology, including the first mechanistic insights of RNA silencing (13) and various mechanistic aspects of RNA splicing (14, 15). In general, cellular extracts are prepared by culturing the targeted cell type up to the desired scale, lysing the cells, and subjecting the lysate to repeated centrifugal cycles to isolate the soluble fraction. Combining the use of reconstituted RNPs within cell extracts with biophysical techniques that allow imaging and tracking of RNP binding interactions can provide a way to discern RNP mechanisms in approximated native conditions, especially in cases where not all components can be made recombinantly.
Designing in vitro experiments that combine single molecule approaches with cell extracts therefore is a practical alternative to studying the complex reaction pathways of cellular machineries in close-to-native cellular environments with a high degree of control. In contrast to single molecule experiments performed using highly purified biomolecules, in these experiments the reaction trajectories of individually labeled biomolecules within cell extracts are followed. This approach has been applied extensively to large RNP systems that cannot (yet) be reconstituted from purified components, such as yeast and human spliceosomes. The strategy overcomes the substantial technical challenges of detailed biochemical RNP characterization in whole cells, while maintaining some important aspects of the native cellular environment such as physiologically relevant compositions that lead to RNA processing. This is a powerful strategy for mapping the molecular mechanisms of low-abundance or highly transient interactions within biomolecular pathways as has been shown for splicing biochemistry over many years and can be applied to many other RNP-processes.
In this review, we survey single molecule strategies for investigating genomic processes in crude or partially reconstituted cell extracts. We start by providing a historical context for the use of cell extracts to approximate RNP assembly and behavior within a cellular-like environment. We subsequently describe the single molecule fluorescence microscopy approaches that have enabled mechanistic understanding of RNP biology in cellular extracts and highlight some recent insights that these approaches have generated. Finally, we conclude with a summary of practical considerations and experimental designs to follow biomolecule trajectories in cell extracts in order to facilitate future discoveries using this approach.
2. STUDYING MULTI-COMPONENT RNP SYSTEMS IN VITRO AND IN LIVE CELLS
2.1. Historical perspective and the need to shift towards studying RNP biochemistry in cellular environments
Historically, in vitro purification has been pivotal in the characterization and evaluation of biomolecular systems. While originally restricted to only the most robust enzymes (16), the sophistication of this approach has matured for over 150 years. Experiments elucidating the biochemistry of RNPs can be traced back to work in the 1950s on the ribosome (17). Piece-by-piece interrogation of ribosome assembly provided the basis for our understanding of the chemical foundations of the world’s most conserved RNP (18). Since those early days, there have been great strides in RNP component structure determination (19, 20), mapping of component interactions (21, 22), descriptions of intramolecular motion (23), and assays of enzymatic activity (24). These biochemical methods provided a methodical approach to understanding the capabilities and properties of multi-component RNP molecular machines like the ribosome.
While these approaches provide a clear window into the nature of RNPs, they are insufficient to explain the scope of interactions in the cellular context. By necessity, in vitro experiments simplify the composition and physicochemical conditions of reactions relative to the complex intracellular milieu. As a result, in many cases they only partially recapitulate in vivo behaviors (25, 26). An alternative is to incorporate the extracted contents of cells that may play a necessary biological role within the system being studied in an otherwise in vitro experiment.
Over time, cell extracts have been used to probe a diverse set of biological questions. One strategy is to use them when the biological process under study is too complex, in terms of biochemical components or knowing their exact concentration for in vitro work. A classic example is the study of effects of sperm nuclei stimulating multiple facets of the mitotic cycle when exposed to extracts from ova in Xenopus laevis (27). More recently, extracts from Drosophila embryos have been used to study the regulation of protein translation through development by repressive granules (28). Another example highlighting the specificity that cell extracts allow is the study of patient-specific misregulation resulting in neurodegeneration by studying amyloid formation in extracts from patient-derived tissue (29). In this instance, patient cell extract was used to collect and form experimentally tractable aggregates for insights into neurodegeneration. Recently, cell extracts have additionally been used for biophysical characterization of protein stability. By mimicking the complexity of the cellular environment, the authors were able to approximate the effect of cellular components showing that the stability of cold shock protein B monotonically increased with extract concentration (30).
The utility of using cell extracts to help investigate RNP biology may become apparent considering these and other examples, yet advances are still not trivial. For instance, mRNAs can interact dynamically with >1,500 RNA-binding proteins throughout their lifetimes with varying affinities. These interaction partners decorate mRNAs in complex temporal orders (10, 31). Consequently, studying specific RNP processes in the backdrop of this complexity remains an ambitious endeavor.
Over the past 10 years, live-cell single molecule fluorescence methods have made great strides towards the goal of understanding close-to-native molecular behavior with developments and increasingly widespread adoption of live-cell single particle tracking, molecular tethering (32, 33) and multi-color colocalization (34–36). Innovations in intracellular site-specific labeling of both RNAs and proteins (35), genetic manipulation methods, and constant improvements in imaging technologies suggest that live-cell single-molecule methods are on their way to become increasingly widely adopted, heralding a new paradigm of intracellular biochemistry (37).
Despite recent advances, intracellular live-cell studies are still challenging, particularly for the study of large multi-component systems. Even with the maturation of CRISPR-based tools for gene editing, creating cell lines with mutations and deletions on a large scale, especially in essential proteins, still remains out of reach. Further, many important RNP complexes such as the ribosome are expressed at high copy numbers, making them challenging to study at single molecule resolution due to optical crowding during microscopic imaging. Moreover, molecular conformational dynamics remain difficult to study in live cells using smFRET due to rapid photobleaching. Given these challenges, combining single molecule fluorescence methods to image and track RNP biology within cell extracts provides a more tractable approach to dissecting RNP mechanisms in a cell-like environment that offers a greater degree of control.
2.2. Cell Extracts: From crude to partially reconstituted
Whole cell extracts range from crude to partially reconstituted, the latter of which involve modifying crude lysates by the addition of purified components to reconstitute the RNP system of interest (Figure 1). The absence of biochemical manipulations to overexpress or isolate components within cell extracts allows for them to potentially retain endogenous RNP concentrations, or at least relative levels, as well as the biomolecular complexity and macromolecular crowding that approximates the cellular environment (38). Crude extract preparations containing genomic alterations such as deletions or additions of specific components are typically achieved by genetically manipulating parental cells (39, 40). For example, in order to characterize the role of small nuclear RNAs in the spliceosome, a number of yeast strains have been created containing genetically engineered cDNA complementary regions that lead to RNase H mediated depletion of the targeted endogenous RNA (41–45). Crude extracts can also be fractionated, thereafter referred to as fractionated extracts, to enrich for components from subcellular compartments, such as nuclear versus cytosolic extracts (46). For example, the recently developed direct analysis of ribosome targeting technology (DART) leverages the use of fractionated whole cell extracts in combination with next generation sequencing to determine the role of 5’ UTR elements on translation (47).
Figure 1:
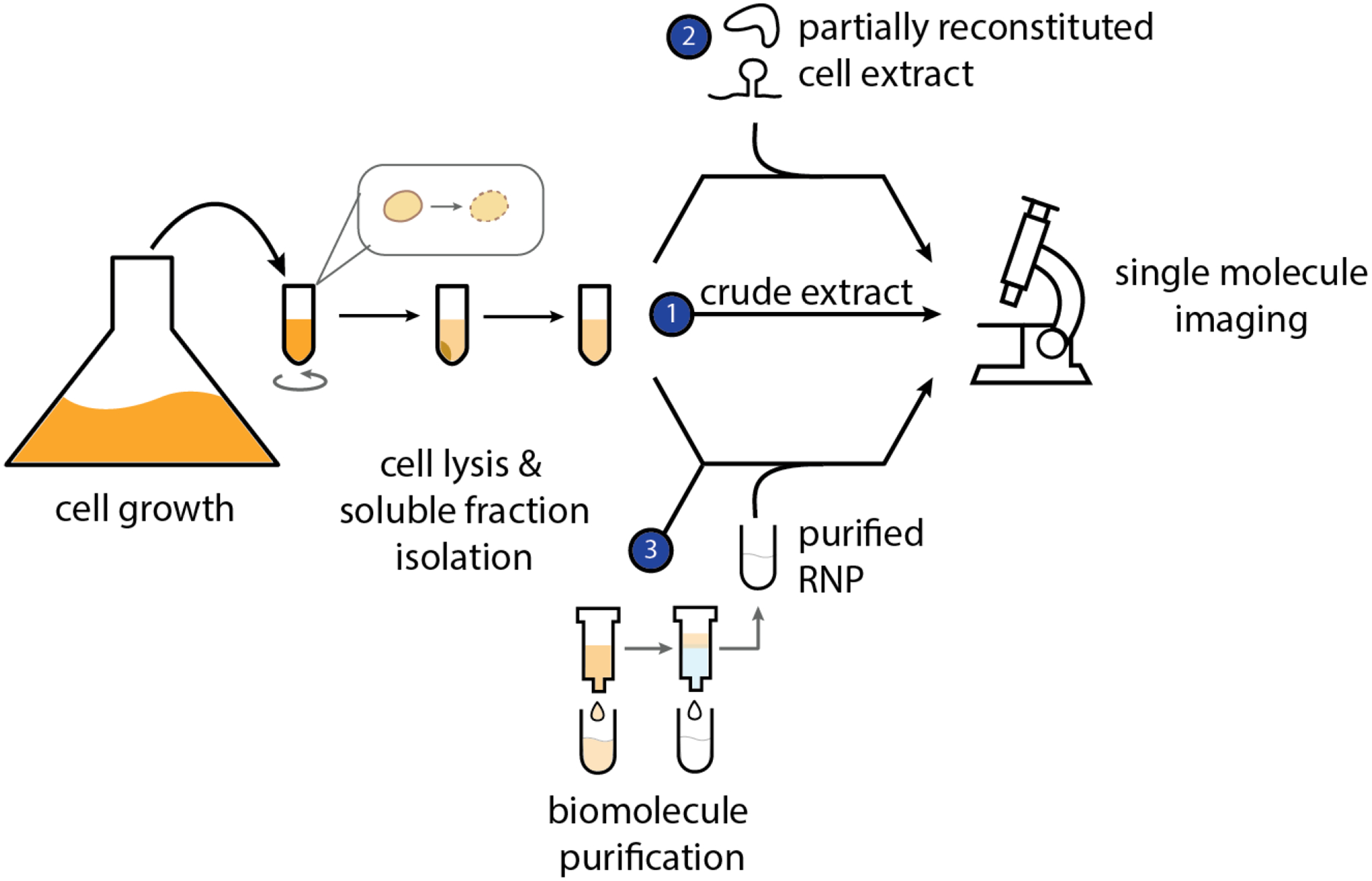
Cell extract versus purified RNP preparations for single molecule characterization. Prior to single molecule microscopy imaging, cell extract is prepared by culturing a cell line of choice to a high density and subsequently subjecting the cells to multiple rounds of centrifugation. (1) The isolated soluble fraction makes up crude cell extract. (2) In order to follow RNP interactions with other biomolecules, crude extracts can be supplemented with purified biomolecules thereby creating partially reconstituted cell extracts. (3) If desired and/or possible, crude extract can be further purified using traditional biomolecule purification strategies (e.g. affinity tags, ion exchange, gel filtration, etc.) to fully isolate and image the RNP of choice in the absence of complex cellular extract components.
