

Abstract
Two children are presented who have a distinct syndrome of multiple buccolingual frenula, a stiff and short fifth finger with small nails, a hypothalamic hamartoma, mild to moderate neurological impairment, and mild endocrinological symptoms. No variant assessed to be pathogenic or likely pathogenic was detected in the GLI3 gene in either child. This syndrome appears to be distinct from the inherited Pallister-Hall syndrome associated with GLI3 variants, which is characterized by hypothalamic hamartoma, mesoaxial polydactyly, and other anomalies. In the individuals described here, manifestations outside of the central nervous system were milder and the mesoaxial polydactyly, which is common in individuals with Pallister Hall syndrome, was absent. Instead, these children had multiple buccolingual frenula together with the unusual appearance of the fifth digit. It remains unclear whether these two individuals represent a separate nosologic entity or if they represent a milder manifestation of one of the more severe syndromes associated with a hypothalamic hamartoma.
INTRODUCTION
The delineation of clinically recognizable syndromes is particularly important in cases where early diagnosis may help to predict potential medical complications as well as for recurrence risk determination. A hypothalamic hamartoma is a congenital malformation that may be asymptomatic or may present with neurological symptoms, such as gelastic seizures, or with endocrinologic complications, such as hypopituitarism (Graham, 1985). The hypothalamic hamartoma may occur in isolation or as part of a multiple anomaly syndrome. One example of the syndromic form of hypothalamic hamartoma is the GLI3-related Pallister–Hall syndrome (PHS, MIM # 146510), which was described initially by Judith Hall and Philip Pallister (Hall, 1980; Clarren, 1980) as a neonatal lethal syndrome associated with hypopituitarism, imperforate anus, pulmonary segmentation anomalies, postaxial polydactyly, small nails, and oral and pharyngeal anomalies, including bifid epiglottis, pharyngeal cleft, and buccal frenula.
As is true for many disorders, the earliest recognized individuals had more severe abnormalities. This led to an unfortunate designation of ‘cerebro-acro-visceral early lethality complex’ or CAVE (Verloes, 1992). With further clinical experience since 1980, it has become clear that GLI3-related PHS has a wide range of severity, with most individuals manifesting only a subset of these anomalies and having a normal lifespan (Biesecker, 1996). The syndrome in these individuals was found to be associated with heterozygosity for a pathogenic variant in the transcription factor GLI3 (Kang, 1997).
Here, we describe two unrelated children with a yet milder congenital hypothalamic hamartoma phenotype, characterized by mild anomalies of the fifth digit without polydactyly and not associated with a variant in the GLI3 gene (Johnston, 2010).
CLINICAL REPORTS
Individual 1:
This male infant was born at term weighing 3.7 kg (50 to 75th centile) after an uncomplicated pregnancy to a 34-year-old G0, P0->P1 mother (individual PH20, Johnston, 2010). On the first day of life, episodes of shallow respiration and cyanosis occurred. On examination, he had a short lingular frenulum, multiple frenula from the buccal membranes to the alveolar ridges, partially clefting the gum line (Figure 1A), clinodactyly of the right fifth finger, camptodactyly of the left fifth finger with a single flexor crease, and a tapered fingertip with a small nail (Figure 1C). His left testicle was not palpable, but his external genitalia and anus were otherwise normal. No laryngeal malformations were identified in a direct mirror examination by an ENT specialist. At 5 months of age, his weight was 21.8 kg (>97th centile), length was 13.7 cm (>97th centile) and head circumference was 53 cm (90-95th centile). The father was 34 years old and was 72 inches tall. The mother was 33 and 68 inches tall. Echocardiography showed a small patent foramen ovale. There was no apparent polydactyly by inspection or visible in radiographs (Figure 2C). The Giemsa-banded karyotype was 46,XY. The spells and congenital anomalies prompted magnetic resonance neuroimaging, which showed a suprasellar, interpeduncular mass in continuity with the hypothalamus, proportionally unchanged on repeated imaging (Figure 2A).
