

Abstract
Diffuse large B-cell lymphoma (DLBCL) can be subdivided into activated B-cell like (ABC) and germinal center B-cell-like (GCB) DLCBL. Self-antigen engagement of B-cell receptors (BCRs) in ABC tumors induces their clustering, thereby initiating chronic active signaling and activation of NF-κB and PI3 kinase. Constitutive BCR signaling is essential in some GCB tumors but primarily activates PI3 kinase. We devised genome-wide CRISPR-Cas9 screens to identify regulators of IRF4, a direct transcriptional target of NF-κB and an indicator of proximal BCR signaling in ABC DLBCL. Unexpectedly, inactivation of N-linked protein glycosylation by the oligosaccharyltransferase-B (OST-B) complex reduced IRF4 expression. OST-B inhibition of BCR glycosylation reduced BCR clustering and internalization while promoting its association with CD22, which attenuated PI3 kinase and NF-κB activation. By directly interfering with proximal BCR signaling, OST-B inactivation killed models of ABC and GCB DLBCL, supporting the development of selective OST-B inhibitors for the treatment of these aggressive cancers.
Keywords: B cell lymphoma, DLBCL, B cell receptor, glycosylation, CD22, TLR9, super-resolution microscopy, glycoproteomics
Introduction
Diffuse large B-cell lymphoma (DLBCL) is an aggressive cancer that accounts for approximately 30% of lymphoma cases. Gene expression profiling revealed two major biological subtypes, termed activated B-cell like (ABC) and germinal center B-cell-like (GCB) DLCBL (1), which have subdivided further by multi-platform genomic profiling into 7 genetic subtypes (2–5). DLBCL tumors frequently rely on constitutive B cell receptor (BCR) signaling to trigger a cascade of downstream kinases and adapters, culminating in NF-κB and/or PI3 kinase activation (6). However, the mechanisms responsible for this pathological BCR signaling differ in the molecular subtypes of DLBCL (7). ABC tumors rely on self-antigen-induced clustering of the BCR, invoking chronic active signaling to downstream survival pathways (8). A genetic subset of ABC DLBCL, termed MCD, is enriched for the MYD88L265P mutant isoform that associates with the BCR and TLR9 in an endolysosomal multiprotein complex – the My-T-BCR – that triggers NF-κB activation (9). Another ABC genetic subtype, BN2, is also BCR-dependent but lacks MYD88L265P and does not depend on TLR9 (9). By contrast, constitutive BCR signaling in GCB DLBCL is antigen-independent and triggers PI3 kinase but not NF-κB signaling (3). Because of these and other mechanistic differences in BCR signaling, therapeutic agents targeting downstream NF-κB and PI3 kinase activation have clinical activity in some DLBCL subtypes but not others (7), prompting us to search for strategies to target proximal BCR signaling directly. Using CRISPR-Cas9 screens, proteomics, and super-resolution microscopy, we discovered a novel, glycosylation-dependent pathway that inhibits oncogenic BCR signaling, providing an unexpected new therapeutic avenue for both ABC and GCB DLBCL.
Results
Genome-wide IRF4-GFP knock-in CRISPR-Cas9 screens
To uncover novel therapeutic vulnerabilities in DLBCL, we employed genome-wide CRISPR-Cas9 screens to identify regulators of IRF4, a direct NF-κB target gene and robust marker of BCR signaling in ABC DLBCL. Specifically, we used CRISPR-Cas9-mediated genome editing to knock in a GFP-P2A-CD34 cassette into the IRF4 locus, creating an IRF4-GFP fusion protein that is expressed under control of endogenous regulatory elements (Fig.S1A). We studied five IRF4-expressing lymphoma lines that we previously engineered for doxycycline-inducible Cas9 expression (9,10), including three ABC DLBCL models (TMD8, HBL1 and RIVA) that express IRF4 in a BCR-dependent fashion and two models of adult T-cell lymphoma (ST1, SU9T01) in which IRF4 is essential but not BCR-dependent (11). We validated expression of the IRF4-GFP fusion proteins by immunoblot (Fig.S1B) and FACS (Fig.S1C), and their nuclear localization by confocal microscopy (Fig.S1D). As expected, treatment of the ABC models with the BTK kinase inhibitor acalabrutinib attenuated IRF4-GFP expression (Fig.S1E).
The IRF4-GFP knock-in lines were transduced with the “Brunello” library of lentiviruses that express single guide RNAs (sgRNAs) targeting most protein-coding genes (12) (Fig.1A). One week after Cas9 induction, the transduced cells were sorted by FACS into GFPhigh and GFPlow subpopulations (Fig.S2A–B) and the abundance of each sgRNA in each subpopulation was determined by high-throughput sequencing (Supplementary table 1). We gauged the relative degree of depletion or enrichment of sgRNAs in each subpopulation using a Z-score-normalized metric termed the CRISPR screen score (CSS), as described (9). Additionally, we defined a “segregation score” as the difference between the GFPhigh and GFPlow CSS values, with negative values identifying genes that reduced IRF4-GFP expression when inactivated (Supplementary table 2). sgRNAs targeting pan-essential genes as well as non-targeting control sgRNAs were neither enriched nor depleted in either subpopulation (Fig.S2C–D), while IRF4 sgRNAs were significantly depleted in IRF4-GFPhigh cells and enriched in IRF4-GFPlow cells, as expected (Fig.1B). In the ABC DLBCL models, knockout of BCR pathway components decreased IRF4 expression as expected, including BCR subunits and BCR-proximal signaling mediators (CD79A, CD79B, BLNK, SYK, BTK, PLCG2), the IκB kinase complex (IKBKB, IKBKG, CHUK), PI3K/mTOR pathway members (PIK3CD, MTOR, RPTOR, RICTOR), and MYD88. Conversely, IRF4 was upregulated by inactivation of negative regulators of BCR signaling (GRB2, SLA, PRKCD) or NF-κB signaling (NFKBIA, TRAF2, CYLD, TNFAIP3 and TNIP1) (Fig.1B). In ATLL models, IRF4 expression decreased upon inactivation of the transcription factor BATF3, as well as signaling mediators in the PI3K-MTOR (PIK3CA, PDPK1, RRAGC, LAMTOR1, LAMTOR2, LAMTOR3, RICTOR) and JAK-STAT (JAK1, STAT3) pathways, whereas inactivation of the BCR or NF-κB pathway components had no effect (Fig.S2E).
Figure 1. IRF4 knock-in CRISPR screens reveal the OST-B complex as a regulator of NF-κB and IRF4.
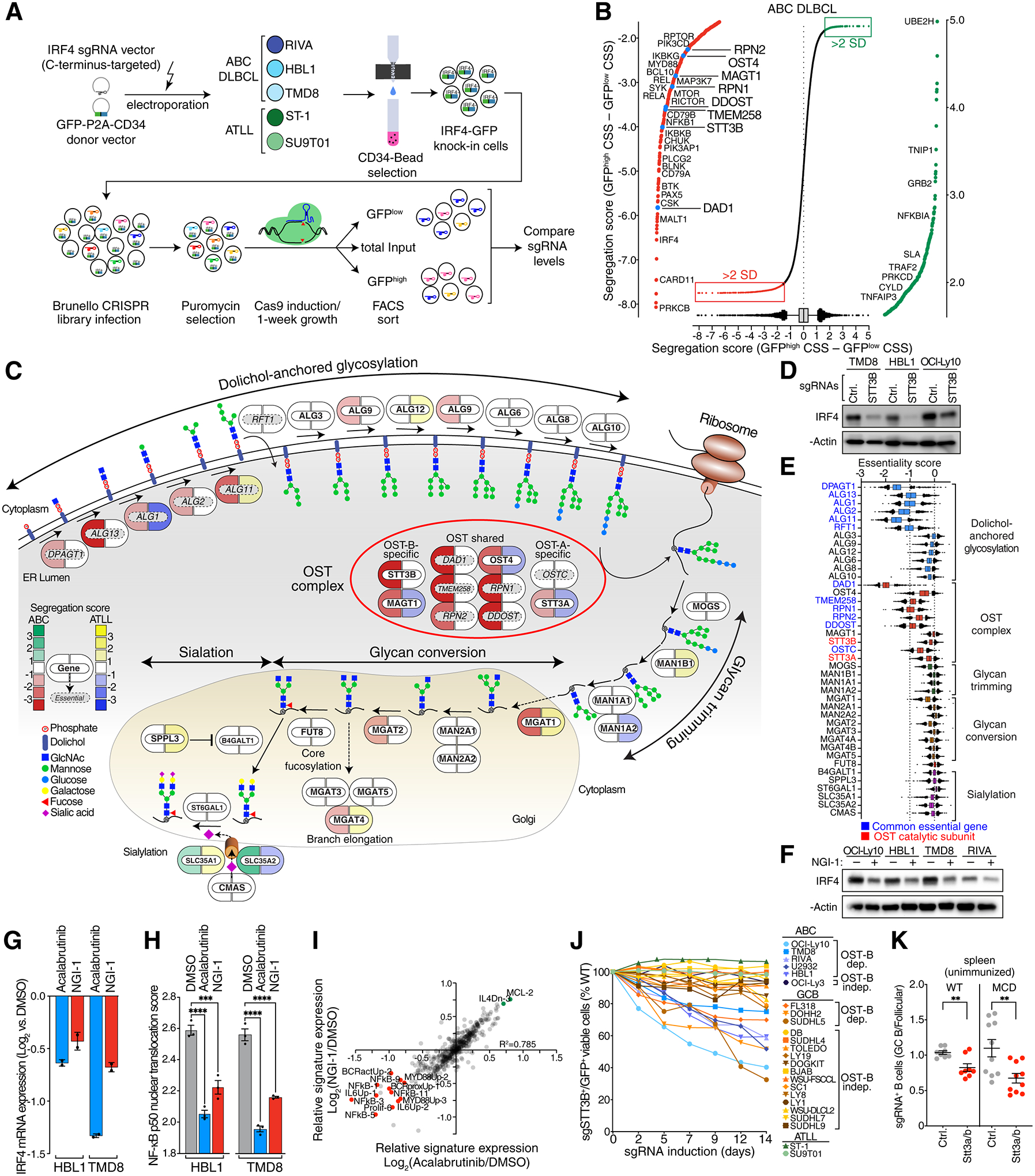
A. Scheme and workflow of IRF4 knock-in CRISPR screens. B. Ranked list of genes by their segregation score (CSS GFPhigh – CSS GFPlow) with their 5–95% percentile (box and whiskers) averaged from three ABC DLBCL lines (TMD8, HBL1 and RIVA). Outliers (>2 SD) with negative segregation scores (decreased IRF4-GFP when inactivated) are highlighted in red and outliers (>2 SD) with positive segregation scores (increased IRF4-GFP when inactivated) in green. Highlighted are OST subunits (blue) as well as regulators of oncogenic BCR, PI3 kinase and NF-κB signaling. CSS: CRISPR screen score. C. Pill diagram of the N-linked glycosylation, fucosylation and sialylation pathway. Icons indicate effects on IRF4 protein levels assessed by IRF4-GFP CRISPR screens in ABC cells (left, red to green) or ATLL cells (right, blue to orange). Panessential genes (by DepMap) are marked in italic/grey. D. Immunoblots for the indicated proteins in three ABC cell lines (TMD8, HBL1 and OCI-Ly-10) with knockout of STT3B. E. DepMap essentiality scores of genes involved in N-linked glycosylation, fucosylation and sialylation. Common essential genes are highlighted in blue, OST catalytic subunits in red. F. Immunoblots for the indicated proteins in four ABC cell lines (OCI-Ly10, HBL1, TMD8 and RIVA) after 24h NGI-1 treatment. G. Relative IRF4 mRNA levels (RNA-seq) in HBL1 and TMD8 cells treated with acalabrutinib (ACAL) or NGI-1 for 24h, compared to DMSO-treated cells (n=2). H. Mean nuclear NF-κB (p50) translocation score assessed by ImageStream flow cytometry in HBL1 and TMD8 cells treated for 16h with DMSO, acalabrutinib (ACAL) or NGI-1. ***P ≤ 0.001, ****P ≤ 0.0001 (one-way ANOVA). Data from three independent replicates. Error bars represent SEM. I. Scatter plot of gene expression signature averages in TMD8 cells treated for 24h with acalabrutinib (x-axis) or NGI-1 (y-axis), relative to DMSO-treated cells. Signatures that decreased (red) or increased (green) by 0.7 log2 fold or more are highlighted. J. Toxicity of STT3B knockout in ABC DLBCL, GCB DLBCL and in adult T-cell leukemia/lymphoma (ATLL) lines. Viable cell numbers are normalized to day 0. Error bars represent SEM. dep.: dependent; indep.: independent. K. Ratio of frequency Stt3a/b sgRNA positive GCBs to Stt3a/b sgRNA positive FoB in wildtype (WT) radiation chimeras or MCD radiation chimeras in unimmunized spleen. Data are pooled from 2 independent experiments representative of 4 with 5 mice per group. **P ≤ 0.01 (unpaired two-tailed t test).
Unexpectedly, IRF4 expression in ABC but not ATLL cells was significantly diminished by inactivation of the N-linked oligosaccharyltransferase (OST) complex (Fig.1B, 1C). OST resides in the endoplasmic reticulum (ER) lumen and is responsible for the transfer of a core glycan (Glc3Man9GlcNAc2) to asparagine residues of membrane and secreted proteins (13). Mammals express two distinct OST complexes, OST-A and OST-B, that share a 6-subunit core (DAD1, OST4, TMEM258, RPN1, RPN2, DDOST) but have paralogous catalytic subunits: STT3A in OST-A and STT3B in OST-B. Additionally, OST-A uniquely includes DC2 (OSTC), which interacts with the ER translocon complex, whereas OST-B uniquely includes MAGT1, which functions as a redox chaperone. Of note, the OST-A and OST-B complexes have non-redundant functions: they modify distinct asparagines in their protein targets (14), presumably because OST-A acts as the nascent target protein translocates to the ER lumen while OST-B acts at a later post-translational step in the ER (15).
