

SUMMARY
Ferroptosis is a regulated form of cell death associated with the iron-dependent accumulation of phospholipid hydroperoxides. Inducing ferroptosis is a promising approach to treat therapy resistant cancer. Ferroptosis suppressor protein 1 (FSP1) promotes ferroptosis resistance in cancer by generating the antioxidant form of coenzyme Q10 (CoQ). Despite the important role of FSP1, few molecular tools exist that target the CoQ-FSP1 pathway. Through a series of chemical screens, we identify several structurally diverse FSP1 inhibitors. The most potent of these compounds, ferroptosis sensitizer 1 (FSEN1), is an uncompetitive inhibitor that acts selectively through on target inhibition of FSP1 to sensitize cancer cells to ferroptosis. Furthermore, a synthetic lethality screen reveals that FSEN1 synergizes with endoperoxide-containing ferroptosis inducers, including dihydroartemisinin, to trigger ferroptosis. These results provide new tools that catalyze the exploration of FSP1 as a therapeutic target and highlight the value of combinatorial therapeutic regimes targeting FSP1 and additional ferroptosis inducers.
eTOC blurb
Hendricks et al. identify a set of structurally diverse small molecules that inhibit FSP1, a CoQ oxidoreductase that suppresses ferroptosis. The most potent FSP1 inhibitor, ferroptosis sensitizer 1 (FSEN1) sensitizes cancer cells to ferroptosis and synergizes with several ferroptosis inducers, including GPX4 inhibitors and endoperoxide-containing molecules such as DHA.
Graphical Abstract
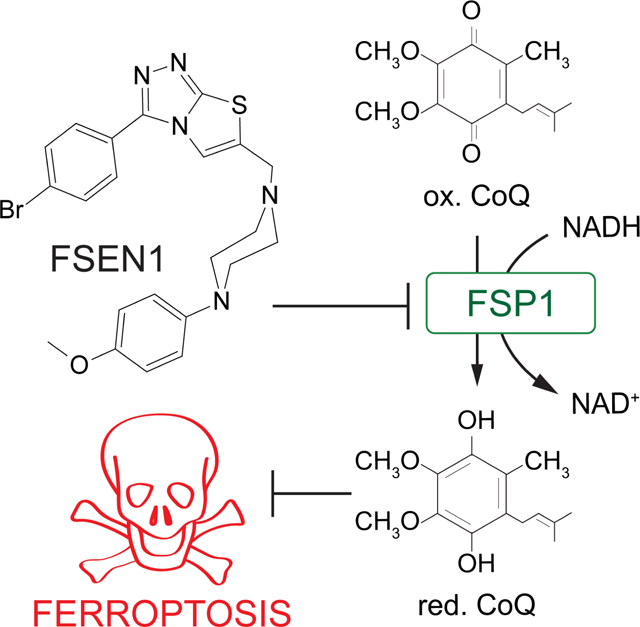
INTRODUCTION
Ferroptosis is an iron-dependent, non-apoptotic form of regulated cell death that is associated with the lethal accumulation of oxidatively damaged phospholipids (e.g., lipid hydroperoxides)1,2. As the ultimate effectors, phospholipid hydroperoxides, or their breakdown products, mediate membrane rupture and ferroptotic cell death1,2. Cellular ferroptosis sensitivity is determined by a variety of metabolic processes, including pathways that regulate labile iron pools and the generation of reactive oxygen species (ROS), the addition and removal of oxidation-sensitive polyunsaturated fatty acids (PUFAs) to and from phospholipids, and ferroptosis defense systems that suppress the accumulation of lipid hydroperoxides1,2. These ferroptosis defense systems function through two mechanisms, the conversion of lipid hydroperoxides into non-toxic lipid alcohols by the glutathione (GSH) peroxidase GPX43 and the generation of endogenous antioxidants that prevent lipid radical propagation, such as the enzymatic regeneration of the reduced form of coenzyme Q10 (CoQ) (i.e., ubiquinol) by ferroptosis suppressor protein 1 (FSP1)4,5 and dihydroorotate dehydrogenase (DHODH)6, tetrabiohydropterin by GTP cyclohydroxylase-1 (GCH1)7,8, and hydropersulfides by enzymatic and non-enzymatic pathways9,10.
Ferroptosis serves as a natural mechanism to restrict cancer proliferation and survival that is engaged by tumor suppressors. For example, the expression of the system antiporter subunit SLC7A11, required for cystine uptake and GSH synthesis, is inhibited by the tumor suppressors BAP111, p5312, and Kelch-like ECH-associated protein 1 (KEAP1)13. Inactivating mutations in these tumor suppressors upregulate SLC7A11 and other genes that promote ferroptosis resistance14. While oncogene-driven reprogramming of cancer cell metabolism addresses the amplified cellular demands for nutrients and energy, the accompanying increases in ROS, and a consequent overreliance upon ferroptosis defense systems for survival, yield potential therapeutic vulnerabilities14–16. Indeed, inhibition of the GSH-GPX4 pathway is effective in killing many cancer cells in vitro and in reducing tumor growth in preclinical models of therapy resistant cancers such as pancreatic ductal adenocarcinoma (PDAC)17,18, clear cell and chromophobe renal carcinomas19,20, triple-negative breast cancer21, MYCN-amplified neuroblastoma22–25, and drug-resistant persister cancer cells that give rise to relapse26,27. Despite these promising findings, some cancer cells are resistant to inhibition of the GSH-GPX4 pathway due to compensation by parallel ferroptosis defense systems. However, in contrast to the GSH-GPX4 pathway28, few molecular tools are available that target the other ferroptosis defense systems such as the CoQ-FSP1 pathway.
FSP1 is an important ferroptosis resistance factor in cancer that compensates for the loss of GPX4 by mediating NAD(P)H-dependent reduction of ubiquinone (oxidized form of CoQ) to ubiquinol (reduced form of CoQ), which in turn acts as a lipophilic antioxidant to prevent lipid peroxidation propagation4,5. FSP1 exhibits high expression in ferroptosis resistant non-small cell lung cancer and its expression correlates with poor patient prognosis29. Genetic disruption of FSP1 sensitizes cancer cells to ferroptosis and impairs tumor growth in a model of KEAP1-deficient lung cancer4,29. The tumor suppressor KEAP1 is an adaptor for the cullin-3 E3 ubiquitin-protein ligase that mediates the proteasomal degradation of nuclear factor erythroid 2-related factor 2 (NRF2), a master transcription factor that regulates the expression of an antioxidant gene program30,31. Thus, inhibitors of FSP1 have potential therapeutic value as anti-cancer monotherapies or when used in combination with ferroptosis inducers such as compounds targeting the GSH-GPX4 pathway. Consistent with this possibility, an inhibitor of FSP1 (iFSP1) sensitized multiple cancer cell lines to ferroptosis triggered by GPX4 inhibition and radiotherapy5,29. iFSP1 provides an important proof of concept, but additional structurally distinct FSP1 inhibitors with improved pharmacokinetic and pharmacodynamic properties are critically needed to fully explore the potential of FSP1 as a therapeutic target.
To address the unmet need for FSP1 inhibitors, we conducted a series of small molecule screens that leverage an in vitro assay of FSP1 CoQ oxidoreductase activity and an orthogonal cell-based assay of FSP1-dependent ferroptosis suppression. These screens identified multiple structurally distinct small molecules that directly inhibit FSP1 CoQ oxidoreductase activity in vitro and sensitize cancer cells to ferroptosis. Moreover, a second screen of U.S. Food and Drug Administration (FDA)-approved and bioactive compounds identified a synergistic relationship of FSP1 inhibition with the endoperoxide-containing drug dihydroartemisinin (DHA) in inducing ferroptosis. Our study provides new molecular tools for the characterization of FSP1 as an anti-cancer therapeutic target and demonstrates the utility of combinatorial treatment regimens targeting FSP1 together with other ferroptosis defense pathways.
RESULTS
Chemical library screen identifies small molecule inhibitors of FSP1 activity
To identify small molecule inhibitors of FSP1, we conducted a chemical screen employing an in vitro assay of FSP1 activity (Figure 1A,B). This assay exploits the change in 355 nm absorbance as NADH is oxidized to NAD+ during the reduction of CoQ1 by recombinant FSP1 (Figure 1B,C). As anticipated, the addition of FSP1 to the reaction mix resulted in a decrease in absorbance over time (Figure 1C). Moreover, the decrease in absorbance was blocked in a dose-dependent manner by iFSP1 (IC50 of 4 μM) (Figure 1C, Figure S1A). These data demonstrate that iFSP1 is a direct FSP1 inhibitor and validate our activity assay as a method to identify FSP1 inhibitors.
Figure 1. Small molecule screens identify FSP1 inhibitors.

A) Flow chart of the chemical screens and experimental validation employed to identify FSP1 inhibitors. B) Schematic of the in vitro activity assay using purified recombinant FSP1. C) FSP1 activity was measured using NADH absorbance in the presence of increase amounts of iFSP1. Mean ± SEM of three technical replicates. D) Scatter plots of 15,000 antibacterial compounds and 100,000 diverse + 5,370 FDA and Bioactive compounds assayed with in vitro absorbance-based assay of FSP1 activity. “Hits” are defined by a normalized absorbance value of <0.267. Dashed lines represent SD of 3-sigma cutoff defined by activity range of vehicle and No Protein control. E) Western blot analysis of H460C Cas9 and GPX4KO cells. F) Representative images of H460 GPX4KO cells treated with vehicle and iFSP1. Dead cells are marked with SYTOX Green. Scale bar represents 50 μm. Images from one of three independent experiments are shown. G) Dose response of iFSP1 induced cell-death in H460C Cas9 and GPX4KO cells. Lethal fraction was calculated by IncuCyte quantification of the ratio of dead cells over the total amount of cells over 24 hr. The mean ± SEM bars represent three biological replicates. H) Lethal fraction (AUC) was calculated in H460C Cas9 and GPX4KO cells incubated with increasing doses of 168 small molecules that inhibited FSP1 in vitro. The heat map reflects n=3 cell biological replicates. I) Lethal fraction was calculated H460C GPX4KO cells incubated with increasing doses of the most potent 19 FSP1 inhibitors and iFSP1. Resupplied, validated small molecules were used. The heat map reflects n=3 cell biological replicates.
