

Abstract
Purpose:
Despite promising preclinical studies, toxicities have precluded combinations of chemotherapy and DNA damage response (DDR) inhibitors. We hypothesized that tumor-targeted chemotherapy delivery might enable clinical translation of such combinations.
Patients and Methods:
In a phase I trial, we combined sacituzumab govitecan, antibody-drug conjugate (ADC) that delivers topoisomerase-1 inhibitor SN-38 to tumors expressing Trop-2, with ataxia telangiectasia and Rad3-related (ATR) inhibitor berzosertib. Twelve patients were enrolled across three dose levels.
Results:
Treatment was well tolerated, with improved safety over conventional chemotherapy-based combinations, allowing escalation to the highest dose. No dose-limiting toxicities or clinically relevant ≥ grade 4 adverse events occurred. Tumor regressions were observed in two patients with neuroendocrine prostate cancer and a patient with small cell lung cancer transformed from EGFR-mutant non-small cell lung cancer.
Conclusions:
ADC-based delivery of cytotoxic payloads represents a new paradigm to increase efficacy of DDR inhibitors.
Introduction
DNA damage response (DDR) refers to the cellular processes that orchestrate sensing, signaling, and repair of DNA damage and resolution of DNA replication problems, maintaining genomic integrity while coordinating these with ongoing physiological processes[1]. Several inhibitors of DDR are FDA-approved or undergoing testing in late-phase clinical trials in various disease settings. Yet, despite provocative results of preclinical studies, simultaneously combining DDR inhibitors with DNA damaging chemotherapies has been problematic owing to substantial overlapping toxicities[2].
Defects in DNA recombinational repair due to BRCA mutations cause genomic instability and lead to significantly elevated risks of multiple cancers[3]. These mutations also render such tumors susceptible to therapeutic approaches, notably poly (ADP-ribose) polymerase (PARP) inhibitors that stall the normal progression of replication forks and cause them to collapse, frequently resulting in double-strand breaks (DSBs). PARP inhibitors administered concurrently with DNA damaging chemotherapeutic agents have revealed improved response rates across tumor types, compared with chemotherapy alone, but also increased toxicity – predominantly myelosuppression – requiring dose reductions or treatment delays in a substantial proportion of patients[4–11]. Inhibitors of several other DDR mediators, such as ATM, Chk1 and Chk2, DNA-PK, WEE1, and ataxia-telangiectasia-mutated and rad3-related (ATR), are actively being tested in clinical trials. Yet, despite two decades of clinical trials, a DDR inhibitor-DNA damaging chemotherapy combination is yet to be approved for clinical use.
ATR kinase is a master regulator of DNA damage response, stabilizing the genome when DNA replication is compromised[12]. ATR is activated in response to a wide range of DNA damage and replication problems, such as those induced by DNA damaging agents including topoisomerase 1 (TOP1) inhibitors and platinum agents, and in proliferating cancer cells by oncogene activation. In turn, ATR activates the cell cycle kinase Chk1 by phosphorylation, which suppresses replication fork initiation and elongation. ATR-mediated S-phase arrest prevents cell division and promotes DNA damage repair, avoiding additional DNA damage and maintaining genomic stability. Small-molecule ATR inhibitors have become attractive as therapeutic agents to target cancer cells under replication stress, increase the effectiveness of chemotherapeutic replication inhibitors, and exploit defects in DNA damage response. But, as with PARP inhibitors, combinations of ATR inhibitors with DNA damaging chemotherapy have proven challenging due to overlapping toxicities, primarily moderate to severe myelosuppression[13–17]. Maximizing the therapeutic window of DDR inhibitor-DNA damaging chemotherapy combinations is therefore a major unmet need.
We hypothesized that tumor-targeted delivery of DNA damaging agents might enable tolerable combinations with DDR inhibitors[18]. Antibody-drug conjugates (ADC), which comprise antibody targeting a cancer cell surface protein, cleavable linker molecule, and cytotoxic payload, can target the delivery of cytotoxic agents to tumor sites while reducing exposure to normal tissues and the associated toxicities[19]. We conducted a phase I clinical trial combining ATR inhibitor berzosertib with sacituzumab govitecan, a trophoblast cell surface antigen-2 (Trop-2) directed ADC that delivers high tumoral concentrations of TOP1 inhibitor SN-38 (Fig. 1A). Patients with DNA repair gene-mutated or high replication stress solid tumors were enrolled since such tumors are particularly susceptible to ATR and TOP1 inhibition[20, 21]. The primary end point was identification of the maximum tolerated dose (MTD) of the combination. Efficacy and pharmacodynamics were secondary end points. SN-38-induced DNA damage and its repair by berzosertib was examined in hair follicles by quantifying phosphorylation of the histone H2AX (γH2AX). Whole exome and transcriptome sequencing and Trop-2-immunohistochemistry (IHC) of pre-treatment tumors were used to assess predictors of tumor response.
Figure 1.