In ABC but not ATLL cells, all subunits of the OST-B complex were essential for high IRF4 expression (Fig.1C). Compared with the OST-B-specific subunits STT3B and MAGT1, knockout of the OST-A-specific subunits STT3A and OSTC had less effect on IRF4 expression. Some pathway enzymes involved in dolichol glycosylation upstream of the OST complexes also contributed to IRF4 expression, whereas enzymes acting downstream of the OST complexes to modify the N-linked glycan had little if any effect on IRF4 expression. These data suggest that IRF4 expression is primarily sensitive to removal of the entire glycan chain that is attached by OST-B to its target proteins. Immunoblot analysis of ABC cells transduced with an sgRNA for STT3B confirmed its requirement for full IRF4 expression (Fig.1D).
Since N-linked glycosylation contributes to diverse biological processes, we examined whether OST components were essential for the proliferation and/or survival of cell line models derived from diverse developmental lineages using the DepMap database. While some N-linked glycosylation pathway components were pan-essential across the DepMap cell lines, the catalytic subunits STT3A and STT3B were not essential in most non-lymphoid lines, perhaps due to partial redundancy in their function (Fig.1E).
To validate our results pharmacologically, we used the OST inhibitor NGI-1 that preferentially inhibits STT3B, with a lesser effect on STT3A (16,17). NGI-1 decreased IRF4 protein levels (Fig.1F) and consistently downregulated IRF4 mRNA levels in ABC lines, as did the BTK inhibitor acalabrutinib (Fig.1G). Since NF-κB directly transactivates IRF4 in ABC cells, we tested whether OST inhibition altered NF-κB activation. As measured by imaging flow cytometry, the nuclear translocation of the NF-κB subunit p50 was significantly decreased by NGI-1 treatment of ABC cells (Fig.1H). Gene expression signature analysis revealed a strong correlation between the signatures altered in expression by treatment of ABC cells with NGI-1 or acalabrutinib, including signatures of NF-κB activity and proximal BCR signaling, supporting the notion that OST inhibition impairs chronic active BCR signaling in ABC DLBCL (Fig.1I).
To assess the role of OST-B more broadly, we evaluated the toxicity of STT3B inactivation in a panel of 22 ABC and GCB DLBCL lines. In 5 ABC lines, knockout of STT3B was toxic (Fig.1J, S3A). Notably, inactivation STT3B had no effect on the viability of the ABC line OCI-Ly3, in which a gain-of-function CARD11 mutant isoform strongly activates NF-κB, rendering cells independent of BCR signaling (18,19). Some GCB DLBCLs rely on constitutive BCR signaling but unlike ABC tumors, the downstream consequence is typically activation of PI3 kinase and not NF-κB (7). A survey of 16 GCB lines revealed 3 for which STT3B knockout was toxic, which we operationally term “OST-dependent”, and 13 that were less affected, which we term “OST-independent” (Fig.1J, S3A). Double knockout of STT3A and STT3B was more toxic for OST-dependent lines than single knockout of STT3B, suggesting a partially non-redundant role for these two catalytic subunits (Figs.S3B).
MCD DLBCL is thought to be derived from germinal center B cells arising spontaneously in the spleen, based on “MCD” mice engineered to model the genetic abnormalities of this genetic subtype (20,21). To study the role of the OST complex in spontaneous splenic germinal centers in vivo, we analyzed radiation chimeras reconstituted with bone marrow from MCD mice or control mice engineered to conditionally express Cas9 in germinal center B cells (Fig.S4A). Transduction of bone marrow with sgRNAs targeting Stt3a and Stt3b (21) decreased spontaneous splenic germinal centers in both mouse genotypes (Fig.1K, Fig.S4B–D), suggesting a critical role of the OST complex in the putative cell of origin of MCD DLBCL.
BCR glycosylation regulates its function
To investigate the mechanisms responsible for OST-dependency in lymphoma, we identified proteins that are glycosylated by OST-A and OST-B and are also critical for lymphoma cell viability, which we term the “essential glycoproteome”. We performed mass spectrometry-based glycoproteomics in STT3A- or STT3B-deficient HBL1 and TMD8 ABC cells and quantified global changes in N-linked glycosylation sites (glycosites, Supplementary table 3). We compared glycosite changes with the degree to which each glycoprotein was essential for lymphoma cell growth and survival in CRISPR-Cas9 screens (9) (Fig.S5A; Supplementary table 4,5). As expected, most OST-A or OST-B dependent glycoproteins were integral membrane proteins, the majority of which were not essential for the proliferation and survival of ABC cells (Fig.S5A). Among the essential genes, two glycosites in CD79B and one in CD79A were reduced in abundance by STT3B but not STT3A inactivation (Fig.2A, S5A). Conversely, three glycosites on TLR9 were reduced in STT3A but not STT3B knockout cells. Similarly, NGI-1 treatment of HBL1 and TMD8 ABC lines reduced glycosylation of these glycosites but additionally diminished the glycosylation of two glycosites in IgM, suggesting that their glycosylation may depend on both STT3A and STT3B (Fig.2B, S5B, Supplementary table 6).
Figure 2. OST-mediated BCR glycosylation regulates its plasma membrane clustering and signaling.
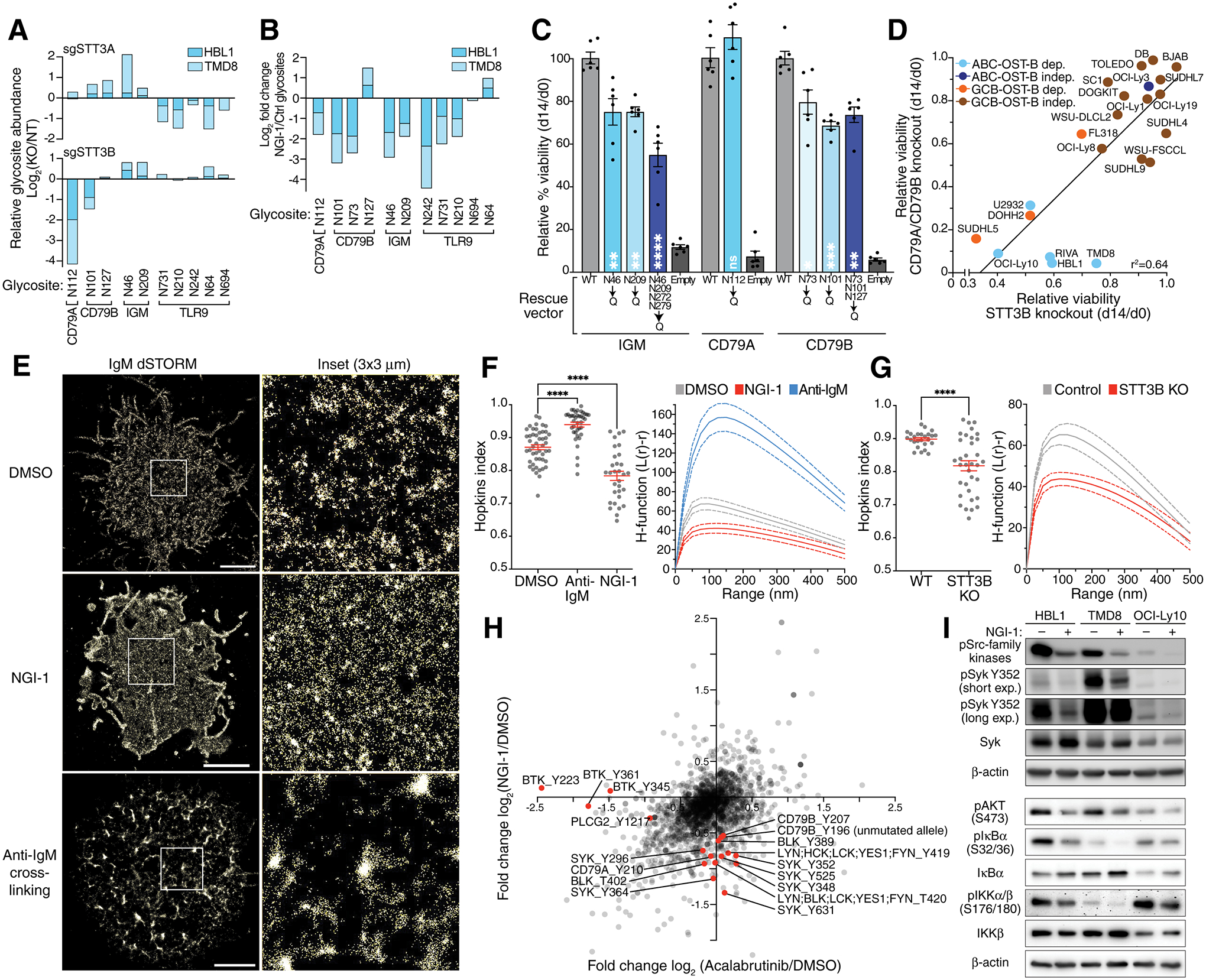
A. Relative glycosite log2 fold changes of the B cell receptor components (CD79A, CD79B and IGM) and TLR9 in HBL1 and TMD8 cells after knockout of STT3A (upper panel) or STT3B (lower panel) assessed by quantitative mass spectrometry. B. Relative glycosite log2 fold changes of the B cell receptor components (CD79A, CD79B and IGM) and TLR9 in HBL1 and TMD8 cells after treatment with NGI-1 assessed by quantitative mass spectrometry. C. Relative rescue capacity (d14 viability/d0 viability) of IgM, CD79A or CD79B wildtype or the indicated mutant Isoforms following knockout of endogenous IgM, CD79A or CD79B. Mean of six replicates (± SEM) is displayed. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001 (one-way ANOVA) compared to WT. D. Scatter plot of toxicity of sgRNAs targeting STT3B on the x-axis compared to the mean toxicity of sgRNAs targeting CD79A and CD79B (y-axis) in the indicated cell lines. E. Reconstructed dSTORM single molecule localizations in TMD8 cells stained with an anti-IgM antibody. White square area is shown magnified (right). Scale bar is 3μm. Representative image of 3 or more experiments. F. Quantification of the IgM distribution assessed by dSTORM in using the Hopkin’s index (left panel) and the H-function (right panel) in TMD8 cells treated for 15 min with a full-length anti-IgM antibody or for 24h with NGI-1 as indicated. Error bars (Hopkins index) and dotted lines (H-function) denote the SEM. Data are pooled from three independent experiments with at least 9 cells per experiment. ****P ≤ 0.0001 (one-way ANOVA). G. Quantification of the IgM distribution assessed by dSTORM in using the Hopkin’s index (left panel) and the H-function (right panel) in TMD8 cells after knockout of STT3B. Error bars (Hopkins index) and dotted lines (H-function) denote the SEM. Data are pooled from three independent experiments with at least 9 cells per experiment. ****P ≤ 0.0001 (one-way ANOVA). H. Changes in global phosphotyrosine abundance in HBL1 and TMD8 cells treated for 16h with acalabrutinib (x-axis) or NGI-1 (y-axis). I. Immunoblots using the indicated antibodies in HBL1, TMD8 and OCI-Ly10 cells treated with NGI-1 as indicated for 16h.
Immunoblot analysis demonstrated that inactivation of STT3B decreased the apparent molecular weight of CD79A and CD79B, consistent with deglycosylation, while STT3A inactivation alone had no apparent effect (Fig.S5C). However, inactivation of both STT3A and STT3B decreased the apparent molecular weight further, suggesting that both catalytic subunits participate in the glycosylation of these proteins. Likewise, knockdown of both STT3A and STT3B was required to produce a detectable shift in the molecular weight of IgM. NGI-1 treatment of three ABC lines decreased the apparent molecular weights of all three BCR subunits (Fig.S5D). Treatment of cell lysates with Peptide:N-glycosidase F (PNGase F), which cleaves the entire N-linked glycan at its attachment to asparagine, further decreased the molecular weight of CD79A and CD79B, suggesting that NGI-1 does not fully deglycosylate these proteins. Conversely, IgM appeared to be fully deglycosylated by NGI-1 treatment by this criterion (Fig.S5D). Together, these data suggests that both OST-A and OST-B contribute to the glycosylation of BCR subunits, with OST-B being dominant in CD79A and CD79B glycosylation and OST-A dominant in TLR9 glycosylation.
To demonstrate that glycosylation of BCR subunits plays an essential role in ABC cells, we created mutant isoforms of CD79A, CD79B and IgM in which particular asparagine glycosite residues were changed to glutamine. We tested whether these mutant isoforms could rescue the survival of cells in which the corresponding genes were inactivated using an sgRNA that targets the endogenous but not ectopic alleles. Relative to wild type IgM, the N46Q and N209Q isoforms were significantly less effective, and a quadruple mutant isoform (N46Q, N209Q, N272Q, N279Q) was further compromised (Fig.2C). Likewise, CD79B N73Q and N101Q as well as a triple mutant isoform (N73Q, N101Q, N127Q) were significantly inferior to wild type CD79B in their rescue ability, whereas the CD79A N112Q isoform was equivalent to wild type CD79A. Importantly, these mutant isoforms trafficked to the plasma membrane equivalently as judged by FACS analysis (Fig.S5E).
The essential role of N-linked glycosylation of the BCR in survival of ABC cells suggested that the effect of OST inhibition might be greatest in DLBCL models that rely upon constitutive BCR signaling for survival. Indeed, in a survey of ABC and GCB lines, we observed a good correlation (r2=0.64) between the toxicity of CD79A or CD79B inactivation and the toxicity of STT3B inactivation (Fig.2D, S5F). Of note, the IC50 values of NGI-1 in HBL1 and TMD8 ABC cells were 4–7-fold lower than the IC50 in the lung cancer line PC-9, which was previously reported to be OST-dependent (16) (Fig.S5G). Together, these findings highlight the exceptional dependence of DLBCL cells with constitutive BCR signaling on the OST complex, particularly OST-B.
One hypothesis we entertained was that OST inhibition might interfere with the trafficking of the BCR to the plasma membrane, as is the case for the epidermal growth factor receptor in NGI-1-treated lung cancer lines (16). However, FACS analysis revealed no reduction in CD79A, CD79B or IgM levels on the cell surface upon OST inhibition (Fig.S6A). In fact, surface BCR levels increased after 24 hours of NGI-1 treatment, potentially due to increased CD79A and CD79B mRNA expression at this time point (Fig.S6B) and/or to reduced BCR internalization (see below).
