

Abstract
The epidemics caused by the influenza virus are a serious threat to public health and the economy. Adding appropriate adjuvants to improve immunogenicity and finding effective mucosal vaccines to combat respiratory infection at the portal of virus entry are important strategies to boost protection. In this study, we exploited a novel type of core/shell protein nanoparticle consisting of influenza NP as the core and NA1-M2e or NA2-M2e fusion proteins as the coating antigens by SDAD heterobifunctional crosslinking. ISCOMs/MPLA adjuvants further boosted the NP/NA-M2e SDAD protein nanoparticle-induced antigen-specific antibodies and cellular immune responses, which provided complete protection against influenza viral challenges when administered intramuscularly. The ISCOMs/MPLA-adjuvanted protein nanoparticles were delivered through the intranasal route to validate the application as mucosal vaccines. ISCOMs/MPLA-adjuvanted nanoparticles induced significantly strengthened antigen-specific antibody responses, cytokine-secreting splenocytes in the systemic compartment, and higher levels of antigen-specific IgA and IgG in the local mucosa. Meanwhile, significantly expanded lung resident memory (RM) T and B cells (TRM/BRM) and alveolar macrophages population were observed in ISCOMs/MPLA-adjuvanted nanoparticle-immunized mice with a 100% survival rate after homogeneous and heterogeneous H3N2 viral challenges. Taken together, ISCOMs/MPLA-adjuvanted protein nanoparticles could improve strong systemic and mucosal immune responses conferring protection in different immunization routes.
Keywords: influenza virus, protein nanoparticles, adjuvant, mucosal vaccine, intranasal delivery
1. Introduction
Influenza A virus has been recognized as one of the most threatening respiratory pathogens that could cause acute morbidity and mortality and heavy economic burdens, especially in flu epidemics or occasional flu pandemics.[1] Although vaccination has been proven to be an effective method to prevent or reduce influenza viral infection during annual flu seasons, the selection of vaccine strains depends mainly on circulating viral surveillance and prediction, and mismatched strains could significantly impair vaccine efficiency. Meanwhile, the production of the current quadrivalent influenza vaccine is based on the time-consuming chicken egg or cell culture systems and is not suitable for urgent uses when a pandemic strain is identified. Thus, new vaccine technologies such as mRNA or protein nanoparticle vaccines that are easily manufactured and quality-controlled are promising alternatives for developing a universal influenza vaccine.[2, 3]
We have focused our studies on different types of protein nanoparticle vaccines against both influenza A and influenza B viral infections. We demonstrated that the double-layered M2e and full-length hemagglutinin (HA) or HA stalk protein nanoparticles and M2e-NA protein nanoparticles induced immune protection against both homologous and heterologous influenza A viral infections.[4-6] The nanoparticles fabricated using influenza B virus-derived nucleoprotein (NP) and HA stalk displayed cross-protection against influenza B viruses from two lineages.[7] The fabrication of our previous protein nanoparticles is based on homobifunctional DTSSP crosslinking, more controlled and efficient fabrication methods are worth investigating to improve the nanoparticle formulations. In addition, combining appropriate adjuvants with protein nanoparticle vaccines will be a convenient way to further increase immune responses and protective effectiveness. For example, we found the combination of toll-like receptor 4 (TLR4) ligand- Monophosphoryl lipid A (MPLA) in double-layered NP and neuraminidase (NA) nanoparticles enhanced Th1 immune responses, as well as increased cross-protections against different influenza viral infections.[8]
Besides aluminum salts, only a few adjuvants have been approved for human usage by the FDA in the past seventy years.[3] Combined adjuvant systems have been evaluated and approved for vaccine usage in humans to exploit the advantages of different adjuvants and enhance a more comprehensive immune response. For example, Adjuvant System 4 (AS04), a combination of MPLA and aluminum salt, was approved in HPV vaccine formulation (Cervarix). A liposomal formulation (Adjuvant system AS01) including MPLA and a synthetic saponin QS21 has been approved for malaria and recombinant zoster vaccine vaccines (RTS and RZV). In the AS04 adjuvant formulation, aluminum stimulates relatively low cellular immunity and induces primarily humoral immune responses.[9] ISCOMs (Immune-stimulating complexes) matrix, described as early as in the 1980s, are cage-like self-assembled nanoparticles containing Quil A, cholesterol, and DOPC, which have been demonstrated to promote both humoral and cellular immune responses, including cytotoxic T cells (CTLs).[10, 11] The ISCOM concept-based Matrix-M adjuvant, composed of saponin extracts, cholesterol, and phospholipids, was applied in the Novavax COVID-19 Vaccine to elicit robust CD4 T cell, a Th1 biased responses in both pre-clinical and clinical studies.[12-14] In addition, other small molecules like TLR agonists and cGAMP, which function as pathogen-associated molecular patterns (PAMPs) to stimulate innate immune responses, have also been recognized and studied as effective vaccine adjuvants.[9] Therefore, exploring appropriate combinations of adjuvants is vital to enhance immune responses and guide immune directions.
In this study, a heterobifunctional crosslinker, NHS-SS-Diazirine, succinimidyl 2-((4,4'-azipentanamido) ethyl)-1,3'-dithiopropionate, designated as SDAD, was utilized to conjugate the influenza M2e-NA fusion protein on the surface of NP core, and a liposome-based adjuvant containing MPLA and ISCOMs was included as an adjuvant mixture for the novel SDAD protein nanoparticles. The intramuscular immunization of the adjuvanted protein nanoparticles induced significantly improved antigen-specific antibody and cellular immune responses and provided efficient protection against homologous and heterologous influenza viral challenges. The MPLA/ISCOM adjuvant combination also significantly improved the immunogenicity and protection efficiency of protein nanoparticles delivered via the intranasal route. The results emphasize the importance of supplementing appropriate adjuvants to improve the immunogenicity and mucosal immune responses of vaccines in mucosal immunizations.
2. Results
2.1. Generation and characterization of SDAD-crosslinked protein nanoparticles.
We synthesized a novel kind of protein nanoparticles using an SDAD crosslinker containing an amine-reactive N-hydroxysuccinimide (NHS) ester and a photoactivatable diazirine ring at the termini. The diagram for the fabrication process is displayed in Figure 1A. Briefly, through the reactive NHS arm, we conjugated the SDAD onto the surface of the influenza NP protein nanoparticle (NP nanos) that was generated by ethanol desolvation. Following purification of the SDAD-conjugated nanoparticle (NP nanos-SDAD), we further conjugated M2e-NA1 or M2e-NA2 fusion proteins onto the surface of the NP nanos-SDAD by ultraviolet exposure under 350nm. The sizes of the resulting NP/M2e-NA1 (core/shell) and NP/M2e-NA2 nanoparticles were 200 nm to 250 nm by dynamic light scattering (DLS) analysis (Figure 1B). The content of the NP core to the coating proteins in the nanoparticles was analyzed by the Coomassie blue staining and Western blot with ratios ranging from 1.5 to 2.1 (Figure 1C). The nanoparticle size and average ratio of activated NA in total SDAD-conjugated nanoparticles were comparable to the previous DTSSP-conjugated protein nanoparticles (Figure S1A, S1B, and S1C). The resultant nanoparticles exhibit a spherical shape by transmission electron microscopy (TEM) (Figure 1D). Unlike self-assembled nanoparticles, the uncontrolled orientation of the shell protein deposited on the nanoparticle cores and the unorganized structures of particle cores themselves constituted the nanoparticles with indistinguishable core/shell structures under TEM.[15, 16] The hetero-bifunctional nature of SDAD prevents crosslinking between particles and between M2e-NA proteins in the coating solution. Unconjugated M2e-NA proteins in the supernatant remain intact and can be used for a subsequent batch of nanoparticle fabrication after pelleting the nanoparticles. Therefore, the SDAD conjugation significantly increases the utilization of the initial proteins, which could be one restricting factor for future industrial applications.
