

Abstract
Understanding the timing and spectrum of genetic alterations that contribute to the development of pancreatic cancer is essential for effective interventions and treatments. The aim of this study was to characterize somatic ATM alterations in non-invasive pancreatic precursor lesions and invasive pancreatic adenocarcinomas from patients with and without pathogenic germline ATM variants. DNA was isolated and sequenced from the invasive pancreatic ductal adenocarcinomas and precursor lesions of patients with a pathogenic germline ATM variant. Tumor and precursor lesions from these patients as well as colloid carcinoma from patients without a germline ATM variant were immunolabeled to assess ATM expression. Among patients with a pathogenic germline ATM variant, somatic ATM alterations, either mutations and/or loss of protein expression, were identified in 75.0% of invasive pancreatic adenocarcinomas but only 7.1% of pancreatic precursor lesions. Loss of ATM expression was also detected in 31.0% of colloid carcinomas from patients unselected for germline ATM status, significantly higher than in pancreatic precursor lesions (pancreatic intraepithelial neoplasms (P = 0.0013); intraductal papillary mucinous neoplasms, P = 0.0040) and pancreatic ductal adenocarcinoma, (P = 0.0076) unselected for germline ATM status. These data are consistent with the second-hit to ATM being a late event in pancreatic tumorigenesis.
Keywords: Pancreatic, Cancer, ATM, Immunohistochemistry, Sequencing, Inherited, Pathogenic, Variant, Somatic, DNA
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a lethal disease with a 5-year survival rate of just 11% (1). The incidence of PDAC in the United States has increased, with more than 55,000 people diagnosed with the disease in 2021. Patients with PDAC often harbor germline pathogenic variants in pancreatic cancer susceptibility genes (2–4), and clinical care guidelines now recommend offering patients with PDAC and their first-degree relatives germline genetic testing (5,6). Furthermore, individuals with a strong family history of PDAC or individuals with a known pathogenic germline variant in a pancreatic cancer susceptibility gene are recommended to consider annual surveillance once they reach age-criteria (7,8).
ATM codes for a member of the phosphoinositide 3-kinase-related protein kinase (PIKK) family and the protein is a crucial component of DNA damage response that ultimately plays a role in cell death, cell survival, cell cycle arrest, apoptosis, and DNA repair (9,10). Approximately 2–3% of patients with PDAC and 1.6% of patients with surgically-resected intraductal papillary mucinous neoplasm (IPMN) (11), a pancreatic cancer precursor lesion, carry a pathogenic germline ATM variant (12–18). Importantly, individuals with a pathogenic germline ATM variant have a significantly increased risk of developing PDAC. We recently estimated age-specific risk of pancreatic cancer in 130 pancreatic cancer kindreds with a pathogenic germline ATM variant using a modified segregated analysis and found that the cumulative risk of pancreatic cancer was 1.1% (95% CI 0.8%-1.3%) by age 50, 6.3% (95% CI 3.9%-8.7%) by age 70, and 9.5% (95% CI 5.0%-14.0%) by age 80 (19). These cumulative risk estimates are consistent with a previously published odds ratio of pancreatic cancer in pathogenic germline ATM variant carriers of 5.71 (95% CI, 4.38–7.30) compared to publicly available gnomAD control data (4).
PDAC develops from non-invasive precursor lesions that include pancreatic intraepithelial neoplasia lesions (PanINs) and IPMNs. Studying these precursor lesions is important to further our understanding of pancreatic tumorigenesis. In particular, understanding the timing and spectrum of genetic alterations that contribute to the development of PDAC is essential for effective interventions and treatments. For example, cancers from patients with germline and/or somatic mutations that lead to defects in homology-directed repair or mismatch repair may be more susceptible to poly(ADP-ribose) polymerase 1 inhibitors and immunotherapy respectively (20–22). Similarly, the loss of ATM-mediated DNA repair in neoplastic cells may increase their susceptibility to ionizing radiation and ATR inhibitors. In vitro studies of multiple pancreatic cancer cell lines demonstrated significant radiosensitivity after shRNA knockdown of ATM expression and we have reported significant pathologic response to radiation therapy in a patient with pancreatic cancer and a pathogenic germline ATM variant (23). Additionally, studies of pancreatic and prostate cancer cell lines indicate ATM loss increases sensitivity to ATR inhibitors (24–26). Furthermore, preclinical in vitro and in vivo studies have shown that inhibitors of PARP, ATR, and DNA-PK act synergistically to target pancreatic cancer cells with ATM loss and may improve outcomes when used as maintenance therapy after FOLFIRINOX (folinic acid, fluorouracil, irinotecan, and oxaliplatin) induction therapy (27,28).
A recent study of cancers arising in patients with pathogenic germline ATM variants found that 17 of 19 (89.5%) evaluable cancers had either somatic loss of heterozygosity (LOH) encompassing the variant or additional somatic mutations in ATM (29). The important role of ATM in pancreatic tumorigenesis is further supported by an in vivo study indicating that loss of ATM expression accelerates pancreatic cancer formation (30). However, the timing of ATM inactivation is still poorly understood.
In this study we characterized the timing and spectrum of somatic ATM alterations in invasive adenocarcinomas and precursor lesions from patients with PDAC and a pathogenic germline ATM variant using a targeted gene panel and immunohistochemistry (IHC). We also explored the role of ATM loss in pancreatic tumorigenesis in precursor lesions and invasive carcinoma, stratified by histologic subtype, in patients unselected for germline ATM status using immunohistochemical labeling (IHC).
Materials and methods
Ethics approval
The study was reviewed and approval obtained from the Johns Hopkins Institutional Review Board.