Figure 1:
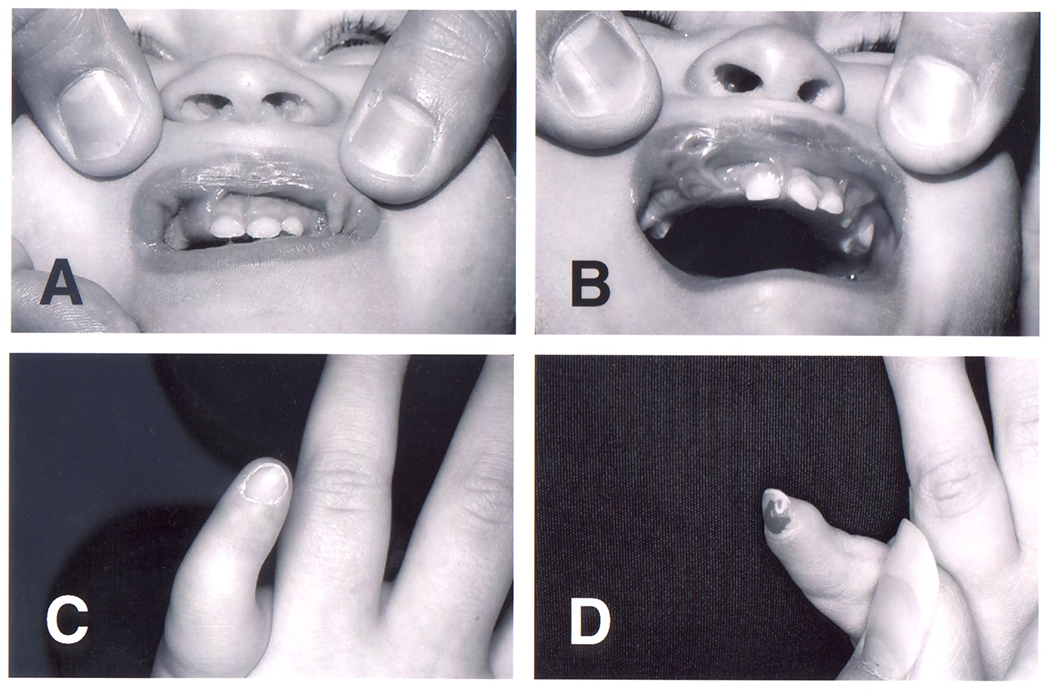
Multiple frenula from the lips to the alveolar ridge in individuals 1 (A) and 2 (B). Note additional lip pits especially in individual 2 (B). Fifth fingers of the left hand in individual 1 (C) and 2 (D). Note the pronounced shortness, small nails, and stiff interphalangeal joints.
Figure 2:
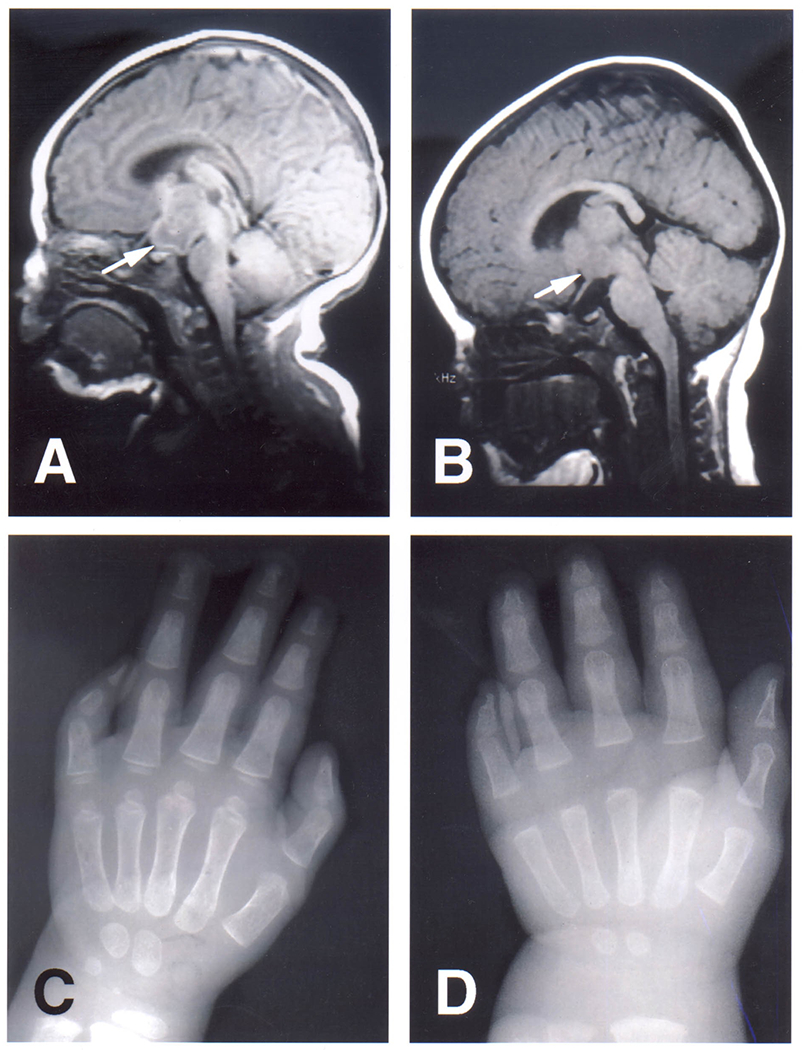
Sagittal T1-weighted MRI of the brain in individual 1 (A) at the age of 2 weeks and in individual 2 (B) at the age of 7 months. Note the interpeduncular mass in continuity with the hypothalamus (arrows). The masses are isointense to grey matter. X-rays of the left hands of individual 1 (C) at the age of 2 years and of individual 2 (D) at the age of 7 months. Note the pronounced shortness of especially the middle and distal phalanx of the fifth finger. The middle and distal phalanx in individual 2 appear to show symphalangism.