Figure 1. Fabrication and characterization of SDAD-crosslinked protein nanoparticles.

(A) Schematic of the NHS-diazirine crosslinking reactions. (B) Size distributions of SDAD protein nanoparticles. (C) Nanoparticle characterization by Coomassie blue staining and Western blot. (D) SDAD nanoparticle TEM images. Bone marrow-derived dendritic cells (BMDCs) were stimulated with protein nanoparticles with and without various adjuvant combinations. (E) The expression of CD40, CD80, and CD86 on CD11c+ BMDCs by flow cytometry. TNF-α (F), IL-6 (G), IL-12 (H), IL-1β (I) and IFN-α (J) secretions from BMDCs cultured supernatant after stimulations with protein nanoparticles in combination with different adjuvants. The histograms were presented as mean ± SEM. Statistical significance was analyzed by F-test and two-tailed t-test. *p < 0.05; **p < 0.01; ***p < 0.001. (E)–(J) display the results of one experiment that is representative of two independent experiments.
Dendritic cells (DCs) are important innate immune cells to participate in antigen processing, presentation, and immune regulations. DC maturation and cytokine secretion are indicators of the potential initiation of antigen-specific immune responses.[17, 18] Our previous studies found that protein nanoparticles predominantly induced Th2-biased immune responses versus Th1 cell type.[8] Supplementing appropriate adjuvants with protein nanoparticles could improve the antigen immunogenicity and orchestrate the immune responses. Therefore, we supplemented SDAD protein nanoparticles with different adjuvant combinations and determined the maturation and cytokine secretions of bone marrow-derived dendritic cells (BMDCs) after stimulation by the SDAD nanoparticles with or without ISCOMs, ISCOMs/cGAMP, or ISCOMs/MPLA adjuvants. As shown in Figure 1E, ISCOMs adjuvanted SDAD nanoparticles significantly induced the DC maturation-marker expression including CD40, CD80 and CD86 with enhanced median fluorescence intensities (MFI) compared with the untreated control, and supplementing cGAMP or MPLA with ISCOMs further increased the expression of CD40, CD80, and CD86 (Figure 1E). Tumor necrosis factor-α (TNF-α) is a well-recognized factor for the maintenance of DC maturation and survival.[19, 20] The cytokines secreted from DCs are important regulators for T helper cell fates. DC-secreted interleukin-12 (IL-12) is a hallmark of inducing the Th1 responses, while IL-6 modulates Th2-oriented immune differentiation.[21, 22] The nanoparticles alone stimulated increased TNF-α expression in supernatant compared with the controls (Figure 1F). Consistent with the surface marker expression, nanoparticles adjuvanted with ISCOMs/cGAMP and ISCOMs/MPLA provoked significantly increased IL-6 and TNF-α generation compared with other groups, while adding ISCOMs alone only slightly increased TNF-α levels (Figure 1G and 1F). ISCOMs and MPLA were reported as the inducers of IL-1β [23-25] and the expression of IL-1β promoted the secretion of IL-12.[26] We observed that the combination of ISCOMs/MPLA uniquely stimulated higher expressions of IL-1β and IL-12 compared with slightly increased IL-1β expression by ISCOMs alone or ISCOMs/cGAMP incubations (Figure 1I and 1H), In contrast, ISCOMs/cGAMP preferentially induced IFN-α expression (Figure 1J). The specific induction of IFN-α was related to cGAMP-activated STING-mediated type I IFN production.[27, 28] The activation of BMDCs with SDAD nanoparticle formulations in vitro suggested the potential induction of effective immune responses in vivo.
2.2. Immune responses induced by SDAD protein nanoparticle immunizations.
We evaluated the immunogenicity of adjuvanted SDAD nanoparticles in mice. Different SDAD nanoparticle vaccine formulations included NP/M2e-NA2 nanoparticles (NA2 nano), NP/M2e-NA1 & NP/M2e-NA2 nanoparticles (NA1+NA2 nano), and ISCOMs/MPLA-adjuvanted NA1+NA2 nano (NA1+NA2 nano with Adj). We collected mice sera three weeks after the prime and boost vaccination to analyze antigen-specific antibody responses by ELISA assays. SDAD-crosslinked NP/M2e-NA elicited significantly increased M2e, NA1, and NA2-specific IgG antibody levels three weeks post the prime and boost immunization (Figure S2 and 2A). The boost immunization greatly improved the antigen-specific IgG1 and IgG2a titers (Figure 2B-2D). The combination of MPLA and ISCOMs displayed attractive adjuvant effects with considerably increased M2e, NA1, and NA2-specific IgG (Figure 2A), IgG1, and IgG2a antibody (Figure 2B-2D) titers after boost immunizations. Notably, compared to the nanoparticle alone group, the addition of MPLA/ISCOMs adjuvants significantly enhanced IgG2a levels with a more balanced IgG2a and IgG1, which suggested the regulation of immunological profiles by this adjuvant formulation.
Figure 2. Immune responses induced by intramuscular immunization of different protein nanoparticle vaccines.

Sera were collected and analyzed three weeks post-boost immunization. (A) M2e, NA1, and NA2 specific IgG levels after boost. (B), (C) and (D) NA2, NA1, and M2e specific IgG isotypes after boost. Groups of mice were euthanized at week four post the boosting immunization. The spleens and bone marrow were collected and processed into single-cell suspensions for analysis. (E) and (F) NA-specific IL-4 and IFN-γ-secreting cells in spleens. (G), (H) and (I) Aichi-, PR8- and M2e- specific antibody-secreting cells in the bone marrow. The histograms were presented as mean ± SEM. Statistical significance was analyzed by F-test and two-tailed t-test. *p < 0.05; **p < 0.01; ***p < 0.001. The results show one experiment with two replicates for each sample. Mice groups were n = 5.
We euthanized the mice four weeks post-boost vaccination and evaluated the cellular immune responses in spleens and bone marrows by ELISpot. As shown in Figures 2E and 2F, there were significantly increased and comparable numbers of IL-4 and IFN-γ-secreting splenocytes after NA2 stimulation in all nanoparticle-immunized mouse groups in comparison with the naïve mice. Supplementing MPLA/ISCOM adjuvants significantly enhanced the virus- (Aichi and PR8) and M2e peptide-specific IgG-secreting cells in bone marrow (Figure 2G-2I), consistent with the strengthened antibody responses observed in adjuvanted nanoparticles-immunized groups. These results suggested that the MPLA/ISCOMs remarkably improved the immunogenicity of the protein nanoparticles in intramuscular immunization.
2.3. Protection against influenza viral infections.
To determine the protective efficacy induced by SDAD protein nanoparticle immunization, different mouse groups were challenged with a dose of 5× LD50 A/Aichi (H3N2), 5× LD50 of rVet (H5N1), or 3× LD50 of A/Philippine (H3N2) four weeks post-boosting immunization and the body weights were monitored for fourteen days. Although mice receiving the NA2 nano immunization displayed a 100% survival rate (Figures 3B and 3D) during the A/Aichi and A/Philippine influenza viral infections, they lost substantial body weight with symptoms from days 3 to 7 (Figure 3A and 3C). On the other hand, the adjuvanted nanoparticle group had fewer symptoms and recovered quickly during all the viral infections (Figure 3A, 3C, and 3E). After NA homologous A/Aichi and rVet challenges, the mixed NA2 and NA1 nano-immunized mice showed a 90% survival rate which was less than the NA2 nano group (Figure 3B and 3F). These results demonstrated that the MPLA and ISCOMs adjuvant combination could enhance the protective efficacy of protein nanoparticle immunization to fight against homologous and heterologous influenza viral infection.
Figure 3. Protective efficacy against homologous and heterologous influenza viral challenge.