While crude and fractionated cell extracts have proven useful in providing mechanistic understanding of various RNA processes, the protocols to prepare them can be complex and arduous, often leading to functional variation amongst batches. Moreover, there is limited ability to control biomolecular concentration of specific components or ensure their functionality. Partially reconstituted cell extracts serve as an alternative to crude cell extracts that gets around some of these limitations (48–50). These systems are created by carefully mixing a small number of purified components with crude or fractionated cell extracts. Controlled biomolecular additions are enabled by transcription or translation (50). Small molecule and buffer exchanges are also possible through dialysis and depletion of specific components, achieved by immunodepletion or degron tagging (51–55).
Extracts containing RNP complexes of interest depleted of a specific component offer a useful strategy to characterize the function of that missing component. Such an approach using Xenopus cell extract partially reconstituted with CMG (Cdc45–MCM2–7–GINS) helicase complex has been employed to uncover novel DNA replication and repair mechanisms. For example, DNA replication of templates containing DNA-protein cross-links (DPC) in the presence of CMG-reconstituted extracts immunodepleted of the accessory helicase RTEL1 or the DNA-dependent protease SPRTN revealed the role of these proteins in a DPC repair mechanism in which the replication fork triggers DPC proteolysis (56, 57). In another example, auxin-inducible degron tags were used for targeted depletion of the kinetochore motor protein, Kip3, from cell extracts to characterize its role in kinetochore attachment to microtubules through both single molecule microscopy and ensemble experiments (58).
Like all experimental model systems, both crude and reconstituted cell extracts are associated with a number of limitations, such as limited control of component concentration, that may complicate the study of certain biological problems (Table 1). Another important consideration is the possibility of batch-to-batch composition variability incurred during cell extract preparation. Despite precisely optimized protocols, subtle changes in the cell culture growth conditions, as well as in the lysis and post-lysis clearing steps, could lead to the differential loss or inactivation of components between preparations. As a result, it is critical to implement quality control measures for each preparation such as activity assays, western blots and mass spectrometry analysis to ensure that the components of interest are present and active in the extract prior to using it in the desired experiments. Similarly, it is advisable to implement such quality control measures over longer periods of storage and, possibly, to re-optimize the experimental conditions (i.e., temperature, time, buffer composition, etc). Despite these limitations, cell extracts offer an important set of capabilities that can be powerful when combined with single molecule techniques, as exemplified by the discoveries that this approach has enabled as detailed below. To facilitate future experimental designs, Table 1 summarizes the scope and limitations of single-molecule experiments in cell extracts, relative to both in vitro reconstituted and live-cell systems.
Table 1.
A comparison of the scope and limitations of experiments in cell extract with in vitro reconstituted and in-cell single-molecule systems.
| Experimental consideration | In vitro reconstitution | In live cells (in cellulo) | In cell extract |
|---|---|---|---|
| Temperature of measurement | Can be independently varied | Confined to physiological temperatures | Similar to in vitro, can be independently varied within optimized range |
| Concentration of component | Can be independently varied | Can be altered by manipulating gene expression characteristics | Can be extended beyond in cellulo by doping in purified components |
| Purity of component | Highly pure, but involves time-consuming purification steps | Complex mixture | Similar to in cellulo, but affords local “pull-down” to enrich for specific components (51–54) |
| Macromolecular Crowding | Can be simulated | Has intrinsic crowding | Can be simulated |
| Equilibrium measurements | Long-term equilibration is possible | Non-equilibrium | Similar to in vitro |
| Experimentally tractable (long term imaging) | Oxygen scavenging systems can enhance imaging times | Short term imaging only | Similar to in vitro |
| Small molecule testing and/or exchange | Yes. Highly controllable | Possible, but limited within the constraints of maintaining cell viability | Similar to in vitro, but retaining much of the complexity of the cell |
3. COMBINING SINGLE MOLECULE FLUORESCENCE MICROSCOPY AND PARTIALLY RECONSTITUTED CELL EXTRACTS
3.1. Fluorescence microscopy tools for single molecule imaging in cellular extracts
Single molecule experiments with cellular extracts require the same tools as classical in vitro single molecule experiments. While there are potential differences in sample viscosity, turbidity, autofluorescence, and non-specific interactions associated with cellular extracts, the basic measurement tenets are the same. The techniques discussed in this review all use total internal reflection (TIRF) microscopy. While TIRF systems vary in terms of laser lines, microscopes, and cameras, these technical variations are outside of the scope of this review and have been reviewed extensively elsewhere (59–61). More specifically, here we focus on techniques with surface-immobilized biomolecules as these have been well established for use in the characterization of RNP complex assemblies within cellular extracts. Confocal techniques for solution measurements are, thus far, underutilized for experiments in cellular extracts (62, 63).
Surface immobilization techniques can facilitate observation of individual biological complexes for extended periods of time (11). This begins with passivating an optical surface (e.g., microscope slide) while adding specific attachment points for molecules of interest. Next, the cellular extracts and fluorescently labeled biomolecules are introduced to the surface. The surface is then illuminated with laser light and the behavior of individual labeled molecules can be observed. The observable behaviors include, but are not limited to conformational changes via FRET, co-localization, and molecular interactions with antibody surface captured interaction partners (Figure 2).
Figure 2:
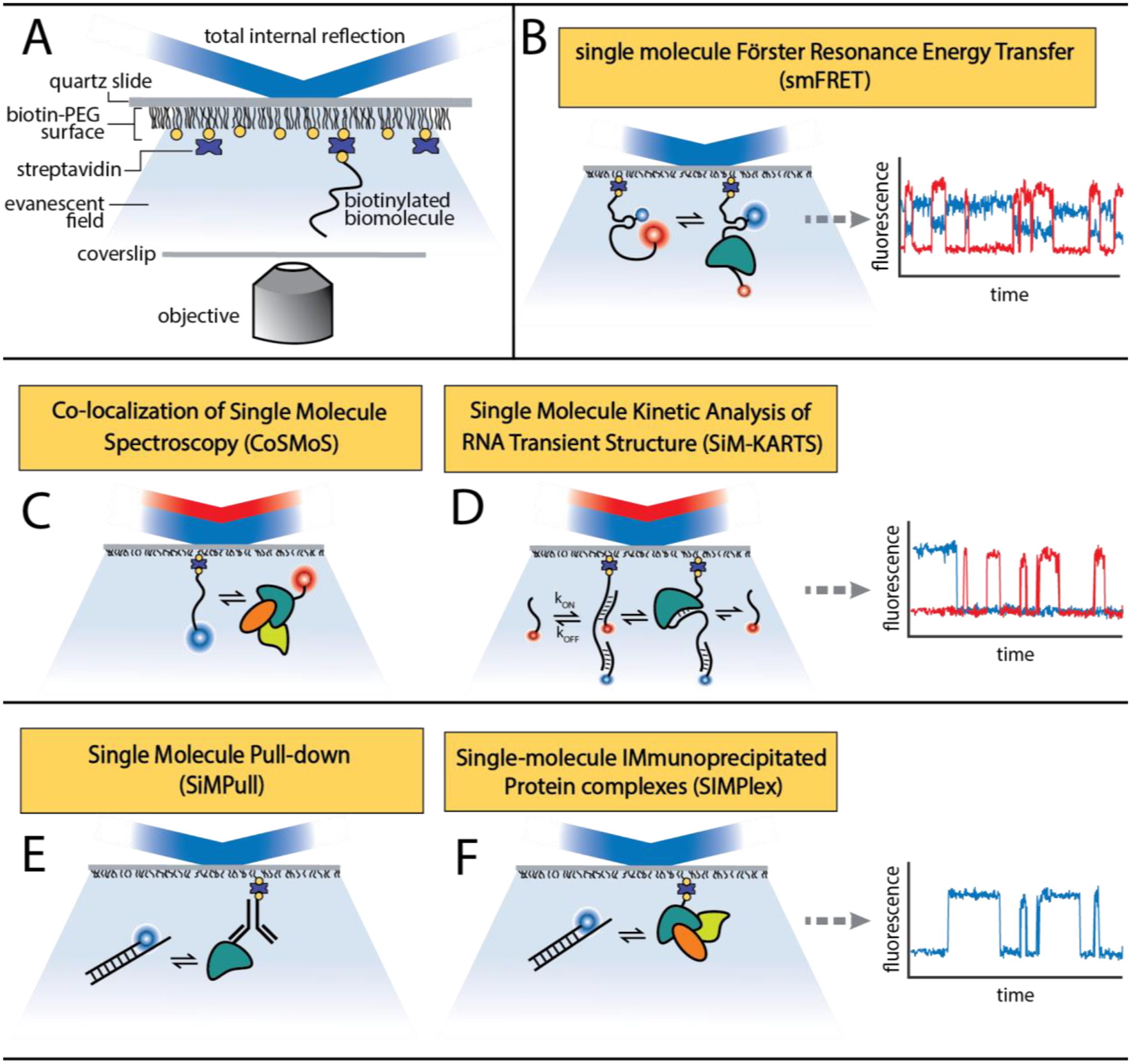
Single molecule fluorescence microscopy experiments. (A) Single molecule microscopy experiments, allow for the trajectories and interactions of single biotinylated biomolecules immobilized on a slide surface to be recorded in the background of complex cellular lysates over time through the excitation of strategically placed fluorophores. (B) In single molecule single molecule FRET experiments, anticorrelated fluorescence signals between two fluorophores report on the interactions occurring between two biomolecules within certain relative distances. In fluorescent colocalization experiments such as (C) CoSMoS and (D) SiM-KARTS (144, 145), repeated rounds of interactions between two or more fluorescently labeled biomolecules is tracked over time to determine binding kinetics and extract information about the accessibility of certain nucleic acid regions with RNPs. Single molecule pull-down experiments such as SiMPull and SIMPlex report on the interaction of between biomolecules and either (E) a protein or (F) protein complex surface captured through a biotinylated antibody.