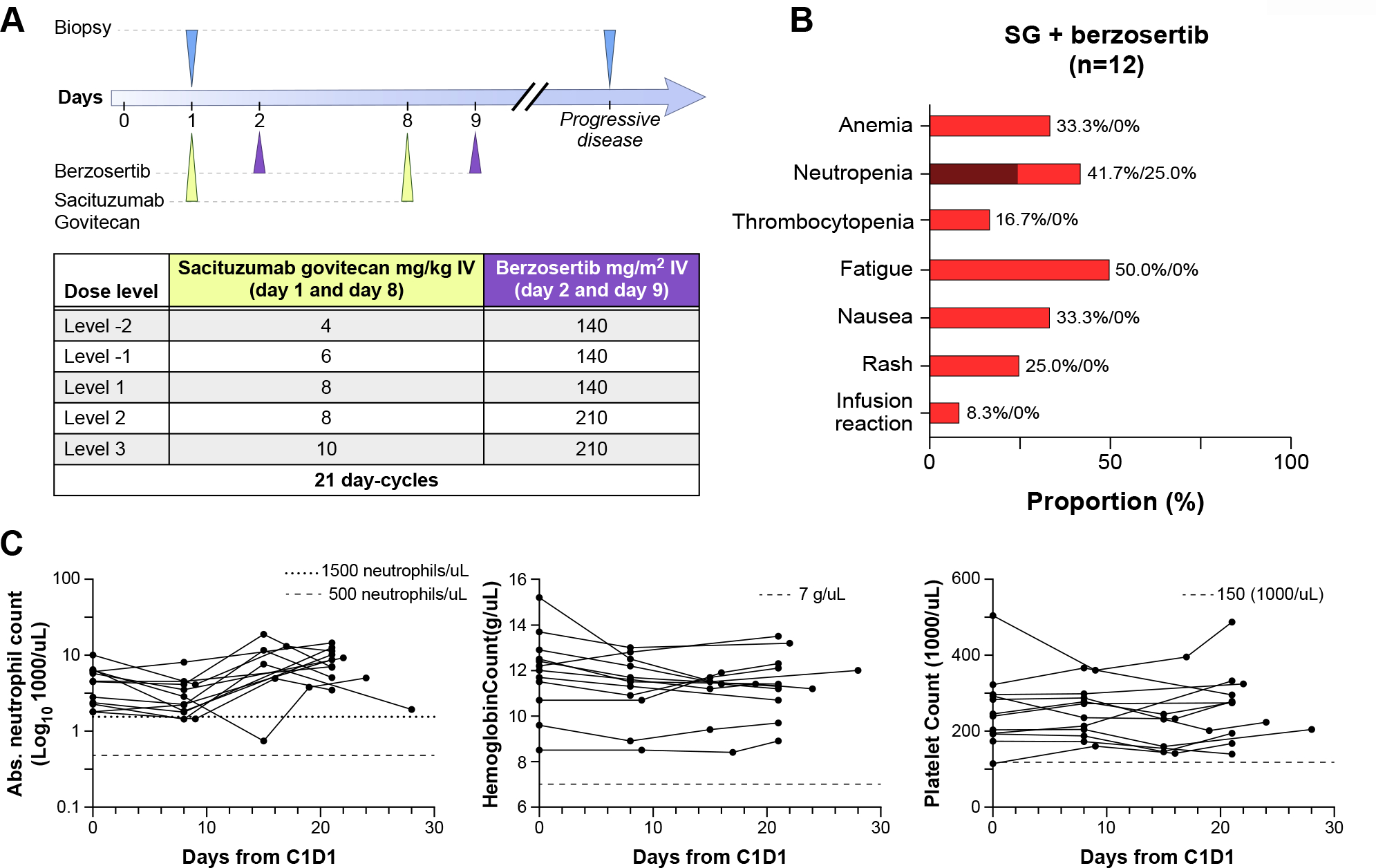
Trial design and safety of sacituzumab govitecan and berzosertib combination. A: Trial design and dose escalation schema. B: Adverse events, labeled as red for grade 1–2 and dark red for grade 3–4[25]. C: Blood cell count trends for all patients across cycle 1, showing minimal hematologic toxicity related to treatment.
Methods
Study Design and Eligibility
This study is a phase I/II, single arm, open label trial enrolling patients with histologically or cytologically confirmed advanced solid tumors with progression on at least one prior chemotherapy. Eligible patients were 18 years or older with an Eastern Cooperative Oncology Group (ECOG) performance status of 0,1 or 2. Subjects must not have received chemotherapy or undergone major surgery within 2 weeks of and radiotherapy within 24 hours prior to cycle 1 day 1. All participants had to have adequate renal, hepatic and bone marrow function at the time of enrollment. Patients with asymptomatic brain metastases or treated brain metastases without evidence of progression or hemorrhage for at least 2 weeks after local treatment were permitted. Participants must have been off any systemic corticosteroids for the treatment of brain metastasis for at least 7 days prior to enrollment. The study protocol was approved by the NIH Institutional Review Board and written informed consent was obtained before patients were enrolled in the study (Clinicaltrials.gov identifier NCT04826341). The trial was carried out in accordance with International Conference on Harmonization Good Clinical Practice (ICH GCP) and the United States (US) Code of Federal Regulations (CFR) applicable to clinical studies.
Treatment
Eligible patients received sacituzumab govitecan on days 1 and 8, and berzosertib on days 2 and 9, both administered intravenously, in 21-day cycles (Fig. 1A). A 3+3 dose escalation schema was used, with increasing doses of both agents administered across three dose levels. The highest planned dose level consisted of the full approved dose of sacituzumab govitecan 10 mg/m2 and berzosertib 210 mg/m2, the dose associated with pharmacodynamic activity in patients with advanced solid tumors[22, 23]. Pegfilgrastim was administered prophylactically on day 9 of each cycle. Treatment was continued until disease progression or development of intolerable side effects.
Outcomes
The primary objective was to determine the MTD of the combination of sacituzumab govitecan and berzosertib. Pharmacokinetics (PK), pharmacodynamics, objective response rate (ORR), and exploratory assessment of biomarkers of response were additional objectives.
Assessment
Adverse events (AEs) were reported in accordance with the National Cancer Institute Common Terminology Criteria for AEs version 5.0. Patients were monitored with weekly labs during cycle 1, and prior to day 1 and day 8 treatment on subsequent cycles. A maximum of 2 dose reductions per patient were permitted, after which treatment would be discontinued. The MTD was defined as the dose level at which no more than 1 of up to 6 participants experience dose limiting toxicity (DLT) during one cycle of treatment, and the dose below that at which at least 2 (of ≤6) participants had DLT. Dose limiting toxicities were defined as: grade 4 neutropenia lasting >7 days despite growth factor support, febrile neutropenia, grade 3 neutropenia with infection, grade 3 or 4 thrombocytopenia with significant bleeding or requiring platelet transfusion, grade 4 thrombocytopenia lasting >7 days, grade 3 or 4 toxicity to organs other than bone marrow, excluding nausea, vomiting, fatigue, and excluding mucositis and diarrhea if patients have not received optimal supportive therapy, and cardiac toxicities as defined in the supplemental materials. Tumor response and progression were evaluated with computerized tomography scans after every 2 cycles (every 6 weeks) using Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1.
Statistical Analysis
Toxicities were identified at each dose level and reported by type and grade. Comparisons between pharmacokinetic data and toxicity were conducted using the Mann-Whitney U test or the Kruskal Wallis test, as appropriate. Additional statistical methods used in pharmacokinetic analysis are included in the supplementary data. Descriptive methods were used for the other analyses.
Pharmacokinetics
Blood samples for PK were collected from each patient before, during, and after treatment with berzosertib and sacituzumab govitecan (Fig. S2). Quantification of SN-38, SN38-G, and berzosertib were performed using a bioanalytical assay that was developed and validated by the NCI Clinical Pharmacology Program, per FDA guidance[24]. A noncompartmental analysis was used to estimate patient-specific PK parameters for total berzosertib, SN-38, and SN-38G. The maximum plasma concentration (Cmax) and time to Cmax (Tmax) were recorded as observed values. The area under the curve extrapolated to time infinity (AUCINF) was calculated using the log-linear trapezoidal rule and the elimination rate (lambda z). The UGT1A1 (TA)n repeat polymorphism (rs3064744) was tested via fluorescent polymerase chain reaction.
