

Abstract
Background:
Senescent hepatocytes accumulate in parallel with fibrosis progression during NASH. The mechanisms that enable progressive expansion of nonreplicating cell populations and the significance of that process in determining NASH outcomes are unclear. Senescing cells upregulate thrombomodulin-protease activated receptor-1 (THBD-PAR1) signaling to remain viable. Vorapaxar blocks the activity of that pathway. We used vorapaxar to determine if and how THBD-PAR1 signaling promotes fibrosis progression in NASH.
Methods:
We evaluated the THBD-PAR1 pathway in liver biopsies from NAFLD patients. Chow fed mice were treated with viral vectors to over-express p16 in hepatocytes and induce replicative senescence. Effects on the THBD-PAR1 axis and regenerative capacity were assessed; the transcriptome of p16 over-expressing hepatocytes was characterized and we examined how conditioned medium from senescent but viable (dubbed ‘undead’) hepatocytes reprograms hepatic stellate cells. Mouse models of NASH caused by genetic obesity or Western diet/CCl4 were treated with vorapaxar to determine effects on hepatocyte senescence and liver damage.
Results:
Inducing senescence up-regulates the THBD-PAR1 signaling axis in hepatocytes and induces their expression of fibrogenic factors, including hedgehog ligands. Hepatocyte THBD-PAR1 signaling increases in NAFLD and supports sustained hepatocyte senescence that limits effective liver regeneration and promotes maladaptive repair. Inhibiting PAR1 signaling with vorapaxar interrupts this process, reduces the burden of ‘undead’ senescent cells, and safely improves NASH and fibrosis despite ongoing lipotoxic stress
Conclusion:
The THBD-PAR1 signaling axis is a novel therapeutic target for NASH because blocking this pathway prevents accumulation of senescing but viable hepatocytes that generate factors that promote maladaptive liver repair.
Keywords: Senescence, fatty liver disease, fibrosis, thrombomodulin, protease-activated receptor
Introduction
NAFLD afflicts one out of three adults and can cause cirrhosis and liver cancer, potentially lethal liver diseases. There are no effective medicines to prevent or treat NAFLD, cirrhosis, or liver cancer. Hence, NAFLD is driving the health and economic burden of liver disease. In order to develop effective interventions for NAFLD, its pathogenesis must be better understood. It is known that NAFLD-related cirrhosis and liver cancers mainly occur in the subgroup of NAFLD patients who both develop nonalcoholic steatohepatitis (NASH, an inflammatory type of liver damage caused by hepatocyte lipotoxicity) and mount maladaptive repair responses, thereby destining themselves for progressive liver damage. The mechanisms that determine whether lipotoxicity triggers liver degeneration, as opposed to liver regeneration, are unknown (1, 2).
Lipotoxic hepatocytes exhibit metabolic stress-related pathologies (mitochondrial malfunction, DNA damage and telomere attrition) that trigger cell senescence programs (3, 4, 5). These programs encompass a spectrum of responses that on one extreme, temporarily prevent wounded hepatocytes from replicating until their damaged components are repaired, and on the opposite end of the spectrum, promote irreversible senescence and eventual demise of more seriously compromised hepatocytes. Senescing hepatocytes themselves remain metabolically active and critically regulate the programs that determine their fate by generating repertoires of autocrine and paracrine factors (dubbed Senescence Associated Secretory Phenotypes, SASPs) that orchestrate wound healing responses that control tissue inflammation and scarring (6, 7). While transient accumulation of senescent hepatocytes can occur when liver regeneration is effective, senescent hepatocytes accumulate in parallel with fibrosis progression during NASH, linking sustained hepatocyte senescence with maladaptive repair responses that eventuate in lethal liver damage. Therefore, blocking sustained hepatocyte senescence might be an effective therapeutic strategy to prevent progressive liver degeneration in patients with NASH (8, 9).
We recently discovered that many types of senescing cells, including hepatocytes, upregulate their expression of thrombomodulin (THBD), an integral membrane protein that binds thrombin to modulate its coagulant activities. Unexpectedly, we found that survival of senescent cells is maintained by a THBD-driven mechanism that promotes the assembly of a membrane-tethered signaling complex that recruits thrombin, activates protein C, cleaves protease activated receptor-1 (PAR-1) on a specific arginine residue (R46) and then stabilizes this receptor-ligand complex in early endosomes to perpetuate PAR-1 signaling that antagonizes the pro-apoptotic actions of the SASP. Importantly, we demonstrated that pharmacologic agents that inhibit this process, including vorapaxar (an FDA-approved PAR-1 antagonist), reduce senescent cell viability (10). It is not known whether this THBD-PAR-1 signaling axis becomes activated in hepatocytes during NAFLD. If this occurs, vorapaxar could provide a tool to reduce the burden of senescent hepatocytes that accumulate in livers during chronic lipotoxic stress and thereby, enable us to evaluate the role of sustained hepatocyte senescence in the pathogenesis of NAFLD-related liver degeneration.
Herein we report studies of liver biopsy samples from large cohorts of NAFLD patients with a spectrum of liver fibrosis, a genetically obese mouse model of NASH with little liver fibrosis, and a diet-induced mouse model of NASH with advanced fibrosis. Healthy chow fed mice were also treated with viral vectors to over-express p16 specifically in hepatocytes to induce replicative senescence, and effects on the THBD-PAR-1 axis and regenerative capacity were assessed. The results show that inducing senescence up-regulates the THBD-PAR1 signaling axis in hepatocytes and that hepatocyte THBD-PAR1 signaling increases in NAFLD and supports sustained hepatocyte senescence that limits effective liver regeneration and promotes maladaptive repair. Importantly, inhibiting PAR-1 signaling with vorapaxar interrupts this process, reduces the burden of senescent cells, and safely improves hepatic steatosis, inflammation and fibrosis despite ongoing lipotoxic stress, identifying the THBD-PAR1 signaling axis as a novel therapeutic target for NASH.
Materials and Methods
Human liver RNA-seq analysis
We analyzed a publicly available GSE213623 RNA sequencing dataset from a cohort of 368 liver biopsies that were classified into Healthy, NASH with F0/F1, F2, or F3/F4 fibrosis (11). DESeq2 was used to normalize gene counts and identify differential expressed genes (12). The p values were adjusted for multiple test correction using Benjamini and Hochberg’s method. The correlations of CDKN1A (P21), CDKN2A (P16), THBD and PAR1 expression against ballooning score, fibrosis stage, serum aspartate aminotransferase (AST) or portal inflammation were estimated using Pearson’s correlation coefficient with R (13). Gene Set Enrichment Analysis (GSEA) was performed to identify significant enriched pathways using R package fgsea (14).
Human liver immunohistochemistry
Liver biopsies were collected from another group of human subjects (n=15) with biopsy-proven NAFLD (n=5) or NASH (n=10) and archived in the Duke University Health System (DUHS) NAFLD Clinical Database and Biorepository. The DUHS NAFLD Clinical Database and Biorepository is approved by the DUHS Institutional Review Board (Pro00005368). Details about the DUHS NAFLD Clinical Database and Biorepository are provided in Supplementary Methods. A summary of patient characteristics in the present study is detailed in Supplementary Table 1. Immunohistochemistry (IHC) for p21 (ab73958, Abcam) was performed using HRP-conjugated anti-rabbit (K4003, DAKO) secondary antibody, and chromogenic detection was performed using the DAKO Envision System with DAB substrate (DAKO Corporation) according to the manufacturer’s protocol. Tissue sections were counterstained with Hematoxylin Gill N°1 (Sigma Aldrich). The IHC for p21 was scored by counting the number of positive hepatocyte nuclei per mm of biopsy length, or by using Image J software to quantitate the percentage of positive area or positive cells per field. To measure the strength of the linear relationship between IHC parameters and H&E histologic scores, we used Pearson’s correlation coefficient.
Mice
Animal care and surgical procedures were conducted in compliance with an approved Duke University IACUC protocol (A200-21-09) as specified in the “Guide for the Care and Use of Laboratory Animals” published by the National Research Council. Mice were housed in a barrier facility on 12 h:12 h light cycle with free access to water and standard chow diet (Rodent diet 20, 5033; Picolab, St. Louis, MO) except when specified otherwise. Mice were monitored daily and weighed weekly. At sacrifice, blood was collected; liver was harvested and liver slices were formalin-fixed for paraffin embedding, OCT compound snap-freezing in 2-Methylbutane for cryosectioning, or snap frozen in liquid nitrogen for RNA and protein analysis.