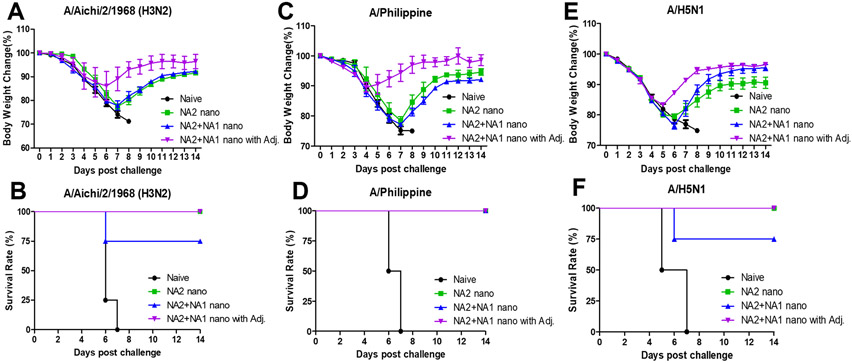
Groups of mice were challenged with A/Aichi, A/Philippine, and A/H5N1 influenza viruses at week four post-boosting immunization. Mouse body weight was monitored for 14 days. The body weight changes after (A) A/Aichi (H3N2), (C) A/Philippine (H3N2), and (E) rViet (A/H5N1) virus infections. (B), (D), and (F) The survival rates after different viral infections. The data were presented as mean ± SEM. Mice groups were n = 5.
2.4. Adjuvants improved the immunogenicity of protein nanoparticles during intranasal vaccination and induced protection against viral infection.
Besides systemic immunity, the induction of strong mucosal immune responses in local tissues is also crucial for impeding respiratory viral infection. To explore the possibility of protein nanoparticles as mucosal vaccines, we intranasally primed and boosted immunized mice with M2e-NA2 protein nanoparticles with or without the ISCOM/MPLA adjuvant. One group of mice was immunized with ISCOM/cGAMP-adjuvanted protein nanoparticles (Nanos+ISCOM/cGAMP) as a control. Compared to the naïve and protein nanoparticle alone (Nanos) groups, mice immunized with adjuvanted protein nanoparticles showed significantly increased NA2 and M2e-specific IgG antibodies three and seven weeks post-vaccination, respectively (Figure 4A and 4C). ISCOM/cGAMP-adjuvanted protein nanoparticle immunization induced higher antigen-specific antibody levels than ISCOM/MPLA-supplemented nanoparticles (Nanos+ISCOM/MPLA) (Figure 4A and 4C). For NA2- and M2e-specific IgG isotypes, ISCOM/MPLA- or ISCOM/cGAMP-adjuvanted nanoparticle immunization elicited more robust IgG1 antibody levels than IgG2a (Figure 4B and 4D). ISCOM/cGAMP was a more powerful supplement to stimulate increased IgG1 and IgG2a than ISCOM/MPLA adjuvant (Figure 4B and 4D). These results demonstrated that supplementing ISCOM/MPLA or ISCOM/cGAMP adjuvant combinations to protein nanoparticles could significantly improve the immunogenicity of protein nanoparticles during intranasal immunization.
Figure 4. Adjuvants improve the humoral immune responses and protection efficiency against influenza viral infection after intranasal immunization.

Groups of mice were intranasally immunized at a three-week interval with different protein nanoparticle formulations with and without ISCOMs/MPLA or ISCOMs/cGAMP adjuvant combinations. Mouse sera were collected and analyzed three weeks post-primary and boosting immunization. (A) NA2 specific IgG levels after prime and boost. (B) NA2-specific IgG1 and IgG2a levels. (C) M2e-specific IgG levels after prime and boost. (D) M2e-specific IgG1 and IgG2a levels. Groups of immunized mice were challenged with 5x LD50 of A/Aichi/2/1968 H3N2 influenza virus. (E) Body weight changes after viral infection. (F) The mice survival rate. The data were presented as mean ± SEM. The Mice groups were n = 5.
To determine the protective efficiency against viral infection, mice were challenged with 5× LD50 A/Aichi four weeks post the intranasal boosting immunizations and monitored for fourteen days. During the initial five days post-challenge, the ISCOM/cGAMP-adjuvanted protein nanoparticle immunized group showed more body weight loss than other groups. The body weight recovered from day 6 with a 75% of survival rate (Figure 4E and 4F). In comparison, the protein nanoparticles adjuvanted with ISCOM/MPLA provided complete protection against homologous A/Aichi infection with an average of 12% of body weight loss in the fourteen days (Figure 4E and 4F). The body weight curve of protein nanoparticle-vaccinated mice displayed a similar pattern to the naïve group except one mouse survived in this group during the challenge study with a 20% of survival rate (Figure 4E and 4F). These data indicated that supplementing adjuvants in protein nanoparticles during intranasal immunization significantly improved the immunogenicity of antigens and ISCOMs/MPLA adjuvanted protein nanoparticles provided better protection against homologous influenza viral infection.
2.5. Intranasal immunization of ISCOMs/MPLA-adjuvanted nanoparticles increased systematic cellular immune responses.
To further elucidate the immune responses induced by ISCOMs/MPLA adjuvanted nanoparticles, the cytokine-secreting cells in the immune system were analyzed. IL-2 has been reported as an important cytokine to mediate the differentiation of naïve T cells into T helper 1 (Th1) and T helper 2 (Th2) effector cells, while IFN-γ and IL-4 are essential cytokines secreted from Th1 and Th2 cells to play major roles in antiviral immunity and humoral immunity, respectively.[29, 30] In this study, groups of mice were intranasally immunized with protein nanoparticles with or without ISCOMs/MPLA to determine the above cytokine secretion in splenocytes one month post-boosting immunization. Mice immunized with ISCOMs/MPLA adjuvanted nanoparticles (Nano with Adj.) have significantly increased numbers of NA2- (Figure 5A), NP peptide- (Figure 5C), and Aichi virus- (Figure 5D) specific IFN-γ, IL-2 and IL-4 secreting splenocytes compared with nanoparticles alone (Nanos) or naïve groups. The observation of increased IL-4-secreting cells in the spleen after adjuvanted nanoparticle immunization was consistent with the remarkable enhancement of antigen-specific antibody responses in Figure 4. Meanwhile, significantly increased NA2-specific antibody-secreting cells (ASCs) were observed in the spleen and bone marrow cells (Figure 5B). These results suggested that intranasal immunization of ISCOMs/MPLA-adjuvanted protein nanoparticles could elicit strong Th1 and Th2 immune responses.
Figure 5. Cellular immune responses after intranasal immunization.

Mice were intranasally immunized twice at a three-week interval with protein nanoparticles with or without ISCOMs/MPLA. Spleens and bone marrow were collected one month post the boosting immunization and analyzed by ELISpot assays. (A) IFN-γ, IL-2, and IL-4 secreting splenocytes after purified NA2 protein stimulation. (B) NA2-specific antibody-secreting cells in the bone marrow. (C) IFN-γ, IL-2, and IL-4 secreting splenocytes after NP peptides stimulation. (D) IFN-γ, IL-2, and IL-4 secreting splenocytes after stimulation of A/Aichi virus. The histograms were presented as mean ± SEM. Statistical significance was analyzed by F-test and two-tailed t-test. *p < 0.05; **p < 0.01; ***p < 0.001. The results show one experiment with three independent replicates for each sample. Mice groups were n = 3.
