

Summary:
The pulmonary surfactant system of the lung is a lipid and protein complex, which regulates the biophysical properties of the alveoli to prevent lung collapse, and the innate immune system in the lung. Pulmonary surfactant is a lipoprotein complex consisting of 90% phospholipids and 10% protein, by weight. Two minor components of pulmonary surfactant phospholipids, phosphatidylglycerol (PG) and phosphatidylinositol (PI), exist at very high concentrations in the extracellular alveolar compartments. We have reported that one of most dominant molecular species of PG, palmitoyl-oleoyl phosphatidylglycerol (POPG) and PI inhibit inflammatory responses induced by multiple Toll-like receptors (TLR2/1, TLR3, TLR4, and TLR2/6) by interacting with subsets of multiprotein receptor components. These lipids also exert potent anti-viral effects against RSV and influenza A, in vitro, by inhibiting virus binding to host cells. POPG and PI inhibit these viral infections in vivo, in multiple animal models. Especially noteworthy, these lipids markedly attenuate SARS-CoV-2 infection including its variants. These lipids are natural compounds that already exist in the lung, and thus are less likely to cause adverse immune responses by hosts. Collectively, these data demonstrate that POPG and PI have strong potential as novel therapeutics for applications as anti-inflammatory compounds and preventatives, as treatments for broad ranges of RNA respiratory viruses.
Keywords: Pulmonary surfactant phospholipids, Toll-like receptors, anti-inflammatory treatment, Respiratory RNA viruses, Antivirals
1. Introduction
The pulmonary surfactant system of the lung is a lipid and protein complex, secreted at the air-liquid interface of alveolar compartments.1,2 It mainly consists of phospholipids and lesser amounts of protein. Pulmonary surfactant contains specific proteins designated SP-A, SP-B, SP-C and SP-D. SP-A and SP-D are water soluble, whereas SP-B and SP-C are extraordinarily hydrophobic. One major role of pulmonary surfactant is to stabilize the biophysical properties of the alveoli at the end of the respiratory cycle, thereby preventing disruption of gas exchange. SP-A and SP-D are highly oligomeric proteins (consisting of 18mers or 12mers with assembled molecular sizes ranging from, 650 kDa - 106 kDa) comprised of smaller primary translation products (28kDa-~40kDa). It has been well established that the functions of the water-soluble surfactant proteins, SP-A and SP-D play critical roles in the innate immune system of the lung for host defense.2–4 The major components of surfactant are phospholipids and their biophysical roles have been well established.5,6 We have reported that two minor components of surfactant phospholipids, phosphatidylglycerol (PG) and phosphatidylinositol (PI), have very potent inhibitory innate immune regulatory activities, which are due to blocking activation of mutiple Toll-like receptors (TLR2/1, TLR3, TLR4 and TLR2/6).2,7–10 We have also discovered that these lipids have anti-viral effects against broad ranges of respiratory RNA viruses, including repiratory syncytial virus (RSV),11–13 influenza A viruses (IAV)14,15 and SARS-CoV-2.2 In this review, we will describe, 1) fundamental properties of pulmonary surfactant, 2) anti-inflammatory mechanisms of action of surfactant phospholipids in innate immunity of the lung, especially focusing on inhibitory effects on TLR related inflammation, and lastly, 3) the mechanisms of action of surfactant phospholipids against different respiratory viruses. We will also discuss our perspectives on the future applications of the aforementioned phospholipds as potential thereupeutics and preventatives for inflammatory lung diseases.
2. The basics of pulmonary surfactant
2– 1). Pulmonary surfactant composition, structure, and biological activities
Pulmonary surfactant is a mixture of lipids and proteins, consisting of 90% phospholipids and 10% proteins (Fig. 1). Phosphatidylcholine (PC) is the most abundant molecular class of phospholipid, and dipalmitoylphosphatidylcholine (DPPC), is the dominant molecular species within the PC class. In biophysical studies, DPPC is the most active constituent within the mixture for reducing surface tension.1,2,16 As shown in Fig. 1, there are other molecular species within the PC class including 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (16:0–18:1 PC) (POPC), and 1-palmitoyl, 2-palmitoleoyl phosphatidylcholine (16:0, 16:1 PC) (PPPC). The minor anionic phospholipids are comprised of phosphatidylglycerol (PG) and phosphatidylinositol (PI). These lipids exist in extracellular compartments of aloveoli at very high concentraions as compared to other organs.2,16 Both PG and PI, are necessary to support hydrophobic surfactant protein (SP-B and SP-C), adsorption into surfactant films.1,16 The interactions beween SP-B/SP-C, and these phospholipids, not only stabilize, but also maintatin surfactant membrane structure during dynamic size changes within the alveolus.1,16 The hydrophilic proteins, SP-A and SP-D have been extensively investigated for their roles in the innate immunity of the lung2,3 and critically correlated with multiple respiratory diseases.2,17 Both SP-A and SP-D are highly oligomeric lectins that interact with various pathogens including bacteria, fungi, and viruses.3,18 These proteins support lung defense by enhancing and regulating: 1) Opsonization of pathogens to facilitate recognition by phagocytes and macrophages, 2) regulating inflammatory mediator production (e.g., TNF-alpha and nitric oxide (NO) secretion by immune cells), 3) clearance of apoptotic cells and DNA by alveolar macrophage and monocytic cells.3,18 Both SP-A and SP-D directly bind to pathogens, or substituents of microorganism surfaces (e.g., LPS and high mannose oligosaccharides) to promote their clearance.3,18 Additionally, these proteins have direct antimicrobial effects, exerted by increasing permeability of bacteria and inhibiting fungal growth.19,20 SP-A and SP-D also play an important role in the transition to adaptive immunity from innate immunity of the lung.3,18 Both proteins have different effects on dendritic cells (DCs). SP-A inhibits DC maturation by modulating expression of cell surface markers3 whereas SP-D promotes antigen presentation by DCs.21 For T cells, both proteins inhibit T-cell proliferation. Taken together, these effects on DCs and T cells, result in regulating excessive innate immune responses induced by pathogens, and prevention of excessive lung damage.3
Figure1: Surfactant composition.

Pulmonary surfactant consists of phospholipids (~90%) with phosphatidylcholine (PC) and phosphatidylglycerol (PG) and phosphatidylinositol (PI) as the major molecular classes in that order. About 10% of pulmonary surfactant is comprised of proteins that are either hydrophobic, (SP-B and SP-C) or hydrophilic and highly oligomeric (SP-A and SP-D) (adapted figure from ref 2).
As stated above, major components of pulmonary surfactants are phospholipids 2,16,22 (Fig. 1). We have accumulated evidence that the minor anionic surfactant phospholipids, phosphatidylglycerol (PG) and phosphatidylinositol (PI), exert anti-inflammatory effects by suppressing activation of mutiple TLRs and disrupting infections by mutiple respiratory RNA viruses.2,10,16 PG and PI share structual similarities, harboring polar headgroups with multiple hydroxyl substituents 2,16,22–24 (see Fig. 2). Phosphatidylglycerol (PG) is an important structural constituent of membranes of bacteria, plants and mammalian cells, and in some cases, serves as a major component.24 PG is found as a consituent of most Gram-negative bacterial inner membranes (Kdo2-Lipid A) (Fig. 2).25 Phosphatidylinositol is also an important lipid as a membrane constituent in both fungi and mammalian cells.24 Both PG and PI share structural similarity as shown in Fig.2. We have reported that these lipids have similar efficacies as antagonists against inflammation resulting from activation of multiple TLRs.2,9
Figure 2: Structural similarity of PG, PI and KdO2 lipid A.

The structure of PG, PI and KdO2 Lipid A are shown. The schematic figures for PG and PI highlight their structural similarity, especially in the polar headgroups. PtdO is a shorthand abbreviation for the phosphatidyl radyl harboring a diacylglycerol backbone coupled via phosphodiester linkage to the polar headgoups.2,10
2– 2). Pulmonary surfactant and lung diseases
Pulmonary surfactant plays important roles regulating and maintaining lung homeostasis.2,16,17 Dysfunctions of pulmonary surfactant are connected to numerous lung diseases.17 Impairments of pulmonary surfactant in pulmonary diseases are mainly related to; 1) alteration of pulmonary surfactant levels (e.g., respiratory distress syndrome of newborn (RDS), acute respiratory distress syndrome (ARDS) and interstitial lung diseases (ILD)), 2) genetic disorders of the pulmonary surfactant systems (e.g., pulmonary alveolar proteinosis (PAP), familial interstitial pneumonia, chronic pulmonary microlithiasis), and 3) gene polymorphisms of collectins (SP-A and SP-D) (e.g., asthma, viral infections).17,22 Especially, pulmonary surfactant phospholipids are key elements in the pathogenesis of interstitial lung diseases (ILDs) (e.g., idiopathic pulmonary fibrosis (IPF), sarcoidosis, hypersensitivity pneumonitis) and RDS/ARDS.22,26 Alveolar type II cells (AT2) are, the primary sources of pulmonary surfactant phospholipids, and maintain homeostasis of pulmonary surfactant in the alveolus.1,2,22,24 IPF is a chronic progressive disease of unknown etiology, with a poor prognosis.17,26,27 In IPF, the loss and dysfuntion of AT2 cells leads to reduction of pulmonary surfactant synthesis and hyperplasia of AT2 cells, resulting in imbalance of alveolar niche homeostasis.26,27 The dysfunction of AT2 cells also alters pulmonary surfactant phospholipid metabolism, increasing release of arachidonic acid and lysophospholipids, to promote lung fibrosis.22,26,27 In some studies, the composition of surfactant phospholipids changed with increasing sphingomyelin and PI, and reducing PG, and levels of various PC subspecies, in the BALF recovered from IPF patients.17,26
RDS usually impacts pre-term infants, because of immature AT2 cells, which cause insuffcient pulmonary surfactatnt production in the lung, and alteration of the composition of surfactant phosholipids and proteins recovered in BALF.17,27 Surfactant replacement therapy can bring signifnicant benefits to improve RDS patient outcomes.22,27 The surfactant formulations need a specific balance of phospholipids and proteins.22,28,29 There is a new generation of synthetic clinical pulmonary surfactant composed of two phospholipid species, dipalmitoyl phosphatidylcholine (DPPC) and palmitoyl-oleoyl phosphatidylglycerol (POPG), and animal-derived SP-B and SP-C that have significant potential for clinical usage.29
ARDS has many etiologies for its causes, and is mainly initiated by severe infections and sepsis.2 The surfactant phospholipid compositions were dramatically alterd by reductions in PC and PG, and concommitant increases of PI, phosphatidylethanolamine (PE) and phosphatidylserine (PS), in BALF of ARDS patients.30 These changes are similar to changes in RDS patients.22,26 The COVID19 pandemic has increased case numbers for ARDS, with high mortality rates. Severe COVID19 disease causes fibrotic changes as one major complication, resulting in a poor prognosis.31 The ACE2 receptor for SARS-CoV-2 is highly expressed in AT2 cells, and SARS-CoV-2 infection causes severe damage to AT2 cells,32–34 which leads to disruption of the pulmonary surfactant system.
COVID19 related ARDS and RDS share similarities in their clinical features that impair lung compliance and create intravascular coagulation and microthrombi.34–36 SARS-CoV-2 infection changes surfactant composition and impairs pulmonary surfactant in COVID19 patients.37–40 The reduction of PCs, and the increase of PE and PS were observed in the plasma of COVID19 patients and correlated to outcome of patients.37–40 The DPPC levels in the BALF of COVID19 patients were also reduced.38 Based on these data, several clinical trials of surfactant treatments for COVID19 are ongoing, but the benefits of surfactant therapy for COVID19 are not yet clear.37 If we can demonstrate the purified phospholipid treatments are beneficial, the simplicity of these formulations, makes their use more attractive than the complexity of artificial surfactants.
Collectively, dysfunction and impairment of pulmonary surfactant phospholipids are strongly correlated to pathophysiology of a variety of respiratory diseases.