In vivo model of hepatocyte senescence
C57BL/6J 3-month-old male mice (Jackson Laboratories, Bar Harbor, ME) were injected with AAV8-TBG-eGFP-P16 (Vector Biolabs) or control vector, AAV8-TBG-eGFP (Vector Biolabs) via tail vein injection. One week later, 70% partial hepatectomy was performed. Mice were sacrificed before PH (0h) and at 48 or 72h after PH to obtain liver tissue (n = 3 mice/group/time point) or primary hepatocytes (n = 3 mice/group/time point).
Murine NASH models and Vorapaxar treatment
Genetic obesity model: 3-month-old male mice homozygous or heterozygous (het) for the spontaneous obese mutation, Leptinob, (Jackson Laboratories, Bar Harbor, ME) were fed standard chow diet (15). Diet-induced model: C57BL/6J 3-month-old male mice were fed a normal chow diet, or Western Diet containing 21.1% fat, 41% Sucrose, and 1.25% Cholesterol by weight (Teklad diets, TD. 120528) and a high sugar solution (23.1 g/L d-fructose (Sigma-Aldrich, F0127) and 18.9 g/L d-glucose (Sigma-Aldrich, G8270)). CCl4 (Sigma-Aldrich, 289116-100ML) at the dose of 0.2 μl (0.32 μg)/g of body weight or its vehicle (corn oil) was injected intra-peritoneally once per week, starting with diet administration and continued for 12 weeks (16). Vorapaxar stock solution was made by diluting 25 mg of vorapaxar (Sellekchem, S8067) in 2 ml of DMSO. A working solution was prepared daily by diluting the stock in saline to get a final concentration of 3 mg/kg. WD-CCl4 and OB mice received Vorapaxar (3 mg/kg) (n=10 for WD-CCl4 and n=6 for OB) or vehicle (n=10 for WD-CCl4 and n=6 for OB) daily via oral gavage for the last 4 weeks before sacrifice.
Other methods are detailed in Supplemental Materials & Methods
Results
Senescence markers and THBD-PAR1 correlate with severity of NASH in humans.
To determine whether THBD and PAR1 associate with senescence burden in NAFLD, we performed RNA-seq analysis in 368 liver biopsies from patients suspected of having NAFLD. Liver histology in this previously described cohort (11) showed only minor nonspecific changes in 69 individuals but demonstrated a spectrum of hepatic steatosis, inflammation and fibrosis in the remaining 299 patients. Overall, 98% of this NAFLD cohort has a NAS ≥4, 36% has F2 fibrosis and 32% has F3–4 fibrosis, demonstrating that most of these NAFLD patients have fibrosing NASH. GSEA revealed that the senescent gene signature is highly enriched in the patients with NAFLD versus individuals without NAFLD (Fig 1A). Moreover, among the group with NAFLD, the senescent signature is more enriched in those with advanced fibrosis than those with mild fibrosis (Fig 1B). Consistent with these results, we found that transcripts of two standard senescence markers, p21 and p16, are up-regulated in NAFLD (Fig 1C) and positively correlate with fibrosis stage (Fig 1D). Immunostaining further revealed that these cell cycle inhibitors are mainly up-regulated in hepatocyte nuclei (Fig 1E) and showed that numbers of p21(+) hepatocytes significantly correlate with the severity of hepatocyte ballooning, portal inflammation, overall NAS and fibrosis stage (Fig 1F and Suppl Table 1), independently corroborating prior smaller studies (9, 17) which had suggested that senescent hepatocytes accumulate during NAFLD and correlate with disease severity. Hepatocytes in healthy livers are known to express the G protein-coupled receptor for thrombin (i.e., PAR-1) and PAR-1 mRNA expression is similar in livers with and without NAFLD, as well as across the spectrum of liver fibrosis in the NAFLD cohort (Figs 1G, H). In contrast, expression of THBD mRNA is significantly increased in livers with, versus without, NAFLD. Further, among NAFLD patients, THBD mRNA expression increases with fibrosis severity (Figs 1G, H). Our recent work shows that binding of thrombin to membrane-tethered THBD protein stabilizes a protein complex that includes THBD and PAR-1 (10). Consistent with this, immunostaining for THBD and PAR-1 demonstrates striking accumulation of these proteins in subsets of hepatocytes in NAFLD livers with advanced fibrosis (Fig 1I). Thus, NAFLD livers with NASH and advanced fibrosis accumulate the greatest numbers of senescent hepatocytes, have the highest expression of THBD, and accumulate the most THBD/PAR1 positive cells, supporting the concept that THBD-PAR1 signaling may play a role in maintaining hepatocyte senescence during chronic lipotoxic stress.
Figure 1. Senescence markers and THBD-PAR1 correlate with severity of NASH in humans.
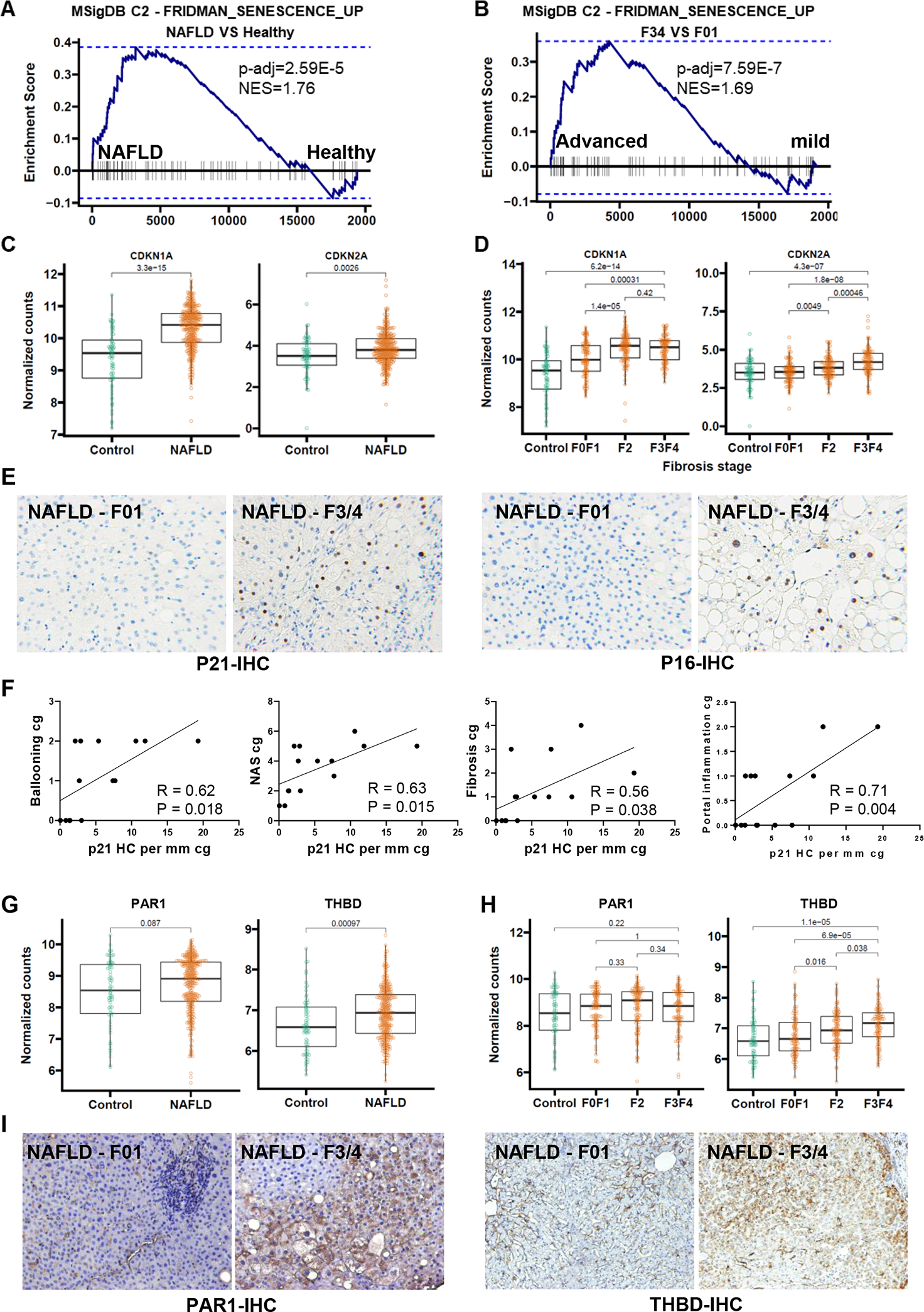
Genome set enrichment analsysis of GSE213623 RNAseq dataset, which includes 368 liver biopsies from healthy controls or patients with NAFLD and F0/F1, F2, or F3/F4 fibrosis (A and B). CDKN1A (P21) and CDKN2A (P16) mRNA levels based on NAFLD diagnosis (C) and fibrosis stage (D). Representative micrographs (100X magnification) of P21 and P16 immunostaining from NAFLD livers with mild (F0–1) or advanced (F3–4) stage fibrosis (E). Pearson correlation measuring the strength of the linear relationship between +P21 hepatocytes and Ballooning, Fibrosis, NAS Score and Portal Inflammation in liver biopsies from a validation NAFLD cohort (Supplementary Table 1) (F). PAR1 and THBD mRNA levels based on NAFLD diagnosis (G) and fibrosis stage (H). Representative micrographs (100X magnification) of PAR1 and THBD immunostaining in human NAFLD livers with mild (F0–1) or Advanced (F3–4) NAFLD fibrosis (I). RNAseq cohort n=368 patients; validation cohort n = 15 patients.
