

Abstract
Well-designed placebo-controlled clinical trials are critical to the development of novel treatments for epilepsy, but their design has not changed for decades. Patients, clinicians, regulators, and innovators all have concerns that recruiting for trials is challenging, in part, due to the static design of maintaining participants for long periods on add-on placebo when there are an increasing number of options for therapy. A traditional trial maintains participants on blinded treatment for a static period (e.g., 12 weeks of maintenance), during which participants on placebo have an elevated risk of sudden unexpected death in epilepsy compared to patients on an active treatment. Time-to-event trials observe participants on blinded treatment until a key event occurs (e.g., post-randomization seizure count matches pre-randomization monthly seizure count). In this manuscript, we review the evidence for these designs based on re-analysis of prior trials, one published trial that used a time-to-2nd seizure design, and experience from an ongoing blinded trial. We also discuss remaining concerns regarding time-to-event trials. We conclude that, despite potential limitations, time-to-event trials are a potential promising mechanism to make trials more patient-friendly and reduce placebo exposure, which are urgent needs to improve safety and increase recruitment to trials.
Keywords: prerandomization seizure count, sudden unexpected death in epilepsy (SUDEP), recruitment, seizure cycling, research roundtable for epilepsy (RRE)
1. Introduction
The dual goals of clinical trials for epilepsy are to establish efficacy while maintaining safety. In traditionally designed clinical trials, participants keep a seizure diary during a pre-randomization baseline period of 4- to 8-weeks, followed by a blinded post-randomization phase that can include titration plus 12 or more weeks of maintenance (Figure 1).[1] To be eligible for these trials, participants frequently are required to have a high baseline seizure frequency (at least 4 seizure per month).[2, 3] For placebo-controlled adjunctive efficacy trials of antiseizure medications (ASMs), participants’ baseline ASMs are unchanged during the entire study so that the only change in seizure treatment is the introduction of active drug or placebo at randomization. In the traditional design, patients remain on the same dose of the same ASMs during 1 month before entry, 1–2 months of baseline, and 3 or more months in the treatment phase, for a total of at least 5 months. In clinical practice, medicine changes would be made over this interval. Maintenance of ineffective treatment can cause harm. In a review of mortality for participants enrolled in epilepsy trials, participants in the placebo arm had a 5.8 times greater risk of sudden unexpected death in epilepsy (SUDEP) compared to participants taking effective treatments.[4] In infants, a higher seizure burden has been required until recently to limit trial duration.[5, 6] Investigators and families were reluctant to expose participants with this high seizure burden to placebo when the studies’ ASM usually was available off label. Therefore, there is a need to reduce participant exposure to placebo and other ineffective treatments.
Figure 1:
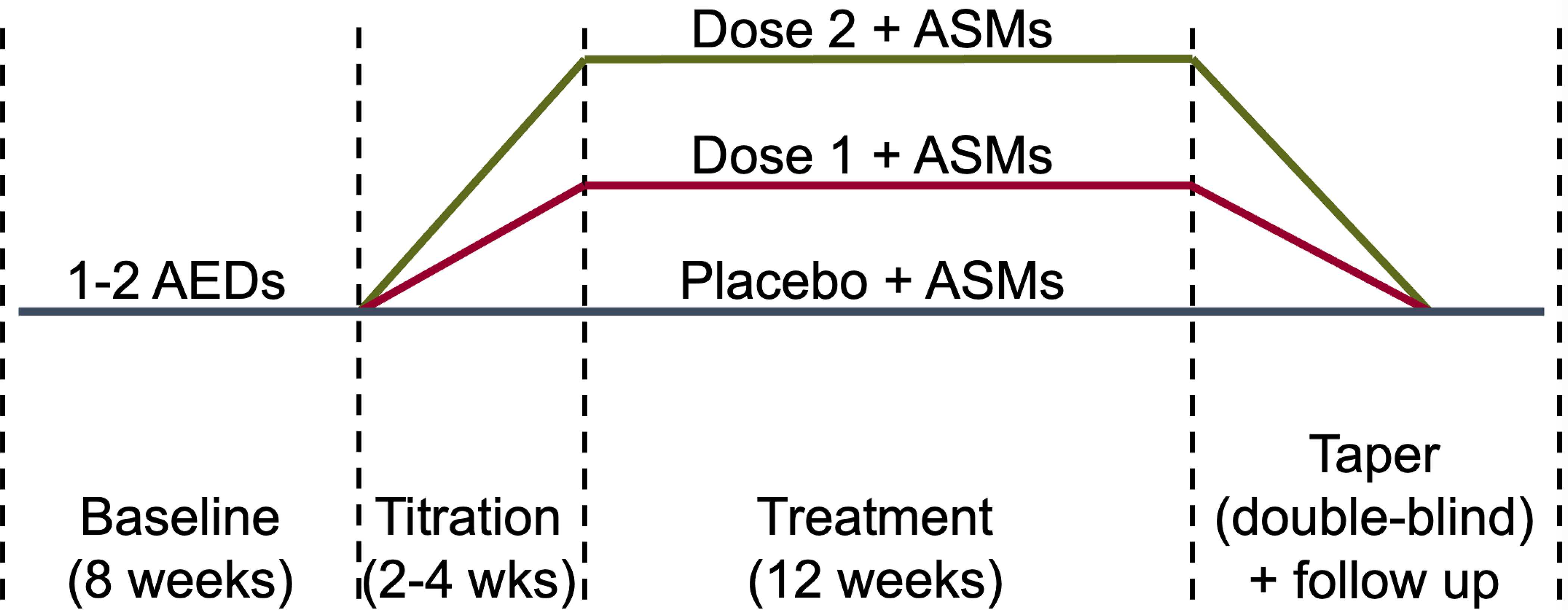
Typical timeline of a double-blind placebo-controlled parallel trial for a treatment of epilepsy. Abbreviations: antiseizure medications (ASMs), weeks (wks).
Reducing this risk of harm could improve recruitment. The time needed to recruit participants has been increasing over time, in part, because each trial site typically recruits 1 to 3 participants instead of the 7 to 15 recruited into trials years ago.[7–10] Limited participant numbers from each site make it difficult to statistically evaluate for site-level sources of variability. Also, the rate of placebo-response substantially varies by country, which can lead to substantially different effect sizes.[11] Therefore, there is a critical need to improve participant recruitment.