Because of the discovery of the hypothalamic hamartoma, endocrine studies were initiated at two months of age. The results were suggestive of hypogonadotropic hypogonadism with elevated serum prolactin (60.5 ng/ml, norm: less than 20 ng/ml), and low FSH (0.5 U/L, norm: <1-7.1), LH (1 U/L, norm: <1-33), and testosterone (42 ng/dl, norm: 75-400) at the time of the normal neonatal surge. At age 3 8/12 years, he developed symptoms and laboratory evidence of mild secondary hypothyroidism (free T4 index = 4.2, norm: 0.9-2.3 ng/L) and hypoadrenalism (AM cortisol of 4.9 g/dl, norm: <1-35) and was treated with hydrocortisone and L-thyroxine supplementation. At 2 years 5 months, absence of the left testicle was confirmed by peritoneoscopy. Linear growth had accelerated to the 95th centile at 6 months of age and remained stable (~97th centile) up to age 12 years (height 160 cm; weight 60.5 kg).
On neurological evaluation, there was mild axial hypotonia in infancy and delay in expressive language, which resolved with speech therapy. Gelastic seizures and complex partial seizures commenced at 6 weeks of age and were controlled initially with anticonvulsant medication. However, he had breakthrough of partial seizures with secondary generalization, which were not controlled with different medications. He had a surgical removal of the hypothalamic hamartoma with better control of his seizures at age 15 years.
His parents and a younger sister were normal by examination and cranial MRI studies. There is no known consanguinity in the family and no history of other, similarly affected individuals.
Individual 2
This female infant was born at 37 weeks gestation to a 42-year-old G1, P0->1 mother after in vitro fertilization (individual PH19, Johnston, 2010). The pregnancy was complicated by the mother having surgery for an isolated cerebellar hemangioblastoma. At birth, multiple bucco-alveolar frenula were noted on both the upper and lower jaw, partly clefting the gum line (Figure 1B). Both fifth fingers were small, the left more so than right, with stiff interphalangeal joints and a small nail on the left (fig 1D). There was partial (out to the first interphalangeal joint) cutaneous syndactyly between the second and third toes on the right foot. No internal anomalies were detected on radiological investigations There was no polydactyly in the radiographs of each hand (Figure 2D).
Because of her phenotypic resemblance to Individual 1, neuroimaging was carried out at 7 months of age, which showed a midline mass extending from the hypothalamus (Figure 2B). This mass remained proportionally unchanged on repeated imaging. At 18 months her weight was 9.5 kg (10th centile), height was 80.5 cm (50th centile), and head circumference was 45.6 cm (10th centile). The father was 45 years old; his height was 67 inches. The mother was 44 and 62 inches tall. Endocrine studies showed only mildly elevated prolactin on two occasions (95 and 33 ng/ml, norm: 5-25 ng/ml) and normal thyroid and adrenal function. She showed initially mild axial hypotonia, but by age 24 months she was developmentally age appropriate. She continued to do well without seizures at the age of 14 years. Her height was 155 cm (25th centile) and weight 61 kg (75th centile). She had no neurologic problems or endocrine deficiencies. She has never had problems in learning. Her parents had no hypothalamic lesions on brain neuroimaging studies. They were not known to be related.