3. Anti-inflammatory properties of anionic pulmonary surfactant phospholipids in the lung
Toll-like receptors (TLRs) were identified as pathogen related receptors (PRRs) that recognize pathogen associated molecular patterns (PAMPs).41 TLRs play a critical role to initiate innate immune responses against numerous pathogens, including bacteria, fungi, and viruses.41 As stated above, we have accumulated significant evidence that the minor pulmonary surfactant phospholipids, POPG and PI have broad anti-inflammatory effects by inhibiting activation of multiple TLR, 2,7–10; and regulating innate immunity within the lung. As shown in Fig. 3, both POPG and PI have inhibitory effects on TLR2/1, TLR3, TLR4 and TLR2/6 that result in attenuation of inflammatory mediator secretions. In this section, we will describe the mehanisms of action of these lipids upon inflammation induced by TLR activations.2
Figure 3: The mechanisms of POPG and PI antagonism against multiple TLR signaling pathways.

POPG and PI inhibit multiple TLR induced signaling pathways including TLR4, TLR2/1. TLR2/6 and TLR3. POPG and PI prevent TLR4 induced inflammation (e.g., LPS) by preventing the TLR4 co-receptors CD14 and MD2 from forming multi-protein signaling platforms. POPG and PI inhibit the recognition of Pam3CysK4 by TLR2/1 and MALP2 by TLR2/6 complexes. These lipids also antagonize proinflammatory cytokine production by TLR3 that exists in endosome.2
3– 1). Anionic pulmonary surfactant phospholipids inhibit inflammatory responses induced by TLR4 activation.
Lipopolysaccharide (LPS), also known as Gram-negative bacteria endotoxin, is recognized by TLR4 via sequential processes. First, LPS engages LPS-binding protein (LBP) which subsequently transfers it to cluster of differentiation 14 (CD14) and subsequently myeloid differentiation factor 2 (MD2). These events result in formation of the activated heterooligomer of LPS/ CD14/MD2/TLR4 as shown in Figs. 3 and 5E.2,41
Figure 5. POPG and PI disrupt TLR4 induced inflammation by binding to LPS binding sites of CD14.


We determined POPG and PI affinity to bind to CD 14 using phospholipids solid phase binding assay. We used DPPC as a negative control lipid. The lipids were coated onto ELISA plate (1.25nomol/well) and CD14 was added to the wells at various concentrations. The binding reaction was determined by anti CD14 antibody. Panel A) shows the binding interaction of the lipids and CD14. POPG and PI bound to CD14 with high affinity and a dose dependent manner. DPPC did not bind to CD 14 very weakly. To determine the binding sites of POPG and PI, we performed the competition assays. CD14 was incubated with monoclonal antibodies that recognize LPS specific binding sites in CD14 (biG14, RDIg, MEM-18 and biG2)7,10 We used mouse IgG (mIgG) as negative control, and CD14 alone binding was shown as control. The mixture of CD14 and each monocolnal antibody was preinculated for 1hr at 37°C and then added into lipid solid phase coated with POPG or PI (1.25nmol/well) for 1hr. The binding of CD14 or CD14+ antibody mixtures to the lipids were developed by using anti-CD14 antibody, and signals were measured at 490nm absorbance. CD14 alone binding is indicated as 100%. Panel B) shows the binding interaction of the lipids and MD2. POPG binds to MD2 with high affinity, but PI has low binding affinity to MD2. The CD14 or CD14+ antibody mixture binding interaction is shown as panel C) in POPG solid phase, and panel D) in PI solid phase. Data are shown as mean ± SE and * indicates p<0.05 by unpaired tailed t-test.7,10 These figures were adapted from ref 7 Panel E) is a schematic figure to represent the inhibitory mechanisms of actions of POPG and PI on TLR4 via disrupting TLR4/CD14 and TLR4/MD2 complex.
The major TLR4 expressing cells are myeloid cells 41 (e.g., monocytes, macrophages, granulocyte). TLR4 can initiate innate immune responses via both the MyD88-dependent pathway with NF-κ B and AP-1 activation 2,41 (Fig. 3), and the MyD88-independent pathway involving the adaptor proteins TIR-domain-containing adaptor inducing interferon-β (TRIF) and the TRIF-related Adaptor Molecule (TRAM). TRAM-TRIF signals result in activation of the transcription factor, Interferon Regulatory Factor-3 (IRF3) to induce expression of Type1 interferons (IFN alpha/beta) (Fig. 3).41 Previous work reported that some of the pulmonary surfactant phospholipids can inhibit inflammation induced by LPS or pathogens.2 PGs, PI and cardiolipin (CL) inhibited LPS-induced macrophage activation7 by antagonizing TLR activation.42
We detemined the potencies of POPG and PI on inflammation, induced by LPS stimulation, using human primary alveolar macrophages. The cells were pretreated with or without phospholipids (200μg/ml) following challenge with LPS from E.Coli 0111:B4 (10ng/ml), for 6hrs. Culture supernatants were used for quantification of TNF-α by ELISA. LPS challenge induced significant secretion of TNF-α at the level of ~40ng/ml by human primary alveolar macrophages. Both POPG and PI treatment markedly attenuated TNF-α production induced by LPS, whereas DPPC failed to have a significant effect upon TNF-α secretion (data are shown as mean ± SD (pg/ml), * indicates, p<0.05, (Fig. 4A).7,10
Figure 4: POPG and PI inhibit TNF-α production via multiple TLR activations at 6hr.

POPG or PI inhibit induced TNF-α produced from human alveolar macrophages by activation of, A) TLR4 with LPS (10ng/ml)7,10 B) TLR2/1 with Pam3CysK4 (1μg/ml), C) TLR2/6 with MALP2 (10ng/ml)8,9 and D) TLR3 with polyI:C (10μg/ml). The TNF-α production was measured by ELISA at 6hrs following stimulation with TLR agonists.7,10 Data are shown as mean ± SD (pg/ml), * indicates p<0.05, § indicates p<0.01, §§ indicates p<0.001 by paired t-test.
Next, we investigated the mechanism by which POPG and PI disrupt LPS induced inflamatory responses.7,43 We examined the effects of POPG and PI on CD14 which is required for TLR4 activation, by quantifying interactions with solid phase phospholipids.7,11 Both POPG and PI bound to CD14 protein with high affinity, in a dose-dependent manner (Fig. 5A). In contrast, DPPC showed non-specific and relatively weak binding to CD14 ( Fig. 5A).7 To detemine which specific sites within CD14 interact with the lipids, we incubated CD14 protein with specific monoclonal antibodies (biG14, RDIg, Big14 and MEM-18, biG2 ) that recognize LPS binding sites at specific domains within the protein.7,44 The monoclonal antibodies biG14 and MEM-18 inhibited the binding interaction of POPG and CD14 by 40% and 50% respectively7 (Fig. 5C). Coincubation with biG14 and MEM-18 was slightly more potent for preventing CD14 binding to POPG by 60% (Fig. 5C). The results were quite similar for biG14 and MEM-18 antagonism for the CD14 binding interaction to PI (Fig. 5D). MD-2 is required for TLR4 activation by LPS, because LPS directly binds to CD14 and MD-2, but not TLR4.41,44 We also tested the binding activities of POPG and PI to MD-2. POPG had very strong binding to MD-2; whereas PI did not (Fig. 5B).7,10
Collectively, POPG directly binds to LPS binding sites of CD14 and MD-2 resulting in antagonism of TLR4 activation as shown in Fig. 5E. The direct binding affinities appear to be good criteria for predicting the inhibitory effects on TLR4 activation. These data also demonstrate that POPG and PI inhibit LPS induced TLR4 activation by non-identical mechanisms.7
3– 2). POPG inhibits TLR4 related inflammatory cascades induced by LPS stimulation
We examined the impact of POPG on TLR 4 related intracelluar signaling pathway induced by LPS stimulation of the human macrophage cell line, U937.7,10 As shown in Fig. 3, LPS activates TLR4 related inflammatory pathways including Myeloid differentiation primary response 88 (MyD88), to activate IRAKs (interleukin 1 receptor associated kinases). The phosphorylation of MAP kinases (ERK, JNK, p38); or activation of kappa B kinase (IKK) inactivates IkBa, these events initiate transcription factor activation, (NFkB or AP-1) to produce inflamatory mediators releasing proinflammatory cytokines, chemokines and eicosanoids.10,41,45 U937 cells were stimulated with LPS (10ng/ml) at various time points (15mins, 30min and 60mins), and cell lysates were processed to quantify MAP kinases, and/or IκBα, by immunoblotting. LPS challenge induced strong phosphorylation of MAP kinases (ERK, JNK, p38) after 15–30 mins of stimulation and these signals weakened at 60mins. Pretreament with POPG, at 30 mins before LPS stimulation completely abolished phosphorylation of MAP kinases and IκBα.7,10
3– 3). POPG inhibits LPS induced lung injury in an vivo model in mice.
We also detemined the effect of POPG in LPS-induced lung injury using an in vivo model in mice. Mice were challenged with LPS intratracheally using a MicroSprayer.7 LPS challenge induced significant proinflammatory cytokine production including TNF-α, KC and MIP-2, recovered in the BALF of mice at 18hrs after challenge.7 Both POPG and PI treatment markedly attenuated these mediator productions in BALF by 50~80%. We also injected LPS intravenously via the tail vein (50μg/200μl of PBS) and introduced POPG (50μg in 20μl of PBS) intratracheally at the same time. BALF were harvested at 3hrs after LPS challenge. POPG treatment also significantly reduced LPS-induced proinflammatory cytokine levels in BALF.7 DPPC failed to have an impact on these cytokine productions. These data clearly show that both POPG and PI inhibited LPS-induced inflammation in the lung, in vivo, in mice. There are several reports that reduced levels of PG in the lung, might contribute to develop, or cause, lung diseases, especially ARDS17,27,22,26 perhaps because of insufficient suppression of inflammation by TLR4 activation.17,27
In summary, POPG and PI suppress the inflammatory responses induced by TLR4 activation by interacting with CD14 or CD14/MD2. These lipids also attenuated LPS induced lung inflammation in an in vivo model in mice. The data suggest that POPG and/or PI might be useful to treat ALI/ARDS patients.
3– 4). Pulmonary surfactant phospholipids inhibit effects induced by multiple TLR activations
The findings stated above, led us to investigate whether pulmonary surfactant phospholipids antagonize other TLR activations. We examined the potency of POPG and PI using human primary macrophages, with stimulation by multiple cognate TLR agonists. For TLR2/1, we used Pam3CysK4, for TLR3, we used poly I:C, and for TLR2/6, we used the Mycoplasma fermentans derived ligand, (MALP2).2 The cells were challenged with each TLR agonist alone, or with TLR agonists in the presence of phospholipids for 6hrs. The culture supernatants were harvested to quantify TNF-α secretion, as an outcome of inflammation (Fig. 4). As previously stated, both POPG and PI inhibited TNF-α production by TLR4 activation, with LPS. POPG also markedly reduced TNF-α secretion induced by Pam3Cys by 98% (Fig. 4B), MALP2 by 85% (Fig.4C) and poly I:C by 96% (Fig. 4D). We also quantified lipid potencies against multiple TLRs using the bronchial epithelial cell line, BEAS-2B.2 The cells were challenged with TLR agonists including flagellin, as a TLR5 agonist, both in the presence and absence of phospholipids (POPG, PI and POPC at 200μg/ml) for 48hrs. Culture supernatants were harvested to quantify IL-8 production as an inflammatory outcome. Each TLR agonist induced significant IL-8 release into the media (6000 to 10,000pg/ml) as shown in Fig. 6. Both POPG and PI markedly reduced IL-8 production induced by Pam3Cys (Fig. 6A), MALP-2 (Fig. 6B), and poly I:C (Fig. 6C). POPC failed to inhibit IL-8 production induced by these TLR agonists. Interestingly, these lipids did not affect flagellin induced IL-8 secretion (Fig. 6D). The data demonstrate POPG and PI are effective antagonists against multiple TLRs. This antagonism is not a consequence of either cytotoxicity, or non-specific inhibitory processes. 2
Figure 6: Pulmonary surfactant phospholipids broadly antagonize IL-8 production induced by multiple TLR agonists in human bronchial epithelial cells.

The human bronchial epithelial cell line, BEAS-2B, was stimulated with TLR agonists, A) Pam3Cys4K (1mg/ml)for TLR2/1 (Pam), B) MALP2 (10ng/ml) for TLR2/6 (MALP2), C) poly I:C (0.1mg/ml) for TLR3 (PolyI:C), and D) flagellin (10ng/ml) for TLR5 (Fla) with or without of POPG, PI or POPC (200mg/ml each lipid). IL-8 production was quantified by ELISA at 48hrs after challenge with agonists. Data are shown as mean ± SD, §§ indicates p<0.001 by paired t-test.2 Figures were adapted from ref 2.