Time-to-event (TTE) design clinical trials require post-randomization observation only until a target “event” has occurred, thereby potentially reducing harm. Withdrawal to monotherapy trials were performed as TTE trials, but these studies carried a high risk of harm since participants were withdrawn to a “pseudoplacebo,” defined as an inadequate ASM dose. The “event” consisted of “escape” criteria that resulted in 85% of participants on pseudoplacebo reaching an endpoint before 112 days: (1) post-randomization doubling of monthly seizure frequency; (2) seizure clusters defined by doubling of the highest consecutive 2-day seizure rate; (3) a new, more severe, seizure type; or (4) clinically significant prolongation of generalized tonic-clonic seizures.[12, 13] More recently, proposals for TTE trials have evaluated the time-to-Nth seizure counts including the 1st seizure, 2nd seizure, and the individually determined average monthly pre-randomization seizure count (PSC).[14] After reaching this “event” of interest, participants can switch to the open-label extension or withdraw from the study. In traditionally designed trials, more than 80% of participants opted for open-label extension therefore the risk of withdrawing effective treatment is low.[15–28] Treatment efficacy can be measured using Cox-proportional hazards models, proportion of failures, and the traditional primary efficacy endpoints of percent reduction in seizure frequency and 50% responder rate.[29] These latter primary efficacy endpoints are calculated to the “event” of interest, similar to the last observation carried forward method for addressing missing data that is used within trials commonly.
In this review, we provide an up-to-date overview of the TTE design by summarizing the existing evidence on the TTE design for ASM evaluation, presenting experience from an ongoing TTE designed trial, and reporting a discussion of remaining concerns from multiple stakeholders who attended the 2022 Research Roundtable for Epilepsy (RRE).
2. Existing Evidence about TTE Trials
The evidence supporting time-to-Nth trials focuses on re-analysis of traditionally designed trials for adjunctive ASMs plus the trial of lacosamide that used the endpoint of time-to-2nd primary generalized tonic-clonic (PGTC) seizure.[14, 29–31] Most retrospective evidence supports the time-to-PSC design where each participant is observed until their seizure count post-randomization exceeds their individual baseline average monthly seizure count (Figure 2). As compared to the time-to-1st and 2nd seizure designs, time-to-PSC encourages at least a month of observation post-randomization, which can reduce sensitivity to seizure clusters and monthly seizure cycles. In the re-analysis of fenfluramine for Dravet syndrome a substantial portion of participants on placebo reached time-to-PSC between 30 to 50 days after randomization (Figure 3).[32] Figure 4 uses idealized simulated seizure diaries designed to match adult patients with medication refractory epilepsy to illustrate the expected time-to-PSC based on the degree of response to treatment. Due to a placebo effect, the median time-to-PSC is longer than 28-days for participants with no change (100% of baseline seizure frequency).
Figure 2:
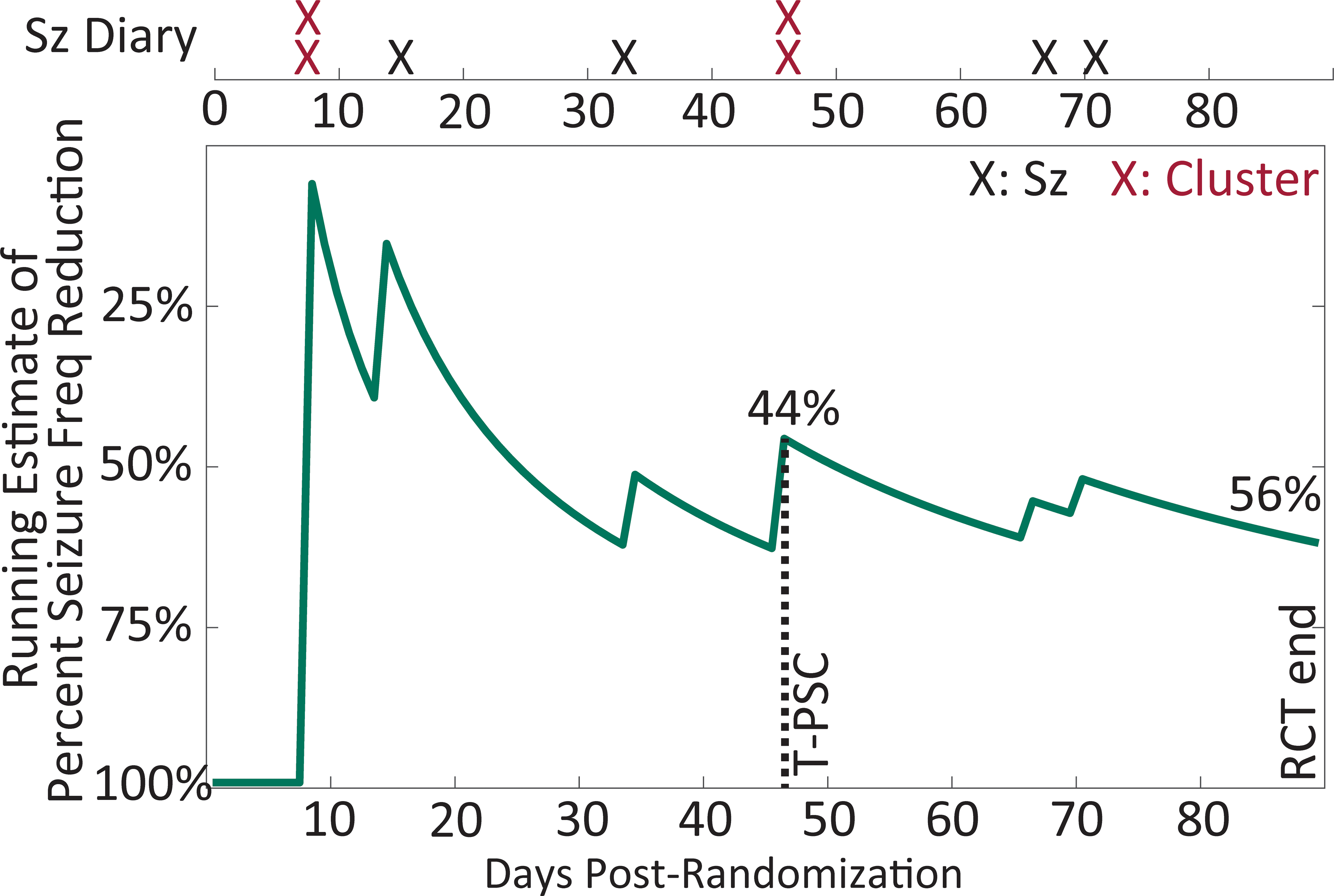
A hypothetical illustration of how the estimate of seizure frequency (seizure freq) stabilizes over the course of the typical 12-week randomized controlled trial (RCT) for a simulated participant randomized to placebo who had 6 seizures (Sz) in a 4-week baseline. The simulation was using the methods of Goldenholz and colleagues (Ann Clin Trans Neurol 2017). The time-to-event endpoint of time-to-prerandomization monthly seizure count (T-PSC) is illustrated at 48 days, with an interim seizure frequency reduction of 44%, compared to a 56% reduction when treatment was continued through 12-weeks. Comparison to other historical escape criteria is illustrated by highlighting seizure clusters.