Over the last two decades, a number of single molecule modalities have been established to investigate multi-domain complexes under near-native conditions in various contexts. One approach is to immobilize exogenous macromolecules (typically protein or oligonucleotides) to surfaces that nucleate biological complex formation. This is a powerful tool that can be used to enrich a surface with numerous complexes. When used in conjunction with labeled endogenous components (e.g., SNAP tag fluorescent labeled protein), such as in the colocalization single-molecule spectroscopy (CoSMoS) technique (64), one can measure how different species colocalize within biomolecular complexes.
While the classic biotin/avidin surface attachment chemistry is still popular for simpler systems, immunoprecipitation has been recently used to great effect for pulling down biological machinery directly from bulk lysate. SiMPull and SIMPlex (51, 65, 66) methodologies utilize biotinylated antibodies to pulldown a biomolecule of choice from the lysate. These immunoprecipitation-based approaches make it possible to study endogenous proteins of interest without the need to add exogenous proteins.
As exemplified by the biological contributions highlighted in Sections 3.2 – 3.4, the common principles underlying all these methods are:
-
Enrichment of target molecules:
In the case of reconstitution experiments, proteins or complexes of interest are purified based on affinity principles after heterologous expression in bacteria and/or eukaryotic systems of choice. RNAs may be synthesized by chemical synthesis or in vitro transcription. Alternatively, RNAs or proteins may be overexpressed in transcription/translation-competent extracts.
-
Surface proximal immobilization:
These strategies use various high affinity non-covalent molecular interactions, such as biotin-streptavidin binding or antibody-antigen interactions to tether a molecule or complex of interest to a surface; typically, PEG (polyethylene glycol) passivated glass or quartz. This has two-fold utility: 1) it allows performing single-molecule total internal reflection fluorescence (TIRF) microscopy to selectively excite target molecules proximal to the surface, and 2) allows individual, tethered molecules or complexes to be followed over time (seconds-to-tenth of minutes), enabling the detection of multiple interaction events and/or extraction of kinetic information.
3.2. Insights from the spliceosome
One RNP complex that has been extensively studied using single molecule fluorescence experiments in whole cell extract is the spliceosome (64, 67, 68). Unlike molecular machines that adopt an enzymatically active complex which remains compositionally intact through multiple rounds of catalysis on a given substrate, spliceosomes assemble stepwise onto targeted pre-mRNA splice sites forming pre-catalytic complexes. These are then remodeled into catalytically active spliceosomes that further transition through several compositionally distinct states on the catalytic path in a process involving dozens of transiently acting factors. The large number of components (both RNA and proteins) that assemble and transition through each stage of the substrate splicing cycle precludes reconstitution of this RNP machine from purified materials. The size, dynamics, and poorly understood regulatory pathways of the spliceosome machinery also poses enormous technical challenges for using in vivo experiments to extract molecular level understanding of spliceosomal transitions. Thus, in vitro single molecule fluorescence microscopy experiments using partially reconstituted cell extracts have been implemented to provide insights about spliceosome assembly and RNA conformational dynamics at various points during the pre-mRNA splicing reaction.
A number of single molecule colocalization experiments have been implemented to investigate multiple aspects of spliceosome assembly. Hoskins et al. used yeast cell extract with genetically engineered SNAP-tags on various splicing components to follow the kinetics of spliceosome assembly on a model pre-mRNA substrate with multi-wavelength CoSMoS (69). Real-time kinetic analysis of the single molecule trajectories revealed the dynamic and reversible nature of spliceosome subcomplex associations during step-wise assembly. In a later study, yeast cell extracts were used in single molecule CoSMoS and CoSMoS-FRET experiments to follow how pre-mRNA dynamics were coupled to spliceosome assembly and activation (70). The data suggest a model in which the spliceosome machinery coordinates the physical proximity of the splice sites to ensure that splicing chemistry takes place only once the catalytic complex is fully assembled.
Single molecule co-localization studies using cell extracts from mammalian cell lines have also advanced our understanding of spliceosome complex composition and mechanisms of human spliceosomes. For example, Cherny et al. quantified the photobleaching events exhibited by immobilized spliceosomal complexes pulled down from HEK293 cell extracts to determine the number of polypyrimidine tract-binding proteins within single spliceosome complexes (71). Given that human pre-mRNAs are often alternatively spliced, others have used similar approaches to characterize alternative splice site selection mechanisms. For example, Hodson et al. followed the colocalization of U1 snRNPs with pre-mRNA substrate containing alternative splice sites to show that, although multiple U1 snRNPs initially bind the pre-mRNA, only a single U1 is bound after the ATP-dependent transition to complex A during spliceosomal assembly (72). More recently, single molecule CoSMoS experiments with HEK293 cell extracts revealed insights about cross-intron and cross-exon interactions in human pre-spliceosome assembly (73). In these experiments, Braun, et al. fluorescently labeled spliceosome subcomplexes in order to follow their assembly with model pre-mRNA substrates (Figure 3A). They observed that pre-mRNA binding of the subcomplex that recognizes the 5’ splice site (U1) was not affected by the presence of the subcomplex that recognizes and binds 3’ splice sites (U2). In contrast, they found that U2 binding is strongly accelerated when U1 is bound at the 5’ splice site either upstream (cross-intron) or downstream (cross-exon) revealing an alternative kinetic pathway for U2 recruitment during pre-spliceosome assembly that may be key to exon definition in the multi-intron containing pre-mRNAs that predominate within human genomes (Figure 3B).
Figure 3:
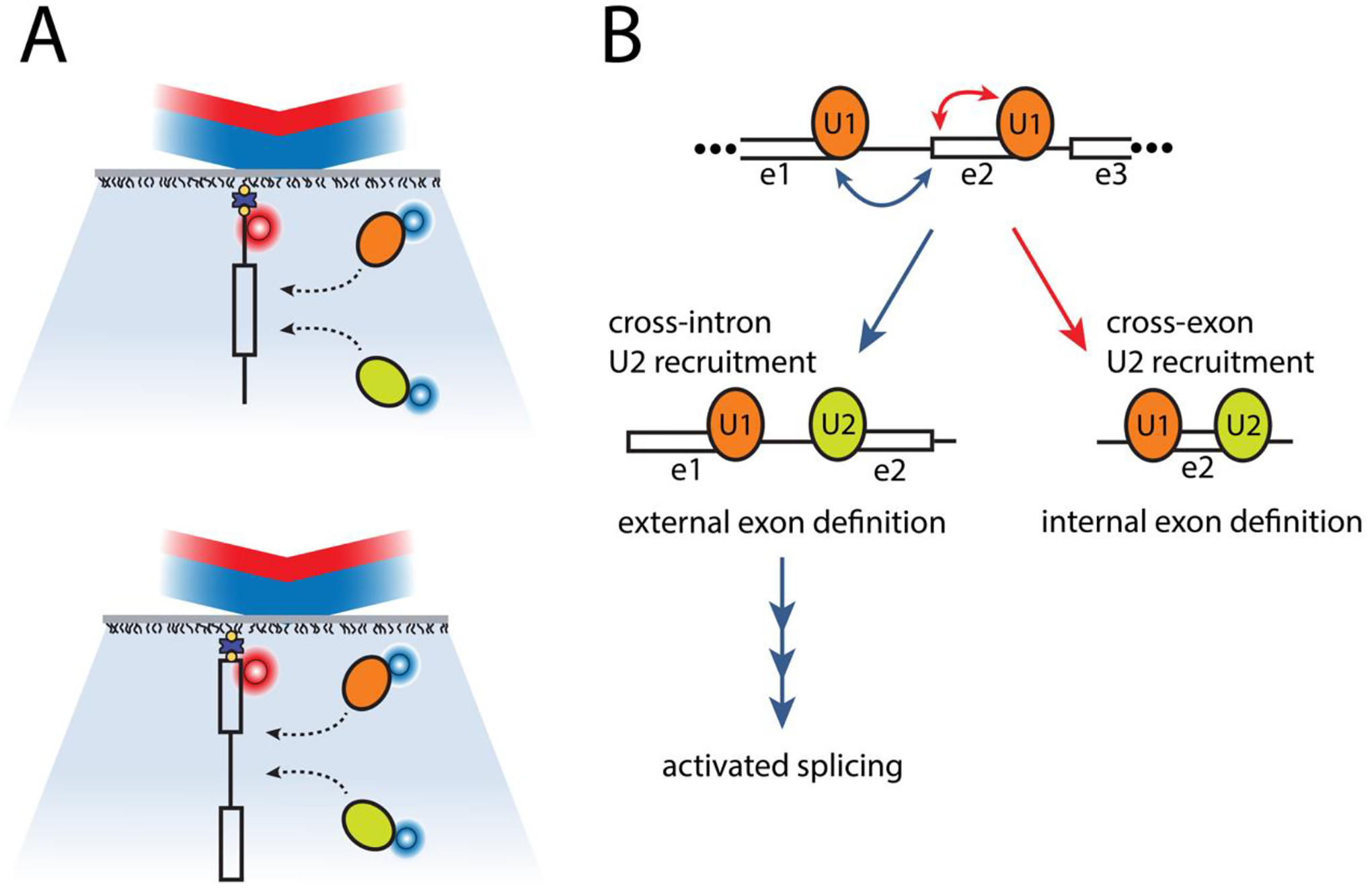
CoSMoS experiments reveal cross-intron and cross-exon cooperation in human pre-spliceosome assembly (73). (A) Braun et al. assembled pre-spliceosome complexes using mammalian cell extracts containing fluorescently labeled spliceosome subcomplexes (orange and green ovals). Using CoSMoS experiments, they follow these complexes’ interactions with RNA substrates containing either (top) an intron flanked by two partial exons or the reverse (bottom), an exon flanked by two partial introns. (B) The results reveal that the binding dynamics of subcomplex U2 are significantly altered based on the presence of U1 at adjacent 5’ splice sites in either direction (upstream or downstream). This U1-U2 pre-spliceosome cooperation mechanism may be one way that spliceosomes distinguish internal exons within multi-exon containing pre-mRNAs and may serve as a kinetic checkpoint along the pathway towards activated spliceosome assembly.