Correlative Studies
The study design incorporated optional research biopsies before treatment and at disease progression, when safe and feasible. Pre-treatment biopsies performed any time prior to trial enrollment were also collected in lieu of an immediate pre-treatment biopsy. Exploratory studies included Trop-2 immunohistochemistry (IHC; Abcam Cat# ab214488, RRID: AB_2811182), matched germline (blood) and somatic (tumor) whole exome sequencing (WES), and tumor RNA-sequencing (RNA-seq). WES and RNA-seq were performed by the NCI Laboratory of Pathology using Comprehensive Oncologic Molecular Pathology and Sequencing Service (NCI-COMPASS) as described in Table S1. Gene Set Enrichment Analysis (GSEA) analysis was done using previously reported signatures [25–27]. GSEA enrichment scores were computed using R studio version 4.1.0 (R Foundation for Statistical Computing), the GSVA R/Bioconductor package[28], and gene sets obtained from MSigDB with default parameters[26].
Data Availability
The human sequence data generated in this study are not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author. Other data generated in this study are available within the article and its supplementary data files.
Results
Patients
A total of 12 patients with advanced solid tumors were enrolled in the phase I trial between September 2021 and August 2022 (Table 1). The median age was 51 years (range 33–78), and all patients had an Eastern Cooperative Oncology Group (ECOG) performance score of 0–2. Seven patients were male and five were female. Eight patients (66.7%) were white and four patients (33.3%) Asian. Study representativeness in comparison with the population is described Table S2. All patients had previously received cytotoxic chemotherapy; five (41.7%) patients had received TOP1 inhibiting chemotherapy with topotecan or irinotecan and seven (58.3%) patients had received immune checkpoint blocking therapies. Patients had not received ATR inhibitors previously. Enrollment was enriched for two patient groups: those with solid tumors harboring DNA repair mutations, and those with high-grade neuroendocrine tumors known to have high replication stress[21, 25, 29]. Seven patients had high-grade neuroendocrine cancers. Three of these patients had small cell lung cancer (SCLC), including one patient with SCLC transformed from EGFR-mutant non-small cell lung cancer (NSCLC). Two patients had histologically confirmed neuroendocrine prostate cancer (NEPC); one patient with neuroendocrine transformation from prostate adenocarcinoma following androgen deprivation therapy and another patient diagnosed de novo with small cell prostate cancer. Two patients had high grade neuroendocrine carcinoma of unknown primary. Most of the remaining patients had advanced solid tumors with DNA repair mutations, including two patients with BRCA-mutated colon adenocarcinoma, two patients with pancreatic adenocarcinoma, one harboring a somatic ATM mutation, and a patient with germline TP53-mutated adrenocortical carcinoma (Table S1). Tumor Trop-2 expression was not a requirement for enrollment.
Table 1.
Patient Characteristics N = 12 (%)
| Gender | |
|---|---|
| Female | 5 (41.7) |
| Male | 7 (58.3) |
| Median (range) age in years | 51 (Range 33–78) |
| Eastern Cooperative Oncology Group performance status | |
| 0 | 2 (16.7) |
| 1 | 9 (75) |
| 2 | 1 (8.3) |
| Race | |
| White | 8 (66.7) |
| Asian | 4 (33.3) |
| Diagnosis | |
| Small cell lung cancer | 3 (25) |
| Neuroendocrine prostate cancer | 2 (16.7) |
| Colorectal cancer | 2 (16.7) |
| Pancreatic cancer | 2 (16.7) |
| Neuroendocrine carcinoma of unknown primary | 2 (16.7) |
| Adrenocortical carcinoma | 1 (8.3) |
| Type of prior therapy | |
| Chemotherapy | 12 (100) |
| Topoisomerase I inhibitor | 5 (41.7) |
| Immunotherapy | 7 (58.3) |
| No. of prior systemic treatment | |
| 1 | 2 (16.7) |
| 2 | 4 (33.3) |
| 3 | 3 (25) |
| 4 | 1 (8.3) |
| >4 | 2 (16.7) |
Safety
All patients received at least one cycle of treatment, with 11 of 12 patients receiving at least two cycles, and were evaluable for safety (Fig. 1B). One patient stopped therapy prior to cycle 2 due to early clinical progression. A median of four cycles were administered, with a range of 1 to 9 cycles. One patient remains on treatment at the time of data cutoff. No dose-limiting toxicities were observed, allowing for escalation to the highest dose level: sacituzumab govitecan 10 mg/kg and berzosertib 210 mg/m2.
Lymphopenia was the most common treatment-related adverse event (TRAE), affecting 10 (83.3%) patients, grade 3 in four patients and grade 4 in one patient (Table 2). There were no other grade 4 TRAEs. Five (41.7%) patients experienced neutropenia, including three (25%) patients with grade 3 neutropenia. No patients required dose reductions per protocol due to TRAEs, though one patient had days 8 and 9 treatment delayed by a week every cycle due to neutropenia not meeting criteria for dose adjustment, based on clinical judgement and patient preference. There were no instances of neutropenic fever or infection. Grade 1 or 2 anemia occurred in four patients (33.3%) and grade 1 thrombocytopenia occurred in two (16.7%) patients. No blood or blood product transfusions were required. Gastrointestinal side effects, including diarrhea, nausea, and anorexia were observed in 6 (50%), 4 (33.3 %), and 3 (25%) patients, respectively, but were mostly grades 1 or 2. There was one case of grade 3 diarrhea which improved with supportive therapy. Alopecia and acneiform rash occurred in four (33.3%) and three (25%) patients, respectively. One patient experienced a grade 1 infusion reaction with berzosertib but was able to complete subsequent cycles without further events. Overall, there was no association observed between severity of TRAEs and doses administered. Blood counts of all patients throughout cycle 1 of therapy are shown in Fig. 1C, showing the minimal hematologic toxicity of therapy.
Table 2.