Patient selection
Twenty-four patients (P1–P24) with surgically resected pancreatic cancer and pathogenic germline ATM variants previously identified in research studies were collected from the Johns Hopkins Hospital, Memorial Sloan Kettering Cancer Center, University of Pittsburgh Medical Center, and Radboud University Medical Center Nijmegen. All patients included in the study had formalin-fixed paraffin embedded (FFPE) tissue available that included invasive carcinoma, PanINs, or IPMNs, suitable DNA sequencing (15 of 24 patients) and/or immunostaining (24 of 24 patients). The histomorphology and clinicopathologic features of 16 of these patients were reported previously (23).
Pathologic review
Hematoxylin and eosin (H&E) stained slides were reviewed FFPE resection specimens and tissue microarrays (TMAs). Invasive carcinomas and precursor lesions, including pancreatic intraepithelial neoplasia (PanIN) and intraductal papillary mucinous neoplasm (IPMN), were identified for laser capture microdissection and IHC. Invasive pancreatic carcinomas were classified by histologic subtype according to World Health Organization criteria (31). Neoplastic precursor lesions were graded for dysplasia based on consensus recommendations (32).
Laser capture microdissection of invasive carcinoma and precursor lesions
Ten to twenty serial tissue sections from FFPE tissue blocks were cut onto membrane slides (Carl Zeiss Microscopy, LLC, White Plains, NY, USA, catalog no. 415190–9041-000) at 10-μm thickness. Deparaffinization and staining were performed as described previously (33). Sixteen invasive carcinoma samples were microdissected from 14 patients. Morphologically and regionally distinct precursor lesions were microdissected, including one IPMN region and 13 PanIN regions from 8 patients (Table 1). Microdissection was performed using a Leica LMD7000 laser-capture microscope (Leica Microsystems, Wetzlar, Hesse, Germany).
Table 1.
Clinicopathologic information for patients with a pathogenic germline ATM variant
| ID | Pathogenic germline ATM variant | Age (yr) | Sex | Race | Histologic subtype | Neoadjuvant therapy* | Pathologic stage | Pathologic response | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Genomic variant~ | Transcript variant^ | Protein variant+ | ||||||||
| P1 | chr11:108173655-108173656_AG>A | c.5396delG | p.Ser1799MetfsTer8 | 50–59 | Male | White | Ductal adenocarcinoma | No | pT1c pN1 | N/A |
| P2 | chr11:108224608-108224608_G>A | c.8786+1G>A | Splicing | 50–59 | Male | White | Ductal adenocarcinoma | No | pT1a pN0 | N/A |
| P3 | chr11:108188128-108188129_TT>T | c.6228delT | p.Leu2077PhefsTer5 | 60–69 | Female | No data | Ductal adenocarcinoma | No | pT1c pN0 | N/A |
| P4 | chr11:108115702-108115702_C>T | c.850C>T | p.Gln284Ter | 70–79 | Male | White | Ductal adenocarcinoma | No | pT2 pN1 | N/A |
| P5 | chr11:108098354-108098354_G>A | c.3G>A | p.Met1Ile | 60–69 | Male | White | Ductal adenocarcinoma | No | pT2 pN2 | N/A |
| P6 | chr11:108121755-108121757_AGA>A | c.1564_1565delGA | p.Glu522IlefsTer43 | 60–69 | Male | White | Ductal adenocarcinoma | No | pT2 pN0 | N/A |
| P7 | chr11:108206605-108206605_C>T | c.8185C>T | p.Gln2729Ter | 70–79 | Female | No data | Ductal adenocarcinoma | No | pT2 pN1 | N/A |
| P8 | chr11:108214064-108214074_ATTTCAGTGCC>A | c.8395_8404delTTTCAGTGCC | p.Phe2799LysfsTer4 | <50 | Male | White | Colloid Carcinoma | No data | pT1a pN0 | N/A |
| P9 | chr11:108202604-108202604_A>C | c.7630–2A>C | Splicing | 50–59 | Male | White | Colloid Carcinoma | No | pT3 pN2 | N/A |
| P10 | chr11:108181032-108181032_C>T | c.5908C>T | p.Gln1970Ter | 70–79 | Female | Other | Adenosquamous carcinoma | No | pT1c pN0 | N/A |
| P11 | chr11:108202604-10820260477_A>T | c.7630–2A>T | Splicing | 60–69 | Female | White | Ductal adenocarcinoma | Gemcitabine + nab-paclitaxel | ypT2 ypN2 | Poor |
| P12 | chr11:108100051-108100051_G>A | c.331+1G>A | Splicing | 60–69 | Female | White | Ductal adenocarcinoma | Gemcitabine + nab-paclitaxel | ypT2 ypN2 | Poor |
| P13 | chr11:108121561-108121561_C>T | c.1369C>T | p.Arg457Ter | 70–79 | Female | White | Ductal adenocarcinoma | Gemcitabine + nab-paclitaxel | ypT2 ypN2 | Moderate |
| P14 | chr11:108121755-108121757_AGA>A | c.1564_1565delGA | p.Glu522IlefsTer43 | <50 | Male | White | Ductal adenocarcinoma | FOLFIRINOX | ypT2 ypN2 | Poor |
| P15 | chr11:108198361-108198385_TTCTTATACAGAACAATCCCAGCCT>T | c.6976–10_6989delTCTTATACAGAACAATCCCAGCCT | Splicing | 70–79 | Female | White | Ductal adenocarcinoma | No | pT2 ypN0 | N/A |
| P16 | chr11:108235935-108235935_C>T | c.8977C>T | p.Arg2993Ter | 70–79 | Female | Other | Ductal adenocarcinoma | Gemcitabine + paclitaxel | ypT1c ypN2 | Poor |
| P17 | chr11:108128198-108128198_T>G | c.2251–10T>G | Splicing | 70–79 | Male | No data | Ductal adenocarcinoma | No data | pT1N0 | N/A |
| P18 | chr11:108121755-108121757_AGA>A | c.1564_1565delGA | p.Glu522Ilefs*43 | 70–79 | Female | No data | Colloid Carcinoma | No data | pT1 | N/A |
| P19 | chr11:108196885-108196886_A>AA | c.6908dupA | p.Glu2304Glyfs*69 | 60–69 | Female | No data | Ductal adenocarcinoma | No data | No data | N/A |
| P20 | chr11:108122615-108122616_AA>A | c.1660delA | p.Thr554Argfs*2 | 60–69 | Female | No data | Ductal adenocarcinoma | No data | pT3N0 | N/A |
| P21 | chr11:108205825-108205825_C>T | c.8140C>T | p.Gln2714Ter | 50–59 | Female | No data | Ductal adenocarcinoma | No data | No data | N/A |
| P22 | chr11:108216545-108216545_C>T | c.8494C>T | p.Arg2832Cys | 60–69 | Female | No data | Ductal adenocarcinoma | No data | No data | N/A |
| P23 | chr11:108123641-108123641_T>G | c.1898+2T>G | Splicing | 60–69 | Female | No data | Ductal adenocarcinoma | No data | pT3N0 | N/A |
| P24 | chr11:108153571-108153576_TTTATT>T | c.3712_3716delTTATT | p.Leu1238Lysfs*6 | 70–79 | Female | No data | Ductal adenocarcinoma | No data | No data | N/A |
Genomic variant shown based on GRCh37 version of the human genome.