2.6. Mucosal immune responses and protection against heterologous viral infection.
Besides the induction of potent cellular immune responses in the systemic compartment, an efficient mucosal vaccine should also stimulate the production of secretory IgA (sIgA), which is critical in protecting against viruses on mucosal surfaces.[31, 32] Mucosal IgA has been demonstrated to protect against respiratory viruses, including SARS-CoV, MERS-CoV, and influenza.[33-36] Here, we intranasally immunized mice as previously described and determined the antigen-specific IgG and IgA levels in mucosal fluids, including bronchoalveolar lavage fluid (BALF) and nasal washes one month post the boosting immunization. Significantly increased levels of NA2-specific IgA were detected in the nasal and BALF washes from ISCOMs/MPLA adjuvanted nanoparticles intranasally immunized mice. However, only background signals could be observed from mucosal samples derived from Nanos and naïve groups (Figure 6A and 6B). Similarly, there were also greatly enhanced NA2-specific IgG in the nasal and BALF washes from the adjuvanted nanoparticle-immunized group compared with the Nanos and naïve groups (Figure 6C and 6D). Meanwhile, we observed improved Aichi virus-specific IgA and IgG antibody responses in the mucosal samples from adjuvanted nanoparticles immunized mice (Figure 6E and 6F), although the signals of virus-specific IgA were lower than the NA2 -specific IgA in Figure 6A. Furthermore, the immunized mice were also challenged with 3x LD50 of A/Philippine one month post the boosting vaccination to evaluate the protective efficiency against heterologous virus infection. As shown in Figure 6G, ISCOMs/MPLA-adjuvanted nanoparticles intranasally immunized mice maintained their initial body weights fourteen days after virus inoculation, while the naïve mice and the nanoparticles alone immunized mice indistinguishably lost the body weights below 75% of initial body weights within nine days post-infection. Therefore, the ISCOMs/MPLA adjuvant combination could significantly improve the mucosal antibody immune responses of the protein nanoparticles in intranasal delivery and provide better protection against heterogeneous influenza viral infection.
Figure 6. Mucosal immune responses and protection against heterologous influenza viral infection.

Mice were intranasally immunized with NP/M2e-NA2 protein nanoparticles with and without ISCOMs/MPLA. The nasal and BALF washes were collected one month post-boosting intranasally immunization to determine the IgA and IgG levels by ELISA. (A) and (C) NA2-specific IgA and IgG levels in nasal washes. (B) and (D) NA2-specific IgA and IgG levels in BALFs. (E) Aichi-specific IgA and IgG in BALFs. (G) The body weight changes after A/Philippine (H3N2) influenza viral challenge. The data were presented as mean ± SEM. Statistical significance was analyzed by F-test and two-tailed t-test. *p < 0.05; **p < 0.01; ***p < 0.001. The Mice groups were n = 3.
2.7. Recruitment of lymphocytes in localized pulmonary tissue after ISCOMs/MPLA adjuvanted nanoparticles intranasal immunization.
In addition to the antibody responses in the mucosa, the immune cells in mucosal tissues also play critical roles in protecting against respiratory viral infection. Increasing evidence in recent years has supported the positive functions of lung-resident memory T and B cells in response to bacterial and viral infections.[37, 38] In this study, we found that there were significant increases in percentages of CD8+CD44+CD69+CD103+ (Figure 7A), CD4+CD44+CD69+CD103+ (Figure 7B), and CD4+CD44+CD69+CD103− (Figure 7C) resident memory T cell populations (TRM) in lungs from the ISCOMs/MPLA-adjuvanted nanoparticles intranasally immunized mice (Figure 7D). Compared with the Nanos or naïve group, ISCOMs/MPLA-adjuvanted nanoparticles elicited increased percentages of CD19+B220−IgD−IgM−CD38+CD69+ (Figure 7E) lung-resident memory B cell populations (BRM) (Figure 7G). Although CD38 is often used to distinguish memory from naïve B cell subset in mice,[39] we also observed an induction of CD19+B220−IgD−IgM−CD38−CD69+ cell population in lungs of the adjuvanted nanoparticle immunized mice (Figure 7F and 7G).
Figure 7. Cell populations in localized pulmonary tissue.

The lung tissues were collected and processed one month post the boosting intranasally immunization to determine the inside cell populations. (A) The percentages of the CD69+CD103+ population in CD8+CD44+ T cells. (B) and (C) The percentages of CD69+CD103+ and CD69+CD103− populations in CD4+CD44+ T cells. (D) The representative gating methods for (A), (B), and (C). (E) and (F) The frequencies of CD38+CD69+ and CD38−CD69+ populations in CD19+B220−IgD−IgM− cells. (G) The representative gating method for (E) and (F). (H) The percentage of CD64+CD24− population in CD11c+CD11b− cells. (I) The gating method for results of (H). The cells in BALFs were collected for analysis. (J) and (K) The percentage of CD4+CD44+ and CD8+CD44+ cells in BALFs. The histograms were presented as mean ± SEM. Statistical significance was analyzed by F-test and two-tailed t-test. *p < 0.05; **p < 0.01; ***p < 0.001. The Mice groups were n = 3.
Alveolar macrophage is another kind of immune cell critical in protecting against influenza A viral infection.[40] Adjuvanted protein nanoparticle immunization stimulated higher percentages of CD11c+CD11b−CD64+CD24− alveolar macrophages in lungs (Figure 7H and 7I). The cells from BALF were separated and analyzed for the expression of CD44 on CD4 and CD8 T cells and increased CD4+CD44+ and CD8+CD44+ populations were detected (Figure 7J and 7K). As shown in Figure 7, intranasal immunization of the protein nanoparticle alone could not promote the establishment of lung-resident memory cells or the induction of alveolar macrophages in the lung. These data might suggest that ISCOMs/MPLA-adjuvant enhanced antigen uptake and presentation in the respiratory tract leading to the activation of CD4 and CD8 T cells, which would promote the establishment and maintenance of pulmonary TRM and BRM in local tissues.
3. Discussion
The nanoparticle size is critical in optimizing the delivery routes and immunogenicity of protein nanoparticle vaccines. However, no agreement exists regarding the optimum nanoparticle size range generating the best immune responses in different circumstances.[41, 42] In our previous studies, we found that our DTSSP-crosslinked protein nanoparticles around 200nm could be efficiently drained to and retained in inguinal LNs and spleens after intramuscular injection and induced strong humoral and cellular immune responses.[4, 5, 43] Similarly, robust immune responses were observed after intramuscular injection of the SDAD-crosslinked protein nanoparticles in this study. However, when applied through the intranasal route, the protein nanoparticles could not induce significant immune reactions, which might be due to the less efficiency of penetrating the mucus surface or uptaking by local antigen-presenting cells (APCs). Thus, it is necessary to include appropriate adjuvant to boost the uptake and process of antigens by APCs during mucosal immunization.
Supplementing ISCOM/MPLA and ISCOM/cGAMP with nanoparticles could stimulate strong immune responses in intranasal immunization. Various mucosal adjuvants have been reported to boost the antigen-induced immune response, such as bacterial toxins, TLR agonists, cytokines, and chemokines.[31] ISCOMs have been used in different studies as an effective mucosal adjuvant to improve immune response.[44, 45] MPLA has been used with other molecules in different adjuvant systems to boost immune responses. When we formulated these adjuvant combinations with our protein nanoparticle vaccines, we found that ISCOMs/MPLA combination provided better protection against influenza viral infection, although less improvement in antibody responses compared with the ISCOMs/cGAMP combination. ISCOMs and cGAMP have been studied as potent mucosal adjuvants when applied alone or in combinations with other adjuvants,[46-49] while MPLA takes its advantage as a ligand of TLR4 that is abundantly expressed on the surface of DCs and is essential for DCs activation.[50-52] cGAMP elicited signaling activities through cytosolic receptors and showed better adjuvanticity when delivered to inside cells.[53] This indicated the ISCOMs/MPLA combination could provide stimulations to DCs more directly and efficiently. Meanwhile, ISCOMs/MPLA-adjuvanted protein nanoparticles induced a more balanced and robust Th1 and Th2 immune response no matter the immunization route. These results indicated the superiority of ISCOMs/MPLA as a potent adjuvant with protein nanoparticles. In parallel with the development of novel universal vaccines, the incorporation of appropriate adjuvants into the mature vaccine formulations will shortly be the quickest method to strengthen the immunogenicity and improve the protective efficiency of original vaccines.