In the primary screen, the effect of 120,370 small molecules on FSP1 activity was analyzed using the in vitro FSP1 activity assay (Figure 1A,D). This screen identified 1,120 candidate FSP1 inhibitors (Figure 1D). Duplicate analyses of the candidate inhibitors were performed for validation (Figure S1B). A control lacking FSP1 protein was included to identify small molecules that altered absorbance independently of FSP1 (Figure S1C). This series of validation steps yielded 323 inhibitors. Finally, triplicate 10-point dose response analyses were performed to determine the in vitro potency of 168 selected FSP1 inhibitors. 26 of these compounds have a lower IC50 than iFSP1 (< 4 μM) and 11 compounds have an IC50 below 100 nM.
Small molecule inhibitors of FSP1 trigger cell death in a cancer cell model
To determine whether the 168 FSP1 inhibitors are able to inhibit FSP1 in cells, we developed an orthogonal cell-based assay of FSP1 activity. This assay uses NCI-H460 KEAP1 mutant lung cancer cells expressing mCherry (H460C) in which GPX4 was knocked out using CRISPR-Cas9 (H460C GPX4KO) or which expressed Cas9 as a control (H460C Cas9) (Figure 1E). mCherry was used as a live cell marker, which together with the SYTOX Green cell death marker can be used to calculate the fraction of dead cells (i.e., lethal fraction32,33). To validate this assay, H460C Cas9 cells and H460C GPX4KO cells were treated with iFSP1 (Figure 1F,G). iFSP1 selectively triggered cell death in H460C GPX4KO cells, but not in the H460C Cas9 cells (Figure 1F,G). These data demonstrate that this assay can be used to characterize the ability of FSP1 inhibitors to inhibit FSP1 and induce ferroptosis in cancer cells.
Employing this assay, we analyzed the amount of cell death induced by 168 compounds that inhibited FSP1 oxidoreductase activity in vitro (Figure 1A,D) in the H460C Cas9 and H460C GPX4KO cells (Figure 1H). H460C Cas9 cells were included to identify any small molecules that kill cells through a ferroptosis-independent mechanism. Triplicate 10-point dose response analyses were performed, and cell death was measured using fluorescence time-lapse imaging (Figure 1H). ~50 of the FSP1 inhibitors induced cell death in the H460C GPX4KO cells (Figure 1H), but not the H460C Cas9 cells, indicating that these compounds are synthetic lethal with GPX4KO and are not generally toxic to cells.
19 of the most potent compounds were tested again using validated compounds (>95% purity) in 20-point dose response analyses in both the in vitro (Figure S2 and S3) and cell-based FSP1 assays (Figure 1I). As observed in our primary and follow up screens, these 19 compounds directly inhibited purified FSP1 in vitro (Figure S2 and S3) and triggered cell death in the H460C GPX4KO cells, but not the H460C Cas9 cells (Figure 1I). We named these validated FSP1 inhibitors – ferroptosis sensitizer 1–19 (FSEN1–19). The structures of FSEN1–19, their IC50 for inhibition of purified FSP1 activity, and their EC50 for triggering cell death in H460C GPX4KO cells are shown in Figure 2. These compounds can be organized into seven groups of structurally related compounds. The largest group of compounds (Group 1, red box) share a disubstituted [1,2,4] triazolo-thiazole core scaffold structure (Figure 2).
Figure 2. Multiple structurally distinct FSP1 inhibitor scaffolds.

The structures of FSEN1–19 are shown with their calculated IC50 values from end-point assays measuring inhibition of FSP1 activity in vitro (n=2 independent biological replicates) and their calculated EC50 values from lethal fraction (AUC) quantification in H460C GPX4KO cells (n=3 cell biological replicates).
FSEN1 is an uncompetitive inhibitor of FSP1
Amongst the newly identified FSP1 inhibitors, FSEN1 exhibited the highest potency in triggering cell death in the H460C GPX4KO cells (EC50 = 69.363 nM) (Figure 2). To test the specificity of FSEN1, we examined its ability to inhibit NQO1, another CoQ oxidoreductase that has been implicated in ferroptosis34. In contrast to FSP1 (Figure 3A), FSEN1 had no effect on the CoQ oxidoreductase activity of NQO1 (Figure 3B), indicating that FSEN1 does not generally inhibit CoQ oxidoreductases and that FSEN1 exhibits selectivity towards FSP1.
Figure 3. Mechanisms of inhibition of FSP1 by FSEN1.

A,B) Purified recombinant FSP1 (A) and NQO1 (B) CoQ1 oxidoreductase activities were measured in the presence of increasing concentrations of FSEN1. Data was normalized to the slopes calculated from DMSO and No Protein controls. IC50 values displayed in nM were calculated from a non-linear regression curve fit. Data are mean ± SEM bars (n=3 independent biological replicates). C,D) Michaelis-Menten and Line Weaver-Burk plots of FSP1 treated with increasing concentrations of NADH in the presence of vehicle or FSEN1. 10 μM CoQ-Coumarin was used as the co-substrate for FSP1, and reduced CoQ-Coumarin fluorescence was measured as a read-out of enzymatic product formation. Error bars represent linear regression standard error of initial rates taken from three biological replicates. Vmax and Km were calculated from the non-linear regression curve fit. E,F) Michaelis-Menten and Line Weaver-Burk plots of FSP1 treated with increasing concentrations of CoQ-Coumarin in the presence of vehicle or FSEN1. 200 μM NADH was included as a co-substrate for FSP1, and reduced CoQ-Coumarin fluorescence was measured as the read-out of enzymatic product formation. Error bars represent linear regression standard error of initial rates (n=3 independent biological replicates). Vmax and Km were calculated from the non-linear regression curve fit.
To understand the mechanism of FSP1 inhibition by FSEN1, enzyme kinetics were analyzed using the in vitro FSP1 activity assay in the presence of increasing amounts of its substrates, NADH (Figure 3C,D) and a fluorescent ubiquinone analogue CoQ-coumarin (Figure 3E,F). As FSEN1 concentrations increased, we observed a decrease in Kcat values (Figure 3C,E), and the slopes of the Lineweaver-Burke plots were parallel (Figure 3D,F). We observed a similar effect of FSEN1 on the Vmax of FSP1 using CoQ1 as a substrate (Figure S4), but this assay was unable to resolve the Km due to limitations in its sensitivity. These findings reveal that FSEN1 is an uncompetitive inhibitor of FSP1. Thus, these data suggest that FSP1 requires binding to its substrates NADH and CoQ first in order to be permissive for FSEN1 binding, which then yields the inactive complex.
FSEN1 triggers ferroptosis in cancer cells by inhibiting FSP1
To characterize the cell death induced by FSEN1 treatment, we further examined its effects on H460C Cas9 lung cancer cells. Treatment with FSEN1 sensitized H460C Cas9 cells to cell death induced by two GPX4 inhibitors, RSL3 and ML162 (Figure 4A–C). A checkerboard dose-response matrix of FSEN1 (0–15 μM) and RSL3 (0–15 μM) provided further evidence of synthetic lethality and indicated that the minimal doses that achieve maximal synergy for inducing ferroptosis in H460C Cas9 cells are 0.55 μM FSEN1 together with 0.55 μM RSL3 (Figure 4B). Dying cells exhibited characteristic morphologies of ferroptotic cells and the cell death was blocked by ferrostatin-1 (Fer-1) (Figure 4C). Consistent with FSEN1 sensitizing these cells to ferroptosis, cell death induced by the co-treatment of FSEN1 and RSL3 was blocked by incubation with known ferroptosis inhibitors, including the radical trapping antioxidants idebenone, Fer-1, and tocopherol, and the iron chelator deferoxamine (DFO) (Figure 4D, Figure S5E). In contrast, the apoptosis inhibitor Z-VAD-FMK and necroptosis inhibitor Nec1s had no effect (Figure 4D, Figure S5E). Furthermore, treatment with FSEN1 sensitized cells to lipid peroxidation, as measured using the fluorescent lipid peroxidation reporter dye BODIPY C11 (Figure 4E). These findings demonstrate that FSEN1 sensitizes H460 lung cancer cells to lipid peroxidation and ferroptosis induced by GPX4 inhibition.
Figure 4. FSEN1 is synthetic lethal with GPX4 inhibitors and sensitizes cancer cells to ferroptosis through inhibition of FSP1.

A) Dose response of RSL3 and ML162-induced cell death in H460C Cas9 cells co-treated with 1 μM FSEN1 and 2 μM Fer-1 where indicated. Data are mean ± SEM (n=3 cell biological replicates). B) Heat map represents the fraction of viable H460C Cas9 cells co-treated with increasing doses of FSEN1 and RSL3 (n=3 cell biological replicates). C) Representative images of H460C Cas9 cells co-treated with 1.6 μM RSL3, 1 μM FSEN1 and 2 μM Fer-1 as indicated. Scale bar = 200 μm. Images from one of three independent experiments are shown. D) Lethal Fraction (AUC) of 5 μM RSL3-induced cell death in H460C Cas9 cells co-treated with 1 μM FSEN1 together with the indicated inhibitors of ferroptosis (Fer-1 [2 μM], DFO [100 μM], idebenone [10 μM], tocopherol [10 μM]), apoptosis (Z-VAD [10 μM]), and necroptosis (Nec1s [1 μM]). Data are mean ± SEM (n=3 cell biological replicates). **p<0.01 by one-way ANOVA with Dunnett’s multiple comparisons test. E) Representative flow cytometry histogram (left panel) and quantification (right panel) of H460C Cas9 cells treated with 200 nM RSL3 and/or 1 μM FSEN1 and labeled with the lipid peroxidation sensor BODIPY 581/591 C11. Green fluorescence intensity was analyzed by flow cytometry with the FITC channel. Data are mean ± SD (n=3 cell biological replicates). *p<0.05, ***p=0.0003 by one-way ANOVA with Tukey’s multiple comparisons test. F) Dose response of RSL3-induced cell death in H460C FSP1KO cells co-treated with 1 μM FSEN1. Data are mean ± SEM (n=3 cell biological replicates). G) H460C spheroids were treated with vehicle, 5 μM FSEN1, and 5 μM RSL3 as indicated. Representative images from one of 20 independent experiments are shown. Scale bar represents 100 μm. The total intensity of the SYTOX green signal for each spheroid was quantified and the mean ± SEM plotted (n=20 independent spheroids).