Figure 3:
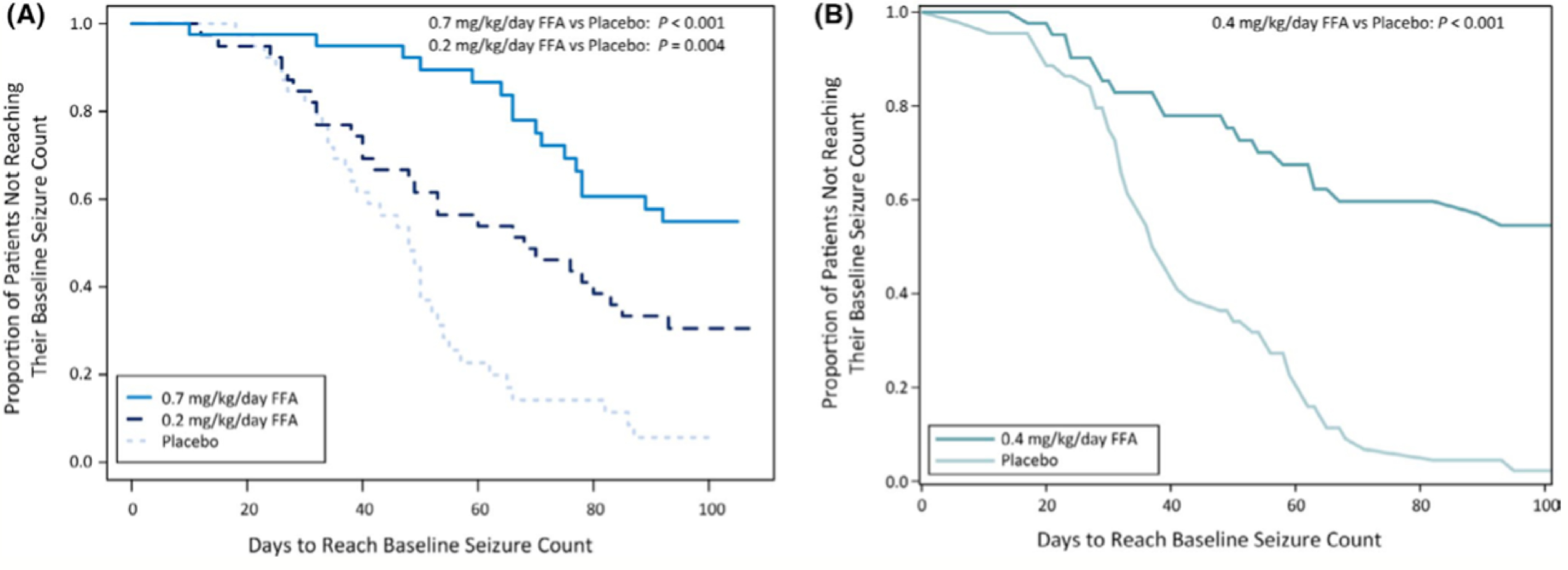
Time to pre-randomization monthly seizure count for participants with Dravet Syndrome enrolled in the fenfluramine (FFA) trial. Reproduced from Sullivan et al. Epilepsia 2021.
Figure 4:
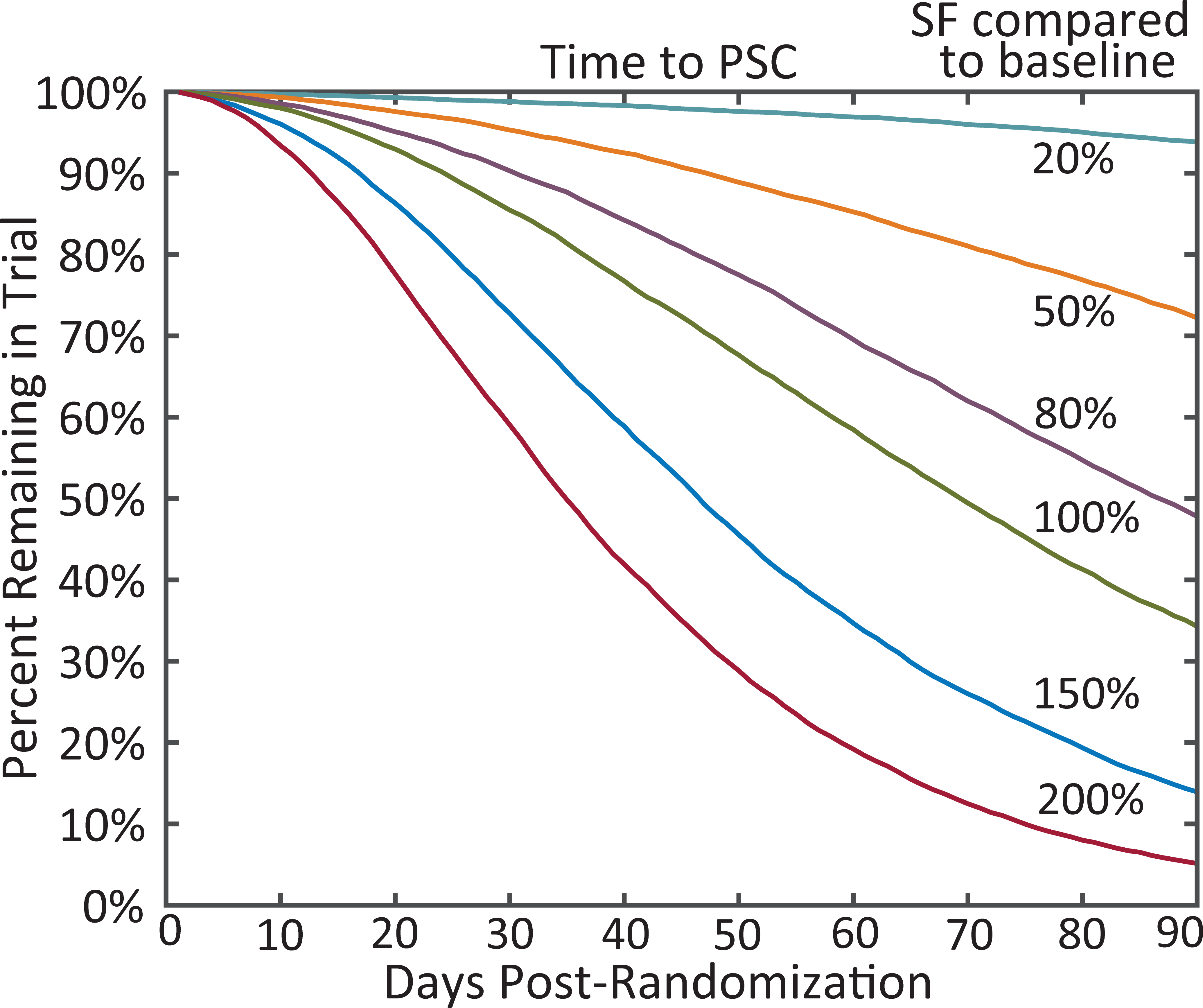
The expected time to pre-randomization monthly seizure count (PSC) for participants with simulated seizure frequency (SF) response to an antiseizure medication. The 100% curve reflects no change; 200% reflects doubling of SF; and 50% reflects halving of SF. Simulations based on Goldenholz et al. Ann Clin Trans Neurol 2017.