We next used super-resolution microscopy to test the hypothesis that deglycosylation of the BCR might alter its clustering on the surface of ABC cells (19,22). ABC cells were stained with an anti-IgM Fab conjugated to Alexa Fluor 647 and imaged using direct stochastic optical reconstruction microscopy (dSTORM) in total internal reflection fluorescence (TIRF) mode. The rendered localizations were analyzed by two different metrics: the Hopkins index (23), which measures the clustering tendency of the receptors and ranges from 0.5 (random) to 1.0 (fully clustered), and the H-function, a nearest neighbor metric derived from Ripley’s K function that estimates clustering tendency and cluster size (23,24). IgM was indeed highly clustered on the ABC cell surface (Fig.2E) and NGI-1 treatment dispersed these clusters, as judged by the Hopkins index and the H-function (Fig.2F). As expected, BCR crosslinking using an anti-IgM antibody increased clustering (Fig. 2E–F). Notably, BCR surface clustering also decreased significantly in ABC cells in which STT3B had been inactivated genetically, implicating OST-B as a key regulator of chronic active BCR signaling in ABC DLBCL (Fig.2G).
To investigate the consequence of BCR reorganization on downstream signaling, we measured tyrosine phosphorylation changes after treatment with NGI-1 or acalabrutinib using quantitative proteomics (Fig.2H, Supplementary table 7). Treatment with NGI-1 but not acalabrutinib decreased phosphorylation of the CD79A and CD79B ITAM tyrosines as well as tyrosines of several BCR-proximal kinases, including Src-family kinases (LYN, BLK, FYN) and SYK, which we confirmed by immunoblot analysis (Fig.2H–I, S7A). Unlike acalabrutinib, NGI-1 did not alter phosphorylation of BTK and PLCγ2 (Fig.2H, S7B), suggesting that the altered clustering of the deglycosylated may primarily affect SYK-dependent PI3 kinase activation. Accordingly, NGI-1 treatment decreased the PI3 kinase-dependent phosphorylation of AKT (Fig.2I). Nonetheless, NGI-1 treatment also suppressed downstream NF-κB activation, as judged by decreased phosphorylation of IκB kinase (IKK) and IκBα, with a consequent increase in IκBα abundance (Fig.2I). This observation may be explained, in part, by the fact that PI3 kinase inhibition in these ABC models attenuates IKK activation at endolysosomal membranes in an mTOR-dependent fashion (9). Together, these data support a model in which diminished clustering of the deglycosylated BCR in the plasma membrane reduces proximal BCR signaling and engagement of downstream oncogenic pathways.
Recruitment of CD22 to the deglycosylated BCR
We hypothesized that the altered distribution of the deglycosylated BCR in the plasma membrane might affect its association with other membrane proteins, potentially impeding signaling. To explore this possibility, we expressed a CD79A-BioID2 fusion protein in ABC cells, thereby permitting the promiscuous biotinylase BioID2 to biotinylate proteins in the vicinity of the BCR, which we detected by quantitative mass spectrometry (Supplementary table 8). In cells treated with NGI-1 or DMSO, proximal BCR signaling proteins (e.g., SYK, BTK, PLCG2, BLNK) were biotinylated equivalently as were STT3B and other OST subunits, presumably in the endoplasmic reticulum (Fig.3A). NGI-1 treatment increased biotinylation of the BCR subunits CD79A, CD79B and the immunoglobulin kappa light chain (IGK) relative to DMSO treatment, possibly reflecting the altered clustering and/or conformation of the deglycosylated BCR. Most notably, however, the BCR in NGI-1-treated cells was more associated with CD22, a B cell-specific surface receptor that negatively regulates BCR signaling by recruiting SHP-1 (PTPN6), GRB2 and SHIP-1 (INPP5D) (25,26). Separately and consistently, in ABC cells expressing a CD22-BioID2 fusion, NGI-1 treatment increased the biotinylation of CD79A, CD79B and IGK, again suggesting an induced proximity of CD22 with the deglycosylated BCR (Fig.3A, Supplementary table 9).
Figure 3. CD22 recruitment to the deglycosylated BCR.
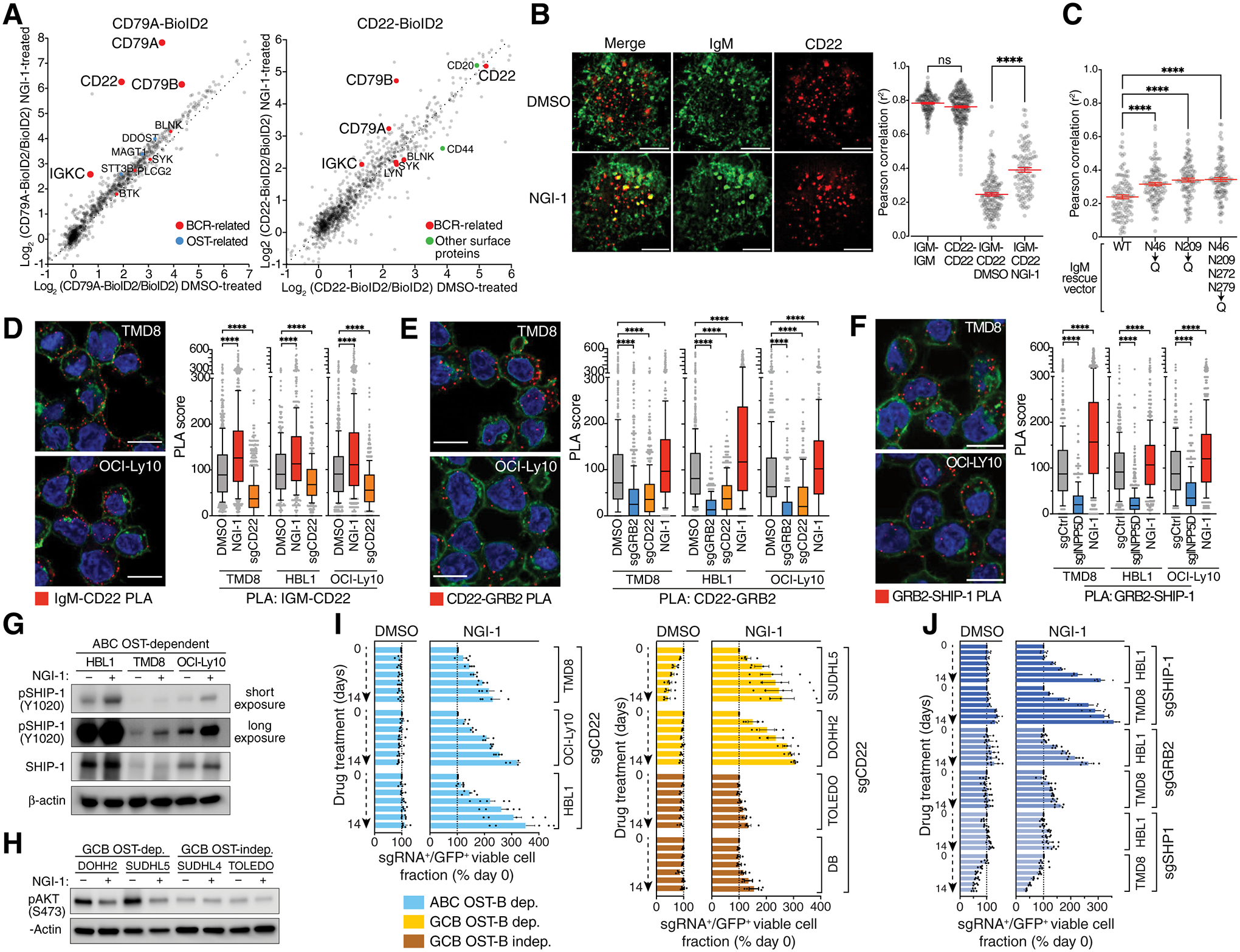
A. Left, CD79A-BioID2 interactomes in TMD8 cells treated for 24h with DMSO (x-axis) or NGI-1 (y-axis). OST subunits (blue) and BCR-related proteins (red) are indicated. Right, CD22-BioID2 interactomes in HBL1 cells treated for 24h with DMSO (x-axis) or NGI-1 (y-axis). B. Left, Dual color instant structured illumination microscopy (iSIM) images of TMD8 cells stained for IgM (Alexa-488, green) and CD22 (PE, red) treated for 24h with DMSO or NGI-1. Scale bar: 5μm. Right, Mean Pearson correlation coefficients of dual color iSIM images in TMD8 cells stained with the indicated antibodies and treated for 24h with NGI-1 as indicated. ****P ≤ 0.0001 (one-way ANOVA). Error bars represent SEM. ns, non-significant. C. Mean Pearson correlation coefficients of dual color iSIM images in TMD8 IgM asparagine mutant isoforms as indicated after knockout of endogenous IgM stained with IgM-Alexa488 and CD22-PE antibodies. ****P ≤ 0.0001 (one-way ANOVA). Error bars represent SEM. D., E., F. Left, Representative images from 3 independent PLA assays for the indicated protein-protein interactions showing PLA puncta (red), wheat-germ agglutinin (green) and DAPI (blue) staining. Scale bar: 10μm. Right, PLA scores for the indicated PLA pairs in TMD8, HBL1 and OCI-Ly10 cells. Box and whiskers: 10–90% percentile. ****P ≤ 0.0001 (one-way ANOVA). G., H. Immunoblots using the indicated antibodies in the indicated cell lines treated for 16h with NGI-1 as indicated. I., J. Competitive growth experiment in which the indicated cell lines expressing GFP and the indicated sgRNAs were mixed with GFP– wildtype cells in DMSO or NGI-1 as indicated and following GFP over two weeks. Bars display relative GFP+ cells normalized to day 0. Mean of at least three independent experiments. Error bars represent SEM.
To support this hypothesis further, we used super-resolution microscopy to visualize the colocalization of the BCR and CD22. Specifically, we localized IgM and CD22 in the plasma membrane by dual-color structured illumination microscopy (iSIM) (27) and calculated the Pearson correlation coefficient of their locations. The self-correlations of IgM-IgM and CD22-CD22 controls were high (r2=0.75–0.8), as expected, and the IgM-CD22 correlation was relatively low (r2=0.24) in DMSO-treated cells (Fig.3B, S8A). NGI-1 treatment significantly increased the IgM-CD22 correlation (r2=0.39, p<0.0001), in accord with the previous CD79A and CD22 proximity labelling analysis (Fig. 3B, Fig.S8A). Importantly, mutation of the N46 and N209 glycosites in IgM increased the spatial correlation of IgM and CD22 (Fig.3C), in keeping with the decreased ability of these mutant IgM isoforms to sustain the survival of ABC DLBCL cells.
Upon interaction with the BCR, the CD22 ITIM motif is phosphorylated by proximal BCR tyrosine kinases, thereby enabling the recruitment of negative regulators, including the protein phosphatase SHP1, the lipid phosphatase SHIP-1, and the SHIP-1 adapter GRB2. We used proximity ligation assays (PLAs) to examine these protein-protein interactions in DLBCL lines upon OST inhibition. As expected, NGI-1 treatment of three ABC lines increased the interaction of IgM and CD22 (Fig.3D). In turn, NGI-1 increased the association of CD22 with both GRB2 (Fig.3E) and SHIP-1 (Fig.3F). Following recruitment to the BCR, SHIP-1 is phosphorylated on Y1020, which is required for its attenuation of PI3 kinase signaling. NGI-1 treatment of ABC lines increased SHIP-1 Y1020 phosphorylation and concomitantly decreased in AKT phosphorylation (Fig.3G). Similarly, NGI-1 reduced AKT phosphorylation in OST-dependent, but not OST-independent, GCB lines (Fig.3H) and promoted the nuclear translocation of the AKT target FoxO1 (Fig.S8B), demonstrating that PI3 kinase inhibition is a shared consequence of OST inhibition in BCR-dependent ABC and GCB DLBCL models.
To test whether CD22 itself is glycosylated by the OST complex, we treated cells with NGI-1, or genetically deleted STT3A or STT3B, and blotted for CD22. NGI-1 altered the observed molecular weight of CD22 but the inactivation of either STT3A or STT3B alone did not produce a size shift, suggesting that OST-A and OST-B glycosylate CD22 in a redundant fashion (Fig.S8C). Since inactivation of OST-B alone was sufficient to reduce BCR signaling and survival of ABC lymphoma cells, these results suggest that CD22 deglycosylation is unlikely to contribute to these phenotypes.
To evaluate the functional consequence of CD22 recruitment to the BCR, we tested whether CD22 inactivation would reduce the toxic effect of NGI-1 in DLBCL cells. We transduced Cas9-expressing DLBCL lines with a vector co-expressing sgCD22 and GFP and tracked the proportion of sgCD22+/GFP+ cells over time in the presence of NGI-1 or DMSO. In cells treated with NGI-1 but not DMSO, the proportion of CD22 knockout cells increased over time, indicating that CD22 was required for the toxic effect of NGI-1 (Fig.3I). A similar outgrowth of CD22 knockout cells was observed in NGI-1-treated, OST-dependent GCB lines but not in OST-independent GCB lines (Fig.3I). Given that CD22 can recruit the negative regulators GRB2, SHP1, and SHIP-1, we tested whether these proteins were required for NGI-1 toxicity. We transduced two ABC lines with vectors co-expressing GRB2, SHP1, or SHIP-1 sgRNAs and GFP, treated with NGI-1 or DMSO, and monitored the sgRNA+/GFP+ fraction over time. With NGI-1 treatment, SHIP-1 and GRB2 knockout cells were selectively expanded, but SHP1 knockout cells were not (Fig.3J). Thus, signaling by the deglycosylated, reorganized BCR is limited by CD22/GRB2/SHIP-1-mediated attenuation of PI3 kinase activity.
OST inhibition impairs My-T-BCR formation
The My-T-BCR multiprotein supercomplex acts as a central signaling hub in the MCD subtype of ABC DLBCL bearing the MYD88L265P mutation (9). Using ABC lines with MYD88L265P (TMD8, OCI-Ly10, HBL1), we visualized the My-T-BCR using PLA assays for IgM-TLR9 (Fig.4A), MYD88-MALT1 (Fig.4B) and IgM-pIκBα (Fig.4C) interactions. In all three assays, NGI-1 treatment reduced My-T-BCR formation. The reduction in IgM-TLR9 association by NGI-1 was particularly notable since BTK inhibition did not affect this interaction (Fig.4A), as previously described (9).
Figure 4. BCR deglycosylation disrupts My-T-BCR-dependent survival in MCD DLBCL.