Treatment-related adverse events (TRAE)
| TRAE | Any Grade, n (%) | Grade 3, n (%) | Grade 4, n (%) |
|---|---|---|---|
|
| |||
| Lymphopenia | 10 (83.3) | 4 (33.3) | 1 (8.3) |
| Leukopenia | 6 (50.0) | 2 (16.7) | 0 |
| Diarrhea | 6 (50.0) | 1 (8.3) | 0 |
| Fatigue | 6 (50.0) | 0 | 0 |
| Neutropenia | 5 (41.7) | 3 (25) | 0 |
| Anemia | 4 (33.3) | 0 | 0 |
| Nausea | 4 (33.3) | 0 | 0 |
| Alopecia | 4 (33.3) | 0 | 0 |
| Rash | 3 (25.0) | 0 | 0 |
| Anorexia | 3 (25.0) | 0 | 0 |
| Thrombocytopenia | 2 (16.7) | 0 | 0 |
| Vomiting | 2 (16.7) | 0 | 0 |
The following grade 1 TRAEs each occurred in 1 patient: dysgeusia, dyspepsia, bloating, dizziness, hypotension, infusion reaction, constipation
Pharmacodynamics
Hair follicles were obtained from eight patients to assess γH2AX foci formation as a marker of DNA DSBs [30, 31]. Three patients were unable to provide hair samples due to alopecia from prior chemotherapy and in one patient, hair follicles were not suitable for the assay (no anagen hairs). There was substantial increase in the number and intensity of γH2AX foci in hair follicles following sacituzumab govitecan, indicating more DNA DSBs. Hair follicles on day 2 after berzosertib infusion showed reduced γH2AX signals in several patients, suggesting reduced ATR-dependent phosphorylation of H2AX following berzosertib. The impact of berzosertib on DSBs induced by sacituzumab govitecan may have been affected by variable sampling time points post berzosertib (30 to 180 minutes), and by biological variability such as age and genetic makeup. Detailed methods and results are described in Figure S1.
Pharmacokinetics
PK of sacituzumab govitecan and berzosertib were assessed in all 12 patients spanning up to 2 cycles of therapy (Fig. S2) and showed the expected patterns of Cmax, clearance, and AUC, after accounting for sparse sampling performed here and the known variability of these measures [22, 23, 32, 33]. No association was found between TRAEs and UGT1A1 (TA)n polymorphism [34]. Patients experiencing fatigue and lymphopenia had higher SN-38G Cmax (P<0.01) and SN-38 AUC (P=0.016) respectively (Fig. S3). Higher grades of diarrhea were also non-significantly related to higher berzosertib half-life.
Efficacy
Confirmed partial responses per RECIST v1.1 were observed in two of 12 patients, one with NEPC (Patient 1) and the other with SCLC transformed from EGFR-mutated NSCLC (Patient 8; Fig. 2A–C), lasting 33 weeks and 17 weeks respectively. One patient with de novo NEPC (Patient 11) had a minor response on CT scan, but a notable metabolic response on positron emission tomography (PET) imaging, maintained at 31 weeks at the time of data cutoff. Of note, both patients who had partial responses had neuroendocrine tumors that arose via histologic transformation from adenocarcinoma. Patient 1 who had initially been diagnosed with high-risk prostate adenocarcinoma, which later transformed to NEPC while on treatment with abiraterone, and progressed after carboplatin, etoposide, and atezolizumab had 42.3% tumor shrinkage as the best response (Fig. 2D). Patient 8 with metastatic lung adenocarcinoma transformed to SCLC had a partial response as shown in Fig. 2E. At diagnosis, her tumor carried an EGFR exon 19 deletion, and she was treated with osimertinib, followed by progression with transformation to SCLC about 8 months later. She was then treated with multiple lines of therapy including carboplatin, etoposide, and osimertinib, paclitaxel, and a clinical trial of olaparib and durvalumab, prior to initiation of this therapy. Both patients had platinum-resistant disease, and neither patient continued use of prior targeted therapy (abiraterone or osimertinib) while on trial. Patient 11, with platinum-resistant de novo NEPC had a metabolic response on PET scan (Fig. 2F). Patient 5 with pancreatic adenocarcinoma had prolonged stable disease for 24 weeks. Efficacy was observed across all dose levels.
Figure 2.
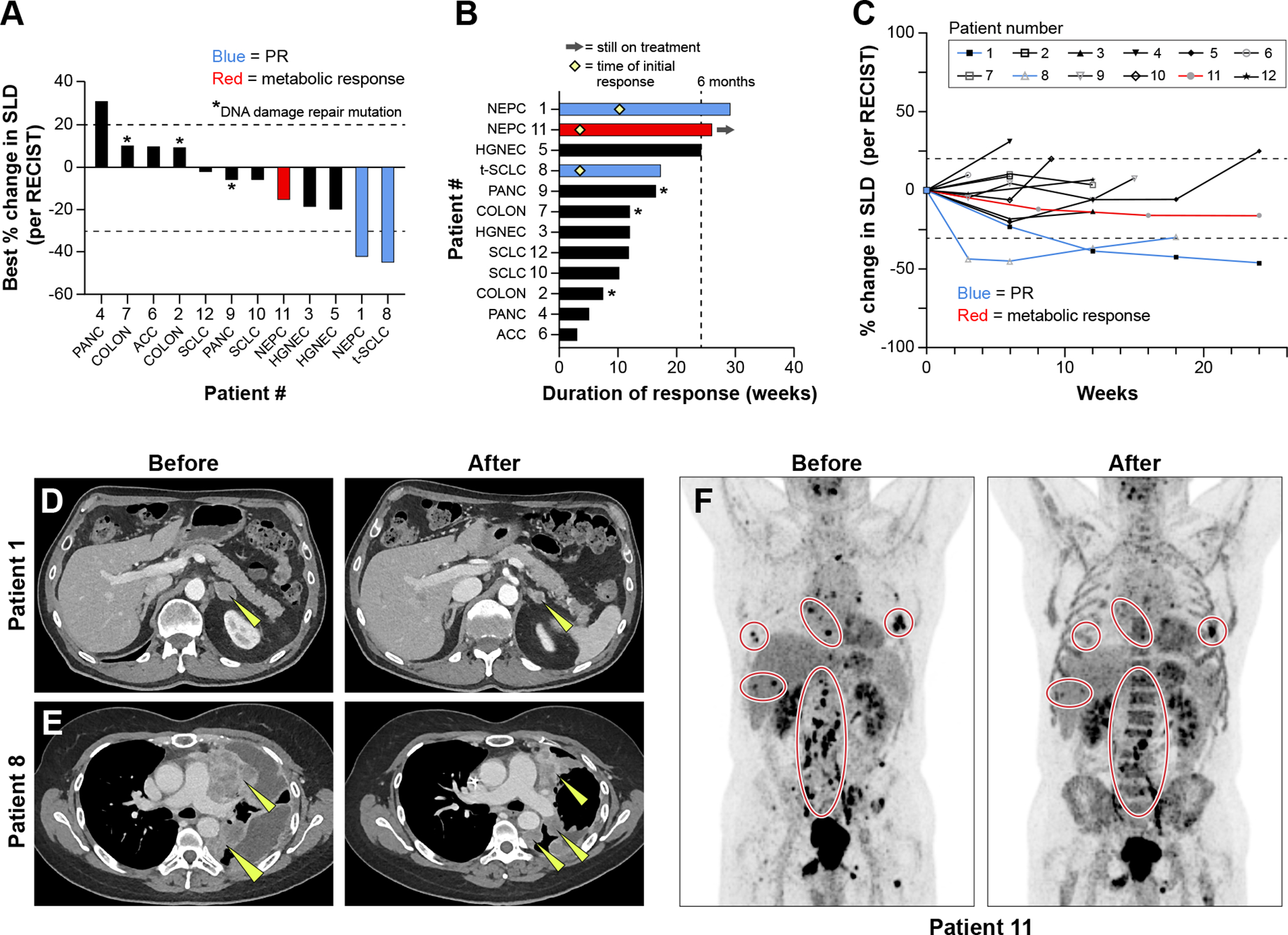
Efficacy of sacituzumab govitecan and berzosertib combination. A: Tumor responses based on maximum change in tumor dimensions from baseline. Each bar represents a patient’s tumor response, corresponding with the assigned patient number. Patients who experienced a partial response per RECIST criteria are annotated in blue, and those with a metabolic response on PET imaging are annotated in red. B: Efficacy based on duration of response, including timing of partial responses, indicated with a yellow diamond, when applicable. C: Efficacy based on change in tumor dimensions from baseline over time. D. Patient 1 with NEPC experienced a partial response, with yellow arrows annotating target lesions. E: Patient 8 with SCLC transformed from EGFR-mutated NSCLC experienced a partial response with a decrease in size of multiple lung masses, as indicated by yellow arrows. F: Patient 11 with de novo NEPC had a metabolic response to therapy across many metastatic sites as shown on PET imaging.