Although mucosal vaccines have been recognized and proved to be an ideal method to trigger immune responses at the primary infection sites, there is only one licensed influenza mucosal vaccine for human use.[54] The complex properties and structures of the nasal cavity and respiratory tract are naturally challenging obstacles to vaccine delivery.[55] Only the vaccine candidates that penetrate the mucus layer, translocate into antigen-presenting cells and escape antigen clearance will have the chance to stimulate downstream immune responses.[56] In our in vitro studies, we observed that ISCOMs/MPLA adjuvanted protein nanoparticles facilitated BMDCs maturation and stimulated multi-cytokines secretion from BMDCs over ISCOMs alone or ISCOMs/cGAMP. This finding indicated that ISCOMs/MPLA might further stimulate substantial DC maturation and cytokine secretion to help the protein nanoparticle uptake and antigen presentation by antigen-presenting cells, as we have seen that DCs could efficiently internalize the protein nanoparticles in previous studies.[7, 57] The influence of ISCOMs/MPLA on the retention, internalization, and translocation of antigens by immune cells on mucosal surfaces will be further determined.
Our study provided the first evidence that ISCOMs/MPLA adjuvanted protein nanoparticles could elicit strong mucosal immune responses and the accumulation of lung resident memory cells in the local respiratory tracts. With the understanding of features and functions of lung tissue-resident memory cells in recent years, TRM and BRM are realized as attractive targets for vaccine design.[56, 58-61] The unique effector functions of TRM cells in restricting the reinfections of various respiratory pathogens at the first exposure sites of mucosal surfaces have been extensively studied during the last decade.[62, 63] Influenza viral infection could elicit lung BRM and plasma cells,[64, 65] and the lung BRM responded rapidly to localized ASCs following viral challenge.[65] Although site-specific vaccination such as intranasal or pulmonary vaccination could increase the chances for antigen encounter and are more likely to elicit TRM and BRM in lungs,[65, 66] ours and most emerging studies demonstrated that intradermal, intramuscular, and intranasal immunizations of mRNA vaccines could induce pulmonary resident memory T cells.[27, 67, 68] Therefore, the heterologous immunizations of systemic prime and mucosal boost will be another new way for next-generation mucosal vaccines.[69-71]
In summary, we have developed a novel influenza SDAD protein nanoparticle vaccine and found that the ISCOMs/MPLA adjuvant combination could significantly enhance the immunogenicity and protective effectiveness induced by protein nanoparticles after intramuscular and intranasal immunizations. This work highlights the importance of applying adjuvants in mucosal vaccine formulations. The ISCOMs/MPLA-adjuvanted protein nanoparticles have the potential to be used as mucosal vaccines alone or in combination with other vaccines to improve mucosal immunity and protection in the future.
4. Materials and Methods
Ethics statement
This study was carried out strictly according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Georgia State University animal facility is fully accredited by the American Association for Assessment and Accreditation of Laboratory Animal Care. The animal studies in this work were approved by the Georgia State University Institutional Animal Care and Use Committee under protocol No. A20027.
Fabrication and characterization of SDAD cross-linking protein nanoparticles.
NP core nanoparticles were generated by ethanol desolvation. Briefly, 200 μg of NP protein solution was mixed with four times the volume of ethanol (100%, 1ml−1 min) while stirring for 20 min. The pellet was resuspended in PBS (300 μl) after centrifugation at 20,000xg for 20 min and sonication with 40%-amp, 3 sec on and 3 sec off. A ten-fold molar excess of SDAD ((NHS-SS-Diazirine) (succinimidyl 2-((4,4'-azipentanamido)ethyl)-1,3'-dithiopropionate)), purchased from Thermo Scientific, was added to the NP core nanoparticle solution and stirred for 30 min followed by adding Tris-HCl (20 μl, 1 M, PH 8.0) buffer to quench the reaction for 5 min. The SDAD-conjugated NP core nanoparticles (NP Nano-SDAD) were washed with DPBS and centrifuged at 20,000xg for 20 min to remove any excessive SDAD. M2e-NA1 or M2e-NA2 proteins (200 μg) were mixed with NP Nano-SDAD, incubated, and stirred for 1 hour under the 365 nm UV light (UVP UVL-4 UV Lamp, Analytik Jena US). The nanoparticles were pelleted by centrifugation at 20,000xg for 20min. The pellet was resuspended and sonicated in DPBS (200 μl) for further characterization. The concentration of protein nanoparticles was measured by Micro BCA Protein Assay Kit (Thermo Scientific), and the total yield was calculated as total protein input/ total output ×100%. The individual content of NA and NP proteins was characterized by 10% SDS-PAGE followed by coomassie blue staining and western blot. The nanoparticle sizes and morphology were determined by dynamic light scattering analysis (Malvern Zetasizer) and transmission electron microscopy imaging (JEOL 100 CX-II TEM).
Mouse immunization and challenge.
Groups of mice received primary and boosting immunizations of 10 μg of NP/M2e-NA2 nanoparticles (NA2 nano), mixed NP/M2e-NA2, NP/M2e-NA1 nanoparticles (NA2+NA1 nano), or mixed nanoparticles with ISCOMs/MPLA adjuvants (NA2+NA1 nano with Adj.) by intramuscular injection at three weeks intervals. To generate ISCOMs, a lipid film of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) (Avanti Polar Lipids) and cholesterol (Sigma) obtained by centrifugal drier (Vacufuge Plus, Eppendorf) were solubilized and sonicated in sterile water and then mixed and vortexed with QuilA (InvivoGen). The ISCOMs/MPLA combination comprised MPLA (2 μg, Avanti Polar Lipids) and ISCOMs (16 μg) with a 5:1:2 ratio of QuilA: cholesterol: DOPC. One group of naïve mice was included as controls.
For the intranasal immunization, mice were intranasally immunized with 30 μl of vaccine formulations including 10 μg of NP/M2e-NA2 nanoparticles (Nanos), NP/M2e-NA2 nanoparticles with ISCOMs/MPLA (Nanos+ISCOMs/MPLA or Nanos+Adj.), or NP/M2e-NA2 nanoparticles with ISCOMs/cGAMP containing ISCOMs (16 μg) and cGAMP (5 μg) (Nanos+ISCOM/cGAMP). cGAMP was purchased from InvivoGen. Four weeks post boosting immunization, mice were intranasally challenged with 5× LD50 of A/Aichi/1968 (A/Aichi, H3N2), 3× LD50 of A/Philippines/1986 (A/Phili, H3N2) or 3× LD50 of reassortant Viet (rViet, H5N1). The body weights were measured, and mice conditions were monitored for fourteen days after infection.
Antigen-specific antibodies.
The sera were collected three weeks post-primary and secondary immunizations, and the antigen-specific antibody levels in the mouse sera were measured by enzyme-linked immunosorbent assay (ELISA). Briefly, the ELISA plates (Nunc, Maxisorp) were coated with 4 μg−1 ml of NA1, NA2 purified proteins, M2e peptides (Synpeptide, SLLTEVETPT) or whole inactivated A/Aichi virus overnight and blocked in PBST with 2% BSA. After 1 hour of incubation, 50 μg of sera dilutions were added and incubated for 2 hours at 37°C. Plates were washed and incubated with goat anti-mouse IgG-, IgG1- or IgG2a-HRP secondary antibodies (SouthernBiotech) for 1 hour, and the plates were read by BioTek Microplate Reader at 450 nm after reacting with TMB and stopping by H2SO4 (1m). Mice nasal washes and BALFs were collected one month post-intranasally boosting immunization. The NA2- and Aichi- specific IgA levels were measured using HRP conjugated goat anti-mouse IgA antibody (SouthernBiotech).
Bone marrow dendritic cell maturation and cytokine secretion.