To determine if FSEN1 sensitizes cells to ferroptosis by inhibiting FSP1, we tested the effect of FSEN1 on RSL3-induced ferroptosis in a H460C cell line in which we knocked out FSP1 using CRISPR-Cas9 (H460C FSP1KO cells). H460C FSP1KO cells were greatly sensitized to RSL3-induced cell death (Figure 4F). Importantly, although FSEN1 sensitized H460C Cas9 cells to RSL3-induced cell death, treatment of the H460C FSP1KO cells with FSEN1 did not result in any additional sensitization to RSL3-induced cell death (Figure 4F). These data indicate that the effects of FSEN1 are due to on target inhibition of FSP1 and not through inhibition of other ferroptosis resistance factors, including other CoQ oxidoreductases implicated in ferroptosis such as NQO134.
High cell densities and cell-cell interactions promote ferroptosis resistance by inducing multiple signaling pathways, including the NF2–YAP35 and TAZ-EMP1-NOX436 pathways, underscoring the importance of testing ferroptosis sensitivity in 3-dimensional (3-D) tumor models. Spheroids are 3-D aggregates of cancer cells that more closely reflect key characteristics of solid tumor biology, including cell-cell interactions, hypoxia, drug penetration, and interactions with deposited extracellular matrix37. Importantly, similar to our 2-D culture experiments, RSL3 and FSEN1 synergized to trigger cell death in H460 cells grown in the 3-D spheroids (Figure 4G). Together, these findings indicate that FSEN1 sensitizes H460 lung cancer cells to ferroptosis in both 2-D and 3-D culture models.
FSEN1 sensitizes multiple cancer cell lines of different tissue origins to ferroptosis
To examine the role of FSP1 in suppressing ferroptosis in different types of cancer, we measured the impact of FSEN1 on RSL3-induced cell death in a panel of cancer cell lines of various tissue origins, including lung (A549N), breast (HCC1143C), liver (Huh7C), glial (T98GN), bone (U 2-OSN), connective (HT-1080N), lymphoid (RL, SUDHL5), and skin (A375, SKMEL28, 501-MEL) (Figure 5A,B and Figure S5A,B). FSEN1 sensitized all cancer cells to RSL3-induced ferroptosis to varying extents, and in all cases the cell death was rescued by co-treatment with Fer-1. These findings indicate that FSEN1 can sensitize multiple cancer cell lines of different origins to ferroptosis induced by GPX4 inhibition. Notably, FSEN1 had a particularly large sensitizing effect on ferroptosis induction in A549 lung cancer cells (Figure 5A). A549, and the H460 cells used in our initial screens, are both lung cancer cell lines with KEAP1 mutations, which results in NRF2-dependent upregulation of FSP129. The prominent role of FSP1 in protecting A549 and H460 cells from ferroptosis correlated with high FSP1 protein levels and low GPX4 levels relative to other cancer cell lines (Figure S5C,D). It was also notable that FSEN1 induced a small amount of ferroptosis in A375 melanoma cells in the absence of an RSL3 co-treatment (Figure 5B), indicating that A375 cells have a particularly strong dependence on FSP1 for ferroptosis suppression. The amount of sensitization imbued by FSEN1 likely depends on the expression levels of FSP1 and ferroptosis related factors, such as GPX4, DHODH, GCH1, ACSL3, ACSL4, and others.
Figure 5. FSEN1 sensitizes cancer cells from different origins to ferroptosis.

A) Dose response of RSL3-induced cell death in the indicated cancer cell lines treated in the presence and absence of 1 μM FSEN1 and 2 μM Fer-1 as indicated. Data are mean ± SEM (n=3 cell biological replicates). B) Quantification of cell death in melanoma cell lines treated as in (A), calculated as SYTOX green positive object per mm2. Data are mean ± SEM (n=2 cell biological replicates).
Analysis of FSEN1 metabolic stability, in vivo pharmacokinetics, and inhibition of mouse FSP1
To test the in vitro metabolism and in vivo disposition of FSEN1, we measured FSEN1 metabolic stability and pharmacokinetics in mice, respectively. Following a single 20 mg/kg intraperitoneal (I.P.) dose of FSEN1 to mice (n=5), the median (interquartile range; IQR) maximum plasma concentration was 97.6 ng/mL (71.0–123.0), while the median (IQR) elimination half-life was 8.0 hours (6.1 – 8.8) (Figure S6A). FSEN1 also exhibited low intrinsic clearance (CLint) in mouse liver microsomes (11.54 μL/min/mg protein), further indicating adequate metabolic stability (Figure S6B). Collectively these data indicate that FSEN1 may be suitable for in vivo experiments in mice, such as studies examining the impact of FSEN1 on the growth of tumor xenografts.
To examine the ability of FSEN1 to inhibit mouse FSP1 (mFSP1), we expressed mFSP1 in the H460C FSP1KO cells. As expected, mFSP1 overexpression suppressed RSL3 induced ferroptosis (Figure S6C–E). Treatment with 1 μM FSEN1 had no effect on the ferroptosis sensitivity of the FSP1KO cells nor the FSP1KO cells overexpressing mFSP1 (Figure S6D). In addition, increasing the amount of FSEN1 up to 15 μM also had no effect on the sensitivity of FSP1KO cells overexpressing mFSP1 to RSL3 (Figure S6F). Furthermore, FSEN1 had no effect on the CoQ1 oxidoreductase activity of purified mFSP1 in vitro (Figure S6G). Together, these data indicate that despite the high amount of amino acid similarity between human and mouse FSP1, FSEN1 is selective to human FSP1 and does not inhibit the oxidoreductase activity of mFSP1. Future studies examining the two proteins may facilitate the identification of mutations that influence FSEN1 inhibition of human and mouse FSP1.
FSP1 and dihydroartemisinin treatment synergize to trigger ferroptosis in cancer cells
As ferroptosis suppression is mediated by several different pathways, it is perhaps not surprising that FSP1 inhibition alone is not sufficient to induce ferroptosis in most cancer cell lines. Furthermore, while FSEN1 is synthetically lethal with GPX4 inhibitors such as RSL3 (Figure 4A), the in vivo efficacy of RSL3 is known to be limited due to low solubility and poor pharmacokinetics28. Therefore, we conducted a synthetic lethal screen to identify compounds that eliminate cancer cells specifically in the absence of FSP1. H460C Cas9 and H460C FSP1KO cells were treated for 24 h with a library of 5,370 compounds that includes 1,200 FDA approved drugs and 4,170 bioactive compounds, and lethal fraction was quantified by time-lapse fluorescence imaging (Figure 6A,B). RSL3 was included as a positive control. As expected, RSL3 was cytotoxic for the H460C FSP1KO cells but not the H460C Cas9 cells (Figure 6B), validating the ability of our screening approach to detect synthetic lethal relationships.
Figure 6. FDA library screens identify DHA as inducer of ferroptosis in FSP1KO cells.

A) Western blot analysis of H460C Cas9 and FSP1KO cell lysates and screen schematic. H460C Cas9 and FSP1KO cells were incubated with 40 μM compounds and lethal fraction was quantified. B) Scatter plot of the lethal fraction (LF) for each compound in H460C Cas9 and H460C FSP1KO cells (n=1 cell biological replicate). Red dotted line represents the cutoff defined by the RSL3 positive control. C) Dose response validation of lethal fraction over time comparing H460C Cas9 and FSP1KO cells treated with 40 μM DHA. D) Representative IncuCyte images of H460C Cas9 and FSP1KO cells treated with DHA. Scale bar represents 50 μm. Images from one of two independent experiments are shown. E) The chemical structures of DHA and FINO2. F,G) Dose response of DHA and FINO2-induced cell death in H460C Cas9 (F) and FSP1KO (G) cells co-treated with vehicle or 1 μM FSEN1. Data are mean ± SEM bars (n=2 biological replicates). H,I) Dose response of DHA-induced cell death in H460C cells co-treated with vehicle (H) or 1 μM FSEN1 (I) together with the indicated inhibitors of ferroptosis (Fer-1 [2 μM], DFO [100 μM], idebenone [10 μM], tocopherol [10 μM]), apoptosis (Z-VAD [10 μM]), and necroptosis (Nec1s [1 μM]). Data are mean ± SEM (n=2 cell biological replicates).
Several compounds selectively induced cell death in the FSP1KO cells, including DHA (Figure 6B–D). DHA is a sesquiterpene lactone compound (Figure 6E) that is the active metabolite of an FDA approved drug (artesunate), which has been widely used as an antimalarial, and that has also been explored as an anti-cancer therapeutic38. The endoperoxide bridge within DHA (Figure 6E) is known to react with ferrous iron and stimulate the formation of toxic free radicals38. In addition, DHA was recently shown to induce ferroptosis in multiple cancer cell lines39–42. Similar to DHA, the ferroptosis inducer FINO2 contains an endoperoxide bridge (Figure 6E) that oxidizes iron, increases lipid peroxidation, and induces ferroptosis43. FSEN1 treatment and FSP1 KO strongly sensitized H460C cells to cell death induced by both DHA and FINO2 (Figure 6F,G). FSEN1 had no additional sensitizing effect in the FSP1KO cells (Figure 6F,G), indicating that the ability of FSEN1 to sensitize cells to DHA and FINO2 induced cell death is due to its on target inhibition of FSP1. DHA and FSEN1 treatment together induced cell death (DHA EC50 = 21.3 μM) that was strongly suppressed by ferroptosis inhibiting radical trapping antioxidants (Fer-1, tocopherol, idebenone) and completely suppressed by DFO (Figure 6H,I).