3– 5). The mechanism of action of POPG against TLR3 activation
Previous publications, have documented that bronchial epithelial cells express high levels of TLR3, including BEAS-2B cells.46,47 We next focused on the mechanism by which POPG antagonizes TLR3 induced inflammation, using BEAS-2B cells, which can be stimulated by poly I:C, as shown in Fig. 6C. To determine the site of action of POPG on TLR3 activation by poly I:C, we employed BODIPY-PG to visualize the intracellular co-localization with Rhodamine labeled poly I:C (Rho-pIC) by confocal microscopy. BEAS-2B cells were incubated with Rho-poly I:C (1μg/ml) and BODIPY-PG (50μg/ml) in liposomes containing 2% of the fluorescent lipid, BODIPY-PG, for 48hrs. Following washing with PBS, the cells were fixed, and processed for analysis by confocal microscopy. As shown in Fig. 7A and 7B, both BODIPY-PG and Rho-poly I:C were internalized and distributed among organelles of BEAS-2B cells. The higher magnification images demonstrate that BODIPY-PG and Rho-poly I:C co-localized within intracellular compartments of the cells (7D and 7E). The merged images (Fig. 7C and 7F) showed that BODIPY-PG and Rho-poly I:C significantly colocalized in subdomains of the cytosolic compartment. We also tested whether Rho-poly I:C and BODIPY-PG maintain the TLR3 antagonism of IL-8 secretion by BEAS-2B cells. Rho-poly I:C (1μg/ml) induced significant IL-8 secretion, which was nearly identical to unlabeled poly I:C. In addition, BODIPY-PG (50μg/ml) markedly inhibited Rho-poly I:C induced IL-8 production by ~80% (unpublished data, as personal communication from Voelker and Numata-Nakamura Labs, National Jewish Health, Denver, CO).
Figure 7. Rhodamine-Poly I:C and Fluorescent PG colocalize in same intracellular compartment of BEAS-2B cells.

Panel A) shows the internalization of BODIPY-POPG and Panel B) shows Rhodamine polyI:C (RhoPIC) internalization in BEAS-2B cells. Panels D), E) and F) are the images at higher magnifications from Panel A)-C). Panel C) and F) shows the colocalization of BODIPY-PG and RhoPIC in intracellular compartments.
These data demonstrate that POPG inhibits TLR3 induced inflammation by colocalizing with the TLR3 agonist, intracellularly.
4. The mechanisms of actions of pulmonary surfactant phospholipids against multiple respiratory viral infections
The finding of anti-inflammatory effects of POPG and PI with respect to inhibition of the activation of multiple TLRs led us to investigate the anti-viral effects of these lipids. As shown in Fig.4–6, these lipids are very potent inhibitors of TLR4-induced proinflammatory cytokine production by directly binding to MD2 and CD14.2,7 CD14 and TLR4 also play critcal roles for RSV and influenza A infections as previously reported.48,49 RSV is one of the most common causes of serious lower respiratory infection in both young children and older adults.50,51,52 Recently, clinical trials of promising vaccines for RSV, have been ongoing, but currently there is no vaccine for healthy infants.53 Palivizumab [Synagis] is a monoclonal antibody against the F (fusion) - protein of RSV, and used as a prophylaxis, and its indications are very limited for pediatric patients, and are restricted to, 1) preterm infants, 2) patients with bronchopulmonary dysplasia (BPD) and 3) patients with congenital heart disease (CHD) at high risk for RSV infection.52,54 There is currently no approved mediince for the treatment of RSV infection.51,52 In 2022, RSV started to surge earlier than usual, and resulted in higher than average numbers of hospitalized cases; and also affected older children compared to previous seasonal outbreaks.55 Because of mask mandates and home isolations due to COVID19 pandemic, there was a reduction in general immunity as a consequence of reduced exposures to RSV in infants and young children. This phenomena has been called “immune debt”.56,57 The lack of usual population immunity might be a contributing factor to severe RSV illnesses.56 Especially, in the context of the COVID19 pandemic, new strategies and inteventions for RSV infection, are urgently needed.
4– 1). Pulmonary surfactant phospholipids, POPG and PI, inhibit RSV infection by disrupting binding between virus and host cells
We hypothetized POPG and PI can inhibit RSV infection by blocking CD14/TLR4 activation. To determine the antiviral activity of POPG against RSV in vitro, we treated HEp-2 cells either with, or without POPG, (200μg/ml or 500μg/ml suspended in agarose/well) at 2hrs after RSV challenge. The RSVA2, lab strain, was used in these experiments. RSV plaque formation was visualized by neutral red staining 11 (Fig. 8A). Treatment with POPG significantly reduced plaque numbers in a dose dependent manner, but POPC failed to alter RSV plaque numbers as complete plaques (data are shown as mean ± SD (plaque numbers/well), RSV: 153.8 ± 23.2, RSV + POPG-200 μg: 5.9 ± 5.7, RSV + POPG-500 μg: 1.0 ± 1.3, RSV + POPC-200 μg: 149.2 ± 22.9 (Fig. 8C), §§ represents, p<0.001). Under careful observations, we found unusual regions in the plaque assays (indefinite plaques) (Fig. 8B–2). Following staining with anti RSV antibody, the unusual regions, were defined as indefinite plaques, and turned out to be RSV infected cells (Fig. 8B–4) yielding plaques, of much smaller diameter. Typical plaques were created by cell lysis and presented with the appearance of bullseyes, with clear areas being surrounded with brown immunostaining for RSV (Fig. 8B–1 and 8B–3). We quantified indefinite plaque numbers as shown in Fig. 8C. Treaments with higher concentrations of POPG reduced the numbers of indefinite plaques in a dose-dependent manner (data are shown as mean ± SD (indefinite plaque numbers/well), RSV + POPG-200 μg: 78.8 ± 29.1, RSV + POPG-500 μg: 44.3 ± 11.0, *indicates, p<0.05 in comparison to RSV complete plaque numbers). The results show that POPG can suppress viral spreading in HEp2 cells monolayers even after viral infection was estalished.
Figure 8. POPG inhibits RSV spread in HEp2 cells after infection was established.

HEp2 cells were challenged with RSV at a multiplicity of infection (m.o.i) of 10−4 for 2 h. Subsequently, the cells were washed and next overlayed with 0.3% agarose prepared in tissue culture medium either with, or without, or 200, or 500 μg/mL phospholipid (RSV, RSV+POPG200, RSV+POPG500, RSV+POPC200). Plaque numbers were quantified by staining with neutral red at day 6. Plaques in each condition were visualized with neutral red staining (panel A upper row) or were detected by immunostaining with antibody for RSV F protein11 (panel A lower row). Panel B shows images under microscopy with neutral red (panel B-1 and B-2), or with anti-RSV antibody (panel B-3 and B-4) 11. The quantitative data for plaque numbers are shown in Fig. 8C and the data are shown as mean ± SD, §§ indicates p<0.001 by paired t-test.
In addition, we examined how POPG inhibits RSV infection even after RSV infection was established. CD14 and TLR4 are critical for RSV infection establishment, as previoulsy reported.2,48 We studied whether 1) RSV can directly bind to CD14, and 2) POPG treatment can impact RSV binding to CD14 using a solid phase CD14 binding assay. The data demonstrate that direct binding of RSV to CD14 was inhibited by POPG treatment (Fig. S1A, and S1B). We also determined the efficacy of POPG to prevent RSV binding to the host cells, by quantifying bound RSV, using anti-RSV antibody and FACS. Treatment with POPG inhibited RSV binding to host cells and reduced mean fluorescence intensity (M.F.I) by 70% (Fig. S1C and S1D).11 There was no cytotoxic effect of POPG to the cells, as determined by 3H-leucine incorporation into total cellular proteins.11
We also determed the effcacy of PI, which shares similar structural features with POPG (see Fig. 2). PI bound to RSV directly, in a dose dependent manner, and with high affinity similar to POPG.13 PI interrupted the viral binding to HEp2 cells, as previoulsy reported.13 PI did not show any toxicity or direct virucidal effects. 2,13
Collectively, the minor sufactant phospholipids, POPG and PI, inhibit RSV infection and replication by inhibiting RSV binding to the host cells without any direct virucidal effects 2,11.
4– 2). Both POPG and PI inhibit RSV infection in an in vivo model in mice, when used prophylactically
Next, we examined the efficacies of POPG and PI against RSV infection an in vivo model in mice. BALB/c mice (6–8-week-old) were infected with an intranasal RSV inoculum (1X107pfu/mouse), either in the presence, or absence of POPG (400μg total/mouse). After 5 days, mice were harvested and the whole left lung was processed for quantitative plaque assay, to determine viral loads. Simultaneous treatment with POPG markedly reduced RSV plaque numbers by a factor of 300 (the data are shown as mean ± SD (plaque numbers/left lung), RSV alone (RSV): 4041.7 ± 2294.0, RSV + POPG-400μg (RSV + PG400): 13.5 ± 12.0, §§ indicates, p<0.001, Fig. 9A). We determined the prophylaxis effect of POPG against RSV, also using an in vivo model. As previously reported, the turnover rate of surfactant in the lung depends on the respiratory rate.58 We quantified the half-life of added POPG in mouse lung by following the turnover of the stable isotope, d5POPG.12 The POPG half-life in the lung of the mouse was approximately ~75mins. Based on this data, we introduced POPG into mice, intranasally, 45mins before RSV challenge. Prophylaxis treatments with POPG attenuated RSV plaque numbers in a dose-dependent manner (data are shown as mean ± SD (plaque numbers/left lung), RSV alone (RSV): 4041.7 ± 2294.0, RSV + prohylaxis treatment with POPG 800μg (RSV+pRPG800): 1726.7 ± 1077.3, RSV + prohylaxis treatment with POPG 1600μg (RSV + pRPG1600): 1317.6 ± 698.4, and RSV + prohylaxis treatment with POPG 3000μg (RSV + pRPG3000): 663.6 ± 518.2, §§ represents, p<0.001, Fig. 9A). The effect of POPG against RSV infection in the lung, correlated with histological changes, as shown in images and histological scores (inset into panels of Fig. 9B). This scoring system ranges from 0–26 and scoring performed by pathologists blinded to the study.12 Histologically, POPG treatments obviously reduced, inflammatory cell infiltration in alveoli and damage of bronchi, by RSV infection, in a dose-dependent manner (data are shown as mean ± SD; RSV alone (RSV): 11.4 ± 2.6, RSV + simultaneous treatment with POPG 400μg/mouse: RSV + POPG400: 6.9 ± 2.4, or with 800 μg/mouse RSV + pRPG800: 10.0 ± 3.4,or with 1600 μg/mouse RSV + pRPG1600: 9.2 ± 2.9, and with 3000 μg/mouse RSV + pRPG3000: 4.8 ± 2.5, Fig. 9B) 11. The POPG 3000 μg treament alone, did not cause noteworthy lung damage, and histological score changes, compared to sham-infected group (CONL).12
Figure 9. POPG and PI inhibit RSV infection and pulmonary injury in mice with pre-treatment.