SLD: sum of the total diameter of target lesions
PR: Partial Response per RECIST criteria
PANC = pancreatic adenocarcinoma, COLON = colon adenocarcinoma, ACC = adrenocortical carcinoma, SCLC = small cell lung cancer, NSCLC = non-small cell lung cancer, t-SCLC = SCLC transformed from NSCLC, NEPC = neuroendocrine prostate cancer, HGNEC = high grade neuroendocrine carcinoma
Correlative studies
All 12 patients had tumor sampled immediately before the trial (n=6) or had archival samples before an earlier course of therapy (n=6). RNA-seq was performed on eight tumors from seven patients, including one responder. Two patients did not consent to sequencing and RNA quality from archival tumors precluded sequencing in three cases. One patient had biopsies from two locations profiled. As an exploratory analysis, tumor RNA-seq of one RECIST responder (Patient 1) and a patient with prolonged stable disease for 24 weeks (Patient 5), deemed to have derived “clinical benefit”, were compared with five non-responding tumors. Gene set enrichment analysis (GSEA) showed that tumors from patients with clinical benefit were enriched for pathways related to replication stress and proliferation and were de-enriched for immune and inflammatory pathways (Fig. 3A). GSEA for the Hallmarks of Cancer showed that tumors from patients with clinical benefit were enriched for pathways related to genomic instability (Fig. 3B), consistent with tumors deriving clinical benefit harboring a replication stress phenotype[21, 25, 27].
Figure 3.
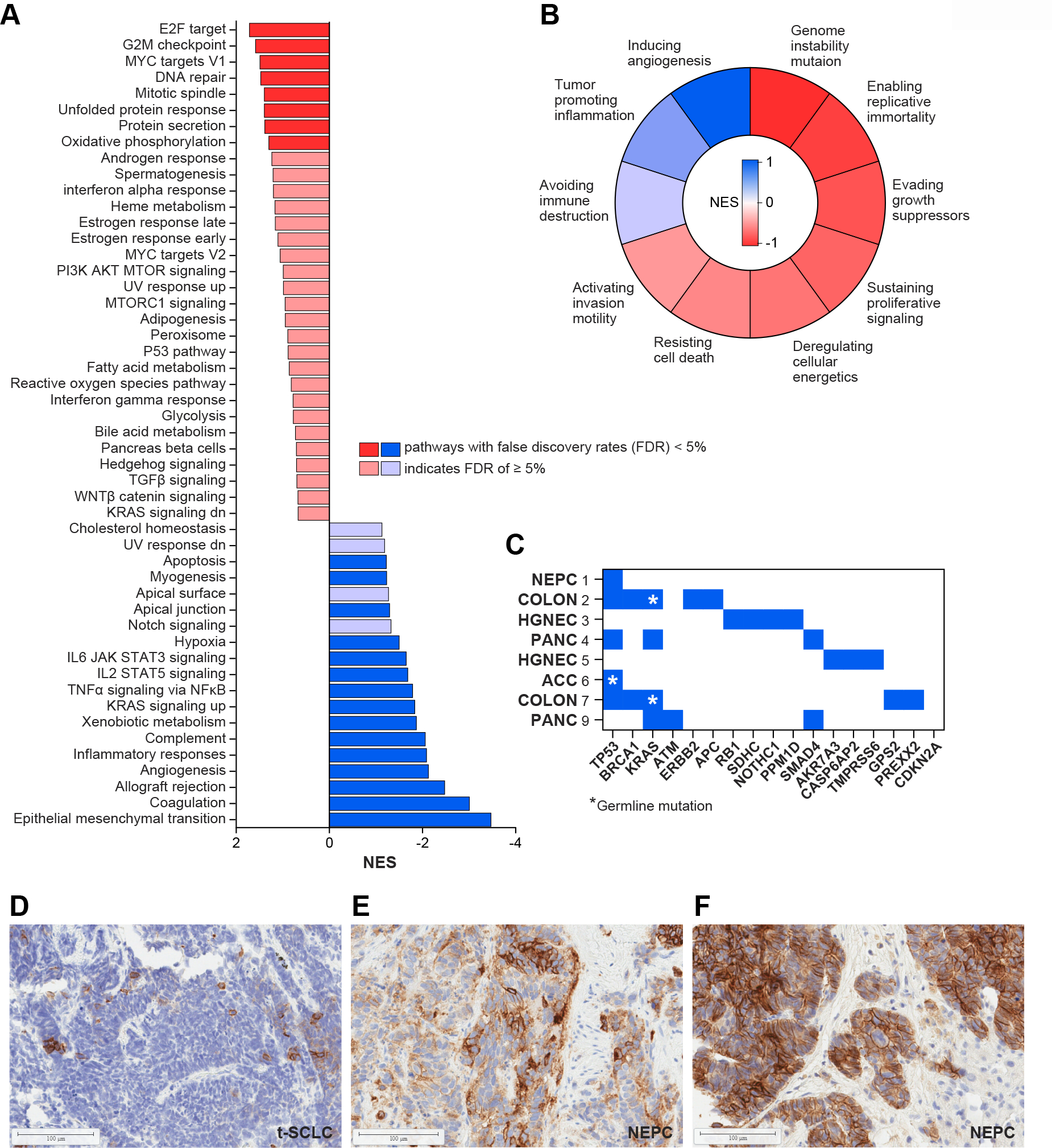
Predictors of benefit to sacituzumab govitecan and berzosertib combination A: RNA sequencing of tumors from patients with clinical benefit (n=2) and without clinical benefit (n=5). GSEA using the Hallmark signature showed that tumors with clinical benefit were enriched for genes associated with replication stress and proliferation and were de-enriched for immune and inflammatory pathways. B: GSEA using the Hallmarks of Cancers signature showing that tumors with clinical benefit were enriched for pathways associated with genome instability. C: Summary of whole exome sequencing results. Blue squares indicate presence of a gene mutation in tumor tissue, with germline mutations indicated by a yellow asterisk. D. Trop-2 IHC from Patient 8 with SCLC transformed from EGFR-mutated NSCLC showed minimal expression. Patients 11 (E) and 1 (F) with NEPC both had IHC with high Trop2 expression.