In addition to providing insights about spliceosome complex assembly, smFRET experiments have revealed insights about the conformational dynamics of substrate pre-mRNAs captured bound within splicing complexes (74, 75). Alongside these measurements, computational methodologies such as single molecule clustering analysis (SiMCAn) have enabled computational sorting and identification of kinetic signatures associated with various steps of the splicing cycle (76). Characterization of the kinetics of such RNA rearrangements and how those are correlated to spliceosomal transitions and interacting protein partners that mediate those transitions continue to expand our understanding of the kinetic proofreading mechanisms that underlie the tightly controlled regulation of spliceosomal transitions and splice site selection (77, 78).
3.3. Revealing dynamic aspects of eukaryotic transcriptional activation
Similar to splicing, the transcription of RNA is a highly dynamic process requiring the temporally controlled coordination of many RNPs to tune transcriptional fate. In eukaryotes, the transcription of protein-coding genes is carried out by RNA polymerase II (RNAP II), while transcription of ribosomal and transfer RNAs are carried out by RNAPs I and III, respectively. A large number of transcription factors interact with each RNAP to control transcription initiation, elongation, and termination.
While the identities and biochemical properties of the essential eukaryotic transcriptional factors are known, the dynamics of their interactions remain largely unknown. For example, it is known that the process of turning on eukaryotic gene expression by RNAP II is preempted by a highly coordinated assembly of a minimum of six general transcription factors (GTFs: TFIIA, TFIIB, TFIID, TFIIE, TFIIF, and TFIIH) with Mediator and RNAP II into a pre-initiation complex (PIC) at the promoter just upstream of the coding region (79, 80). It has also been shown that transcriptional activators and co-activators increase the transcription of particular genes through interactions with DNA elements such as enhancer or upstream activator sequences (UAS) found upstream of the transcription start site (81, 82). Yet many questions remain regarding how these transcriptional activators are recruited to transcription complexes, the effect of activators and co-activators on PIC assembly, and how they facilitate transitions between PIC assembly and productive elongation to stimulate transcription.
Single molecule studies of eukaryotic transcription initiation have enabled important mechanistic insights on PIC assembly. Using fully reconstituted PIC from purified GTFs and RNAP II, single molecule experiments have revealed previously averaged out aspects of the assembly dynamics. For instance, Zhang et al. used immobilized transcription templates and fluorescently labeled GFTs, show that TFIIB interreacts transiently with the promoter and undergoes a transient-to-stable transition only in the presence of RNAP II (83). Others have used optical or magnetic tweezers with reconstituted transcription reactions to show the role of TFIIH in ATP-dependent expansion of the transcription bubble prior to RNAP II-driven promoter escape (84, 85). More recently, PIC assembly dynamics has been followed in cell extracts using CoSMoS in order to determine the impact of the rich repertoire of transcription factors present in cell lysates on transcription initiation (86). Using fluorescently labeled DNA template containing UAS ± promoter, and cell lysates with fluorescently labeled RNAP II, TFIIF, TFIIE, or TFIIH, Baek et al. observed that RNAP II associated with UAS with the same kinetic “on” rate whether the promoter was present or not (87). Similarly, they observed that the general transcription factors TFIIE and TFIIF also associated with the UAS independently of the promoter, but the TFIIH did not. Together their findings support a model in which some GTFs and RNAP II can be recruited to UAS, potentially through activator interactions, in order to enhance PIC assembly and thereby increase transcription of the downstream gene (Figure 4). In a complementary study, Nguyen and colleagues employed live-cell single particle tracking experiments to follow the nuclear dynamics of ten PIC components in budding yeast (88). Their findings support a highly dynamic model in which the Mediator and TFIID complexes stimulate the “trapping” of other GTFs in radially limited zones in order to promote PIC assembly. Together these findings show how single molecule studies using cell extracts can complement and bridge gaps in the mechanistic understanding of dynamic RNP processes recapitulated from fully reconstituted systems versus within the rich cellular milieu of in vivo conditions.
Figure 4:
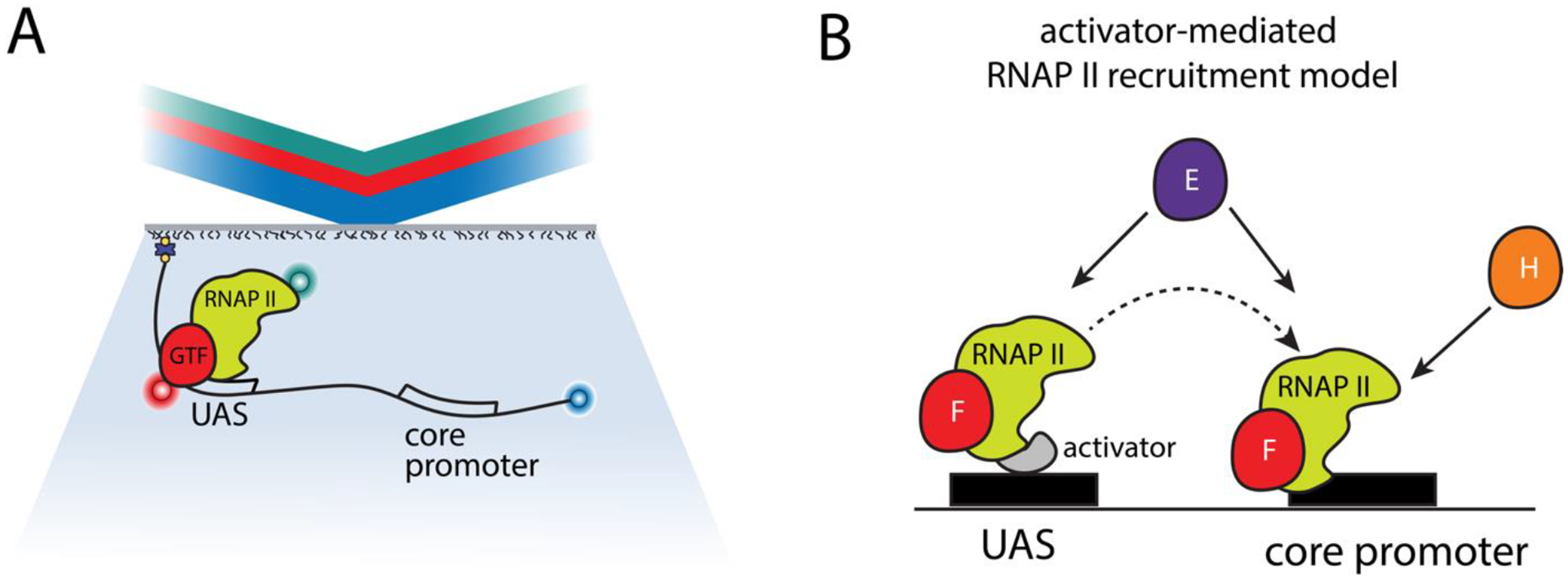
Emerging PIC assembly model. A) In a recent study, Baek et al. performed three-color CoSMoS experiments using mammalian cell extracts to following the association of RNAP II and general transcription factors TFIIF, TFIIE, or TFIIH with a DNA promoter template (87). B) The data support an emerging model whereby RNAP II associated with TFIIF and/or TFIIE binds primarily at the enhancer (UAS) through UAS bound transcription activators. From there, partially assembled PIC can be transferred to the core promoter for binding with TFIIH and additional GTFs for complete PIC assembly, in a branched assembly pathway.
3.4. Functional, reconstituted cell-free systems to interrogate gene silencing mechanisms
Cell extracts have also been the cornerstone of miRNA research, enabling mechanistic interrogation of miRNA biogenesis (100–102, 4), miRISC assembly (103–107) and miRISC-mediated gene regulation (108–115) at the ensemble level. miRNAs are evolutionarily conserved small (21–24 nucleotides) non-coding RNAs which guide miRNA-induced silencing complexes (miRISC) to fine tune gene expression through translational repression and/or mRNA degradation (89). These complexes are composed of Argonaute (Ago), miRNA, and mRNA-resident miRNA response elements (MREs). miRNAs typically bind MREs via incomplete complementarity, requiring only a short (7–8 nucleotide) “seed” (perfect) match. Consequently, one miRNA can regulate many mRNAs (90) and, conversely, a single mRNA can be regulated by many miRNAs (91), resulting in a complex gene regulatory network that controls 70% of all mammalian mRNAs. Although the basic mechanism of miRNA-mediated gene silencing is widely accepted, it is still unclear how the ~200,000 miRNA molecules of the cell find, bind, and regulate their targets, which are interspersed among a pool of ~7,000,000 mRNA molecules (92). Moreover, mRNAs typically contain multiple MREs, be it for miRNAs of similar or distinct sequences (93), and whether all these MREs are simultaneously occupied i.e. stoichiometric aspects, for optimal regulation is unclear.