When this time-to-PSC design was used, sufficient seizures were observed so that the efficacy conclusions were unchanged, despite shortening observation duration. When comparing percent reduction in seizure frequency and 50% responder rate at time-to-PSC to the full-length trial, there was more than 90% and 80% correspondence on an individual participant level, respectively.[29] Using simulated data, the time-to-PSC outcome had similar power and total trial cost to the percent reduction in seizure frequency, and both approaches were superior to 50% responder rate.[33]
The only published prospective time-to-Nth seizure trial evaluated the efficacy of lacosamide for PGTC seizures.[31] After the 6-week titration period, participants were observed until the 2nd PGTC seizure. Around 25% of participants on lacosamide and 50% of participants on placebo had experienced the 2nd PGTC seizure before 12 weeks. By 24 weeks, 45% of participants on lacosamide and 67% of participants on placebo had had experienced the 2nd PGTC seizure.
There are unique challenges and proposals for TTE designs in pediatrics. The seizure frequency in infantile epilepsy can be higher than adult epilepsy. Auvin and colleagues evaluated if “placebo”-observation could be reduced further by applying TTE both in the baseline and post-randomization phase.[34] In their re-analysis of trials for levetiracetam and lacosamide in children, they proposed three baseline duration categories: (1) at least daily seizures observed for 7 days, (2) at least weekly seizures for 14 days, and (3) at least 3 seizures per month for 28 days.[30] After enrollment, participants were evaluated at 7, 14, and 28 days. If the participant had sufficient baseline seizures at day 7, then blinded randomization would start. If baseline seizure frequency was lower, then the participant would continue baseline until either qualifying for baseline category (2), or baseline category (3), or having insufficient baseline seizures for enrollment. After randomization, participants who qualified for these categories were followed until the pre-randomization seizure count matched the post-randomization seizure count. When analyzed using a stratified Cox-proportional hazards model, the efficacy of lacosamide and levetiracetam were re-demonstrated, and the effect sizes were similar to the original trials’ analysis.
3. Experience from an ongoing clinical study
We provide insights from an ongoing TTE study for adults with focal-onset seizures. In a proof-of-concept study, Dr. van der Geyten and colleagues required at least 28 days of post-randomization observation. If baseline seizure count was exceeded during the 1st 28 days, or any subsequent moving 28-day window, the participant would either complete the trial or enter the open label extension. If this event was not reached, then participants would take blinded treatment for up to 12 weeks.
In their preliminary analysis, they observed substantial variability in baseline seizure frequency, seizure cycling, and unanticipated missing data (Figure 5). They observed cycles of seizures where some participants had 2 to 3 weeks of seizure freedom, followed by a few days with a cluster of seizures. The relationship between each individual participant’s seizure cycle and the phases of the trial could substantially impact the conclusions of the trial. In one example participant, the blinded treatment lengthened the interval between seizures but the total number of seizures within each cluster remained the same. In another example participant, the baseline was variable: the first 4 weeks included 15 seizures whereas the subsequent 4 weeks included 4 seizures. After randomization, the patient had 14 seizures in 12 weeks, representing a 69% improvement from 15 seizures during the first 4-week baseline, but a 17% worsening from 4 seizures during the second 4-week baseline period. At this time the study was blinded, so it was unknown whether the participant received placebo or active treatment. It was theoretically possible that the participant had spontaneously transitioned to a lower seizure frequency state between the first and second month of baseline. When in that lower seizure frequency state, the blinded treatment had no substantial impact upon that participant’s seizure frequency. This natural variability in seizure frequency also has been described in re-analysis of other trials [35] and could substantially impact the conclusions of a trial with a short baseline like that proposed by Auvin and colleagues.[34] This natural variability in seizure frequency could be addressed with a longer baseline, but a longer baseline period would incur a similar increase in SUDEP due to more exposure to a placebo equivalent.[4]
Figure 5:
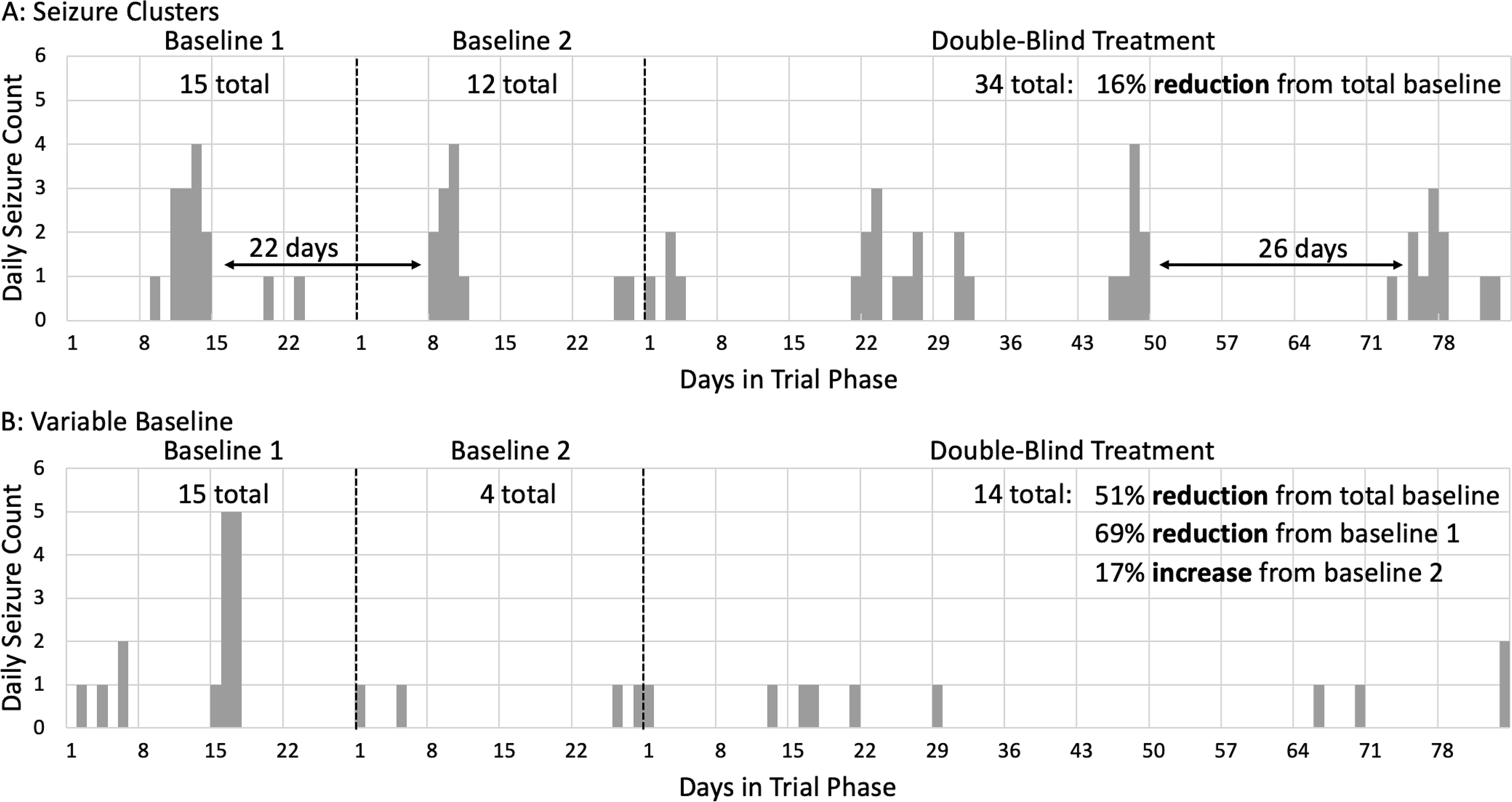
Individual seizure diaries from blinded adult participants in an ongoing time-to-event clinical trial for focal-onset seizures. (A) Periodicity in seizures with brief periods of seizures and semi-regular seizure free intervals. (B) Variability in the seizure frequency in the first versus second baseline period.