Bone marrow dendritic cells (BMDCs) were isolated and cultured at ten million cells per milliliter in the completed RPMI 1640 medium (cRPMI) with GM-CSF (20 ng−1 ml) as previously described [72]. Briefly, fresh culture medium with GM-CSF was supplemented at day 2 after initial culture and then continually cultured for another three days. The BMDCs were the non-adherent and loosely adherent cells in the culture, which were collected by centrifugation at 250x g for 8 min and then seeded at 1 million cells per well into 24-well plates for the following stimulations.
Fabricated SDAD protein nanoparticles and the adjuvants were diluted with cRPMI. The BMDCs were stimulated with 4 μg−1 ml of nanoparticles and nanoparticles with 4 μg−1 ml of different adjuvant combinations separately overnight. At 24 hours post simulations, the cell culture supernatant was harvested to analyze cytokine secretion. The cell pellet was resuspended in a staining buffer (PBS with 2% of FBS) and used for the cell staining. As previously described, to determine the expressions of IL-1β, IL-6, TNF-α, IFN-α, and IL-12 in BMDC-cultured supernatant, the ELISA plates were coated with 4 μ−1 ml of cytokine-specific captured antibodies at 4°C overnight. Fifty microliters of supernatant were added into each well and incubated at 37°C for 2 hours followed by the incubation of individually biotin-conjugated detection antibodies and HRP-conjugated streptavidin [27]. The standard curves for each cytokine were generated respectively. To characterize the BMDCs maturation, we incubated the collected cells with Zombie Aqua™ dye (Zombie Aqua™ Fixable Viability Kit, Biolegend) to distinguish dead/live cells and then stained with anti-mouse CD11c-APC (BD Biosciences), CD40-PE, CD80-FITC, and CD86-APC/Cy7 surface antibodies. After thorough washes, the cells were analyzed by BD LSRFortessa™ Cell Analyzer. The standards and other antibodies used in this assay were purchased from Biolegend.
Cellular immune responses.
Mice were euthanized four weeks after immunization. Single spleen and bone marrow cell suspensions were prepared in cRPMI media for enzyme-linked immunosorbent spot (ELISPOT) assays [73]. The 96-well filter plates (Millipore) were coated overnight with 4 μg/ml of anti-mouse IL-2, IL-4, or IFN-γ capture antibodies (Biolegend). Then one million splenocytes were seeded with stimulators containing 4 μg−1 ml of NA1 and NA2 purified proteins, whole inactivated A/Aichi viruses, or NP peptides (BEI Resources, NR-2611) after blocking. The cells were cultured at 37°C for 48 h and incubated with biotin-conjugated IL-2, IL-4, or IFN-γ detection antibodies and HRP-conjugated streptavidin (BioLegend). After KPL True Blue substrate (SeraCare) staining, the colonies were measured by Bioreader-6000-E (BIOSYSTEM). To determine ASCs in spleens and bone marrow, one million cells were seeded into filter plates which were coated with 4 μg−1 ml of A/Aichi, A/PR8 whole inactivated viruses, M2e peptides or NA2 purified proteins. The antibodies secreted from cells were detected with goat anti-mouse IgG-HRP antibodies. The spots were stained with KPL Trueblue and counted by Bioreader-6000-E.
Flow cytometry.
BALFs and Lung tissues were collected one month after intranasally boosting immunization. The cells in BALFs were isolated by centrifugation at 500x g for 10 min and stained with anti-mouse CD45-PE, CD4-Percp, CD8-FITC, CD44-BV421, CD16/32 (BD Biosciencs) antibodies and Zombie NIR™ dye (Zombie NIR™ Fixable Viability Kit, Biolegend) for T cell population analysis by flow cytometry. Lungs tissues were processed with 1 mg/ml of Collagenase type 4 (Worthington Biochemical) and 30 μg−1 ml of DNase I (Sigma-Aldrich) in RPMI 1640 media at 37°C for 30 mins followed by grinding through a 70-μm cell strainer and centrifuged at 1500 rpm for 5 min at 4°C. After discarding the supernatant, cells were washed twice with the staining buffer. To analyze the different cell populations, we divided cells into three parts and stained them with different combinations of antibodies as below: (1) anti-mouse CD45-PE, CD4-Percp, CD8-FITC, CD103-APC, CD44-BV421, CD69-PE/Cy7, CD16/32 antibodies, and Zombie NIR™ dye; (2) anti-mouse CD19-APC, B220-AF700, IgD-FITC, IgM-PE/Cy7, CD69-PE, CD38-Pacific Blue, CD16/32 antibodies and Zombie NIR™ dye; (3) anti-mouse CD45-PE, CD11c-Percp/Cy5.5, CD11b-PE/Cy7, CD64-APC, CD24-BV510 (BD Biosciences), CD103-FITC, CD16/32 antibodies, and Zombie NIR™ dye. The other antibodies used in this assay were purchased from Biolegend. The cells were recorded by BD LSRFortessa™ Cell Analyzer.
Statistical analysis
The data were displayed by means with the standard error of the mean (SEM). The p values were assigned by calculating F-test and a two-tailed Student’s t-test between two groups. p values less than 0.05 were statistically significant.
Supplementary Material
Acknowledgments
This study was supported by the US NIH/National Institute of Allergy and Infectious Diseases under Grants R01AI101047 and R01AI143844 to B.-Z.W. This work was performed in part at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant No. ECCS-2025462). The content in this study is solely our responsibility and does not necessarily represent the official views of the funders.
Footnotes
Declaration of interests
The authors declared no financial or commercial conflict of interest.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Reference
- 1.Harrington WN; Kackos CM; Webby RJ, Exp Mol Med 2021, 53 (5), 737–749. DOI 10.1038/s12276-021-00603-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei CJ; Crank MC; Shiver J; Graham BS; Mascola JR; Nabel GJ, Nat Rev Drug Discov 2020, 19 (4), 239–252. DOI 10.1038/s41573-019-0056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu W; Dong C; Wei L; Wang BZ, Pharmaceutics 2021, 13 (1). DOI 10.3390/pharmaceutics13010068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng L; Mohan T; Chang TZ; Gonzalez GX; Wang Y; Kwon YM; Kang SM; Compans RW; Champion JA; Wang BZ, Nat Commun 2018, 9 (1), 359. DOI 10.1038/s41467-017-02725-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y; Deng L; Gonzalez GX; Luthra L; Dong C; Ma Y; Zou J; Kang SM; Wang BZ, Adv Healthc Mater 2020, 9 (2), e1901176. DOI 10.1002/adhm.201901176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma Y; Wang Y; Dong C; Gonzalez GX; Song Y; Zhu W; Kim J; Wei L; Wang BZ, Nanomedicine 2022, 40, 102479. DOI 10.1016/j.nano.2021.102479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song Y; Zhu W; Wang Y; Deng L; Ma Y; Dong C; Gonzalez GX; Kim J; Wei L; Kang SM; Wang BZ, Biomaterials 2022, 287, 121664. DOI 10.1016/j.biomaterials.2022.121664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y; Dong C; Ma Y; Zhu W; Gill HS; Denning TL; Kang SM; Wang BZ, Nanomedicine 2023, 47, 102614. DOI 10.1016/j.nano.2022.102614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pulendran B; P SA; O'Hagan DT, Nat Rev Drug Discov 2021, 20 (6), 454–475. DOI 10.1038/s41573-021-00163-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sjolander A; Cox JC; Barr IG, J Leukoc Biol 1998, 64 (6), 713–23. DOI 10.1002/jlb.64.6.713. [DOI] [PubMed] [Google Scholar]