GSEA: gene set enrichment analysis
NES: normalized enrichment score
SCLC: small cell lung cancer
NSCLC: non-small cell lung cancer
t-SCLC: transformed SCLC
Eight patients had matched germline blood and somatic WES performed successfully (Fig. 3C; Table 3; Table S1). Those without sequencing either did not consent, as noted above, or lacked adequate tumor DNA. Two patients, both with colon cancer, carried germline BRCA1 mutations. One patient with pancreatic cancer was found to have a somatic ATM mutation. A patient with adrenocortical carcinoma had a germline TP53 mutation. Gene alterations considered pathogenic or likely pathogenic are described with the corresponding sequencing test in Figure S1. None of the patients with DNA repair mutations had tumor responses.
Table 3.
Demographics by Patient
| No. | Demographics | Tumor Type (DDR Mutation) | No. of Prior Regimens | Best Response |
|---|---|---|---|---|
|
| ||||
| Dose Level 1: | ||||
|
| ||||
| 1 | White Male, Age 73 | Neuroendocrine prostate | >5 | PR |
| 2 | White Male, Age 44 | Colorectal (BRCA1) | 3 | PD |
| 3 | White Female, Age 50 | High grade neuroendocrine carcinoma | 1 | SD |
|
| ||||
| Dose Level 2: | ||||
|
| ||||
| 4 | Asian Male, Age 52 | Pancreatic | 3 | PD |
| 5 | White Male, Age 72 | High grade neuroendocrine carcinoma | 1 | SD |
| 6 | White Female, Age 50 | Adrenocortical carcinoma (p53/Li Fraumeni) | >5 | PD |
|
| ||||
| Dose Level 3 | ||||
|
| ||||
| 7 | White Female, Age 33 | Colorectal (BRCA1) | 4 | SD |
| 8 | Asian Female, Age 49 | Transformed SCLC | 2 | PR |
| 9 | White Male, Age 69 | Pancreatic (ATM) | 2 | SD |
| 10 | Asian Male, age 78 | SCLC | 2 | SD |
| 11 | White Male, age 71 | Neuroendocrine Prostate | 2 | SD* |
| 12 | Asian Female, Age 51 | SCLC | 3 | SD |
SD per RECIST, but with metabolic response on PET scan
DDR: DNA damage response; PR: partial response; SD: stable disease; PD: progressive disease; SCLC: small cell lung cancer. Best response is evaluated by response evaluation criteria in solid tumors version 1.1.
Tumor samples also were evaluated for Trop-2 expression by IHC (Fig. 3D–F). Tumors of both NEPC patients highly expressed Trop-2, correlating with clinical benefit from therapy. However, patients with SCLC and other high-grade neuroendocrine cancers had low to no expression. Trop-2 expression was also low in EGFR-transformed SCLC with a partial response. Tumor from a patient with BRCA1-mutated colon cancer showed high expression but did not respond to trial therapy.
Discussion
Genomic instability is a characteristic of most cancer cells. Cancers rely on several surveillance mechanisms to maintain genomic integrity. As such, a synthetic lethal combination of DNA-damaging agents and DDR inhibitors should have widespread utility across many cancers that rely on these mechanisms. However, overlapping normal tissue toxicities from these drug classes have precluded clinical translation of such combinations to date. Tumor-targeted delivery of DNA damaging agents represent one potential approach for achieving safe combinations of these agents. In a phase I study, we examined the tolerability of a combination of ATR inhibitor berzosertib and sacituzumab govitecan, an ADC that delivers high concentrations of TOP1 inhibitor SN-38 to tumors expressing Trop-2. The combination was well tolerated with no dose limiting toxicities, which allowed dose escalation to the highest planned dose level. Additionally, the combination resulted in lower rates of hematologic toxicity compared to the combination of berzosertib and conventional TOP1 inhibitor topotecan (Fig. S4)[25], though a head-to-head comparison would be needed to draw definitive conclusions. The study provides proof-of-concept for ADC-based delivery of cytotoxic payload as a new therapeutic paradigm to extend the benefit of DDR inhibitors, with minimal added toxicities. Principles of this combination strategy are relevant to clinical development of several potent and specific ADCs targeting an increasingly diverse number of targets, many of them carrying TOP1 inhibitor payloads.
Here, we show that combining sacituzumab govitecan with berzosertib is feasible and tolerable, with no patients requiring dose reductions or blood product transfusions. These safety outcomes are on par with studies of single-agent sacituzumab govitecan, where grade ≥ 3 neutropenia was reported in about one-third of patients[35]. While targeted delivery of cytotoxic payload is expected to provide an advantage over systemic chemotherapy, there is little data assessing combination therapy with ADCs and DDR pathway-targeted agents. A recent case report from the phase 1b SEASTAR study found that although concurrent treatment of the PARP inhibitor rucaparib and the sacituzumab govitecan provided promising antitumor activity, neutropenia was dose-limiting[13]. These dose-limiting events can be explained in part by the limited selectivity of rucaparib for PARP1 over PARP2, which is essential for the survival of hematopoietic and stem progenitor cells. The continuous dosing of rucaparib may also have contributed to myelosuppression. PARP1-selective inhibitors with improved safety and tolerability compared with first-generation PARP1/2 inhibitors, currently in clinical development have less myelosuppressive effects and may enable more tolerable PARP-TOP1 inhibitor combinations[36]. The therapeutic index of ADC-based combinations with DDR inhibitors including PARP and ATR inhibitors and others may be further widened using the “gapped” schedule wherein the DDR inhibitor is administered following a gap or interval after the ADC administration which would allow for systemic clearance of the cytotoxic payload while it is still retained in the tumor[18]. Advances in the design of linker and conjugation chemistry will also enable development of newer generation ADCs with better tumor specificity.