Inducing senescence in hepatocytes up-regulates hepatocyte expression of THBD-PAR1 and inhibits liver regeneration.
To more directly evaluate the relationship between hepatocyte senescence and THBD-PAR1 signaling we treated healthy mice with AAV8-TBG-eGFP-p16 vectors to over-express p16 selectively in hepatocytes. RT-PCR-ISH (Reverse Transcript polymerase chain reaction-in situ hybridization) confirmed that this approach selectively increases p16 in hepatocytes (Fig 2A). Over-expressing p16 in hepatocytes does not induce liver injury as liver histology (Fig 2B) and serum levels of ALT, albumin and bilirubin (Fig 2C) are similar in mice treated with AAV8-TBG-eGFP-p16 and control (AAV8-TBG-eGFP) vectors. However, increasing p16 expression in hepatocytes massively increases staining for beta-galactosidase activity, a marker of senescence, and promotes striking accumulation of THBD and PAR-1-positive hepatocytes (Fig 2D). Thus, the aggregate data confirm that senescing hepatocytes up-regulate their expression of THBD and PAR1, similar to many other senescent cell types (10). To evaluate the functional significance of senescent hepatocyte accumulation in intact liver tissue, the regenerative response to 70% partial hepatectomy (PH) was compared in mice that had received control- versus p16- vectors. Consistent with p16’s acknowledged function as a cyclin-dependent kinase inhibitor, mice selectively over-expressing p16 in hepatocytes demonstrated less recovery of liver mass and significantly reduced hepatocyte proliferation 48–72 h after PH, the time window when hepatocyte replication is significantly increased in control mice (Figs 2E, F). Similar liver enrichment with senescent cells and inhibition of regeneration occurs in p16-overexpressing mice following acute CCl4-induced liver injury (Suppl Fig 1). Together, these preclinical data show that the accumulation of senescent hepatocytes compromises effective hepatic regeneration. Thus, progressive increases in the burden of senescing hepatocytes may contribute to maladaptive repair and explain the correlations we noted among THBD (a factor that maintains the survival of senescent cells), numbers of senescent hepatocytes, and liver disease severity in human NAFLD.
Figure 2. Inducing senescence in hepatocytes up-regulates hepatocyte expression of THBD-PAR1 and inhibits liver regeneration.
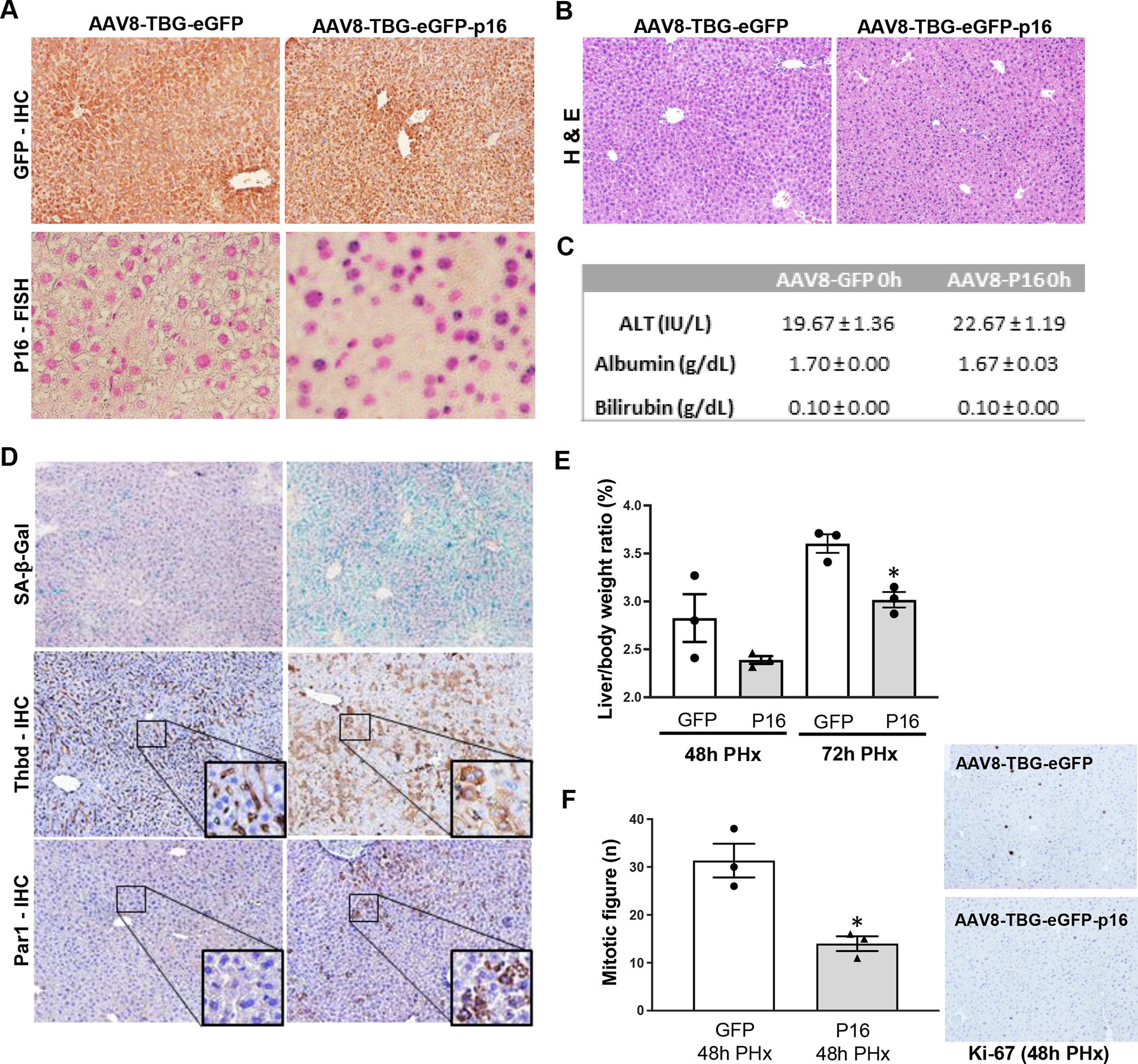
AAV8-TBG-eGFP-P16 (P16) or its control vector, AAV8-TBG-eGFP (GFP), were administered to C57BL/6 wild-type mice via tail vein injection. (A) Representative micrographs of GFP immunohistochemistry and P16-RT-PCR-ISH (Reverse Transcript polymerase chain reaction-in situ hybridization), (B) Hematoxylin & Eosin staining in liver tissue. (C) Serum biochemical parameters (ALT, Albumin and Bilirubin). (D) SA-β-Gal staining and THBD- and PAR1- immunostaining of liver sections from GFP or P16 over-expressing mice. One week after the AAV injection, mice underwent 70% PHx and were sacrificed 48h or 72h later. (E) Liver to body weight ratios from GFP and P16 over-expressing mice after PHx. (F) Markers of hepatocyte proliferation after PHx in liver sections from GFP and P16 over-expressing mice. Results shown as MEAN +/− SEM (n=3 mice/group/time, *p<0.05). Micrographs 100X magnification.
THBD-PAR1 axis enables senescent hepatocyte accumulation during NAFLD.
Because correlation does not prove causality, further research is needed to determine if (and how) THBD’s pro-senescence actions might help to drive NAFLD progression. Our prior work demonstrated that THBD maintains the survival of senescing cells by stabilizing a membrane-tethered protein complex that irreversibly cleaves PAR-1 on a specific arginine residue (R46) to selectively activate PAR-1 pro-survival signaling (10). Those studies also showed that vorapaxar, an FDA-approved drug that inhibits PAR-1 cleavage at R46, reduces the survival of senescing cells without compromising the viability of cells that retain proliferative capacity. Therefore, we tested whether vorapaxar could limit accumulation of senescent hepatocytes and prevent progressive liver damage in two mouse models of fibrosing NASH.