Transcript variant shown based on NM_000051.3.
Protein variant shown based on NP_000042.3.
FOLFIRINOX – folinic acid, fluorouracil, irinotecan, and oxaliplatin.
DNA extraction
DNA from laser capture microdissected tissue (invasive carcinoma or precursor lesions) or four to six 4-μm sections of FFPE blocks containing duodenum (non-neoplastic tissue) was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen, Germantown, MD, USA, catalog no. 56404) and deparaffinization solution (Qiagen, catalog no. 19093) as previously described (11). Extracted DNA was quantified with the Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) using the Qubit 1x dsDNA BR Assay Kit (Thermo Fisher Scientific, catalog no. Q32853). Sufficient microdissected neoplastic DNA for sequencing was available from 15 of the 24 patients.
Targeted gene sequencing
DNA sequence libraries for each sample were prepared using the Nextera Flex for Enrichment Kit protocol (Illumina, San Diego, CA, USA, catalog no. 20025524). Sequence libraries were PCR amplified before amplification with a custom Illumina AmpliSeq panel according to the manufacturer’s specifications (supplementary material, Table S1). Fragment size and yields of amplified libraries were determined using an Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA, catalog no. G2939BA) and a Qubit 3.0 Fluorometer (Thermo Fisher Scientific) using the Qubit 1x dsDNA HS Assay Kit (Thermo Fisher Scientific, catalog no. Q33230). Amplified libraries were sequenced using an Illumina MiSeq genome analyzer and the MiSeq Reagent Kit v2 (500-cycles) (Illumina, catalog no. MS-102–2003) configured to produce 2 × 150 bp paired-end reads.
Analysis of genetic data
Sequence reads were aligned to the human genome hg19 using Burrows-Wheeler Aligner (BWA) (SourceForge, San Diego, CA, USA) (34). Somatic mutation and germline variant calling was performed using the Genome Analysis Toolkit (GATK) Mutect2 and haplotype caller pipelines (Broad Institute, Cambridge, MA, USA) (35,36). Mutations were filtered with GATK FilterMutectCalls (Broad Institute). Default settings were used. Somatic mutations and germline variants were annotated with ANNOVAR (GitHub, San Francisco, CA, USA) (37). Somatic mutations were visually inspected in IGV (Broad Institute) (38).
Immunohistochemistry
IHC for the ATM protein was performed as previously described using an anti-ATM primary antibody (Abcam, Cambridge, UK, catalog no. 32420, 1:100 dilution) and 4-μm tissue slides sectioned from archived FFPE tissue blocks or tissue microarrays (39). Tissue slides from patients with surgically resected PDAC and a pathogenic germline ATM variant included 25 invasive carcinoma samples from 22 patients, 4 IPMN samples from 4 patients, and 24 PanIN samples from 12 patients (Table 1). Tissue microarrays (TMAs) were generated from patients unselected for germline AT status and grouped by lesion type. TMAs contained samples from 105 patients with IPMN (80 low-grade IPMN and 47 high-grade IPMN samples; 94 non-intestinal-type and 15 intestinal-type samples), 51 patients with PanIN (53 samples), and 29 patients with colloid carcinoma (30 samples). Nuclear ATM labeling was scored by board-certified pathologists (D.H. and L.D.W.). ATM was determined to be lost when >90% of neoplastic cells lacked positive labelling, as described previously (40).
Statistical analysis
Prism6 (GraphPad, Boston, MA, USA) was used for all statistical analyses. To assess differences in ATM loss between different pancreatic lesions, a Fisher’s exact test was used. A p value <0.05 was considered significant. As the TMAs included multiple samples for some patients, the most severe ATM characterization for a given lesion type, for example loss of ATM, was used for each patient when conducting statistical analyses.
Results
Characteristics of patients with germline ATM alterations
For patients with germline ATM alterations, clinicopathologic and ATM variant data are detailed in Table 1. Patients were 62.5% female (15 of 24 patients), predominantly white (12 of 14 patients with race data; 85.7%) and had a median age range of 60–69 years. The most common cancer histological type among patients was infiltrating ductal adenocarcinoma (20 of 24 patients; 83.3%). Colloid carcinoma was diagnosed in three patients (12.5%) and adenosquamous carcinoma in one patient (4.2%). Of the 15 patients with information on receipt of neoadjuvant therapy, 5 received neoadjuvant chemotherapy (33.3%) including gemcitabine and nab-paclitaxel (three patients), gemcitabine and paclitaxel (one patient), and FOLFIRINOX (one patient). ATM germline variants were classified as pathogenic or likely pathogenic based on American College of Medical Genetics guidelines. Pathogenic or likely pathogenic variants included six nonsense variants (25.0%), eight frameshift deletions (33.3%), one frameshift insertion (4.2%), seven splicing variants (29.2%), and two missense variants (8.3%).