To characterize the synergy between DHA, RSL3, and FSEN1, we performed a checkerboard dose-response matrix of DHA (0–80 μM) and RSL3 (0–15 μM) in the presence of increasing doses of FSEN1 (0, 0.05, 0.5, 5 μM), and quantified synergy potency using a computational zero interaction potency (ZIP) modeling system44 (Figure 7A). The ZIP synergy scoring system defines compound synergy as a value >10, additive effects −10<0<10, and antagonistic effects <−10 (Figure 7A). The combinations of FSEN1 and RSL3 (Mean ZIP score = 38.28) and FSEN1 and DHA (Mean ZIP score = 26.45) exhibited strong synergy (Figure 7B,C and Figure S7). In contrast, DHA and RSL3 showed little synergy (Mean ZIP score = 4.11) (Figure 7B,C and Figure S7) and a stronger synergy score was observed with all three compounds (Mean ZIP score = 52.69) (Figure 7D). Together, these data indicate that FSP1 inhibition increases cancer cell sensitivity to endoperoxide-containing ferroptosis inducers, including DHA. Moreover, these findings demonstrate the potential value of combinatorial therapeutic strategies that combine FSP1 inhibitors with additional ferroptosis inducers to overcome the multitude of cancer ferroptosis defenses.
Figure 7. FSEN1 is synergistic with DHA.

A) Overview of the experimental design and analysis of synergy for two and three drug combinations. B) Heat maps of the fraction of viable cells in H460C Cas9 cells co-treated with vehicle and varying doses of FSEN1 (0.05, 0.5, 5 μM) in the presence of increasing concentrations of DHA and RSL3. Data are mean (n=3 cell biological replicates). C) 3D-dose response landscape plots illustrating the distribution and depth of synergy between the different drug combinations and doses tested in H460C Cas9 cells with the corresponding average ZIP synergy scores and p-values. D) Multi-drug synergy bar plots of H460C Cas9 cells illustrating mean ZIP synergy scores for all possible drug combinations tested. Data are mean ± SEM (n=3 cell biological replicates). P-values represents the significance of the difference between the estimated average synergy score over the whole dose-response matrix and 0% inhibition under the null hypothesis of non-response.
DISCUSSION
Induction of ferroptosis has emerged as a promising strategy to treat therapy-resistant cancer14. Despite the discovery of multiple cellular ferroptosis defense systems2,14, current ferroptosis inducers are mostly limited to the GSH-GPX4 pathway, impeding the study and assessment of other ferroptosis regulators as therapeutic targets. Here, we describe the discovery and characterization of structurally distinct small molecule FSP1 inhibitors that directly inhibit FSP1 activity and sensitize cancer cells to ferroptosis. The most potent of these new FSP1 inhibitors, FSEN1, is an uncompetitive FSP1 inhibitor that is synthetic lethal with GPX4 inhibitors and endoperoxide-containing ferroptosis inducers, including FINO2 and DHA, the active metabolite of the FDA approved drug artesunate.
Given that FSP1 expression correlates with resistance to GPX4 inhibitors across many cancer cell lines4,5, it is likely that FSEN1 will be useful for sensitizing many cancer types to ferroptosis inducers that target the GSH-GPX4 axis. Indeed, FSEN1 sensitized several cancer cell lines of different tissue origins bearing different oncogenic mutations to RSL3. The variable amount of sensitization FSEN1 confers on distinct cancer cell lines indicates that some cancers are more reliant on FSP1 for ferroptosis suppression than others, likely reflecting the contribution of other ferroptosis defense pathways or differences in phospholipid composition and ROS production. Strong sensitization was observed in H460 and A549 cancer cells, which are both lung cancer cell lines with KEAP1 mutations. KEAP1 regulates a canonical pathway that mediates the ubiquitin-dependent proteasomal clearance of NRF2, a master transcriptional factor that governs expression of antioxidant factors, including FSP129. We find that these cell lines have high FSP1 and low GPX4 protein levels, suggesting a means to stratify cancers that will respond more strongly than others to FSP1 inhibition based upon FSP1 and GPX4 levels. A similar increased effectiveness of DHODH inhibition on ferroptosis induction in cancer cells with low GPX4 was previously observed6. However, there are many factors that govern cellular sensitivity to ferroptosis and that while analysis of FSP1 and GPX4 levels may be predictive in some cells, this may not be the case in all cancer cells and additional regulators will need to be considered.
FSP1 inhibition alone was not sufficient to trigger ferroptosis in most of the cell types under the culture conditions that we examined here, except for the A375 melanoma cells. It is worth noting that FSP1 KO slowed tumor growth in an in vivo xenograft model of KEAP1 deficient lung cancer29, indicating that loss of FSP1 activity is sufficient to induce ferroptosis under some in vivo conditions. This may reflect the unique contribution of the in vivo tumor microenvironment and higher levels of PUFAs and ROS. Importantly, the observation that FSP1 KO impairs tumor growth in vivo raises the possibility that small molecule FSP1 inhibition may be effective as a monotherapy for specific cancers. The result also highlights the importance of future studies to explore FSP1 inhibition in in vivo cancer models. Although FSEN1 does not inhibit mFSP1, FSEN1 exhibits good pharmacokinetics and metabolic stability in mice and it may be useful for in vivo models that employ human cancer cells (i.e., tumor xenografts).
Our findings are consistent with the utility of FSP1 inhibition in combinatorial therapeutic regimes together with inhibitors of the GSH-GPX4 pathway. It is likely that FSP1 inhibition will sensitize cancer cells to standard of care treatments that trigger ROS generation and ferroptosis such as radiotherapy45,46, photodynamic therapy47,48, and immunotherapy49. Indeed, FSP1 KO and iFSP1 treatment sensitize cancer cells to radiotherapy29. Along these lines, we discover that FSP1 inhibition synergizes with DHA to induce ferroptosis. DHA is the active metabolite of a commonly used antimalarial (artesunate), it has been explored as an anticancer drug38, and several recent studies indicate that DHA is capable of triggering ferroptosis39–42. The endoperoxide bridge within DHA reacts with and oxidizes iron, leading to the production of reactive oxygen species and lipid hydroperoxides38. In H460 lung cancer cells, DHA induced minimal amounts of ferroptosis on its own, but its ability to induce ferroptosis was greatly enhanced by co-treatment with FSEN1. It remains possible that DHA operates through multiple mechanisms, oxidizing iron and triggering lipid peroxide formation and indirectly inhibiting GPX4 similar to what was reported for the endoperoxide ferroptosis inducer FINO243. Our findings highlight the potential of endoperoxide containing compounds (e.g., FINO243, FINO350, DHA) as therapeutic agents to induce ferroptosis by increasing iron oxidation, lipid peroxidation, and potentially GPX4 inhibition. Finally, it is worth noting that while the best known role of FSP1 is in the suppression of ferroptosis, FSP1 has also been implicated in the maintenance of glycolysis in muscle and brown adipocytes through its role in regenerating NAD+51,52. Thus, FSEN1 may also be lethal in cancer types that depend on glycolysis or may synergize with therapeutics that target glycolytic pathways.
In summary, our findings identify a set of structurally distinct FSP1 inhibitors that are effective in overcoming ferroptosis resistance in several cancer cell lines. Future studies will be necessary to determine whether these FSP1 inhibitors are effective in preclinical mouse models of cancer, as single agent therapeutics under certain conditions, and in sensitizing cancer cells to other treatments that increase ROS and lipid peroxidation, including ferroptosis induced by photodynamic therapy, radiation, and immunotherapy.
LIMITATIONS OF THE STUDY
Our findings indicate that FSEN1 directly inhibits recombinant human FSP1 activity in vitro and inhibits FSP1 suppression of ferroptosis in cultured cancer cells. Although the in vivo pharmacokinetics and metabolic stability of FSEN1 suggest that FSEN1 is suitable for in vivo studies, it remains to be determined whether FSEN1 inhibits FSP1 in vivo and whether FSEN1 treatment has any effect on tumor growth. Furthermore, because FSEN1 is selective for human FSP1, it will not be useful for studying the role of mouse FSP1 or tumor growth in Genetically Engineered Mouse Models (GEMM) models. It will be important to determine if other FSP1 inhibitors discovered in this study inhibit mouse FSP1 and to determine their impact in preclinical models of tumor growth.
SIGNIFICANCE
Emerging evidence suggests that induction of ferroptosis has value for the treatment of therapy resistant cancer. FSP1 functions as a key cellular defense factor that promotes ferroptosis resistance in cancer. Here, we report the discovery of several structurally diverse FSP1 inhibitors that increase the sensitivity of cancer cells to ferroptosis in vitro. The most potent of these inhibitors, FSEN1 synergizes with multiple ferroptosis inducers and exhibits pharmacokinetics and metabolic stability suitable for in vivo studies. Our findings provide valuable chemical inhibitors of FSP1 that will be useful for studying cellular mechanisms of ferroptosis and exploring the potential of FSP1 as a therapeutic target in cancer.
STAR METHODS
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, James Olzmann (olzmann@berkeley.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and Culture Conditions
H460 (HTB-177, gender: male), parental cells were obtained from ATCC. A375 (gender: female), HCC1143 (gender: female), and Huh7 (gender: male) parental cells were all obtained from UC Berkeley Cell Culture Facility. All cell lines were expanded for one passage then frozen down in multiple aliquots used for all subsequent experiments. A549N (gender: male), HCC1143C, NCI-H460C, NCI-H460C GPX4KO and FSP1KO lines were cultured in RPMI1640 with l-glutamine (Corning, Cat# 10–040CV). HT-1080N (gender: male), U-2 OSN (gender: female), T98GN (gender: male), Huh7C, and A375 cells were cultured in DMEM with l-glutamine and without sodium pyruvate (Corning, Cat# 10–017-CM). 501-MEL (gender: female) and SKMEL28 (gender: male) cells were cultured in DMEM high glucose with l-glutamine and without sodium pyruvate (Corning, Cat# 10–017-CMR). Cells (HT-1080N, U-2OSN, T98GN)32,53 were generated through stable expression of nuclear-localized mKate2 (denoted by an additional superscript ‘N’). All media were supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific and Gemini Bio Products), and all cell lines were grown at 37 °C with 5% CO2. All cell lines were tested for mycoplasma and were not authenticated.