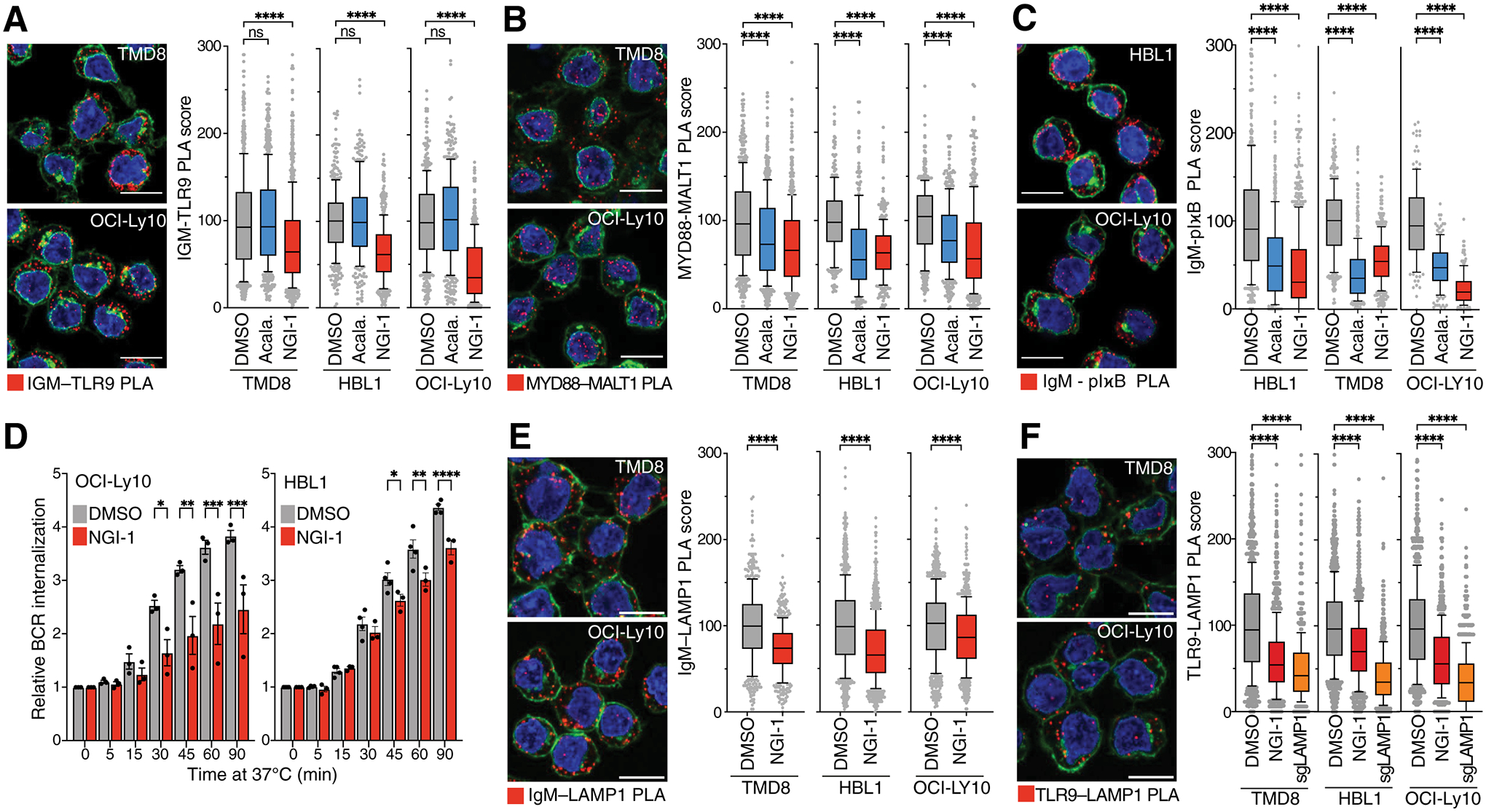
A., B., C. Left, PLA images of A. IgM with TLR9, B. MYD88 with MALT1, C. IgM with pIκBα. Representative images of 3 independent experiments with PLA puncta are shown in red, wheat-germ agglutinin in green and DAPI in blue. Scale bar: 10μm. Right, PLA scores of the indicated PLA pairs in TMD8, HBL1 and OCI-Ly10 cells. Box and whiskers: 10–90% percentile. ****P ≤ 0.0001 (one-way ANOVA). D. Mean (± SEM) relative BCR internalization over 90 mins at 37°C in OCI-Ly10 cells (left panel) or HBL1 cells (right panel) treated with NGI-1 for 24h as indicated. ****P ≤ 0.0001 (one-way ANOVA). *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001 (one-way ANOVA). Error bars represent SEM of at least three independent experiments. E., F. Left, PLA images of E. IgM with LAMP1 and F. TLR9 with LAMP1 in TMD8, HBL1 or OCI-Ly10 cells. Representative images of 3 independent experiments with PLA puncta are shown in red, wheat-germ agglutinin in green and DAPI in blue. Scale bar: 10μm. Right, PLA scores of the indicated PLA pairs in TMD8, HBL1 and OCI-Ly10 cells. Box and whiskers: 10–90% percentile.
Notably, My-T-BCR formation, as assessed by IgM-TLR9 and MYD88-MALT1 PLAs, was also reduced in cells expressing IgM isoforms with mutation in the essential glycosites N46 and N209 (Fig.S9A). In contrast, My-T-BCR formation was not affected by loss of CD22 (Fig.S9B), suggesting that CD22 primarily regulates the BCR-dependent activation of PI3 kinase at the plasma membrane.
We hypothesized that the defective My-T-BCR assembly in OST-inhibited cells was due, in part, to disrupted trafficking of the deglycosylated BCR from the plasma membrane to the endolysosomal compartment. To measure BCR internalization, we coupled an anti-IgM Fab fragment to the “pHrodo” dye, a fluorogenic pH sensor dye that increases its fluorescence in acidic surroundings. We labeled cell surface BCR with anti-IgM Fab-pHrodo on ice and measured the appearance of intracellular BCR over time following warming to 37°C. NGI-1 significantly reduced BCR internalization in three ABC lines (Fig.4D, S9C) and, as predicted, knockout of CD22 (Fig.S9C) had no effect. Concomitantly, the BCR abundance in endolysosomes was reduced as judged by a PLA assay for the association of IgM with the late endosomal marker LAMP1 (Fig.4E).
Given the decreased association of IgM and TLR9 in NGI-1-treated cells, we additionally suspected that deglycosylation of TLR9 might limit the trafficking of TLR9 to endolysosomes. Indeed, the association of TLR9 with LAMP1 was decreased in NGI-1-treated ABC cells by PLA (Fig.4F). Thus, the mechanism by which OST inhibition reduces NF-κB and impairs survival of the MCD subtype of ABC DLBCL is likely due, in part, to compromised assembly of the My-T-BCR.
OST inhibition synergizes with targeted therapies
We next identified cellular pathways that synergize or antagonize the ability of NGI-1 to kill lymphoma cells using genome-wide drug modifier CRISPR screening. To do so, we transduced TMD8 ABC cells with the Brunello sgRNA expression library and selected for transduction with puromycin. After one week of growth, separate cultures were treated with either DMSO or NGI-1 at the approximate IC50 for two additional weeks, at which time the sgRNA prevalence in each culture was determined by next generation sequencing (Supplementary table 10).
STT3B and STT3A as well as other members of the OST-complex (MAGT1, OST4, TMEM258, DDOST, OSTC) scored highly as synergy hits in this screen (Fig.5A, Supplementary table 11), supporting the view that NGI-1 has high on-target activity against OST. Conversely, inactivation of members of the ALG gene family (ALG12, ALG9, ALG6, ALG5, ALG2, ALG24, ALG10B) abrogated the effect of NGI-1, presumably because the encoded enzymes catalyze the synthesis of the dolichol-anchored glycan and act “upstream” of the OST complex in the N-linked glycosylation cascade (Fig. 1C). Inactivation of several components of the BCR-dependent NF-κB pathway synergized with NGI-1 (CARD11, BCL10, MALT1, PLCG2, LYN) and conversely, inactivation of negative regulators of this pathway promoted NGI-1 resistance (TNFAIP3, TNIP1, CD22, GRB2). Likewise, inactivation of components of the PI3 kinase/mTOR pathway synergized with NGI-1 (PIK3CD, PIK3AP1, RICTOR, LAMTOR3), while inactivation of negative regulators of this pathway reduced the effect of NGI-1 (PTEN, INPP5D). Notably, these screen results are concordant with our demonstration that CD22, GRB2 and INPP5D (SHIP-1) are mechanistically responsible for the inhibition of proximal BCR signaling by NGI-1.
Figure 5. OST inhibition synergizes with targeted therapies.
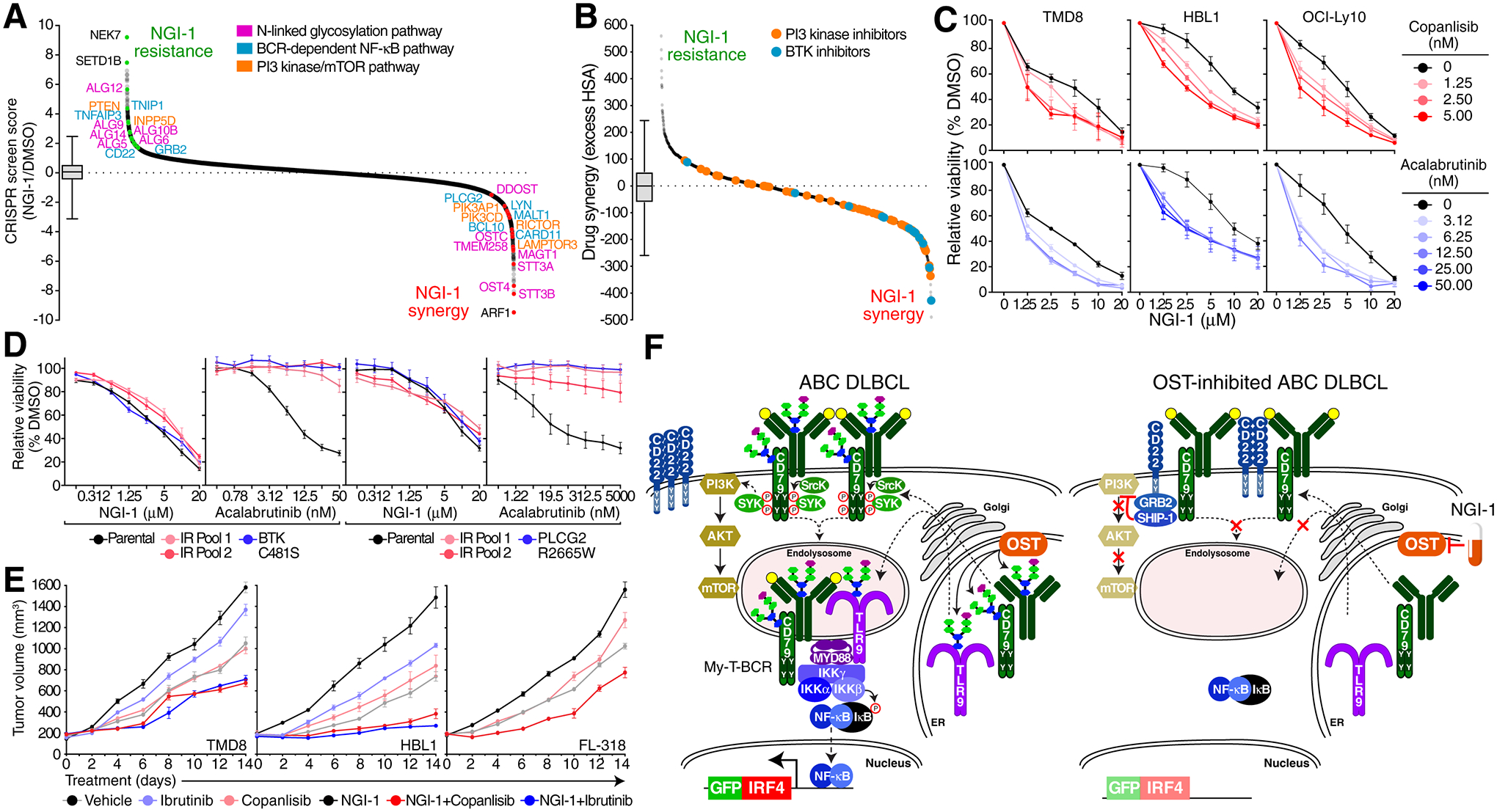
A. Ranked curve (CRISPR screen score) of an NGI-1 drug modifier CRISPR screen in TMD8 cells treated with NGI-1 vs. DMSO. Hits in the N-linked glycosylation pathway are highlighted in pink, in the BCR/NF-κB pathway in blue and in the PI3K/mTOR pathway in orange. Box and whiskers: 1–99% percentile. B. Ranked curve of drug synergy/antagonism in high throughput combinatorial drug screens in TMD8 ABC cells of NGI-1 in combination with the MIPE v. 5.0 small molecule library. BTK inhibitors (red) and PI3K inhibitors (blue) are highlighted. Boxplot represents 1–99% percentile. C. MTS proliferation assays for TMD8, HBL1 or OCI-Ly10 cells treated with vehicle (black) and indicated doses of copanlisib (red) or acalabrutinib (blue) with the indicated doses of NGI-1 (x-axis). D. MTS proliferation assays for BTK inhibitor resistant TMD8 or HBL1 cells treated with indicated doses of NGI-1 (x-axis). IR, ibrutinib resistant. E. Growth of TMD8, HBL1 or FL318 xenografts in NSG mice treated with the indicated drugs. F. Model of OST regulation of oncogenic signaling in lymphoma.