Immunohistochemical analysis of invasive carcinoma and precursor lesions from patients with germline ATM alterations
To determine the prevalence of loss of ATM expression, we immunolabeled histological slides containing invasive carcinoma, PanIN, and/or IPMN from 24 patients. In total, 25 invasive carcinoma samples from 22 patients, 24 PanIN samples (all low-grade) from 12 patients, 4 IPMN samples (3 low-grade and 1 high-grade) each from a different patient were characterized (Table 1). Loss of ATM expression was observed in invasive carcinoma samples from 17 of 22 patients (77.3%) (Table 2). Loss of ATM expression was also observed in 1 of 4 IPMNs (25.0%). Of note, the IPMN sample with loss of ATM expression was the only precursor lesion analyzed with high-grade dysplasia (Figure 1). We found that ATM expression was intact for PanINs, all low-grade, that were assessed (Table 2). Loss of ATM expression was statistically more prevalent in invasive carcinoma compared to PanIN (Fisher’s exact test; P < 0.001).
Table 2.
Samples from patients with a pathogenic germline ATM variant available for immunolabeling and targeted sequencing
| ID | Number of immunostained samples | Number of sequenced samples | ||||
|---|---|---|---|---|---|---|
| PanIN* | IPMN# | Invasive carcinoma | PanIN* | IPMN# | Invasive carcinoma | |
| P1 | 2 | - | 1 | 1 | - | - |
| P2 | 5 | 1 | - | 3 | 1 | 1 |
| P3 | 2 | 1 | - | - | - | 1 |
| P4 | 1 | - | 1 | - | - | 1 |
| P5 | 1 | - | 2 | - | - | 2 |
| P6 | 3 | - | 1 | 1 | - | 1 |
| P7 | 1 | - | 1 | - | - | 1 |
| P8 | - | 1 | 1 | - | - | - |
| P9 | 1 | - | 2 | 1 | - | 2 |
| P10 | - | - | 2 | - | - | 2 |
| P11 | - | - | 1 | - | - | 1 |
| P12 | 2 | - | 1 | 1 | - | - |
| P13 | 1 | - | 1 | 1 | - | - |
| P14 | - | - | 1 | - | - | 1 |
| P15 | 4 | - | 1 | 4 | - | 1 |
| P16 | - | - | 1 | - | - | 1 |
| P17 | 1 | 1 | - | - | - | |
| P18 | - | 1H | 1 | - | - | - |
| P19 | - | - | 1 | - | - | - |
| P20 | - | - | 1 | - | - | - |
| P21 | - | - | 1 | - | - | - |
| P22 | - | - | 1 | - | - | - |
| P23 | - | - | 1 | - | - | - |
| P24 | - | - | 1 | - | - | - |
| Total | 24 | 4 | 25 | 12 | 1 | 15 |
PanIN - Pancreatic intraepithelial neoplasm. IPMN - intraductal papillary mucinous neoplasm.
PanINs were all low-grade.
IPMNs were all low-grade except where indicated by H.
Figure 1. ATM IHC images of PDAC and pancreatic cancer precursor lesions.
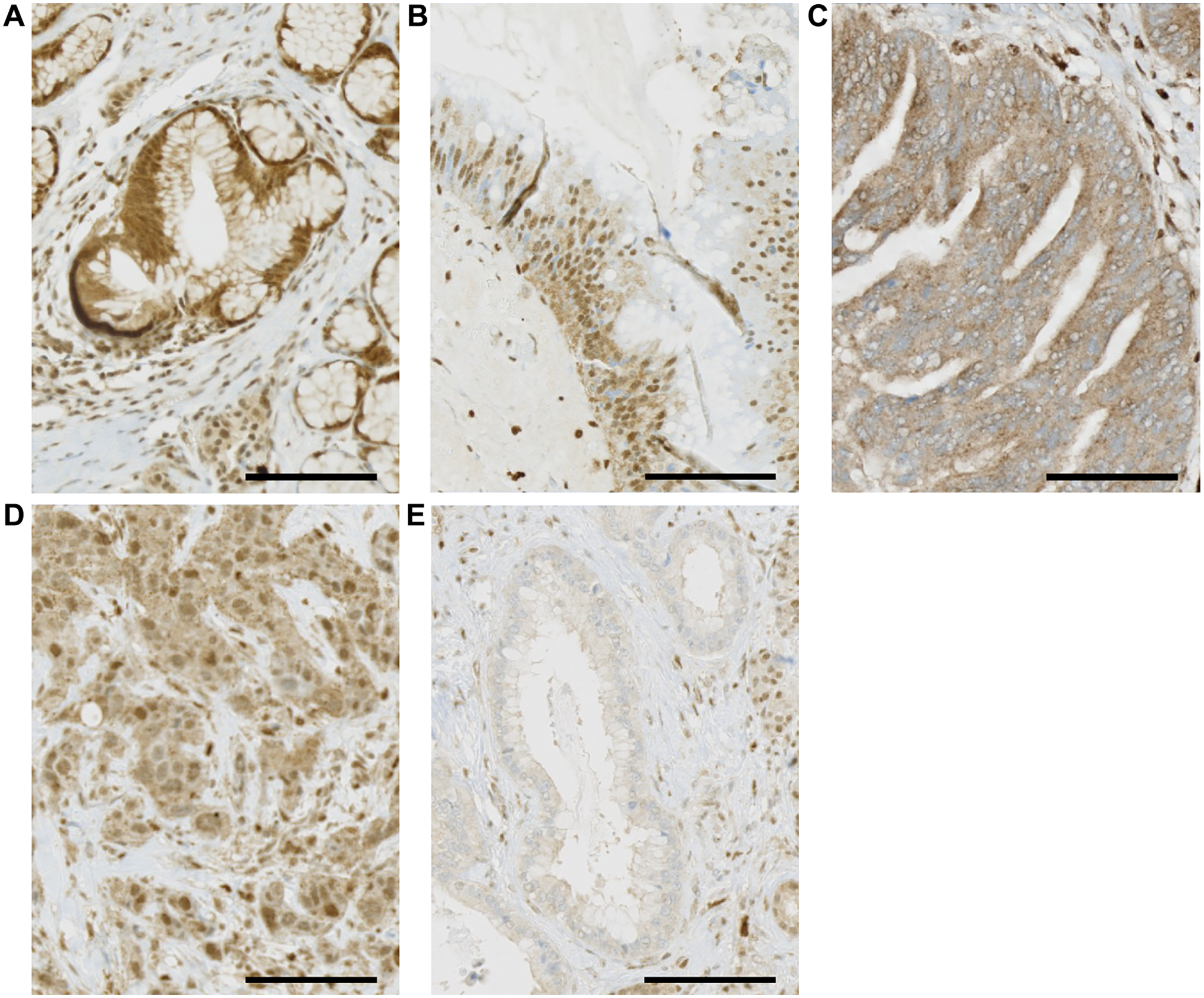
Black scale bar, 100 μm. (A) PanIN showing retention of nuclear ATM staining. (B) Low-grade IPMN showing retention of nuclear ATM staining. (C) High-grade IPMN showing loss of nuclear ATM staining. (D) PDAC showing retention of nuclear ATM staining. (E) PDAC showing loss of nuclear ATM staining.
Targeted sequencing of invasive carcinoma and precursor lesions
Invasive carcinomas, PanINs, and/or IPMN from 15 patients were sequenced using the custom amplicon panel covering the coding regions of ATM, KRAS, GNAS, TP53, CDKN2A, and SMAD4 (supplementary material, Table S1). In total, 15 invasive carcinoma samples from 12 patients, 12 PanIN samples from 7 patients, and 1 IPMN sample were sequenced (Table 1).