In vitro single-molecule microscopy of purified Ago proteins has revealed lateral (1-dimensional) diffusion and augmented hybridization upon seed recognition as major mechanisms of MRE recognition by minimal si/miRISC on sparsely populated, tethered individual mRNAs (94). Recently, smFRET measurements utilizing site specifically-labeled human Ago2 (hAgo2) immunopurified from mammalian lysates have revealed mechanistic details of guide and target RNA loading by hAgo2 (95). Rather than finding a distinct conformational state for the ternary RNP consisting of hAgo2, guide, and target RNA, the authors observed various distinct and conformationally dynamic states required for regulatory activity. Experimental research on small DNA-guided prokaryotic Ago analogs (96) and the theoretical treatment of protein-nucleic acids interactions (97, 98) have suggested that short intersegmental transfers, i.e., 3-dimensional hopping, in addition to 1-dimensional scanning, may make target search more efficient.
While these efforts are significant, they do not recapitulate 1) the complexity of miRISC, a large (> 1 MDa) RNP with numerous proteins that act in concert to mediate gene regulation and 2) the crowded confines of the cell. In fact, supplementing purified Ago with TNRC6B, a miRISC scaffold protein for tertiary regulators, leads to phase separation of Ago-TNRC6B-mRNA complexes in vitro and this phenomenon is correlated with accelerated mRNA deadenylation (99).
Most RISC-active cell extracts have been derived from Drosophila or C. elegans embryos and human cell culture, requiring the meticulous optimization of cell lysis (108–111, 113). While commercial cell-free extracts recapitulate gene silencing, they are incompetent in miRNA biogenesis and miRISC assembly (114–116), often involving non-physiological molecular manipulation, such as pre-annealing of small RNAs with mRNAs prior to extract addition. Cell-free transcription/translation extracts have the potential to overcome these limitations by expressing key miRISC factors such as AGO, TNRC6A and DICER at near physiological levels to recapitulate all facets of the miRNA pathway. The future use of such functional, reconstituted cell extracts for in vitro single-molecule imaging of miRISC, in combination with intracellular single-miRNA imaging (12, 117, 118) may resolve the long-standing questions pertaining to target search and miRNA-binding stoichiometry, critical aspects to consider for the pharmacokinetics of potential anti-miRNA drugs.
4. CONCLUSIONS AND FUTURE DIRECTIONS
Many of the biological insights into pre-mRNA splicing, transcription and gene regulation discussed here were uncovered by combining the use of cell extracts and single-molecule fluorescence microscopy. These examples demonstrate the feasibility and underexplored potential of this approach, yet so far target only few of the many complex RNP genomic maintenance systems spanning the central dogma of molecular biology. We hope that the works highlighted here serve to inspire and encourage many more researchers to consider exploring cell extracts as a way to bridge recombinant in vitro and in cellulo experimental inquiries. To further facilitate the use of this approach, here we additionally describe some universal practical considerations for applying specifically single molecule fluorescence microscopy to cell extracts.
4.1. Component concentration considerations
Investigation of any biological sample of interest employing single-molecule fluorescence microscopy requires the mitigation of several practical challenges. One of these challenges is exceeding the necessary affinity threshold between interacting partners. In vitro single molecule spectroscopy is performed at highly diluted concentration of the fluorescently labeled species to facilitate the observation to single molecule events (119). This means that to study the biological interactions of two or more partners, a low dissociation constant (KD) of those interactions is required.
Reconstituted cell-free systems support the introduction and/or removal of biomolecules, as previously described (Section 2.2). Careful addition or removal of components with minimal change to the near-cellular composition of the extract is a useful strategy for characterizing the role of specific biomolecules in a given RNP mechanism. Conversely, an alternative to adding recombinantly expressed components that is emerging as a powerful additional tool to study RNP systems is to express proteins of interest in translation-competent extracts (120). However, this is associated with some degree of variability in protein expression across those pathways that are highly sensitive to experimental conditions (121).
4.2. Fluorescent labeling of proteins
Since all single-molecule fluorescence microscopy techniques depend on a fluorescence readout, another challenge that must be met is the incorporation of one or more chromophore-labeled biomolecule. In keeping with the scope of this article, here we limit this discussion to strategies particularly suitable for cell extracts. A more exhaustive description of labeling strategies for single molecule experiments was recently published elsewhere (122).
In general, fusion of a fluorescent protein via standard molecular cloning methods represents a straightforward method to site-specifically introduce a fluorophore into the protein of interest (POI). For some fluorescent proteins such as GFP or GFP-derivatives, size of the fluorescent protein and poor photophysical properties often render it unsuitable for single molecule observation (123, 124). As a result, care must be taken to ensure that the fluorescent reporter chosen for single molecule measurements meets the suitable criteria of size, brightness, and lifetime required for the chosen experimental design.
A flexible strategy for introducing chromophores suitable for single molecule applications is to fuse genetically encoded self-labeling protein tags (Halo-, Snap-, and CLIP-tags) to a POI. Cell extracts derived from cell lines containing these tagged genes can then be incubated with an organic dye of desired emission wavelength for site-specific attachment to the POI (125–127). Furthermore, the covalent organic dye is buried in the active site of the self-labeling protein tag and is thereby protected from the environment with respect to photophysical perturbation, resulting in a non-environmentally affected, and potentially enhanced, fluorescence readout (128, 129). A potential concern of such chimeric systems is the significant size of the protein tag (~20–30 kDa) that may potentially perturb aspects of the system of interest, especially in multi-component RNPs (128). Additionally, precise distance measurement and/or structural dynamics may be complicated due to the size of the tag and potential dye orientation effects. These limitations make this system mainly suitable for colocalization and super resolution microscopy studies, as well as stoichiometry measurements (128).
For applications in which a recombinantly expressed and fluorescently labeled protein is added to the cell extract, an alternative organic fluorophore-to-protein attachment strategy is to hijack the reactivity of amino acid side chains. Initial studies relied on the reactivity of primary amino moieties, e.g., lysine side chain within proteins and N-hydroxysuccinimidyl (NHS) ester functionalized fluorescent probes (130). The high abundance of lysine residues often found on protein surfaces makes site-specifically labeling very challenging (131). To improve the site-specificity, the less abundant cysteine amino acid may be utilized using thiol-maleimide chemistry (132). This labeling strategy comes at the expense of needing to mutate other cysteines to achieve specificity within POIs where they are abundantly present. In cases where this need is untenable, an alternative is to site-specifically incorporate an unnatural amino acid into the POI that can subsequently be bioorthogonally labeled (133–135). Initially, this strategy was only applicable for bacterial proteins or eukaryotic proteins recombinantly expressed in bacteria. More recently, it has been expanded to POIs in mammalian cell lines (136). While this strategy circumvents the limitation of site-specifically labeling proteins with high lysine and cysteine abundance, it comes with additional challenges of its own, briefly summarized below.
The limitations of site-specifically labeling of proteins using incorporation of non-natural amino acids can be summarized as four challenges: (i) expression of functionalized protein containing a non-natural amino acid; (ii) potential functional and structural integrity change of the POI upon functionalization; (iii) change in the functional integrity of the POI due to attachment of the fluorescence dye; and (iv) difference in fluorescent dye properties caused by the changing physicochemical microenvironment of the dye upon labeling. Despite these hurdles, this technique allows for very precise control over the labeling site and for incorporation of multiple dyes, a unique feature that can be used to address structural dynamics (137–139). To overcome photophysical perturbation of the dye, a long and flexible linker (~20 Å) was developed that increases a dye’s mobility and reduces hydrophobic and electrostatic interactions between the POI and the dye (140). Together with progress in modeling of fluorophores at specific attachment sites and as well as recent emergence of biochemical workflows to identify suitable labeling sites, these challenges are becoming more easily surmountable (141–143). One might therefore predict that cell extracts will further expand as a tool that allows the dissection of RNP biology at the single molecule level.
Funding Information
This work was supported by NIH R35 grant GM131922 to N.G.W., an NIH IRACDA fellowship on grant K12 GM111725 and NIH K99 MOSAIC grant GM144735 to E. C. D., and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Project-ID 449562930 to A.S..
Contributor Information
Elizabeth Duran, Single Molecule Analysis Group, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA.
Andreas Schmidt, Single Molecule Analysis Group, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA.
Robb Welty, Single Molecule Analysis Group, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA.
Ameya P. Jalihal, Department of Biology, University of North Carolina, Chapel Hill, NC 27517, USA
Sethuramasundaram Pitchiaya, Michigan Center for Translational Pathology, Department of Pathology, Department of Urology, Michigan Medicine, Ann Arbor, MI 48109-1055, USA.
Nils G. Walter, Single Molecule Analysis Group, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA.