An important clinical activity signal was noted in patients with NEPC and SCLC transformed from EGFR-mutant NSCLC, tumor types marked by lineage plasticity, i.e. a shift in phenotype from one committed developmental pathway to another and an aggressive clinical course. Lineage plasticity in cancer, exemplified by the histological transformation of adenocarcinomas to aggressive neuroendocrine cancers, was initially described in lung cancers harboring EGFR mutations[37], and was subsequently reported in many other settings, including prostate cancer[38]. Lineage plasticity has been proposed as a source of intratumoral heterogeneity and of tumor adaptation to adverse tumor microenvironments including exposure to targeted anticancer treatments[39]. NEPCs demonstrate prevalent expression of Trop-2 [40], and overexpression of Trop-2 in preclinical NEPC models led to significantly decreased androgen receptor expression, while inducing lineage plasticity, neuroendocrine features, and metastases [41]. The striking responses, albeit in a small number of patients who had undergone extensive previous therapies, provides compelling rationale to rigorously investigate the effect of this combination in tumors that have acquired neuroendocrine transformation by lineage plasticity.
A notable aspect of the trial was the deep characterization of pretreatment tumor samples using paired somatic and germline exome, RNA-seq, and Trop-2 IHC. These analyses were performed on fresh tissue when a pre-treatment biopsy was deemed low-risk and feasible, otherwise archival tissue was used. Notably, Trop-2 expression by IHC was observed on two of the three responding tumors, but Trop-2 was also expressed on several non- responding tumors, with a high degree of intratumoral heterogeneity across both groups. While Trop-2 is overexpressed across a variety of epithelial cancers, whether its expression predicts benefit from sacituzumab govitecan is poorly understood. In triple negative breast cancer, greater efficacy was observed among patients with medium or high Trop-2 expression, yet patients of all Trop-2 expression subgroups benefited from treatment with sacituzumab govitecan, as compared with physician’s choice of therapy[42]
Responses were not seen in four patients with mutations that increase the reliance on ATR signaling to avoid death due to replication stress; two patients with BRCA mutated tumors, one patient with ATM mutated and another patient with a germline TP53 mutated tumors. All four patients had advanced, heavily pre-treated tumors which may explain the lack of tumor responses. Two patients with BRCA1 mutations had colon adenocarcinoma, which despite the mutations may not have BRCA-associated tumor pathogenesis that is therapeutically actionable[32]. Recent clinical trials with ATR inhibitors have also not shown the expected responses in tumors with DNA repair mutations, underlining the importance of further optimization of biomarkers for this class of drugs[43, 44].
There are important limitations to this study due to the small patient cohort and few available specimens for translational studies. In terms of treatment efficacy, it is unclear if tumor responses are related to synergy between the two agents or to sacituzumab govitecan alone. Sacituzumab govitecan monotherapy has led to tumor responses in SCLC, and is being studied in metastatic castrate-resistant prostate cancer (NCT03725761)[45]. Results of exploratory studies beyond the primary endpoint of safety should be cautiously interpreted given the small sample size. This is particularly true for tumor RNA-seq which uses only two “responders” for comparison, one of whom had clinical benefit but did not achieve PR. Though exploratory, these results provide a framework for expanding these data with samples from the ongoing phase II trial. Larger patient cohorts will also be needed to generate more meaningful correlations between PK and toxicity.
In summary, DDR inhibitors are highly synergistic with chemotherapeutic DNA repair targeted agents, but the synergy exists for toxicities as well as efficacy. Using ATR inhibitor as a paradigm, we provide early clinical evidence supporting the hypothesis that tumor-targeted delivery of a TOP1 inhibitors provides a safety advantage over systemic chemotherapy, and that such combinations could provide durable clinical benefit for patients with tumors under replication stress. Ongoing phase II studies are examining the combination of berzosertib and sacituzumab govitecan in patients with SCLC, PARP inhibitor resistant tumors, and in extrapulmonary small cell cancers (NCT04826341).
Supplementary Material
Statement of Significance.
DNA damage response inhibitors are highly synergistic with chemotherapeutic agents, but the synergy exists for toxicities as well as efficacy. We provide proof-of-concept that tumor-targeted delivery of cytotoxic agent provides safety advantage over conventional chemotherapy, and that such combinations provide durable anti-tumor efficacy. Our data support the broad investigation of cytotoxic payloads delivered via ADCs in combination with DDR inhibitors.
Acknowledgements
This work was supported by the intramural programs of the Center for Cancer Research, National Cancer Institute (ZIA BC 011793).
This project has been funded in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN2612008000031 (BK). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
We are grateful to Drs. Michael Kruhlak, Langston Lim, and Andy Tran (Confocal Microscopy Core Facility, CCR, NCI, NIH) for expert technical assistance in confocal microscopy.
This research was supported by EMD Serono (CrossRef Funder ID: 10.13039/100004755), who provided berzosertib free of charge. The healthcare business of Merck KGaA, Darmstadt, Germany reviewed the manuscript for medical accuracy only before submission. The authors are fully responsible for the content of this manuscript, and the views and opinions described in the manuscript reflect solely those of the authors.
Footnotes
Conflict of Interest Statement
AT report research funding to the institution from the following entities: EMD Serono Research & Development Institute; AstraZeneca; Tarveda Therapeutics; Immunomedics and Prolynx. The remaining authors declare no potential conflicts of interest.