In addition, they observed challenges with missing data, where seizure data was unavailable for up to 45 days for some participants from Ukraine who were impacted by geopolitical events. In one participant, the monthly seizure frequency was lower than baseline prior to the missing data, and higher than baseline after the missing data. Therefore, the time to exceed monthly baseline seizure count was hypothesized to occur sometime within the missing data. Additionally, the moving 28-day window of seizure counts was theoretically and practically concerning when the 28-days spanned the period of missing data. If the missing data were excluded, then either the 28-day windows could include seizure counts more than 1 month apart or participants would need another 28-days of available data before being considered to switch to open label extension or complete participation.
4. Remaining points of discussion
Seizures and seizure frequency are dynamic and can be individually unique. Our current statistical models of seizure counts use averages over longer periods to reduce seizure variability from errors in patient-reported seizure counts, clustering, seizure cycling, and natural variability in seizure frequency.[35–38] These complex patterns in seizure diaries may be enhanced in a TTE trial. For example, seizure clusters likely average out across 12-weeks, but an early seizure cluster in a TTE trial may lead to a false determination of non-response. In addition, the “event” could occur during the titration phase. For this reason, it is advisable to “start the clock” during maintenance. Even in the early maintenance, delayed onset of efficacy could lead to false determinations of non-response. These limitations in a TTE design could be addressed by a composite endpoint requiring a minimum duration of observation followed by a TTE endpoint. However, a long minimum duration of observation would perpetuate the very problems that TTE seeks to address.
In an ideal trial, presumptive participants would have a long-term high-quality seizure diary before enrollment to address this natural variability and avoid the need for a prospective baseline. While seizure diary apps may provide quality diaries, most trials require at least 4 weeks of prospective baseline including at least 1 to 4 seizure(s) to supplement 4 to 12 weeks of historical baseline.[21, 22, 31, 39, 40] This may be due to the poor association between seizure diaries and objectively measured seizures using devices.
The inherent limitation in the reliability of participant-provided seizure diaries applies both to current trial methods and TTE trials. Re-analysis of prior RCTs with a time to pre-randomization seizure count design demonstrated that the TTE design reproduced the group-level efficacy conclusions, effect size, and individual-level efficacy in more than 80% of participants, therefore this TTE design measures the outcome of improvement of seizures similarly to the traditional design. It is important that both trial designs require that seizures be countable. This design would be inappropriate for trials of, for example, generalized absence epilepsies or epileptic spasms in the context of infantile spasm syndrome where video-EEG monitoring may be required to accurately count seizures.[41, 42] An alternative TTE endpoint could be lack of seizure freedom as measured by clinical observation plus video-EEG monitoring at scheduled intervals.
In addition to shorter observation, a TTE design trial observes fewer seizures which, intuitively, could reduce statistical power. However, simulations and re-analyses of prior trials have demonstrated similar or improved statistical power of a TTE design to traditional analyses.[33] In pediatric epilepsies like the application of time-to-PSC used to evaluate the efficacy of fenfluramine for Dravet syndrome, baseline seizure frequencies were high enough that numerous seizures were observed prior to the TTE endpoint (median 26/month).[32] In contrast, the perampanel trial for primary generalized tonic-clonic seizures included seizure frequencies as low as 1 per month (median 2.6/month) and the TTE reanalysis redemonstrated efficacy.[29] These two applications reflect epilepsies that are particularly suitable for a TTE design because of the high risk of SUDEP, clear identifiable clinical correlate of myoclonic or generalized tonic-clonic seizures, and sufficient baseline seizure frequency.
In adult epilepsy, it is becoming increasingly challenging to recruit patients with at least weekly seizures, the traditional minimum seizure frequency for trials. Insufficient seizure frequency of less than 3 per month occurred in at least 55% of patients and was the most common reason for ineligibility for clinical trials (Table 1).[43] Therefore, there’s high interest in lowering these minimum seizure frequency requirements, as has been done in four prior trials.[18, 31, 39, 44] Lowering these minimums requires efficacy to be determined with both fewer pre- and post-randomization seizures.[2, 3] TTE designs reduce the number of post-randomization seizures, thereby potentially less statistical power for low seizure frequencies. For example, there are edge effects where a single seizure that occurs either just before or just after the 12-week maintenance period could substantially change a participant’s percent reduction in seizure frequency and 50% response rate (e.g., 3 baseline seizures over 12 weeks compared to 1 post-randomization seizure in 12 weeks [67% reduction, responder] versus 2 post-randomization seizures in 12 weeks [33% reduction, non-responder]). Increased variability means reduced statistical power. Therefore, expanding trials to patients with fewer seizures both with and without a TTE design could necessitate increased participants or longer observation. It is unclear if the financial costs of that may be outweighed by a shorter recruitment phase due to a profound increase in patient eligibility.
Table 1:
Proportion of patients with focal- or generalized-onset medication resistant epilepsy meeting NINDS 2011 workshop inclusion criteria for clinical trials seen at New York University’s epilepsy clinic in a 3-month period in 2013. Patients with symptomatic and combined focal- and generalized-onset epilepsy were excluded.
| Number (%) | Focal | Generalized |
|---|---|---|
|
| ||
| Total number | 144 | 29 |
| Eligible for clinical trials | 19 (13%) | 1 (3%) |
| Reason for exclusion | ||
|
| ||
| Seizure frequency < 3/month | 84 (58%) | 16 (55%) |
| Age (< 16 years) | 18 (13%) | 10 (37%) |
| Recent ASM change (< 4 weeks) | 27 (19%) | 4 (14%) |
| Unstable medical or psychiatric condition | 11 (8%) | 1 (3%) |
| Inability to maintain seizure diary | 10 (7%) | 0 |
| More than 3 concurrent ASMs | 8 (6%) | 1 (3%) |
This uncertainty regarding statistical power of TTE trials may have been why the time-to-2nd PGTC seizure trial for lacosamide used a 24-week maintenance phase, even though the efficacy results were nearly identical at 12- and 24-weeks.[31] If this uncertainty is not resolved, an overpowered trial may result in more participant-years on placebo by either observing participants on placebo for longer or randomizing more participants to placebo. While an increase of 1,000 participant-years on placebo would be expected to contribute to 6 additional deaths from SUDEP,[4] participants who had not reached the TTE endpoint would be expected to have fewer seizures and thereby a lower risk of SUDEP.[45] The potential for overpowered trials due to increased uncertainties would counteract some of the original goals of TTE trials to reduce total participant-years on placebo and ineffective treatment.