REFERENCES
- 1.Doudna JA, and Charpentier E (2014) The new frontier of genome engineering with CRISPR-Cas9. Science. 346, 1258096. [DOI] [PubMed] [Google Scholar]
- 2.Adli M (2018) The CRISPR tool kit for genome editing and beyond. Nat. Commun 9, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilkinson ME, Charenton C, and Nagai K (2020) RNA Splicing by the Spliceosome. Annu. Rev. Biochem 89, 359–388 [DOI] [PubMed] [Google Scholar]
- 4.O’Brien J, Hayder H, Zayed Y, and Peng C (2018) Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol 10.3389/fendo.2018.00402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ozata DM, Gainetdinov I, Zoch A, O’Carroll D, and Zamore PD (2019) PIWI-interacting RNAs: small RNAs with big functions. Nat. Rev. Genet 20, 89–108 [DOI] [PubMed] [Google Scholar]
- 6.An H, de Meritens CR, and Shelkovnikova TA (2021) Connecting the “dots”: RNP granule network in health and disease. Biochim. Biophys. Acta Mol. Cell Res 1868, 119058. [DOI] [PubMed] [Google Scholar]
- 7.Rhine K, Skanchy S, and Myong S (2022) Single-molecule and ensemble methods to probe RNP nucleation and condensate properties. Methods 197, 74–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S, Kim S, Chang HR, Kim D, Park J, Son N, Park J, Yoon M, Chae G, Kim Y-K, Kim VN, Kim YK, Nam J-W, Shin C, and Baek D (2021) The regulatory impact of RNA-binding proteins on microRNA targeting. Nat. Commun 12, 5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warf MB, and Berglund JA (2010) The role of RNA structure in regulating pre-mRNA splicing. Trends Biochem. Sci 35, 169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serebrov V, and Moore MJ (2016) Single Molecule Approaches in RNA-Protein Interactions. Adv Exp Med Biol 907, 89–106 [DOI] [PubMed] [Google Scholar]
- 11.Roy R, Hohng S, and Ha T (2008) A practical guide to single-molecule FRET. Nat Methods 5, 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pitchiaya S, Androsavich JR, and Walter NG (2012) Intracellular single molecule microscopy reveals two kinetically distinct pathways for microRNA assembly. EMBO Rep 13, 709–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wightman B, Ha I, and Ruvkun G (1993) Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855–862 [DOI] [PubMed] [Google Scholar]
- 14.Padgett RA, Hardy SF, and Sharp PA (1983) Splicing of adenovirus RNA in a cell-free transcription system. Proc. Natl. Acad. Sci. U. S. A 80, 5230–5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruskin B, Krainer AR, Maniatis T, and Green MR (1984) Excision of an intact intron as a novel lariat structure during pre-mRNA splicing in vitro. Cell 38, 317–331 [DOI] [PubMed] [Google Scholar]
- 16.Payen A (1838) Mémoire sur la composition du tissu propre des plantes et du ligneux. C R Geosci 7, 1052–1056 [Google Scholar]
- 17.Schachman HK, Pardee AB, and Stanier RY (1952) Studies on the macro-molecular organization of microbial cells. Arch Biochem Biophys 38, 245–260 [DOI] [PubMed] [Google Scholar]
- 18.Moore PB (1988) The ribosome returns. Nature 331, 223–227 [DOI] [PubMed] [Google Scholar]
- 19.Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, and Yonath A (2000) Structure of functionally activated small ribosomal subunit at 3.3 angstroms resolution. Cell 102, 615–623 [DOI] [PubMed] [Google Scholar]
- 20.Wimberly BT, Brodersen DE, Clemons WM Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, and Ramakrishnan V (2000) Structure of the 30S ribosomal subunit. Nature 407, 327–339 [DOI] [PubMed] [Google Scholar]
- 21.Korostelev A, Ermolenko DN, and Noller HF (2008) Structural dynamics of the ribosome. Curr Opin Chem Biol 12, 674–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noller HF, Lancaster L, Zhou J, and Mohan S (2017) The ribosome moves: RNA mechanics and translocation. Nat Struct Mol Biol 24, 1021–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dashti A, Schwander P, Langlois R, Fung R, Li W, Hosseinizadeh A, Liao HY, Pallesen J, Sharma G, Stupina VA, Simon AE, Dinman JD, Frank J, and Ourmazd A (2014) Trajectories of the ribosome as a Brownian nanomachine. Proc Natl Acad Sci U A 111, 17492–17497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monro RE (1967) Catalysis of peptide bond formation by 50 S ribosomal subunits from Escherichia coli. J Mol Biol 26, 147–151 [DOI] [PubMed] [Google Scholar]
- 25.Perez CE, and Gonzalez RL Jr (2011) In vitro and in vivo single-molecule fluorescence imaging of ribosome-catalyzed protein synthesis. Curr Opin Chem Biol 15, 853–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu RA, Dagdas YS, Yilmaz ST, Yildiz A, and Collins K (2015) Single-molecule imaging of telomerase reverse transcriptase in human telomerase holoenzyme and minimal RNP complexes. Elife 10.7554/eLife.08363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lohka MJ, and Masui Y (1983) Formation in vitro of sperm pronuclei and mitotic chromosomes induced by amphibian ooplasmic components. Science 220, 719–721 [DOI] [PubMed] [Google Scholar]
- 28.Avilés-Pagán EE, Hara M, and Orr-Weaver TL (2021) The GNU subunit of PNG kinase, the developmental regulator of mRNA translation, binds BIC-C to localize to RNP granules. eLife 10, e67294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, and Diamond MI (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welte H, and Kovermann M (2020) Insights into Protein Stability in Cell Lysate by 19F NMR Spectroscopy. ChemBioChem 21, 3575–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh G, Pratt G, Yeo GW, and Moore MJ (2015) The Clothes Make the mRNA: Past and Present Trends in mRNP Fashion. Annu Rev Biochem 84, 325–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonneveld S, Verhagen BMP, and Tanenbaum ME (2020) Heterogeneity in mRNA Translation. Trends Cell Biol 30, 606–618 [DOI] [PubMed] [Google Scholar]
- 33.Biswas J, Liu Y, Singer RH, and Wu B (2019) Fluorescence Imaging Methods to Investigate Translation in Single Cells. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a032722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cialek CA, Koch AL, Galindo G, and Stasevich TJ (2020) Lighting up single-mRNA translation dynamics in living cells. Curr Opin Genet Dev 61, 75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt A, Gao G, Little SR, Jalihal AP, and Walter NG (2020) Following the messenger: Recent innovations in live cell single molecule fluorescence imaging. Wiley Interdiscip Rev RNA 11, e1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boersma S, Khuperkar D, Verhagen BMP, Sonneveld S, Grimm JB, Lavis LD, and Tanenbaum ME (2019) Multi-Color Single-Molecule Imaging Uncovers Extensive Heterogeneity in mRNA Decoding. Cell 178, 458–472.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walter NG (2019) Biological Pathway Specificity in the Cell-Does Molecular Diversity Matter? Bioessays 41, e1800244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellis RJ (2001) Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci 26, 597–604 [DOI] [PubMed] [Google Scholar]
- 39.Ilagan JO, and Jurica MS (2014) Isolation and accumulation of spliceosomal assembly intermediates. Methods Mol. Biol. Clifton NJ 1126, 179–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Folco EG, and Reed R (2014) In vitro systems for coupling RNAP II transcription to splicing and polyadenylation. Methods Mol. Biol. Clifton NJ 1126, 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Keefe RT, Norman C, and Newman AJ (1996) The Invariant U5 snRNA Loop 1 Sequence Is Dispensable for the First Catalytic Step of pre-mRNA Splicing in Yeast. Cell 86, 679–689 [DOI] [PubMed] [Google Scholar]
- 42.McPheeters DS, Fabrizio P, and Abelson J (1989) In vitro reconstitution of functional yeast U2 snRNPs. Genes Dev 3, 2124–2136 [DOI] [PubMed] [Google Scholar]
- 43.Fabrizio P, McPheeters DS, and Abelson J (1989) In vitro assembly of yeast U6 snRNP: a functional assay. Genes Dev 3, 2137–2150 [DOI] [PubMed] [Google Scholar]
- 44.Pan ZQ, and Prives C (1988) Assembly of functional U1 and U2 human-amphibian hybrid snRNPs in Xenopus laevis oocytes. Science 241, 1328–1331 [DOI] [PubMed] [Google Scholar]
- 45.Stark MR, and Rader SD (2014) Complementation of U4 snRNA in S. cerevisiae splicing extracts for biochemical studies of snRNP assembly and function. Methods Mol. Biol. Clifton NJ 1126, 193–204 [DOI] [PubMed] [Google Scholar]
- 46.Holden P, and Horton WA (2009) Crude subcellular fractionation of cultured mammalian cell lines. BMC Res. Notes 2, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niederer RO, Rojas-Duran MF, Zinshteyn B, and Gilbert WV (2022) Direct analysis of ribosome targeting illuminates thousand-fold regulation of translation initiation. Cell Syst 13, 256–264.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perez JG, Stark JC, and Jewett MC (2016) Cell-Free Synthetic Biology: Engineering Beyond the Cell. Cold Spring Harb. Perspect. Biol 10.1101/cshperspect.a023853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kelwick RJR, Webb AJ, and Freemont PS (2020) Biological Materials: The Next Frontier for Cell-Free Synthetic Biology. Front. Bioeng. Biotechnol 10.3389/fbioe.2020.00399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laohakunakorn N, Grasemann L, Lavickova B, Michielin G, Shahein A, Swank Z, and Maerkl SJ (2020) Bottom-Up Construction of Complex Biomolecular Systems With Cell-Free Synthetic Biology. Front. Bioeng. Biotechnol 10.3389/fbioe.2020.00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aggarwal V, and Ha T (2014) Single-molecule pull-down (SiMPull) for new-age biochemistry: methodology and biochemical applications of single-molecule pull-down (SiMPull) for probing biomolecular interactions in crude cell extracts. BioEssays News Rev. Mol. Cell. Dev. Biol 36, 1109–1119 [DOI] [PubMed] [Google Scholar]
- 52.Lee H-W, Ryu JY, Yoo J, Choi B, Kim K, and Yoon T-Y (2013) Real-time single-molecule coimmunoprecipitation of weak protein-protein interactions. Nat. Protoc 8, 2045–2060 [DOI] [PubMed] [Google Scholar]