6–8 weeks old BALB/c mice were infected with 107 pfu of RSV using intranasal inoculation in either the absence, or presence, of phospholipids. Mice were harvested on day 5, post-infection. Panel A) shows viral loads in the whole left lung in each group, virus alone (RSV), Virus + POPG, simultaneous treatment (400μg/inoculum) (RSV+PG400), and virus + prophylaxis treatment with POPG 800μg/mouse (pRPG800), POPG 1600μg/mouse (pRPG1600), and POPG 3000μg/mouse (pRPG3000). Mice were treated with POPG 45mins before viral challenge. Panel B) shows histological images of the lungs with each condition, with histological scores (mean ± SD) inset within the images. Panel C) shows viral burden of the whole left lung either with, or without, PI treatment: virus alone (RSV), Virus + PI simultaneous treatment (300μg/inoculum) (RSV + PI), and virus + prophylaxis treatment with PI 600μg/mouse (RSV + proRPI), PI treatment alone (600μg/mouse) (PI). Mice were inoculated with PI, 2hrs before viral challenge. Panel D) shows histological images of each condition with histology score inset into each image (mean ± SD). Data are shown as mean ± SD, §§ indicates p<0.001. Statistical analysis by one-way Anova and unpaired t-test. The figures and images were adapted from ref 11–13
We tested PI efficacy against RSV infection using the same in vivo model in mice as described above.13 We quantified the half-life of PI in mouse by the same method as for d5-POPG,12,13 and quantified the PI by a combination of thin layer chromatography and lipid phosphorus measurements. The half-life of PI in the mouse lung was ~6hrs, a turnover rate significantly slower than POPG.13 We treated mice with PI (600 μg/mouse) using intranasal prophylaxis, 2hrs before RSV challenge. As shown in Fig. 9C, the simultaneous treatment with PI 300μg/mouse) and RSV challenge, significantly reduced viral load in the lung by a factor of 40 (the data are shown as mean ± SD (plaque numbers/left lung), RSV alone (RSV): 2000.0 ± 1037.7, RSV + PI 300μg (RSV + PI): 57.5 ± 65.0, §§ indicates, p<0.001, Fig. 9C). The prophylaxis treatment with PI markedly reduced viral load in the lung by a factor of ~10 (RSV alone (RSV): 2000.0 ± 1037.7, RSV + prophylaxis treatment with PI 600μg (RSV + proPI): 375.0 ± 471.8, §§ indicates, p<0.001, Fig. 9C). In histopathology images, the PI treatment significantly reduced inflammatory changes and damage to bronchi by RSV infection, and the histopathology score was the same as uninfected levels (data are shown as mean ± SD; sham infected (CONL): 1.8 ± 1.1, RSV alone (RSV): 8.9 ± 3.1, RSV + simultaneous treatment with PI 300μg/mouse (RSV + PI): 2.7 ± 1.3, treamtent with 300μg of PI alone (PI) : 2.6 ± 1.8 ).
One limitation using the in vivo model in mice, is the rapid turnover rate of these lipids, which are related to the mouse respiratory rate of up to 300/min.58 To examine the critical effects of POPG and PI as post-infection treaments for viral infection, we need to examine the antiviral effects of the lipids in larger animal models (e.g., non-human primates, or ferrets).
In summary, both POPG and PI strongly inhibit RSV infection in an in vivo model in mice as prophylaxis. It is also noteworthy, that in vitro, the lipids halt the spread of RSV from infected to uninfected cells. Because of the half-life in the lung of these lipids, PI has a longer time window for effectiveness than POPG.
4– 3). POPG inhibits infection by clinical isolates of RSV and recombinant variants of RSV
In most of our studies, we used RSV-A2, which is a common lab strain of the virus. In addition, we also examined the efficacy of POPG against clinical isolates from 5 patients and a genetically engineered recombinant virulent variants of RSV. We used plaque assays to determine whether POPG can inhibit RSV clinical isolates and RSV variant infections. The recombinant RSV variants were generated based on modification of the F genes, that enhances F protein’s roles in airway mucous secretion and airway hyper reactivity (AHR).59 rA2-line19F causes higher viral burden, increasing IL-13 levels, and more mucous secretion in mice in comparison of rA2-A2 F and rA2 Long F variants.59 We quantified RSV plaque numbers with or without treatments with lipids (POPG 200μg/ml, or PC 200μg/ml, using these recombinant variants. POPG treatment was exceptionally potent at reducing plaque formation for each clinical isolate by 105 - 106 fold (see Table 1). For recombinant RSV variants, POPG efficacy against these viruses was also quite potent and inhibited plaque formation by 105 – 107 fold (see Table 1). POPC failed to have an effect on these clinical isolates and recombinant RSV variant infections.
Table 1.
POPG inhibits infection by clinical isolates and recombinant variants of RSV.
| RSV Strain | Titer (pfu/ml) | Plaques w/ POPG | Plaques w/ POPC |
|---|---|---|---|
| JX069798.1 | 8.3±4.7(E+05) | 0 | 3.0±3.5(E+05) |
| JX069803.1 | 4.8±0.4(E+06) | 0 | 0.5±0.1(E+06) |
| JX069801.1 | 1.1±0.2(E+07) | 2.6±3.6(E+02) | 0.4±0.2(E+07) |
| JX069800.1 | 1.1±0.1(E+07) | 3.0±4.2(E+02) | 0.5±0.2(E+07) |
| JX069799.1 | 5.3±0.5(E+04) | 0 | 0.7±0.3(E+04) |
| rA2-A2 F | 1.2±0.4(E+07) | 0.5±0.01(E+02) | 0.8±0.02(E+07) |
| rA2- line19F | 3.0±0.7(E+06) | 0 | 3.0±0.2(E+06) |
| rA2 Long F | 6.2±0.2(E+06) | 0 | 1.6±0.6(E+06) |
| wtRSV A2 | 2.7±1.1(E+07) | 0 | 1.3±0.3(E+07) |
We determined the antiviral efficacy of POPG against five clinical isolates of RSV (designated by GenBank accession numbers), and recombinant strains of RSV (rA2-A2F, rA2–19F, and rA2 Long F) in comparison to RSV-A2.12 The titer of each strain is shown as PFU/ml, and the efficacies of POPG and POPC (200μg/ml) on each strain are stated as the viral titers by plaque assays. Data are shown as means ± SD from two independent experiments.
These data demonstrate that POPG, not only inhibits RSV-A2 lab strains, but also various RSV subtypes of clinical isolates. Such findings provide important information to support the clinical application of POPG for patients in the future.
4– 5). The minor surfactant anionic phospholipids antagonize inflammation and infections induced by different subtypes of influenza A virus
Influenza A virus requires the formation of contacts between the sialic acid receptor binding amino acids present in HA proteins to initiate infection.60,61 Additional evidence reveals that TLR4 plays critical roles for influenza A infection, and its pathogenesis.2,49,62 Uncontroled TLR4 activation invites systemic sepsis, and elicits viral induced lung injury. TLR4−/− mice were highly tolerant to infection by mouse-adapted influenza, A/PR/8/34 (PR8).2,49,62 A TLR4 antagonist inhibits LPS induced inflammatory cytokine production and prevents lethal influenza infection in an in vivo model in mice.2,49,62 Therefore, the phamacologcial approach to control TLR4 signalling pathway is very important to treat acute lung injury by pathogens.2,49,62 The roles of TLR4 in the pathognesis of influenza A have been reported using animal models.49,63 As described above, POPG and PI are very potent antagonists of TLR4 activation.7,9 Based on these data and evidence, we initiated studies of the antiviral effects of POPG and PI against influenza A infection.
4– 6). POPG and PI antagonize various serotypes of influenza A infection, in vitro
To determine the potencies of POPG and PI against influenza A infection, we used BEAS-2B cells, and challenges with influenza A (Philippines 82/H3N2) (H3N2-IAV).14 The cells were challenged by virus at m.o.i = 2 either with, or without lipids (POPG or PC) at a concentration of 200 μg/ml. The lipids were introduced into the culture medium at the same time as the viral challenge. POPG treatment siginificantly reduced IL-8 secretion induced by H3N2-IAV challenge, by 90%, at 48hrs post-infection. We also examined the antiviral effect of POPG using Madin-Darby canine kidney (MDCK cells) 64 which are an ideal cell line to propagate influenza A virus. We challenged MDCK cells with H3N2-IAV at m.o.i = 0.5, either with, or without, these lipids (POPG, or PC) at concentrations of 200 – 1000 μg/ml. Using tissue culture lysates, we quantified hemagglutinin (HA) mRNA expression levels at 36hrs, after viral challenges. The simultaneous treatment with POPG strongly reduced HA mRNA expression by 80–90% in a dose-dependent manner. We also quantified neuraminidase (NA) and matrix protein (MP1) by immunoblotting at 36hrs after viral challenge. Treatment with POPG at 1000 μg/ml inhibited NA and MP1 protein expression by 80% and 75%, respectively.14 POPC treament failed to reduce either HA mRNA expression, or NA/M1 protein expression elicited by H3N2-IAV infection.
We examined the antiviral effects of these lipids against the (H1N1)pdm09 (pH1N1-IAV) that caused a worldwide pandemic in 2009.15,65 The aforementioned strain continues to appear as a a significant component within the seasonal infleunza viruses, and has an important impact upon human public health.66 To determine the antagonism of POPG and PI against pH1N1-IAV, we challenged MDCK cells with pH1N1-IAV at m.o.i = 5 × 10−3 for 2hrs. Following viral adsorption for 2hrs, POPG and PI (at 1 mg/ml) were added into tissue culture medium. Total cell cultures were harvested and used for quantitative plaque assays. POPG and PI significantly reduced plaque numbers, compared pH1N1-IAV alone, at 2hrs after viral adsorption and internalization were established; by 80% and 99%2, 15 respectively. We also repeated the same experiments using the human epithelial cell line, A549. Both POPG and PI inhibited NA and M1 protein expression by pH1N1-IAV infection in A549 cells by 70–80%.2, 15
In summary, POPG and PI strongly inhibit infection by different strains of influenza A viruses (H3N2-IAV and pH1N1-IAV), in a dose-dependent manner, in vitro. These lipids significantly antagonized pH1N1-IAV infection even after viral adsorption and the initial infection were established.
4– 7). Antiviral mechanisms of POPG and PI against Influenza A infection
We investgated how POPG and PI inhibit H3N2-IAVand pH1N1-IAV infection in vitro. Consistent with our previous findings regarding the anti RSV effects of these lipids, both POPG and PI directly bound to IAV and intefered with the viral attachement to the host cells.2,11–13 First, we evaluated direct virus binding, using phospholipid solid phases.2,11–13 Briefly, the lipids were precoated onto ELISA plates, and serial dilutions of influenza A viral inocula were added to the lipid precoated wells. Viral binding to the lipids was quantified by ELISA using anti-influenza A virus antibodies. POPG bound to H3N2-IAV with high affinity and in a saturable manner.14 Such properties were not found for the control lipid POPC. We also quantified H3N2-IAV binding to MDCK cells by using immunoblotting of M1protein (MP). H3N2-IAV bound to MDCK cells in a viral dose-dependent, and high affinity reaction. POPG strongly inhibited H3N2-IAV binding to MDCK cells in a ipid concentration-dependent reaction.14
We performed similar studies using pH1N1-IAV.2,14 Both POPG and PI bound pH1N1-IAV with high affinity and in a saturable manner.15 These properties were not reproduced with the control lipid, POPC. We also quantified pH1N1-IAV attachment to host cells using MDCK and human A549 cells, and immunoblotting for M1 protein.15 The pH1N1-IAV virions attached to MDCK cells with high affinity, and in a virion concentration-dependent manner. Both POPG and PI prevented pH1N1-IAV binding to MDCK and A549 cells.2,15
Further data demonstrated that POPG and PI do not exert a direct virudical effects. Data supporting such conclusions were generated from experiments in which we incubated pH1N1-IAV (1X105 pfu) and POPG (1mg/ml), or PI (1mg/ml), at 37oC for 1hr followed by serial dilutions (x 10−3−10−5) for plaque assays. If the lipids have direct virucidal effects, the plaque numbers should be reduced by the lipid pre-treatments. There were no reductions in plaque numbers by the lipid pretreatments.2,15
These data demonstrate that both POPG and PI bind to H3N2-IAV and pH1N1-IAV directly, and with high affinity. The lipids prevent viral attachment to the host cells, resulting in prevention of influenza A virus infection.
4– 8). Endogeneous minor, anionic pulmonary surfactant phospholpids inhibit influena A virus infecton in several animal models
We critically tested the antiviral effects of POPG and PI against influenza A virus infection in an in vivo model in mice. We challenged mice with pH1N1-IAV, which continues to be a clinically important strain circulating during the annual flu season.66 Mice were challenged with pH1N1-IAV (100pfu/mouse) either with, or without lipid (POPG 3 mg/mouse, PI 600 μg/mouse) and harvested and quantified at day 6 after viral challenge. Virus and the lipid were introduced into mice simultaneously by intranasal inoculation. Left lungs were processed for quantifying viral loads by plaque assays. In vivo lipid dosing in mice was determined empirically.2 Simultaneous treatment with POPG reduced pH1N1-IAV replication in the lung by a factor of 100 (data are shown as mean ± SD (plaque numbers x 106 pfu / left lung): pH1N1-IAV challenged group (pH1N1): 4.37 ± 2.49, pH1N1-IAV + POPG treatment (pH1N1 + PG): 0.04 ± 0.13, §§ indicates, p<0.001, Fig. 10A). We also quantified the potency of PI in this model. The PI treatment (600 μg/mouse) was also introduced into mice intranasally with virus. Viral loads in the left lung were strongly inhbited by PI treatment (plaque numbers (x 106 pfu / left lung): pH1N1-IAV challenged group (pH1N1): 2.63 ± 0.69, pH1N1-IAV + PI treatment (pH1N1 + PI): 0.14 ± 0.25, §§ indicates, p<0.001, Fig. 10B). These data demonstrate that both lipids dramatically inhibit pH1N1-IAV infection and replication in the lung, in vivo, in mice. Mice can tolerate high dose of POPG (up to 3mg/mouse) without adverse effect.15
Figure 10: POPG and PI inhibit pH1N1-influenza A viral infection and rescue from lethal infection in an in vivo model in mice.