We chose ob/ob mice as a model of genetic obesity-related NAFLD. Ob/ob mice harbor a naturally-occurring loss-of-function mutation in the gene that encodes the satiety factor, leptin, and thus, they are hyperphagic and progressively develop obesity-related diseases, including type 2 diabetes and NAFLD (18). Similar to human livers with obesity-related steatosis, fatty ob/ob livers exhibit impaired liver regeneration (19). However, they do not develop significant liver fibrosis because leptin is important for optimal myofibroblastic activation of hepatic stellate cells. Analysis of a publicly available RNA-seq data set (GSE167264) revealed that both Thbd and Par1 mRNAs are significantly upregulated in livers of ob/ob mice compared to lean, ob heterozygous littermate controls (Fig 3A and Suppl Fig 2A). Interestingly, immunostaining indicates that although Thbd and Par1 proteins are mainly expressed by sinusoidal cells in the healthy livers of lean mice, both proteins are strongly upregulated in subset of hepatocytes of obese ob/ob mice (Fig 3B), recapitulating the staining pattern for these proteins in human livers with advanced NAFLD fibrosis (Fig 1I) and complementing previous reports that leptin deficiency and other obesogenic factors promote accumulation of senescent cells in many tissues, including liver (20). Treating morbidly obese, adult ob/ob mice with vorapaxar for 4 weeks virtually eliminates hepatic parenchymal staining for THBD/PAR-1 (Fig 3B and Suppl Fig 2B), consistent with evidence that vorapaxar disrupts Thbd-Par1 signaling that is necessary to maintain cell senescence (10). To determine the direct effects of vorapaxar on ob/ob hepatocyte senescence, primary hepatocytes isolated from ob/ob mice were also treated with various doses of vorapaxar. Vorapaxar causes dose-dependent suppression of the cell cycle inhibitors, p16 and p21 in ob/ob hepatocytes (Fig 3C). Thus, the aggregate data indicate that vorapaxar depletes populations of senescing hepatocytes that typically accumulate in ob/ob livers. Remarkably, while vorapaxar does not decrease obesity in ob/ob mice, it significantly decreases liver/body ratio and slightly decreases liver weight (Fig 3D and Suppl Fig 2C). Ob/ob livers with a lower burden of senescent hepatocytes also exhibit dramatically improved hepatic steatosis and reduced markers of liver fibrosis (staining for Sirius and alpha smooth muscle actin) and macrophage accumulation (F4/80 staining) (Figs 3E, F). Together, these findings prove that obesogenic factors cannot sustain hepatocyte senescence when the THBD-PAR-1 signaling axis is disrupted and suggest that reducing the burden of senescing hepatocytes promotes recovery from NAFLD, even in the context of chronic obesity.
Figure 3. THBD-PAR1 axis enables senescent hepatocyte accumulation during NAFLD.
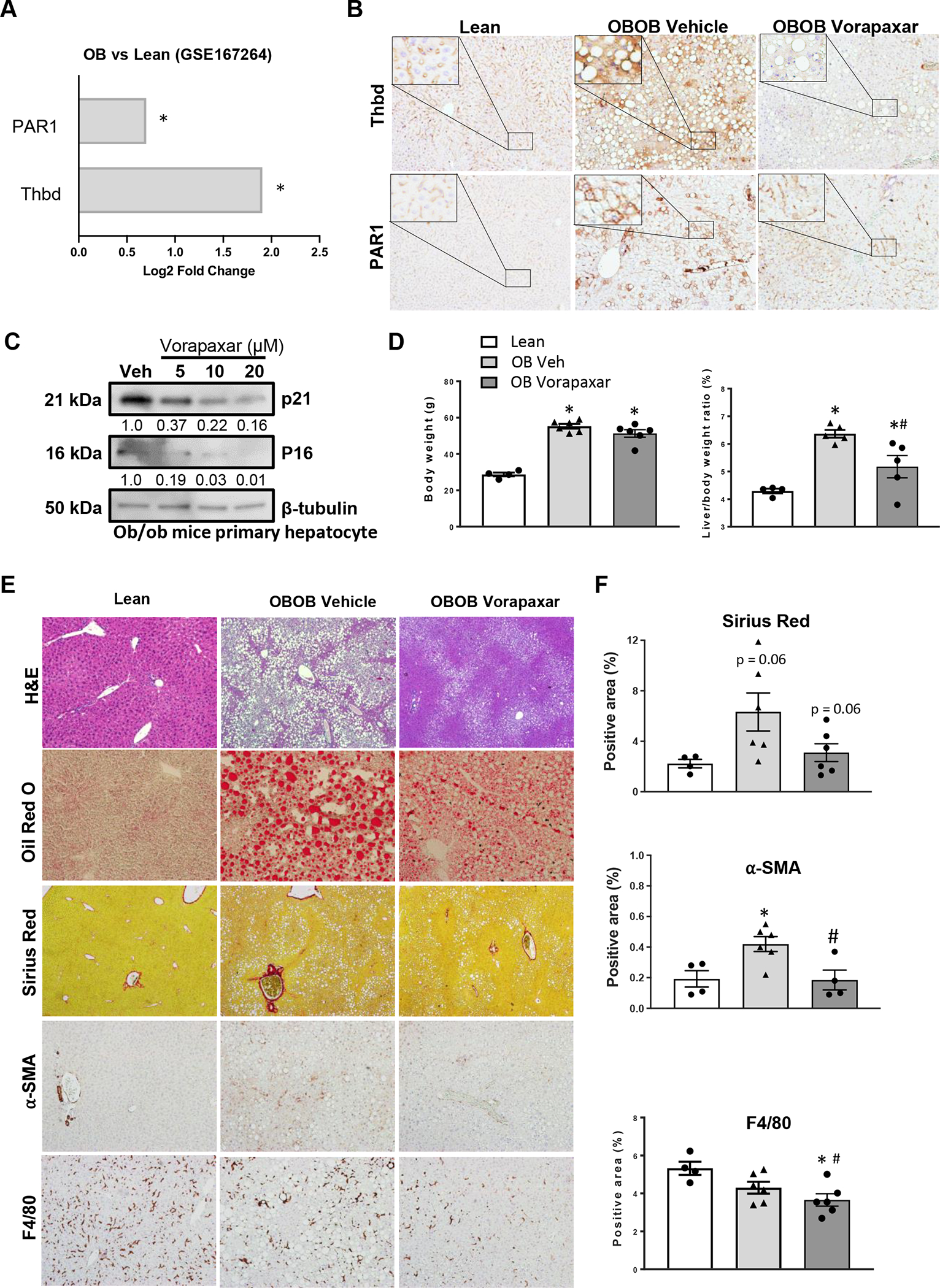
(A) Hepatic expression of THBD and PAR1 was examined in a published RNA-seq dataset (GSE167264), and data graphed as Log2 fold change in obese (ob/ob) vs lean mice *p<0.05. In separate experiments, Vorapaxar or its vehicle control (saline) was administered daily by oral gavage to ob/ob mice for 4 weeks before blood and liver tissue were harvested. Results are compared to age- and sex-matched lean ob/+ littermate controls. (B) Representative photomicrographs of liver sections stained for THBD and PAR1. (C) Dose-dependent effect of vorapaxar on p16 and p21 expression in freshly isolated primary hepatocytes from ob/ob mice. Relative expression of p16 and p21 was normalized to loading control β-tubulin. (D) Body weights and liver/body weight ratios. (E) Liver sections were stained to assess effects of vorapaxar on histology (H&E), fat accumulation (Oil Red O), fibrosis severity (Sirius Red and α-SMA), and inflammation (F4/80 staining). Representative photomicrographs and morphometric quantitation (F). Results shown as MEAN +/− SEM (n=6 mice/group, *p<0.05 vs lean, #p<0.05 vs ob/ob vehicle).
Suppression of THBD-PAR1 axis improves advanced NAFLD.