References
- 1.Harper JW and Elledge SJ, The DNA damage response: Ten years after. Molecular Cell, 2007. 28(5): p. 739–745. [DOI] [PubMed] [Google Scholar]
- 2.Pilie PG, et al. , State-of-the-art strategies for targeting the DNA damage response in cancer. Nature Reviews Clinical Oncology, 2019. 16(2): p. 81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lord CJ and Ashworth A, BRCAness revisited. Nature Reviews Cancer, 2016. 16(2): p. 110–120. [DOI] [PubMed] [Google Scholar]
- 4.Hendrickson AEW, et al. , A Phase I Clinical Trial of the Poly(ADP-ribose) Polymerase Inhibitor Veliparib and Weekly Topotecan in Patients with Solid Tumors. Clinical Cancer Research, 2018. 24(4): p. 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LoRusso PM, et al. , Phase I Safety, Pharmacokinetic, and Pharmacodynamic Study of the Poly(ADP-ribose) Polymerase (PARP) Inhibitor Veliparib (ABT-888) in Combination with Irinotecan in Patients with Advanced Solid Tumors. Clinical Cancer Research, 2016. 22(13): p. 3227–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samol J, et al. , Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Investigational New Drugs, 2012. 30(4): p. 1493–1500. [DOI] [PubMed] [Google Scholar]
- 7.Kummar S, et al. , Phase I Study of PARP Inhibitor ABT-888 in Combination with Topotecan in Adults with Refractory Solid Tumors and Lymphomas. Cancer Research, 2011. 71(17): p. 5626–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen EX, et al. , A Phase I study of olaparib and irinotecan in patients with colorectal cancer: Canadian Cancer Trials Group IND 187. Investigational New Drugs, 2016. 34(4): p. 450–457. [DOI] [PubMed] [Google Scholar]
- 9.Rajan A, et al. , A Phase I Combination Study of Olaparib with Cisplatin and Gemcitabine in Adults with Solid Tumors. Clinical Cancer Research, 2012. 18(8): p. 2344–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhawan MS, et al. , Differential Toxicity in Patients with and without DNA Repair Mutations: Phase I Study of Carboplatin and Talazoparib in Advanced Solid Tumors. Clinical Cancer Research, 2017. 23(21): p. 6400–6410. [DOI] [PubMed] [Google Scholar]
- 11.Bendell J, et al. , Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann Oncol, 2015. 26(4): p. 804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeman MK and Cimprich KA, Causes and consequences of replication stress. Nat Cell Biol, 2014. 16(1): p. 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas A, et al. , Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. Journal of Clinical Oncology, 2018. 36(16): p. 1594–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konstantinopoulos PA, et al. , Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: a multicentre, open-label, randomised, phase 2 trial. Lancet Oncology, 2020. 21(7): p. 957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas A, et al. , Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell, 2021. 39(4): p. 566–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choudhury AD, et al. , A phase 2 study of berzosertib (M6620) in combination with carboplatin compared with docetaxel in combination with carboplatin in metastatic castration-resistant prostate cancer. Journal of Clinical Oncology, 2021. 39(15). [Google Scholar]
- 17.Pal SK, et al. , Effect of Cisplatin and Gemcitabine With or Without Berzosertib in Patients With Advanced Urothelial Carcinoma A Phase 2 Randomized Clinical Trial. Jama Oncology, 2021. 7(10): p. 1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas A and Pommier Y, Targeting Topoisomerase I in the Era of Precision Medicine. Clin Cancer Res, 2019. 25(22): p. 6581–6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas A, Teicher BA, and Hassan RT, Antibody-drug conjugates for cancer therapy. Lancet Oncology, 2016. 17(6): p. E254–E262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Josse R, et al. , ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res, 2014. 74(23): p. 6968–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi N, et al. , Replication stress defines distinct molecular subtypes across cancers. Cancer Res Commun, 2022. 2(6): p. 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.M6620 Investigator’s Brochure, v8.0, in Project #201923. 2017.
- 23.Sacituzumab Govitecan Investigator’s Brochure. 2022.
- 24.Kim Y, et al. , A phase II study of pembrolizumab and paclitaxel in refractory extensive disease small cell lung cancer. Journal of Clinical Oncology, 2018. 36(15). [Google Scholar]
- 25.Thomas A, et al. , Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell, 2021. 39(4): p. 566–579.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liberzon A, et al. , The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst, 2015. 1(6): p. 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alcala N, et al. , Integrative and comparative genomic analyses identify clinically relevant pulmonary carcinoid groups and unveil the supra-carcinoids. Nat Commun, 2019. 10(1): p. 3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanzelmann S, Castelo R, and Guinney J, GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics, 2013. 14: p. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas A and Pommier Y, Small cell lung cancer: Time to revisit DNA-damaging chemotherapy. Science Translational Medicine, 2016. 8(346): p. 346fs12–346fs12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fong PC, et al. , Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med, 2009. 361(2): p. 123–34. [DOI] [PubMed] [Google Scholar]
- 31.Ivashkevich A, et al. , Use of the gamma-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett, 2012. 327(1–2): p. 123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Middleton MR, et al. , Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine +/− cisplatin in patients with advanced solid tumours. Br J Cancer, 2021. 125(4): p. 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reck M, et al. , Antiangiogenic therapy for patients with aggressive or refractory advanced non-small cell lung cancer in the second-line setting. Lung Cancer, 2018. 120: p. 62–69. [DOI] [PubMed] [Google Scholar]
- 34.Karas S and Innocenti F, All You Need to Know About UGT1A1 Genetic Testing for Patients Treated With Irinotecan: A Practitioner-Friendly Guide. JCO Oncol Pract, 2022. 18(4): p. 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ocean AJ, et al. , Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer, 2017. 123(19): p. 3843–3854. [DOI] [PubMed] [Google Scholar]
- 36.Yap T, I.S., Schram A, et al. First in class, first in human trial of the next generation PARP1-selective inhibitor AZD5305 in patients (pts) with BRCA1/2, PALB2 or RAD51C/D mutations. in American Association for Cancer Research Annual Meeting. 2022. Virtual. [Google Scholar]
- 37.Sequist LV, et al. , Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Science Translational Medicine, 2011. 3(75). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aggarwal R, et al. , Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. Journal of Clinical Oncology, 2018. 36(24): p. 2492–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quintanal-Villalonga A, et al. , Lineage plasticity in cancer: a shared pathway of therapeutic resistance (Mar, 10.1038/s41571-020-0340-z, 2020). Nature Reviews Clinical Oncology, 2020. 17(6): p. 382–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeLucia DC, et al. , Regulation of CEACAM5 and Therapeutic Efficacy of an Anti-CEACAM5-SN38 Antibody-drug Conjugate in Neuroendocrine Prostate Cancer. Clinical Cancer Research, 2021. 27(3): p. 759–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu EC, et al. , Trop2 is a driver of metastatic prostate cancer with neuroendocrine phenotype via PARP1. Proceedings of the National Academy of Sciences of the United States of America, 2020. 117(4): p. 2032–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bardia A, et al. , Biomarker analyses in the phase III ASCENT study of sacituzumab govitecan versus chemotherapy in patients with metastatic triple-negative breast cancer. Ann Oncol, 2021. 32(9): p. 1148–1156. [DOI] [PubMed] [Google Scholar]
- 43.Shah PD, et al. , Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol Oncol, 2021. 163(2): p. 246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Owonikoko TK, et al. , Phase 2 Study of Talazoparib in Patients With Homologous Recombination Repair-Deficient Squamous Cell Lung Cancer: Lung-MAP Substudy S1400G. Clin Lung Cancer, 2021. 22(3): p. 187–194 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gray JE, et al. , Therapy of Small Cell Lung Cancer (SCLC) with a Topoisomerase-I–inhibiting Antibody–Drug Conjugate (ADC) Targeting Trop-2, Sacituzumab Govitecan. Clinical Cancer Research, 2017. 23(19): p. 5711–5719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The human sequence data generated in this study are not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author. Other data generated in this study are available within the article and its supplementary data files.