Generation of cell lines
NCI-H460C FSP1KO lines were generated by infection with lentiCRISPR v2-Blast (Addgene plasmid no. 83489) virus, and NCI-H460C GPX4KO lines were generated by infection with lentiCRISPR v2-Hygro (Addgene plasmid no. 98291) virus, as previously described2. Cells expressing sgSAFE-mCherry (denoted by an additional superscript ‘C’) were generated from the respected parental cells via transduction with the lentiviral sgRNA expression vector with mCherry pMCB320 (Addgene plasmid no. 89359) virus, which directs the expression of cytosolic sgSAFE-mCherry. Polyclonal sgSAFE-mCherry expressing cells were selected for using puromycin or hygromycin respectively. H460C polyclonal pools were selected puromycin and further enriched using FACS (UC Berkeley shared FACS Facility). H460C FSP1KO + mFSP1 OE lines were generated by infection with pLenti-CMV-Hygro-DEST (Addgene plasmid no. 17454) containing mFSP1.
METHOD DETAILS
Plasmids
For protein expression, FSP1(WT), lacking the ATG start codon were inserted into the pET-His6-TEV vector (Addgene plasmid no. 29653), as previously described2. For protein expression of NQO1, NQO1(WT) lacking the ATG start codon was generated by PCR amplification of NQO1 from an NQO1-GFP pcDNA5/FRT/TO plasmid generated in our previous study2. For protein expression of mFSP1, mFSP1(WT) lacking the ATG start codon was generated by PCR amplification of mFSP1 (NM_153779.2, OMu13438D) from mammalian expression cloning vector, pcDNA3.1+/C-(K)-DYK (GenScript, Cat# SC1200). The NQO1(WT) and mFSP1(WT) amplicon was inserted into the pET-His6-TEV vector (Addgene plasmid no. 29653), C-terminal to the His6-TEV tag using restriction enzyme-independent fragment insertion by polymerase incomplete primer extension. Cloning of full length mFSP1(WT) expression plasmid was generated by PCR amplification of mFSP1 (NM_153779.2, OMu13438D) from mammalian expression cloning vector, pcDNA3.1+/C-(K)-DYK (GenScript, Cat# SC1200). The mFSP1(WT) amplicon was inserted into the pENTR1A-GFP-N2 (Addgene plasmid no. 19364) vector using restriction enzyme-independent fragment insertion by polymerase incomplete primer extension, followed by Gateway recombination cloning (Thermo Fisher Scientific, Cat# 12535–029) into pLenti-CMV-Hygro-DEST (Addgene plasmid no. 17454). Specific primers can be found in Table S1.
Chemicals and reagents
Reagents used in this study include: RSL3 (Cayman Chemical, Cat# 19288), Fer-1 (Cayman Chemical, Cat# 17729), Idebenone (Cayman Chemical, Cat# 15475), DFO (Cayman Chemical Cat# 14595), ML162 (Cayman Chemical, Cat# 20455), ZVAD(OMe)-FMK (Cayman Chemical, cat.# 14463), Necrostatin-1 (Cayman Chemical Cat# 11658), DHA (Selleck Chemical Cat# S2290), Puromycin (Thermo Fisher Scientific, Cat# A1113802), SYTOX Green Dead Cell Stain (Thermo Fisher Scientific, Cat# S34860), Polybrene (Sigma-Aldrich, Cat# TR-1003), Coenzyme Q1 (Sigma-Aldrich, Cat# C7956), Resazurin (Thermo Fisher Scientific, Cat# R12204), Bodipy C11 (Thermo Fisher Scientific, Cat# D3861) and NADH (Millipore Sigma, Cat# 481913). A 100,000-member compound Diverse Library and a 15,000 compound Antibacterial Library were obtained from ChemDiv. The 1,200 compound FDA approved, and 4,170 compounds Bioactive libraries were obtained from TargetMol. Compounds were stamped into 384 well Non-Binding Surface (NBS) plates (Corning, Cat# CLS3640) using a Cybio Well Vario liquid handler (Analytik-Jena, Germany).
Small molecule screen for FSP1 inhibitors / Cell death analysis
For the small molecule drug screen, 0.5 μL of compounds were stamped into 384 well NBS plates (Corning, Cat# CLS3640) in dose response diluted 2-fold with a high of 2 mM. 60 mL of purified His tagged FSP1(WT) protein at 50 nM was prepared and 12.5 μL of this protein solution was aliquoted into 384 well NBS-plates (Corning, Cat# CLS3640) containing compound and allowed to incubate for 30 min at room temperature. After 30 min incubation 12.5 μL of reaction buffer (1 mM NADH, 800 μM CoQ1) was added into the 384 well NBS-plates containing protein and compounds for a final concentration of 25 nM His tagged FSP1(WT) protein, 500 μM NADH (Millipore Sigma, Cat# 481913) and 400 μM CoQ1 (Sigma-Aldrich, Cat# C7956). Each well contained a 25 μL mixture and 40 μM compound in the primary screen. All aliquots for in-vitro drug screen were completed with an Analytik-Jena Cybio Well Vario liquid handler. 2 mM compounds were stored in 100% DMSO in 384 well plates. Wells were homogenized with a Bioshake 3000 ELM orbital shaker at 2,400 rpm for 45 sec and condensates were allowed to settle for 30 min before scanning. Measurements were taken at 355 nm with a EnVision 2104 multilabel plate reader (PerkinElmer). No Protein control data was used for background subtraction prior to upload to CCDvault for normalization to DMSO vehicle control wells. Compounds with confirmed data that have normalized absorbance values of less than 0.164 were chosen for dose response. Wells that exhibit three standard deviations from the untreated sample and a No Protein control were selected for dose-response screening. The same His-tagged FSP1(WT) protein were tested against candidate drugs using a 10-point serial dilution starting at 40 μM using the same procedure.
For the cell-based screen, cells were seeded in triplicate at a density of 1000–1500 cells per 50 μL per well in black 384-well plates (Thermo Fisher Scientific, Cat# 142761) and (Corning, Cat# CLS3985) 24 hr before start of imaging. After 24 hr, an additional 50 μL of drug infused medium containing 30 nM SYTOX Green Dead Cell Stain was carefully placed into the wells on top of existing medium. The plates were immediately transferred to an IncuCyte S3 imaging system (Essen Bioscience) enclosed in an incubator set to 37°C and 5% CO2. One image per well were captured in the phase, green, and red channels every 1.5 or 3 hr over a 24 hr period, and the ratio of SYTOX Green-positive objects (dead cells) to SYTOX Green-positive plus Sg-SAFE mCherry-positive objects (total cells) was quantified using S3 image analysis software (Essen Bioscience). For each treatment condition, the SYTOX-to-’SYTOX+mCherry’-object ratio was plotted against the 24 hr imaging interval, the Area Under the Curve (AUC) was calculated and the average AUC was plotted using Prism (GraphPad). To calculate the half-maximal effective concentration (EC50) values, the AUC curve was fit to a variable slope function comparing response to drug concentration.
Lipid peroxidation assay
Cells seeded in a 6-well plate were treated with 200 nM RSL3 for 5 hr and washed once with DPBS containing calcium and magnesium (Gibco, Cat# 14040117). Cells then were incubated in DPBS containing 5 μM BODIPY 581/591 C11 (Invitrogen, Cat# D3861) at 37°C for 10 min and washed 3x with DPBS without calcium or magnesium (Gibco, Cat# 14190144). Cells were detached from the plate with trypsin, and green fluorescence was analyzed by flow cytometry (>10,000 cells) on a BD LSRFortessa. Data were analyzed using FlowJo.
Spheroid / 3D cell culture
2500 NCI-H460 cells with 100 μL full serum RPMI media were seeded in 96-well Black/Clear Round Bottom Ultra-Low Attachment Spheroid Microplate (Corning, Cat# 4515). Cells were incubated at 37°C for 30 min before another 100 μL RPMI media containing 2% Matrigel (Corning, Cat# 354234) was added. Plates were centrifuged at 750x g for 15min and grew at 37°C for 2 days. For FSEN1 and RSL3 treatment, 100 μL RPMI media was slowly removed without disturbing the spheroid. Another 100 μL RPMI media containing 1% Matrigel, 60 nM SYTOX Green dye, 10 μM FSEN1 or 10 μM RSL3 or both was added back into each well. Spheroids were imaged in IncuCyte S3 with a 10x objective.
FDA and Bioactive Library Screen, FSP1KO + mFSP1 overexpression and Synergy experiments
For the FDA and Bioactive Library Screen, FSP1KO + mFSP1 overexpression and Synergy experiments, cells were seeded at a density of 1500 cells per 25 μL per well in black 384-well plates (Thermo Fisher Scientific, Cat# 142761), (Corning, Cat# CLS3985), and (Grenier Bio, Cat# 781091) 24 hr before start of imaging. After 24 hr, an additional 25 μL of drug infused medium containing 30 nM SYTOX Green Dead Cell Stain was carefully placed into the wells on top of existing medium. The plates were immediately transferred to an IncuCyte S3 imaging system (Essen Bioscience) enclosed in an incubator set to 37°C and 5% CO2. One image per well were captured in the phase, green, and red channels every 4 hr over a 24 hr period, and the ratio of SYTOX Green-positive objects (dead cells) to SYTOX Green-positive plus mCherry-positive objects (total cells) was quantified using S3 image analysis software (Essen Bioscience). For each treatment condition, the SYTOX-to-’SYTOX+mCherry’-object ratio was plotted against the 24 hr imaging interval, the Area Under the Curve (AUC) was calculated, and the average AUC was plotted as a function of drug concentration (for example, RSL3) using Prism (GraphPad). To calculate the half-maximal effective concentration (EC50) values, the AUC curve was fit to a variable slope function comparing response to drug concentration.
Compound preparation for synergy experiments
Vehicle, RSL3 and DHA combinations were stamped in grid format 1:1 at 4x concentrations. The compounds were then diluted with SYTOX green infused media containing either vehicle, 0.10 μM, 1 μM, or 10 μM FSEN1 to the appropriate 2x concentration (PlateOne, Cat# 1884–2410). Compounds were then homogenized with a Bioshake 3000 ELM orbital shaker at 2,400 rpm for 45 sec prior to treatment.