As noted above, myofibroblastic activation of hepatic stellate cells is impaired in ob/ob mice and myofibroblasts derived from hepatic stellate cells (MF-HSC) are critical determinants of maladaptive repair responses that eventuate in cirrhosis (21). Our analysis of human livers with NAFLD demonstrate that NASH livers with advanced (F3–4) fibrosis exhibit the strongest induction of THBD transcripts, greatest accumulation of THBD/PAR-1 positive hepatocytes, and most senescent hepatocytes (Fig 1). Therefore, it is important to determine if inhibiting THBD-PAR1 signaling is able to clear senescing hepatocytes and improve liver damage in livers with NASH and severe fibrosis. Hence, we repeated the vorapaxar experiments in wild type mice while treating them with an established protocol that combines western diet (WD) with intermittent injections of low dose CCl4 to induce NASH cirrhosis (16). Mice develop significant fibrosing NASH after 8 weeks on this protocol and at that time point, we randomized them to receive daily injections of either vehicle or vorapaxar and treated them with WD/CCl4 for an additional 4 weeks (Fig 4A). At the time of sacrifice, advanced bridging fibrosis/micronodular cirrhosis was evident in vehicle treated mice. In contrast, fibrosis was significantly milder and regenerative nodules were larger in the vorapaxar-treated group (Figs 4B, C). Fibrosis improvement is accompanied by decreased staining for αSMA and F4/80, consistent with reduced accumulation of MF-HSC and inflammatory macrophages, respectively (Figs 4B, C). Conversely, staining for CD163, a marker of anti-inflammatory/fibrinolytic macrophages is increased (Figs 4B, C). Importantly, the suppression of fibro-inflammatory repair responses is accompanied by reduced expression of THBD, PAR-1 (Figs 4D, E) and senescent markers, as indicated by hepatic parenchymal staining for beta galactosidase and western blotting for p16 and p21 (Figs 4D, F, G and Suppl Fig 3B). Together, these results confirm that accumulation of senescing hepatocytes can be reduced during metabolic stress by blocking THBD-PAR1 signaling. Importantly, they also provide novel support for the concept that reducing the burden of senescing hepatocytes prevents maladaptive repair responses that would otherwise trigger progressive liver fibrosis.
Figure 4. Suppression of THBD-PAR1 axis improves advanced NAFLD.
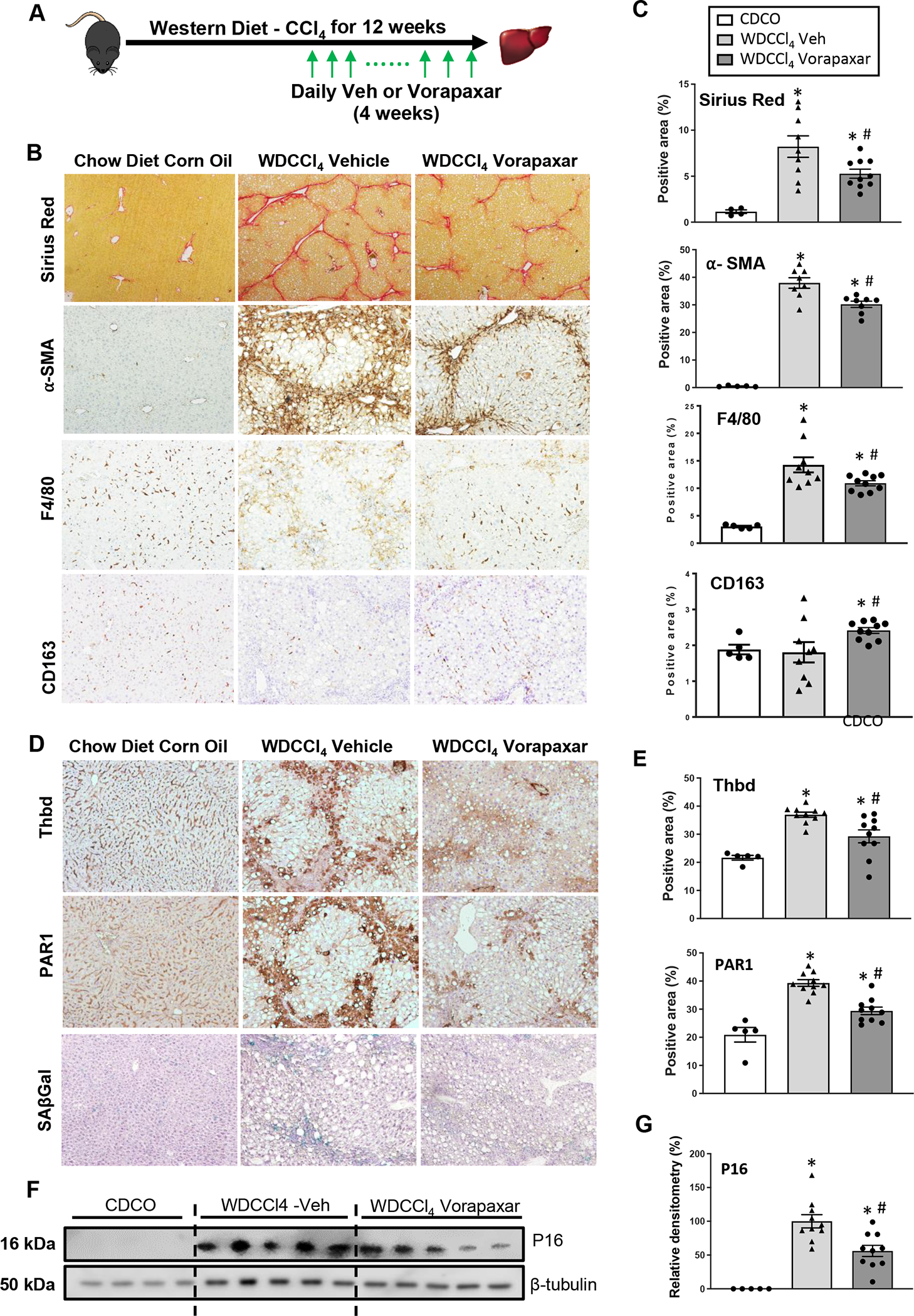
(A) Vorapaxar or its vehicle control (saline) was administered daily by oral gavage to WD+CCl4 mice during the last 4 weeks of diet. (B and C) Liver fibrosis (Sirius Red staining and α-SMA), hepatic inflammation (F4/80 and CD163 staining) and (D-G) senescence markers (Thbd, PAR1, SAβGal and P16) were assessed by quantitative morphometry of stained liver sections or densitometry of liver immunoblots. Results shown as MEAN +/− SEM (n=10 mice/group, *p<0.05 vs CDCO (chow diet plus corn oil vehicle), #p<0.05 vs WD+CCl4 vehicle).
Senescent hepatocytes secrete Shh to promote liver fibrosis and inflammation.
To more directly determine the paracrine effects of senescent hepatocytes on liver repair we obtained fresh conditioned medium (CM) from cultures of Huh7 cells that had been treated with either vehicle or palbociclib, an inhibitor of cyclin dependent kinases that is commonly used to induce senescence (22), and screened for effects on hepatic stellate cell activation using LX2 cells as targets (Fig 5A). Compared to CM from control Huh7 cells, CM from senescent Huh7 cells significantly up-regulated several makers of myofibroblastic stellate cell activation in LX2 cells (Fig 5B). To determine whether the secretome of senescent primary hepatocytes is also pro-fibrogenic and identify potential paracrine mechanisms involved, we isolated primary hepatocytes from wild type mice that were either treated with AAV8-TBG-GFP-p16 vectors or control (AAV8-TBG-GFP) vectors. RNA was obtained from some of the freshly isolated cells for RNA-seq analysis and the remainder were cultured overnight to obtain conditioned medium (Fig 5C). Compared to CM from control hepatocytes, CM from p16-overexpressing hepatocytes elicited greater myofibroblastic trans-differentiation in LX2 cells (Fig 5D) and thus, had similar paracrine pro-fibrogenic actions as the senescent Huh7 cells (Fig 5B).
Figure 5. Senescent hepatocytes secrete Shh to promote liver fibrosis and inflammation.