Balb/c mice were challenged with pH1N1-IAV (100pfu/mouse) by intranasal inoculation either with, or without, phospholipids (POPG 3mg/mouse, PI 600μg/mouse), added simultaneously and harvested at day 6.
Panel A) Viral burdens in whole left lung were determined by plaque assays, either with or without POPG treatment. The groups are virus alone (pH1N1), or virus + PG (pH1N1 + PG).
Panel B) Viral burdens in whole left lung were determined by plaque assays either with, or without, PI treatment.
The groups are virus alone (pH1N1), or virus + PI (pH1N1 + PI). In panel A) and B), Data are shown as mean ± SD, §§ indicates, p<0.001 by unpaired t-test.
Panel C) Survival ratio of mice with lethal infection with pH1N1-IAV. 100% of mice were dead by day 10 post- infection (closed circle). The POPG treatment prevents lethal infection of mice by 100% (closed square).
Panel D) Body weight charts are shown for each group as 1) sham infected group (CONT, closed rhomboid), 2) viral infection alone (pH1N1, closed circle), 3) virus+ POPG treatment (3mg/mouse) (pH1N1+PG, closed square), and 4) POPG treatment alone (POPG, gray square). Data are shown as mean ± SE.
The figures were adapted and modified from ref 15.
We also investigated POPG efficacy for protection against lethal infection by pH1N1-IAV. Mice were challenged with LD100 dose of pH1N1-IAV (i.e., 1000 pfu/mouse) either with, or without, POPG (3 mg/mouse) using intranasal inoculation. Mice without POPG treatment (pH1N1) started to die within 6 days, and those not succumbing, lost significant weight, starting 72hrs after infection; and all animals were dead at day 10 (Fig. 10C and Fig.10D). All animals treated with virus and POPG survived until day 12 (Fig. 10C). POPG treatment alone (shown as: PG) and virus + PG treatment (pH1N1 + PG) lost weight starting at day 1 (Fig. 10D), but they recovered weight at day 2 and gained weight until day 12.
Taken together, both POPG and PI significantly inhibit pH1N1-IAV infection in an in vivo model in mice. POPG treatment also protects against lethal infection by pH1N1-IAV in mice.
There are limitations to the use of small mammalian animal models to test therapeutic interventions using these lipids. Mice have very high respiratory rates and the turnover of POPG, and PI are relatively rapid as previously reported.12,13 We also examined POPG potency against pH1N1-IAV infection in ferrets.15 Ferrets are widely used as animal models, and are considered the best model, especially for influenza studies to investigate horizontal transmissibility and pathogenicity.67 The ferret model with influenza infection can provide similar clinical symptoms to humans (e.g., sneezing, nasal discharge, fever, weight loss). Nasal, pharyngeal, lavage and tissue samples can be used to determine viral loads throughout the entire infection period. We challenged ferrets with pH1N1-IAV (at 1 × 106 pfu/ferret) either with, or without, POPG (5 mg/ferret) administered intranasally.15 Nasal and pharyngeal swabs were collected every day until day 4 to measure viral loads. The animals were euthanized at day 4, and lavage was performed for cell counting and plaque assays. Using samples of BALF and pharyngeal swabs, the POPG treatment reduced pH1N1-IAV plaque numbers by ~80%.15 Following lavage, the lungs were processed for histological analysis 15. pH1N1-IAV caused inflammatory cell accumulations in alveoli, alveolar wall damage, and lung collapse (Fig. 11A). The treatment with POPG significantly prevented lung damage and inflammation induced by pH1N1-IAV infection (Fig. 11A) and reduced histopathology scores (data are shown as mean ± SD, pH1N1-IAV challenged alone group (pH1N1): 7.9 ± 2.9, pH1N1-IAV with POPG treament (pH1N1 + PG): 1.7 ± 1.3, * represents, p<0.05, Fig. 11B).
Figure 11. POPG treatment markedly attenuates inflammation and lung damage induced by pH1N1-IAV infection in ferrets.

Panel A) shows histology of lung sections in each group, sham group, sham treated (CONL), virus alone (pH1N1), virus + POPG treatment (5mg/ferret) (pH1N1 + PG) and POPG treatment alone (PG).
Panel B) shows the histopathology score of each group as mean ± SD, * indicates p<0.05 by unpaired t-test. The images and data are adapted from ref 15.
In summary, these data demonstrate that POPG and PI are very potent inhibitors of pH1N1-IAV infection in an in vivo model in mice. POPG also has significant protective effects against lethal infection by pH1N1-IAV. An in vivo model of pH1N1-IAV infection in ferrets will be useful to determine the pharmacokinetics of the lipids and serve as a valuable model approach, to critically test the application of lipids for post-infection treatments.
4– 9). Anionic pulmonary surfactant phospholipids inhibit infection and replication of SARS-CoV-2 and its variants
From our findings of the antiviral effects of POPG and PI, we can conclude that they have antiviral effects against multiple enveloped RNA viruses.2,10 These lipids can directly bind to some viruses and subsequently prevent their binding to host cells.2 The aforementioned evidence led us to investigate whether these lipids could also inhibit SARS-CoV-2 infection. Coronavirus disease 2019 (COVID-19) has emerged and caused a worldwide pandemic since 2020.68 Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the pathogen of cause of COVID19.69 COVID-19 continues to overwhelm health care systems in multiple countries and accounts for more than 656 million identified cases and 6.6 million deaths worldwide.68 SARS-CoV-2 spread is facilitated by person-to-person transmission, and appears more transmissible than its cousin, SARS-CoV, discovered in 2003.70 Genetic variants of SARS-CoV-2 have continued to emerge and expand since the end of 2020.68 More lethal and transmissible genetic variants of SARS-CoV-2 continued to emerge and expand, since the end of 2020.68 The 3 major variants in late 2020, alpha (B.1.1.7), beta (B.1.351), and gamma (P.1) were replaced by delta (B.1.617.2) in the middle of 2021.68 These variants have higher transmissibility, contribute to more severe diseases, and reduce the effectiveness of vaccines.68,71 W.H.O have approved 8 vaccines for global usage as of May 2022. In the USA, FDA has approved three different types of vaccines, mRNA, viral vector, and protein subunit vaccines.71 Because of the new variants surging, vaccine formulations need to be updated to maintain sufficient efficacies, and FDA has started the shift to use bivalent vaccine for annual future vaccination.72,73
Guidelines for treatments of COVID19 have been published by NIH.74 Treatment regimens are selected based on the severity of COVID19, and the risk factors of patients (e.g., age, cardiovascular diseases, obesity). Neutralizing antibodies are one choice for treatment, but some have lost efficacy against omicron variants,74,75 Oral antiviral medicines are useful to prevent severe COVID19 and hospitalizations in elderly.76,77 But there are reasons to be concerned about their use. Drug interactions with antivirals are a significant concern, for their usage, as well as COVDI19 rebound phenomena.78 Even after COVID19 patients overcome the acute COVID19 phase, there is accumlating evidence of cases that continue to suffer from post COVID19 symptoms.79 The SARS-CoV-2 variants still remain a global threat, puting many lives in danger.71,79 Prevention and early interventions with antiviral therapeutics are key to reducing severe COVID19. Unfortunately, there are many issues that impede both approaches, including vaccine hesitancy, drug interactions, and medical care expenses. Simpler preventatives and antivirals than those currently available are still neeed.
To examine whether POPG, or PI inhibit SARS-CoV-2 infection, we challenged VeroE6 cells (with high ACE2 receptor content) that have high expression of the angiotensin I converting enzyme 2 receptor (ACE2).80 The cells were pretreated with lipids (1mg/ml) for 30mins and then challenged with SARS-CoV-2 (USA, WA/2020) (m.o.i=0.01). Culture media were collected at 24hr post infection and processed for quantitative plaque assays to measure viral loads. Viral loads were significantly reduced by treatment with POPG and PI, by 64% and 80% respectively (data in Fig. 12 shown as mean ± SD of % of SARS-CoV2 infection as 100%; SARS-CoV-2 + POPG: 36.2 ± 21.6 (%), SARS-CoV-2 + PI: 19.7 ± 16.2 (%), SARS-CoV-2 + PC:84.1 ± 17.5 (%) ) 2 (Fig.12B). SARS -CoV2 infection caused cell lysis at 72hr, and cells exhibited large nuclei and enlarged cytosolic compartment, all typical morphological changes associated with apoptosis (Fig. 12A). The treatment with POPG, or PI prevented these SARS-CoV-2 infection-mediated changes (Fig. 12A). Cell viability assays in the VeroE6 cells revealed no cytotoxicity by POPG, PI or PC 2.
Figure 12. POPG and PI inhibit SARS-CoV-2 infection of VeroE6 cells and human primary differentiated tracheal cells in air-liquid interface cultures.
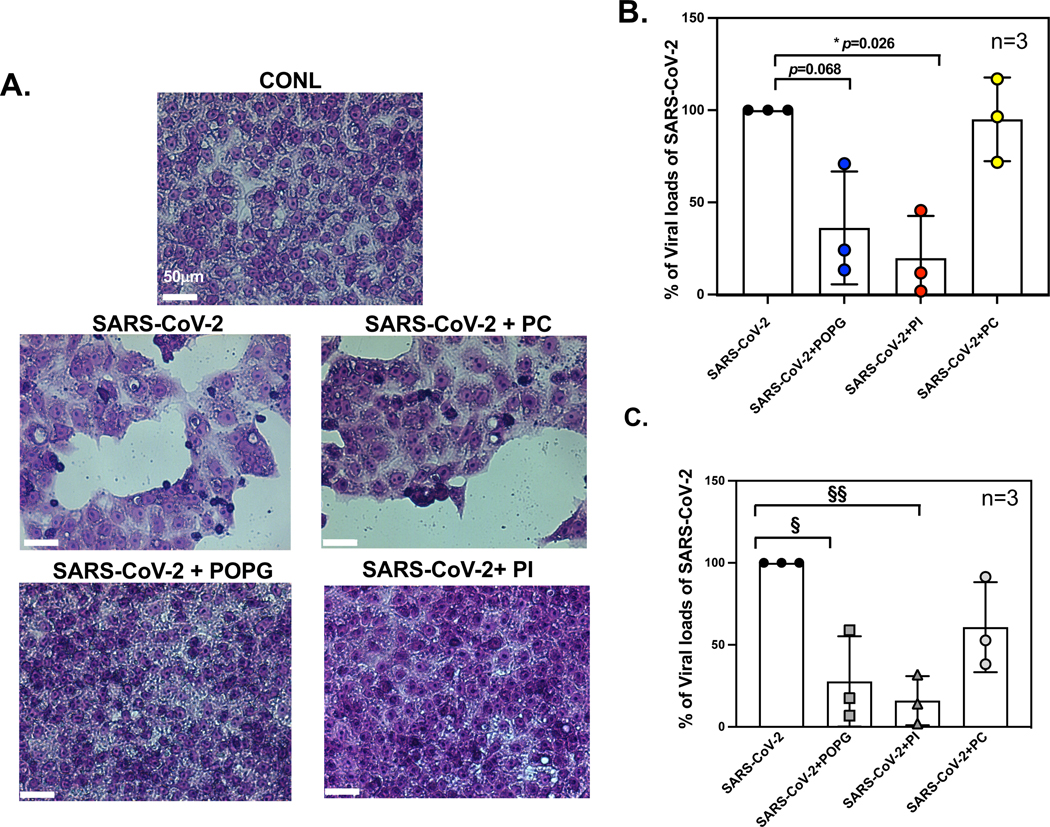
Panel A) Histological images in Vero E6 cell monolayers with Hematoxylin-Eosin staining of 1) untreated (CONL), 2) SARS-CoV-2 challenged, alone (SARS-CoV-2), 3) SARS-CoV-2 challenge and pretreated with POPG (SARS-CoV-2+POPG), 4) SARS-CoV-2 challenge + PI (SARS-CoV-2+PI) and 5) SARS-CoV-2 challenge +POPC (SARS-CoV2 +PC). The cells were pretreated with lipids for 30 mins, prior to the time of infection. The white bar indicates 50 μm.2 SARS-COV-2 infection causes significant cell damage, cell lysis, and cell hypertrophy and hypochromic changes consistent with apoptosis. Panel B) viral loads in VeroE6 cells quantified by plaque assays. POPG and PI reduced viral loads by 65% and 80%, respectively. The figure and images were adapted from2 Panel C) shows viral mRNA expression of SARS-CoV-2, using human primary differentiated tracheal cells, in ALI with viral challenge alone, or with viral challenge +lipid treatments; virus +POPG (SARS-CoV-2 + POPG), virus+PI (SARS-CoV-2 + PI), and virus +POPC (SARS-CoV-2 + PC). POPG and PI treatment reduced viral mRNA expression by 72% and 84%, respectively. The data are shown as mean ± SD from three different cell donors. § indicates p<0.01, §§ indicates p<0.001 by paired t-test. The data and figures are from published abstract, Am J Respir Crit Care Med 2022;205: A1196, Am J Respir Crit Care Med 2023, in press.