Protein expression and purification
Expression vectors were transformed into LOBSTR-BL21 (DE3) competent cells (Kerafast, Cat# EC1002) and LB broth was inoculated for overnight growth at 37°C while shaking at 280 rpm. The following day, the cultures were diluted 1:100 into 1L LB broth and allowed to grow at 37°C while shaking at 280 rpm until the cultures reached an optical density at 600 nm (OD600) of 0.6, measured by NanoDrop One (Thermo Fisher Scientific, Cat# 13–400-518). The cultures were then induced with 0.7 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and grown at 30°C while shaking at 280 rpm overnight. Cultures were then centrifuged for 45 min at 4°C and 4500 rpm to pellet. 1L Bacterial pellets were resuspended in 25 mL lysis buffer (50 mM KH2PO4, 300 mM KCl, 10% glycerol, 30 mM imidazole, 1 mg/mL lysozyme, and 1 mM PMSF, pH 8.0) and incubated at 37°C while shaking at 280 rpm for 10 min. The resuspended cells were lysed by passing through a microfluidizer (Microfluidics model: LM10) 3× at 15,000 PSI and collected in an ice-bath-chilled beaker. Lysate was then ultracentrifuged for 30 min at 4°C and 50,000 x g. All subsequent purification steps were carried out at 4°C. The supernatant was passed through an Econo-Pac disposable chromatography column (Bio-Rad Cat: 732–1010) packed and equilibrated with 1 mL HisPur Ni-NTA agarose resin (Thermo Fisher Scientific, Cat# 88221) and washed 4× with 2 mL EQ buffer (50 mM KH2PO4, 300 mM KCl, 10% glycerol, and 30 mM imidazole, pH 8.0). Bound proteins were eluted from Ni-NTA resin bed by gravity flowing 5 mL elution buffer (50 mM KH2PO4, 300 mM KCl, 10% glycerol, 250 mM imidazole, pH 8.0) through the column. Using an Amicon Ultra-15 centrifugal filter (Sigma-Aldrich, SKU: UFC910008), the eluted proteins were buffer exchanged into SEC buffer (50 mM HEPES, 100 mM KCl, 2 mM DTT, pH 8.0), spiked with 15% glycerol and 10 mM DTT and further concentrated to 3 mL total volume. Protein sample was then further purified by gel filtration through a HiLoad 16/600 Superdex 75 Pg size exclusion chromatography column (Sigma-Aldrich, SKU: GE28–9893-33) in SEC buffer using a GE Akta Pure FPLC. Fractions were collected in 1.6 mL aliquots and combined based on purity, visualized by Coomassie staining after separation in SDS-page. Combined fractions were then concentrated to 2 mg/mL and snap-frozen in 50 μL aliquots using liquid nitrogen. Protein concentration was determined using a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Cat# PI23227).
FSP1 Kinetics (CoQ1 and CoQ-Coumarin)
To measure FSP1 kinetics, FSEN1 was dissolved and diluted in DMSO, and recombinant purified FSP1 was diluted in PBS. 0.5 μL FSEN1 and 12.5 μL FSP1 were mixed and incubated for 30 min in a NBS polystyrene 384-well plate (Corning, Cat# CLS3640) at RT. Vehicle control wells included 0.5 μL DMSO and were absent of any FSEN1. NADH (Millipore Sigma, Cat# 481913) was then dissolved and diluted in PBS and added to the plate wells. CoQ-Coumarin (Cayman Chemical, Cat# 29554) was dissolved in DMSO, diluted in PBS, and added to the plate wells to start the reaction. For NADH kinetics the final well volume was 25 μL, and final concentrations were 25 nM FSP1, 10 μM CoQ-Coumarin, 1–50 μM NADH, and 50–500 nM FSEN1. For CoQ-Coumarin kinetics the final well volume was 25 μL, and final concentrations were 6.25 nM FSP1, 1–30 μM CoQ-Coumarin, 200 μM NADH, and 12.5–125 nM FSEN1. Reduced CoQ-Coumarin fluorescence (Ex: 405 nm, Em: 475 nm) was measured as read-out of enzymatic product formation on a kinetic cycle with an interval time of 15 sec at RT. All data was acquired using a Tecan Spark. For CoQ1 final well volume was 25 μL, and final concentrations were 25 nM FSP1, 200–500 μM CoQ1, 500 μM NADH, and 0.05–1 μM FSEN1. NADH Absorbance (355 nm) was measured as an inverse read-out of enzymatic product formation on a kinetic cycle with an interval time of 3 min at RT. All data was acquired using a Tecan infinite M1000.
FSP1 and NQO1 % Activity Curves
To measure in vitro activity of FSP1 and NQO1 for IC50 calculation, recombinant purified FSP1 and NQO1 were diluted in PBS. FSEN1 was dissolved and diluted in DMSO, and 0.5 μL FSEN1 and 12.5 μL FSP1 or NQO1 respectively, were mixed on an orbital shaker for 45 sec at 450 rpm and incubated for 30 min in in a NBS polystyrene 384-well plate (Corning, Cat# CLS3640) at RT. NADH (Millipore Sigma, Cat# 481913) was dissolved and diluted in PBS, and CoQ1 (Sigma-Aldrich, Cat# C7956) was dissolved in DMSO and diluted in PBS. Diluted NADH and CoQ1 were combined into a 2X reaction mix and added to the plate wells to start the reaction. Final well volume was 25 μL, and final concentrations were 12.5 nM FSP1, 400 μM CoQ1, 500 μM NADH, and 0.01–20 μM FSEN1. NADH Absorbance (355 nm) was measured as an inverse read-out of enzymatic product formation on a kinetic cycle with an interval time of 2.5 min at RT. All data was acquired using a Tecan infinite M1000. Raw data from three biological replicates were then plotted and initial slopes were calculated using a linear regression in prism (GraphPad). All rates were normalized to vehicle and No Protein controls where the highest and lowest slope values were used as 0 and 100%. Normalized values were then plotted as a function of FSEN1 concentration in log scale, and prism (GraphPad) was used to perform a non-linear regression curve fit for log (inhibitor) vs. normalized (variable) slopes. IC50 values of FSEN1 for FSP1 and NQO1 were obtained from this non-linear regression. Specific activity rates were determined for human and mouse (Mus musculus) FSP1 (FSP1 and mFSP1) by measuring NADH Absorbance (355 nm) over time, and similar specific activities were utilized. As such, 125 nM mFSP1 was used to match the specific oxidoreductase activity of 12.5 nM FSP1. Raw data for mFSP1 activity was acquired and analyzed with identical methods to acquisition and analysis of FSP1 to determine percent activity.
Western blotting
Cells were washed two times with PBS prior to lysis in 1% SDS. Samples were then sonicated for 30 sec and incubated for 5 min at 100°C. Protein concentrations were determined using the bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, Cat# PI23227), and equal amounts of protein by weight were combined with 1× Laemmli buffer, separated on 4–20% polyacrylamide gradient gels (Bio-Rad Laboratories) and transferred onto nitrocellulose membranes (Bio-Rad Laboratories). Membranes were washed in PBS with 0.1% Tween-20 (PBST) and blocked in PBST containing 5% (w/v) dried milk or 5% (w/v) bovine serum albumin (BSA) for 30 min. Membranes were incubated for 24 hr in PBST containing 5% BSA (Akron Biosciences) and primary antibodies. After washing with PBST, membranes were incubated at room temperature for 30 min in 5% BSA and PBST containing fluorescent secondary antibodies. Immunoblots were imaged on a LI-COR imager (LI-COR Biosciences).
The following blotting reagents and antibodies were used: anti-AMID (Santa Cruz Biotechnology, Cat# sc-377120), anti-AIFM2 (LS bio, Cat# LS-C382008–50), anti-β-actin (Santa Cruz Biotechnology, Cat# sc-47778), anti-GPX4 (Abcam, Cat# ab125066), anti-ACSL4 (Sigma-Aldrich, Cat# SAB2701949), anti-GAPDH (EMD Millipore Corp, Cat# MAB374), anti-rabbit IRDye800 conjugated secondary (LI-COR Biosciences, Cat# 926–32211) and anti-mouse Alexa Fluor 680 conjugated secondary (Invitrogen, Cat# A32723).
In Vivo Pharmacokinetics
Gonadally intact adult male BALB/c mice (n=5) weighing 20–25 gm were used to characterize the in vivo pharmacokinetics of FSEN1. Mice were group-housed, five per cage, in a temperature-controlled (22°C) vivarium on a 12 hr/12 hr light/dark cycle with ad libitum access to food and water. Each mouse received a single 20 mg/kg FSEN1 dose administered (I.P.) in a 20% DMSO, 20% Ethanol, 20% PEG40, 40% PBS solution. Whole blood samples were collected via retro-orbital bleeds at 0.5, 1, 2, 4, 8 and 24 hr post-dose. Samples were processed to plasma and FSEN1 concentrations were quantitated using liquid chromatography with tandem mass spectrometry (LC-MS/MS). FSEN1 standards were prepared in blank mouse plasma and used to generate an external calibration curve using linear regression to plot the peak area ratio versus concentration with 1/x weighting (r2 ≥ 0.99) over the analytically reportable range (9.8 – 313 ng/mL). Pharmacokinetic data were analyzed using non-compartmental methods. All animal procedures were conducted in strict adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of California San Diego Institutional Animal Care and Use Committee (IACUC).
Metabolic stability in mouse liver microsomes
FSEN1 was incubated at a concentration of 2 μM in 0.5 mg/mL mouse liver microsomes (Xenotech, Cat# M1000). The reaction mixture contained 1.0 mM NADPH. Incubations were performed in triplicate in a final volume of 1.5 mL at 37°C. The incubation system without NADPH was used as a negative control, while midazolam (2 μM) was used as a positive control. Incubation was stopped at 0, 5, 15, 30 and 45 min by taking an aliquot of the reaction mixture and adding it to cold acetonitrile containing the analytical internal standard. Peak area of each analyte was determined by LC-MS/MS and used to calculate % remaining. The natural log (ln) of % remaining was plotted versus incubation time and the elimination rate constant (k) was calculated from linear regression. Half-life (t1/2) was calculated as ln2/k. Intrinsic clearance (CLint, μL/min/mg protein) was calculated as ln2/t1/2 x [volume of incubation (μL)/microsomal protein in incubation (mg)].