Our NGI-1 drug modifier screen raised the possibility that drugs targeting the BCR-dependent NF-κB pathway and the PI3 kinase/mTOR pathway might cooperate with OST inhibitors in killing DLBCL cells. To test this, we performed a high-throughput matrix drug screen in TMD8 ABC cells combining dose titrations of NGI-1 versus dose titrations of 2,450 mechanistically annotated compounds in the oncology-focused MIPE (v 5.0) library (28) (Supplementary table 12). Indeed, the combination of NGI-1 with multiple BTK and PI3 kinase inhibitors produced synergistic (more than additive) killing of ABC cells (Fig.5B, Fig.S10A, B). As confirmation of these screen results, we demonstrated that NGI-1 synergized with acalabrutinib and copanlisib in downregulating IRF4, as judged by our IRF4-GFP reporter system (Fig.S10C), and in killing of 3 ABC lines (Fig.5C). Resistance to BTK inhibitors can be due to mutations in BTK or PLCG2 or to an epigenetic rewiring of BCR signaling (29). Interestingly, in HBL1 and TMD8 cells rendered resistant to BTK inhibition by either genetic or epigenetic alterations, NGI-1 was as toxic as in parental cells, consistent with its BTK-independent mode of action (Fig.5D). Finally, we evaluated NGI-1 in OST-B-dependent ABC (HBL1, TMD8) and GCB (FL-318) xenograft models (Fig.5E). In all three models, tumor formation was reduced by NGI-1 monotherapy and the addition of ibrutinib or copanlisib increased this effect, without overt signs of systemic toxicity.
Discussion
By combining functional genomics, proteomics, and super-resolution microscopy, we uncovered a previously unknown mechanism by which N-linked glycosylation regulates BCR signaling. Our data suggest a model in which glycosylation of the BCR by OST-B is necessary for proximal BCR signaling and activation of the PI3 kinase pathway in BCR-dependent ABC and GCB DLBCL cells (Fig.5F). Additionally, glycosylation is required for effective trafficking of the BCR and TLR9 to endolysosomes where they assemble the My-T-BCR supercomplex, which coordinates NF-κB activation in the MCD genetic subset of ABC DLBCL (2,3,9). This model and its supporting data reveal N-linked glycosylation, specifically by OST-B complex, as an unanticipated and mechanistic novel therapeutic target in DLBCLs that rely on BCR signaling. Since inhibition of OST-B was sufficient to compromise BCR signaling and kill BCR-dependent DLBCL models, selective small molecule inhibitors of OST-B could have therapeutic potential in these aggressive cancers.
Although OST inhibition altered the glycosylation of many membrane and secreted proteins in lymphoma cells, we demonstrated an essential role for BCR glycosylation in several assays: 1) Mutation of particular glycosites in the BCR subunits IgM and CD79B impaired their ability to sustain survival of ABC DLBCL cells; 2) IgM glycosite mutations enabled association of the BCR with the negative regulator CD22; 3) Formation of the My-T-BCR supercomplex was compromised in cells expressing IgM glycosite-mutant isoforms, thereby attenuating BCR-dependent NF-κB signaling. Previously, the N46 glycosite of IgM was shown to be critical for pre-BCR formation with the surrogate light chain λ5 (30). However, IgM N46Q mutant isoforms functioned differently in ABC cells since they did not impair the expression of the BCR on the cell surface.
BCR deglycosylation was associated with decreased clustering in the plasma membrane and enhanced association with CD22, which recruited SHIP-1, a negative regulator of PI3 kinase signaling. CD22 contains an extracellular lectin domain that binds to α2,6 sialic acid (31), which is attached to glycan chains by the sialyltransferase ST6Gal1 (31). In normal mouse B cells, CD22 is largely, but not completely, confined to homotypic multimer islands (32,33), but in mice lacking ST6Gal1, CD22 is more mobile and able to impair BCR activation (31,34,35). However, we do not believe this mechanism contributes to the loss of BCR signaling by OST inhibition in lymphomas since neither ST6Gal1 inactivation, nor inactivation of other components of the sialic acid machinery reduced BCR signaling. Moreover, inhibition of OST-B alone was not sufficient to deglycosylate CD22 but was sufficient to inhibit proximal BCR signaling and decrease lymphoma survival. Rather, our data suggests a BCR-centered model in which disruption of the BCR microcluster architecture is necessary to allow CD22 to attenuate BCR signaling.
Broad inhibition of all N-linked glycosylation by the DPAGT1 inhibitor tunicamycin (36) invokes an unfolded protein response (UPR) in the endoplasmic reticulum and consequently is toxic for most cells and not therapeutically useful. Conversely, inactivation of N-linked glycosylation downstream of the OST complexes did not block constitutive BCR signaling in DLBCL models. Instead, we focused on the therapeutic potential of OST inhibition that is afforded by the fact that the OST catalytic subunits STT3A and STT3B have distinct biological functions and are not pan-essential. STT3A, acting at the translocon, glycosylates proteins co-translationally and is responsible for the majority (~70%) of N-linked glycosylation (14). In contrast, STT3B glycosylates a smaller proportion of N-linked glycosites in a post-translational fashion (15,37). Although, knockdown of STT3A induces the UPR, as judged by increase BiP protein expression, STT3B knockdown does not (15), consistent with the lack of toxicity of STT3B knockout in BCR-independent DLBCL lines, ATLL lines, and in DepMap cell lines.
Development of a selective STT3B inhibitor, which has been shown to be feasible (17), would have several attractive features that distinguish it from currently available therapeutic agents for DLBCL. Our data suggest that an STT3B inhibitor would have activity in all DLBCLs that rely on constitutive BCR signaling for survival, unlike BTK inhibitors that are primarily active in ABC but not GCB DLBCL (38). STT3B inactivation disrupts BCR clustering, thereby attenuating BCR-proximal Src-family and Syk kinases and consequently impairing PI3 kinase activation in both ABC and GCB models. In ABC DLBCL, PI3 kinase potentiates NF-κB activation by activating mTORC1, which is co-located with the My-T-BCR supermolecular complex in endolysosomes (9). This could explain our observation that OST inhibition was able to kill ABC lines that had acquired genetic or epigenetic resistance to BTK inhibition. PI3 kinase signaling also maintains the survival of GCB DLBCLs that have a constitutive, “toncogenic” form of BCR signaling. Toncogenic BCR signaling, unlike chronic active BCR signaling in ABC DLBCL, utilizes CD19/CD81 to recruit and activate PI3 kinase but does not activate NF-κB (9,39). By targeting the BCR directly, OST inhibition reduces oncogenic PI3 kinase signaling in both ABC and GCB DLBCL while avoiding the on-target toxicities of existing PI3 kinase inhibitors that interfere with this important pathway systemically (40). As with other targeted therapies being developed for DLBCL, a selective OST-B inhibitor would not be expected to induce durable complete responses in most patients as monotherapy. However, we showed that the OST inhibitor NGI-1 synergized with the BTK inhibitor ibrutinib and the PI3 kinase inhibitor copanlisib to kill DLBCL models in vitro and restrain xenograft growth in vivo. Beyond DLBCL, PI3 kinase is constitutively activated in other germinal center-derived lymphomas, including follicular lymphoma (41) and Burkitt lymphoma (42), suggesting that a selective OST-B inhibitor might have broad activity against B-cell non-Hodgkin lymphoma.
More generally, our work reveals that N-linked glycosylation can regulate the function of plasma membrane receptors by controlling their spatial organization and their association with other membrane proteins. Thus, OST inhibition may be harnessed for the treatment of lymphomas and other malignant (16) or non-malignant diseases that depend on pathological receptor signaling.
Methods
Cell Culture
Lymphoma cell lines were engineered with an ecotropic retroviral receptor, a tet-repressor (9,19) and a doxycycline inducible Cas9(9). Despite DOGKIT and BJAB being frequently classified as Burkitt lymphoma cell lines, they were classified in the present study as GCB DLBCL cell lines due to the lack of a MYC translocation (BJAB) and the presence of a BCL2 translocation and lack of Burkitt associated genetic alterations as ID3, TCF3, CCND3 (DOGKIT). All cells were grown in advanced RPMI medium with 5% fetal bovine serum (FBS) + 1% penicillin/streptomycin and 2 mM L-Glutamine except for A549 and PC-9, where 10% FBS was used. HEK293FT and HEK293T cells were grown in Dulbecco’s modified Eagle medium (DMEM) with 10% FBS, 1% penicillin/streptomycin and 2 mM L-glutamine. Cells were tested regularly for mycoplasma using the MycoAlert assay (Lonza) and cells were profiled by DNA fingerprinting: Known copy number variant regions were amplified by PCR and compared to a database. A549 and PC-9 cells were STR profiled (LabCorp cell line authentication). WSU-DLCL2 (RRID:CVCL_1902), HBL1 (RRID:CVCL_4213), RIVA (RRID:CVCL_1885), DOHH2 (RRID:CVCL_1179), SC-1 (RRID:CVCL_1888), WSU-FSCCL (RRID:CVCL_1903), DOGKIT (RRID:CVCL_2023) and BJAB (RRID:CVCL_5711) were acquired from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). SUDHL4 (RRID:CVCL_0539), SUDHL5 (RRID:CVCL_1735), SUDHL9 (RRID:CVCL_4379) and SUDHL7 (RRID:CVCL_4380) were obtained from Mark Raffeld (NCI, Bethesda, MD). TMD8 (RRID:CVCL_A442) was obtained from Shuji Tohda (Tokyo University) and U-2932 (RRID:CVCL_1896) was obtained from Gunilla Enblad (Uppsala University). OCI-Ly10 (RRID:CVCL_8795), OCI-Ly1 (RRID:CVCL_1879), OCI-Ly8 (RRID:CVCL_8803), OCI-Ly3 (RRID:CVCL_8800) and OCI-Ly9 (RRID:CVCL_QW65) were obtained from Hans Messner (Ontario Cancer Institute). TOLEDO (RRID:CVCL_3611) was obtained from Craig Jordan. A549 and PC-1 were gifted from Ji Luo (NCI, Bethesda, MD), FL318 was gifted from Kojo Elenitoba-Johnson (University of Pennsylvania).
Generation of GFP knock-in cell lines
To generate GFP tags in the endogenous IRF4 gene, we used a two-plasmid system exploiting non-homology directed repair mechanisms as described previously (43). Briefly, we used pLKO.1 puro (RRID:Addgene_8453). As sgRNA vector and modified the donor vector by switching the hygromycin sequence with a truncated CD34 sequence lacking the internal signaling domain, to knock-in a cassette containing a GFP-P2A-CD34 sequence. Before electroporation, Cas9 protein expression was induced with doxycycline (0.2 μg/ml) and cells were electroporated with a total of 1 μg plasmid DNA using the Neon electroporation system (Thermo Fisher) with previously optimized settings according to manufacturer’s instructions. After recovery from electroporation, cells were selected with CD34 microbeads (Miltenyi Biotec) and afterwards sorted on a MA900 cell sorter (Sony) for GFP positive cells to achieve a pure population.
Virus production
Lentivirus was made in HEK293FT cells (RRID:CVCL_6911) by transfecting with psMD2.G (Addgene RRID: Addgene_12259) and psPAX2 (Addgene, RRID: Addgene_12260) gag/pol and envelope plasmids using OptiMEM medium (Life Technologies) and the TransIT293 transfection reagent (Mirus) following manufacturer’s instructions. Lentiviral particles were harvested on three following days and concentrated using Lenti-X concentrator (TaKaRa).
For retrovirus production, HEK293T cells (RRID:CVCL_0063) were transfected with TransIT293 transfection reagent with pHIT-60/full length GALV or pHIT-60/EA6x3* helper plasmids and retroviral supernatant was harvested after 24–48h. To infect lymphoma cell lines, polybrene (1 μg/ml) was added to viral supernatant and cells were spin-infected by centrifugation (90 min, 2500 rpm).
sgRNA single and double knockouts
sgRNA sequences were cloned into pLKO.1-Puro-GFP, pLKO.1 puro or pLKO.1-Neo-BFP using standard cloning techniques as described previously (9). Lymphoma cell lines were infected with concentrated virus and 3 days after transduction doxycycline was added to induce Cas9 expression. For double knockouts of STT3A and STT3B, cells were transfected at the same time with an excess of virus with a sgRNA against STT3A co-expressing GFP and with a sgRNA against STT3B co-expressing BFP or vice versa. Three days after transduction, doxycycline was added to induce Cas9 expression and one week after induction cells were sorted for double positive (BFP+ and GFP+) cells and immediately used for further downstream applications.
The following sgRNAs were used in this study: sgCtrl.: TAAAGCAGAAGAATATACAG; sgSTT3A: GCTGCGCAGGTAATCCACAA; sgSTT3B: AAGAAAGACACCCAAGTCGT; sgCD79A: GCCACTCACGGCGCACGCGG; sgCD79B: CCCCGCAGGATTCAGCACCT; sgCD22: ATTCATACCGGGTAACACTG; sgGRB: ACGAGCTGAGCTTCAAAAGG; sgPTPN6: CCAGGGTGGACGCTACACAG; sgINPP5D: GCTGTTGACGAACCCTAAGG; sgIgM: GCTTCCCATCAGTCCTGAGA; sgLAMP1: GTGGACAAGTACAACGTGAG.
CRISPR-Screens
For genome-scale CRISPR/Cas-9 screens, we used the pooled Brunello CRISPR knockout library (RRID:Addgene_73178). Screens were performed as described previously (9). Briefly, lentiviral particles were made by transfecting HEK293FT cells with Brunello library plasmid, psPAX2 and pMD2.G using OptiMEM and the TransIT293 (Mirus Bio) reagent. Supernatant was harvested on three subsequent days and concentrated 1:40 using LentiX concentrator (Takara).
Lymphoma cell lines were infected in duplicates at an MOI of 0.3 and a coverage of ~500x. After puromycin selection, a d0 cell pellet was harvested and doxycycline (0.2 μg/ml) was added. Cells were passaged every 2–3 days until day 21.
For IRF4 knock-in CRISPR Screens, endogenously IRF4-GFP tagged cells were infected as described above, except for RIVA where 3 biological replicates were transduced. At day 6–8 after doxycycline induction, each biological replicate was split into two technical replicates and sorted for the top and bottom 5% of GFP. In addition, an unsorted total, “input” pellet was collected.
For NGI-1 drug modifier screens, after one week in culture (to allow for adequate genetic deletion,) targeted inhibitors were added at an approximate IC50 concentration and cells were further passaged for two additional weeks maintaining NGI-1 in the culture.