For trials of ultra-rare epilepsies where a critical challenge for the trial is the small number of eligible participants, TTE designs as described here may be inappropriate because, due to limited sample size, trials may need to focus on N-of-1 designs, crossover designs, non-seizure outcomes (e.g., cognitive outcomes), and other methods to obtain as much information as possible from each participant. For urgent situations such as treatment of acute repetitive seizures or status epilepticus, there may be alternative “events” or escape criteria including time to cessation of motor seizures in convulsive status epilepticus, seizure-free withdrawal of general anesthesia in super refractory status epilepticus, or use of a rescue treatment in patients with recurrent repetitive seizures or status epilepticus.
Clinical trials for epilepsy aim to address more than seizures; they also establish safety as well as secondary outcomes.[46] An issue with the TTE design is the potential for reduced observation in the placebo arm of trials that may lead to reduced placebo-controlled adverse effect information. The majority of dose-dependent adverse effects occur early, but idiosyncratic and longer-term adverse effects may take longer to evaluate.[47–51] By having fewer participants on placebo, it’s harder to estimate the background rate of more rare idiosyncratic adverse effects. While these longer-term adverse effects typically are established in open-label extension and phase 4 studies, a minimum duration of blinded observation could observe some of the medium-term adverse effects without substantially increasing placebo-exposure. Further studies of the timing of adverse effects are needed to evaluate this potential limitation of TTE trials.
A broader theme of modern clinical trials for epilepsy includes the exploration of disease modifying treatments for epilepsy. In these treatments, the primary outcome may focus on cognitive function, functional independence, and quality of life. Improvements in non-seizure outcomes could be observed even when seizure frequency is not improved, as suggested by prior work addressing comorbid depression in patients with epilepsy.[52] Changes in these functional outcomes also are slower than changes in seizure frequency, therefore TTE trials focused on seizures alone may be underpowered to observe these additional benefits of treatment. Ideas to address these dual outcomes included an initial TTE trial to evaluate efficacy for seizure, followed by a longer blinded period where patients would be allowed to change non-trial ASMs to optimize seizure control as the non-seizure efficacy outcomes would be evaluated.
5. Conclusion
TTE trials potentially establish the efficacy of novel treatments for epilepsy, while also reducing exposure to ineffective treatments. This design could improve recruitment by reducing participant risk. There is building evidence from re-analysis of prior trials and emerging evidence from trials that implemented this design that the time-to-PSC endpoint may accomplish this without reducing statistical power. However, due to a reliance on a comparatively small number of seizures, these TTE designs may have reduced power if participants with lower seizure frequencies are included in future trials. TTE trials also primarily focus on seizure counts and have reduced placebo-controlled observation for adverse effects or non-seizure efficacy outcomes (e.g., cognition). Additionally, the TTE design does not address known challenges in the analysis of seizure counts within clinical trials including contributions of clustering, seizure cycles, and delayed onset of treatment efficacy. These uncertainties may lead to the initial trials implementing a TTE design to be overpowered by including more participants and lengthening the duration of observation, which counteracts the goals of the TTE design to reduce exposure to placebo and ineffective treatments. Despite these limitations, TTE designed trials may address the urgent needs to increase participant recruitment and improve safety by reducing placebo exposure.
Highlights.
Time to Event designs can reduce participant risk of exposure to ineffective treatments and thereby improve recruitment.
Trial re-analyses suggest Time to Event endpoints can demonstrate the efficacy of treatments in substantially less time.
Time to Event endpoints may be impacted by seizure cycling, clusters, missing data, inaccurate seizure diaries, and low seizure counts.
Time to Event designs may limit the duration of placebo-controlled observation for adverse events.
6. Acknowledgements
This manuscript is a summary of the “hot topic” from the 2022 Research Roundtable for Epilepsy (RRE), which is an annual meeting that involves representatives from the scientific community, pharmaceutical and device companies, patient advocacy groups, and regulatory agencies such as the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), and Health Canada.[46] The intent of the RRE is to address issues in therapy development for epilepsy and seizures. RRE consists of a main topic and a “hot topic”, which is a timely issue that the community should address. In 2022, the RRE selected “time to event” trials as the hot topic due to concerns with ability to recruit for trials in participants with focal onset seizures, and concerns with prolonged placebo exposure when there are an increasing number of therapies. Therefore, this summary reflects both a review of the evidence as well as a statement based on discussion from the multiple stakeholders in clinical trials for epilepsy. We thank all the participants in the Research Roundtable for Epilepsy for their contributions including representatives from multiple stakeholder groups including patients, patient advocates, industry, regulators, and researchers.
Dr. Kerr’s research time was supported by NIH R25 NS089450, NIH U24NS107158, and the American Epilepsy Society.