QUANTIFICATION AND STATISTICAL ANALYSIS
Lethal fraction scoring was performed using Microsoft Excel 16.71 (Microsoft Corporation, Redmond, WA). Lethal fraction, in vitro, and in vivo assay plotting and curve fitting were performed using Prism 9.5.1 (GraphPad Software, La Jolla, CA). Flow cytometry data were processed using FlowJo (V10.6.1) (FlowJo LLC, Ashland, OR). Synergy scoring and plotting were performed in RStudio version 2022.07.0 Build 548 using the SynergyFinder R package available from Bioconductor. Figures were assembled using Adobe Illustrator (Adobe Systems, San Jose, CA). Statistical details of experiments and statistical tests used can be found in the main text, figure legends, and STAR Methods.
Supplementary Material
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal Anti-AMID | Santa Cruz Biotechnology | Cat# sc-377120 |
| Mouse monoclonal Anti-β-actin | Santa Cruz Biotechnology | Cat# sc-47778 |
| Rabbit polyclonal Anti-GPX4 | Abcam | Cat# ab125066 |
| Rabbit polyclonal Anti-ACSL4 | Sigma-Aldrich | Cat# SAB2701949 |
| Rabbit polyclonal Anti-AIFM2 | LS Bio | Cat# LS-C382008-50 |
| Mouse monoclonal Anti-GAPDH | EMD Millipore Corp | Cat# MAB374 |
| Bacterial and virus strains | ||
| LOBSTR-BL21 (DE3) competent cells | Kerafast | Cat# EC1002 |
| Stbl3 competent cells | Macrolab at UC Berkeley | N/A |
| NEB 5DH-α | NEB | Cat# C278H |
| Biological samples | ||
| Mouse microsomes | Xenotech | Cat# M1000 |
| Chemicals, peptides, and recombinant proteins | ||
| RSL3 | Cayman Chemical | Cat# 19288 |
| Fer-1 | Cayman Chemical | Cat# 17729 |
| Idebenone | Cayman Chemical | Cat# 15475 |
| Deferoxamine mesylate (DFO) | Cayman Chemical | Cat# 14595 |
| ML162 | Cayman Chemical | Cat# 20455 |
| ZVAD(OMe)-FMK | Cayman Chemical | Cat# 14463 |
| Necrostatin-1 | Cayman Chemical | Cat# 11658 |
| Dihydroartemisin (DHA) | Selleck Chemical | Cat# S2290 |
| Puromycin | Thermo Fisher Scientific | Cat# A1113802 |
| SYTOX Green Dead Cell Stain | Thermo Fisher Scientific | Cat# S34860 |
| Polybrene | Sigma-Aldrich | Cat# TR-1003 |
| Coenzyme Q1 | Sigma-Aldrich | Cat# C7956 |
| CoQ-Coumarin | Cayman Chemical | Cat# 29554 |
| Resazurin | Thermo Fisher Scientific | Cat# R12204 |
| Bodipy C11 | Thermo Fisher Scientific | Cat# D3861 |
| NADH | Millipore Sigma | Cat# 481913 |
| Small Molecule Library | ChemDiv | N/A |
| FDA-Approved Drug Library | TargetMol | Cat# L4200 |
| Bioactive Compound Library | TargetMol | Cat# L4000 |
| Hygromycin | Thermo Fisher Scientific | Cat# J67371.XF |
| Bleomycin | Thermo Fisher Scientific | Cat# J60727.MCR |
| Recombinant FSP1 (Human) | This Study | N/A |
| Recombinant FSP1 (Mouse) | This Study | N/A |
| Critical commercial assays | ||
| CellTiter Glo Luminescent Cell Viability Assay | Promega | Cat# G7572 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# PI23227 |
| PCR Cloning System with Gateway® Technology | Thermo Fisher Scientific | Cat# 12535-029 |
| Experimental models: Cell lines | ||
| Human: NCI-H460 | ATCC | HTB-177 |
| Human: A375 | UC Berkeley Cell Culture Facility | N/A |
| Human: HCC1143 | UC Berkeley Cell Culture Facility | N/A |
| Human: Huh7 | UC Berkeley Cell Culture Facility | N/A |
| Human: NCI-H460C | This Study | N/A |
| Human: A549N | Forcina et al. 2022 | N/A |
| Human: HCC1143C | Li et al. 2022 | N/A |
| Human: NCI-H460C GPX4KO | This Study | N/A |
| Human: NCI-H460C FSP1KO | This Study | N/A |
| Human: HT-1080N | Forcina et al. 2022 | N/A |
| Human: U-2OSN | Forcina et al. 2022 | N/A |
| Human: T98GN | Forcina et al. 2022 | N/A |
| Human: Huh7C | This Study | N/A |
| Human: 501-MEL | Kind gift from Imanol Arozarena Martinicorena at Navarrabiomed | N/A |
| Human: SKMEL28 | Kind gift from Imanol Arozarena Martinicorena at Navarrabiomed | N/A |
| Human: NCI-H460C FSP1KO mFSP1 OE | This Study | N/A |
| Experimental models: Organisms/strains | ||
| Balb/C mice | Envigo | N/A |
| Oligonucleotides | ||
| See Table S1 | ||
| Recombinant DNA | ||
| pET-His6-TEV vector | Addgene | Cat# 29653 |
| hFSP1-pET-His6-TEV vector | Bersuker et al. 2019 | N/A |
| mFSP1-pET-His6-TEV vector | This paper | N/A |
| NQO1-pET-His6-TEV vector | This paper | N/A |
| GenEZ ORF Clone: Aifm2_OMu13438D_pcDNA3.1+/C-(K)-DYK | GenScript | Cat# SC1200 |
| CSII-prEF1a-mCherry-3xNLS | Addgene | Cat# 125262 |
| mCherry pMCB320 | Addgene | Cat# 89359 |
| pENTR1A-GFP-N2 | Addgene | Cat# 19364 |
| mFSP1-pENTR1A | This paper | N/A |
| pLenti CMV mFSP1 Hygro | This paper | N/A |
| Software and algorithms | ||
| Prism 9 | GraphPad Software | N/A |
| R Studio | Posit Software, PBC | https://posit.co/download/rstudio-desktop/ |
| Microsoft Excel | Microsoft Corporation | N/A |
| FlowJo | Becton, Dickinson & Company | N/A |
| SynergyFinder R package | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/synergyfinder.html |
| Oligonucleotides | ||
| His-pET-hNQO1 I-PIPE primer Forward 5’- CTG TAC TTC CAA TCC AAT ATT GTC GGC AGA AGA GCA CTG ATC −3’ | This paper | N/A |
| His-pET-hNQO1 I-PIPE primer Reverse 5’- TCG GAT CCG TTA TCC ACT TCC TCA TTT TCT AGC TTT GAT CTG GTT GTC −3’ | This paper | N/A |
| His-pET-hNQO1 V-PIPE primer Forward 5’ CAG ATC AAA GCT AGA AAA TGA GGA AGT GGA TAA CGG ATC CGA −3’ | This paper | N/A |
| His-pET-hNQO1 V-PIPE primer Reverse 5’- CAG TGC TCT TCT GCC GAC AAT ATT GGA TTG GAA GTA CAG −3’ | This paper | N/A |
| His-pET-mFSP1 I-PIPE primer Forward 5’- CTG TAC TTC CAA TCC AAT ATT GGG TCC CAG GTC TCG GTG GAT AC −3’ | This paper | N/A |
| His-pET-mFSP1 I-PIPE primer Reverse 5’ TCG GAT CCG TTA TCC ACT TCC TTA CGG TGG AGA CTG CCG CAT −3’ | This paper | N/A |
| His-pET-mFSP1 V-PIPE primer Forward 5’ ATG CGG CAG TCT CCA CCG TAA GGA AGT GGA TAA CGG ATC CGA ATT C −3’ | This paper | N/A |
| His-pET-mFSP1 V-PIPE primer Reverse 5’- GT ATC CAC CGA GAC CTG GGA CCC AAT ATT GGA TTG GAA GTA CAG G −3’ | This paper | N/A |
| mFSP1 I-PIPE primer Forward 5’- AAG GAA CCA ATT CAG TCG ACT GGA TGC CAC CAT GGG GTC C −3’ | This paper | N/A |
| mFSP1 I-PIPE primer Reverse 5’- GTC TAG ATA TCT CGA GTG CGG CCG CTT CAC GGT GGA GAC T −3’ | This paper | N/A |
| mFSP1 V-PIPE primer Forward 5’- AGT CTC CAC CGT GAA GCG GCC GCA CTC GAG ATA TCT AGA C −3’ | This paper | N/A |
| mFSP1 V-PIPE primer Reverse 5’- GGA CCC CAT GGT GGC ATC CAG TCG ACT GAA TTG GTT CCT T −3’ | This paper | N/A |
Highlights.
Identification of structural diverse small molecule inhibitors of FSP1
FSEN1 is an uncompetitive FSP1 inhibitor that sensitizes cancer cells to ferroptosis
FSEN1 synergizes with GPX4 inhibitors and endoperoxide-based ferroptosis inducers
ACKNOWLEDGEMENTS
This research was supported by grants from the National Institutes of Health (R01GM112948 to J.A.O. and 1R01GM122923 to S.J.D.), American Cancer Society (Research Scholar Award RSG-19-192-01 to J.A.O. and RSG-21-017-01-CCG to S.J.D.), and Melanoma Research Alliance (Award 620458 to J.A.O.). J.A.O. is a Chan Zuckerberg Biohub investigator and Miller Institute Professor. J.M.H. was supported by a University of California Cancer Research Coordinating Committee (CRCC) predoctoral fellowship. C.E.D. was supported by a Cayman Biomedical Research Institute (CABRI) Undergraduate Fellowship. The project was supported by the UC Berkeley Drug Discovery center within the Henry Wheeler Center for Emerging and Neglected Diseases with support from the Sergey Brin Family Foundation.