METHODS
DNA Isolation, PCR, and Sequencing: DNA was isolated from whole blood using the salting out method (PureGene, Qiagen, Germantown, MD, USA) described in the manufacturer’s instructions. Exome sequence analysis was performed using the Nimblegen SeqCap EZ Library + UTR (Nimblegen/Roche Sequencing, Pleasanton, CA, USA) with the HiSeq 2500 sequencer (Illumina, San Diego, CA, USA). Image analyses and base calling were performed using RTA 1.18.64 and Casava 1.8.2. Reads were aligned to the NCBI Genome browser reference genome GRCh37, hg19 with Novoalign (Novocraft Technologies, Selangor, Malaysia). Samples were sequenced to sufficient coverage such that at least 85% of the targeted exome was called with high quality variant detection (reported as genotype at every callable position). Genotypes were called with only those sequence bases with Phred base qualities of at least Q20 via Most Probable Genotype3 (MPG) and an MPG score of ≥10 (Teer, 2010). Germline analyses of variants were performed and filters were applied with the VarSifter Next-Gen variant analysis software v1.7 (Teer, 2012). For filtering purposes both an autosomal-dominant mode of inheritance using the de novo trio model or an autosomal-recessive mode of inheritance were used as primary filters. Coding and splice site variants were further filtered based on frequency in gnomAD v2.1.1. Mosaic analysis of variants in GLI3 was performed with LoFreq v2.1.3.1 (Wilm, 2012).
RESULTS
Analyses of the exome data did not identify putative causative germline variants in GLI3 for either proband, confirming prior sanger sequence analysis results (Johnston, 2010). A variant of unknown significance was identified in GLI3 in Individual 2, NM_000168.6:c.3611C>G; p.Pro1204Arg, inherited from the unaffected father. Mosaicism analyses for variants in GLI3 coding sequence (average read depth 82) also failed to detect putative causative variants, however, with an average read depth of 82 it should be noted that detection of variants with a low variant allele fraction (<0.05) would likely not have been detected. Exome-wide analyses of germline variants, including analyses of variants in SMO previously implicated in individuals with syndromic hypothalamic hamartoma (Le, 2020; Green, 2022), also failed to identify any likely causative variants. Three variants consistent with de novo inheritance, and absent from gnomAD v2.1.1, were identified in Individual 1 and were classified as variants of uncertain significance (FOSB, chr19:45974199G>C. NM_006732.3:c.439G>C, p.(Glu147Gln); GBP5, chr1: 89729487_89729489del, NM_052942.5:c.1294_1296del, p.(Asn432del); GIMAP6, chr7:150327209G>T, NM_024711.6(GIMAP6):c.22C>A, p.(Gln8Lys). No variants that passed filters and were consistent with de novo inheritance were identified in Individual 2.
DISCUSSION
The Pallister-Hall syndrome (PHS) phenotype was described in 1980 by Judith Hall, Philip Pallister, and coworkers (Hall, 1980; Clarren, 1980) as a lethal syndrome of multiple congenital anomalies associated with a hypothalamic mass that had the histological features of a hamartoma. The associated craniofacial anomalies included cleft palate, cleft epiglottis and larynx, and multiple buccal frenula. The reported hand anomalies included postaxial or mesoaxial poly- or oligodactyly, small nails, cutaneous syndactyly, and clino- and camptodactyly. Renal malformations, imperforate anus, aganglionic megacolon, abnormal lung lobulation, and congenital heart disease were common associated internal malformations. Unrecognized panhypopituitarism and/or adrenal hypoplasia was thought to have resulted in high early mortality (Hall, 1980; Clarren, 1980). These original severely affected individuals showed considerable overlap with other multiple congenital anomaly syndromes, such as the oral-facial-digital syndromes (OFDS, in particular type VI, Varadi-Papp), hydrolethalus syndrome, Smith-Lemli-Opitz syndrome type II, some of the short-rib polydactyly syndromes, and the McKusick-Kaufmann syndrome, all of which have included some individuals with hypothalamic masses, before molecular confirmation was available for some of these conditions (reviewed in Verloes, 1992). This degree of overlap has complicated the nosological discussion in this group of disorders considerably (Verloes, 1995; Neri, 1995).
Less severely affected individuals with PHS have been reported since the initial description, with autosomal dominant transmission apparent in many families. These cases showed insertional and postaxial polysyndactyly, mostly in combination with the hypothalamic malformation, but with normal cognitive development and few, if any, additional malformations, such as imperforate anus (reviewed in Biesecker and Graham, 1996). Isolated growth hormone deficiency and precocious puberty have been the most common endocrinological abnormalities in mild PHS individuals; panhypopituitarism has also been described (Feuillan, 2001). Of note, no buccal or lingual frenula have been reported in any of these individuals, whereas laryngeal malformations have been common (Ondrey, 2000).