Across all samples, target regions were sequenced to a mean depth of 204X (range: 60 – 451X), with 83.1% (range: 42.5 – 92.2%) of target bases covered at a minimum of 50X. Coverage was similar for invasive carcinoma (217X mean coverage; 84.2% at 50X), precursor lesions (194X mean coverage; 85.5% at 50X), normal tissue samples (200X mean coverage; 80.0% at 50X) (supplementary material, Table S 2).
Somatic mutations identified in sequenced samples are presented in Figure 2 and supplementary material, Table S 3. Somatic mutations in ATM were identified in 6 of 15 sequenced invasive carcinoma samples (40.0%) representing 5 of 12 patients (41.7%). Similarly, and 1 of 12 sequenced PanIN samples (8.3%) had a somatic mutation in ATM. No ATM somatic mutations were identified in the single sequenced IPMN sample. ATM somatic mutations identified included truncating mutations (nonsense, frameshift indel, and splicing; 3 of 7 mutations; 42.9%) and missense mutations (4 of 7 mutations; 57.1%).
Figure 2. OncoPrint diagram showing immunolabeling and somatic mutation status for invasive carcinoma, PanIN, and IPMN samples in patients with a pathogenic germline ATM variant.

ATM expression status (ATMIHC) determined by immunolabeling. Lost ATM expression – blue square. Retained ATM expression - red square. Not assessed – white rectangle. Genetic alterations in ATM, KRAS, GNAS, TP53, and SMAD4 determined by targeted next generation sequencing. Missense somatic mutations – green square. Truncating somatic mutations – black square. No mutation identified – grey rectangle. Not assessed – white rectangle. Samples grouped by histologic type.
Hotspot KRAS mutations were frequently identified in invasive carcinomas (13 of 15 samples; 86.7%), PanINs (11 of 12 samples; 91.7%), and the IPMN (1 of 1 sample; 100%). While most sequenced samples had only one KRAS somatic mutation, the single sequenced IPMN sample (P2) had both p.G12D and p.Q61H somatic mutations (supplementary material, Table S 2). The spectrum of KRAS somatic mutations identified included p.G12D (8 of 26 KRAS mutations; 30.8%), p.G12R (7 of 26; 26.9%), p.G12V (6 of 26; 23.1%), p.G13D (1 of 26; 3.8%), p.Q61H (4 of 26; 15.4%). Somatic mutations in GNAS, TP53, and SMAD4 were infrequent in sequenced invasive carcinoma and precursor samples (Figure 2; supplementary material, Table S2). No somatic mutations in CDKN2A were identified, however, we did not assess copy number alterations or chromosomal alterations in sequenced samples.
We did not observe co-occurrence of TP53 somatic mutations with either ATM somatic mutation or loss of ATM expression (Figure 2). In one patient (P5), with two sequenced invasive carcinoma samples, somatic mutations in ATM and SMAD4 were identified in both samples. Notably the ATM somatic mutation (NM_000051:c.7629+1G>T) was the same in each sample, while the two SMAD4 somatic mutations were different (NP_005350.1:p.H132N and NP_005350.1:p.R361T) (supplementary material, Table S3). These data suggest a common origin for both sequenced carcinomas, where the somatic mutation in ATM occurred before the somatic mutations in SMAD4.