- 53.Srinivasan S, Hazra JP, Singaraju GS, Deb D, and Rakshit S (2017) ESCORTing proteins directly from whole cell-lysate for single-molecule studies. Anal. Biochem 535, 35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee YK, Low-Nam ST, Chung JK, Hansen SD, Lam HYM, Alvarez S, and Groves JT (2017) Mechanism of SOS PR-domain autoinhibition revealed by single-molecule assays on native protein from lysate. Nat. Commun 8, 15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shetty A, Reim NI, and Winston F (2019) Auxin-inducible degron system for depletion of proteins in S. cerevisiae. Curr. Protoc. Mol. Biol 128, e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Larsen NB, Gao AO, Sparks JL, Gallina I, Wu RA, Mann M, Räschle M, Walter JC, and Duxin JP (2019) Replication-Coupled DNA-Protein Crosslink Repair by SPRTN and the Proteasome in Xenopus Egg Extracts. Mol. Cell 73, 574–588.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sparks JL, Chistol G, Gao AO, Räschle M, Larsen NB, Mann M, Duxin JP, and Walter JC (2019) The CMG Helicase Bypasses DNA-Protein Cross-Links to Facilitate Their Repair. Cell 176, 167–181.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Torvi JR, Wong J, Serwas D, Moayed A, Drubin DG, and Barnes G (2022) Reconstitution of kinetochore motility and microtubule dynamics reveals a role for a kinesin-8 in establishing end-on attachments. eLife 10.7554/eLife.78450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oheim M, Salomon A, Weissman A, Brunstein M, and Becherer U (2019) Calibrating Evanescent-Wave Penetration Depths for Biological TIRF Microscopy. Biophys. J 117, 795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Niederauer C, Blumhardt P, Mücksch J, Heymann M, Lambacher A, and Schwille P (2018) Direct characterization of the evanescent field in objective-type total internal reflection fluorescence microscopy. Opt. Express 26, 20492–20506 [DOI] [PubMed] [Google Scholar]
- 61.Oheim M (2016) TIRF (Total Internal Reflection Fluorescence). in eLS, pp. 1–8, John Wiley & Sons, Ltd, 10.1002/9780470015902.a0022505 [DOI] [Google Scholar]
- 62.Currie M, Thao C, Timerman R, Welty R, Berry B, Sheets ED, and Heikal AA (2015) Multiscale diffusion of a molecular probe in a crowded environment: a concept. in Ultrafast Nonlinear Imaging and Spectroscopy III, pp. 60–75, SPIE, 9584, 60–75 [Google Scholar]
- 63.Mateu-Regué À, Christiansen J, Bagger FO, Hellriegel C, and Nielsen FC (2021) Unveiling mRNP composition by fluorescence correlation and cross-correlation spectroscopy using cell lysates. Nucleic Acids Res 49, e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, and Moore MJ (2008) Visualizing the splicing of single pre-mRNA molecules in whole cell extract. RNA 14, 170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fareh M, Yeom K-H, Haagsma AC, Chauhan S, Heo I, and Joo C (2016) TRBP ensures efficient Dicer processing of precursor microRNA in RNA-crowded environments. Nat. Commun 7, 13694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Park S, Zeng L, Jain A, and Ha T (2018) Toward Single-Cell Single-Molecule Pull-Down. Biophys J 115, 283–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoskins AA, Gelles J, and Moore MJ (2011) New insights into the spliceosome by single molecule fluorescence microscopy. Curr Opin Chem Biol 15, 864–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ray S, Widom JR, and Walter NG (2018) Life under the Microscope: Single-Molecule Fluorescence Highlights the RNA World. Chem. Rev 118, 4120–4155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, and Moore MJ (2011) Ordered and dynamic assembly of single spliceosomes. Science 331, 1289–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, and Moore MJ (2013) Single-molecule colocalization FRET evidence that spliceosome activation precedes stable approach of 5′ splice site and branch site. Proc. Natl. Acad. Sci 110, 6783–6788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cherny D, Gooding C, Eperon GE, Coelho MB, Bagshaw CR, Smith CWJ, and Eperon IC (2010) Stoichiometry of a regulatory splicing complex revealed by single-molecule analyses. EMBO J 29, 2161–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hodson MJ, Hudson AJ, Cherny D, and Eperon IC (2012) The transition in spliceosome assembly from complex E to complex A purges surplus U1 snRNPs from alternative splice sites. Nucleic Acids Res 40, 6850–6862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Braun JE, Friedman LJ, Gelles J, and Moore MJ (2018) Synergistic assembly of human pre-spliceosomes across introns and exons. Elife 10.7554/eLife.37751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abelson J, Blanco M, Ditzler MA, Fuller F, Aravamudhan P, Wood M, Villa T, Ryan DE, Pleiss JA, Maeder C, Guthrie C, and Walter NG (2010) Conformational dynamics of single pre-mRNA molecules during in vitro splicing. Nat Struct Mol Biol 17, 504–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Krishnan R, Blanco MR, Kahlscheuer ML, Abelson J, Guthrie C, and Walter NG (2013) Biased Brownian ratcheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nat. Struct. Mol. Biol 20, 1450–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blanco MR, Martin JS, Kahlscheuer ML, Krishnan R, Abelson J, Laederach A, and Walter NG (2015) Single Molecule Cluster Analysis dissects splicing pathway conformational dynamics. Nat Methods 12, 1077–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Semlow DR, Blanco MR, Walter NG, and Staley JP (2016) Spliceosomal DEAH-Box ATPases Remodel Pre-mRNA to Activate Alternative Splice Sites. Cell 164, 985–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beier DH, Carrocci TJ, van der Feltz C, Tretbar US, Paulson JC, Grabowski N, and Hoskins AA (2019) Dynamics of the DEAD-box ATPase Prp5 RecA-like domains provide a conformational switch during spliceosome assembly. Nucleic Acids Res 47, 10842–10851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hahn S (2004) Structure and mechanism of the RNA polymerase II transcription machinery. Nat. Struct. Mol. Biol 11, 394–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schier AC, and Taatjes DJ (2020) Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev 34, 465–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Petrenko N, and Struhl K (2021) Comparison of transcriptional initiation by RNA polymerase II across eukaryotic species. eLife 10.7554/eLife.67964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morse RH (2022) Function and dynamics of the Mediator complex: novel insights and new frontiers. Transcription 13, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Z, English BP, Grimm JB, Kazane SA, Hu W, Tsai A, Inouye C, You C, Piehler J, Schultz PG, Lavis LD, Revyakin A, and Tjian R (2016) Rapid dynamics of general transcription factor TFIIB binding during preinitiation complex assembly revealed by single-molecule analysis. Genes Dev 30, 2106–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tomko EJ, Fishburn J, Hahn S, and Galburt EA (2017) TFIIH generates a six-base-pair open complex during RNAP II transcription initiation and start-site scanning. Nat. Struct. Mol. Biol 24, 1139–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fazal FM, Meng CA, Murakami K, Kornberg RD, and Block SM (2015) Real-time observation of the initiation of RNA polymerase II transcription. Nature 525, 274–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rosen GA, Baek I, Friedman LJ, Joo YJ, Buratowski S, and Gelles J (2020) Dynamics of RNA polymerase II and elongation factor Spt4/5 recruitment during activator-dependent transcription. Proc. Natl. Acad. Sci 117, 32348–32357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baek I, Friedman LJ, Gelles J, and Buratowski S (2021) Single-molecule studies reveal branched pathways for activator-dependent assembly of RNA polymerase II pre-initiation complexes. Mol. Cell 81, 3576–3588.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nguyen VQ, Ranjan A, Liu S, Tang X, Ling YH, Wisniewski J, Mizuguchi G, Li KY, Jou V, Zheng Q, Lavis LD, Lionnet T, and Wu C (2021) Spatiotemporal coordination of transcription preinitiation complex assembly in live cells. Mol. Cell 81, 3560–3575.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jonas S, and Izaurralde E (2015) Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet 16, 421–433 [DOI] [PubMed] [Google Scholar]
- 90.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, and Rajewsky N (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63 [DOI] [PubMed] [Google Scholar]
- 91.Global microRNA level regulation of EGFR-driven cell-cycle protein network in breast cancer (2012) Mol. Syst. Biol 8, 570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Milo R, Jorgensen P, Moran U, Weber G, and Springer M (2010) BioNumbers—the database of key numbers in molecular and cell biology. Nucleic Acids Res 38, D750–D753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Agarwal V, Bell GW, Nam J-W, and Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Salomon WE, Jolly SM, Moore MJ, Zamore PD, and Serebrov V (2015) Single-Molecule Imaging Reveals that Argonaute Reshapes the Binding Properties of Its Nucleic Acid Guides. Cell 162, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Willkomm S, Jakob L, Kramm K, Graus V, Neumeier J, Meister G, and Grohmann D (2022) Single-molecule FRET uncovers hidden conformations and dynamics of human Argonaute 2. Nat. Commun 13, 3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cui TJ, Klein M, Hegge JW, Chandradoss SD, van der Oost J, Depken M, and Joo C (2019) Argonaute bypasses cellular obstacles without hindrance during target search. Nat. Commun 10, 4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Halford SE, and Marko JF (2004) How do site‐specific DNA‐binding proteins find their targets? Nucleic Acids Res 32, 3040–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cui TJ, and Joo C (2019) Facilitated diffusion of Argonaute-mediated target search. RNA Biol [online] https://www.tandfonline.com/doi/abs/10.1080/15476286.2019.1616353 (Accessed May 18, 2022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sheu-Gruttadauria J, and MacRae IJ (2018) Phase Transitions in the Assembly and Function of Human miRISC. Cell 173, 946–957.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Förstemann K, Horwich MD, Wee L, Tomari Y, and Zamore PD (2007) Drosophila microRNAs Are Sorted into Functionally Distinct Argonaute Complexes after Production by Dicer-1. Cell 130, 287–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Miyoshi T, Takeuchi A, Siomi H, and Siomi MC (2010) A direct role for Hsp90 in pre-RISC formation in Drosophila. Nat. Struct. Mol. Biol 17, 1024–1026 [DOI] [PubMed] [Google Scholar]