Primary human airway epithelial cells, nasal epithelial cells, and bronchial epithelial cells, all highly express ACE2, the major receptor for SARS-CoV-2, and also express the transmembrane serine protease (TMPRSS2) that is crucial for SARS-CoV-2 cell entry.80 These features are especially true for nasal epithelial cells, which serve as a gateway cell population for virus entry and subsequent propagation, eventually expanding the infection into the gas exchange regions of the lungs.32 We determined the potencies of POPG and PI against SARS-CoV-2 using human primary differentiated trachea cells in air-liquid interface (ALI) culture. The cells were differentiated for 21–24days. Differentiated cells were pretreated with lipids at the apical surface for 16hrs (10mg/ml of POPG, 4mg/ml of PI and 10mg/ml of PC). Cells were then challenged with SARS-CoV-2 of 2X104 pfu/ well for 48hrs, and subsequently harvested after removal of the apical medium, washed with DPBS and lysed with lysis buffer. The lysate was processed for RNA extraction. We determined the viral burden by qRT-PCR. SARS-CoV-2 mRNA expression increased 105-fold compared to sham infected cultures (the data from published abstract, Am J Respir Crit Care Med 2022;205: A1196). The treatment with POPG or PI reduced viral mRNA expression by 72% and 84 % respectively (Fig. 12C). We also used ALI cultures of human primary nasal epithelial cells from healthy participants to determine the antiviral effects of these lipids against SARS-CoV-2. The cells were differentiated for ~24 days in ALI and treated with these lipids for 16hr before identical viral challenge, cell lysis and harvesting, and mRNA extraction as described above. For primary human nasal epithelial cells, SARS-CoV2 replication was 106-fold higher compared to sham infected cells. Both POPG and PI significantly reduced viral mRNA expression, by 75% and 80%, respectively (the data from published abstract, Am J Respir Crit Care Med 2022;205: A1196, Am J Respir Crit Care Med 2023 in press). Next, we examined the antiviral effects of POPG and PI against the SARS-CoV-2 variants B.1.1.7 and B1.351, which were dominant subtypes in 2020 and early of 2021.68 Treatment with POPG, or PI, reduced viral mRNA expression of B1.351 by 50% and by 65%, respectively. Both POPG and PI also inhibited viral mRNA expression of B1.1.7, by 88% and 90%, respectively (the data from published abstract, Am J Respir Crit Care Med 2022;205: A1196). Additionally, we examined efficacies of these lipids against B.1.529 (delta variant) and B.1.617.2 (omicron variant). The B.1.617.2 variant replicated in trachea cells with a 104-fold increase of mRNA (n=4), whereas the B.1.1.529 variant replicated with a 109-fold increase of mRNA. Especially noteworthy, the phospholipid treatment markedly reduced viral mRNA expression, by 80% with POPG and 85% with PI; against the B.1.1.529 variant. In addition, the lipid efficacy at reducing viral mRNA was 99% for POPG and 97% for PI against the B.1.1.529 variant (the data from published abstracts, Am J Respir Crit Care Med 2022;205: A1196, Am J Respir Crit Care Med 2023, in press).
In summary, both POPG and PI, but not PC, inhibit SARS-CoV-2 replication in ALI-differentiated human primary trachea cells and nasal epithelial cells. Notably, PG and PI also caused very potent inhibition of the SARS-CoV-2 variants B.1.1.7, B.1.351, B.1.529 (delta variant) and B.1.617.2 (omicron variant). These findings indicate that POPG and PI have strong potential as antiviral therapeutics against SARS-CoV-2 and its numerous variants.
5. Future directions
In this review, we highlighted the anti-inflammatory effects, and antiviral effects of minor pulmonary surfactant phospholipids; and emphasized their activity against problematic RNA viruses. We have investigated the mechanisms and the efficacies of resident pulmonary surfactant phospholipids against inflammation elicited by multiple TLRs and multiple RNA respiratory viruses, including newly emergent RNA viruses such as SARS-CoV-2 and its variants.2
As stated in this review, both POPG and PI inhibit LPS induced inflammation via TLR4 activation by directly binding to CD14 and MD2 that disrupts TLR4/CD14/MD2 complex formation.7 POPG also inhibits LPS-induced lung injury/inflammation and inflammatory mediators production (e.g., TNF-α, KC and MIP-2) in an in vivo model in mice.7 Additionally, these lipids antagonize proinflammatory cytokine production and inflammatory mediator production, including arachidonic acid (AA) secretion and eicosanoid production, elicited by TLR2/1, TLR/2/6 agonist stimulation (e.g., lipoprotein of mycoplasma pneumonia) ; and proinflammatory cytokine production from human primary alveolar macrophages10 and human bronchial epithelial cells. The constellation of inflammatory mediators includes TNF-α, IL-6 and IL-8.2 The excessive production of proinflammatory cytokines (cytokine storm) contributes to the pathogenesis of acute lung injury (ALI)/ARDS.81 TLR4 and TLR2 contribute to development of ARDS.82 TLR4 also plays a critical role in initiating ARDS, and in in vivo models mimicking acute respiratory diseases. It is noteworthy that blocking TLR4 activation with a neutralizing antibody, attenuated LPS-induced lung injury.83 TLR3 also contributes to hyperoxia-induced ARDS.82 Multiple TLRs, ,especially TLR4, TLR2 and TLR3 have been implicated in development of ARDS.82 Based on our findings regarding broad inhibitory effects of POPG and PI against multiple TLR activations, these lipids are ideal compounds to test for treating ALI/ARDS patients, who have complex etiologies for the causes of their acute illnesses.81,82
CD14 and TLR4 play important roles in establishments of RSV and influenza A infections.48,49 We extended our findings to determine whether POPG and PI prevent RSV and influenza A infection using different strains (e.g., H3N2-IAV, pH1N1-IAV). These lipids directly bind to RSV and influenza A viruses to disrupt the initial virion interactions with host cells.2,11–15
Recently, we reported that PI inhibits Rhinovirus A infection of differentiated human primary airway epithelial cells.84 Rhinovirus is a non-enveloped virus, and PI does not bind to RV-A16 directly, and does not to inhibit viral attachment to host cells or internalization, either.84 In this paper, PI treatment altered RV-A16 induced expression of phosphatidylinositol 4-kinase (PI4K), acyl-CoA-binding domain-containing proteins (ACBD) and low-density lipoprotein receptor (LDLR) genes that are necessary in the formation of replication organelles (ROs) required for RV replication in host cells.84 The anti-viral mechanisms of PI against RV-A16 are quite different from enveloped respiratory viruses, like RSV and influenza A.
Lastly, we have determined the antiviral effects of POPG and PI against SARS-CoV-2 and its variants using Vero E6 cells and differentiated human airway epithelial cells.2 Both POPG and PI markedly inhibit SARS-CoV-2 infection and replication in VeroE6 cells and human primary airway epithelial cells2. These lipids also inhibit multiple SARS-CoV-2 variant replication. Among the most important issues we are facing regarding SARS-CoV-2 are the intrinsically high mutation rates of RNA viruses, which continually re-invite vaccine breakthrough events.71–73 This situation is exacerbated by the ineffectiveness of neutralizing antibodies,71–73,75 and possible drug resistance against antiviral medicines.15,85,86 At least one major reasons for severe COVID19 comes from a cytokine storm that causes excessive production of proinflammatory cytokines (e.g., IL-1β, TNF-α, IL-6 and IL-8).2 These events cause lung damage that leads to ALI/ARDS.76,77 Our data demonstrate that POPG and PI have excellent potential to be applied as anti-viral and anti-inflammatory agents for treating COVID19.2,10
Both POPG and PI have several benefits to have strong potentials as novel antivirals and anti-inflammatory compounds because they are, 1) existing as natural pulmonary surfactant phospholipids in the lung already and are non-immunogenic, 2) chemically stable and easy to keep, 3) inexpensive, and 5) easy to apply and use provide for clinical usage. We confirmed and reported that there was no toxic effect of POPG nor PI at the doses that we used with both in vitro and in vivo experiments.2,7,10,11,15 The turnover speed of pulmonary surfactant phospholipid depends on respiratory rate.12,13 The application for therapeutic usage of these lipids, the intranasal usage via aquas form or lyophilized powder will be ideal. According to our pharmacokinetics analysis of the lipids in vivo and previous publications 2, these lipids can be used at 2–3 times a day, for 3–5 days as antiviral and preventatives for viral infections.
The bulk of our work has used mice, hamsters, and ferrets to understand the protective effects of PI and POPG against inflammation and viral infection. For applications to humans, we must consider possible delivery methods for the lipids. The most desirable route is via inhalation to ensure uniform distribution of the lipids in the respiratory tissues. At the outset, we rule out using systemic vascular approaches. Current methods in use for respiratory therapeutics are: 1) dry powder inhalation, 2) aerosol inhalation, and 3) nebulization-inhalation. Each of these modalities has its own set of advantages and disadvantages. Our work with small mammals allowed us the freedom to introduce aqueous suspensions of liposomes into the airways. Mice are obligate nose breathers, and this ensured relatively uniform distribution of lipids, within the airways and parenchyma. Dry powder inhalation offers the most concentrated delivery option, but the dose limits may require multiple rounds of delivery. Aerosol inhalation is most similar to our current delivery methods for small mammals and the efficacy is well established by our experimentation. Nebulization is an attractive approach, but we do not have any data for this modality. Intrinsically, the lipids we use are quite resistant to mechanical, but not necessarily chemical formulations that facilitate generation of minute particles. Ultimately, the most useful approach for phospholipid therapeutic delivery must be determined empirically.
6. Summary
The minor anionic pulmonary surfactant phospholipids, POPG and PI, exhibit anti-inflammatory effects as antagonists for multiple TLRs. These lipids also have strong efficacies as antivirals against multiple respiratory RNA enveloped viruses such as RSV, Influenza A viruses, and SARS-CoV- 2. PI also has a potent effect against non-enveloped virus (human rhinovirus A). These lipids also have very strong potential to be applied as anti-inflammatory compounds for acute lung injury, induced by cytokine storms, including severe COVID19.
Supplementary Material
Supplemental Figure S1. POPG directly binds to RSV and disrupts binding interaction between RSV and HEp2 cells.
Panel A) shows, RSV binds to CD14 with high affinity and in a dose dependent manner. The binding interactions between RSV and CD14 are determined with an HRP-conjugated anti-RSV antibody and measured by A490 absorbance. Panel B) shows that POPG disrupts the binding interaction between RSV and CD14, in a dose-dependent manner. CD14 is captured on an ELISA plate, and RSV with or without POPG is subsequently added to the CD14 solid phase. The binding interaction between RSV and CD 14 is determined by anti RSV antibody. Panel C) HEp2 cells are incubated with 1) no virus (NO RSV, black line), 2) RSV alone (RSV, green line) 3) RSV and POPG (200mg/ml) (RSV+POPG, red line), and 4) RSV+POPC 200mg/ml (RSV+POPC, blue line) and RSV binding to the cells is determined by anti RSV antibody detection. The signals are detected by FACS. Panel D) Quantitative data of FACS from 3 independent experiments are shown. The data are shown as mean ± SD, §§ indicates p<0.001. Statistical analysis is performed by paired t-test. N.S indicate not significant. The data and figures are adapted from ref 11.