DNA of cell pellets was isolated using QIAamp DNA Blood Maxi kits or QIAamp DNA Mini kit (Qiagen). Libraries were amplified following a nested PCR strategy with the previously described primers(9). Briefly, in the first PCR, the sgRNA regions were amplified and in the second PCR, sequencing adapters were added. After amplification, libraries were size selected and purified with an E-Gel SizeSelect II agarose gel (Invitrogen). DNA was quantified using the Qubit dsDNA high-sensitivity assay (Invitrogen), sequenced on an Illumina NextSeq 2000 sequencer (Illumina) and reads were aligned using Bowtie2 (v. 2.2.9, RRID:SCR_016368).
To compare the changes in sgRNA counts for a given gene, log2 fold changes of normalized read counts for individual sgRNAs were calculated and fold changes of different sgRNAs for a given gene were averaged. Of the total of 77,441 sgRNAs, 11,740 sgRNAs were excluded from the analysis due to poor performance in many essential gene experiments (3,9,11). Furthermore, sgRNAs with low read counts at d0 due to low representation in the library were excluded.
Averaged log2 fold changes were then converted into a z-score, which is referred to as the CRISPR screen score (CSS). The CSS in GFPhigh sorted samples was subtracted from the CSS of GFPlow sorted samples for each gene, which was defined as segregation score.
To calculate the essential glycoproteome in TMD8 cells, data from a previously published CRISPR dropout screen were used (9). Toxicity data from the Cancer Dependency Map portal (DepMap; RRID:SCR_017655) were available from https://depmap.org/portal/.
Competitive growth assays and toxicity assays
For toxicity assays, cells were transduced with an excess of virus with sgRNAs targeting the indicated genes co-expressing either BFP or GFP. Three days after transduction, doxycycline was added (d0) and GFP or BFP, respectively, were followed by flow cytometry over a period of 2 weeks with measurements every 2–3 days. For double knockout toxicity assays, cells were transfected at the same time with an excess of virus with a sgRNA co-expressing GFP and with a sgRNA co-expressing BFP against the indicated genes. After three days, doxycycline was added (d0) and double positive cells were followed as for single knockout toxicity assays.
For competitive growth assays, cells stably expressing a non-targeting control were mixed with cells stably carrying the indicated sgRNAs co-expressing GFP in a 5:1 ratio to aim for 20% GFP positive cells. After mixing (d0), cells were treated with either DMSO, NGI-1 (2.5 μM for OCI-Ly10 and TMD8 and 5 μM for HBL1) or acalabrutinib (10 nM for OCI-Ly10, 15 nM for TMD8, 50 nM for HBL1) and GFP positive cells were monitored over 2 weeks with measurements every 2–3 days.
Data was acquired on a BD Calibur flow cytometer using CellQuest Pro (v. 6.0, RRID:SCR_014489) or a Beckman Coulter cytoflex S flow cytometer using the CytExpert software (RRID:SCR_017217). Further data analysis was done in FlowJo (v. 10, RRID:SCR_008520). GFP and/or BFP positive cells were normalized to their respective d0 percentage.
Knockout-rescue experiments
TMD8 cells were first retrovirally engineered to ectopically express wildtype IgM amplified from TMD8 cells as described previously (8) or different IgM asparagine mutants, all carrying a silent mutation in the PAM sequence of an sgRNA (GCTTCCCATCAGTCCTGAGA). Second, the endogenous IgM gene was targeted with a sgRNA co-expressing GFP and GFP levels were followed over a period of two weeks to monitor the rescue of the ectopically expressed mutants.
RNA-seq
HBL1 and TMD8 were treated 3h, 6h and 24h with acalabrutinib (15 nM for TMD8 and 50 nM for HBL1), NGI-1 (2.5 μM for TMD8 and 5 μM for HBL1) or Copanlisib (20 nM for TMD8 and 30 nM for HBL1). RNA was extracted using the AllPrep kit (Qiagen) and RNA libraries were prepared using the TruSeq V3 chemistry (Illumina) according to the manufacturer’s protocol. Sequencing of libraries was done on a NovaSeq S1 with a read length of 2×100 bp. Alignment to the human genome (hg19) was done using STAR-aligner. Normalized reads were Log2 transformed to calculate Digital Gene Expression values. RNA-seq datasets have been deposited to the GEO database (accession number GSE212337).
Gene expression signature analysis
Gene expression signatures were taken from the SignatureDB database (44). Gene expression estimates were generated from RNAseq data as previously described (2). Fold change in signature expression was calculated by averaging the log2 fold change between the treated samples versus the control over all genes in the signature.
Flow Cytometry
Cells were washed in PBS and stained for 30 min at room temperature with the following antibodies as indicated: anti-IgM-AlexaFluor-488 (Jackson Immunoresearch; cat. 109–547-043, RRID:AB_2337855), anti-IgM-AlexaFluor-647 (Jackson Immunoresearch; cat. 109–607-043, RRID:AB_2337904), anti-CD79A-PE (BioLegend; cat. 333504, RRID:AB_1089073), anti-CD79B-647 (BioLegend; cat. 341409, RRID:AB_2632946). Cells were washed twice and analyzed on a BD Calibur flow cytometer using CellQuest Pro (v. 6.0, RRID:SCR_014489) or a Beckman Coulter cytoflex S flow cytometer using the CytExpert software (RRID:SCR_017217). Mean fluorescence intensities were calculated in FlowJo (v. 10, RRID:SCR_008520).
Imaging flow cytometry (ImageStream)
Two million cells of the indicated cell lines were treated with 100 nM copanlisib, 5 μM NGI-1or 100 nM acalabrutinib. Cells were prepared for analysis using the Amnis NF-κB Translocation kit (Luminex Corporation) following manufacturer’s instructions. Briefly, cells were washed in PBS, fixed for 10 min with fixation buffer, washed in assay buffer and stained with an anti-human NF-κB (p50) antibody conjugated to Alexa Fluor 488 or an anti-FoxO1 antibody conjugated to Alexa Fluor 647 (CST; cat. #2880, RRID:AB_2106495), for 30 min at room temperature. Cells were then washed in assay buffer, resuspended in fixation buffer, and stained with DAPI (1 μg/ml). Cells were analyzed on Image Stream at 40x amplification with medium speed. A nuclear translocation score was calculated by calculating a log normalized Pearson correlation coefficient between the nuclear DAPI channel and the p50/FoxO1 channel, respectively.
High-throughput drug screens
High throughput drug screens were performed as described previously (45). In brief, 500 cells per well in 5 μl of media were plated in a 1,536 well plate (solid bottom white Greiner Bio-one) with stainless steel gasket lids (Kalypsis). Drugs were acoustically dispensed into wells with an Echo 550 acoustic liquid handler (Labcyte). A 5-point concentration range of each drug in the MIPE library in 1:4 dilutions was tested against a 5-point concentration range of NGI-1 (constant 1:4 dilutions) resulting in 6×6 blocks. The plates were incubated for 48h at 37 °C with 5% CO2 under 95% humidity. After 48h, 3 μl of CellTiter Glo cell viability assay (Promega) was added and incubated for 15 min at room temperature. Luminescence was read using a Viewlux imager (PerkinElmer) with 10 seconds exposure time.
MTS proliferation assays
Lymphoma cell lines were seeded in triplicates at 8,000 cells/well in 96-well plates in 250 μl total volume. NGI-1 and acalabrutinib (SelleckChem) were dissolved in DMSO, Copanlisib (SelleckChem) was dissolved in 5% trifluoroacetic acid and equal volumes of drugs at the indicated concentrations were added. Cells were cultured for 96h, and relative growth was measured with the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega) following manufacturer’s instructions. Absorbance was measured on a Tecan Infinite 200 Pro plate reader. Absorbance values from media only wells were subtracted from reads and normalized to vehicle reads.
Immunoblotting
Cells were washed in ice cold PBS and lysed in ice cold RIPA buffer (1% NP-40, 0.5% deoxycholate, 150 mM NaCl, 50 mM Tris at pH 7.5–7.8, 5 mM NaF, 0.5% NP40, 1 mM sodium vanadate, complete protease inhibitor cocktail (Roche)) for 10–30 min on ice. Lysates were cleared by centrifugation (17,000xg, 4°C) for 15 min and protein concentrations were determined using the Pierce 660nm assay (Thermo Fisher) supplemented with Ionic Detergent Compatibility Reagent (Thermo Fisher) according to manufacturer’s instructions. As indicated, cleared lysates were treated with PNGase F (New England Biolabs) according to manufacturer’s instructions. Equal protein amounts were boiled for 10 min in Laemmli sample buffer (BioRad) supplemented with β-mercaptoethanol. Samples were run on a Mini Protean TGX Precast Gel (4–15%) (BioRad), transferred to a PVDF membrane, blocked in 5% milk in TBST and incubated with as indicated the following primary antibodies in 5% milk overnight: anti-IRF4 (Cell Signaling Technology, CST; cat. #4964, RRID:AB_10698467), anti-phospho-IKKα/β Ser176/180 (CST; cat. #2697, RRID:AB_2079382), anti-IKKβ (CST; cat. #8943, RRID:AB_11024092), anti-IKKβ (CST; cat. #8943, RRID:AB_11024092), anti-β-actin (CST; cat. #3700, RRID:AB_2242334), anti-STT3A (Santa Cruz Biotechnology, SCB; cat. sc-100796, RRID:AB_2302814), anti-STT3B (abcam; cat. ab122351, RRID:AB_11133042), anti-phospho-IκBα Ser32/36 (CST; cat. #9246, RRID:AB_2267145), anti-IκBα (CST; cat. #4814, RRID:AB_390781), anti-phospho-AKT S473 (CST; cat. #4060, RRID:AB_2315049), anti-phospho-SRC Y416 (CST; cat. #2101, RRID:AB_331697), anti-phospho-SYK Y352 (CST; cat. #2717, RRID:AB_2218658), anti-SYK (CST; cat. #13198, RRID:AB_2687924), anti-phospho PLCγ2 Y1217 (CST; cat. #3871, RRID:AB_2299548), anti-PLCγ2 (CST; cat. #3872, RRID:AB_2299586), anti-phospho-BTK Y223 (CST; cat. #87457, RRID:AB_2800102), anti-BTK (CST; cat. #8547, RRID:AB_10950506), anti-CD79A (CST; cat. #13333, RRID:AB_2798183), anti-CD79B (CST; cat. #96024, RRID:AB_2800254), anti-IgM (SCB; cat. sc-53347, RRID:AB_672096), anti-SHIP-1 (CST; cat. #2727, RRID:AB_2126136), anti-phospho-SHIP-1 Y1020 (CST; cat. #3941, RRID:AB_2296062). Membranes were washed 3x in TBST, probed with secondary antibodies (anti-rabbit-HRP or anti-mouse-HRP (Cell Signaling Technology)) 1:10,000 in 5 % milk, washed again 3x in TBST and imaged on a BioRad ChemiDoc imaging system.
Imaging of IRF4-GFP knock-in cells
IRF4-GFP engineered cell lines were plated on a poly-L-lysine coated 15-well μ-Slide Angiogenesis ibiTreat chamber slide (Ibidi) and allowed to adhere for 30 min at 37 °C. Cells were washed with PBS, fixed for 10 min with 4% paraformaldehyde (Electron Microscopy Sciences) and stained with a wheat germ agglutinin Alexa-647 conjugate (5 μg/ml) for 15 min at room temperature. Next, cells were washed twice in PBS and permeabilized in ice cold methanol for 15 min at −20 °C, washed again, and blocked in Duolink Blocking buffer (Sigma) for 1h at room temperature. Cells were stained with conjugated CD34-PE antibody (BioLegend; cat. 343606, RRID:AB_1732008) for 1h at room temperature and imaged on a Zeiss LSM 880 confocal microscope. Images were prepared with FIJI 1.53f51 for display purposes.
Proximity ligation assays
Cell lines with indicated knockouts or drug treatments (acalabrutinib: 10 nM for OCI-Ly10, 15 nM for TMD8 and 50 nM for HBL1; NGI-1: 2.5 μM for OCI-Ly10/TMD8 and 5 μM for HBL1) were prepared as described above (“Imaging of IRF4-GFP knock-in cells”), except that a wheat germ agglutinin Alexa-488 conjugate (5 μg/ml) was used. PLA staining was done as previously described(9,46). After blocking, cells were incubated with the following primary antibodies in the listed dilutions in Duolink Antibody Diluent (Sigma) at 4 °C overnight: anti-LAMP1 (SCB; cat. sc-20011, RRID:AB_626853), anti-SHIP-1 (SCB; cat. sc-8425, RRID:AB_628250), anti-MyD88 (SCB; cat. sc-136970, RRID:AB_2146721), anti-MALT1 (SCB; cat. sc-28246, RRID:AB_2139156), anti-GRB2 (CST; cat. #3972, RRID:AB_10693935), anti-TLR9 (CST; cat. #2254, RRID:AB_2256024), anti-CD22 (BioLegend; cat. 302502, RRID:AB_314264), anti-IgM (Jackson Immunoresearch; cat. 109–005-129, RRID:AB_2337543). On the next day, cells were washed twice in a large volume of PBS. Ligation and amplification steps were done according to manufacturer’s protocol. After amplification, cells were washed in buffer B (Sigma) and mounted in Prolong Gold mounting media with DAPI (abcam). Cells were imaged on a Zeiss LSM 880 confocal microscope and images were prepared using FIJI 1.53f51 (RRID:SCR_002285). To calculate the PLA score, PLA spots were enumerated using Blobfinder 3.2 (RRID:SCR_015788) and counts were normalized to the average spot counts in control cells. Boxplots were plotted in GraphPad Prism (v.9, RRID:SCR_002798) with whiskers representing the 10–90% percentile.