- 102.Betancur JG, and Tomari Y (2012) Dicer is dispensable for asymmetric RISC loading in mammals. RNA 18, 24–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yoda M, Kawamata T, Paroo Z, Ye X, Iwasaki S, Liu Q, and Tomari Y (2010) ATP-dependent human RISC assembly pathways. Nat. Struct. Mol. Biol 17, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kawamata T, and Tomari Y (2011) Native Gel Analysis for RISC Assembly. in Argonaute Proteins: Methods and Protocols (Hobman TC, and Duchaine TF eds), pp. 91–105, Methods in Molecular Biology, Humana Press, Totowa, NJ, 10.1007/978-1-61779-046-1_7 [DOI] [PubMed] [Google Scholar]
- 105.Iwasaki S, Kobayashi M, Yoda M, Sakaguchi Y, Katsuma S, Suzuki T, and Tomari Y (2010) Hsc70/Hsp90 Chaperone Machinery Mediates ATP-Dependent RISC Loading of Small RNA Duplexes. Mol. Cell 39, 292–299 [DOI] [PubMed] [Google Scholar]
- 106.Azuma-Mukai A, Oguri H, Mituyama T, Qian ZR, Asai K, Siomi H, and Siomi MC (2008) Characterization of endogenous human Argonautes and their miRNA partners in RNA silencing. Proc. Natl. Acad. Sci 105, 7964–7969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kawamata T, Seitz H, and Tomari Y (2009) Structural determinants of miRNAs for RISC loading and slicer-independent unwinding. Nat. Struct. Mol. Biol 16, 953–960 [DOI] [PubMed] [Google Scholar]
- 108.Fukao A, Mishima Y, Takizawa N, Oka S, Imataka H, Pelletier J, Sonenberg N, Thoma C, and Fujiwara T (2014) MicroRNAs Trigger Dissociation of eIF4AI and eIF4AII from Target mRNAs in Humans. Mol. Cell 56, 79–89 [DOI] [PubMed] [Google Scholar]
- 109.Wakiyama M, Ogami K, Iwaoka R, Aoki K, and Hoshino S (2018) MicroRNP-mediated translational activation of nonadenylated mRNAs in a mammalian cell-free system. Genes Cells 23, 332–344 [DOI] [PubMed] [Google Scholar]
- 110.Wakiyama M, Kaitsu Y, and Yokoyama S (2006) Cell-free translation system from Drosophila S2 cells that recapitulates RNAi. Biochem. Biophys. Res. Commun 343, 1067–1071 [DOI] [PubMed] [Google Scholar]
- 111.Wakiyama M, Takimoto K, Ohara O, and Yokoyama S (2007) Let-7 microRNA-mediated mRNA deadenylation and translational repression in a mammalian cell-free system. Genes Dev 21, 1857–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, and Filipowicz W (2005) Inhibition of Translational Initiation by Let-7 MicroRNA in Human Cells. Science 309, 1573–1576 [DOI] [PubMed] [Google Scholar]
- 113.Mathonnet G, Fabian MR, Svitkin YV, Parsyan A, Huck L, Murata T, Biffo S, Merrick WC, Darzynkiewicz E, Pillai RS, Filipowicz W, Duchaine TF, and Sonenberg N (2007) MicroRNA Inhibition of Translation Initiation in Vitro by Targeting the Cap-Binding Complex eIF4F. Science 317, 1764–1767 [DOI] [PubMed] [Google Scholar]
- 114.Wang B, Doench JG, and Novina CD (2007) Analysis of microRNA effector functions in vitro. Methods 43, 91–104 [DOI] [PubMed] [Google Scholar]
- 115.Wang B, Love TM, Call ME, Doench JG, and Novina CD (2006) Recapitulation of Short RNA-Directed Translational Gene Silencing In Vitro. Mol. Cell 22, 553–560 [DOI] [PubMed] [Google Scholar]
- 116.Wang B, Yanez A, and Novina CD (2008) MicroRNA-repressed mRNAs contain 40S but not 60S components. Proc. Natl. Acad. Sci 105, 5343–5348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pitchiaya S, Heinicke LA, Park JI, Cameron EL, and Walter NG (2017) Resolving Subcellular miRNA Trafficking and Turnover at Single-Molecule Resolution. Cell Rep 19, 630–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pitchiaya S, Mourao MDA, Jalihal AP, Xiao L, Jiang X, Chinnaiyan AM, Schnell S, and Walter NG (2019) Dynamic Recruitment of Single RNAs to Processing Bodies Depends on RNA Functionality. Mol. Cell 74, 521–533.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Holzmeister P, Acuna GP, Grohmann D, and Tinnefeld P (2014) Breaking the concentration limit of optical single-molecule detection. Chem Soc Rev 43, 1014–1028 [DOI] [PubMed] [Google Scholar]
- 120.Lo Gullo G, Mattossovich R, Perugino G, La Teana A, Londei P, and Benelli D (2019) Optimization of an In Vitro Transcription/Translation System Based on Sulfolobus solfataricus Cell Lysate. Archaea 2019, e9848253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hunter DJB, Bhumkar A, Giles N, Sierecki E, and Gambin Y (2018) Unexpected instabilities explain batch-to-batch variability in cell-free protein expression systems. Biotechnol. Bioeng 115, 1904–1914 [DOI] [PubMed] [Google Scholar]
- 122.Ha T, Kaiser C, Myong S, Wu B, and Xiao J (2022) Next generation single-molecule techniques: Imaging, labeling, and manipulation in vitro and in cellulo. Mol. Cell 82, 304–314 [DOI] [PubMed] [Google Scholar]
- 123.Thorn K (2017) Genetically encoded fluorescent tags. Mol Biol Cell 28, 848–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gust A, Zander A, Gietl A, Holzmeister P, Schulz S, Lalkens B, Tinnefeld P, and Grohmann D (2014) A starting point for fluorescence-based single-molecule measurements in biomolecular research. Molecules 19, 15824–15865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Blackstock D, and Chen W (2014) Halo-tag mediated self-labeling of fluorescent proteins to molecular beacons for nucleic acid detection. Chem Commun 50, 13735–13738 [DOI] [PubMed] [Google Scholar]
- 126.Cole NB (2013) Site-specific protein labeling with SNAP-tags. Curr Protoc Protein Sci 73, 30.1.1–30.1.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liss V, Barlag B, Nietschke M, and Hensel M (2016) Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Sci. Rep 10.1038/srep17740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Banaz N, Mäkelä J, and Uphoff S (2019) Choosing the right label for single-molecule tracking in live bacteria: side-by-side comparison of photoactivatable fluorescent protein and Halo tag dyes. J Phys Appl Phys 52, 064002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Erdmann RS, Baguley SW, Richens JH, Wissner RF, Xi Z, Allgeyer ES, Zhong S, Thompson AD, Lowe N, Butler R, Bewersdorf J, Rothman JE, St Johnston D, Schepartz A, and Toomre D (2019) Labeling Strategies Matter for Super-Resolution Microscopy: A Comparison between HaloTags and SNAP-tags. Cell Chem Biol 26, 584–592.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nanda JS, and Lorsch JR (2014) Labeling a protein with fluorophores using NHS ester derivitization. Methods Enzym 536, 87–94 [DOI] [PubMed] [Google Scholar]
- 131.Carugo O (2008) Amino acid composition and protein dimension. Protein Sci 17, 2187–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kim Y, Ho SO, Gassman NR, Korlann Y, Landorf EV, Collart FR, and Weiss S (2008) Efficient site-specific labeling of proteins via cysteines. Bioconjug Chem 19, 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ryu Y, and Schultz PG (2006) Efficient incorporation of unnatural amino acids into proteins in Escherichia coli. Nat. Methods 3, 263–265 [DOI] [PubMed] [Google Scholar]
- 134.Chin JW (2017) Expanding and reprogramming the genetic code. Nature 550, 53–60 [DOI] [PubMed] [Google Scholar]
- 135.Lang K, and Chin JW (2014) Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem Rev 114, 4764–4806 [DOI] [PubMed] [Google Scholar]
- 136.Gust A, Jakob L, Zeitler DM, Bruckmann A, Kramm K, Willkomm S, Tinnefeld P, Meister G, and Grohmann D (2018) Site-Specific Labelling of Native Mammalian Proteins for Single-Molecule FRET Measurements. ChemBioChem 19, 780–783 [DOI] [PubMed] [Google Scholar]
- 137.Lee KJ, Kang D, and Park H-S (2019) Site-Specific Labeling of Proteins Using Unnatural Amino Acids. Mol Cells 42, 386–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee TC, Kang M, Kim CH, Schultz PG, Chapman E, and Deniz AA (2016) Dual Unnatural Amino Acid Incorporation and Click-Chemistry Labeling to Enable Single-Molecule FRET Studies of p97 Folding. Chembiochem 17, 981–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang K, Schmied WH, and Chin JW (2012) Reprogramming the genetic code: from triplet to quadruplet codes. Angew Chem Int Ed Engl 51, 2288–2297 [DOI] [PubMed] [Google Scholar]
- 140.Sánchez-Rico C, von Voithenberg LV, Warner L, Lamb DC, and Sattler M (2017) Effects of Fluorophore Attachment on Protein Conformation and Dynamics Studied by spFRET and NMR Spectroscopy. Chem. - Eur. J 23, 14267–14277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kalinin S, Peulen T, Sindbert S, Rothwell PJ, Berger S, Restle T, Goody RS, Gohlke H, and Seidel CAM (2012) A toolkit and benchmark study for FRET-restrained high-precision structural modeling. Nat Methods 9, 1218–1225 [DOI] [PubMed] [Google Scholar]
- 142.Nagy J, Grohmann D, Cheung ACM, Schulz S, Smollett K, Werner F, and Michaelis J (2015) Complete architecture of the archaeal RNA polymerase open complex from single-molecule FRET and NPS. Nat. Commun 10.1038/ncomms7161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schmidt A, Altincekic N, Gustmann H, Wachtveitl J, and Hengesbach M (2018) The Protein Microenvironment Governs the Suitability of Labeling Sites for Single-Molecule Spectroscopy of RNP Complexes. ACS Chem Biol 13, 2472–2483 [DOI] [PubMed] [Google Scholar]
- 144.Rinaldi AJ, Lund PE, Blanco MR, and Walter NG (2016) The Shine-Dalgarno sequence of riboswitch-regulated single mRNAs shows ligand-dependent accessibility bursts. Nat. Commun 7, 8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Welty R, Schmidt A, and Walter NG (2023) Probing Transient Riboswitch Structures via Single Molecule Accessibility Analysis. in RNA Structure and Dynamics (Ding J, Stagno JR, and Wang Y-X eds), pp. 37–51, Methods in Molecular Biology, Springer US, New York, NY, 10.1007/978-1-0716-2687-0_4 [DOI] [PMC free article] [PubMed] [Google Scholar]