Pathogenic variants in the GLI3 gene were found initially in two large families with autosomal dominantly inherited PHS (Kang, 1997). Variants in GLI3 can also cause GLI3-related Greig cephalopolysyndactyly syndrome (MIM #175700), autosomal dominantly inherited GLI3-related postaxial polydactyly type A/B (MIM #174200) and GLI3-related preaxial polydactyly type IV (MIM #174700), all of which have polydactyly in common (Johnston, 2005). However, many of these individuals with apparently “isolated” polydactyly or polysyndactyly and associated GLI3 variants have not been examined for asymptomatic hypothalamic hamartomas, bifid epiglottis, or mild heart or kidney defect. Therefore, the distinction of syndromic from non-syndromic polydactyly in association with GLI3 variants is unclear (Biesecker, 2006). Isolated hypothalamic hamartomas have been shown to have somatic pathogenic variants in GLI3, which is consistent with germline variants causing autosomal dominantly inherited PHS (Saitsu, 2016). Recently, nine individuals have been described with features suggestive of autosomal recessively inherited PHS with bi-allelic variants in SMO (Le, 2020; Green, 2022). The reported individuals had diverse phenotypes including hypothalamic hamartoma although this was an inconsistent finding (5/9). Other reported features included postaxial polydactyly (9/9), microcephaly (3/9), dysmorphic facial features (5/9), cardiac defects (3/9) and shortening of long bones (3/9). Of note one individual was reported to have gingival frenula. These individuals do not meet the proposed clinical diagnostic criteria defined by a hypothalamic mass in combination with insertional polydactyly, most commonly of the third or fourth digit, as necessary for the clinical diagnosis of PHS in an index case (Biesecker, 1996). The diagnostic criteria proposed by Verloes et al (Verloes, 1995) emphasized, also, proximal synostosis of the axial metacarpals with mesoaxial polydactyly, short terminal phalanges, and small nails of several digits as necessary criteria for the diagnosis of PHS.
In contrast to individuals with typical PHS with autosomal dominant inheritance, the two individuals reported here presented with a hypothalamic hamartoma in combination with multiple buccal frenula and a short fifth finger with small nails and stiff interphalangeal joint, but no evidence of polydactyly or significant syndactyly. There were no laryngeal malformations. The clinical course was complicated by difficult-to-control seizures, hypogonadotropic hypogonadism, central hypothyroidism, and ACTH deficiency in the first individual and no neurological and only mild endocrinological abnormalities in the second. In contrast to the common endocrine finding of GH deficiency associated with hamartomas, both these individuals were growing well at their most recent examination. Since the parents were unaffected clinically and radiologically, there was no evidence for an autosomal dominant mode of transmission, although a de novo mutation in a dominantly acting gene is a possibility. However, a screen for GLI3 variants in the two individuals reported here did not identify pathogenic or likely pathogenic variants (Johnston, 2010). Additionally, analyses of exome data for mosaic variants in GLI3 did not identify any likely pathogenic variants in peripheral blood. It is important to recognize that the molecular pathogenesis of GLI3-related PHS is one of a frameshift or nonsense variant in a delimited region of the gene that does not induce nonsense-mediated decay, which upregulates expression of a protein that mimics the GLI3 repressor (PMID 20672375). Therefore, we reason that a non-coding variant of GLI3 that was not detected or recognized by our sequencing experiments is unlikely although we cannot rule this out and genome analysis could allow for further investigation of this possibility. As well, exome sequencing did not identify any good candidate variants in other genes including SMO and CPLANE1. This raises the question of where these two individuals belong from a nosological point of view.
In addition to PHS (the severe original description as well as the milder autosomal dominant form) there are other rare forms of syndromic hypothalamic hamartoma (Biesecker, 2003). Amongst the oro-facial-digital syndromes it has been seen in the type CPLANE1-related oral-facial-digital syndrome VI (OFD VI, Varadi-Papp, MIM# 277170)) (Stephan, 1994). Rarely it may be seen in Bardet Biedl syndrome (Diaz, 1991) and associated with SOX2 pathogenic variants (Kelberman, 2006). More recently bi-allelic variants in SMO have been identified in individuals with syndromic features including hypothalamic hamartoma (Le, 2020). Thus, there is clearly evidence for considerable genetic heterogeneity for syndromic hamartoma. We suggest that it is highly unlikely that the individuals described by us have undetected biallelic SMO variants.