Instant structured illumination microscopy
TMD8 cells were treated overnight with 2.5 μM NGI-1 or DMSO control and allowed to adhere on poly-L-lysine coated (PLL), glass-bottomed μ-Slide 8 well chambers (Ibidi) for 10 min at 37 °C. Cells were then fixed for 10 min with 4% paraformaldehyde (PFA, Electron Microscopy Sciences), washed with PBS and stained with the following antibodies: anti-CD22-PE (Miltenyi; cat. 130–123-245, RRID:AB_2802011), anti-CD22-AlexaFluor-647 (BioLegend; cat. 302518, RRID:AB_2716168), anti-IgM-AlexaFluor-488 (Jackson Immunoresearch; cat. 109–547-043, RRID:AB_2337855), anti-IgM-AlexaFluor-647 (Jackson Immunoresearch; cat. 109–607-043, RRID:AB_2337904). Instant structured illumination microscopy was performed on an in-house, custom build microscope with a VT-iSIM (VisiTech International) module based on a Nikon Eclipse Ti base body equipped with a 405-nm, 488-nm, 561-nm and a 640-nm laser (Coherent OBIS) as described previously(47,48). Imaging was done with a 100×/1.49 oil-immersion objective (N.A. = 1.49) and images were collected with a sCMOS camera (ORCA-Fusion BT; Hamamatsu) with 100–200 ms exposure time. The microscope, lasers, and the camera were controlled through NIS-Elements (Nikon, USA). Images were analyzed in MATLAB (The MathWorks, RRID:SCR_001622) using the “corr2” function in MATLAB image processing toolbox.
dSTORM
For dSTORM, TMD8 cells were treated overnight with 2.5 μM NGI-1 or for 10 min with 2 μg/ml anti-IGM Fab (Jackson Immunoresearch) and washed in ice cold PBS. Next, cells were stained for 20 min on ice with an anti-human IgM-Fab fragment (anti-IgM-AlexaFluor-647, Jackson Immunoresearch; cat. 109–607-043, RRID:AB_2337904), washed twice with PBS and plated on PLL coated coverslips (Thorlabs CG15XH1 - Precision Cover Glasses) for 10 min at 37 °C to promote adherence. Cells were then fixed with 4% PFA for 10 min at room temperature and washed twice with PBS.
For imaging, PBS was exchanged to freshly prepared dSTORM imaging buffer (50 mM Tris pH 8.0, 10 mM NaCl, 10% glucose, 50 mM β-mercaptoethylamine (Sigma), 4500 U catalase (Sigma), 0.5 mg/ml glucose oxidase (Sigma)). Imaging was done on a custom build microscope with a Nikon Eclipse Ti base and a Nikon N-STORM module equipped with a 100×/1.49 oil-immersion objective (N.A. = 1.49), a multi-band bandpass filter (405/488/561/633 BrightLine quad-band bandpass filter, Semrock) and a piezo z-stage (ASI). Images were acquired with a sCMOS camera (Prime 95B, Teledyne Photometrics) in near TIRF mode using a N-STORM module (Nikon). Acquisition at 640 nm and activation at 405 were simultaneously and 6100 frames were collected with an exposure time of 20 ms. Microspheres (FluoSpheres Carboxylate-Modified Microspheres, 0.2 μm, Thermo Fisher) were used as fiducials to correct for sample drift.
Images were rendered in FIJI (v. 2.3.0, RRID:SCR_002285) using the GDSC SMLM plugin. Localizations in the raw data were fitted using a 2D Gaussian model of the point spread function with a maximum likelihood estimation fit solver and a minimum photon count of 150. We analyzed localizations in a rectangular region of interest of 5 μM side length at the center of each cell sparing out the borders due to potentially different cluster characteristics with two different metrics describing the clustering tendency of a given set of localizations. First, the Hopkins index (23), expressing the clustering tendency of the molecules ranging from 0 indicated a fully dispersed distribution, 0.5 representing a random distribution and 1.0 representing a fully clustered distribution. Second, the H-function, the first derivate from Ripley’s K function, a nearest neighbor metric allowing to estimate clustering tendency and cluster size (23,24). Data processing and analysis were done in MATLAB (RRID:SCR_001622) using custom written codes.
Mass spectrometry
To metabolically label cells for mass spectrometry, we used “stable isotope labeling with amino acids in cell culture” (SILAC) as described previously (49). Cells were grown for two weeks in RPMI medium (Silantes) lacking lysine and arginine with 10% dialyzed FCS, and 1% penicillin/streptomycin (Life technologies). Medium was supplemented with amino acids were “Light” medium contained regular amino acids (Sigma), “Medium” medium contained 13C614N4-L-arginine/L-lysine-4,4,5,5-D4 and “Heavy” 13C615N4-L-arginine/13C615N2-L-lysine (Eurisotop).
Non-SILAC samples were labeled with “tandem mass tag” (TMT)-labels (ThermoFisher) following manufacturer’s instructions. Briefly, protein concentrations were determined using the Pierce 660 nm kit (Thermo Fisher). 100 μg protein per sample was reduced using TCEP and alkylated with iodoacetamide. Next, proteins were precipitated using acetone, tryptic digested and labeled with the TMT tags.
To profile the global proteome SILAC labeled cells were washed two times in ice cold PBS and lysed in RIPA buffer (1% NP-40, 0.5% deoxycholate, 150 mM NaCl, 50 mM Tris at pH 7.5–7.8, 5 mM NaF, 0.5% NP40, 1 mM sodium vanadate, complete protease inhibitor cocktail (Roche)) for 10–30 min on ice. Lysates were cleared by centrifugation (17,000xg, 4°C) for 15 min. SILAC labeled cleared protein lysates were separated by SDS-PAGE, stained with Coomassie blue, and cut into 23 slices. Proteins in each slice were subjected to tryptic digestion.
Eluted peptides (in 2% acetonitrile/0.05% TFA) were separated on an HPLC system (UltiMate 3000 RSLCnano, Thermo Fisher) and analyzed using Orbitrap Q Exactive series or Orbitrap Fusion mass spectrometers (Thermo Fisher).
BioID2
BioID2 fusions were cloned using Gibson cloning as described previously (9). In brief, BioID2 was PCR amplified from purchased Addgene plasmids (Addgene; cat. 80899, RRID:Addgene_80899) with an 8X GSGGG (for N-terminal) or a 10X GSGGG (for C-terminal) into the StuI site of a modified pBMN vector backbone (Addgene; cat. 1736, RRID:Addgene_1736), where GFP was exchanged with LYT2 (pBMN-IRES-LYT2) as described previously (50). The resulting plasmids (N-terminus: pBMN-BioID2–8Xlinker-MCS-IRES-LYT2 and C-terminus: pBMN-MCS-10Xlinker-BioID2-IRES-LYT2) allowed the insertion of any open reading frame of interest using the SnaBI cutting site. Using Gibson cloning, the following primers were used to create BioID2-fusions: CD79A C-term Forward: CTGCCGGATCCGAATTCTACCCCACCATGCCTGGGGGTCCAGGAG; CD79A C-term Reverse: CCCGACCCGCCTCCACCTACCGGCTTCTCCAGCTGGACATC; CD22 C-term forward: CTGCCGGATCCGAATTCTACCCCACCATGCATCTCCTCGGCCCCTG;
CD22 C-term reverse: CCCGACCCGCCTCCACCTACATGTTTGAGGATCACATAGTCCACATTTTCTTG;
Constructs were retrovirally expressed in the indicated cell lines and purified using LYT2 dynabeads (Invitrogen). Cells were grown in SILAC medium and treated as indicated. 24h before lysis, 50 μM biotin was added to culture. Cells were counted and equal cell numbers of “Light”, “Medium” and “Heavy” labeled cells were pooled, washed twice, lysed in RIPA buffer, and cleared by centrifugation (see “Mass Spectrometry” section). Cleared lysates were incubated for 2–4h with 75 μl of high-capacity streptavidin beads (Thermo Fisher) under rotating conditions at 4°C. Beads were washed for at least three times and proteins were eluted by boiling in 4X LDS sample buffer (Invitrogen) supplemented with NuPage reducing agent (Invitrogen). Mass spectrometry was carried out as described above.
Glycoproteomics
Site-specific N-linked glycoproteomic analyses were carried out as described previously (51). Briefly, indicated cell lines were washed twice with PBS and lysed in 4% SDS (w/v) in 50 mM HEPES buffer (pH 7.5) containing 1X Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific) and sonicated. Lysates were cleared by centrifugation and protein amounts were determined using Pierce BCA Protein Assay Kit (Thermo Scientific). Recovered proteins were reduced and alkylated using 10 mM TCEP and 20 mM iodoacetamide and then incubated with pre-washed Sera-Mag SpeedBeads containing 1:1 mixed hydrophilic and hydrophobic beads (GE Healthcare) at a bead-to-protein ratio of 10:1 (wt/wt) in 70% (vol/vol) acetonitrile (ACN). Subsequently, supernatant was removed, and beads were washed three times with 95% (vol/vol) ACN. Next, beads were resuspended in 50 mM HEPES (pH 8.0) with trypsin (1:40 of enzyme-to-protein ratio) and incubated overnight (37 °C) at 1000 rpm in a ThermoMixer. The resulting peptides were collected and labeled with TMTpro reagents (Thermo Scientific) according to the manufacturer’s instruction.
Glycopeptides were enriched by using spin columns self-packed with Ultimate Hydrophilic Interaction Liquid Chromatography (HILIC) Amphion II beads (5 μm, Welch). Briefly, we passed the TMT-labeled peptides through pre-equilibrated spin columns five times and washed three times with 75% (vol/vol) ACN and 1% TFA. Bound glycopeptides were eluted with 0.1% TFA and vacuum dried. Enriched glycopeptides were then subjected to basic reverse-phase HPLC (Agilent 1100) fractionation using an XBridge C18 column (3.5 μm particles, 1.0 mm × 150 mm, Waters), and each fraction was analyzed using the Glyco-SPS-MS3 method on an Orbitrap Fusion Lumos.
Raw files were processed using GlycoBinder as previously described (51). Glycopeptides were identified using pGlyco 2 based on a reviewed human protein database (SwissProt, October 2020, 20,370 entries) with the limitation of a maximum of two missed cleavages and 10 and 20 ppm of mass tolerance for precursors and fragment ions, respectively. Cysteine carbamidomethylation and TMTpro labeling on peptide N-termini and lysine residues were set as fixed modifications. Methionine oxidation was set as a variable modification. GlycoBinder determined and reported the quantification of glycosylation sites, glycan compositions, and glycoforms. Log2 fold changes were normalized to global proteome data.
Phosphoproteomics
Tyrosine-phosphorylated peptides were enriched using an IP-based method as described previously (52). In brief, 100 million cells were lysed in 5 ml urea lysis buffer (20 mM HEPES pH 8.0, 9 M urea, 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate), reduced using DTT, alkylated using iodoacetamide and digested using trypsin. Peptides were purified using Sep-Pak tC18 cartridges (Waters) and phosphorylated peptides were immuno-precipitated using the Phospho-Tyrosine Rabbit mAb Kit (Cell Signaling). Enriched phospho-peptides were subjected to mass spectrometry as described above.
Processing of LC-MS data
Raw MS output files were analyzed using MaxQuant (53) (version 1.6.5.0, Max Planck Institute of Biochemistry, RRID:SCR_014485). For protein identification and quantitation, spectra were searched against the Uniprot human reference proteome and a database of 245 common contaminants using the Andromeda search engine (54). The mass tolerance was set to 6 and 20 ppm, minimal length of tryptic peptides was 7 amino acids with a false discovery rate of 1% on peptide and protein level. Carbamidomethylation of cysteine was defined as a fixed modification, N-terminal acetylation and oxidation of methionine were set as variable modifications. In case of SILAC experiments, at least two ratio counts were required for protein quantitation. Further data processing was done using Perseus (version 1.6.0.7, MPI of Biochemistry, RRID:SCR_015753).
B-cell receptor internalization assay
To measure BCR internalization, pHrodo Red (Thermo Fisher) fluorogenic dye conjugated to avidin was mixed with an anti-IgM Fab biotin conjugate (anti-IgM-Biotin, Jackson Immunoresearch; cat. 109–067-043, RRID:AB_2337645), following manufacturer’s instructions except that an excess of dye (molar ratio 5:1 dye to IgM Fab) was used. The pHrodo red dye is non-fluorogenic outside the cell but gains fluorescence if the surrounding pH becomes more acidic. After complex formation, cells were stained 30 min on ice, washed three times with ice-cold PBS, warmed up to 37 °C and fluorescence was immediately measured on a Beckman Coulter cytoflex S flow cytometer. Data was analyzed using FlowJo (v. 10, RRID:SCR_008520) and fluorescence was normalized to time 0.
Radiation Chimeras
For transduction of bone marrow, Aicdacre/+ Rosa26LSLCas9/+ (wildtype) or MCD-allele expressing (Aicdacre/+ Rosa26LSLCas9/LSL-BCL2 Myd88L252P/+ Cd79bY195H/+ Prdm1f/f (21) donor mice were injected intravenously with 3 mg 5-fluorouracil (Sigma). Bone marrow was collected after 4 days and cultured in DMEM containing 15% (v/v) FBS, antibiotics (penicillin (50 IU/ml) and streptomycin (50 mg/ml); Cellgro) and 10 mM HEPES, pH 7.2 (Cellgro), supplemented with IL-3, IL-6 and stem cell factor (at concentrations of 20, 50 or 100 ng/ml, respectively; Peprotech). Cells were ‘spin-infected’ twice with MRIA retrovirus expressing non-targeting sgRNA (5’-AGCAGCGTCTTCGAGAGTG-3’) or Stt3a (5’-AAGGTGGTACGTAACGATGG-3’ and 5’-TACCATGTAGAAATAAGCGA-3’) and Stt3b (5’-AGGGTACATATCTCGGTCAG-3’ and 5’-CGACAGCATGCAGACGACCG-3’) sgRNA that also expressed the fluorescent reporter Ametrine at days 1 and 2 and transferred into irradiated B6-Ly5.1/Cr mice (Charles River Laboratories) that were at least 6 weeks of age on day 3. Hosts were lethally irradiated with 900 rad in split doses and reconstituted by tail vein injection with at least 3×106 transduced bone marrow (BM) cells. Animals were analyzed at least 7 weeks after reconstitution. Spleens were mashed trough a 70 μM cell strainer to obtain single cell suspensions and stained with the following antibodies and analyzed on a on a CytoFLEX LX flow cytometer (Beckman Coulter): Fixable Viability Dye eFluor 780 (ebiosciences), BV786-conjugated anti-B220 (RA3–6B2; BioLegend, cat. 103245, RRID:AB_11218795), BUV395-conjugated anti-CD4 (RM4–5; BD, cat. 740208, RRID:AB_2734761), Pacific blue–conjugated GL7 (GL-7; BioLegend, cat. 144614, RRID:AB_2563292), BV650-conjugated anti-IgD (11– 26c.2a; BioLegend, cat. 405721, RRID:AB_2562731), PerCP Cy5.5 anti-CD38 (90; BioLegend, cat. 303522, RRID:AB_893314), PE-Cy7-conjugated anti-Fas (Jo2; BD, cat. 557653, RRID:AB_396768), Alexa Flour 700-conjugated anti-CD45.2 (104; BioLegend, cat. 109822, RRID:AB_493731). All experiments were performed in accordance with guidelines under protocols approved by the National Cancer Institute Animal Care and Use Committee (NCI-ACUC).