Footnotes
Conflict of Interest Disclosures & Ethical Publication:
Dr. Kerr writes review articles for Medlink Neurology and has consulting agreements with SK Life Science, Janssen, Biohaven Pharmaceutical, and Radius Health. Dr. French receives salary support from the Epilepsy Foundation and for consulting work and/or attending Scientific Advisory Boards on behalf of the Epilepsy Study Consortium for Aeonian/Aeovian, Alterity Therapeutics Limited, Anavex, Arkin Holdings, Angelini Pharma S.p.A, Arvelle Therapeutics, Inc., Athenen Therapeutics/Carnot Pharma, Autifony Therapeutics Limited, Baergic Bio, Biogen, Biohaven Pharmaceuticals, BioMarin Pharmaceutical Inc., BioXcel Therapeutics, Bloom Science Inc., BridgeBio Pharma Inc., Camp4 Therapeutics Corporation, Cerebral Therapeutics, Cerevel, Clinical Education Alliance, Coda Biotherapeutics, Corlieve Therapeutics, Eisai, Eliem Therapeutics, Encoded Therapeutics, Encoded Therapeutics, Engage Therapeutics, Engrail, Epalex, Epihunter, Epiminder, Epitel Inc., Equilibre BioPharmaceuticals, Greenwich Biosciences, Grin Therapeutics, GW Pharma, Janssen Phamaceutica, Jazz Pharmaceuticals, Knopp Biosciences, Lipocine, LivaNova, Longboard Pharmaceuticals, Lundbeck, Marinus, Mend Neuroscience, Marck, NeuCyte Inc., Neumirna Therapeutics, Neurocrine, Neuroelectives USA Corporation, Neuronetics Inc., Neuropace, NxGen Medicine Inc., Ono Pharmaceutical Co., Otsuka Pharmaceutical Development, Ovid Therapeutics Inc., Paladin Labs, Passage Bio, Pfizer, Praxis, Pure Tech LTY Inc., Rafa Laboratories Ltd, SK Life Sciences, Sofinnova, Stoke, Supernus, Synergia Medical, Takeda, UCB Inc., Ventus Therapeutics, Xenon, Xeris, Zogenix, Zynerba. Dr. French also has received research support from the Epilepsy Study Consortium (Funded by Andrews Foundation, Eisai, Engage, Lundbeck, Pfizer, SK Life Science, Sunovion, UCB, Vogelstein Foundation), the Epilepsy Study Consortium/Epilepsy Foundation (Funded by UCB), GW/FACES, and NINDS. She is on the editorial board of Lancet Neurology and Neurology Today. She is Chief Medical/Innovation Officer of the Epilepsy Foundation. She has received travel reimbursement related to research, advisory meetings, or presentation of results at scientific meetings from the Epilepsy Study Consortium, the Epilepsy Foundation, Angelini Pharma S.p.A., Clinical Education Alliance, NeuCyte, Inc., Neurocrine, Praxis, and Xenon. Stéphane Auvin is Deputy Editor for Epilepsia. He has served as consultant or gave lectures for Angelini, Biocodex, Eisai, Encoded, Grintherapeutics, Jazz Pharmaceuticals, Neuraxpharm, Orion, Nutricia, Proveca, UCB Pharma, Vitaflo, Xenon, Zogenix. He has been investigator for clinical trials for Eisai, Marinus, Proveca, Takeda, UCB Pharma and Zogenix. Dr. Van der Geyten is an employee of Janssen Research & Development, a division of Janssen Phamaceutica, Belgium, and holds stock in Johnson & Johnson companies. Dr. Kenney is employed full-time at Xenon Pharmaceuticals as Chief Medical Officer. Dr. Novak is a full-time employee of Janssen Research and Development. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. Dr. Fountain has received clinical trial grants to the University of Virginia from UCB, SK Lifesciences, Xenon, Neurelis, Medtronic, and InSightec and is an independent director and holds stock at Acumen Pharmaceuticals and Hexokine Therapeutics, and consults and receives stock options at Shackleton Pharma. Dr. Grzeskowiak was an employee of the Epilepsy Foundation. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References:
- 1.Romero J and Goldenholz DM, Statistical efficiency of patient data in randomized clinical trials of epilepsy treatments adds value. Epilepsia, 2020. 61(10): p. 2323–2324. [DOI] [PubMed] [Google Scholar]
- 2.French JA, et al. , Designing a new proof-of-principle trial for treatment of partial seizures to demonstrate efficacy with minimal sample size and duration-a case study. Epilepsy Res, 2013. 106(1–2): p. 230–6. [DOI] [PubMed] [Google Scholar]
- 3.Goldenholz DM, et al. , Monte Carlo simulations of randomized clinical trials in epilepsy. Ann Clin Transl Neurol, 2017. 4(8): p. 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryvlin P, Cucherat M, and Rheims S, Risk of sudden unexpected death in epilepsy in patients given adjunctive antiepileptic treatment for refractory seizures: a meta-analysis of placebo-controlled randomised trials. Lancet Neurol, 2011. 10(11): p. 961–8. [DOI] [PubMed] [Google Scholar]
- 5.Mahmoud AA, et al. , Ineffectiveness of topiramate and levetiracetam in infantile spasms non-responsive to steroids. Open labeled randomized prospective study. Neurosciences (Riyadh), 2013. 18(2): p. 143–6. [PubMed] [Google Scholar]
- 6.Novotny E, et al. , Randomized trial of adjunctive topiramate therapy in infants with refractory partial seizures. Neurology, 2010. 74(9): p. 714–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faught E, et al. , Randomized, controlled, dose-ranging trial of carisbamate for partial-onset seizures. Neurology, 2008. 71(20): p. 1586–93. [DOI] [PubMed] [Google Scholar]
- 8.Krauss GL, et al. , Safety and efficacy of adjunctive cenobamate (YKP3089) in patients with uncontrolled focal seizures: a multicentre, double-blind, randomised, placebo-controlled, dose-response trial. Lancet Neurol, 2020. 19(1): p. 38–48. [DOI] [PubMed] [Google Scholar]
- 9.Krauss GL, et al. , Randomized phase III study 306: adjunctive perampanel for refractory partial-onset seizures. Neurology, 2012. 78(18): p. 1408–15. [DOI] [PubMed] [Google Scholar]
- 10.French JA, et al. , Efficacy and safety of extended-release oxcarbazepine (Oxtellar XR) as adjunctive therapy in patients with refractory partial-onset seizures: a randomized controlled trial. Acta Neurol Scand, 2014. 129(3): p. 143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.French JA, et al. , Adjunctive perampanel for refractory partial-onset seizures: randomized phase III study 304. Neurology, 2012. 79(6): p. 589–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.French JA, et al. , Lamotrigine XR conversion to monotherapy: first study using a historical control group. Neurotherapeutics, 2012. 9(1): p. 176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.French JA, et al. , Historical control monotherapy design in the treatment of epilepsy. Epilepsia, 2010. 51(10): p. 1936–43. [DOI] [PubMed] [Google Scholar]
- 14.French JA, et al. , Time to prerandomization monthly seizure count in perampanel trials: A novel epilepsy endpoint. Neurology, 2015. 84(20): p. 2014–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein P, et al. , A randomized, double-blind, placebo-controlled, multicenter, parallel-group study to evaluate the efficacy and safety of adjunctive brivaracetam in adult patients with uncontrolled partial-onset seizures. Epilepsia, 2015. 56(12): p. 1890–8. [DOI] [PubMed] [Google Scholar]
- 16.Kwan P, et al. , Adjunctive brivaracetam for uncontrolled focal and generalized epilepsies: results of a phase III, double-blind, randomized, placebo-controlled, flexible-dose trial. Epilepsia, 2014. 55(1): p. 38–46. [DOI] [PubMed] [Google Scholar]
- 17.Biton V, et al. , Brivaracetam as adjunctive treatment for uncontrolled partial epilepsy in adults: a phase III randomized, double-blind, placebo-controlled trial. Epilepsia, 2014. 55(1): p. 57–66. [DOI] [PubMed] [Google Scholar]