Buccolingual or oral frenula are also seen in a variety of conditions, including most of the oro-facial-digital syndromes (Toriello, 1993; Neri, 1995), EVC-related Ellis-van Creveld Syndrome (MIM #225500) and EVC-related Weyer syndrome, and short-rib/polydactyly syndrome Beemer-Langer type (MIM #269860). Of these disorders, only the OFD VI phenotype has been reported with a hypothalamic hamartoma. However, for OFD VI, the cerebellar malformation (molar tooth malformation) and the typical Y-shaped metacarpal (osseous syndactyly) are more consistent findings. Neither of these findings was present in the individuals described here. Interestingly a single individual with bi-allelic variants in SMO has been described with gingival frenula although this individual was not reported to have a hypothalamic hamartoma (Le, 2020). Kantaputra (2003) reported a newly recognized syndrome of oral frenula with symphalangism and postaxial polydactyly, phenotypic features distinct from the individuals reported here. Guimiot et al (2009) reported individuals with large diencephalic hamartomas in association with widespread and severe CNS findings and facial clefting. Two of the individuals they reported also had dysplastic nails. The deformity of the fifth finger was a distinctive physical finding in each of affected infants reported here. The characteristics are: a shortened, stiff finger with clinodactyly and a tapered tip; there was limited flexion of the distal interphalangeal joint; the nail in one child (Figure 1D) extended beyond the tip of the finger. This appears to be a rare deformity, as no infants with a short, stiff fifth finger with an abnormal nail were identified in the active surveillance of 161,252 newborn infants (McGuirk, 2001). A similar stiffness of the fifth finger has been observed in some infants exposed in utero to chorionic villus sampling. However, those children usually have stiffness or a distal deficiency of other fingers as well (Golden, 2003).
The precise nosological status of the individuals reported here must remain open, based on current evidence. They are clearly distinct from the typical autosomal dominantly-inherited GLI3-related PHS and do not conform to the published clinical diagnostic criteria (Biesecker, 1996). While molecular analysis of the GLI3 gene did not identify pathogenic variants in either of the two individuals, we cannot exclude them with certainty. However, as noted above, given the mechanism of disease, we believe that non-coding GLI3 variants are very unlikely the cause of the anomalies in these two individuals. The individuals reported here also do not conform to one of the alternate conditions associated with syndromic hypothalamic hamartoma. As an alternative possibility, we suggest that they have a distinct mild syndromal hypothalamic hamartoma syndrome, which could be inherited in an autosomal recessive or autosomal dominant pattern. As exome sequence analyses did not identify likely causative variants in either individual, consideration could be given to genome sequencing allowing for interrogation of non-coding regions as well as more complete coverage of the exome. Gene identification in the other syndromic hypothalamic hamartoma syndromes will be required to investigate whether these individuals could represent the mild end of a spectrum of such a more severe syndrome. Both the multiple buccal frenula and the peculiar abnormality of the fifth finger are quite unusual in isolation, and in combination constitute a recognizable pattern. Early clinical recognition of this syndrome in an otherwise asymptomatic individual is important to anticipate potential endocrinological and neurological complications.
In summary, we describe two infants with syndromic hypothalamic hamartomas, with digital anomalies distinct from GLI3-related PHS and an absence of other malformations characteristic of that syndrome. In addition, neither of these individuals have variants in GLI3 assessed as pathogenic or likely pathogenic, nor was a candidate variant determined by exome sequencing in another gene. We suggest that this syndrome is clinically and molecularly distinct from PHS and comprises a recognizable pleiotropic malformation syndrome.
ACKNOWLEDGEMENTS
The authors thank the families for their participation in this work. Exome sequence analyses were performed at the NIH Intramural Sequencing Center. Mosaic sequence analyses were performed by Henoke Shiferaw. We appreciate their assistance.
FUNDING
JJJ and LGB are supported by the Intramural Research Program of the National Human Genome Research Institute, NIH Grant HG200328-16.
CONFLICT OF INTEREST
LGB is a member of the Illumina Corp. medical ethics board, receives research funding from Merck, Inc., and is a compensated editor for Cold Spring Harbor Laboratory Press.
DATA AVAILABILITY
The exome data will be deposited with dbGAP per NIH policy.