Xenografts
All animal experiments were approved by the National Cancer Institute Animal Care and Use Committee (NCI-ACUC) in accordance with NCI-ACUC guidelines under approved protocols (METB054). Female NSG (non-obese diabetic/severe combined immunodeficient/common gamma chain deficient) mice (RRID: IMSR_JAX: 005557) were obtained from NCI Fredrick Biological and used for experiments between 6–8 weeks of age. 10 million TMD8 or HBL1 cells were washed with PBS, mixed 1:1 with Matrigel (Corning) and subcutaneously injected. Drug treatments were started at an average tumor volume of 200 mm3. Ibrutinib (SelleckChem) was prepared in 50% DMSO/PBS (v/v) and administered at 5 mg/kg daily i.p. NGI-1 (SelleckChem) was prepared in 10% DMSO, 40% PEG300, 5% Tween80 and 45% PBS and administered i.p. at 15 mg/kg every other day. Copanlisib (SelleckChem) was prepared in 20%PEG400 and 80% acidified water and administered i.p. at 5 mg/kg every other day. Combinations of drugs were given at the same concentration and schedule as single agents. Tumor volumes were determined every other day by measuring the size in two orthogonal dimensions. Tumor volume was calculated with the equation: tumor volume = (length × width2)/2. Drug randomization or blinding was not possible.
Statistical analysis
Graphpad Prism (v.9, RRID:SCR_002798) was used to calculate statistical significance and p-values ≤0.05 were generally considered significant with asterisks indicating significance: ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. NS, not significant. Data are presented as the mean ± standard deviation or the mean ± standard error of the mean as described in the figure legends. Data were analyzed by one-way ANOVA, two-way ANOVA, or student’s t-test.
Supplementary Material
Significance statement:
Diffuse large B-cell lymphoma depends on constitutive B cell receptor activation and signaling. There are currently no therapeutics that target the BCR directly and attenuate its pathological signaling. Here, we unraveled a therapeutically exploitable, OST-B-dependent glycosylation pathway that drives BCR organization and proximal BCR signaling.
Acknowledgements
Supported by the Intramural Research Program of the National Institutes of Health (NIH), and the Center for Cancer Research, National Cancer Institute (NCI). S.S. was supported by a grant from the German Research Foundation (Deutsche Forschungsgemeinschaft [DFG]) (SCHE 2120/1-1). F.S. was supported by the Wilhelm Sander-Foundation (2015.138.1) and the Deutsche Forschungsgemeinschaft (SCHN1166/4-1). We thank all members of the Staudt, Young and Muppidi laboratories for helpful suggestions and discussions.
Footnotes
COI statement: The authors declare no potential conflicts of interests.
Data availability
All relevant sequencing data generated and used in this study have been deposited in the NCBU Gene Expression Omnibus (GEO) with the accession number GSE212337. Proteomic data supporting the findings of the study have been deposited in the PRIDE database with accession PXD036363 and PXD038951. All other data supporting the findings of the study are available upon request and source data is provided within this paper.
References
- 1.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000;403:503–11. [DOI] [PubMed] [Google Scholar]
- 2.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 2018;378(15):1396–407 doi 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020;37(4):551–68 e14 doi 10.1016/j.ccell.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 2018;24(5):679–90 doi 10.1038/s41591-018-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lacy SE, Barrans SL, Beer PA, Painter D, Smith AG, Roman E, et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a Haematological Malignancy Research Network report. Blood 2020;135(20):1759–71 doi 10.1182/blood.2019003535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov 2013;12(3):229–43 doi 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Young RM, Phelan JD, Wilson WH, Staudt LM. Pathogenic B-cell receptor signaling in lymphoid malignancies: New insights to improve treatment. Immunol Rev 2019;291(1):190–213 doi 10.1111/imr.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc Natl Acad Sci U S A 2015;112(44):13447–54 doi 10.1073/pnas.1514944112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 2018;560(7718):387–91 doi 10.1038/s41586-018-0290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishio T, Kumar S, Shimono J, Daenthanasanmak A, Dubois S, Lin Y, et al. Genome-wide CRISPR screen identifies CDK6 as a therapeutic target in adult T-cell leukemia/lymphoma. Blood 2022;139(10):1541–56 doi 10.1182/blood.2021012734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakagawa M, Shaffer AL 3rd, Ceribelli M, Zhang M, Wright G, Huang DW, et al. Targeting the HTLV-I-Regulated BATF3/IRF4 Transcriptional Network in Adult T Cell Leukemia/Lymphoma. Cancer Cell 2018;34(2):286–97 e10 doi 10.1016/j.ccell.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 2016;34(2):184–91 doi 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zufferey R, Knauer R, Burda P, Stagljar I, te Heesen S, Lehle L, et al. STT3, a highly conserved protein required for yeast oligosaccharyl transferase activity in vivo. EMBO J 1995;14(20):4949–60 doi 10.1002/j.1460-2075.1995.tb00178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cherepanova NA, Venev SV, Leszyk JD, Shaffer SA, Gilmore R. Quantitative glycoproteomics reveals new classes of STT3A- and STT3B-dependent N-glycosylation sites. J Cell Biol 2019;218(8):2782–96 doi 10.1083/jcb.201904004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruiz-Canada C, Kelleher DJ, Gilmore R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 2009;136(2):272–83 doi 10.1016/j.cell.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Sambrooks C, Shrimal S, Khodier C, Flaherty DP, Rinis N, Charest JC, et al. Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nat Chem Biol 2016;12(12):1023–30 doi 10.1038/nchembio.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rinis N, Golden JE, Marceau CD, Carette JE, Van Zandt MC, Gilmore R, et al. Editing N-Glycan Site Occupancy with Small-Molecule Oligosaccharyltransferase Inhibitors. Cell Chem Biol 2018;25(10):1231–41 e4 doi 10.1016/j.chembiol.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008;319(5870):1676–9. [DOI] [PubMed] [Google Scholar]
- 19.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010;463(7277):88–92 doi nature08638 [pii] 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol 2015;15(3):172–84 doi 10.1038/nri3814. [DOI] [PubMed] [Google Scholar]
- 21.Pindzola GM, Razzaghi R, Tavory RN, Nguyen HT, Morris VM, Li M, et al. Aberrant expansion of spontaneous splenic germinal centers induced by hallmark genetic lesions of aggressive lymphoma. Blood 2022;140(10):1119–31 doi 10.1182/blood.2022015926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Havranek O, Xu J, Kohrer S, Wang Z, Becker L, Comer JM, et al. Tonic B-cell receptor signaling in diffuse large B-cell lymphoma. Blood 2017;130(8):995–1006 doi 10.1182/blood-2016-10-747303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Leiderman K, Pfeiffer JR, Wilson BS, Oliver JM, Steinberg SL. Characterizing the topography of membrane receptors and signaling molecules from spatial patterns obtained using nanometer-scale electron-dense probes and electron microscopy. Micron 2006;37(1):14–34 doi 10.1016/j.micron.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 24.Kiskowski MA, Hancock JF, Kenworthy AK. On the use of Ripley’s K-function and its derivatives to analyze domain size. Biophys J 2009;97(4):1095–103 doi 10.1016/j.bpj.2009.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otipoby KL, Draves KE, Clark EA. CD22 regulates B cell receptor-mediated signals via two domains that independently recruit Grb2 and SHP-1. J Biol Chem 2001;276(47):44315–22 doi 10.1074/jbc.M105446200. [DOI] [PubMed] [Google Scholar]
- 26.Poe JC, Fujimoto M, Jansen PJ, Miller AS, Tedder TF. CD22 forms a quaternary complex with SHIP, Grb2, and Shc. A pathway for regulation of B lymphocyte antigen receptor-induced calcium flux. J Biol Chem 2000;275(23):17420–7 doi 10.1074/jbc.M001892200. [DOI] [PubMed] [Google Scholar]
- 27.Heintzmann R, Huser T. Super-Resolution Structured Illumination Microscopy. Chem Rev 2017;117(23):13890–908 doi 10.1021/acs.chemrev.7b00218. [DOI] [PubMed] [Google Scholar]
- 28.Lin GL, Wilson KM, Ceribelli M, Stanton BZ, Woo PJ, Kreimer S, et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci Transl Med 2019;11(519) doi 10.1126/scitranslmed.aaw0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaffer AL 3rd, Phelan JD, Wang JQ, Huang D, Wright GW, Kasbekar M, et al. Overcoming Acquired Epigenetic Resistance to BTK Inhibitors. Blood Cancer Discov 2021;2(6):630–47 doi 10.1158/2643-3230.BCD-21-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ubelhart R, Bach MP, Eschbach C, Wossning T, Reth M, Jumaa H. N-linked glycosylation selectively regulates autonomous precursor BCR function. Nat Immunol 2010;11(8):759–65 doi 10.1038/ni.1903. [DOI] [PubMed] [Google Scholar]
- 31.Hennet T, Chui D, Paulson JC, Marth JD. Immune regulation by the ST6Gal sialyltransferase. Proc Natl Acad Sci U S A 1998;95(8):4504–9 doi 10.1073/pnas.95.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han S, Collins BE, Bengtson P, Paulson JC. Homomultimeric complexes of CD22 in B cells revealed by protein-glycan cross-linking. Nat Chem Biol 2005;1(2):93–7 doi 10.1038/nchembio713. [DOI] [PubMed] [Google Scholar]
- 33.Gasparrini F, Feest C, Bruckbauer A, Mattila PK, Muller J, Nitschke L, et al. Nanoscale organization and dynamics of the siglec CD22 cooperate with the cytoskeleton in restraining BCR signalling. EMBO J 2016;35(3):258–80 doi 10.15252/embj.201593027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collins BE, Smith BA, Bengtson P, Paulson JC. Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nat Immunol 2006;7(2):199–206 doi 10.1038/ni1283. [DOI] [PubMed] [Google Scholar]
- 35.Grewal PK, Boton M, Ramirez K, Collins BE, Saito A, Green RS, et al. ST6Gal-I restrains CD22-dependent antigen receptor endocytosis and Shp-1 recruitment in normal and pathogenic immune signaling. Mol Cell Biol 2006;26(13):4970–81 doi 10.1128/MCB.00308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hou H, Sun H, Lu P, Ge C, Zhang L, Li H, et al. Tunicamycin potentiates cisplatin anticancer efficacy through the DPAGT1/Akt/ABCG2 pathway in mouse Xenograft models of human hepatocellular carcinoma. Mol Cancer Ther 2013;12(12):2874–84 doi 10.1158/1535-7163.MCT-13-0201. [DOI] [PubMed] [Google Scholar]
- 37.Shrimal S, Trueman SF, Gilmore R. Extreme C-terminal sites are posttranslocationally glycosylated by the STT3B isoform of the OST. J Cell Biol 2013;201(1):81–95 doi 10.1083/jcb.201301031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med 2015;21(8):922–6 doi 10.1038/nm.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfeifer M, Grau M, Lenze D, Wenzel SS, Wolf A, Wollert-Wulf B, et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A 2013;110(30):12420–5 doi 10.1073/pnas.1305656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shouse G, Danilova OV, Danilov AV. Current status of phosphoinotiside-3 kinase inhibitors in blood cancers. Curr Opin Oncol 2022;34(5):540–5 doi 10.1097/CCO.0000000000000871. [DOI] [PubMed] [Google Scholar]
- 41.Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med 2014;370(11):1008–18 doi 10.1056/NEJMoa1314583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012;490(7418):116–20 doi 10.1038/nature11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thone FMB, Kurrle NS, von Melchner H, Schnutgen F. CRISPR/Cas9-mediated generic protein tagging in mammalian cells. Methods 2019;164–165:59–66 doi 10.1016/j.ymeth.2019.02.018. [DOI] [PubMed] [Google Scholar]
- 44.Shaffer AL, Wright G, Yang L, Powell J, Ngo V, Lamy L, et al. A library of gene expression signatures to illuminate normal and pathological lymphoid biology. Immunol Rev 2006;210:67–85. [DOI] [PubMed] [Google Scholar]
- 45.Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci U S A 2014;111(6):2349–54 doi 10.1073/pnas.1311846111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alam MS. Proximity Ligation Assay (PLA). Curr Protoc Immunol 2018;123(1):e58 doi 10.1002/cpim.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.York AG, Chandris P, Nogare DD, Head J, Wawrzusin P, Fischer RS, et al. Instant super-resolution imaging in live cells and embryos via analog image processing. Nat Methods 2013;10(11):1122–6 doi 10.1038/nmeth.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu Y, Kumar A, Smith C, Ardiel E, Chandris P, Christensen R, et al. Reflective imaging improves spatiotemporal resolution and collection efficiency in light sheet microscopy. Nat Commun 2017;8(1):1452 doi 10.1038/s41467-017-01250-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Corso J, Pan KT, Walter R, Doebele C, Mohr S, Bohnenberger H, et al. Elucidation of tonic and activated B-cell receptor signaling in Burkitt’s lymphoma provides insights into regulation of cell survival. Proc Natl Acad Sci U S A 2016;113(20):5688–93 doi 10.1073/pnas.1601053113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011;470(7332):115–9 doi 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fang P, Ji Y, Silbern I, Doebele C, Ninov M, Lenz C, et al. A streamlined pipeline for multiplexed quantitative site-specific N-glycoproteomics. Nat Commun 2020;11(1):5268 doi 10.1038/s41467-020-19052-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilke AC, Doebele C, Zindel A, Lee KS, Rieke SA, Ceribelli M, et al. SHMT2 inhibition disrupts the TCF3 transcriptional survival program in Burkitt lymphoma. Blood 2022;139(4):538–53 doi 10.1182/blood.2021012081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26(12):1367–72 doi 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 54.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 2011;10(4):1794–805 doi 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant sequencing data generated and used in this study have been deposited in the NCBU Gene Expression Omnibus (GEO) with the accession number GSE212337. Proteomic data supporting the findings of the study have been deposited in the PRIDE database with accession PXD036363 and PXD038951. All other data supporting the findings of the study are available upon request and source data is provided within this paper.