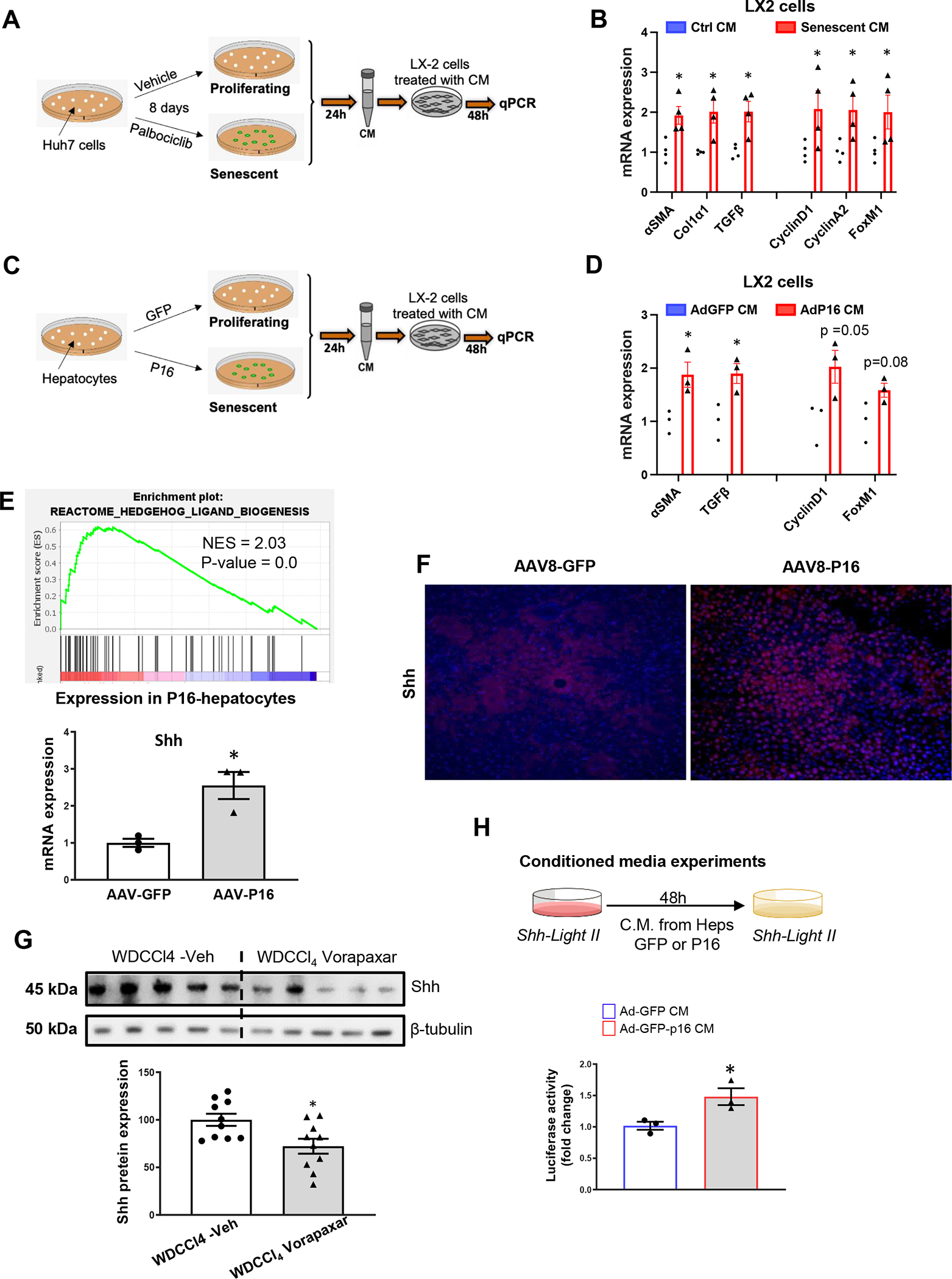
(A) Huh7 cells were treated with Palbociclib to induce senescence; conditioned media (CM) was harvested and added to LX2 cells for 48h. (B) CM from senescent Huh7 cells upregulates expression of proliferative and fibrogenic markers in LX2 cells. (C) Primary hepatocytes were freshly isolated from mice treated with AAV8-TBG-eGFP-P16 (P16) or its control vector, AAV8-TBG-eGFP (GFP) and cultured for 24h to obtain CM. (D) CM from P16-overexpressing hepatocytes upregulated expression of proliferative and fibrogenic markers in LX2 cells. (E) GSEA of RNA seq data from P16 over-expressing hepatocytes reveals enrichment for the Hedgehog ligand biogenesis gene signature. (E) q-PCR and (F) immunostaining confirm that Shh is upregulated in the P16-overexpressing hepatocytes. (G) Shh protein expression in livers of WD-CCl4 mice treated with vorapaxar or its vehicle. (H) CM harvested from GFP control or P16-overexpressing hepatocytes was added to the Shh-Light II cells and luciferase activity was evaluated. Results shown as MEAN +/− SEM (n=4 assays/group, *p<0.05 vs GFP or ctrl CM; n= 10 mice/group, *p<0.05 vs WDCCl4+Veh).
As expected, GSEA demonstrated that the transcriptome of p16 over-expressing hepatocytes is significantly enriched with gene signatures for senescence (e.g., stabilization of p53, G1-S DNA damage check points, oncogene-induced senescence, oxidative stress-induced senescence) and for genes encoding the SASP. Gene signatures for cell stress (e.g., cell response to starvation, cell response to chemical stress), stress-related kinase cascades (e.g., MAPK6_MAPK4_signaling, jnk/c-jun kinase phosphorylation and activation), and their targets (e.g., ATF2 pathway, AP1 pathway) are also up-regulated significantly (Suppl Fig 3A and Suppl Tables 2, 3, 4). Stress-related kinases and AP1 have been reported to regulate expression of THBD (23, 24). These pathways are also known to promote synthesis of Hedgehog ligands (3, 25) and Hedgehog ligand biogenesis is one of the top 25 Reactomes in p16 over-expressing hepatocytes (Fig 5E). To confirm that induction of THBD (Fig 2D) and other stress responses that are triggered by over-expressing p16 in hepatocytes are accompanied by increased expression of Sonic hedgehog (Shh), we compared expression of Shh mRNAs in hepatocytes harvested from p16-overexpressing mice and controls. Levels of Shh transcripts are significantly higher in p16-overexpressing hepatocytes (Fig 5E) and immunostaining confirmed significant hepatocyte accumulation of Shh protein (Fig 5F). Western blot analysis indicates that Shh is also strongly expressed in livers of WD-CCl4-treated mice and demonstrates that Shh levels are significantly reduced by vorapaxar (Fig 5G), an intervention that decreases the liver burden of senescent hepatocytes by specifically inhibiting THBD-mediated activation of PAR1 (Figs 4D–G). To test if hedgehog ligand activity is also increased by p16 overexpression, we screened CM from cultures of primary hepatocytes that had been harvested from mice that were either treated with p16- or control- vectors and tested for evidence of paracrine hedgehog signaling activity in hedgehog-responsive target cells that report hedgehog-driven transcriptional activity (dubbed ShhLight cells) (Fig 5H). Compared to CM from control hepatocytes, CM from p16-overexpressing hepatocytes elicited greater hedgehog reporter gene activity in ShhLight cells (Fig 5H). Thus, the aggregate data demonstrate that hepatocyte senescence evokes stress responses and up-regulates hepatocyte production of biologically active hedgehog ligands. Previous work from us and others indicates that hepatocyte-derived hedgehog ligands help to orchestrate hepatic accumulation of MF-HSC that drive maladaptive repair during NASH (15).
Discussion
NAFLD progresses when hepatocytes that succumb to lipotoxic injury are not replaced efficiently. Although several inherited and acquired factors that increase the risk for maladaptive repair and progressive liver degeneration have been identified, the mechanisms involved remain poorly understood. Herein, we demonstrate that human livers with NAFLD become progressively enriched with senescence-related gene expression programs as damage worsens, and identify strongly positive correlations between the accumulation of senescent hepatocytes and severity of liver injury, inflammation and fibrosis. Importantly, our studies provide new evidence that hepatic expression of thrombomodulin (THBD) increases in parallel with senescent hepatocyte accumulation and fibrosis severity in human NAFLD. This finding is noteworthy because THBD was recently shown to interact with protease-activated receptor 1 (PAR1) to promote signaling that maintains the viability of senescent cells, and an FDA-approved drug that specifically disrupts this signaling, vorapaxar, has been identified (10). Here we report that vorapaxar reduces the burden of senescent hepatocytes in mice with either genetic- or diet-induced NASH. Remarkably, loss of senescent hepatocytes in vorapaxar-treated mice is accompanied by regression of liver inflammation and fibrosis, despite persistence of the metabolic stressors that drives NASH progression in vehicle-treated mice. These findings implicate senescent hepatocytes as critical determinants of disease progression in NASH.
Our studies also identify the stimuli that induce THBD-PAR1 signaling in NASH by demonstrating that selectively enforcing expression of p16 in hepatocytes promotes hepatocyte accumulation of THBD and PAR1. P16 inhibits cyclin dependent kinases that are required for cell cycle progression (26) and we demonstrated that it accumulates in hepatocyte nuclei during NASH. When we over-expressed p16 in otherwise healthy mice, hepatocytes became positive for the senescence marker, SA-β-gal, and liver regeneration was inhibited after partial hepatectomy. Senescent cells are known to have increased stress signaling (27) and GSEA of RNA-seq data from p16-overexpressing senescing hepatocytes in our study demonstrated the expected up-regulation of stress kinase cascades and induction of down-stream transcription factors that are known to promote THBD expression (23, 24). Pan et al recently identified the mechanism whereby THBD stabilizes PAR1 signaling by demonstrating that THBD suppress the expression of NEED4L, an E3 ubiquitin ligase that would otherwise target activated PAR1 for proteasomal degradation (28). Persistent PAR1 activity, in turn, maintains cell survival by preventing activation of apoptosis effector caspases, such as caspase 3 (29, 30).