7. Acknowledgement
We deeply appreciate Dr. Judith Storch (Rutgers university, NJ, USA) for providing valuable advice and insights for this manuscript.
This work was supported by Clinical innovator Award Program (CIA 160010), Flight Attendant Medical Research Institute, Inc. (FAMRI) (MN), U19 -Al 125357 (DRV, MN), NIH P01 HL132821 (DRV, MN) NIH GM 118819 (DRV, MN), and National Emphysema Foundation (MN).
8. Funding resources and supports
This work was supported by CLINICAL INNOVATOR AWARD PROGRAM (CIA 160010), Flight Attendant Medical Research Institute, Inc. (FAMRI) (MN), NIH U19-AI 125357 (DRV, MN), NIH P01 HL132821 (DRV, MN), NIH GM118819 (DRV, MN), National Emphysema Foundation (MN).
Abbreviations
- ALI
acute lung injury
- ARDS
Acute respiratory distress syndrome
- ACBD3
Acyl CoA-binding domain 3
- ALI
Air liquid interface culture
- ACE2
angiotensin I converting enzyme 2
- AA
arachidonic acid
- ABCA gene
ATP-binding cassette transporter gene
- BALF
Bronchoalveolar lavage fluid
- CL
Cardiolipin
- JNK
c-Jun N-terminal kinases (JNKs)
- CXCL11
C-X-C motif chemokine 11
- DGE
Differential gene expression
- DPPC
dipalmitoyl-phosphatidylcholine
- TIRAP
domain-containing adaptor proteins
- ERK
extracellular signal-regulated kinase
- TGN
Golgi and trans-Golgi network
- pH1N1-IAV
H1N1 A/California/07/2009
- H3N2-IAV
H3N2-IAV (Philippines 82/H3N2)
- PI4K
human phosphatidylinositol 4-kinases
- RV-16
human rhinovirus A16
- d5-POPG
1-hexadecanoyl-2-(9Z-octadecenoyl)-sn-glycero-3-phospho-(1’-rac-glycerol-1’,1’,2’,3’,3’-d5) (ammonium salt)
- PI4KB
phosphatidylinositol 4-kinase IIIβ
- IL-6
Interleukin-6
- IL-8
Interluekin-8
- IRAK
interleukin-1 receptor (IL-1R) associated kinase
- IFITM1
Interferon-induced transmembrane protein 1
- IFITM2
Interferon-induced transmembrane protein 2
- IFITM3
Interferon-induced transmembrane protein 3
- IRF1
Interferon regulatory factor 1
- IRF7
Interferon regulatory factor 7
- IFN-λ
interferon lambda
- KC
keratinocytes-derived chemokine
- LDL
low-density lipoprotein
- MIP2
macrophage-inflammatory protein-2
- MKP1
mitogen-Activated Protein Kinase Phosphatase 1
- MD2
myeloid Differentiation factor 2
- MDA5
melanoma differentiation-associated protein 5
- m.o.i
multiplicity of infection
- OAS1
2’−5’-oligoadenylate synthetase 1
- OSBP
oxysterol-binding protein
- POPG
palmitoyl-oleoyl-phosphatidylglycerol
- POPC
palmitoyl-oleoyl-phosphatidylcholine
- PAMPs
pathogen-associated molecular patterns
- PRRs
pattern recognition receptors
- PA
phosphatidic acid
- PS
phosphatidylserine
- PI
phosphatidylinositol
- PtdO
moiety
- AECs
primary human trachea cells
- RO
replication organelle
- RSV
respiratory syncytial virus
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- SP-A
surfactant protein A
- SP-B
protein B
- SP-C
surfactant protein C
- SP-D
surfactant protein D
- COVID-19
the coronavirus disease 2019
- TLRs
toll-like receptors
- TNF-α
tumor necrosing factor-alpha
- VLDLR
very-low-density lipoprotein receptor
References
- 1.Echaide M, Autilio C, Arroyo R, Perez-Gil J. Restoring pulmonary surfactant membranes and films at the respiratory surface. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2017;1859(9):1725–1739. [DOI] [PubMed] [Google Scholar]
- 2.Numata M, Voelker DR. Anti-inflammatory and anti-viral actions of anionic pulmonary surfactant phospholipids. Biochim Biophys Acta Mol Cell Biol Lipids. Jun 2022;1867(6):159139. doi: 10.1016/j.bbalip.2022.159139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. Jan 2005;5(1):58–68. doi: 10.1038/nri1528 [DOI] [PubMed] [Google Scholar]
- 4.Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. Jan 2015;16(1):27–35. doi: 10.1038/ni.3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clements JA, Nellenbogen J, Trahan HJ. Pulmonary surfactant and evolution of the lungs. Science. Aug 7 1970;169(3945):603–4. doi: 10.1126/science.169.3945.603 [DOI] [PubMed] [Google Scholar]
- 6.Clements JA. Surfactant in pulmonary disease. New England Journal of Medicine. 1965;272(25):1336–1337. [DOI] [PubMed] [Google Scholar]
- 7.Kuronuma K, Mitsuzawa H, Takeda K, et al. Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2. J Biol Chem. Sep 18 2009;284(38):25488–500. doi: 10.1074/jbc.M109.040832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kandasamy P, Zarini S, Chan ED, Leslie CC, Murphy RC, Voelker DR. Pulmonary surfactant phosphatidylglycerol inhibits Mycoplasma pneumoniae-stimulated eicosanoid production from human and mouse macrophages. J Biol Chem. Mar 11 2011;286(10):7841–7853. doi: 10.1074/jbc.M110.170241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kandasamy P, Numata M, Berry KZ, et al. Structural analogs of pulmonary surfactant phosphatidylglycerol inhibit toll-like receptor 2 and 4 signaling. J Lipid Res. Jun 2016;57(6):993–1005. doi: 10.1194/jlr.M065201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voelker DR, Numata M. Phospholipid regulation of innate immunity and respiratory viral infection. J Biol Chem. Mar 22 2019;294(12):4282–4289. doi: 10.1074/jbc.AW118.003229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Numata M, Chu HW, Dakhama A, Voelker DR. Pulmonary surfactant phosphatidylglycerol inhibits respiratory syncytial virus-induced inflammation and infection. Proc Natl Acad Sci U S A. Jan 5 2010;107(1):320–5. doi: 10.1073/pnas.0909361107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Numata M, Nagashima Y, Moore ML, et al. Phosphatidylglycerol provides short-term prophylaxis against respiratory syncytial virus infection. J Lipid Res. Aug 2013;54(8):2133–2143. doi: 10.1194/jlr.M037077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Numata M, Kandasamy P, Nagashima Y, Fickes R, Murphy RC, Voelker DR. Phosphatidylinositol inhibits respiratory syncytial virus infection. J Lipid Res. Mar 2015;56(3):578–587. doi: 10.1194/jlr.M055723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Numata M, Kandasamy P, Nagashima Y, et al. Phosphatidylglycerol suppresses influenza A virus infection. Am J Respir Cell Mol Biol. Apr 2012;46(4):479–87. doi: 10.1165/rcmb.2011-0194OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Numata M, Mitchell JR, Tipper JL, et al. Pulmonary surfactant lipids inhibit infections with the pandemic H1N1 influenza virus in several animal models. J Biol Chem. Feb 7 2020;295(6):1704–1715. doi: 10.1074/jbc.RA119.012053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Rodriguez E, Perez-Gil J. Structure-function relationships in pulmonary surfactant membranes: from biophysics to therapy. Biochim Biophys Acta. Jun 2014;1838(6):1568–85. doi: 10.1016/j.bbamem.2014.01.028 [DOI] [PubMed] [Google Scholar]
- 17.Milad N, Morissette MC. Revisiting the role of pulmonary surfactant in chronic inflammatory lung diseases and environmental exposure. Eur Respir Rev. Dec 31 2021;30(162)doi: 10.1183/16000617.0077-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartshorn KL. Role of surfactant protein A and D (SP-A and SP-D) in human antiviral host defense. Front Biosci (Schol Ed). Jan 1 2010;2(2):527–46. doi: 10.2741/s83 [DOI] [PubMed] [Google Scholar]
- 19.McCormack FX, Gibbons R, Ward SR, Kuzmenko A, Wu H, Deepe GS, Jr. Macrophage-independent fungicidal action of the pulmonary collectins. J Biol Chem. Sep 19 2003;278(38):36250–6. doi: 10.1074/jbc.M303086200 [DOI] [PubMed] [Google Scholar]
- 20.van Rozendaal BA, van Spriel AB, van De Winkel JG, Haagsman HP. Role of pulmonary surfactant protein D in innate defense against Candida albicans. J Infect Dis. Sep 2000;182(3):917–22. doi: 10.1086/315799 [DOI] [PubMed] [Google Scholar]
- 21.Borron P, McCormack FX, Elhalwagi BM, et al. Surfactant protein A inhibits T cell proliferation via its collagen-like tail and a 210-kDa receptor. Am J Physiol. Oct 1998;275(4):L679–86. doi: 10.1152/ajplung.1998.275.4.L679 [DOI] [PubMed] [Google Scholar]
- 22.Agudelo CW, Samaha G, Garcia-Arcos I. Alveolar lipids in pulmonary disease. A review. Lipids Health Dis. Jun 3 2020;19(1):122. doi: 10.1186/s12944-020-01278-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borges-Araujo L, Fernandes F. Structure and Lateral Organization of Phosphatidylinositol 4,5-bisphosphate. Molecules. Aug 26 2020;25(17)doi: 10.3390/molecules25173885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agassandian M, Mallampalli RK. Surfactant phospholipid metabolism. Biochim Biophys Acta. Mar 2013;1831(3):612–25. doi: 10.1016/j.bbalip.2012.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Lorenzo F, Duda KA, Lanzetta R, Silipo A, De Castro C, Molinaro A. A Journey from Structure to Function of Bacterial Lipopolysaccharides. Chem Rev. Oct 26 2022;122(20):15767–15821. doi: 10.1021/acs.chemrev.0c01321 [DOI] [PubMed] [Google Scholar]
- 26.Katzen J, Beers MF. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J Clin Invest. Oct 1 2020;130(10):5088–5099. doi: 10.1172/JCI139519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tlatelpa-Romero B, Cázares-Ordoñez V, Oyarzábal LF, Vázquez-de-Lara LG. The Role of Pulmonary Surfactant Phospholipids in Fibrotic Lung Diseases. International Journal of Molecular Sciences. 2022;24(1):326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeon GW. Surfactant preparations for preterm infants with respiratory distress syndrome: past, present, and future. Korean J Pediatr. May 2019;62(5):155–161. doi: 10.3345/kjp.2018.07185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Echaide M, Autilio C, Lopez-Rodriguez E, Cruz A, Perez-Gil J. In Vitro Functional and Structural Characterization of A Synthetic Clinical Pulmonary Surfactant with Enhanced Resistance to Inhibition. Sci Rep. Jan 28 2020;10(1):1385. doi: 10.1038/s41598-020-58248-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fessler MB, Summer RS. Surfactant Lipids at the Host-Environment Interface. Metabolic Sensors, Suppressors, and Effectors of Inflammatory Lung Disease. Am J Respir Cell Mol Biol. May 2016;54(5):624–35. doi: 10.1165/rcmb.2016-0011PS [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herrero R, Sanchez G, Lorente JA. New insights into the mechanisms of pulmonary edema in acute lung injury. Ann Transl Med. Jan 2018;6(2):32. doi: 10.21037/atm.2017.12.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muus C, Luecken MD, Eraslan G, et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat Med. Mar 2021;27(3):546–559. doi: 10.1038/s41591-020-01227-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sungnak W, Huang N, Becavin C, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. May 2020;26(5):681–687. doi: 10.1038/s41591-020-0868-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhatt RM, Clark HW, Girardis M, Busani S. Exogenous pulmonary surfactant in COVID-19 ARDS. The similarities to neonatal RDS suggest a new scenario for an ‘old’ strategy. BMJ Open Respir Res. Sep 2021;8(1)doi: 10.1136/bmjresp-2020-000867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet. Aug 14 2021;398(10300):622–637. doi: 10.1016/S0140-6736(21)00439-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grasselli G, Tonetti T, Protti A, et al. Pathophysiology of COVID-19-associated acute respiratory distress syndrome: a multicentre prospective observational study. Lancet Respir Med. Dec 2020;8(12):1201–1208. doi: 10.1016/S2213-2600(20)30370–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji J, Sun L, Luo Z, et al. Potential Therapeutic Applications of Pulmonary Surfactant Lipids in the Host Defence Against Respiratory Viral Infections. Front Immunol. 2021;12:730022. doi: 10.3389/fimmu.2021.730022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Islam A, Khan MA. Lung transcriptome of a COVID-19 patient and systems biology predictions suggest impaired surfactant production which may be druggable by surfactant therapy. Sci Rep. Nov 10 2020;10(1):19395. doi: 10.1038/s41598-020-76404-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bollag WB, Gonzales JN. Phosphatidylglycerol and surfactant: A potential treatment for COVID-19? Med Hypotheses. Nov 2020;144:110277. doi: 10.1016/j.mehy.2020.110277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richmond BW, Dela Cruz CS. Adding Insult to Injury: Does COVID-19 Promote Acute Respiratory Distress Syndrome by Inhibiting Surfactant? Am J Respir Crit Care Med. Jan 1 2023;207(1):5–6. doi: 10.1164/rccm.202208-1549ED [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. May 27 2011;34(5):637–50. doi: 10.1016/j.immuni.2011.05.006 [DOI] [PubMed] [Google Scholar]