Footnotes
DECLARATION OF INTERESTS
J.A.O. is a member of the scientific advisory board for Vicinitas Therapeutics. S.J.D. is a member of the scientific advisory board for Ferro Therapeutics and Hillstream BioPharma, Inc. J.S. is a member of the board of directors for Zenith Therapeutics and a scientific advisor to Lyterian Biosciences and Organos. S.J.D., J.A.O., J.M.H., E.W., J.S., C.E.D., and K.B. have ferroptosis-related patent applications.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang X, Stockwell BR, and Conrad M. (2021). Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22, 266–282. 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doll S., Freitas FP., Shah R., Aldrovandi M., da Silva MC., Ingold I., Goya Grocin A., Xavier da Silva TN., Panzilius E., Scheel CH., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 6.Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, et al. (2021). DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590. 10.1038/s41586-021-03539-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, Merl-Pham J, Bao X, Anastasov N, Kössl J, et al. (2020). GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci 6, 41–53. 10.1021/acscentsci.9b01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, Molina H, Garcia-Bermudez J, Pratt DA, and Birsoy K. (2020). Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol 16, 1351–1360. 10.1038/s41589-020-0613-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barayeu U, Schilling D, Eid M, Xavier da Silva TN, Schlicker L, Mitreska N, Zapp C, Gräter F, Miller AK, Kappl R, et al. (2022). Hydropersulfides inhibit lipid peroxidation and ferroptosis by scavenging radicals. Nat Chem Biol. 10.1038/s41589-022-01145-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Z, Khodade VS, Chauvin J-PR, Rodriguez D, Toscano JP, and Pratt DA (2022). Hydropersulfides Inhibit Lipid Peroxidation and Protect Cells from Ferroptosis. J. Am. Chem. Soc 144, 15825–15837. 10.1021/jacs.2c06804. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, Sirohi K, Li X, Wei Y, Lee H, et al. (2018). BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol 20, 1181–1192. 10.1038/s41556-018-0178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, Baer R, and Gu W. (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anandhan A, Dodson M, Schmidlin CJ, Liu P, and Zhang DD (2020). Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell Chem Biol. 10.1016/j.chembiol.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lei G, Zhuang L, and Gan B. (2022). Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer 22, 381–396. 10.1038/s41568-022-00459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Labrie M, Brugge JS, Mills GB, and Zervantonakis IK (2022). Therapy resistance: opportunities created by adaptive responses to targeted therapies in cancer. Nat Rev Cancer 22, 323–339. 10.1038/s41568-022-00454-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez R, Schreiber SL, and Conrad M. (2022). Persister cancer cells: Iron addiction and vulnerability to ferroptosis. Molecular Cell 82, 728–740. 10.1016/j.molcel.2021.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee H-J, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM, et al. (2020). Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89. 10.1126/science.aaw9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, Sajjakulnukit P, Myers A, Thurston G, Hou SW, et al. (2021). GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun 12, 4860. 10.1038/s41467-021-24859-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou Y., Palte MJ., Deik AA., Li H., Eaton JK., Wang W., Tseng Y-Y., Deasy R., Kost-Alimova M., Dančík V., et al. (2019). A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun 10, 1617. 10.1038/s41467-019-09277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, Hobeika CS, Khabibullin D, Yu D, Filippakis H, Alchoueiry M, Tang Y, Lam HC, Tsvetkov P, Georgiou G, et al. (2022). Hypersensitivity to ferroptosis in chromophobe RCC is mediated by a glutathione metabolic dependency and cystine import via solute carrier family 7 member 11. Proc. Natl. Acad. Sci. U.S.A 119, e2122840119. 10.1073/pnas.2122840119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Timmerman LA, Holton T, Yuneva M, Louie RJ, Padró M, Daemen A, Hu M, Chan DA, Ethier SP, van ‘t Veer LJ, et al. (2013). Glutamine Sensitivity Analysis Identifies the xCT Antiporter as a Common Triple-Negative Breast Tumor Therapeutic Target. Cancer Cell 24, 450–465. 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alborzinia H, Flórez AF, Kreth S, Brückner LM, Yildiz U, Gartlgruber M, Odoni DI, Poschet G, Garbowicz K, Shao C, et al. (2022). MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat Cancer 3, 471–485. 10.1038/s43018-022-00355-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alborzinia H, Chen Z, Yildiz U, Freitas FP, Vogel FCE, Varga J, Batani J, Bartenhagen C, Schmitz W, Büchel G, et al. (2022). Selenocysteine metabolism is a targetable vulnerability in MYCN -amplified cancers (Cancer Biology) 10.1101/2022.05.17.492172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Floros KV, Cai J, Jacob S, Kurupi R, Fairchild CK, Shende M, Coon CM, Powell KM, Belvin BR, Hu B, et al. (2021). MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res 81, 1896–1908. 10.1158/0008-5472.CAN-20-1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Y, Yang Q, Su Y, Ji Y, Li G, Yang X, Xu L, Lu Z, Dong J, Wu Y, et al. (2021). MYCN mediates TFRC-dependent ferroptosis and reveals vulnerabilities in neuroblastoma. Cell Death Dis 12, 511. 10.1038/s41419-021-03790-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hangauer MJ., Viswanathan VS., Ryan MJ., Bole D., Eaton JK., Matov A., Galeas J., Dhruv HD., Berens ME., Schreiber SL., et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. 10.1038/nature24297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stockwell BR, and Jiang X. (2020). The chemistry and biology of ferroptosis. Cell Chem Biol 27, 365–375. 10.1016/j.chembiol.2020.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koppula P, Lei G, Zhang Y, Yan Y, Mao C, Kondiparthi L, Shi J, Liu X, Horbath A, Das M, et al. (2022). A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun 13, 2206. 10.1038/s41467-022-29905-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rojo de la Vega M, Chapman E, and Zhang DD (2018). NRF2 and the hallmarks of cancer. Cancer Cell 34, 21–43. 10.1016/j.ccell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sporn MB, and Liby KT (2012). NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer 12, 564–571. 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forcina GC, Conlon M, Wells A, Cao JY, and Dixon SJ (2017). Systematic quantification of population cell death kinetics in mammalian cells. Cell Syst 4, 600–610.e6. 10.1016/j.cels.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inde Z, Rodencal J, and Dixon SJ (2021). Quantification of drug-induced fractional killing using high-throughput microscopy. STAR Protocols 2, 100300. 10.1016/j.xpro.2021.100300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, and Tang D. (2016). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, Chen Z-N, and Jiang X. (2019). Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406. 10.1038/s41586-019-1426-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang W-H, Ding C-KC, Sun T, Rupprecht G, Lin C-C, Hsu D, and Chi J-T (2019). The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep 28, 2501–2508.e4. 10.1016/j.celrep.2019.07.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Unger C., Kramer N., Walzl A., Scherzer M., Hengstschläger M., and Dolznig H. (2014). Modeling human carcinomas: physiologically relevant 3D models to improve anti-cancer drug development. Adv Drug Deliv Rev 79–80, 50–67. 10.1016/j.addr.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 38.Krishna S, Bustamante L, Haynes RK, and Staines HM (2008). Artemisinins: their growing importance in medicine. Trends in Pharmacological Sciences 29, 520–527. 10.1016/j.tips.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Contreras F-X, Basañez G, Alonso A, Herrmann A, and Goñi FM (2005). Asymmetric addition of ceramides but not dihydroceramides promotes transbilayer (flip-flop) lipid motion in membranes. Biophys J 88, 348–359. 10.1529/biophysj.104.050690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin R, Zhang Z, Chen L, Zhou Y, Zou P, Feng C, Wang L, and Liang G. (2016). Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Letters 381, 165–175. 10.1016/j.canlet.2016.07.033. [DOI] [PubMed] [Google Scholar]
- 41.Nie J, Lin B, Zhou M, Wu L, and Zheng T. (2018). Role of ferroptosis in hepatocellular carcinoma. J Cancer Res Clin Oncol 144, 2329–2337. 10.1007/s00432-018-2740-3. [DOI] [PubMed] [Google Scholar]
- 42.Yi R, Wang H, Deng C, Wang X, Yao L, Niu W, Fei M, and Zhaba W. (2020). Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Bioscience Reports 40, BSR20193314. 10.1042/BSR20193314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E, et al. (2018). FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol 14, 507–515. 10.1038/s41589-018-0031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ianevski A, Giri AK, and Aittokallio T. (2020). SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Research 48, W488–W493. 10.1093/nar/gkaa216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, Ajani JA, Xiao Q, Liao Z, Wang H, et al. (2020). The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res 30, 146–162. 10.1038/s41422-019-0263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, Dovas A, Higgins DM, Tan H, Zhang Y, et al. (2020). Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem Biol 15, 469–484. 10.1021/acschembio.9b00939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu T, Liu W, Zhang M, Yu W, Gao F, Li C, Wang S-B, Feng J, and Zhang X-Z (2018). Ferrous-Supply-Regeneration Nanoengineering for Cancer-Cell-Specific Ferroptosis in Combination with Imaging-Guided Photodynamic Therapy. ACS Nano 12, 12181–12192. 10.1021/acsnano.8b05860. [DOI] [PubMed] [Google Scholar]
- 48.Zhu T, Shi L, Yu C, Dong Y, Qiu F, Shen L, Qian Q, Zhou G, and Zhu X. (2019). Ferroptosis Promotes Photodynamic Therapy: Supramolecular Photosensitizer-Inducer Nanodrug for Enhanced Cancer Treatment. Theranostics 9, 3293–3307. 10.7150/thno.32867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. (2019). CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274. 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y-C., Oses-Prieto JA., Pope LE., Burlingame AL., Dixon SJ., and Renslo AR. (2020). Reactivity-Based Probe of the Iron(II)-Dependent Interactome Identifies New Cellular Modulators of Ferroptosis. J. Am. Chem. Soc 142, 19085–19093. 10.1021/jacs.0c06709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen HP, Yi D, Lin F, Viscarra JA, Tabuchi C, Ngo K, Shin G, Lee AY-F, Wang Y, and Sul HS (2020). Aifm2, a NADH Oxidase, Supports Robust Glycolysis and Is Required for Cold- and Diet-Induced Thermogenesis. Mol Cell 77, 600–617.e4. 10.1016/j.molcel.2019.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nguyen HP, Villivalam SD, Jung BC, You D, Lin F, Yi D, Pi A, Ma K, Jung S, Park S-H, et al. (2022). AIFM2 is Required for High-Intensity Aerobic Exercise by Promoting Glucose Utilization. Diabetes, db211114. 10.2337/db21-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Magtanong L, Ko P-J, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, et al. (2019). Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol 26, 420–432.e9. 10.1016/j.chembiol.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.