REFERENCES
- Biesecker LG, & Graham JM Jr. (1996). Pallister-Hall syndrome. J Med Genet. 33:585–589. doi: 10.1136/jmg.33.7.585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker LG (2003). Heritable syndromes with hypothalamic hamartoma and seizures: using rare syndromes to understand more common disorders. Epileptic Disord. 5:235–238 [PubMed] [Google Scholar]
- Biesecker LG, Abbott M, Allen J, Clericuzio C, Feuillan P, Graham J. Mm Jr, Hall J, Kang S, Olney AH, Lefton D, Neri G, Peters K, Verloes A (1996). Report from the workshop on Pallister-Hall syndrome and related phenotypes. Am J Med Genet. 65:76–81. doi: [DOI] [PubMed] [Google Scholar]
- Biesecker LG (2006). What you can learn from one gene: GLI3. J Med Genet. 43:465–469. doi: 10.1136/jmg.2004.029181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarren SK, Alvord EC Jr, Hall JG (1980). Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly--a new syndrome? Part II: neuropathological considerations. Am J Med Genet 7:75–83. doi: 10.1002/ajmg.1320070111 [DOI] [PubMed] [Google Scholar]
- Cleper R, Kauschansky A, Varsano I, Frydman M (1993). Varadi syndrome (OFD VI) or Opitz trigonocephaly syndrome: overlapping manifestations in two cousins. Am J Med Genet 47:451–455. doi: 10.1002/ajmg.1320470402 [DOI] [PubMed] [Google Scholar]
- Feuillan P, Peters KF, Cutler GB Jr, Biesecker LG (2001). Evidence for decreased growth hormone in patients with hypothalamic hamartoma due to Pallister-Hall syndrome. J Pediatr Endocrinol Metab 14:141–149. doi: 10.1515/jpem.2001.14.2.141 [DOI] [PubMed] [Google Scholar]
- Foster MG, Sizonenko PC, Cathiard AM, Bertrand J (1974). Hypophysogonadal function in humans during the first year of life. 1. Evidence for testicular activity in early infancy. J Clin Invest 53:819–828. doi: 10.1172/JCI107621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden CM, Ryan LM, Holmes LB (2003). Chorionic villus sampling: a distinctive teratogenic effect on fingers? Birth Def Res (Part A): Clin Mol Teratol 67:556–562. doi: 10.1002/bdra.10078 [DOI] [PubMed] [Google Scholar]
- Graham J,M Jr, Harris M, Frank JE, Little GA, Klein RZ (1985). Congenital hypothalamic hamartoblastoma syndrome: natural history and genetic implications. Prog Clin Biol Res 200:163–174. [PubMed] [Google Scholar]
- Graham JM, Perl D, O’Keefe T, Rawnsley E, Little GA (1983). Apparent familial recurrence of hypothalamic hamartoblastoma syndrome. (Abstract) Proc. Greenwood Genet Center 2:117–118. [Google Scholar]
- Green TE, Schimmel M, Schubert S, Lemke JR, Bennett MF, Hildebrand MS, Berkovic SF. (2022) Bi-allelic SMO variants in hypothalamic hamartoma: a recessive cause of Pallister-Hall syndrome. Eur J Hum Genet. 30:384–388. doi: 10.1038/s41431-021-01023-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimiot F, Marcorelles P, Aboura A, Bonyhay G, Patrier S, Menez F, Drouin-Garraud V, Icowick V, Eurin D, Garel C, Moirot H, Verspyck E, Saugier-Veber P, Attie-Bitach T, Picone O, Oury JF, Verloes A, Delezoide AL, Laquerrière A. (2009). Giant diencephalic harmartoma and related anomalies: a newly recognized entity distinct from the Pallister-Hall syndrome. Am J Med Genet A 149A:1108–1115. doi: 10.1002/ajmg.a.32859 [DOI] [PubMed] [Google Scholar]
- Hall JG, Pallister PD, Clarren SK, Beckwith JB, Wigglesworth FW, Fraser FC, Cho S, Benke PJ, Reed SD. (1980). Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly--a new syndrome? Part I: clinical, causal, and pathogenetic considerations. Am J Med Genet 7:47–74. doi: 10.1002/ajmg.1320070110 [DOI] [PubMed] [Google Scholar]
- Johnston JJ, Olivos-Glander I, Killoran C, Elson E, Turner JT, Peters KF, Abbott MH, Aughton DJ, Aylsworth AS, Bamshad MJ, Booth C, Curry CJ, and 36 others. (2005). Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. Am J Hum Genet 76: 609–622. doi: 10.1086/429346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Sapp JC, Turner JT, Amor D, Aftimos S, Aleck KA, Bocian M, Bodurtha JN, Cox GF, Curry CJ, Day R, Donnai D, Field M, Fujiwara I, Gabbett M, Gal M, Graham JM, Hedera P, Hennekam RC, Hersh JH, Hopkin RJ, Kayserili H, Kidd AM, Kimonis V, Lin AE, Lynch SA, Maisenbacher M, Mansour S, McGaughran J, Mehta L, Murphy H, Raygada M, Robin NH, Rope AF, Rosenbaum KN, Schaefer GB, Shealy A, Smith W, Soller M, Sommer A, Stalker HJ, Steiner B, Stephan MJ, Tilstra D, Tomkins S, Trapane P, Tsai AC, Van Allen MI, Vasudevan PC, Zabel B, Zunich J, Black GC, Biesecker LG. (2010) Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum Mutat. 31:1142–54. doi: 10.1002/humu.21328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Graham JM Jr, Olney AH, Biesecker LG. (1997). GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nature Genet 15: 266–268. doi: 10.1038/ng0397-266 [DOI] [PubMed] [Google Scholar]