- 42.Mueller M, Brandenburg K, Dedrick R, Schromm AB, Seydel U. Phospholipids inhibit lipopolysaccharide (LPS)-induced cell activation: a role for LPS-binding protein. J Immunol. Jan 15 2005;174(2):1091–6. doi: 10.4049/jimmunol.174.2.1091 [DOI] [PubMed] [Google Scholar]
- 43.Kelley SL, Lukk T, Nair SK, Tapping RI. The crystal structure of human soluble CD14 reveals a bent solenoid with a hydrophobic amino-terminal pocket. The Journal of Immunology. 2013;190(3):1304–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JI, Lee CJ, Jin MS, et al. Crystal structure of CD14 and its implications for lipopolysaccharide signaling. J Biol Chem. Mar 25 2005;280(12):11347–51. doi: 10.1074/jbc.M414607200 [DOI] [PubMed] [Google Scholar]
- 45.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol 5: 461. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol. Sep 2004;31(3):358–64. doi: 10.1165/rcmb.2003-0388OC [DOI] [PubMed] [Google Scholar]
- 47.Koarai A, Sugiura H, Yanagisawa S, et al. Oxidative stress enhances toll-like receptor 3 response to double-stranded RNA in airway epithelial cells. Am J Respir Cell Mol Biol. Jun 2010;42(6):651–60. doi: 10.1165/rcmb.2008-0345OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurt-Jones EA, Popova L, Kwinn L, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. Nov 2000;1(5):398–401. doi: 10.1038/80833 [DOI] [PubMed] [Google Scholar]
- 49.Shirey KA, Lai W, Scott AJ, et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature. May 23 2013;497(7450):498–502. doi: 10.1038/nature12118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Wang X, Blau DM, et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in children younger than 5 years in 2019: a systematic analysis. Lancet. May 28 2022;399(10340):2047–2064. doi: 10.1016/S0140-6736(22)00478-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Griffin MR. A Challenge to Respiratory Syncytial Virus Illness in Adults. Mass Medical Soc; 2022. p. 2427–2428. [DOI] [PubMed] [Google Scholar]
- 52.Simoes EA, DeVincenzo JP, Boeckh M, et al. Challenges and opportunities in developing respiratory syncytial virus therapeutics. J Infect Dis. Mar 15 2015;211 Suppl 1(Suppl 1):S1–S20. doi: 10.1093/infdis/jiu828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmoele-Thoma B, Zareba AM, Jiang Q, et al. Vaccine Efficacy in Adults in a Respiratory Syncytial Virus Challenge Study. N Engl J Med. Jun 23 2022;386(25):2377–2386. doi: 10.1056/NEJMoa2116154 [DOI] [PubMed] [Google Scholar]
- 54.Griffin MP, Yuan Y, Takas T, et al. Single-Dose Nirsevimab for Prevention of RSV in Preterm Infants. N Engl J Med. Jul 30 2020;383(5):415–425. doi: 10.1056/NEJMoa1913556 [DOI] [PubMed] [Google Scholar]
- 55.Centers for Disease Control and Prevention. Increased Respiratory Virus Activity, Especially Among Children, Early in the 2022–2023 Fall and Winter Vol. (CDCHAN-00479). CDC Health Alert Network. 2022. Accessed January 25, 2023. https://emergency.cdc.gov/han/2022/han00479.asp [Google Scholar]
- 56.Terliesner N, Unterwalder N, Edelmann A, et al. Viral infections in hospitalized children in Germany during the COVID-19 pandemic: Association with non-pharmaceutical interventions. Frontiers in Pediatrics. 2022;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Binns E, Koenraads M, Hristeva L, et al. Influenza and respiratory syncytial virus during the COVID‐19 pandemic: Time for a new paradigm? Pediatric pulmonology. 2022;57(1):38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hallman M, Merritt TA, Bry K. The fate of exogenous surfactant in neonates with respiratory distress syndrome. Clin Pharmacokinet. Mar 1994;26(3):215–32. doi: 10.2165/00003088-199426030-00005 [DOI] [PubMed] [Google Scholar]
- 59.Moore ML, Chi MH, Luongo C, et al. A chimeric A2 strain of respiratory syncytial virus (RSV) with the fusion protein of RSV strain line 19 exhibits enhanced viral load, mucus, and airway dysfunction. J Virol. May 2009;83(9):4185–94. doi: 10.1128/JVI.01853-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kamiki H, Murakami S, Nishikaze T, et al. Influenza A Virus Agnostic Receptor Tropism Revealed Using a Novel Biological System with Terminal Sialic Acid Knockout Cells. J Virol. Aug 10 2022;96(15):e0041622. doi: 10.1128/jvi.00416-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu M, Huang LZX, Smits AA, et al. Human-type sialic acid receptors contribute to avian influenza A virus binding and entry by hetero-multivalent interactions. Nat Commun. Jul 13 2022;13(1):4054. doi: 10.1038/s41467-022-31840-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perrin-Cocon L, Aublin-Gex A, Sestito SE, et al. TLR4 antagonist FP7 inhibits LPS-induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection. Scientific Reports. 2017;7(1):40791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shirey KA, Blanco JCG, Vogel SN. Targeting TLR4 Signaling to Blunt Viral-Mediated Acute Lung Injury. Front Immunol. 2021;12:705080. doi: 10.3389/fimmu.2021.705080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takada K, Kawakami C, Fan S, et al. A humanized MDCK cell line for the efficient isolation and propagation of human influenza viruses. Nat Microbiol. Aug 2019;4(8):1268–1273. doi: 10.1038/s41564-019-0433-6 [DOI] [PubMed] [Google Scholar]
- 65.Novel Swine-Origin Influenza AVIT, Dawood FS, Jain S, et al. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med. Jun 18 2009;360(25):2605–15. doi: 10.1056/NEJMoa0903810 [DOI] [PubMed] [Google Scholar]
- 66.Merced-Morales A, Daly P, Abd Elal AI, et al. Influenza Activity and Composition of the 2022–23 Influenza Vaccine - United States, 2021–22 Season. MMWR Morb Mortal Wkly Rep. Jul 22 2022;71(29):913–919. doi: 10.15585/mmwr.mm7129a1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Belser JA, Eckert AM, Tumpey TM, Maines TR. Complexities in Ferret Influenza Virus Pathogenesis and Transmission Models. Microbiol Mol Biol Rev. Sep 2016;80(3):733–44. doi: 10.1128/MMBR.00022-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.World Health Organization. Weekly epidemiological update on COVID-19 – 4 January 2023. Vol. Edition 124. Emergency Situational Updates. 2023. Accessed 4 January 2023. https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19-−-4-january-2023 [Google Scholar]
- 69.Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. Apr 16 2020;181(2):271–280 e8. doi: 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. Feb 15 2020;395(10223):507–513. doi: 10.1016/S0140-6736(20)30211-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barouch DH. Covid-19 Vaccines - Immunity, Variants, Boosters. N Engl J Med. Sep 15 2022;387(11):1011–1020. doi: 10.1056/NEJMra2206573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Offit PA. Bivalent Covid-19 Vaccines - A Cautionary Tale. N Engl J Med. Feb 9 2023;388(6):481–483. doi: 10.1056/NEJMp2215780 [DOI] [PubMed] [Google Scholar]
- 73.Zou J, Kurhade C, Patel S, et al. Neutralization of BA.4-BA.5, BA.4.6, BA.2.75.2, BQ.1.1, and XBB.1 with Bivalent Vaccine. N Engl J Med. Mar 2 2023;388(9):854–857. doi: 10.1056/NEJMc2214916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.COVID-19 Treatment Guidelines Panel. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. . Vol. Available at https://www.covid19treatmentguidelines.nih.gov/. Accessed January 21, 2023.
- 75.Miller J, Hachmann NP, Collier AY, et al. Substantial Neutralization Escape by SARS-CoV-2 Omicron Variants BQ.1.1 and XBB.1. N Engl J Med. Feb 16 2023;388(7):662–664. doi: 10.1056/NEJMc2214314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guan W-j Ni Z-y, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. New England journal of medicine. 2020;382(18):1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cao B, Wang Y, Wen D, et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N Engl J Med. May 7 2020;382(19):1787–1799. doi: 10.1056/NEJMoa2001282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang L, Berger NA, Davis PB, Kaelber DC, Volkow ND, Xu R. COVID-19 rebound after Paxlovid and Molnupiravir during January-June 2022. medRxiv. 2022; [Google Scholar]
- 79.Al-Aly Z, Xie Y, Bowe B. High-dimensional characterization of post-acute sequelae of COVID-19. Nature. Jun 2021;594(7862):259–264. doi: 10.1038/s41586-021-03553-9 [DOI] [PubMed] [Google Scholar]
- 80.Harcourt J, Tamin A, Lu X, et al. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with Coronavirus Disease, United States. Emerg Infect Dis. Jun 2020;26(6):1266–1273. doi: 10.3201/eid2606.200516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. Dec 3 2020;383(23):2255–2273. doi: 10.1056/NEJMra2026131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. The Journal of Immunology. 2015;194(3):855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jeyaseelan S, Chu HW, Young SK, Freeman MW, Worthen GS. Distinct roles of pattern recognition receptors CD14 and Toll-like receptor 4 in acute lung injury. Infect Immun. Mar 2005;73(3):1754–63. doi: 10.1128/IAI.73.3.1754-1763.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Numata M, Sajuthi S, Bochkov YA, et al. Anionic Pulmonary Surfactant Lipid Treatment Inhibits Rhinovirus A Infection of the Human Airway Epithelium. Viruses. 2023;15(3):747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kiso M, Mitamura K, Sakai-Tagawa Y, et al. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet. Aug 28-Sep 3 2004;364(9436):759–65. doi: 10.1016/S0140-6736(04)16934-1 [DOI] [PubMed] [Google Scholar]
- 86.Hayden FG, Sugaya N, Hirotsu N, et al. Baloxavir Marboxil for Uncomplicated Influenza in Adults and Adolescents. N Engl J Med. Sep 6 2018;379(10):913–923. doi: 10.1056/NEJMoa1716197 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. POPG directly binds to RSV and disrupts binding interaction between RSV and HEp2 cells.
Panel A) shows, RSV binds to CD14 with high affinity and in a dose dependent manner. The binding interactions between RSV and CD14 are determined with an HRP-conjugated anti-RSV antibody and measured by A490 absorbance. Panel B) shows that POPG disrupts the binding interaction between RSV and CD14, in a dose-dependent manner. CD14 is captured on an ELISA plate, and RSV with or without POPG is subsequently added to the CD14 solid phase. The binding interaction between RSV and CD 14 is determined by anti RSV antibody. Panel C) HEp2 cells are incubated with 1) no virus (NO RSV, black line), 2) RSV alone (RSV, green line) 3) RSV and POPG (200mg/ml) (RSV+POPG, red line), and 4) RSV+POPC 200mg/ml (RSV+POPC, blue line) and RSV binding to the cells is determined by anti RSV antibody detection. The signals are detected by FACS. Panel D) Quantitative data of FACS from 3 independent experiments are shown. The data are shown as mean ± SD, §§ indicates p<0.001. Statistical analysis is performed by paired t-test. N.S indicate not significant. The data and figures are adapted from ref 11.