ATM loss in patients unselected for germline ATM status
We next determined the prevalence of ATM loss in precursor lesions and invasive carcinoma in patients with PDAC unselected for ATM status. As we previously published data on the prevalence of ATM loss in invasive carcinoma from 555 patients with PDAC using the same antibody and scoring system (40), we determined the ATM expression status of PanIN and IPMN from 51 and 105 patients unselected for pathogenic germline ATM variants respectively. The prevalence of ATM loss was similar in PanIN (2 of 51 patients; 3.9%) and (9 of 105 patients; 8.6%) IPMN (Fisher’s exact test; P = 0.5058), in low-grade IPMN (5 of 80 patients; 6.3%) and high-grade IPMN (4 of 47 patients; 8.5%) (Fisher’s exact test; P = 0.7251) and intestinal-type IPMN (0 of 15 patients; 0%) and non-intestinal type IPMN (9 of 94; 9.6%) (Fisher’s exact test; P = 0.3563).
We next compared prevalence of ATM loss in pancreatic precursor lesions to our previously published data on ATM loss in 555 patients with PDAC unselected for ATM germline status. We did not identify any statistically significant differences in ATM loss between PanIN (2 of 51 patients; 3.9%) and PDAC between the two groups (67 of 555 patients; 12.1%) (Fisher’s exact test; P = 0.1040), or IPMN (9 of 105 patients; 8.6%) and PDAC (67 of 555 patients; 12.1%) (Fisher’s exact test; P = 0.4038).
We also determined the prevalence of ATM loss by IHC in 29 patients with colloid carcinoma unselected for ATM germline status. The prevalence of ATM loss in patients with colloid carcinoma (9 of 29 patients; 31.0%) was significantly higher compared to usual PDAC (67 of 555 patients; 12.1%) (Fisher’s exact test; P = 0.0076), IPMN (9 of 105 patients; 8.6%) (Fisher’s exact test; P = 0.0040), or PanIN (2 of 51 patients; 3.9%) (Fisher’s exact test; P = 0.0013). These data clearly indicate that loss of ATM is an important aspect of the pathogenesis of colloid carcinomas.
Interestingly, 6 patients had multiple samples assessed that were discordant for ATM expression status (supplementary material, Table S4). In general, discordant expression occurred within samples of the same histological type and grade (one patient with PanIN, one patient with low-grade IPMN, two patients with high-grade IPMN, and one patient with colloid carcinoma) and likely represented clonal heterogeneity. However, one patient had samples of low-grade and high-grade IPMN assessed had ATM loss restricted to the high-grade sample (supplementary material, Figure S1).
Discussion
ATM is a pancreatic cancer susceptibility gene frequently identified by germline genetic testing in patients with PDAC and their relatives (12,14). In carriers of a pathogenic germline ATM variant, the risk of developing PDAC is high, at 9.5% by age 80 years (41). However, despite these clear findings supporting the importance of ATM in the development of PDAC, little is known about the role of ATM in pancreatic tumorigenesis. For example, as ATM is a tumor suppressor gene, the remaining wildtype copy of ATM in individuals with a pathogenic germline ATM variant is predicted to be mutated or lost during tumorigenesis (42). Understanding the prevalence, timing, and type of somatic alterations that result in loss of ATM during development of PDAC is essential to improve clinical surveillance, early detection initiatives, chemoprevention, and targeted therapies.
The prevalence of somatic loss of ATM in invasive carcinoma from patients with PDAC and pathogenic germline ATM variants, either by somatic mutation and/or loss of ATM expression, was 75% (18 of 24 patients; Figure 2 and Table 2). The 25% of patients without an identifiable somatic alteration in ATM in their invasive carcinoma suggests that either the wildtype ATM allele is not somatically altered, or that somatic loss occurred by a mechanism not detected by the approaches used to assess somatic ATM status in this study. Regardless, these data clearly indicate that somatic loss of ATM in invasive carcinoma is common in patients with PDAC and a pathogenic germline ATM variant.
A recent study suggested that somatic mutations in TP53 are mutually exclusive with inactivation of the ATM gene (29). In support of this observation, we did not identify any TP53 somatic mutations in any precursor or invasive carcinoma samples that had either a somatic mutation in ATM or loss of ATM expression (Figure 2), however, this observation was not statistically significant. Analysis of additional samples is necessary to confirm mutual exclusivity of TP53 somatic mutation and somatic ATM mutations or loss of ATM expression in patients with a pathogenic germline ATM variant.
We were also able to determine the prevalence of ATM loss in colloid carcinomas unselected for germline ATM status using IHC. We found that ATM loss was common in colloid carcinomas (9 of 29 samples; 31.0%) and that ATM loss was significantly more prevalent in this subtype of pancreatic cancer compared to PDAC unselected for subtype, PanIN, and IPMN. These data indicate that loss of ATM is an important event in the development of colloid carcinomas which is also supported by our previous observations of an increased prevalence of colloid carcinomas in patients with pathogenic germline ATM variants (23).
Several lines of evidence from our study suggest that loss of ATM is a relatively late event in pancreatic tumorigenesis. Somatic mutations and/or loss of expression of ATM were almost completely restricted to invasive carcinoma in patients with PDAC and a pathogenic germline ATM variant, with only 1 of 28 immunolabeled precursor lesions and 1 of 13 sequenced precursor lesions demonstrating loss of ATM expression or a somatic mutation in ATM respectively (Table 2; Figure 2). The precursor lesion with loss of ATM expression was a high-grade IPMN. All other immunolabeled precursor lesions were low-grade and had intact ATM expression. Of note, we could not establish whether the high-grade IPMN sampled was a precursor lesion or represented ductal cancerization. The precursor lesion with a somatic mutation in ATM was a PanIN and the mutation resulted in a missense change of unknown functional consequence (p.R832C; Supplementary Table 3). While immunostaining of the PanIN demonstrated retention of ATM expression, invasive carcinoma from the same patient demonstrated loss of ATM expression, indicating a different mechanism for somatic loss of ATM in the patient’s invasive carcinoma compared to their PanIN.