- Kantaputra PN, Pongprot Y, Praditsap O, Pho-iam T, Limwongse C. (2003). A new syndrome of symphalangism, multiple frenula, postaxial polydactyly, dysplastic ears, dental anomalies, and exclusion of NOG and GDF5. Am J Med Genet 120A: 381–385. doi: 10.1002/ajmg.a.20040 [DOI] [PubMed] [Google Scholar]
- Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson IC, Dattani MT. (2006). Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest 116:2442–2455. doi: 10.1172/JCI28658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TL, Sribudiani Y, Dong X, Huber C, Kois C, Baujat G, Gordon CT, Mayne V, Galmiche L, Serre V, Goudin N, Zarhrate M, Bole-Feysot C, Masson C, Nitschké P, Verheijen FW, Pais L, Pelet A, Sadedin S, Pugh JA, Shur N, White SM, El Chehadeh S, Christodoulou J, Cormier-Daire V, Hofstra RMW, Lyonnet S, Tan TY, Attié-Bitach T, Kerstjens-Frederikse WS, Amiel J, Thomas S. (2020) Bi-allelic Variations of SMO in Humans Cause a Broad Spectrum of Developmental Anomalies Due to Abnormal Hedgehog Signaling. Am J Hum Genet. Jun 4;106:779–792. doi: 10.1016/j.ajhg.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuirk CK, Westgate M-N, Holmes LB. (2001). Limb deficiencies in newborn infants. Pediatrics 108:e64–71. doi: 10.1542/peds.108.4.e64 [DOI] [PubMed] [Google Scholar]
- Neri G, Gurrieri F, Genuardi M. (1995). Oral-facial-skeletal syndromes. Am J Med Genet 59:365–368. doi: 10.1002/ajmg.1320590317 [DOI] [PubMed] [Google Scholar]
- Ondrey F, Griffith A, Van Waes C, Rudy S, Peters K, McCullagh L, Biesecker LG. (2000). Asymptomatic laryngeal malformations are common in patients with Pallister-Hall syndrome. Am J Med Genet 94:64–67. doi: [DOI] [PubMed] [Google Scholar]
- Saitsu H, Sonoda M, Higashijima T, Shirozu H, Masuda H, Tohyama J, Kato M, Nakashima M, Tsurusaki Y, Mizuguchi T, Miyatake S, Miyake N, Kameyama S, Matsumoto N. (2016). Somatic mutations in GLI3 and OFD1 involved in sonic hedgehog signaling cause hypothalamic hamartoma. Ann Clin Transl Neurol. 3:356–65. doi: 10.1002/acn3.300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan MJ, Brooks KL, Moore DC, Coll EJ, Goho C. (1994). Hypothalamic hamartoma in oral-facial-digital syndrome type VI (Varadi syndrome). Am J Med Genet 51:131–136. doi: 10.1002/ajmg.1320510209 [DOI] [PubMed] [Google Scholar]
- Teer JK, Bonnycastle LL, Chines PS, Hansen NF, Aoyama N, Swift AJ, Abaan HO, Albert TJ; NISC Comparative Sequencing Program, Margulies EH, Green ED, Collins FS, Mullikin JC, Biesecker LG. (2010). Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res 20:1420–1431. doi: 10.1101/gr.106716.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teer JK, Green ED, Mullikin JC, Biesecker LG. (2012). VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 15;28:599–600. doi: 10.1093/bioinformatics/btr711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriello HV. (1993). Oral-facial-digital syndromes. (1992). Clin Dysmorph 2:95–105. [PubMed] [Google Scholar]
- Verloes A, David A, Ngo L, Bottani A. (1995). Stringent delineation of Pallister-Hall syndrome in two long surviving patients: importance of radiological anomalies of the hands. J Med Genet 32:605–611. doi: 10.1136/jmg.32.8.605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verloes A, Gillerot Y, Langhendries JP, Fryns JP, Koulischer L. (1992). Variability versus heterogeneity in syndromal hypothalamic hamartoblastoma and related disorders: review and delineation of the cerebro-acro-visceral early lethality (CAVE) multiplex syndrome. Am J Med Genet 43:669–677. doi: 10.1002/ajmg.1320430404 [DOI] [PubMed] [Google Scholar]
- Verloes A. (1995). Numerical syndromology: a mathematical approach to the nosology of complex phenotypes. Am J Med Genet 55:433–443. doi: 10.1002/ajmg.1320550 [DOI] [PubMed] [Google Scholar]
- Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. (2012). LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40:11189–11201. doi: 10.1093/nar/gks918 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The exome data will be deposited with dbGAP per NIH policy.