The aforementioned findings support a novel model for NASH progression whereby sublethal lipotoxic injury in hepatocytes forces them to induce adaptive responses that constrain their proliferation but mobilize stress-related signaling that assures their survival (Fig 6). As mentioned earlier, although they have limited inherent regenerative capability, these senescing cells remain metabolically active. In addition to launching autocrine mechanisms to modulate their own phenotype, senescing cells elaborate factors that signal via juxtacrine, paracrine and endocrine mechanisms to shape local and systemic wound healing responses (31). As discussed below, our data support the concept that this senescence associated secreted proteome (SASP) determines the ultimate outcome of hepatocyte lipotoxicity in NASH.
Figure 6. THBD-PAR1 signaling and senescent hepatocyte accumulation promote maladaptive repair of lipotoxic liver damage: Working model.
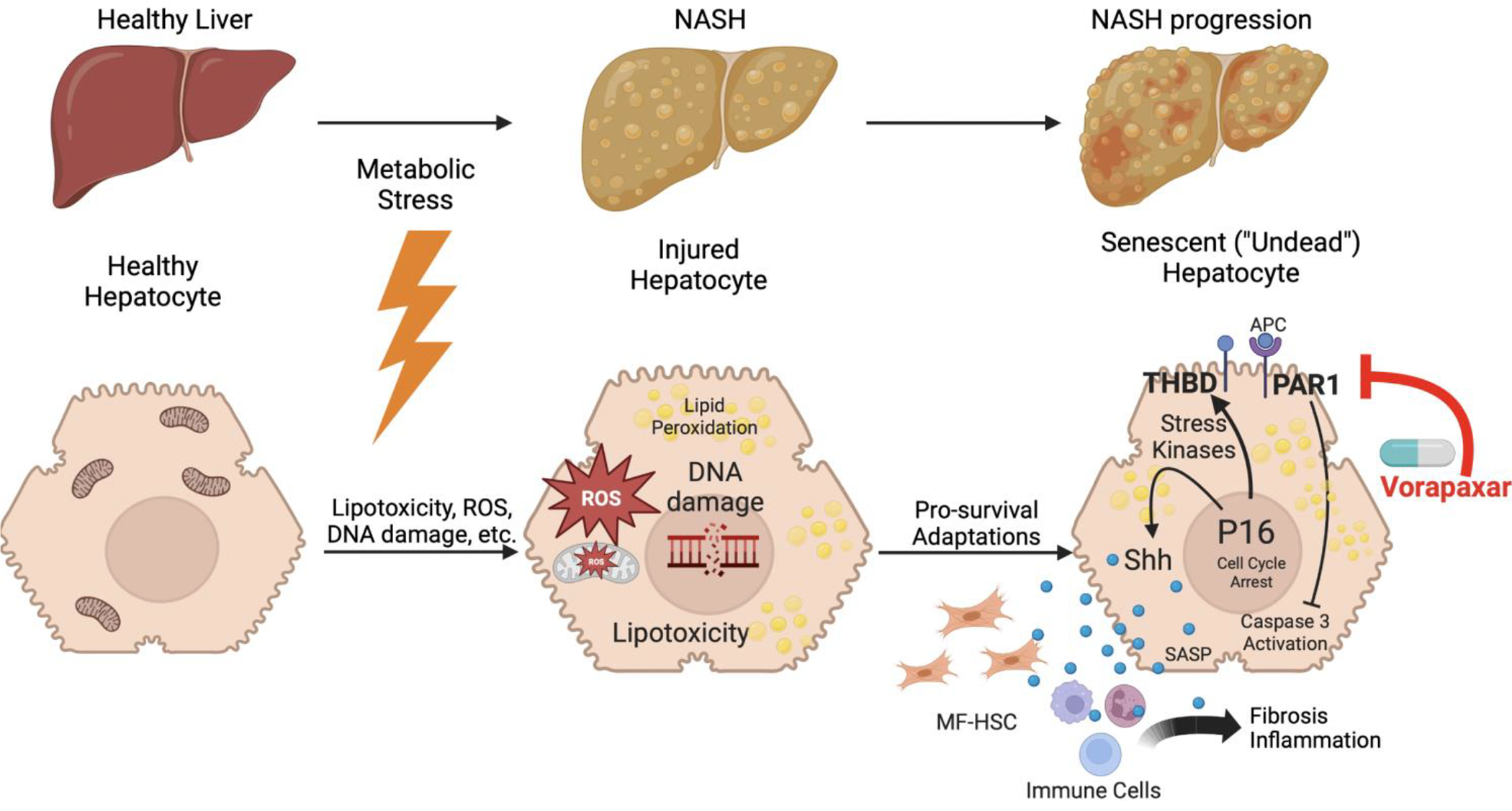
During NAFLD, metabolic stress induces hepatocyte lipotoxicity. Sub-lethal lipotoxic injury forces hepatocytes to induce adaptive responses that constrain their proliferation, but mobilize stress kinase-regulated signaling that assures their survival. Although these injured but viable (i.e., ‘undead’) hepatocytes are non-replicative (i.e., senescent), they accumulate during NASH when THBD-PAR1 signaling and other survival mechanisms that delay the demise of senescing cells become sustained. In turn, the inefficient clearance of senescing cells perpetuates paracrine signaling by their senescence associated secreted proteome (SASP) that drives inflammatory and fibrogenic wound healing responses. Inhibiting PAR-1 signaling with vorapaxar interrupts this process, reduces the burden of ‘undead’ senescent cells, and safely improves NASH and fibrosis despite ongoing lipotoxic stress, identifying the THBD-PAR1 signaling axis as a novel therapeutic target for NASH.
Briefly, our unbiased analysis of RNA seq data from p16 over-expressing hepatocytes identified “Hedgehog ligand biogenesis” as one of the top 25 Reactome pathways in senescing hepatocytes. We validated this observation using qRT-PCR and immunostaining for Sonic hedgehog ligand (SHH). In addition, we performed bioassays to confirm that the secretome of these senescent hepatocytes exhibited hedgehog biological activity and showed that it induced fibrogenic activation of cultured liver cells that are known to be hedgehog-responsive, i.e., hepatic stellate cells. The aggregate findings provide proof for the principle that the hepatocyte SASP can promote fibrogenesis. These results are particularly pertinent to NASH progression because lipotoxic hepatocytes are known to up-regulate their production of SHH and other hedgehog ligands (3, 4). Importantly, hepatocyte expression of hedgehog ligands has been shown to increase in human NASH and accumulation of SHH-positive hepatocytes parallels fibrosis severity in NASH patients (32, 33). Further, gain and loss of function studies in murine models of NASH have proven that hedgehog ligands critically regulate liver inflammation and fibrosis in this disease (34).
Almost two decades ago, researchers studying tissue regeneration in Drosophila demonstrated that injured but still-viable cells (which they dubbed ‘undead’ cells) release hedgehog ligands to trigger compensatory proliferation of neighboring cells that retain regenerative capacity (35). Although those investigations revealed that the ligand-producing cells themselves are unable to regenerate despite exposure to hedgehog ligands, whether these hedgehog-resistant cells express senescence markers was not evaluated. Recently, we showed that when hepatocytes lose their ability to propagate hedgehog ligand-initiated signals, they undergo a process of premature aging and acquire many features of senescing cells, including loss of inherent regenerative capacity and acquisition of the SASP (36, 37). Importantly, we discovered that these ‘hedgehog-resistant’, non-replicative hepatocytes accumulate in human NAFLD livers; are more prevalent in NASH than NAFL; and are most abundant in livers with the most severe fibrosis (36). However, those studies did not identify mechanisms that enable populations of senescing, non-proliferative hepatocytes to expand in injured livers.
The data reported herein suggest that senescing hepatocytes accumulate during NASH because THBD-PAR1 signaling and other stress-related survival mechanisms that delay the demise of senescing cells become sustained. In turn, the inefficient clearance of senescing cells perpetuates paracrine signaling by their SASP that drives inflammatory and fibrogenic wound healing responses. We provide supportive evidence for these concepts by demonstrating that senescing hepatocytes can be cleared from injured livers by disrupting one these senescence survival mechanisms and show that hepatic expression of hedgehog ligands declines and more importantly, that liver inflammation and fibrosis regress, when populations of senescing hepatocytes are depleted. More research is needed to clarify how well-validated genetic and epigenetic regulators of NAFLD susceptibility influence, and/or interact with, senescence programs to determine the fate of livers that are challenged by metabolic stress.