While we did not see a significant difference in prevalence of ATM loss between precursor lesions and PDAC in patients unselected for germline ATM status, the total number of PanIN and IPMN samples included in our study was small and a larger study incorporating more patients with precursor lesions is necessary to elucidate differences in prevalence. Notedly, however, one patient with IPMN with both high-grade and low-grade dysplasia had ATM loss that was restricted to high-grade component (supplementary material, Table S3 and Figure S1). The late biallelic inactivation of ATM in germline ATM alteration carriers is consistent with evidence that patients with pathogenic ATM variants do not have an increase in pancreatic precursor lesions (23).
Finally, we also found that ATM loss was statistically more prevalent in colloid carcinomas compared PDAC unselected for histologic subtype, IPMN, and PanIN from patients unselected for germline ATM status. Furthermore, lack of ATM expression in the more common precursor to colloid carcinoma, intestinal type IPMN, was not detected in this study. This may be due to the small numbers available for study, or potentially, it may reflect the late occurrence of ATM alterations in the progression of IPMN to colloid carcinoma. Given the evidence that ATM loss is a later event in patients with pathogenic germline ATM variants, additional studies are warranted to assess the utility of ATM loss as a biomarker of progression.
In summary, we present an analysis of ATM loss during pancreatic tumorigenesis in patients with a pathogenic germline ATM variant and those unselected for germline ATM status. We find that loss of ATM is a common event in patients with a pathogenic germline ATM variant who develop PDAC, as well as in patients with colloid carcinoma unselected for germline ATM status. We also find that loss of ATM is likely a later event in pancreatic tumorigenesis. These observations may have implications for the use of ATM loss as a biomarker of neoplastic progression and patient care.
Supplementary Material
Table 3.
ATM expression status in invasive carcinoma, IPMN, and PanIN from patients with a pathogenic germline ATM variant as determined by immunolabeling.
| Lesion morphology | Lost | Retained | Total | Percent lost | P value |
|---|---|---|---|---|---|
| PanIN | 0 | 24 | 24 | 0 | <0.0001 |
| IPMN | 1 | 3 | 4 | 25.0 | 0.1048 |
| Invasive carcinoma | 18 | 7 | 25 | 72.0 |
PanIN - Pancreatic intraepithelial neoplasm. IPMN - intraductal papillary mucinous neoplasm. Two-tailed P values (PanIN versus invasive carcinoma and IPMN versus invasive carcinoma) calculated using a Fisher’s exact test.
Acknowledgements
This study was supported by NIH/NCI P50 CA62924 and R00 CA190889; Susan Wojcicki and Dennis Troper, the Sol Goldman Pancreatic Cancer Research Center; the Rolfe Pancreatic Cancer Foundation; the Joseph C. Monastra Foundation; the Gerald O Mann Charitable Foundation (Harriet and Allan Wulfstat, Trustees); The Dutch Cancer Society (Grant 2016-I 10289). We thank Kevin T. Jamouss for help with slide imaging.
Footnotes
Author contributions statement
NJR conceptualized study. CLW, JH, DSK, REB, ADS, MGG, LAAB, RHH, APK, TL, LDW and NJR designed experiments. CLW, JH, DSK, REB, ADS, AD, LZ, MGG, LAAB, RHH, APK, TL and NJR provided resources. RMP, ZJ, DH, VK, CG, LZ, RHH, APK, LDW and NJR curated data. RMP, ZJ, DH, VK, CG, KF, NN, BH, MS, TL, LDW and NJR acquired, analyzed, and interpreted data. RMP and NJR wrote the manuscript. RMP, ZJ, DH, VK, CG, KF, NN, BH, MS, CLW, JH, DSK, REB, ADS, AD, LZ, MGG, LAAB, RHH, APK, TL, LDW and NJR reviewed and edited the manuscript. NJR was responsible for supervision and administration of the study.
SUPPLEMENTARY MATERIAL ONLINE
Figure S1. Low-grade and high-grade IPMN samples from an individual patient discordant for ATM expression
Table S1. Targeted capture regions
Table S2. Summary sequencing statistics for sequenced samples
Table S3. Somatic mutations identified in patient samples
Table S4. Discordant ATM expression in patients unselected for germline ATM status
Data availability statement
Histological images and targeted gene panel data are available on request from the corresponding author in accordance with institutional policies.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin 2021;71:7–33 [DOI] [PubMed] [Google Scholar]
- 2.Shindo K, Yu J, Suenaga M, Fesharakizadeh S, Cho C, Macgregor-Das A, et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J Clin Oncol 2017:JCO2017723502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yurgelun MB, Chittenden AB, Morales-Oyarvide V, Rubinson DA, Dunne RF, Kozak MM, et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet Med 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA 2018;319:2401–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tempero MA, Malafa MP, Al-Hawary M, Behrman SW, Benson AB, Cardin DB, et al. Pancreatic Adenocarcinoma, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2021;19:439–57 [DOI] [PubMed] [Google Scholar]
- 6.Stoffel EM, McKernin SE, Brand R, Canto M, Goggins M, Moravek C, et al. Evaluating Susceptibility to Pancreatic Cancer: ASCO Provisional Clinical Opinion. J Clin Oncol 2018:JCO1801489 [DOI] [PubMed] [Google Scholar]
- 7.Canto MI, Harinck F, Hruban RH, Offerhaus GJ, Poley JW, Kamel I, et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013;62:339–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 2015;110:223–62; quiz 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lavin MF, Scott S, Gueven N, Kozlov S, Peng C, Chen P. Functional consequences of sequence alterations in the ATM gene. DNA Repair (Amst) 2004;3:1197–205 [DOI] [PubMed] [Google Scholar]
- 10.Pizarro JG, Folch J, de la Torre AV, Junyent F, Verdaguer E, Jordan J, et al. ATM is involved in cell-cycle control through the regulation of retinoblastoma protein phosphorylation. J Cell Biochem 2010;110:210–8 [DOI] [PubMed] [Google Scholar]