- 18.Berkovic SF, et al. , Placebo-controlled study of levetiracetam in idiopathic generalized epilepsy. Neurology, 2007. 69(18): p. 1751–60. [DOI] [PubMed] [Google Scholar]
- 19.Babar RK, et al. , Lacosamide add-on therapy for focal epilepsy. Cochrane Database Syst Rev, 2021. 5: p. CD008841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porter RJ, et al. , Retigabine as adjunctive therapy in adults with partial-onset seizures: integrated analysis of three pivotal controlled trials. Epilepsy Res, 2012. 101(1–2): p. 103–12. [DOI] [PubMed] [Google Scholar]
- 21.Halasz P, et al. , Adjunctive lacosamide for partial-onset seizures: Efficacy and safety results from a randomized controlled trial. Epilepsia, 2009. 50(3): p. 443–53. [DOI] [PubMed] [Google Scholar]
- 22.Chung S, et al. , Lacosamide as adjunctive therapy for partial-onset seizures: a randomized controlled trial. Epilepsia, 2010. 51(6): p. 958–67. [DOI] [PubMed] [Google Scholar]
- 23.Naritoku DK, et al. , Lamotrigine extended-release as adjunctive therapy for partial seizures. Neurology, 2007. 69(16): p. 1610–8. [DOI] [PubMed] [Google Scholar]
- 24.Biton V, et al. , Adjunctive lamotrigine XR for primary generalized tonic-clonic seizures in a randomized, placebo-controlled study. Epilepsy Behav, 2010. 19(3): p. 352–8. [DOI] [PubMed] [Google Scholar]
- 25.Biton V, et al. , Double-blind, placebo-controlled study of lamotrigine in primary generalized tonic-clonic seizures. Neurology, 2005. 65(11): p. 1737–43. [DOI] [PubMed] [Google Scholar]
- 26.Brodie MJ, et al. , Efficacy and safety of adjunctive ezogabine (retigabine) in refractory partial epilepsy. Neurology, 2010. 75(20): p. 1817–24. [DOI] [PubMed] [Google Scholar]
- 27.Sachdeo RC, et al. , A double-blind, randomized trial of topiramate in Lennox-Gastaut syndrome. Topiramate YL Study Group. Neurology, 1999. 52(9): p. 1882–7. [DOI] [PubMed] [Google Scholar]
- 28.Biton V, et al. , A randomized, placebo-controlled study of topiramate in primary generalized tonic-clonic seizures. Topiramate YTC Study Group. Neurology, 1999. 52(7): p. 1330–7. [DOI] [PubMed] [Google Scholar]
- 29.Kerr WT, et al. , Time to exceed pre-randomization monthly seizure count for perampanel in participants with primary generalized tonic-clonic seizures: A potential clinical end point. Epilepsia, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson ME, McClung C, and Bozorg AM, Analyses of seizure responses supportive of a novel trial design to assess efficacy of antiepileptic drugs in infants and young children with epilepsy: Post hoc analyses of pediatric levetiracetam and lacosamide trials. Epilepsia Open, 2021. 6(2): p. 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vossler DG, et al. , Efficacy and safety of adjunctive lacosamide in the treatment of primary generalised tonic-clonic seizures: a double-blind, randomised, placebo-controlled trial. J Neurol Neurosurg Psychiatry, 2020. 91(10): p. 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sullivan J, et al. , Fenfluramine significantly reduces day-to-day seizure burden by increasing number of seizure-free days and time between seizures in patients with Dravet syndrome: A time-to-event analysis. Epilepsia, 2022. 63(1): p. 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oliveira A, Romero JM, and Goldenholz DM, Comparing the efficacy, exposure, and cost of clinical trial analysis methods. Epilepsia, 2019. 60(12): p. e128–e132. [DOI] [PubMed] [Google Scholar]
- 34.Auvin S, et al. , Novel study design to assess the efficacy and tolerability of antiseizure medications for focal-onset seizures in infants and young children: A consensus document from the regulatory task force and the pediatric commission of the International League against Epilepsy (ILAE), in collaboration with the Pediatric Epilepsy Research Consortium (PERC). Epilepsia Open, 2019. 4(4): p. 537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romero J, et al. , Natural variability in seizure frequency: Implications for trials and placebo. Epilepsy Res, 2020. 162: p. 106306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karoly PJ, et al. , Cycles in epilepsy. Nat Rev Neurol, 2021. 17(5): p. 267–284. [DOI] [PubMed] [Google Scholar]
- 37.Goldenholz DM, et al. , Simulating Clinical Trials With and Without Intracranial EEG Data. Epilepsia Open, 2017. 2(2): p. 156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elger CE and Hoppe C, Diagnostic challenges in epilepsy: seizure under-reporting and seizure detection. Lancet Neurol, 2018. 17(3): p. 279–288. [DOI] [PubMed] [Google Scholar]
- 39.French JA, et al. , Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy A randomized trial. Neurology, 2015. 85(11): p. 950–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baulac M, et al. , Efficacy, safety, and tolerability of lacosamide monotherapy versus controlled-release carbamazepine in patients with newly diagnosed epilepsy: a phase 3, randomised, double-blind, non-inferiority trial. Lancet Neurol, 2017. 16(1): p. 43–54. [DOI] [PubMed] [Google Scholar]
- 41.Yasumoto S, et al. , Lamotrigine monotherapy for newly diagnosed typical absence seizures in children: A multi-center, uncontrolled, open-label study. Brain Dev, 2016. 38(4): p. 407–13. [DOI] [PubMed] [Google Scholar]
- 42.Knupp KG, et al. , Comparison of Cosyntropin, Vigabatrin, and Combination Therapy in New-Onset Infantile Spasms in a Prospective Randomized Trial. J Child Neurol, 2022. 37(3): p. 186–193. [DOI] [PubMed] [Google Scholar]
- 43.Kerr WT, et al. , Reasons for ineligibility for clinical trials of patients with medication resistant epilepsy. Epilepsia, 2023. [DOI] [PubMed] [Google Scholar]
- 44.Chung SS, et al. , Randomized phase 2 study of adjunctive cenobamate in patients with uncontrolled focal seizures. Neurology, 2020. 94(22): p. e2311–e2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jha A, et al. , Sudden Unexpected Death in Epilepsy: A Personalized Prediction Tool. Neurology, 2021. 96(21): p. e2627–e2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fureman BE, et al. , Reducing placebo exposure in trials: Considerations from the Research Roundtable in Epilepsy. Neurology, 2017. 89(14): p. 1507–1515. [DOI] [PubMed] [Google Scholar]
- 47.Perucca P and Gilliam FG, Adverse effects of antiepileptic drugs. Lancet Neurol, 2012. 11(9): p. 792–802. [DOI] [PubMed] [Google Scholar]
- 48.Tassinari CA, et al. , Double-blind study of vigabatrin in the treatment of drug-resistant epilepsy. Arch Neurol, 1987. 44(9): p. 907–10. [DOI] [PubMed] [Google Scholar]
- 49.Betts T, Waegemans T, and Crawford P, A multicentre, double-blind, randomized, parallel group study to evaluate the tolerability and efficacy of two oral doses of levetiracetam, 2000 mg daily and 4000 mg daily, without titration in patients with refractory epilepsy. Seizure, 2000. 9(2): p. 80–7. [DOI] [PubMed] [Google Scholar]
- 50.Cereghino JJ, et al. , Levetiracetam for partial seizures: results of a double-blind, randomized clinical trial. Neurology, 2000. 55(2): p. 236–42. [DOI] [PubMed] [Google Scholar]
- 51.Porter RJ, et al. , Randomized, multicenter, dose-ranging trial of retigabine for partial-onset seizures. Neurology, 2007. 68(15): p. 1197–204. [DOI] [PubMed] [Google Scholar]
- 52.Boylan LS, et al. , Depression but not seizure frequency predicts quality of life in treatment-resistant epilepsy. Neurology, 2004. 62(2): p. 258–61. [DOI] [PubMed] [Google Scholar]