- 11.Lovgren Bengtsson K; Morein B; Osterhaus AD, Expert Rev Vaccines 2011, 10 (4), 401–3. DOI 10.1586/erv.11.25. [DOI] [PubMed] [Google Scholar]
- 12.Moderbacher CR; Kim C; Mateus J; Plested J; Zhu MZ; Cloney-Clark S; Weiskopf D; Sette A; Fries L; Glenn G; Crotty S, J Clin Invest 2022, 132 (19). DOI ARTN e160898 10.1172/JCI160898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keech C; Albert G; Cho I; Robertson A; Reed P; Neal S; Plested JS; Zhu MZ; Cloney-Clark S; Zhou HX; Smith G; Patel N; Frieman MB; Haupt RE; Logue J; McGrath M; Weston S; Piedra PA; Desai C; Callahan K; Lewis M; Price-Abbott P; Formica N; Shinde V; Fries L; Lickliter JD; Griffin P; Wilkinson B; Glenn GM, New Engl J Med 2020, 383 (24), 2320–2332. DOI 10.1056/NEJMoa2026920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z; Mateus J; Coelho CH; Dan JM; Moderbacher CR; Galvez RI; Cortes FH; Grifoni A; Tarke A; Chang J; Escarrega EA; Kim C; Goodwin B; Bloom NI; Frazier A; Weiskopf D; Sette A; Crotty S, Cell 2022, 185 (14), 2434–2451 e17. DOI 10.1016/j.cell.2022.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyoglu-Barnum S; Ellis D; Gillespie RA; Hutchinson GB; Park YJ; Moin SM; Acton OJ; Ravichandran R; Murphy M; Pettie D; Matheson N; Carter L; Creanga A; Watson MJ; Kephart S; Ataca S; Vaile JR; Ueda G; Crank MC; Stewart L; Lee KK; Guttman M; Baker D; Mascola JR; Veesler D; Graham BS; King NP; Kanekiyo M, Nature 2021, 592 (7855), 623–628. DOI 10.1038/s41586-021-03365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fries CN; Curvino EJ; Chen JL; Permar SR; Fouda GG; Collier JH, Nat Nanotechnol 2021, 16 (4), 1–14. DOI 10.1038/s41565-020-0739-9. [DOI] [PubMed] [Google Scholar]
- 17.Dalod M; Chelbi R; Malissen B; Lawrence T, Embo J 2014, 33 (10), 1104–1116. DOI 10.1002/embj.201488027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mellman I, Cancer Immunol Res 2013, 1 (3), 145–9. DOI 10.1158/2326-6066.CIR-13-0102. [DOI] [PubMed] [Google Scholar]
- 19.Summers deLuca L; Gommerman JL, Nat Rev Immunol 2012, 12 (5), 339–51. DOI 10.1038/nri3193. [DOI] [PubMed] [Google Scholar]
- 20.Maney NJ; Reynolds G; Krippner-Heidenreich A; Hilkensl CMU, Journal of Immunology 2014, 193 (10), 4914–4923. DOI 10.4049/jimmunol.1302929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dienz O; Rincon M, Clin Immunol 2009, 130 (1), 27–33. DOI 10.1016/j.clim.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tait Wojno ED; Hunter CA; Stumhofer JS, Immunity 2019, 50 (4), 851–870. DOI 10.1016/j.immuni.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cibulski SP; Rivera-Patron M; Mourglia-Ettlin G; Casaravilla C; Yendo ACA; Fett-Neto AG; Chabalgoity JA; Moreno M; Roehe PM; Silveira F, Sci Rep 2018, 8 (1), 13582. DOI 10.1038/s41598-018-31995-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duewell P; Kisser U; Heckelsmiller K; Hoves S; Stoitzner P; Koernig S; Morelli AB; Clausen BE; Dauer M; Eigler A; Anz D; Bourquin C; Maraskovsky E; Endres S; Schnurr M, J Immunol 2011, 187 (1), 55–63. DOI 10.4049/jimmunol.1004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casella CR; Mitchell TC, Cell Mol Life Sci 2008, 65 (20), 3231–40. DOI 10.1007/s00018-008-8228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wesa AK; Galy A, Int Immunol 2001, 13 (8), 1053–61. DOI 10.1093/intimm/13.8.1053. [DOI] [PubMed] [Google Scholar]
- 27.Zhu W; Wei L; Dong C; Wang Y; Kim J; Ma Y; Gonzalez GX; Wang BZ, Mol Ther Nucleic Acids 2022, 30, 421–437. DOI 10.1016/j.omtn.2022.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Montoya M; Schiavoni G; Mattei F; Gresser I; Belardelli F; Borrow P; Tough DF, Blood 2002, 99 (9), 3263–3271. DOI DOI 10.1182/blood.V99.9.3263. [DOI] [PubMed] [Google Scholar]
- 29.Zhu J; Yamane H; Paul WE, Annu Rev Immunol 2010, 28, 445–89. DOI 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Garra A, Curr Biol 2000, 10 (13), R492–R494. DOI Doi 10.1016/S0960-9822(00)00556-X. [DOI] [PubMed] [Google Scholar]
- 31.Boyaka PN, J Immunol 2017, 199 (1), 9–16. DOI 10.4049/jimmunol.1601775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corthesy B, Front Immunol 2013, 4. DOI ARTN 185 10.3389/fimmu.2013.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Havervall S; Marking U; Svensson J; Greilert-Norin N; Bacchus P; Nilsson P; Hober S; Gordon M; Blom K; Klingstrom J; Aberg M; Smed-Sorensen A; Thalin C, N Engl J Med 2022, 387 (14), 1333–1336. DOI 10.1056/NEJMc2209651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim MH; Kim HJ; Chang J, PLoS One 2019, 14 (7), e0220196. DOI 10.1371/journal.pone.0220196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheikh-Mohamed S; Isho B; Chao GYC; Zuo M; Cohen C; Lustig Y; Nahass GR; Salomon-Shulman RE; Blacker G; Fazel-Zarandi M; Rathod B; Colwill K; Jamal A; Li Z; de Launay KQ; Takaoka A; Garnham-Takaoka J; Patel A; Fahim C; Paterson A; Li AX; Haq N; Barati S; Gilbert L; Green K; Mozafarihashjin M; Samaan P; Budylowski P; Siqueira WL; Mubareka S; Ostrowski M; Rini JM; Rojas OL; Weissman IL; Tal MC; McGeer A; Regev-Yochay G; Straus S; Gingras AC; Gommerman JL, Mucosal Immunol 2022, 15 (5), 799–808. DOI 10.1038/s41385-022-00511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gould VMW; Francis JN; Anderson KJ; Georges B; Cope AV; Tregoning JS, Front Microbiol 2017, 8, 900. DOI 10.3389/fmicb.2017.00900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barker KA; Etesami NS; Shenoy AT; Arafa EI; Lyon de Ana C; Smith NM; Martin IM; Goltry WN; Barron AM; Browning JL; Kathuria H; Belkina AC; Guillon A; Zhong X; Crossland NA; Jones MR; Quinton LJ; Mizgerd JP, J Clin Invest 2021, 131 (11). DOI 10.1172/JCI141810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Humphries DC; O'Connor RA; Larocque D; Chabaud-Riou M; Dhaliwal K; Pavot V, Front Immunol 2021, 12, 738955. DOI 10.3389/fimmu.2021.738955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ridderstad A; Tarlinton DM, Journal of Immunology 1998, 160 (10), 4688–4695. [PubMed] [Google Scholar]