- 11.Skaro M, Nanda N, Gauthier C, Felsenstein M, Jiang Z, Qiu M, et al. Prevalence of Germline Mutations Associated With Cancer Risk in Patients With Intraductal Papillary Mucinous Neoplasms. Gastroenterology 2019;156:1905–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov 2012;2:41–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takai E, Yachida S, Shimizu K, Furuse J, Kubo E, Ohmoto A, et al. Germline mutations in Japanese familial pancreatic cancer patients. Oncotarget 2016;7:74227–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov 2016;6:166–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaffee KG, Oberg AL, McWilliams RR, Majithia N, Allen BA, Kidd J, et al. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet Med 2018;20:119–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shindo K, Yu J, Suenaga M, Fesharakizadeh S, Cho C, Macgregor-Das A, et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J Clin Oncol 2017;35:3382–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research Network. Electronic address aadhe, Cancer Genome Atlas Research N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017;32:185–203 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith AL, Wong C, Cuggia A, Borgida A, Holter S, Hall A, et al. Reflex Testing for Germline BRCA1, BRCA2, PALB2, and ATM Mutations in Pancreatic Cancer: Mutation Prevalence and Clinical Outcomes From Two Canadian Research Registries. JCO Precision Oncology 2018:1–16 [DOI] [PubMed] [Google Scholar]
- 19.Hsu F-C, Roberts NJ, Childs E, Porter N, Rabe KG, Borgida A, et al. Risk of Pancreatic Cancer Among Individuals With Pathogenic Variants in the ATM Gene. JAMA Oncology 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Reilly EM, Lee JW, Lowery MA, Capanu M, Stadler ZK, Moore MJ, et al. Phase 1 trial evaluating cisplatin, gemcitabine, and veliparib in 2 patient cohorts: Germline BRCA mutation carriers and wild-type BRCA pancreatic ductal adenocarcinoma. Cancer 2018;124:1374–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, De Jesus-Acosta A, Jones S, Maitra A, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther 2011;10:3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutchings D, Jiang Z, Skaro M, Weiss MJ, Wolfgang CL, Makary MA, et al. Histomorphology of pancreatic cancer in patients with inherited ATM serine/threonine kinase pathogenic variants. Mod Pathol 2019;32:1806–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rafiei S, Fitzpatrick K, Liu D, Cai MY, Elmarakeby HA, Park J, et al. ATM Loss Confers Greater Sensitivity to ATR Inhibition Than PARP Inhibition in Prostate Cancer. Cancer Res 2020;80:2094–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunlop CR, Wallez Y, Johnson TI, Bernaldo de Quiros Fernandez S, Durant ST, Cadogan EB, et al. Complete loss of ATM function augments replication catastrophe induced by ATR inhibition and gemcitabine in pancreatic cancer models. Br J Cancer 2020;123:1424–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perkhofer L, Schmitt A, Romero Carrasco MC, Ihle M, Hampp S, Ruess DA, et al. ATM Deficiency Generating Genomic Instability Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Therapy-Induced DNA Damage. Cancer Res 2017;77:5576–90 [DOI] [PubMed] [Google Scholar]
- 27.Gout J, Perkhofer L, Morawe M, Arnold F, Ihle M, Biber S, et al. Synergistic targeting and resistance to PARP inhibition in DNA damage repair-deficient pancreatic cancer. Gut 2021;70:743–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roger E, Gout J, Arnold F, Beutel AK, Muller M, Abaei A, et al. Maintenance Therapy for ATM-Deficient Pancreatic Cancer by Multiple DNA Damage Response Interferences after Platinum-Based Chemotherapy. Cells 2020;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park W, O’Connor CA, Bandlamudi C, Forman D, Chou JF, Umeda S, et al. Clinico-genomic characterization of ATM and HRD in Pancreas Cancer: Application for Practice. Clin Cancer Res 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell R, Perkhofer L, Liebau S, Lin Q, Lechel A, Feld FM, et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat Commun 2015;6:7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bosman FT, World Health Organization., International Agency for Research on Cancer. WHO classification of tumours of the digestive system. Lyon: International Agency for Research on Cancer; 2010. 417 p. p. [Google Scholar]
- 32.Basturk O, Hong SM, Wood LD, Adsay NV, Albores-Saavedra J, Biankin AV, et al. A Revised Classification System and Recommendations From the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. Am J Surg Pathol 2015;39:1730–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hosoda W, Chianchiano P, Griffin JF, Pittman ME, Brosens LA, Noe M, et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. J Pathol 2017;242:16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26:589–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, Lichtenstein L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv 2019:861054 [Google Scholar]
- 36.Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018:201178 [Google Scholar]
- 37.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson JT, Thorvaldsdottir H, Wenger AM, Zehir A, Mesirov JP. Variant Review with the Integrative Genomics Viewer. Cancer Res 2017;77:e31–e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaur H, Salles DC, Murali S, Hicks JL, Nguyen M, Pritchard CC, et al. Genomic and Clinicopathologic Characterization of ATM-deficient Prostate Cancer. Clin Cancer Res 2020;26:4869–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim H, Saka B, Knight S, Borges M, Childs E, Klein A, et al. Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clin Cancer Res 2014;20:1865–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu FC, Roberts NJ, Childs E, Porter N, Rabe KG, Borgida A, et al. Risk of Pancreatic Cancer Among Individuals With Pathogenic Variants in the ATM Gene. JAMA Oncol 2021;7:1664–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971;68:820–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Histological images and targeted gene panel data are available on request from the corresponding author in accordance with institutional policies.