Supplementary Material
Acknowledgements
The authors are grateful to the patients who donated liver tissue for analysis, and to the study coordinators who maintain the Duke NAFLD Biorepository. This work was supported the Duke Endowment (AMD), NIH RO1 DK 077794 (AMD), 2021 AASLD Pinnacle Research Award (KD) and NIH RO1 CA 244564 (X-FW).
Funding information
This work was supported by NIH grants RO1 DK 077794 and the Florence McAlister Professorship of Medicine to Anna Mae Diehl, and 2021 AASLD Pinnacle Research Award to Kuo Du.
Abbreviations
- ALT
Alanine transaminase
- CM
Conditioned medium
- CDKN1A or P21
Cyclin Dependent Kinase Inhibitor 1A
- CDKN2A or P16
Cyclin Dependent Kinase Inhibitor 2A
- GSEA
Gene Set Enrichment Analysis
- PAR1
Protease-activated receptor-1
- SASP
Senescence-associated secretory phenotype
- SA-β-Gal
Senescence associated β galactosidase
- THBD
Thrombomodulin
- NAFLD
Nonalcoholic fatty liver disease
- NASH
Nonalcoholic Steatohepatitis
- RT-PCR-ISH
Reverse Transcript polymerase chain reaction-in situ hybridization
Footnotes
Conflict of interest
Cynthia D. Guy consults for Madrigal, 89 Bio, NGM, AQ13 CymaBay, and Histoindex. Anna Mae Diehl consults/advises for Boehringer Ingelheim and receives research grants paid to her institution from Intercept, Madrigal, Inventiva, Hanmi, NGM Biopharmaceuticals, and Boehringer Ingelheim. Dr. Manal F. Abdelmalek consults/advises 89 Bio, Bristol-MyersSquibb, Hanmi, Intercept, Inventiva, Madrigal, Merck,NGM Bio, Novo Nordisk, SonicIncytes, Theratechnolo-gies, Fishawack Inc., Terra Firma Inc. and receives research grants paid to her institution from Allergan,Boeringher- Ingelheim, Bristol Myers Squibb, Celgene,Durect, Enanta, Enyo, Galmed, Genentech, Gilead,Hanmi, Intercept, Inventiva, Madrigal, Novo Nordisk,Poxel, Target NASH, Viking.
References
- 1.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20. [DOI] [PubMed] [Google Scholar]
- 3.Kakisaka K, Cazanave SC, Werneburg NW, Razumilava N, Mertens JC, Bronk SF, et al. A hedgehog survival pathway in ‘undead’ lipotoxic hepatocytes. J Hepatol. 2012;57(4):844–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rangwala F, Guy CD, Lu J, Suzuki A, Burchette JL, Abdelmalek MF, et al. Increased production of sonic hedgehog by ballooned hepatocytes. J Pathol. 2011;224(3):401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L, Jhaveri R, Huang J, Qi Y, Diehl AM. Endoplasmic reticulum stress, hepatocyte CD1d and NKT cell abnormalities in murine fatty livers. Lab Invest. 2007;87(9):927–37. [DOI] [PubMed] [Google Scholar]
- 6.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482–96. [DOI] [PubMed] [Google Scholar]
- 7.Ferreira-Gonzalez S, Rodrigo-Torres D, Gadd VL, Forbes SJ. Cellular Senescence in Liver Disease and Regeneration. Semin Liver Dis. 2021;41(1):50–66. [DOI] [PubMed] [Google Scholar]
- 8.Bird TG, Muller M, Boulter L, Vincent DF, Ridgway RA, Lopez-Guadamillas E, et al. TGFbeta inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci Transl Med. 2018;10(454). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. 2017;8:15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan CC, Maeso-Díaz R, Lewis TR, Xiang K, Tan L, Liang Y, et al. Antagonizing the irreversible thrombomodulin-initiated proteolytic signaling alleviates age-related liver fibrosis via senescent cell killing. Cell Research. 2023. May 11:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen T, Dalton G, Oh SH, Maeso-Diaz R, Du K, Meyers RA, et al. Hepatocyte Smoothened Activity Controls Susceptibility to Insulin Resistance and Nonalcoholic Fatty Liver Disease. Cell Mol Gastroenterol Hepatol. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Team RC R Foundation for Statistical Computing V, Austria. . R: A language and environment for statistical computing. . 2021. [Google Scholar]
- 14.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A. Fast gene set enrichment analysis. biorxiv. 2019. [Google Scholar]
- 15.Choi SS, Syn WK, Karaca GF, Omenetti A, Moylan CA, Witek RP, et al. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. J Biol Chem. 2010;285(47):36551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol. 2018;69(2):385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aravinthan A, Scarpini C, Tachtatzis P, Verma S, Penrhyn-Lowe S, Harvey R, et al. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J Hepatol. 2013;58(3):549–56. [DOI] [PubMed] [Google Scholar]
- 18.Leclercq IA, Field J, Farrell GC. Leptin-specific mechanisms for impaired liver regeneration in ob/ob mice after toxic injury. Gastroenterology. 2003;124(5):1451–64. [DOI] [PubMed] [Google Scholar]
- 19.Yang SQ, Lin HZ, Mandal AK, Huang J, Diehl AM. Disrupted signaling and inhibited regeneration in obese mice with fatty livers: implications for nonalcoholic fatty liver disease pathophysiology. Hepatology. 2001;34(4 Pt 1):694–706. [DOI] [PubMed] [Google Scholar]
- 20.Schafer MJ, Miller JD, LeBrasseur NK. Cellular senescence: Implications for metabolic disease. Mol Cell Endocrinol. 2017;455:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu C, Tabas I, Schwabe RF, Pajvani UB. Maladaptive regeneration - the reawakening of developmental pathways in NASH and fibrosis. Nat Rev Gastroenterol Hepatol. 2021;18(2):131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bollard J, Miguela V, Ruiz de Galarreta M, Venkatesh A, Bian CB, Roberto MP, et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut. 2017;66(7):1286–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boon RA, Horrevoets AJ. Key transcriptional regulators of the vasoprotective effects of shear stress. Hamostaseologie. 2009;29(1):39–40, 1–3. [PubMed] [Google Scholar]
- 24.Ramachandran A, Ranpura SA, Gong EM, Mulone M, Cannon GM Jr., Adam RM. An Akt- and Fra-1-dependent pathway mediates platelet-derived growth factor-induced expression of thrombomodulin, a novel regulator of smooth muscle cell migration. Am J Pathol. 2010;177(1):119–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papa S, Bubici C. Feeding the Hedgehog: A new meaning for JNK signalling in liver regeneration. J Hepatol. 2018;69(3):572–4. [DOI] [PubMed] [Google Scholar]
- 26.Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2012;130(8):1715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37(5):961–76. [DOI] [PubMed] [Google Scholar]
- 28.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10(6):398–409. [DOI] [PubMed] [Google Scholar]
- 29.Flynn AN, Buret AG. Proteinase-activated receptor 1 (PAR-1) and cell apoptosis. Apoptosis. 2004;9(6):729–37. [DOI] [PubMed] [Google Scholar]
- 30.Molinar-Inglis O, Birch CA, Nicholas D, Orduna-Castillo L, Cisneros-Aguirre M, Patwardhan A, et al. aPC/PAR1 confers endothelial anti-apoptotic activity via a discrete, beta-arrestin-2-mediated SphK1-S1PR1-Akt signaling axis. Proc Natl Acad Sci U S A. 2021;118(49). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumari R, Jat P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front Cell Dev Biol. 2021;9:645593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guy CD, Suzuki A, Zdanowicz M, Abdelmalek MF, Burchette J, Unalp A, et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology. 2012;55(6):1711–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swiderska-Syn M, Suzuki A, Guy CD, Schwimmer JB, Abdelmalek MF, Lavine JE, et al. Hedgehog pathway and pediatric nonalcoholic fatty liver disease. Hepatology. 2013;57(5):1814–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verdelho Machado M, Diehl AM. Role of Hedgehog Signaling Pathway in NASH. Int J Mol Sci. 2016;17(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan Y, Bergmann A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell. 2008;14(3):399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen T, Dalton G, Oh SH, Maeso-Diaz R, Du K, Meyers RA, et al. Hepatocyte Smoothened Activity Controls Susceptibility to Insulin Resistance and Nonalcoholic Fatty Liver Disease. Cell Mol Gastroenterol Hepatol. 2022;15(4):949–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maeso-Diaz R, Dalton GD, Oh S, Du K, Tang L, Chen T, et al. Aging reduces liver resiliency by dysregulating Hedgehog signaling. Aging Cell. 2022;21(2):e13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.