- 40.Schneider C; Nobs SP; Heer AK; Kurrer M; Klinke G; van Rooijen N; Vogel J; Kopf M, PLoS Pathog 2014, 10 (4), e1004053. DOI 10.1371/journal.ppat.1004053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prabha S; Arya G; Chandra R; Ahmed B; Nimesh S, Artif Cells Nanomed Biotechnol 2016, 44 (1), 83–91. DOI 10.3109/21691401.2014.913054. [DOI] [PubMed] [Google Scholar]
- 42.Azharuddin M; Zhu GH; Sengupta A; Hinkula J; Slater NKH; Patra HK, Trends Biotechnol 2022, 40 (10), 1195–1212. DOI 10.1016/j.tibtech.2022.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng L; Chang TZ; Wang Y; Li S; Wang S; Matsuyama S; Yu G; Compans RW; Li JD; Prausnitz MR; Champion JA; Wang BZ, Proc Natl Acad Sci U S A 2018, 115 (33), E7758–E7767. DOI 10.1073/pnas.1805713115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coulter A; Harris R; Davis R; Drane D; Cox J; Ryan D; Sutton P; Rockman S; Pearse M, Vaccine 2003, 21 (9-10), 946–9. DOI 10.1016/s0264-410x(02)00545-5. [DOI] [PubMed] [Google Scholar]
- 45.Vujanic A; Snibson KJ; Wee JL; Edwards SJ; Pearse MJ; Scheerlinck JP; Sutton P, Clin Vaccine Immunol 2012, 19 (1), 79–83. DOI 10.1128/CVI.05265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu KF; Elvander M; Merza M; Akerblom L; Brandenburg A; Morein B, Clin Exp Immunol 1998, 113 (2), 235–43. DOI 10.1046/j.1365-2249.1998.00650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Helgeby A; Robson NC; Donachie AM; Beackock-Sharp H; Lovgren K; Schon K; Mowat A; Lycke NY, J Immunol 2006, 176 (6), 3697–706. DOI 10.4049/jimmunol.176.6.3697. [DOI] [PubMed] [Google Scholar]
- 48.An X; Martinez-Paniagua M; Rezvan A; Sefat SR; Fathi M; Singh S; Biswas S; Pourpak M; Yee C; Liu X; Varadarajan N, iScience 2021, 24 (9), 103037. DOI 10.1016/j.isci.2021.103037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang W; Shi L; Cai L; Wang X; Li J; Li H; Liang J; Gu Q; Ji G; Li J; Liu L; Sun M, Cell Rep 2021, 37 (11), 110112. DOI 10.1016/j.celrep.2021.110112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gregg KA; Harberts E; Gardner FM; Pelletier MR; Cayatte C; Yu L; McCarthy MP; Marshall JD; Ernst RK, mBio 2017, 8 (3). DOI 10.1128/mBio.00492-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fang H; Ang B; Xu X; Huang X; Wu Y; Sun Y; Wang W; Li N; Cao X; Wan T, Cell Mol Immunol 2014, 11 (2), 150–9. DOI 10.1038/cmi.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu T; Matsuguchi T; Tsuboi N; Yajima T; Yoshikai Y, Infect Immun 2002, 70 (12), 6638–45. DOI 10.1128/IAI.70.12.6638-6645.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J; Li P; Yu Y; Fu Y; Jiang H; Lu M; Sun Z; Jiang S; Lu L; Wu MX, Science 2020, 367 (6480). DOI 10.1126/science.aau0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shakya AK; Chowdhury MYE; Tao W; Gill HS, J Control Release 2016, 240, 394–413. DOI 10.1016/j.jconrel.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.LeMessurier KS; Tiwary M; Morin NP; Samarasinghe AE, Front Immunol 2020, 11. DOI ARTN 3 10.3389/fimmu.2020.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lavelle EC; Ward RW, Nat Rev Immunol 2022, 22 (4), 266–266. DOI 10.1038/s41577-021-00599-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Y; Li S; Dong C; Ma Y; Song Y; Zhu W; Kim J; Deng L; Denning TL; Kang SM; Prausnitz MR; Wang BZ, ACS Appl Bio Mater 2021, 4 (6), 4953–4961. DOI 10.1021/acsabm.1c00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng MZM; Wakim LM, Mucosal Immunol 2022, 15 (3), 379–388. DOI 10.1038/s41385-021-00461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee CM; Oh JE, Front Immunol 2022, 13, 953088. DOI 10.3389/fimmu.2022.953088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Savelyeva N; Ottensmeier CH, Blood 2020, 136 (24), 2722–2723. DOI 10.1182/blood.2020007890. [DOI] [PubMed] [Google Scholar]
- 61.Tan HX; Juno JA; Esterbauer R; Kelly HG; Wragg KM; Konstandopoulos P; Alcantara S; Alvarado C; Jones R; Starkey G; Wang BZ; Yoshino O; Tiang T; Grayson ML; Opdam H; D'Costa R; Vago A; Austin Liver Transplant Perfusionist, G.; Mackay LK; Gordon CL; Masopust D; Groom JR; Kent SJ; Wheatley AK, Sci Immunol 2022, 7 (67), eabf5314. DOI 10.1126/sciimmunol.abf5314. [DOI] [PubMed] [Google Scholar]
- 62.Masopust D; Soerens AG, Annu Rev Immunol 2019, 37, 521–546. DOI 10.1146/annurev-immunol-042617-053214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hassert M; Harty JT, Front Immunol 2022, 13, 1039194. DOI 10.3389/fimmu.2022.1039194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Onodera T; Takahashi Y; Yokoi Y; Ato M; Kodama Y; Hachimura S; Kurosaki T; Kobayashi K, Proc Natl Acad Sci U S A 2012, 109 (7), 2485–90. DOI 10.1073/pnas.1115369109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Allie SR; Bradley JE; Mudunuru U; Schultz MD; Graf BA; Lund FE; Randall TD, Nat Immunol 2019, 20 (1), 97–108. DOI 10.1038/s41590-018-0260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McMaster SR; Wein AN; Dunbar PR; Hayward SL; Cartwright EK; Denning TL; Kohlmeier JE, Mucosal Immunol 2018, 11 (4), 1071–1078. DOI 10.1038/s41385-018-0003-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kunzli M; O'Flanagan SD; LaRue M; Talukder P; Dileepan T; Stolley JM; Soerens AG; Quarnstrom CF; Wijeyesinghe S; Ye Y; McPartlan JS; Mitchell JS; Mandl CW; Vile R; Jenkins MK; Ahmed R; Vezys V; Chahal JS; Masopust D, Sci Immunol 2022, 7 (78), eadd3075. DOI 10.1126/sciimmunol.add3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suberi A; Grun MK; Mao T; Israelow B; Reschke M; Grundler J; Akhtar L; Lee T; Shin K; Piotrowski-Daspit AS; Homer RJ; Iwasaki A; Suh HW; Saltzman WM, bioRxiv 2022. DOI 10.1101/2022.03.22.485401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mao T; Israelow B; Pena-Hernandez MA; Suberi A; Zhou L; Luyten S; Reschke M; Dong H; Homer RJ; Saltzman WM; Iwasaki A, Science 2022, 378 (6622), eabo2523. DOI 10.1126/science.abo2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lapuente D; Fuchs J; Willar J; Vieira Antao A; Eberlein V; Uhlig N; Issmail L; Schmidt A; Oltmanns F; Peter AS; Mueller-Schmucker S; Irrgang P; Fraedrich K; Cara A; Hoffmann M; Pohlmann S; Ensser A; Pertl C; Willert T; Thirion C; Grunwald T; Uberla K; Tenbusch M, Nat Commun 2021, 12 (1), 6871. DOI 10.1038/s41467-021-27063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hameed SA; Paul S; Dellosa GKY; Jaraquemada D; Bello MB, NPJ Vaccines 2022, 7 (1), 71. DOI 10.1038/s41541-022-00485-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Helft J; Bottcher J; Chakravarty P; Zelenay S; Huotari J; Schraml BU; Goubau D; Reis e Sousa C, Immunity 2015, 42 (6), 1197–211. DOI 10.1016/j.immuni.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 73.Zhu W; Pewin W; Wang C; Luo Y; Gonzalez GX; Mohan T; Prausnitz MR; Wang BZ, J Control Release 2017, 261, 1–9. DOI 10.1016/j.jconrel.2017.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.